

Abstract
A previously healthy 60-year-old man presented to the hospital with a hemoglobin of 3.5 g/dL. He was diagnosed with severe warm autoimmune hemolytic anemia (wAIHA) with reticulocytopenia on hospital day 1 that was not responsive to steroids, immune globulin, and rituximab. Over a 42-day hospital stay, the patient remained continuously transfusion-dependent with a ninety red cell unit requirement for his refractory disease. He was trialed on therapeutic plasma exchange before ultimately undergoing inpatient splenectomy that led to a response within hours. He remains in complete remission at six months of follow-up.
Background:
wAIHA is caused by IgG antibodies directed against self-erythrocytes causing mostly extravascular hemolysis by splenic macrophages.(1) The condition can be primary (idiopathic) or as in 50–60% of cases (2), secondary to autoimmune disorders, neoplasm, infection, or medications (3). A positive direct antiglobulin test (DAT) in conjunction with clinical history and laboratory evidence of hemolysis makes the diagnosis. Predictors of disease severity include initial presenting hemoglobin ≤ 6 g/dL (4) and the ability of the bone marrow to compensate. Therapies to treat primary wAIHA are driven largely by expert opinion (5,6) with a small number of prospective trials (7,8) and no FDA-approved treatments in 2023. The cornerstone of treatment remains glucocorticoid therapy. Rituximab is increasingly favored in concomitant first-line treatment (9) although the median time to response is three to six weeks. Splenectomy was previously considered a second-line therapy (10) and is effective with long-lasting remission.(9) However, due to perceived tripartite risk of infection, thrombosis and perioperative mortality, it was recommended as third-line treatment status in recent guidelines.(11) Supportive management includes red cell transfusion, intravenous immune globulin, folic acid supplementation, stimulation of bone marrow with recombinant erythropoietin, venous thromboprophylaxis, and vaccination. (1,12)
Objective:
This case highlights the importance of early identification of primary wAIHA, adaptation to its severity, and the use of splenectomy in refractory disease.
Case report:
A healthy 60-year-old male presented to the hospital with jaundice, confusion, progressive fatigue, dark urine, and dyspnea on exertion. Ten days prior, he underwent a root canal procedure and started amoxicillin. Laboratory testing in the emergency department demonstrated a hemoglobin of 3.5 g/dL (reference: 13.2–17.1 g/dL) and a positive direct antiglobulin test for IgG and negative for C3 (as well as a panagglutinin in elution studies), prior to emergent blood cell (RBC) transfusion with the least crossmatch incompatible units. A diagnosis of wAIHA with associated reticulocytopenia (absolute reticulocyte count of 0.002 × 106 cells/uL; reference: 0.023 – 0.140×106 cells/uL) was made and the patient underwent a thorough workup for secondary causes, as shown in Table 1, that was negative.
Table 1.
Diagnostic warm autoimmune hemolytic anemia workup, results, and medical team interpretation of the studies. Recommended lab testing for secondary wAIHA per the Autoimmune Hemolytic Anemia First International Consensus statement are bolded.
| Category | Lab Test | Result | Reference with Units | Date obtained | Date reported | Interpretation |
|---|---|---|---|---|---|---|
| Primary wAIHA | DAT | DAT IgG 2+ DAT C3 negative |
Negative Negative |
D1 | D1 | Warm autoimmune hemolytic anemia |
| SLE/autoimm une | ANA | <1:80 | Negative | D1 | D6 | Negative |
| Lupus anticoagulant | 1.10 | <1.2 | D6 | D8 | Negative | |
| Anticardiolipin antibody | IgG 4.4 IgM 72 |
<10 GPL U/mL <10 MPL U/mL |
D6 | D7 | IgM positive, but IgG negative. Per rheumatology colleagues, IgG usually positive and combined with clinical picture of lack of venous thromboembolism, antiphospholipid syndrome (APS) was not suspected. | |
| Anti-B2gpl antibody | IgG 3.4 IgM 16 |
<7 U/mL <7 U/mL |
D6 | D7 | IgM positive, but as above. | |
| Complement | C3 95 C4 12 |
90–180 mg/dL 10–40 mg/dL |
D6 | D6 | Normal | |
| Lymphoma and other solid tumors | SPEP | Discrete abnormal band measuring 0.2 g/dL present in the gamma region | D1 | D2 | Query bone marrow aspiration and biopsy (MGUS versus more) | |
| IFE | Faint, possibly abnormal band detected in the gamma region in serum and best characterized as IgG kappa. | D1 | D8 | Query bone marrow aspiration and biopsy (MGUS versus more) | ||
| Serum free kappa lambda light chains with ratio | Kappa free light chains 2.97
Lambda free light chains 1.92 Kappa/Lambda free light chains ratio 1.55 |
0.33–1.94 mg/dL 0.57–2.63 mg/dL 0.26–1.65 |
D1 | D1 | Elevated kappa free light chain, but normal ratio. Query bone marrow aspiration and biopsy (MGUS versus more). | |
| immunotyping of B-lymphocytes from peripheral blood | No circulating CD34+ CD117+ blasts detected. Mature myeloid elements demonstrate a normal, although slightly left-shifted, CD10/CD11b/CD13/CD16/C D33 pattern. PNH clone is absent. Granulocytes and monocytes show normal expression of GPI-linked markers CD16, CD24, and CD14 with normal FLAER binding. RBCs show normal expression of CD59. There is no abnormal immunophenotype T cell population suggestive of T-cell lymphoproliferative disease, including no increase in T-LGLs. | D1 | D2 | No evidence of monoclonal non-Hodgkin B cell lymphoproliferative disease. No PNH. No T-cell lymphoproliferative disease. | ||
| CT scan (CAP) | No thoracic or abdomino-pelvic findings, without significant lymphadenopathy. | D1, D4 | D1, D5 | Normal. No evidence of malignancy. | ||
| Bilateral lower extremity venous Doppler ultrasound | No evidence of deep venous thrombosis of the bilateral lower extremities. | D4 | D4 | Normal. No evidence of deep vein thromboses. | ||
| Bone marrow aspiration and biopsy | Hypercellular erythroid-predominant marrow showing maturing trilineage hematopoiesis with erythroid left-shift | D7 | D15 | No evidence of primary bone marrow process. | ||
| Primary immunodefici ency | IgA, IgG, IgM levels | IgM 175 IgA 151 IgG 2320 IgG 1 644 IgG 2 401 IgG 3 40 IgG 4 17.5 IgG total 1137 |
40–230 mg/dL 70–470 mg/dL 700–1600 mg/dL 382–929 mg/dL 242–700 mg/dL 22–176 mg/dL 3.9–86.4 mg/dL 700–1600 mg/dL |
D1 | D1 | Normal levels, IgG elevated in setting of autoantibody. IgG subclasses normal. |
| Infection | HIV, Hepatitis C, Hepatitis B tests | HIV Ab with Ag negative Hepatitis A IgM negative Hepatitis B core IgM negative Hepatitis B surface Ag negative Hepatitis B core Ab negative Hepatitis B surface Ab negative Hepatitis C Ab negative |
Negative Negative Negative Negative Negative ≥12 mIU/mL Negative |
D1 | D1, D2 | Hepatitis B non-immune. No serologic evidence of acute infection. |
| CMV, parvo-B19, EBV IgM, IgG | CMV IgM < 8 CMV IgG <0.20 EBV IgM 21.8 EBV IgG 292 |
<30 AU/mL, <0.59 U/mL <35 U/mL <22 U/mL |
D1 | D1 | Exposed to EBV prior, but no active infection. | |
| Parvovirus DNA PCR negative | Negative | |||||
| Babesia, Ehrlichia, Anaplasma negative; treponema negative | Negative | D9 | D11 | Tickborne disease and syphilis negative. | ||
| Medications | Drug-dependent antibody | Amoxicillin IgG, IgM both negative | Negative | D5 | D18 | No antibodies to amoxicillin, suggesting this was not the trigger. |
Parallel to this extensive workup, the patient was immediately started on treatment, as listed in Table 2. He began steroids (day 1), initially with prednisone 140mg daily (1mg/kg) for one week along with IVIG 1g/kg for two days (days 1 and 2). An erythropoietin (EPO) level was drawn and recombinant EPO administered on hospital day 3. (12) Further doses were canceled once the endogenous EPO level resulted at 690.6 mU/mL (reference: 3–18 mU/mL). With lack of stabilization of hemoglobin despite upfront steroids, rituximab was initiated on day 4, as seen in Figure 1. The patient’s other cell counts decreased in parallel with the anemia - at its nadir, on day 6, the WBC was 0.5 ×103/uL (reference 4–11×103/uL with ANC 0.15 × 103/uL (reference: 2.0–7.6×103/uL) and platelets 22 ×103/uL (reference: 150–420×103/uL). A bone marrow biopsy done on day 7 was negative. Treatment was then intensified with pulse dose methylprednisolone on day 9 for four days. The WBC, absolute neutrophil, and platelet counts recovered although the anemia persisted. Lab medicine was consulted for a trial of therapeutic plasma exchange (TPE). A central line was placed with the first exchange of 1 plasma volume using 5% albumin replacement fluid (Spectra Optia, Terumo BCT, Lakewood, CO) on hospital day 11 and the second rituximab infusion (cycle 1, week 2) given right after TPE.
Table 2.
Lines of therapy in this patient, organized by date, with details of dosages and dates.
| Therapeutic category | Therapeutic details | Dose (if applicable) | Date |
|---|---|---|---|
| Steroids | Prednisone | 100mg daily | D36- |
| 120mg daily | D31–35 | ||
| 130mg daily | D25–30 | ||
| 140mg daily | D1–8, D19–24 | ||
| 280mg daily | D13–18 | ||
| Methylprednisolone | 1000mg daily | D9–12 | |
| Recombinant erythropoietin | Darbepoetin | 200mcg | D2 |
| Intravenous immunoglobulin (IVIG) | 1g/kg | D1–2 | |
| Rituximab | 375mg/mg2 | 1000mg | D4 |
| 1000mg | D11 | ||
| 1000mg | D18 | ||
| 1000mg | D26 | ||
| Plasmapheresis | 1 session | D11 | |
| Splenectomy | D33 | ||
Figure 1. Hemoglobin during hospitalization.
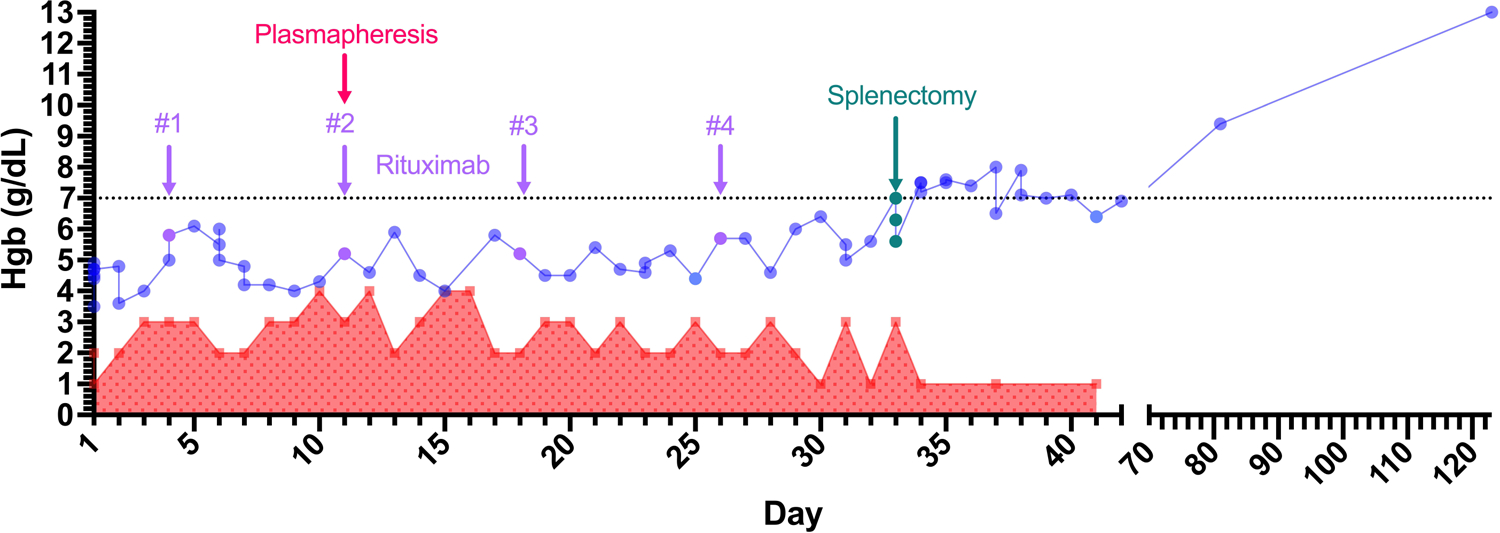
Hemoglobin trend and number of packed red blood cell transfusions over the hospital stay. Interventions labeled with rituximab given in 4 doses (days 4, 11, 18, and 26), one therapeutic plasma exchange session (day 11), and splenectomy (day 33). The patient was discharged on hospital day 42. Outpatient follow-up hemoglobin numbers shown with disrupted x-axis. Patient tapered off steroid therapy outpatient on day 96.
 Hemoglobin
Hemoglobin
 Number of transfusions
Number of transfusions
However on the planned second session of TPE (day 13), the patient fevered. He was diagnosed with a catheter-related E. faecalis bacteremia, treated with ampicillin for six weeks, and the central line was removed on day 15. Further TPE sessions were canceled. His steroid regimen was changed to prednisone 2mg/kg for the following week before return to 1mg/kg on day 19. Two more rituximab infusions were given on day 18 and day 26. While rituximab effectiveness is expected to manifest within weeks, it can take up to several months in this disease. Since the patient continued to require 2–3 RBC units daily to hold at a hemoglobin of 4–5 g/dL despite these interventions, surgery was consulted. The patient underwent a laparoscopic splenectomy on day 33 with no perioperative complications. His hemoglobin stabilized at 6–7 g/dL afterwards with minimal transfusion requirements. He was discharged 42 days after his initial presentation with close hematology follow up. Six months out from discharge, his hemoglobin recovered to 13g/dL and his symptoms resolved completely.
Discussion:
This previously healthy patient’s grave symptoms and critically low hemoglobin and reticulocytopenia on presentation classified him as having severe wAIHA. A near-zero absolute reticulocyte count as well as the rapid progression to pancytopenia were both predictive of poorer clinical outcomes. Recognition of this hematologic emergency necessitated aggressive treatment beyond initial steroid, immune globulin, and recombinant EPO use, which included early initiation of rituximab. Rituximab use first-line was informed by current 2021 expert recommendations (9) and a small randomized controlled trial showing overall response rates of 75% and 31% at 1 year and 63% and 19% at 2 years with and without rituximab use, respectively, with no increase in infectious complications.(8)
While simultaneously escalating treatment, we pursued comprehensive diagnostics due to the severity of disease. This included a bone marrow biopsy, normally recommended with disease relapse after steroid therapy (5), to exclude an underlying bone-marrow limited hematologic malignancy that may not have been identified in the previous peripheral blood testing or CT imaging. This also ensured separately that the small paraprotein identified peripherally was consistent with monoclonal gammopathy of undetermined significance.
With no evidence of response to first-line treatment, we – as initially discussed with the patient and confirmed on daily re-evaluation – pursued TPE as a category III indication for wAIHA per American Society of Apheresis (ASFA) guidelines (13). It is category III given the relatively large volume of distribution of IgG antibodies (unlike largely intravascular IgM) that mediate wAIHA pathophysiology. Unfortunately, our patient suffered from a catheter-associated infection and so we could not complete an empiric trial of at least 2–3 sessions of TPE to assess an effect and avoid splenectomy.
There remains little guidance on the optimal timing of splenectomy, with future research needed to identify the patient population with this disease where pursuing it earlier may be advantageous. The tripartite risk of postoperative infection, thrombosis, and perioperative mortality have contributed to its deprioritization from a second- to third-line treatment in the 2017 British guidelines.(11) However, these concerns predate the modern-day vaccination, thromboprophylaxis, and laparoscopic splenectomy era. Of note, historical data regarding splenectomy is confounded by mixing of primary and secondary wAIHA outcomes, the latter for which splenectomy is less effective. While splenectomy is currently used in fewer than 10% of patients with wAIHA, it remains the most durable treatment (60–90% response rate, sustained remission 75%).(9,14,15)
Our patient ultimately proceeded with laparoscopic splenectomy. While rituximab could have contributed to improvement, the same-day stabilization of his cell counts within hours of surgery with abrogation of the transfusion requirement show that splenectomy remains integral to the therapeutic armamentarium for severe, relapsing or refractory wAIHA in 2023.
Financial Support:
C.W. is funded by the National Heart, Lung, and Blood Institute (T35HL007649). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. G.G. is funded by the Yale Bunker Endowment, and the Yale DeLuca Center for Innovation in Hematology Research.
References:
- 1.Hill A, Hill QA. Autoimmune hemolytic anemia. Hematol Am Soc Hematol Educ Program. 2018. Nov 30;2018(1):382–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalfa TA. Warm antibody autoimmune hemolytic anemia. Hematol Am Soc Hematol Educ Program. 2016. Dec 2;2016(1):690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leger RM, Arndt PA, Garratty G. How we investigate drug-induced immune hemolytic anemia. Immunohematology. 2014;30(2):85–94. [PubMed] [Google Scholar]
- 4.Barcellini W, Fattizzo B, Zaninoni A, Radice T, Nichele I, Di Bona E, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014. Nov 6;124(19):2930–6. [DOI] [PubMed] [Google Scholar]
- 5.U J, W B, Cm B, Ma G, A H, Qa H, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev [Internet]. 2020. May [cited 2023 Mar 19];41. Available from: https://pubmed.ncbi.nlm.nih.gov/31839434/ [DOI] [PubMed] [Google Scholar]
- 6.Go RS, Winters JL, Kay NE. How I treat autoimmune hemolytic anemia. Blood. 2017. Jun 1;129(22):2971–9. [DOI] [PubMed] [Google Scholar]
- 7.Birgens H, Frederiksen H, Hasselbalch HC, Rasmussen IH, Nielsen OJ, Kjeldsen L, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. 2013. Nov;163(3):393–9. [DOI] [PubMed] [Google Scholar]
- 8.Michel M, Terriou L, Roudot-Thoraval F, Hamidou M, Ebbo M, Le Guenno G, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm autoimmune hemolytic anemia in adults (the RAIHA study). Am J Hematol. 2017. Jan;92(1):23–7. [DOI] [PubMed] [Google Scholar]
- 9.Barcellini W, Fattizzo B. How I treat warm autoimmune hemolytic anemia. Blood. 2021. Mar 11;137(10):1283–94. [DOI] [PubMed] [Google Scholar]
- 10.Lechner K, Jäger U. How I treat autoimmune hemolytic anemias in adults. Blood. 2010. Sep 16;116(11):1831–8. [DOI] [PubMed] [Google Scholar]
- 11.Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A, et al. Guidelines on the management of drug-induced immune and secondary autoimmune, haemolytic anaemia. Br J Haematol. 2017. Apr;177(2):208–20. [DOI] [PubMed] [Google Scholar]
- 12.Fattizzo Bruno, Michel Marc, Zaninoni Anna, Giannotta Juri, Guillet Stephanie, Frederiksen Henrik, et al. Efficacy of recombinant erythropoietin in autoimmune haemolytic anaemia: a multicentre international study. Haematologica. 2020. Apr 30;106(2):622–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connelly-Smith L, Alquist CR, Aqui NA, Hofmann JC, Klingel R, Onwuemene OA, et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice – Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Ninth Special Issue. J Clin Apheresis. 2023. Apr;38(2):77–278. [DOI] [PubMed] [Google Scholar]
- 14.Hill QA, Stamps R, Massey E, Grainger JD, Provan D, Hill A, et al. The diagnosis and management of primary autoimmune haemolytic anaemia. Br J Haematol. 2017. Feb;176(3):395–411. [DOI] [PubMed] [Google Scholar]
- 15.Ogbue OD, Bahaj W, Kewan T, Ahmed R, Dima D, Willimas N, et al. Splenectomy outcomes in immune cytopenias: Treatment outcomes and determinants of response. J Intern Med. 2024. Feb;295(2):229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]