

Abstract

Sitting on the interface between biologics and small molecules, peptides represent an emerging class of therapeutics. Numerous techniques have been developed in the past 30 years to take advantage of biological methods to generate and screen peptide libraries for the identification of therapeutic compounds, with phage display being one of the most accessible techniques. Although traditional phage display can generate billions of peptides simultaneously, it is limited to expression of canonical amino acids. Recently, several groups have successfully undergone efforts to apply genetic code expansion to introduce noncanonical amino acids (ncAAs) with novel reactivities and chemistries into phage-displayed peptide libraries. In addition to biological methods, several different chemical approaches have also been used to install noncanonical motifs into phage libraries. This review focuses on these recent advances that have taken advantage of both biological and chemical means for diversification of phage libraries with ncAAs.
1. Introduction
1.1. Peptide Therapeutics
Peptides, or short protein fragments with a molecular weight less than 5000 Da, occupy an intriguing space in drug discovery, as they have the potential to combine the specificity of large biologics (antibodies, proteins, etc.) with the low cost of production, storage, and administration of small molecules. Due to their larger size, peptides are efficient at inhibiting protein–protein interactions, which small molecules generally are ineffective in.1 Despite these advantages, many hindrances have prevented peptide drugs from playing a major role in therapeutics, such as low membrane permeability, poor serum stability, and poor oral bioavailability.2 Initially developed peptide therapeutics were related to natural peptide hormones, such as insulin, oxytocin, vasopressin, and somatostatin.3−7 Primarily, peptide research in the mid- to late-twentieth century focused on different chemical modifications of these hormones for the development of peptide drugs with improved pharmacologic properties. Successful campaigns took advantage of incorporation of a variety of modifications to the peptides to prevent degradation from serum proteases or reduction of the disulfide bonds that were essential to the peptides.8 Recently, advances in synthesis, screening techniques, and delivery methods have allowed for peptides to become promising candidates to treat a wide variety of diseases, and over 52 peptide drugs have been approved in the past two decades.2 A recent drastic market hike of peptide therapeutics is due to the approval of GLP-1 agonists for weight loss. Driven by a huge market size surpassing 13 billion USD in 2023 and a projected annual growth rate of 4.8% (GLP-1 Receptor Agonist Global Market Report 2023, Reportlinker.com), there has been a tremendous burst of different biotech companies launched for the development of further GLP-1 analogs.9−12 Therapeutic peptides have now diversified into multiple classes, such as hormones,13 neurotransmitters,14 ligands,15 and anti-infective agents.16 One of the main advantages of peptides is the ability to easily screen large peptide libraries to identify therapeutic candidates. There are numerous methods to generate peptide libraries that range in diversity from 106 to 1015 peptides, and several excellent reviews for them are available (see Figure 1 for an overview of these techniques and references to recent reviews).17−20 Of these, phage display has emerged as one of the most versatile and easily adaptable techniques over the past three decades. This review will focus on recent developments in techniques to integrate noncanonical amino acids (ncAAs) and other motifs into phage-displayed peptide libraries.
Figure 1.

Overview of techniques for peptide library generation. Several methods are currently available for the generation of large peptide libraries. These include completely synthetic methods (Split and Pool), in vitro methods (mRNA Display), and strictly in vivo methods (SICLOPPS). Phage display gives the possibility to generate libraries in vivo but perform selections in a variety of different conditions. Techniques that are not illustrated in the figure but widely used are yeast display, DNA-encoded peptide library technique, one-bead-one-compound peptide library technique, etc.
1.2. M13 Bacteriophage Structure
Prior to discussing modifications of peptides displayed on bacteriophages, it is important to understand the structure of the bacteriophage itself. While there has been some experimentation using other types of phages for phage display, the majority of studies have taken advantage of filamentous bacteriophage. As the name suggests, filamentous phages consist of long filaments that are approximately 1 μm long and 6 nm wide.21 They have been found to infect various genera of Gram-negative bacteria, with a few reports of filamentous phages infecting Gram-positive bacteria.22 Of the filamentous phages, the most well classified phages are those that infect using the F pilus of Escherichia coli and are known as f1, M13, or fd phages. M13 phages consist of viral proteins encapsulating a circular positive single-stranded DNA (+ssDNA) genome that encodes 9 open reading frames that produce 11 different proteins through the use of alternative start codons.23 In regard to phage display, an important aspect of M13 phage infections is that due to their slow replication and ejection from the cells, they do not lyse their hosts resulting in high phage titers (1013 phages/mL of culture).22,24 They also tolerate a wide variety of structural modifications,25 show high stability in a wide range of temperatures,26 and are very resistant to proteolysis.27 This makes them ideal for the display of peptide and protein libraries for selections against therapeutic targets.
The M13 bacteriophage virion consists of five types of coat proteins encapsulating a circular ssDNA genome (Figure 2A). In wild type M13 phages, the major coat protein is pVIII, with each virion containing roughly 2700 copies surrounding the circular ssDNA. A small, disordered N-terminal domain of pVIII consisting mainly of negatively charged residues is solvent exposed in the virion.28 The rest of the protein comprises an α helix that contains an amphipathic region (residues 5–20), hydrophobic region (residues 21–39), and then a positively charged region (residues 40–49) that interacts with the ssDNA (Figure 2B).28 The virion is held together through hydrophobic interactions between the alpha helices of adjacent pVIII proteins that spiral in a 5-fold symmetry around the encapsulated ssDNA.29 The phosphate backbone of the ssDNA interacts with the positively charged region of pVIII, and the length of the virion is typically dependent upon the length of ssDNA being packaged.22
Figure 2.

Structure of M13 bacteriophage. A) Representation of the M13 virion encapsulating its +ssDNA genome. B) Crystallography of pVIII (PDB: 2C0W) with colors assigned according to functionality of the region and the N- and C-termini labeled. Interactions between the hydrophobic core are responsible for maintaining structural integrity of the capsid. C) Structure of the pIII protein’s N1 (A19-P83, green) and N2 (E109-A235, red) domains (PDB: 1G3P) with a depiction of the full-length encoded protein below. The N1 domain contains a signaling peptide (gray) that is cleaved after incorporation in the membrane. Glycine-rich linkers are depicted as black lines between the three domains. N and C in the crystal structure refer to the N- and C-terminus. D) Cryo-EM of C-terminal domain (D275-K422) within the packaged virion (PDB: 8B3O) demonstrates a helical nature for insertion into the virion. The N- and C-termini in the structure are labeled as N and C.
The virion is then capped on either end by a variety of proteins, with pVII and pIX on the “head” and pIII and pVI on the “feet” of the phage. pVII and pIX consist of about 30 residues each and are the initial proteins (approximately five of each protein) excreted from the cell during packaging.30 There is much less structural information known about these proteins, but it is thought that C-terminal ends recognize the packaging signal in the phage genome to initiate assembly.31 Phage assembly is terminated by the addition of pVI and pIII to the virion. While the interactions between pIII and pVI are not well characterized, there are five copies of each protein, and it is proposed that they maintain the 5-fold axial symmetry that pVIII displays.22 pVI is a moderately sized protein consisting of 112 residues that are mainly hydrophobic and is predicted to have three transmembrane alpha helices.22,30 pIII is significantly larger than the other proteins in the virion and is composed of three different domains (N1, N2, C) that are joined by glycine-rich linkers, along with an N-terminal signaling peptide for localization to the membrane (Figure 2C). N1 and N2 are similarly folded with each containing two antiparallel β-sheets and an α-helix.32,33 Infection into host cells is mediated through the N1 and N2 domains of pIII, while the C domain appears to be involved with interactions to pVI to terminate viral assembly.22 Through recent Cryo-EM studies, it was determined that the C-terminal domain is primarily composed of alpha helices for insertion within the virion (Figure 2D).34 The C-terminal end of the protein also contains a transmembrane region that serves to anchor the protein in the host membrane during assembly.35
1.3. Overview of Phage Display Biopanning
Over the past 40 years, researchers have taken advantage of M13 bacteriophages to genetically encode large peptide and protein libraries. These libraries are then used to select for peptides or proteins to bind to desirable targets through iterative rounds of selection (Figure 3). The packaging mechanism can tolerate large insertions and deletions of the phage ssDNA, as long as the packaging signal and origins of replication remain intact. Thus, modifications to the phage genome through molecular biology techniques allow for the insertion of large, randomized regions into the genome that can be subsequently expressed as a fusion to other phage proteins. Although all phage coat proteins have been exploited to display libraries, the most commonly used proteins for display are pVIII and pIII.22 Displayed peptide sequences usually are spliced into the phage protein sequences just after the signaling peptide domain to ensure that the fusions are incorporated into the membrane prior to virion formation. Peptides displayed using pVIII typically modify the N-terminal end of the protein, which is exterior to the phage virion. Insertions of less than seven amino acids to pVIII is tolerated for virion formation, however, larger libraries necessitate coexpression of wild-type pVIII to generate functional virions.36 Also, when displaying peptides on pVIII one should be aware of potential valency effects, as there are more than 2700 copies per virion. Thus, pIII display has been the preferred method for many different applications.
Figure 3.

Overview of phage display biopanning. Phage-displayed peptide libraries are typically produced using E. coli that are transformed with two plasmids. One plasmid produces a modified phage protein that has a genetically encoded peptide library fused to it, while the other plasmid provides all other necessary phage proteins. These phages are then used to bind to an immobilized target, protein or other biomolecule, to allow for washing of the phages that do not bind. After elution, phages are amplified again in E. coli to repeat the selection until enrichment of peptides in the phage library is observed through sequencing of the phage DNA. After sequencing, the corresponding peptides are synthesized and validated for binding to the target of interest.
There are three different regions that can be exploited to display peptide libraries on pIII: the N-terminus, between the N2 and C-terminal domain, and on the C-terminus. Initially, fusion of peptides to the middle region (between the N2 and C domains) of pIII was explored to avoid adversely affecting phage infectivity.37 While the display of peptides in this location was possible, there was a decrease in infectivity with these clones. Therefore, another system that encoded the peptide library fused to the N-terminus of pIII was subsequently developed.38 Similar to the pVIII fusions, large peptides displayed on the N-terminal domain of pIII can diminish infectivity of the phages and the coexpression of wild-type pIII is necessary alongside these proteins. Alternatively, methods have been developed to cleave the peptide fusion off with trypsin prior to infecting cells, which allows for the expression of large proteins on the N-terminus of pIII.39 Alongside this system, expression of peptides on the C-terminus of pIII has also been explored and proven successful, although this is typically less popular and has been reserved for peptides that require special processing by cytoplasmic enzymes.40 It is worth noting that there are different systems for producing libraries on pIII that are characterized by the location of library insertion. The first developed system, Type 3 display, directly modified the pIII gene in the M13 phage genome to afford a modified pIII with the fused peptide library. This results in every pIII copy containing the modified peptide fusion. However, to circumvent the possibility of the peptide fusion hindering infectivity, other systems that provide wild-type pIII protein have been developed.25 Type 33 display refers to the incorporation of genes encoding both wild-type pIII and the peptide-fusion pIII into the phage genome. Therefore, a mixture of modified and wild-type pIII proteins are displayed on the surface of the virion and infectivity is not reduced for these variants. One major disadvantage of Type 3 and Type 33 display is that they require insertion of the peptide library directly into the phage genome. This can result in the amplification and enrichment of phages that acquire mutations in proteins other than the pIII-library fusion, severely hindering the potential for selections using these libraries.41,42
One way to avoid this undesired enrichment of parasitic phage genomes is to split the phage production into two different plasmids. In Type 3+3 display, a phagemid is used that encodes the peptide-fusion pIII and contains the f1 origin for packaging of the virion. Alongside this, a helper phage genome is also used that encodes for all phage proteins but has a split f1 origin to favor packaging of the phagemid DNA over the helper phage genome.43,44 To select for the presence of each plasmid, they both contain antibiotic resistance casettes, typically for kanamycin (helper phage) and ampicillin (phagemid).45 This system affords less risk of accumulating mutations within non-pIII related proteins in comparison with Type 3 and Type 33 systems. Also, having the library within the phagemid results in numerous advantages including improved pIII expression, increased transformation efficiencies, facile cloning, and inducible promoters.45 However, because the 3+3 system expresses wild-type pIII alongside the modified version, slowly expressing pIII-fusions can have low coverage in the packaged virions.46 To circumvent this, Type 3+0 systems have also been developed, where the helper phage genome produces either no pIII or a dysfunctional pIII.39,46−48 Similar to the Type 3 system, this affords virions where all pIII subunits are modified in the virion. For large libraries that are produced using the Type 3+0 system, functionality of pIII may be reduced, so it is generally required to cleave the modification to pIII prior to infection of cells.46 In this review, most of the systems discussed take advantage of the phagemid-based systems (Type 3+3 or Type 3+0) for expression, although there are a few reports that directly modify pIII on the phage genome.
1.4. Review Outline
Many works have been published that use phage display or other peptide display techniques to identify therapeutic peptides. In this review, we will focus on recent developments for phage-assisted identification of peptides that contain ncAAs and other noncanonical motifs, with a particular focus on consolidating methods to generate modifications on phage. Table 1 summarizes recent reviews on additional topics that are relevant to this work, including overviews of phage display,25,49,50 genetic code expansion,51−54 platforms for genetically encoded libraries,55,56 and peptide therapeutics.8,57−59 This review begins with the application of genetic code expansion to phage-displayed libraries (Section 2), followed by chemical post-translational modifications (Section 3), and it concludes with a general overview of remaining challenges in displaying noncanonical motifs on phages (Section 4).
Table 1. Summary of Recent Relevant Reviews on Peptide Libraries, Genetic Code Expansion, and Peptide Therapeutics.
| Topic | Publication Title | Year | Reference |
|---|---|---|---|
| Historical Accounts of Phage Display | Phage Display | 1997 | (25) |
| Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture) | 2019 | (49) | |
| Harnessing Evolution to Make Medicines (Nobel Lecture) | 2019 | (50) | |
| Genetic Code Expansion | Expanding and Reprogramming the Genetic Code | 2017 | (51) |
| Reprogramming the Genetic Code | 2021 | (52) | |
| Pyrrolysyl-tRNA Synthetase: An Ordinary Enzyme but an Outstanding Genetic Code Expansion Tool | 2014 | (53) | |
| Update of the Pyrrolysyl-tRNA Synthetase/tRNAPyl Pair and Derivatives for Genetic Code Expansion | 2023 | (54) | |
| Genetically Encoded Libraries | Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery | 2020 | (55) |
| Expanding the Chemical Diversity of Genetically Encoded Libraries | 2020 | (56) | |
| Cyclic Peptides | Phage Selection of Cyclic Peptides for Application in Research and Drug Development | 2017 | (57) |
| Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges | 2019 | (58) | |
| Peptide Therapeutics | Peptide-based drug discovery: Current status and recent advances | 2023 | (59) |
| Trends in Peptide Drug Discovery | 2021 | (8) |
2. Applications of Genetic Code Expansion to Phage Displayed Peptides
2.1. Overview of Orthogonal Genetic Code Expansion
In nature, there are typically only 20 canonical amino acids (cAAs) that are incorporated into proteins through translation of the genetic code. This provides significant limitations to the chemical diversity that can be encoded in phage libraries. Twenty cAAs are classified into several categories based on their side chains as positively or negatively charged, polar, hydrophobic including both small aliphatic and large aromatic, and special cases including cysteine, glycine and proline. Chemical groups that are typically observed in approved small molecule drugs such as halides, nitro, alkyne, and nonimidazole heteroaromatic rings are missing in cAAs. However, studies over the past 50 years have looked to expand the genetic code to allow for the ability to generate proteins and peptides containing novel moieties with interesting chemistries.
Genetic code expansion by amber suppression was pioneered by several laboratories in the 1970s and later expanded by the Schultz Lab in the 1990s, where chemically synthesized tRNAs were used to recognize and incorporate an ncAA at the amber stop codon (UAG) site in an mRNA.60 An important advantage is that translation is stopped when there is no incorporation at the amber codon, thus preventing background proteins from being expressed. Although being limited to in vitro systems, this technique has since been generalized to encode for a wide variety of amino acids by taking advantage of flexizymes, or RNA molecules that serve as catalysts for tRNA acylation.61 Applications of flexizymes to peptide display have recently been reviewed thoroughly, and any readers interested in this should be directed to the review by Suga et al.18 Nevertheless, limitations still exist with in vitro suppression of amber codons, especially considering proteins and systems that require cellular environments for processing, such as with phage display.
Further work by Peter Schultz and others led to the ability for in vivo genetic code expansion in E. coli, greatly expanding the scope of amber suppression techniques. For in vivo translation to be successful, several key components must be present in the system.62 The ncAA needs to be able to be transported into the cell, but it cannot be recognized by endogenous translational machinery. Also, the tRNA/tRNA synthetase pair for the ncAA must be orthogonal to the endogenous system so that no cAAs are incorporated at the desired site and no ncAAs are incorporated at undesired codons. Interestingly, a Methanococcus jannaschii tRNATyr/tyrosyl-tRNA synthetase pair (MjTyrRS) was identified and evolved to orthogonally suppress amber codons in E. coli with high selectivity for ncAAs.62 This allowed for adaptable in vivo translation of ncAAs in E. coli and also presented a system to evolve novel tRNA/tRNA synthetase pairs for new ncAAs. Other tRNA/tRNA synthetase systems that were explored for genetic code expansion in E. coli include Pyrococcus horikoshii tRNALys/lysyl-tRNA synthetase and even E. coli tRNATrp/tryptophanyl-tRNA synthetase by swapping them with Saccharomyces cerevisiae tRNATrp/tryptophanyl-tRNA synthetase.63,64
Shortly after using tRNATyr/MjTyrRS as an artificial system for the genetic encoding of ncAAs, a naturally occurring tRNA/synthetase pair was discovered in Methanosarcina barkeri and other Methanosarcina species for encoding pyrrolysine (Pyl) at the amber codon.65−67 It was found that the tRNA synthetase (Pyrrolysyl-tRNA synthetase or PylRS) recognized a unique tRNA (tRNAPyl) gene (PylT) that had a CUA anticodon (Figure 4).68,69 PylRS contains an N-terminal domain that serves to recognize tRNAPyl and a C-terminal domain that catalyzes the charging of tRNAPyl with Pyl. The original PylRS demonstrated promiscuous substrate selectivity and was found to charge tRNAPyl with and therefore encode for a variety of ncAAs that were pyrrolysine analogs or structurally distinct derivatives.70−72 This promiscuity is aided by both the lack of an editing domain and that the interactions between PylRS and Pyl are primarily driven through hydrophobic interactions. Also, PylRS has been shown to have promiscuity to even incorporate α-hydroxy derivatives of α-amino acids due to its low interactions with the α-amine of Pyl.73,74 Further modifications to PylRS have allowed for increased promiscuity and diversity of ncAAs that can be encoded using the tRNAPyl/PylRS pair, with currently hundreds of different ncAAs able to be incorporated.53 Due to its unique structures, tRNAPyl/PylRS is largely orthogonal to many commonly used lab cells including E. coli, S. cerevisiae, and mammalian cells, enabling easy transfer of evolved tRNAPyl/PylRS pairs between different cellular systems. For the aforementioned reasons, tRNAPyl/PylRS has become the dominant system for the genetic code expansion.
Figure 4.

Genetic code expansion using pyrrolysyl tRNA synthetase. A) In amber suppression, pyrrolysyl tRNA synthetase (PylRS), wild type or evolved, charges its cognate tRNA (tRNAPyl) with an ncAA. PylRS and its mutants have been evolved to recognize more than 200 ncAAs to charge tRNAPyl. B) The ncAA on the charged tRNAPyl is then incorporated into a peptide at the ribosome by suppression of an amber (UAG) codon on the corresponding mRNA.
Although there have been elegant systems developed to evolve substrates for tRNA-synthetases using fluorescence-based sorting75 and phage-assisted continuous evolution,76 inherent limitations in ncAA diversity still exist. All evolution systems for orthogonal translation require the ncAA-tRNA complex to be a substrate of the ribosome, which prevents many different amino acids from being efficiently incorporated into peptide chains. While the ribosome can tolerate incorporation of α-hydroxy derivatives of α-amino acids and N-methyl amino acids, α,α-disubstituted amino acids, β-amino acids, and d-amino acids are poor substrates for translation.77 Additional backbone modifications, such as N-alkylation, can also hinder efficient translation at the ribosome.78 Alongside modifications to the backbone, there are limitations to the side chains as well when using orthogonal translation systems. For example, tRNAs loaded with negatively charged amino acids have lower affinities for elongation factor Tu (EF-Tu), which initially hindered the development of a system to incorporate phosphoserine.79,80 Large side chains may also prevent binding to the tRNA synthetase and/or ribosomal pocket. In spite of these limitations, there is still a wide variety of amino acids that have been efficiently encoded into proteins using amber suppression systems. In the past ten years, researchers have also begun adapting the versatility of the genetic code expansion technique to phage display and have taken advantage of it in a variety of applications. A general overview of these applications is given in Figure 5.
Figure 5.

Overview of ncAA incorporation into phage libraries. A variety of advantageous moieties have been installed into phage libraries through genetic code expansion via amber suppression. These include the ability to direct selections toward active sites by incorporation of ligands into ncAAs, introduction of metal-chelating and reversibly covalent ncAAs, proximity-driven macrocyclization with cysteines, and introduction of clickable handles into phage libraries.
2.2. Initial Studies for ncAA Incorporation into Phage Displayed Proteins
The first reported genetically encoded ncAA incorporated into phage libraries was selenocysteine (Sec), where Sandman et al. took advantage of the endogenously expressed selABCD operon to incorporate Sec at an opal codon (Table 2).81 In this study, phages were successfully isolated that were dependent upon Sec for incorporation by integrating the selenocysteine insertion sequence (SECIS) and an opal-containing library upstream of gIII in the phage genome. While the incorporation of Sec gives access to novel reactivities, using the endogenous expression system limits the ability to encode other ncAAs. Shortly after this landmark study, Tian et al. significantly increased the diversity of ncAAs able to be displayed in phage libraries by taking advantage of mutant MjTyrRS/tRNATyr pairs to orthogonally translate five different ncAAs into phage-displayed peptides (MeY, AzF, AcF, BpF, and NpA; Table 2).82 In this study, they were able to demonstrate phage expression that was dependent on ncAA incorporation by fusing an amber-containing streptavidin binding peptide (SBP) to the N-terminal domain of pIII. Several different ncAAs that were used could be incorporated at amber codons through specifically engineered tRNA synthetases based on the original MjTyrRS, and each system is described in Table 2.62,83−86 One interesting application from this work was the ability to incorporate p-azido-phenylalanine (AzF, Table 2) into phage proteins, which allows for potentially clickable modifications to phage libraries (Figure 5).82
Table 2. Genetic Incorporation of ncAAs into Phage Libraries.


Despite successful ncAA integration into phage-displayed libraries in the early 2000s, the application of these libraries to identify ligands for certain targets has been limited due to the inability to outcompete production of wild-type phage that do not contain amber codons within their genome. Although amber suppression systems are optimized to efficiently encode ncAAs, they still produce less protein than those containing strictly sense codons due to competition with termination machinery. This results in lower pIII production, and subsequently lower phage yields for libraries containing ncAAs. Thus, any phage libraries that contained a mixture of sense-only and amber-containing clones would be heavily biased toward the sense-only clones. One innovation that helped to counter this was the development of hyperphages, helper phages that contain a nearly complete deletion of gIII in their genome, that gave the ability to create large phagemid libraries completely dependent upon amber suppression for phage production.46 Additionally, the Schultz Lab demonstrated that the bias for sense-only codons could be lessened by increasing pIII production through expressing phages with higher concentrations of ncAA and prolonged expression times at lower temperatures (30 °C).87 These discoveries allowed for the generation of a sulfotyrosine (SY, Table 2) containing antibody library through genetic code expansion using an MjTyrRS mutant to incorporate SY into amber codons of a randomized CDR3 region and subsequent identification of SY containing scFv antibodies that selectively bound to gp120.87,88 Similarly, introduction of boronic acids (BoF, Table 2) into a phage-displayed scFv library by using a mutant MjTyrRS afforded the ability to identify proteins that potently bind geminal diols and gave the possibility to develop antibodies that interact with glycans.89,90 Other interesting examples of ncAA incorporation into phage libraries involved using an MjTyrRS mutant to incorporate bipyridyl-alanine (BpyF, Table 2) into a randomized disulfide-cyclized peptide library of six amino acids and also into zinc-finger peptides.91−93 In doing this, they identified peptide ligands that bound to Ni2+ with sub micromolar affinity (Table S1, Peptides 1 and 2) and also developed novel iron(II)-finger proteins.92,93 Follow-up studies with these libraries may allow for the evolution of metallopeptide catalysts and other beneficial probes that take advantage of metals for activity. While these initial cases all demonstrate the ability and potential of genetic code expansion in phage libraries, they were reliant on incorporation of already strong ligands into the ncAAs for successful selections. Until recently, phage-assisted identification of de novo peptides that contained ncAAs with modest affinities for targets was hindered by parasitic peptide sequences that lacked amber codons and outcompeted ncAA-containing libraries.
2.3. Development and Application of Amber-Obligate Peptide Libraries
Although amber suppression techniques were first applied to phage-displayed libraries in the early 2000s, there were only a handful of examples of them successfully employed in selections in the following decade. One of the main complications was due to the inability to isolate amber-containing phage libraries from parasitic sense-only libraries. In 2020, Tharp et al. reported a strategy to enrich for amber-containing peptides in vivo that took advantage of superinfection immunity of filamentous phages (Figure 6).94 Because expression of pIII results in interactions with the TolA receptor, any cells that express pIII cannot be infected by other filamentous phages that are dependent upon TolA for entry.95,96 This allows for the ability to isolate libraries that contain amber codons by initiating pIII expression in the absence of amber suppression machinery and subsequent infection with a helper phage to give E. coli with the amber-containing libraries kanamycin resistance.94 All other libraries (sense-only) that express pIII can then be eliminated through selection with kanamycin.94 This differentiation of amber-containing and sense-only clones affords the ability to enrich amber codons at any stage in a phage selection. Thus, ncAAs that give modest affinity to protein targets can be used without concern for inadvertently enriching parasitic sense-only clones. Additionally, the amber enrichment technique facilitates the generation of libraries that contain randomly positioned amber codons, rather than it being fixed in one position. This gives increased diversity of phage libraries and higher potential in identifying potent ligands.
Figure 6.

Generation of amber obligate phage libraries through superinfection immunity selection. Plasmids expressing pIII-library fusions are induced for expression without amber suppressing mechanisms, resulting in sense-only clones expressing pIII and preventing infection by a helper phage CM13. Cells are then selected for kanamycin resistance and plasmids extracted, isolating the clones that lack pIII expression. After transformation and phage expression in amber suppressing cells (DH5α), amber obligate libraries are generated.
Tharp et al. took advantage of the newly developed amber-obligate library to successfully incorporate six novel ncAAs into phage libraries.94 Four phenylalanine derivatives (oClF, mClF, mBrF, and mCF3F, Table 2) and two lysine derivatives (BuK and CrK, Table 2) were incorporated using the previously developed PhdRS and BuKRS mutants of PylRS, respectively.97−99 Initial selections against TEV protease and streptavidin using these amber-obligate phage libraries and the phenylalanine derivatives demonstrated the robust ability to isolate ncAA-containing peptide ligands for proteins even in the absence of a strong warhead to direct binding (Table S1, Peptides 3–6).94 In addition to this, they also demonstrated a phage-assisted active site-directed ligand evolution (PADLE) technique (Figure 5), with BuK directing binding of the peptide library toward the active site of sirtuin 2, which ultimately resulted in low nanomolar inhibitors of the protein (Table S1, Peptides 7 and 8).94 This technique shows promise especially in discovering ligands/inhibitors for proteins that recognize post-translational modifications. PADLE has recently been applied to different epigenetic readers, including histone deacetylase 8 (HDAC8)100 and ENL-YEATS.101 For the development of HDAC8 inhibitors, an acetyl-lysine analog (Aoda, Table 2) was incorporated into phage libraries using a previously developed AcKRS mutant of PylRS and subsequently used to develop sub nanomolar isoform-selective inhibitors (Table S1, Peptide 9).100,102 In the application of PADLE to develop ENL inhibitors, BuK-containing phage were panned against the ENL YEATS domain and low nanomolar inhibitors of the protein’s interaction with an acetylated peptide were discovered (Table S1, Peptides 10 and 11).101 Given the success in identifying ligands for multiple epigenetic readers, we envision PADLE can be applied to a wide variety of proteins that recognize post-translational modifications for the identification of selective probes and inhibitors.
While the development of amber-obligate libraries was a revolutionary technique for ncAA incorporation in phage libraries, there were still a few aspects of the system that hindered its application. The first of these is that the amber enrichment procedure was tedious and required multiple rounds of amplification/electroporation. This process magnifies the limitation of electroporation on library size and may introduce biases in the libraries themselves. Also, the use of a three-plasmid expression system appeared to give lower phage yields in comparison to two-plasmid systems. To combat these issues, a novel amber-encoding helper phage, CMa13ile40, has been recently developed that both shortens the amber enrichment process and results in higher phage yields.103 CMa13ile40 was developed by insertion of a Methanomethylophilus alvus PylRS/tRNAPyl cassette into the intergenic region of helper phage CM13d3. In addition to shortening the amber enrichment process, the new helper phage also demonstrated higher specificity for packaging of the phagemid-containing library in comparison to commercially available helper phages.103 In this work, two additional ncAAs were incorporated into phage libraries, BocK and AllocK (Table 2), and the influence of different ncAAs on selection was demonstrated with the isolation of peptides with low micromolar affinity for the membrane-bound E3 ligase ZNRF3 (Table S1, Peptides 12–14). Although the use of CMa13ile40 significantly shortens the amber enrichment process, development of new helper phage derivatives for incorporation of other amino acids will be necessary for expanded applications.
2.4. Genetically Encoded Macrocyclic Peptide Libraries
In addition to integrating ncAAs within phage-displayed peptide libraries to direct selections, there have been a few studies that have taken advantage of ncAAs to generate genetically encoded cyclic peptide libraries (Figure 5). Interest in applying cyclic peptides as therapeutics has risen recently due to increased stability and potency in comparison to their linear counterparts.104 Typically, phage-displayed peptide libraries are linear, resulting in limitations to their application for therapeutic development. Therefore, incorporation of ncAAs that have intrinsic reactivities to catalyze cyclization of peptides on the displayed libraries gives great potential for identifying therapeutic lead compounds. One challenge in the incorporation of reactive ncAAs is the fine-tuning of the reactivity of the ncAA—it must remain unreactive until incorporation into phage libraries and then react with the incorporated peptides. If the ncAA is too reactive, it will be modified before incorporation into libraries and have difficulty being encoded. The first study that successfully used an ncAA to cyclize peptide libraries on phage took advantage of an acryloyl-lysine (AcrK, Table 2) to react with an N-terminal cysteine flanking a peptide library.105 The AcrK was genetically encoded using a mutant PylRS (PrKRS) that had been previously evolved from AcKRS.99 Biotin pulldown assays indicated approximately 60% efficiency in displaying cyclic peptide libraries and that the cyclization was dependent upon the N-terminal cysteine.105 The cyclic peptide library was subsequently used to identify cyclic peptide inhibitors for TEV protease and HDAC8, with each selection resulting in peptides with low micromolar affinity for each protein (Table S1, Peptides 15–17).105
Shortly after the development of phage-displayed cyclic peptide libraries using AcrK, an additional strategy (MOrPH-PhD) was reported that took advantage of a bromoethyl-tyrosine derivative (O2beY, Table 2) to cyclize a variety of cysteine-containing phage libraries on pIII.106 Prior to incorporation of O2beY into phage, in a separate study Fasan et al. developed an MjTyrRS mutant for O2beY (O2beY-RS) and rigorously characterized its ability to cyclize peptides containing cysteine at different positions relative to O2beY.107 They observed high cyclization efficiencies containing cysteine in the i + 2 to i + 8 position and also showed reactivity in the i – 6 and i – 8 positions.107 This prior validation gave them confidence in producing macrocyclic peptide phage libraries with the cysteines and O2beY at different positions. Interestingly, they observed a dependence in selection efficiency on the library type, as different proteins gave preference to libraries containing cysteine upstream or downstream to the ncAA (Table S1, Peptides 18–33).106 Ultimately, this study was afforded low nanomolar macrocyclic peptide ligands for The Keap1 Kelch Domain (Table S1, Peptides 28 and 29), and low micromolar ligands for the Sonic Hedgehog protein (Table S1, Peptides 32 and 33). These selections favored use of libraries with internal cysteines and an N-terminal O2beY, whereas the selections against streptavidin isolated proteins with O2beY on the C-terminal end (Table S1, Peptides 18–33). This highlights the necessity to create and screen diverse cyclic peptide scaffolds for the development of potent ligands for proteins. In addition to incorporation of O2beY, the same study also incorporated propargyl-tyrosine (OpgY, Table 2) at high efficiency in phage using the developed O2beY-RS.106 Although they did not demonstrate it, this amino acid could potentially be used to generate phage-displayed libraries labeled with pharmacophores or other therapeutically relevant moieties. All-in-all, these two studies highlight the use of ncAAs to genetically encode cyclic peptides on phage and should encourage further adaptions to target other proteins.
2.5. Outlook of Genetic Code Expansion in Phage Display
There have been numerous studies demonstrating the ability to increase the chemical diversity of phage-displayed peptide libraries since its inception in the early 2000s. In spite of the great versatility in amber suppression, there is still a need for improved systems, especially due to the poor stability of the N-terminal domain of PylRS.108,109 Ongoing efforts to improve upon the solubility and stability of the N-terminal domain include directed evolution,109,110 formation of chimeric interspecies PylRSs,108,111,112 and phage-assisted continuous evolution.76,113 In addition to developing novel systems for stabilizing PylRS and tRNA synthetase activities, investigation into new techniques to evolve for the encoding of novel backbones, large side chains, and charged side chains will help overcome current limitations on ncAAs that can be incorporated in orthogonal translation (see Section 2.1 for limitations in ncAA scope). A recent study by Schepartz et al. demonstrated the ability to expand substrate scope to encode for a variety of backbone modifications, such as α-hydroxy, α-thio, and N-formyl amino acids.114 Also, Chin et al. have devloped a novel tRNA display system to identify aminoacyl tRNA synthetases for β-amino acids, α,α-disubstituted-amino acids, and β-hydroxy acids.115 Subsequent integration of these suppression systems into phage display will allow for increased diversity of the displayed peptides and enhanced stability of the identified peptide ligands. Even with the development of highly efficient amber suppression systems, there is still a limitation to only incorporating one ncAA into the phage libraries and other systems will need to be employed to compete with the ability of in vitro translated mRNA displayed libraries that can simultaneously encode multiple ncAAs. Using two orthogonal tRNA/tRNA synthetase pairs in E. coli, the genetic incorporation of two different ncAAs simultaneously in one protein were demonstrated a decade ago.116,117 One can potentially use these cellular systems for the production of phages with multiple different ncAAs, which has been recently demonstrated. One report by Chin et al. for encoding multiple ncAAs into phage took advantage of both amber suppression and quadruplet codons to incorporate a propargyl-tyrosine (OpgY, Table 2) and cyclo-propene containing amino acid (CypK, Table 2), respectively.118 To increase amber suppression and allow for use in reading the quadruplet codon, they employed use of an orthogonal ribosome (riboQ1) that they had previously developed.117 When using riboQ1, they demonstrated enhanced amber suppression and were able to use a helper phage that contained wild-type pIII (CM13) to allow for monovalent antibody expression.118 Coupled with two different orthogonal tRNA synthetase mutants, they were able to achieve double incorporation of CypK and OpgY into an scFv-displayed phage, then demonstrated the ability to label both ncAAs using click reactions.118 This may be a promising system to incorporate multiple ncAAs into phage libraries and afford more diverse peptides. Further diversification of phage libraries using orthogonal translation will require the development of more techniques for genetic code expansion. The Chin Lab has also reported orthogonal translation of three different ncAAs, albeit not applied to phage libraries.119 Additional diversification may require adapting phage display to E. coli containing synthetic genomes, such as Syn61Δ3, to employ use of additional codons other than the amber codon.120,121
3. Chemical Post-translational Modifications of Phage-Displayed Peptides
3.1. Introduction and Initial Studies for Chemical Diversification of Phage Libraries
Alongside the installation of novel chemical moieties through genetic code expansion, several different bioconjugation reactions have been employed to diversify phage-displayed peptides. There have been numerous reactions developed for introducing new functional groups into proteins. While there have been a few studies that nonspecifically label phages with fluorophores or clickable handles (usually on pVIII), this section will be focused on selectively labeling phage-encoded libraries.122−124 The main difficulty in labeling phage libraries revolves around tuning the specificity of the compounds to modify the desired peptide without affecting pIII and other phage coat proteins that are required for proper infectivity. In addition to concerns involving selectivity of chemical modifications, it has also been challenging to directly quantify labels on phage. Especially when libraries are displayed on pIII, the individual proteins are typically at concentrations that are too low to be detected by mass spectrometry. Therefore, several different indirect methods are used to quantify phage modifications, such as ELISA, biotin pulldown, and activity assays. These indirect techniques make it difficult to identify exactly which peptides are being modified on phage and to what degree they have been modified. While these are ongoing issues, there have been several successful campaigns over the past 20 years that selectively modify phage libraries and identify therapeutic peptides via phage display.
Most methods for selectively incorporating unnatural modifications to phage involve taking advantage of N-terminal residues. Because the phage coat proteins typically have N-terminal periplasmic tags that are cleaved during packaging, N-terminal amino acids other than methionine can be easily generated. One of the first techniques to install novel modalities into phage libraries involved native chemical ligation to introduce peptides onto the N-termini of pIII and pVIII.125 By genetically encoding an N-terminal cysteine to pVIII or pIII, they were able to use thiophenol for ligation of peptide chains containing the ncAAs kynurenine and norvaline to each of the phage proteins. Another strategy developed by Carrico et al. selectively modified N-terminal residues using pyridoxal-l-phosphate (PLP) to catalyze a transamination reaction and generate N-terminal ketones (Figure 7A).126 Following generation of a ketone, they were then able to successfully label phage proteins with alkoxyamine-fluorophores. MALDI-MS demonstrated efficient, selective labeling of the N-terminus of pVIII at pH 6.5 in spite of two lysines also being available for modification in pVIII. With the addition of 100 mM aniline, the reaction proceeded to label approximately 80% of pVIII with fluorophores. However, labeling was also observed on all other coat proteins with accessible N-termini (pIII, pVII, and pIX).126 To create productive phage libraries for biopanning, more efficient and selective labeling should be desired.
Figure 7.
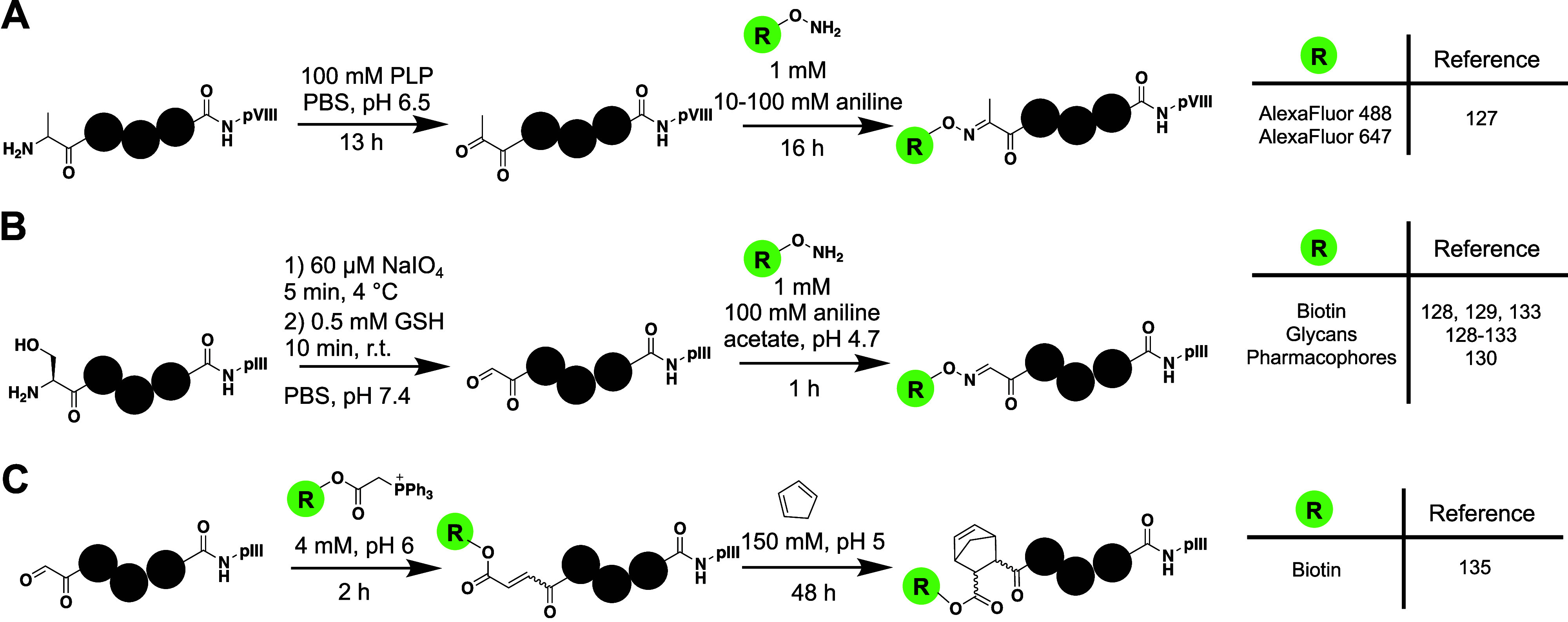
Functionalization of ketones and aldehydes on phage. A) Generation of ketones on phage by transamination allows for functionalization via an oxime ligation with alkoxyamines. B) The N-terminal serine can be converted into glyoxylate by oxidative cleavage with NaIO4 and the afforded aldehyde has high reactivity toward alkoxyamines. The resulting aldehydes have been functionalized with a wide variety of moieties. C) Functionalization of phage-displayed libraries containing N-terminal serines through Wittig and Diels–Alder reactions.
3.2. Oxime-based Ligation into Libraries Containing N-terminal Serines or Threonines
In a similar manner to the generation of N-terminal ketones and subsequent substitution of alkoxyamine derivatives on phage, there have been several studies that have taken advantage of N-terminal serine and threonine to selectively introduce new moieties onto pIII. The N-terminal serine or threonine can be converted into an N-glyoxylate that contains an aldehyde functional group through oxidative cleavage by sodium periodate.127 The Derda Lab pioneered application of this reaction to phage libraries and has thoroughly demonstrated its effectiveness in introducing a variety of unnatural modifications. Introduction of glycopeptide libraries onto phage-displayed peptides has been employed through generation of N-terminal aldehydes, followed by oxime ligation with aminooxy-derivatized glycosides (Figure 7B).128 Elegant biotin pulldown assays allowed for the kinetics of the reaction to be characterized thoroughly on phage. After 5 min of reaction with NaIO4, nearly all phages were converted to the reactive aldehyde intermediate (k = 210 M–1 s–1). Addition of aniline in the subsequent step (100 mM aniline acetate, pH 4.7) increased the oxime ligation nearly 100-fold, resulting in an observed rate on phage of 3.3 M–1 s–1.128 They demonstrated approximately 80% labeling only on phages containing the N-terminal serine and also showed effectiveness in labeling a commercially available heptapeptide library (Ph.D. 7).128 The resulting glycopeptides from this method resemble natural N- and O-linked glycans, giving them promise in identifying peptide ligands for lectins and other carbohydrate-binding proteins.129 The Derda Lab has taken advantage of this technique to guide phage-based selections toward active sites by modifying phage libraries with glycans,130−133 sulfonamides,130 and biotin.130,134 They have also combined these modifications with integration of silent barcodes downstream of the peptide library, allowing for the ability to simultaneously screen libraries containing different N-terminal modifications. In doing this, they have identified peptide ligands with low nanomolar potency for carbonic anhydrase and femtomolar peptides for streptavidin (Table S1, Peptides 34–48). This study also observed selective identification of ligands dependent upon the compound that the N-terminus is functionalized with.130 Libraries modified with glycans show good potential to identify peptides for lectins and other proteins with affinities for oligosaccharides. In particular, an arabinose-modified library was used to isolate peptides with low micromolar affinity for CS-35 Ab, an antibody that binds to an arabinose-containing mycobacterial antigen (Table S1, Peptides 49–52).131 Similar studies using mannose and galactose-modified N-termini have demonstrated the ability to isolate glycopeptides with micromolar affinity for DC-SIGN (an antigen recognition protein in dendritic cells) and phages with affinity for Galectin-3 (Table S1, Peptides 53–56).132,133 The Derda Lab has also used the afforded aldehyde from NaIO4 oxidation to investigate ligands for the Wittig reaction and install dienophiles onto phage libraries that can subsequently be modified through Diels–Alder reactions (Figure 7C).135 One consideration is that when using aniline as catalyst for the oxime ligation, the intermediate Schiff base undergoes quick hydrolysis, resulting in irreversible loss of the original aldehyde.136 To prevent this degradation on phage, Derda et al. developed benzamidoximes that react quickly (k = 40 M–1 s–1) with aldehydes without the addition of aniline.136 Modification of phage libraries through N-terminal aldehydes demonstrates the effectiveness of chemically functionalizing phage; however, the requirement of harsh oxidative conditions and multiple buffer exchanges may be undesired for some studies.
3.3. Post-translational Chemical Modification of Cysteine-Containing Phages
In addition to chemically modifying the N-terminal serine and threonine, there have been several studies that take advantage of cysteines to functionalize phage libraries. There are multiple biocompatible electrophilic reagents that have been developed to react nonspecifically with cysteines in proteins. With the abundance of cysteine being relatively low in proteins, it makes it a good candidate for phage modification even if a reaction is nonselective. However, there are key disulfide bonds in pIII that result in decreased infectivity when modified. Two different techniques have been established to mitigate this issue. One innovation that helped to reduce off-target concerns when modifying cysteines in phage libraries was the development of phages that lack disulfide bonds in pIII. Schmid et al. employed an in vitro evolution strategy to select for proteolytically stable pIII variants, ultimately identifying pIII proteins that lacked disulfide bonds.137 Although the disulfide-free phage showed lower infectivity, it was crucial in allowing for modifications of cysteine-containing peptide libraries. Additionally, the use of immobilized TCEP allows for selective reduction of cysteines within peptide libraries on phage, rather than the wild-type disulfides within pIII.138 Zheng et al. used the immobilized TCEP to selectively reduce cysteines on a CX7C library displayed on pIII, and then selectively reacted the two cysteines with (2-acetyl-4-((2-iodoacetamido)methyl)phenyl)boronic acid (APBA-IA, Figure 8A).139 Incorporation of APBA into the phage library allows for reversible covalent interactions with amines under biological conditions, and they took advantage of this to develop selective peptide antibiotics/probes for amine-presenting bacteria (Table S1, Peptides 57–67).139−141 This study highlights the potential applications of using cysteine-reactive reagents to modify phage, yet more selective reactions should be desired to avoid potentially modifying cysteines within the library or in other phage proteins.
Figure 8.

Modifications of cysteine residues on phage. A) Iodoacetamide derivatives react quickly, but nonspecifically with thiols on phage. These were used to install boronates for reversibly covalent functionalization of phage libraries. B) N-Terminal cysteines can be selectively modified by electron-deficient nitriles, such as Biotin-CBT, that quickly condense to thiazolidines at neutral conditions.
To develop cysteine-modifying reagents with higher selectivity, most studies have looked to differentiate the N-terminal cysteine from an internal cysteine. This was inspired by previous development of cyanobenzothiazole (CBT) reagents that quickly condense (k = 9.19 M–1 s–1) with N-terminal cysteines at physiological conditions to generate thiazolidine adducts to proteins.142 Although primarily focused on generating macrocyclic peptide libraries on phage, two different studies have demonstrated biotin labeling of N-terminal cysteine-containing phages using biotin-CBT (Figure 8B).143,144 An additional study has also demonstrated similar reactivity using biotinylated 2-((alkylthio)(aryl)methylene)malononitrile (TAMM) derivatives that form thiazolidine adducts with the N-terminal cysteine.145 Biotin pulldown assays using these reagents indicated approximately 70% labeling of the peptide libraries.143−145 All of these labeling experiments were performed at neutral pHs in standard phage buffers. Although these only demonstrated biotinylation of peptide libraries, we envision that similar strategies could be used to further functionalize peptide libraries with diverse functional groups, such as glycosides, lipopolysaccharides, or pharmacophores. In comparison to the originally developed oxime ligation techniques, condensation on N-terminal cysteines provides a more straightforward labeling strategy that eliminates buffer exchanges and multiple phage precipitation steps. One strategy has combined the cyanobenzothiazole approach with alkylation of an internal cysteine to have selective double labeling of N-terminal and internal cysteines, respectively.146 The double labeling afforded integration of two covalent warheads to a CX9C library displayed on pIII. To do this, they first installed an APBA-CBT derivative by reacting it with an N-terminal cysteine. Then, they reacted the phage library with an iodoacetamide containing α-cyanoacrylamide to label the internal cysteine residue. This gives the ability to covalently interact with amines and cysteines on target proteins. Through biotin pulldown assays, they demonstrated at least 50% efficiency for labeling the N-terminal residues and nearly complete labeling of the internal cysteine.146 They then used the double-labeled library to select for peptides that bound to TEV protease in the low micromolar range (Table S1, Peptides 68–73). While these are promising avenues for creating new modalities in phage-displayed peptide libraries, most other cysteine-based reactions have been tailored for generating cyclic peptide libraries on phage and this will be discussed in detail in the following sections.
3.4. Post-translational Modifications for Generating Phage-Displayed Cyclic Peptide Libraries
Cyclic peptides show increased therapeutic potential in comparison to their linear counterparts due to several factors, such as having more rigid structures, having possibly enhanced membrane permeability, and being less susceptible proteolysis.104,147−149 Many efforts have attempted to use phage display for the identification of cyclic peptides that bind to therapeutic targets. Initial displayed cyclic peptides libraries were generated in situ through disulfide bond formation that forms spontaneously within the periplasmic space.150,151 However, the use of peptides containing disulfides is potentially problematic, as they are unstable in reducing environments. Because of this, several studies have investigated the development of reagents that create stable cyclic peptide libraries on phage. In addition to creating cyclic peptide libraries on phage, reagents used for cyclization can also be functionalized to display other moieties and install new reactivities to the phage libraries. The majority of reported techniques rely on nonspecifically reacting with cysteines displayed on phage; however, there have been a few recent strategies to develop cyclic peptide libraries using reagents that selectively modify N-terminal amino acids.
The most standard approach involves using alkylating reagents that are composed of highly reactive bis-electrophiles to react with two cysteines on phage libraries, resulting in cyclization of the displayed peptides (Figure 9). A diverse amount of electrophiles are available for labeling cysteine residues on phage. Although they typically react nonspecifically with cysteine residues, the use of phages that contain disulfide-free pIII proteins can allow for modification of the library region without reductions in infectivity.137 Due to the promising abilities of α-helical peptides, several groups developed linkers to chemically synthesize stapled peptides using cysteine alkylating reagents in the early 2010s.152−154 Similarly, the first reported molecules for cyclization of peptides on phage consisted of alkyl and benzyl halides that looked to stabilize alpha-helices on phage libraries.155 Heinis et al. attempted to stabilize α-helices by modifying phages at i and i+4 cysteine residues with α,α′-dibromo-m-xylene (mDBMB, Table 3), trans-1,4-dibromo-2-butene (TDBB), and cis-1,4-dichloro-2-butene (CDCB, Table 3).155 Through circular dichroism studies of a model peptide, they observed efficient formation of α-helices only with those reacted with mDBMB and CDCB. Phage selections using both CDCB and mDBMB then afforded low nanomolar ligands for β-catenin (Table S1, Peptides 74–78). Shortly after this initial study, the Heinis Lab developed several alkyl bromides to create cyclic peptide scaffolds on phage (oDBMB, pDBMB, mDBMP, DBAmB, DBAc, and DBPyD; Table 3). Interestingly, they took advantage of reacting the bis-electrophiles with libraries containing four cysteines to form double-bridged peptides.156 Through comparative phage selections against plasma kallikrein, they observed preferential enrichment of libraries dependent upon the organic linker used in selection, indicating modification of the phage libraries. In doing this, they identified low nanomolar ligands for IL-17 (Table S1, Peptides 79 and 80), an interleukin target that binds to some currently approved antibody drugs.156,157 The same library was also used to identify subnanomolar inhibitors of plasma kallikrein (Table S1, Peptides 81 and 82) with enhanced specificity for the protein.156 These same reagents have also resulted in the identification of potent cyclic peptide inhibitors (Table S1, Peptides 83–93) for other kallikrein-related peptidases (KLK5 and KLK7) using the same four-cysteine library approach.158
Figure 9.

Peptide cyclization on phage by highly reactive bis-electrophiles. Phages have traditionally been cyclized via two cysteines that react with activated halides (Y = Br, Cl, I) through SN2 or SNAr reactions, with the exception of one allene-based derivative. Representative reactive moieties are grouped by reaction type or additional functionalization of the peptides.
Table 3. Symmetrical Bis-Electrophiles for Cyclization of Two-Cysteine Phage Libraries.


All phages were reduced using TCEP prior to cyclization. See reference for specific conditions.
Buffer A: 20 mM NH4CO3, 5 mM EDTA, pH 8.
Buffer B: 50 mM Tris pH 8.5.
Phages reduced with iTCEP for 48 h.
Buffer C: 55 μL of PBS (phage), 36 μL of water, 10 μL of 1 M sodium bicarbonate, pH 8.5.
In addition to traditional SN2 reactions with cysteines on phage, SNAr substitutions have also been successfully employed to cyclize phage displayed libraries with two cysteines (Figure 9). The first of these applications took advantage of a decafluoro-diphenylsulfone (DFS, Table 3) to react with cysteines at extremely quick kinetics with rates upward of 180 M–1 s–1.159 A promising advantage of developing peptide ligands using these perfluoro compounds is their ability to be directly detected for binding and in vivo analysis through 19F NMR.160 Another similar strategy that takes advantage of SNAr reactivity involves using 2,4-difluoro-6-hydroxy-1,3,5-benzenetricarbonitrile (DFB, Table 3) to cyclize phage libraries with two cysteines.161 Although it is slower than the reaction with DFS, the kinetics are sufficient for effective phage labeling with a second order rate constant of 9.2 M–1 s–1. Both linkers have proven useful in the phage-assisted identification of cyclic peptides, with the resulting peptides (Table S1, Peptides 94 and 95) binding with micromolar affinity to human serum albumin and Bcl-xl.160,161
In addition to simply creating macrocyclic peptides on phage, cyclization of peptides with organic linkers also allows for further functionalization of peptide libraries. By incorporation of additional moieties into the linkers, additional properties can be given to the peptide libraries that facilitate selections for certain proteins. In two separate studies, alkylation of cysteines using chloroacetamides (BSBCA, Table 3)138 and bromoacetamides (BSBBA, Table 3)162 has been used to generate light-responsive genetically encoded cyclic peptide libraries by incorporating an azobenzene core within the organic linker. These allow for the potential to create photoswitching peptides that can activate or deactivate in the presence of UV light, which have proven useful in developing probes for a variety of assays. The BSBCA linkers showed only 50% reactivity on the phage and did not produce high affinity peptides in selections with affinities in the range of 300–500 μM for streptavidin (Table S1, Peptides 96–98). However, the BSBBA functionalized library resulted in peptides with low micromolar affinity for streptavidin (Table S1, Peptides 99–102).162 Interestingly, Heinis et al. performed their selection to identify peptides that were activated by UV light, rather than inactivated, which may have contributed to more successful binding and inhibition.162 Photoswitchable libraries have also been made through reactivity with a bis(allenamide) functionalized azobenzene (BSBDA, Table 3). This derivative reacts significantly quicker than the previously established alkyl halide reagents with rates on the order of 16–30 M–1 s–1 and reactions with reduced phages are near completion after only 20 min of incubation.163 Because of its increased reactivity, application of BSBDA to phage selections may result in more efficient production and selection of phage-displayed photoswitchable libraries.
There have been multiple studies that have created unnatural motifs on phage-displayed libraries by using bis-electrophiles that react with cysteine. For example, Derda and co-workers have used dichloro-oxime (DCO, Table 3) derivatives to generate glycan-modified macrocyclic peptide libraries on phage.164 A diketone-containing organic linker has also been used to generate cyclic peptide libraries containing pharmacophores to direct phage selections. To do this, a dichloropentadione (DPD, Table 3) derivative first reacted with two cysteines on the phage to afford cyclic peptide libraries.165 Biotin pulldown assays indicated approximately 75% efficiency for introduction of the diketone to the phage libraries. Following cyclization, the linker was then functionalized with pharmacophores containing hydrazines that can condense with the diketone, ultimately forming cyclic peptides bearing pharmacophores through a diazole linkage.165 Hydrazine reactivity with the diketone proceeds quickly at pH 5.0, with the reaction completing after only 1 h. The pharmacophore-functionalized libraries successfully identified ligands with low nanomolar affinity for carbonic anhydrase (Table S1, Peptides 103 and 104). While this is an excellent display of functionalizing cyclic peptide libraries through macrocyclic linkers, it is hindered by the generation of regioisomers during the hydrazine condensation with diketones. Similar reactions that are symmetric in nature would simplify characterization and identification of hit peptides following selections.
Another interesting design has been the introduction of covalent warheads into phage-displayed cyclic peptide libraries through derivatized macrocyclic linkers. Bogyo et al. introduced vinyl sulfone and diphenylphosphonate into cyclic peptide libraries that were cyclized with a derivatized dichloroacetone linker (DCA-VS and DCA-DPP, respectively; Table 3).166 The afforded DCA-VS and DCA-DPP functionalized libraries were then successfully used to identify inhibitors of TEV protease and FphF, respectively, with IC50 values in the low micromolar range (Table S1, Peptides 105 and 106). In a similar approach by Gao et al., cyclic peptide phage libraries were modified by a dichloro-oxime derivative to contain 2-acetylphenyboronic acids (APBAs) for covalent targeting of lysine residues. The APBA-functionalized library was used to identify peptides that inhibited sortase A at micromolar potency and for identifying peptides to detect the SARS-CoV-2 spike protein (Table S1, Peptides 107–109).167
Although there have been a broad range of modifications employed to generate cyclic peptide libraries through highly reactive bis-electrophiles, they are potentially limited in a few capacities. First, all of the above-mentioned bis-electrophiles are agnostic in their selectivity for cysteines, resulting in them necessarily being symmetrical. This limits functionalization and hinders further diversification of phage libraries. Additionally, the lack of specificity for labeling may result in less efficient display of the desired cyclic peptide libraries, with different modifications occurring elsewhere on the phage proteins. To counter these issues, a few recent studies have looked to take advantage of N-terminal residues for generating cyclic peptide libraries more selectively and allow for asymmetrical designs. The first example of potentially asymmetric labeling involved taking advantage of N-terminal cysteines to selectively modify phage libraries. Wu et al. designed a cyclic peptide linker (ClAc-3, Figure 10) that introduced a chloroacetamide moiety into 2-((alkylthio)(aryl)methylene)malononitrile (TAMM) derivatives.145 In doing this, the TAMM condensation reaction with the N-terminal cysteine directed reactivity to the displayed N-terminal cysteine-containing library. Following this reaction, a proximity-driven cyclization event occurs between an internal cysteine and the chloroacetamide. In this method nonspecific labeling should be minimal, as the rate for chloroacetamide labeling is low (>0.05 M–1 s–1) at neutral pH and no other phage proteins contain N-terminal cysteines. Through biotin pulldown assays, they demonstrated approximately 75% reactivity on phage and observed no significant phage toxicity. Biopanning of a CX9C cyclic peptide library to Bcl-2 resulted in the identification of cyclic peptides with mid nanomolar affinity for the protein (Table S1, Peptides 110–112). The study also identified peptides for MDM2 and Keap1 with nanomolar potency (Table S1, Peptides 113–116). Two similar strategies have also been developed that take advantage of the cyanobenzothiazole (CBT) condensation reaction with N-terminal cysteines to selectively cyclize phage libraries.143,144 These also resulted in similar efficiencies (60–75%) for modifying N-terminal cysteine containing libraries with the CBT derivatives. The main difference in these two involved the internal cysteine reactive moiety—Liu et al. reported using a chloroacetamide-containing CBT (CAmCBT, Figure 10) that reacts irreversibly,143 while Gao et al. functionalized CBT with a reversibly reacting α-cyanoacrylamide (M-a-23, Figure 10).144 Because of its reversible nature, peptides cyclized with the α-cyanoacrylamide did exhibit some free cysteine after cyclization, albeit the reverse reaction occurred at a low rate. To counter this, they demonstrated that the peptides could be synthesized without the nitrile to prevent the reversible reaction, although the nitrile is necessary for phage reactivity.144 M-a-23 was successful in producing macrocyclic peptide ligands for the Keap1 Kelch Domain, Sortase A, and streptavidin (Table S1, Peptides 118–121).144 CAmCBT also proved useful in creating a neutralizing macrocyclic peptide with low micromolar affinity for the Spike protein that prevented SARS-CoV-2 infection (Table S1, Peptide 117).143 An additional strategy has also been reported that takes advantage of an internal cysteine and reductive amination of the N-terminus to generate asymmetric linkers for macrocyclic peptide libraries.168 In this study, Mayer et al. developed two different aldehyde derivatized benzyl bromoacetamides that displayed asymmetric cyclization of libraries consisting of AX8C peptides fused to pIII. The reaction was performed in two steps: 1) alkylation of the cysteine with bromoacetamide 2) followed by reductive amination with NaBH3CN. The library was screened against streptavidin and proved to efficiently identify a peptide with low nanomolar affinity (Table S1, Peptide 122). In comparison to the approach taking advantage of N-terminal cysteines to direct cyclization, this strategy is likely less selective, as lysines within the library could also react via reductive amination. Nonetheless, all of these studies open up new avenues in selective peptide cyclization on phage and allow for asymmetric functionalization of macrocyclic libraries.
Figure 10.

N-Terminal cysteine directed cyclization of phage displayed peptide libraries. Three reagents have been developed that take advantage of condensation with N-terminal cysteines to provide selectivity for cyclization of peptide libraries. aBuffer C: 50 mM PBS, pH 7.4. bBuffer D: 10 mM HEPES, 150 mM NaCl, 10 mM MgCl2, 1 mM KCl, pH 7.4.
3.5. Post-translational Modifications to Generate Bicyclic Peptides
Just as the constraints in monocyclic peptides result in improved pharmacokinetics, the generation of bicyclic peptides as either a fused bicycle or double-bridged peptide adds conformational constraints for even more rigidity in comparison to monocyclic peptides. This subsequently results in improved binding affinities and resistance to proteolysis. In this section, we will review methods for producing bicyclic peptide libraries on phage and look into applications that demonstrate these enhanced properties. The Heinis Lab has developed and characterized several different molecules that modify cysteines on phage to produce bicyclic peptides (Figure 11A). All of these are symmetrical electrophiles that contain activated halides for reaction with cysteines through SN2 or Michael Addition reactions. Initially, a trisymmetric derivative, TBMB (Table 4), was used to create bicyclic peptide libraries by reacting with three cysteines displayed on the phage.169−172 While TBMB demonstrated complete reactivity with a peptide–protein fusion after only 1 h reaction at 30 °C, there was a considerable loss in phage infectivity when incubated with more than 10 μM compound. Considering the highly electrophilic nature of benzyl bromides, this is likely due to reactivity with other nucleophilic residues on the phage coat proteins. Despite this toxicity, low nanomolar peptide inhibitors for kallikrein were developed using TBMB and they demonstrated high selectivity for the bicyclic form (Table S1, Peptide 123).169 Following these initial studies, TBMB has proven to be a linker for a variety of bicyclic peptide sizes, ranging from 3 to 6 amino acids between each cysteine, and the cyclization appears to enhance binding activities in comparison to disulfide-cyclized peptides.170,173 One study used TBMB to identify peptides (Table S1, Peptides 124 and 125) with micromolar affinity for uPA, a serine protease upregulated in many tumors.170 In addition to peptides fused to phage, TBMB has also proven to be effective at displaying bicycles on antibody Fc domains, although some side reactivity was observed that resulted in dimerization of the antibody fragments.174 While TBMB is an effective linker for producing bicyclic peptides on proteins, it appears to be less effective when reacting with phages, only generating the desired bicyclic peptide on 10% of phages and showing considerable toxicity to wild-type phages above 10 μM compound.171 Despite this apparently low reactivity, it has still been successfully used in numerous selections to identify bicyclic peptide ligands for proteins.169,175−179 It may be worth exploring additional ways to verify the reactivity on phage, such as with biotin pulldown assays.
Figure 11.

Generation of phage-displayed bicyclic peptides. A) Trisymmetric linkers containing cysteine-reactive electrophiles were initially developed to create bicyclic peptide scaffolds on phage. B) N-terminal serine selective bicyclic linkers have been recently developed to screen bicyclic peptides through a two-step reaction following oxidation of the N-terminal serine with sodium periodate.
Table 4. Reagents to Generate Bicyclic Peptide Scaffolds on Phage.

Wild-type phage.
Disulfide-free phage mutant.
Buffer A: 20 mM ABC, 5 mM EDTA, pH 8.
n.d.: not determined.
n.t.: No observed toxicity.
There have been several campaigns to improve upon the initially developed bicyclic linker TBMB both by modifying the central core and the electrophilic moieties. Bicyclic peptides exhibit different conformations depending upon the central linker used, so the development of additional linkers is expected give access to novel structures of peptides.180 The first variations looked to generate more hydrophilic cores to promote interactions between the peptide libraries and the target proteins, with the core pointing toward the solvent.178 They developed two hydrophilic linkers, 1,3,5-triacryloyl-1,3,5-triazinane (TATA, Table 4) and N,N′,N′-(benzene-1,3,5-triyl)-tris(2-bromoacetamide) (TBAB, Table 4), that contained hydrophilic cores fused to cysteine reactive groups. As expected, the acrylamide moieties in TATA showed considerably slower reactivity than TBMB or TBAB, with reactions needing 40 μM compound to completely cyclize a model pIII fusion protein.178 Additionally, when using acrylamide derivatives to react with cysteine, it is necessary to remove all of the TCEP in solution before cyclization to prevent a Phospha-Michael addition of TCEP.181 Despite having a similar mode of action to TBMB, there was no observed phage toxicity when incubating disulfide-free phages with 160 μM TATA or TBAB.178 Bicyclic peptides isolated from phage selections using the three molecules (TBMB, TATA, and TBAB) exhibited enhanced activity (up to 1000-fold higher) when cyclized with the linker used during the selection, indicating a structural role for the linkers to play in peptide folding and protein interactions.178 To account for this, structural studies demonstrated efficient hydrogen bonding between the linkers and the peptide backbone and/or amino acid residues.178,180 These additional interactions can potentially stabilize peptides in the bicyclic form and promote affinity to desired targets. With this increased potential and lowered phage toxicity, TATA and TBAB appear to be superior organic linkers for generating bicyclic peptides on phage. Nevertheless, there are some limitations to these two compounds, such as the necessity to have symmetrical linker cores and the nonspecific reactivity of the electrophilic regions. In addition to using a single bridge compound to generate bicyclic peptides from phage libraries containing three cysteines, Heinis et al. have also generated bicyclic peptide libraries using peptides containing four cysteines in combination with either two bis-electrophiles or disulfide bonds.170,171 In one interesting application, generation of a double-bridged peptide library using DBAc (Table 3) afforded identification of proteolytically stable peptides (Table S1, Peptides 126 and 127) with low nanomolar affinities for FXIa and IL-23R.171 This could be an exciting avenue to directly screen peptides that show good stability in vivo.
There are numerous applications of these bicyclic peptide linkers in drug discovery campaigns for a wide variety of targets, demonstrating the particular usefulness of these libraries. Peptides bridged using TBMB, TATA, and TBAB have been isolated in panning against β-catenin and have micromolar affinities (Table S1, Peptides 128–131).175 For the potential treatment of thrombotic diseases, a peptide cyclized using TBMB has been identified that binds the FXIIa with micromolar affinity (Table S1, Peptide 132).176 TBMB has also been used to identify possible antibiotic peptides through screening bicyclic peptides to bind Sortase A from S. aureus, with the optimized peptides giving low micromolar affinity (Table S1, Peptides 133–135).177 Studies have investigated cancer targets and identified bicyclic peptides that bind to oncoproteins serine protease uPA, Her2, Nectin-4, type-1 metalloproteases (MT1-MMP), and EphA2 using TBMB, TBAB, and TATA as linkers, with all of the optimized peptides showing nanomolar affinity for their respective targets (Table S1, Peptides 136–143).178,179,182−184 Bicyclic peptides have also been identified using a methylated TBMB (TBMB-Me, Table 4) to bind with high affinity to proteins associated with inflammatory disorders, such as TNFα (Tables S1, Peptide 144).185 These demonstrate the robustness of these bicyclic peptide libraries and their great potential to impact drug discovery efforts. Currently, the peptides identified for Nectin-4, MT1-MMP, and EphA2 are under clinical trials with Bicycle Therapeutics.
There have been multiple strategies to further optimize linkers for displaying bicyclic peptide scaffolds on phage. To avoid toxicities associated with the TBMB linker, a study was performed to optimize the TCEP reduction and coupling conditions that allowed for incubation using 100 μM TBMB, although there was still considerable toxicity of the linker (100-fold less infectivity). Although TBMB is the only compound that exhibited toxicity when incubated with phages, all three linkers blindly react with cysteines through highly activated electrophilic groups. This necessitates that they have trifold symmetry to avoid complications in peptide synthesis, which limits the potential diversity and modifications that can be introduced to the bicyclic peptides. Some strategies to work around these issues involve taking advantage of reactions that differentiate N-terminal amino acids from their internal counterparts. The first examples of this came from the Derda Lab, where an N-terminal serine was converted into glyoxylate by sodium periodate oxidative cleavage and the resulting aldehyde was reacted with an hydroxyl amine containing a dichloromethyl-benzene (TSL-6, Figure 11B, Table 4) for bicyclization with two internal cysteines.172,186 It may also allow for higher specificity in modifying phage cysteines within the library because less-reactive electrophiles can be catalyzed to react through proximity-driven interactions following the oxime ligation. Because of this, rather than being constricted to using disulfide-free pIII, wild-type pIII is compatible with this linker and higher phage yields can be achieved. Using this technique, the reported display of bicyclic peptides was 41% using a biotin pulldown assay, which is considerably higher than the reported 10% (ELISA assay) with TBMB.171,172 Conditions may still need to be optimized to achieve higher efficiencies of bicyclization. In addition to the improved selectivity of the linker, the displayed peptides also exhibit higher serum stability because they lack an N-terminus that can be recognized by proteases, and a recent report demonstrated the ability to generate proteolytically stable peptides for NODAL that inhibited in the micromolar range (Table S1, Peptide 145).172 A recently reported technique for generating bicyclic peptide scaffolds on phage involves using a compound (BC-Osu) with two chlorooximes and one NHS-ester to react with two cysteines and the N-terminus, respectively.187 The reaction exhibited high efficiency, with the desired bicyclic product forming with 66% and 94% yield in phage pIII proteins modified with ACX7C and AX3CX7C peptides on the N-terminus, respectively.187 While these yields are the highest reported for display of bicyclic peptides, BC-Osu is also limited by reacting nonspecifically. Because of this, it would be expected to modify not only the N-terminus of the library region on pIII, but also N-termini, lysines, and cysteines in other phage coat proteins. Thus, there is still a need to develop efficient, highly selective bicyclic linkers to afford the most useful phage-displayed bicyclic peptide libraries.
3.6. Outlook of Chemically Modified Phage Libraries
As discussed above, there have been many different techniques developed to chemically modify phage displayed peptide libraries. These have primarily been employed to generate novel moieties on phage for directing selections and to produce cyclic peptide scaffolds on the phage. The main challenge with modifying phage displayed libraries revolves around generating reagents that selectively modify the libraries rather than other phage coat proteins. We have highlighted several promising strategies on this front that look to selectively modify N-terminal cysteines and serines to direct the modifications to displayed peptide libraries. While these are efficient techniques, they usually show less than 75% labeling on the displayed peptides. However, this is expected since the labeling reactions are significantly affected by the chemical contexts surrounding the N-terminal cysteine and serine on both kinetics and thermodynamics. Given the extremely large library size, some of the N-terminal cysteines and serines might also be buried in the peptide contexts and therefore not accessible for labeling. Recently, site specific modification of the N-terminal glycine has been reported, which could allow for more quantitative production of pIII with an N-terminal glycine.188 As new bioconjugation approaches develop, they should be considered for use in phage displayed libraries.
Although there have been many approaches developed to modify phage libraries, there is still a need to develop standardized methods to directly characterize the modified products on phage. Usually, concentrations of pIII in phage libraries are in the femtomolar to nanomolar range, making them too dilute for normal mass spectrometry techniques. Because of this, indirect methods are used to quantify modifications on phage, such as ELISA and biotin pulldown assays. While these are innovative techniques that provide good insights into the modifications present, they still leave some ambiguity about the quality of the libraries that are displayed and actual modifications present. This ambiguity makes comparisons of reagents difficult and complicates quality control during biopanning experiments. Thus, novel mass spectrometry techniques allowing quantitative analysis of phage-displayed peptides would help immensely advance new labeling and cyclization methods for phage-displayed libraries.
4. Concluding Remarks for Phage-Display of Noncanonical Motifs
The identification of therapeutic peptides from unmodified phage-displayed libraries is hindered because linear peptides have poor pharmacokinetic properties. The introduction of ncAAs and motifs allows for a more simplistic transition of peptide lead compounds into therapeutics, as they can increase the stability and improve binding toward a desired target. In the past two decades, noncanonical peptides have been produced on phage-displayed libraries through two segregated techniques: genetic code expansion and chemical transformations. Both techniques have afforded highly diverse phage-displayed libraries, ranging from linear libraries containing noncanonical moieties for enhanced phage selections to display of cyclic peptide libraries for enhanced pharmacokinetics. To further diversify phage libraries, it may be worthwhile to combine the two fields. For example, introduction of ncAAs through genetic code expansion can give highly specific reactive handles to selectively label phage libraries and to produce specific cyclic peptide motifs that avoid the ambiguity of labeling natural residues with chemical reagents. This would afford highly diverse libraries that are selectively modified, increasing efficiencies of selections and peptide characterization.
While integration of genetic code expansion with chemically modified phage libraries would result in diverse scaffolds for phage-displayed peptides, there are still limitations to consider with phage display. The first of these involves the limitation regarding initial transformation efficiency—plasmids must be transformed into cells, which usually limits the displayed libraries to 1010 peptides. There has been a recent case that generated a library of 1011 peptides by transformation of diverse peptide scaffolds, but this would be the upper limit for most laboratories.189 One way that may get around this limitation would be the integration of phage-displayed peptides into in vivo evolution systems, such as phage-assisted continuous evolution, that simultaneously mutate and select for peptides with desired activities.190 This would provide essentially unlimited peptides available for screening, as higher diversities are generated as the evolution continues. Additionally, genetic code expansion in phage display is limited to only orthogonal translation systems, as the endogenous translation is necessary for producing all the phage proteins. Because of this, incorporation of ncAAs at sense codons is difficult due to competition with the natural amino acids. Adaption of phage display systems to E. coli with synthetic genomes may help to work around this limitation and further diversify the number of amino acids that can be incorporated. Nonetheless, phage display still is one of the most accessible techniques to develop peptide libraries, and the diversification of phage libraries gives opportunities to identify peptide ligands for various therapeutic targets.
Of the peptide display techniques, mRNA display has also demonstrated similar levels of success in identifying therapeutic peptides over the past 20 years. Although mRNA display can give libraries of up to 1015 peptides along with incorporation of highly diverse amino acids through use of flexizymes, there are several advantages in phage display that one should consider when deciding on a peptide display method. First, phages exhibit high stability in a wide variety of conditions (pH, temperature, proteolytic, etc.), which gives researchers the ability to diversify screening conditions.26 This also allows for the ability to perform selections on whole cells, organoids, tumors, tissues, and in vivo.191−194 In addition to enhanced stability, techniques that take advantage of the phage biology, such as the ability to enrich amber codons throughout the selection, can be very useful tools to enhance selections with ncAAs.94 Phage display also may be more accessible to certain research laboratories, as it requires simple molecular biology techniques in comparison to intricate in vitro translation systems. Display of larger proteins, such as nanobodies, may also be more practical using phage display, as the presence of puromycin in mRNA display could cause truncated protein expression.195 As development of peptide therapeutics has increased in the past 20 years, concurrent innovations in both mRNA display and phage display provide researchers with useful tools for de novo peptide drug discovery. It will be exciting to see future developments and applications as both techniques continue to address their current limitations.
Acknowledgments
This work was supported by the National Institutes of Health (Grant R35GM145351) and Welch Foundation (Grant A-1715) to WRL.
Biographies
J. Trae Hampton earned a BS in Biochemistry at the University of Dallas in 2018. During his undergraduate career, he investigated novel synthetic pathways for silylated tetrahydropyrans as a summer researcher under Prof. Asunción Barbero at the University of Valladolid in Valladolid, Spain. At the University of Dallas, he also studied and developed techniques for purification of phospholipase D under supervision of Prof. Scott Boegeman. Following his undergraduate studies, Trae received his PhD in Chemistry at Texas A&M University in 2023 with Prof. Wenshe Liu as his mentor. During his doctoral studies, he developed novel techniques to enhance phage display of noncanonical amino acids and macrocyclic peptides. Currently, he serves as a postdoctoral fellow at Texas A&M, where he manages the Texas A&M Drug Discovery Center. He continues research on adapting biological methods to generate novel therapeutic techniques.
Prof. Wenshe Ray Liu attained his BS in Chemistry at Peking University in 2000, then proceeded to receive a PhD in Biological Chemistry in 2005 at UC Davis under supervision of Prof. Michael Toney where he investigated enzyme mechanisms of aminotransferases and decarboxylases through X-ray crystallography. He then was a postdoctoral fellow at The Scripps Research Institute under Prof. Peter Schultz and developed new amber suppression techniques for genetic code expansion in bacterial and mammalian cells. Following his postdoctoral studies, he became a professor in the Chemistry Department at Texas A&M University in 2007, where he continued research on genetic code expansion using the pyrrolysine tRNA synthetase and in the development of novel chemical biology probes. In the past nine years, he has shifted his research to focus on drug discovery of cancer and COVID-19 therapeutics using phage display and traditional small molecule approaches. Currently, he is the Harry E. Bovay Chair in the Chemistry Department at Texas A&M and serves as the Director of the Texas A&M Drug Discovery Center. He holds additional appointments in the Departments of Biochemistry and Biophysics, Cell Biology and Genetics, Translational Medical Sciences, and Pharmaceutical Sciences.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrev.4c00004.
Supplementary Table S1 includes the sequences and potencies of top selected peptides in works related to this review (PDF)
Author Contributions
CRediT: Joshua Trae Hampton formal analysis, writing-original draft, writing-review & editing; Wenshe Ray Liu conceptualization, funding acquisition, writing-original draft, writing-review & editing.
The authors declare no competing financial interest.
Special Issue
Published as part of Chemical Reviewsvirtual special issue “Noncanonical Amino Acids”.
Supplementary Material
References
- Petta I.; Lievens S.; Libert C.; Tavernier J.; De Bosscher K. Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol. Ther. 2016, 24, 707–718. 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wang N.; Zhang W.; Cheng X.; Yan Z.; Shao G.; Wang X.; Wang R.; Fu C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 1–27. 10.1038/s41392-022-00904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Vigneaud V.; Ressler C.; Trippett S. THE SEQUENCE OF AMINO ACIDS IN OXYTOCIN, WITH A PROPOSAL FOR THE STRUCTURE OF OXYTOCIN. J. Biol. Chem. 1953, 205, 949–957. 10.1016/S0021-9258(18)49238-1. [DOI] [PubMed] [Google Scholar]
- du Vigneaud V.; Ressler C.; Swan J. M.; Roberts C. W.; Katsoyannis P. G. The Synthesis of Oxytocin. J. Am. Chem. Soc. 1954, 76, 3115–3121. 10.1021/ja01641a004. [DOI] [Google Scholar]
- du Vigneaud V.; Gish D. T.; Katsoyannis P. G.; Hess G. P. Synthesis of the Pressor-Antidiuretic Hormone, Arginine-Vasopressin. J. Am. Chem. Soc. 1958, 80, 3355–3358. 10.1021/ja01546a040. [DOI] [Google Scholar]
- Weckbecker G.; Lewis I.; Albert R.; Schmid H. A.; Hoyer D.; Bruns C. Opportunities in Somatostatin Research: Biological, Chemical and Therapeutic Aspects. Nat. Rev. Drug Discovery 2003, 2, 999–1017. 10.1038/nrd1255. [DOI] [PubMed] [Google Scholar]
- Mathieu C.; Martens P.-J.; Vangoitsenhoven R. One Hundred Years of Insulin Therapy. Nat. Rev. Endocrinol. 2021, 17, 715–725. 10.1038/s41574-021-00542-w. [DOI] [PubMed] [Google Scholar]
- Muttenthaler M.; King G. F.; Adams D. J.; Alewood P. F. Trends in Peptide Drug Discovery. Nat. Rev. Drug Discovery 2021, 20, 309–325. 10.1038/s41573-020-00135-8. [DOI] [PubMed] [Google Scholar]
- Knudsen L. B.; Lau J.. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10 10.3389/fendo.2019.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau J.; Bloch P.; Schäffer L.; Pettersson I.; Spetzler J.; Kofoed J.; Madsen K.; Knudsen L. B.; McGuire J.; Steensgaard D. B.; Strauss H. M.; et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J. Med. Chem. 2015, 58, 7370–7380. 10.1021/acs.jmedchem.5b00726. [DOI] [PubMed] [Google Scholar]
- GLP-1 Agonists Market Development Scope & Potential: 2023–2028; ReportLinker, https://www.reportlinker.com/p06483826/Glucagon-Like-Peptide-1-GLP-1-Agonists-Market-Size-Share-Analysis-Growth-Trends-Forecasts.html (accessed 2024-01-01). [Google Scholar]
- Wang J.-Y.; Wang Q.-W.; Yang X.-Y.; Yang W.; Li D.-R.; Jin J.-Y.; Zhang H.-C.; Zhang X.-F. GLP-1 Receptor Agonists for the Treatment of Obesity: Role as a Promising Approach. Front. Endocrinol. 2023, 14, 1085799. 10.3389/fendo.2023.1085799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henninot A.; Collins J. C.; Nuss J. M. The Current State of Peptide Drug Discovery: Back to the Future?. J. Med. Chem. 2018, 61, 1382–1414. 10.1021/acs.jmedchem.7b00318. [DOI] [PubMed] [Google Scholar]
- Yeo X. Y.; Cunliffe G.; Ho R. C.; Lee S. S.; Jung S. Potentials of Neuropeptides as Therapeutic Agents for Neurological Diseases. Biomedicines 2022, 10, 343. 10.3390/biomedicines10020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport A. P.; Scully C. C. G.; de Graaf C.; Brown A. J. H.; Maguire J. J. Advances in Therapeutic Peptides Targeting G Protein-Coupled Receptors. Nat. Rev. Drug Discovery 2020, 19, 389–413. 10.1038/s41573-020-0062-z. [DOI] [PubMed] [Google Scholar]
- Huan Y.; Kong Q.; Mou H.; Yi H.. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11. 10.3389/fmicb.2020.582779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furka A. Forty Years of Combinatorial Technology. Drug Discovery Today 2022, 27, 103308. 10.1016/j.drudis.2022.06.008. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Wiedmann M. M.; Suga H. RNA Display Methods for the Discovery of Bioactive Macrocycles. Chem. Rev. 2019, 119, 10360–10391. 10.1021/acs.chemrev.8b00430. [DOI] [PubMed] [Google Scholar]
- Tavassoli A. SICLOPPS Cyclic Peptide Libraries in Drug Discovery. Curr. Opin. Chem. Biol. 2017, 38, 30–35. 10.1016/j.cbpa.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Jaroszewicz W.; Morcinek-Orłowska J.; Pierzynowska K.; Gaffke L.; Wȩgrzyn G. Phage Display and Other Peptide Display Technologies. FEMS Microbiol. Rev. 2022, 46, fuab052. 10.1093/femsre/fuab052. [DOI] [PubMed] [Google Scholar]
- Passaretti P.; Khan I.; Dafforn T. R.; Goldberg Oppenheimer P. Improvements in the Production of Purified M13 Bacteriophage Bio-Nanoparticle. Sci. Rep. 2020, 10, 18538. 10.1038/s41598-020-75205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakonjac J.; Bennett N. J.; Spagnuolo J.; Gagic D.; Russel M. Filamentous Bacteriophage: Biology, Phage Display and Nanotechnology Applications. Curr. Issues Mol. Biol. 2011, 13, 51–76. [PubMed] [Google Scholar]
- Russel M.; Linderoth N. A.; Šali A. Filamentous Phage Assembly: Variation on a Protein Export theme1Presented at the Workshop on `Type-4 Pili - Biogenesis, Adhesins, Protein Export, and DNA Import’, Schloss Ringberg, Germany, 26–29 November 1995.1. Gene 1997, 192, 23–32. 10.1016/S0378-1119(96)00801-3. [DOI] [PubMed] [Google Scholar]
- Clokie M. R.; Millard A. D.; Letarov A. V.; Heaphy S. Phages in Nature. Bacteriophage 2011, 1, 31–45. 10.4161/bact.1.1.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. P.; Petrenko V. A. Phage Display. Chem. Rev. 1997, 97, 391–410. 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- Branston S. D.; Stanley E. C.; Ward J. M.; Keshavarz-Moore E. Determination of the Survival of Bacteriophage M13 from Chemical and Physical Challenges to Assist in Its Sustainable Bioprocessing. Biotechnol. Bioprocess Eng. 2013, 18, 560–566. 10.1007/s12257-012-0776-9. [DOI] [Google Scholar]
- Ledsgaard L.; Kilstrup M.; Karatt-Vellatt A.; McCafferty J.; Laustsen A. H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. 10.3390/toxins10060236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin D. A.; Welsh L. C.; Symmons M. F.; Scott W. R. P.; Straus S. K. Molecular Structure of Fd (F1, M13) Filamentous Bacteriophage Refined with Respect to X-Ray Fibre Diffraction and Solid-State NMR Data Supports Specific Models of Phage Assembly at the Bacterial Membrane. J. Mol. Biol. 2006, 355, 294–309. 10.1016/j.jmb.2005.10.048. [DOI] [PubMed] [Google Scholar]
- Caspar D. L. D.; Makowski L. The Symmetries of Filamentous Phage Particles. J. Mol. Biol. 1981, 145, 611–617. 10.1016/0022-2836(81)90549-0. [DOI] [PubMed] [Google Scholar]
- Endemann H.; Model P. Lcoation of Filamentous Phage Minor Coat Proteins in Phage and in Infected Cells. J. Mol. Biol. 1995, 250, 496–506. 10.1006/jmbi.1995.0393. [DOI] [PubMed] [Google Scholar]
- Russel M.; Model P. Genetic Analysis of the Filamentous Bacteriophage Packaging Signal and of the Proteins That Interact with It. J. Virol. 1989, 63, 3284. 10.1128/jvi.63.8.3284-3295.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliger P.; Riechmann L.; Williams R. L. Crystal Structure of the Two N-Terminal Domains of G3p from Filamentous Phage Fd at 1.9 Å: Evidence for Conformational lability11Edited by J. M. Thornton. J. Mol. Biol. 1999, 288, 649–657. 10.1006/jmbi.1999.2720. [DOI] [PubMed] [Google Scholar]
- Lubkowski J.; Hennecke F.; Plückthun A.; Wlodawer A. The Structural Basis of Phage Display Elucidated by the Crystal Structure of the N-Terminal Domains of G3p. Nat. Struct. Biol. 1998, 5, 140–147. 10.1038/nsb0298-140. [DOI] [PubMed] [Google Scholar]
- Conners R.; León-Quezada R. I.; McLaren M.; Bennett N. J.; Daum B.; Rakonjac J.; Gold V. A. M. Cryo-Electron Microscopy of the F1 Filamentous Phage Reveals Insights into Viral Infection and Assembly. Nat. Commun. 2023, 14, 2724. 10.1038/s41467-023-37915-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis N. G.; Model P. An Artificial Anchor Domain: Hydrophobicity Suffices to Stop Transfer. Cell 1985, 41, 607–614. 10.1016/S0092-8674(85)80033-7. [DOI] [PubMed] [Google Scholar]
- Greenwood J.; Willis A. E.; Perham R. N. Multiple Display of Foreign Peptides on a Filamentous Bacteriophage: Peptides from Plasmodium Falciparum Circumsporozoite Protein as Antigens. J. Mol. Biol. 1991, 220, 821–827. 10.1016/0022-2836(91)90354-9. [DOI] [PubMed] [Google Scholar]
- Smith G. P. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Parmley S. F.; Smith G. P. Antibody-Selectable Filamentous Fd Phage Vectors: Affinity Purification of Target Genes. Gene 1988, 73, 305–318. 10.1016/0378-1119(88)90495-7. [DOI] [PubMed] [Google Scholar]
- Baek H.; Suk K.; Kim Y.; Cha S. An Improved Helper Phage System for Efficient Isolation of Specific Antibody Molecules in Phage Display. Nucleic Acids Res. 2002, 30, e18 10.1093/nar/30.5.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J. H.; Moosmeier M. A.; Aumuller T.; Thein M.; Bosma T.; Rink R.; Groth K.; Zulley M.; Siegers K.; Tissot K.; Moll G. N.; Prassler J. Phage Display and Selection of Lanthipeptides on the Carboxy-Terminus of the Gene-3 minor Coat Protein. Nat. Commun. 2017, 8, 1500. 10.1038/s41467-017-01413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matochko W. L.; Cory Li S.; Tang S. K. Y.; Derda R. Prospective Identification of Parasitic Sequences in Phage Display Screens. Nucleic Acids Res. 2014, 42, 1784–1798. 10.1093/nar/gkt1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brammer L. A.; Bolduc B.; Kass J. L.; Felice K. M.; Noren C. J.; Hall M. F. A Target-Unrelated Peptide in an M13 Phage Display Library Traced to an Advantageous Mutation in the Gene II Ribosome-Binding Site. Anal. Biochem. 2008, 373, 88–98. 10.1016/j.ab.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Enea V.; Zinder N. D. Interference Resistant Mutants of Phage F1. Virology 1982, 122, 222–226. 10.1016/0042-6822(82)90395-6. [DOI] [PubMed] [Google Scholar]
- Vieira J.; Messing J.. Production of Single-Stranded Plasmid DNA. In Methods in Enzymology; Recombinant DNA Part D; Academic Press, 1987; Vol. 153, pp 3–11. [DOI] [PubMed] [Google Scholar]
- Qi H.; Lu H.; Qiu H.-J.; Petrenko V.; Liu A. Phagemid Vectors for Phage Display: Properties, Characteristics and Construction. J. Mol. Biol. 2012, 417, 129–143. 10.1016/j.jmb.2012.01.038. [DOI] [PubMed] [Google Scholar]
- Rondot S.; Koch J.; Breitling F.; Dübel S. A Helper Phage to Improve Single-Chain Antibody Presentation in Phage Display. Nat. Biotechnol. 2001, 19, 75–78. 10.1038/83567. [DOI] [PubMed] [Google Scholar]
- Rakonjac J.; Jovanovic G.; Model P. Filamentous Phage Infection-Mediated Gene Expression: Construction and Propagation of the gIII Deletion Mutant Helper Phage R408d3. Gene 1997, 198, 99–103. 10.1016/S0378-1119(97)00298-9. [DOI] [PubMed] [Google Scholar]
- Dueñas M.; Borrebaeck C. A. K. Novel Helper Phage Design: Intergenic Region Affects the Assembly of Bacteriophages and the Size of Antibody Libraries. FEMS Microbiol. Lett. 1995, 125, 317–321. 10.1016/0378-1097(94)00517-U. [DOI] [PubMed] [Google Scholar]
- Smith G. P. Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture). Angew. Chem., Int. Ed. 2019, 58, 14428–14437. 10.1002/anie.201908308. [DOI] [PubMed] [Google Scholar]
- Winter G. Harnessing Evolution to Make Medicines (Nobel Lecture). Angew. Chem., Int. Ed. 2019, 58, 14438–14445. 10.1002/anie.201909343. [DOI] [PubMed] [Google Scholar]
- Chin J. W. Expanding and Reprogramming the Genetic Code. Nature 2017, 550, 53–60. 10.1038/nature24031. [DOI] [PubMed] [Google Scholar]
- de la Torre D.; Chin J. W. Reprogramming the Genetic Code. Nat. Rev. Genet. 2021, 22, 169–184. 10.1038/s41576-020-00307-7. [DOI] [PubMed] [Google Scholar]
- Wan W.; Tharp J. M.; Liu W. R. Pyrrolysyl-tRNA Synthetase: An Ordinary Enzyme but an Outstanding Genetic Code Expansion Tool. Biochim. Biophys. Acta BBA - Proteins Proteomics 2014, 1844, 1059–1070. 10.1016/j.bbapap.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X.; Zhang H.; Shen Y.; Fu X. Update of the Pyrrolysyl-tRNA Synthetase/tRNAPyl Pair and Derivatives for Genetic Code Expansion. J. Bacteriol. 2023, 205, e00385-22 10.1128/jb.00385-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrabi C.; Foster A.; Tavassoli A. Methods for Generating and Screening Libraries of Genetically Encoded Cyclic Peptides in Drug Discovery. Nat. Rev. Chem. 2020, 4, 90–101. 10.1038/s41570-019-0159-2. [DOI] [PubMed] [Google Scholar]
- Iskandar S. E.; Haberman V. A.; Bowers A. A. Expanding the Chemical Diversity of Genetically Encoded Libraries. ACS Comb. Sci. 2020, 22, 712–733. 10.1021/acscombsci.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyle K.; Kong X.-D.; Heinis C. Phage Selection of Cyclic Peptides for Application in Research and Drug Development. Acc. Chem. Res. 2017, 50, 1866–1874. 10.1021/acs.accounts.7b00184. [DOI] [PubMed] [Google Scholar]
- Vinogradov A. A.; Yin Y.; Suga H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. 10.1021/jacs.8b13178. [DOI] [PubMed] [Google Scholar]
- Sharma K.; Sharma K. K.; Sharma A.; Jain R. Peptide-Based Drug Discovery: Current Status and Recent Advances. Drug Discovery Today 2023, 28, 103464. 10.1016/j.drudis.2022.103464. [DOI] [PubMed] [Google Scholar]
- Noren C. J.; Anthony-Cahill S. J.; Griffith M. C.; Schultz P. G. A General Method for Site-Specific Incorporation of Unnatural Amino Acids into Proteins. Science 1989, 244, 182–188. 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- Murakami H.; Ohta A.; Ashigai H.; Suga H. A Highly Flexible tRNA Acylation Method for Non-Natural Polypeptide Synthesis. Nat. Methods 2006, 3, 357–359. 10.1038/nmeth877. [DOI] [PubMed] [Google Scholar]
- Wang L.; Brock A.; Herberich B.; Schultz P. G. Expanding the Genetic Code of Escherichia Coli. Science 2001, 292, 498–500. 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- Anderson J. C.; Wu N.; Santoro S. W.; Lakshman V.; King D. S.; Schultz P. G. An Expanded Genetic Code with a Functional Quadruplet Codon. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7566–7571. 10.1073/pnas.0401517101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italia J. S.; Addy P. S.; Wrobel C. J. J.; Crawford L. A.; Lajoie M. J.; Zheng Y.; Chatterjee A. An Orthogonalized Platform for Genetic Code Expansion in Both Bacteria and Eukaryotes. Nat. Chem. Biol. 2017, 13 (4), 446–450. 10.1038/nchembio.2312. [DOI] [PubMed] [Google Scholar]
- Hao B.; Gong W.; Ferguson T. K.; James C. M.; Krzycki J. A.; Chan M. K. A New UAG-Encoded Residue in the Structure of a Methanogen Methyltransferase. Science 2002, 296, 1462–1466. 10.1126/science.1069556. [DOI] [PubMed] [Google Scholar]
- Burke S. A.; Lo S. L.; Krzycki J. A. Clustered Genes Encoding the Methyltransferases of Methanogenesis from Monomethylamine. J. Bacteriol. 1998, 180, 3432–3440. 10.1128/JB.180.13.3432-3440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul L.; Ferguson D. J.; Krzycki J. A. The Trimethylamine Methyltransferase Gene and Multiple Dimethylamine Methyltransferase Genes of Methanosarcina Barkeri Contain In-Frame and Read-Through Amber Codons. J. Bacteriol. 2000, 182, 2520–2529. 10.1128/JB.182.9.2520-2529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan G.; James C. M.; Krzycki J. A. Pyrrolysine Encoded by UAG in Archaea: Charging of a UAG-Decoding Specialized tRNA. Science 2002, 296, 1459–1462. 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- Polycarpo C.; Ambrogelly A.; Bérubé A.; Winbush S. M.; McCloskey J. A.; Crain P. F.; Wood J. L.; Söll D. An Aminoacyl-tRNA Synthetase That Specifically Activates Pyrrolysine. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12450–12454. 10.1073/pnas.0405362101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polycarpo C. R.; Herring S.; Bérubé A.; Wood J. L.; Söll D.; Ambrogelly A. Pyrrolysine Analogues as Substrates for Pyrrolysyl-tRNA Synthetase. FEBS Lett. 2006, 580, 6695–6700. 10.1016/j.febslet.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T.; Kobayashi T.; Hino N.; Yanagisawa T.; Sakamoto K.; Yokoyama S. Adding L-Lysine Derivatives to the Genetic Code of Mammalian Cells with Engineered Pyrrolysyl-tRNA Synthetases. Biochem. Biophys. Res. Commun. 2008, 371, 818–822. 10.1016/j.bbrc.2008.04.164. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T.; Ishii R.; Fukunaga R.; Kobayashi T.; Sakamoto K.; Yokoyama S. Multistep Engineering of Pyrrolysyl-tRNA Synthetase to Genetically Encode Nε-(o-Azidobenzyloxycarbonyl) Lysine for Site-Specific Protein Modification. Chem. Biol. 2008, 15, 1187–1197. 10.1016/j.chembiol.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Yanagisawa T.; Sakamoto K.; Yokoyama S. Recognition of Non-α-Amino Substrates by Pyrrolysyl-tRNA Synthetase. J. Mol. Biol. 2009, 385, 1352–1360. 10.1016/j.jmb.2008.11.059. [DOI] [PubMed] [Google Scholar]
- Li Y.-M.; Yang M.-Y.; Huang Y.-C.; Li Y.-T.; Chen P. R.; Liu L. Ligation of Expressed Protein α-Hydrazides via Genetic Incorporation of an α-Hydroxy Acid. ACS Chem. Biol. 2012, 7, 1015–1022. 10.1021/cb300020s. [DOI] [PubMed] [Google Scholar]
- Santoro S. W.; Wang L.; Herberich B.; King D. S.; Schultz P. G. An Efficient System for the Evolution of Aminoacyl-tRNA Synthetase Specificity. Nat. Biotechnol. 2002, 20, 1044–1048. 10.1038/nbt742. [DOI] [PubMed] [Google Scholar]
- Bryson D. I.; Fan C.; Guo L.-T.; Miller C.; Söll D.; Liu D. R. Continuous Directed Evolution of Aminoacyl-tRNA Synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. 10.1038/nchembio.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z.; Forster A. C.; Blacklow S. C.; Cornish V. W. Amino Acid Backbone Specificity of the Escherichia Coli Translation Machinery. J. Am. Chem. Soc. 2004, 126, 12752–12753. 10.1021/ja0472174. [DOI] [PubMed] [Google Scholar]
- Pavlov M. Y.; Watts R. E.; Tan Z.; Cornish V. W.; Ehrenberg M.; Forster A. C. Slow Peptide Bond Formation by Proline and Other N-Alkylamino Acids in Translation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 50–54. 10.1073/pnas.0809211106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale T.; Sanderson L. E.; Uhlenbeck O. C. The Affinity of Elongation Factor Tu for an Aminoacyl-tRNA Is Modulated by the Esterified Amino Acid. Biochemistry 2004, 43, 6159–6166. 10.1021/bi036290o. [DOI] [PubMed] [Google Scholar]
- Park H.-S.; Hohn M. J.; Umehara T.; Guo L.-T.; Osborne E. M.; Benner J.; Noren C. J.; Rinehart J.; Söll D. Expanding the Genetic Code of Escherichia Coli with Phosphoserine. Science 2011, 333, 1151–1154. 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandman K. E.; Benner J. S.; Noren C. J. Phage Display of Selenopeptides. J. Am. Chem. Soc. 2000, 122, 960–961. 10.1021/ja992462m. [DOI] [Google Scholar]
- Tian F.; Tsao M.-L.; Schultz P. G. A Phage Display System with Unnatural Amino Acids. J. Am. Chem. Soc. 2004, 126, 15962–15963. 10.1021/ja045673m. [DOI] [PubMed] [Google Scholar]
- Chin J. W.; Santoro S. W.; Martin A. B.; King D. S.; Wang L.; Schultz P. G. Addition of P-Azido-l-Phenylalanine to the Genetic Code of Escherichia Coli. J. Am. Chem. Soc. 2002, 124, 9026–9027. 10.1021/ja027007w. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhang Z.; Brock A.; Schultz P. G. Addition of the Keto Functional Group to the Genetic Code of Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 56–61. 10.1073/pnas.0234824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. W.; Martin A. B.; King D. S.; Wang L.; Schultz P. G. Addition of a Photocrosslinking Amino Acid to the Genetic Code of Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11020–11024. 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Brock A.; Schultz P. G. Adding L-3-(2-Naphthyl)Alanine to the Genetic Code of E. Coli. J. Am. Chem. Soc. 2002, 124, 1836–1837. 10.1021/ja012307j. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Mack A. V.; Tsao M.-L.; Mills J. H.; Lee H. S.; Choe H.; Farzan M.; Schultz P. G.; Smider V. V. Protein Evolution with an Expanded Genetic Code. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17688–17693. 10.1073/pnas.0809543105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. Recombinant Expression of Selectively Sulfated Proteins in Escherichia Coli. Nat. Biotechnol. 2006, 24, 1436–1440. 10.1038/nbt1254. [DOI] [PubMed] [Google Scholar]
- Liu C. C.; Mack A. V.; Brustad E. M.; Mills J. H.; Groff D.; Smider V. V.; Schultz P. G. Evolution of Proteins with Genetically Encoded “Chemical Warheads.. J. Am. Chem. Soc. 2009, 131, 9616–9617. 10.1021/ja902985e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustad E.; Bushey M. L.; Lee J. W.; Groff D.; Liu W.; Schultz P. G. A Genetically Encoded Boronate-Containing Amino Acid. Angew. Chem., Int. Ed. 2008, 47, 8220–8223. 10.1002/anie.200803240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Liu W.; Schultz P. G. A Genetically Encoded Bidentate, Metal-Binding Amino Acid. Angew. Chem. 2007, 119, 9399–9402. 10.1002/ange.200703397. [DOI] [PubMed] [Google Scholar]
- Day J. W.; Kim C. H.; Smider V. V.; Schultz P. G. Identification of Metal Ion Binding Peptides Containing Unnatural Amino Acids by Phage Display. Bioorg. Med. Chem. Lett. 2013, 23, 2598–2600. 10.1016/j.bmcl.2013.02.106. [DOI] [PubMed] [Google Scholar]
- Kang M.; Light K.; Ai H.; Shen W.; Kim C. H.; Chen P. R.; Lee H. S.; Solomon E. I.; Schultz P. G. Evolution of Iron(II)-Finger Peptides by Using a Bipyridyl Amino Acid. ChemBioChem. 2014, 15, 822–825. 10.1002/cbic.201300727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharp J. M.; Hampton J. T.; Reed C. A.; Ehnbom A.; Chen P.-H. C.; Morse J. S.; Kurra Y.; Pérez L. M.; Xu S.; Liu W. R. An Amber Obligate Active Site-Directed Ligand Evolution Technique for Phage Display. Nat. Commun. 2020, 11, 1392. 10.1038/s41467-020-15057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke J. D.; Model P.; Zinder N. D. Effects of Bacteriophage F1 Gene III Protein on the Host Cell Membrane. Mol. Gen. Genet. MGG 1982, 186, 185–192. 10.1007/BF00331849. [DOI] [PubMed] [Google Scholar]
- Click E. M.; Webster R. E. Filamentous Phage Infection: Required Interactions with the TolA Protein. J. Bacteriol. 1997, 179, 6464–6471. 10.1128/jb.179.20.6464-6471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-S.; Fang X.; Chen H.-Y.; Wu B.; Wang Z. U.; Hilty C.; Liu W. R. Genetic Incorporation of Twelve Meta-Substituted Phenylalanine Derivatives Using a Single Pyrrolysyl-tRNA Synthetase Mutant. ACS Chem. Biol. 2013, 8, 405–415. 10.1021/cb300512r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.-S.; Fang X.; Wallace A. L.; Wu B.; Liu W. R. A Rationally Designed Pyrrolysyl-tRNA Synthetase Mutant with a Broad Substrate Spectrum. J. Am. Chem. Soc. 2012, 134, 2950–2953. 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.-J. C.; Wu B.; Raymond J. E.; Zeng Y.; Fang X.; Wooley K. L.; Liu W. R. A Genetically Encoded Acrylamide Functionality. ACS Chem. Biol. 2013, 8, 1664. 10.1021/cb400267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse J. S.; Sheng Y. J.; Hampton J. T.; Sylvain L. D.; Das S.; Alugubelli Y. R.; Chen P.-H. C.; Yang K. S.; Xu S.; Fierke C. A.; Liu W. R. Phage-Assisted, Active Site-Directed Ligand Evolution of a Potent and Selective Histone Deacetylase 8 Inhibitor. Protein Sci. 2022, 31, e4512 10.1002/pro.4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.-H. C.; Guo X. S.; Zhang H. E.; Dubey G. K.; Geng Z. Z.; Fierke C. A.; Xu S.; Hampton J. T.; Liu W. R.. Leveraging a Phage-Encoded Noncanonical Amino Acid: A Novel Pathway to Potent and Selective Epigenetic Reader Protein Inhibitors. ACS Cent. Sci. 2024. Article ASAP. (accessed 2024-04-03). 10.1021/acscentsci.3c01419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umehara T.; Kim J.; Lee S.; Guo L.-T.; Söll D.; Park H.-S. N-Acetyl Lysyl-tRNA Synthetases Evolved by a CcdB-Based Selection Possess N-Acetyl Lysine Specificity in Vitro and in Vivo. FEBS Lett. 2012, 586, 729–733. 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- Hampton J. T.; Cho C.-C. D.; Coleman D. D.; Geng Z. Z.; Chen P.-H. C.; Dubey G. K.; Sylvain L. D.; Xu S.; Liu W. R. An Amber-Encoding Helper Phage for More Efficient Phage Display of Noncanonical Amino Acids. Nucleic Acids Res. 2023, 51, 6566–6577. 10.1093/nar/gkad488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison C. Constrained Peptides’ Time to Shine?. Nat. Rev. Drug Discovery 2018, 17, 531–533. 10.1038/nrd.2018.125. [DOI] [PubMed] [Google Scholar]
- Wang X. S.; Chen P.-H. C.; Hampton J. T.; Tharp J. M.; Reed C. A.; Das S. K.; Wang D.-S.; Hayatshahi H. S.; Shen Y.; Liu J.; Liu W. R. A Genetically Encoded, Phage-Displayed Cyclic-Peptide Library. Angew. Chem., Int. Ed. 2019, 58, 15904–15909. 10.1002/anie.201908713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens A. E.; Iannuzzelli J. A.; Gu Y.; Fasan R. MOrPH-PhD: An Integrated Phage Display Platform for the Discovery of Functional Genetically Encoded Peptide Macrocycles. ACS Cent. Sci. 2020, 6, 368–381. 10.1021/acscentsci.9b00927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bionda N.; Cryan A. L.; Fasan R. Bioinspired Strategy for the Ribosomal Synthesis of Thioether-Bridged Macrocyclic Peptides in Bacteria. ACS Chem. Biol. 2014, 9, 2008–2013. 10.1021/cb500311k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R.; Krzycki J. A. PylSn and the Homologous N-Terminal Domain of Pyrrolysyl-tRNA Synthetase Bind the tRNA That Is Essential for the Genetic Encoding of Pyrrolysine. J. Biol. Chem. 2012, 287, 32738–32746. 10.1074/jbc.M112.396754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Zeng Y.; Wang W. W.; Qiao Y.; Kurra Y.; Liu W. R. Evolving the N-Terminal Domain of Pyrrolysyl-tRNA Synthetase for Improved Incorporation of Noncanonical Amino Acids. ChemBioChem. 2018, 19, 26–30. 10.1002/cbic.201700268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho C.-C.; Blankenship L. R.; Ma X.; Xu S.; Liu W. The Pyrrolysyl-tRNA Synthetase Activity Can. Be Improved by a P188 Mutation That Stabilizes the Full-Length Enzyme. J. Mol. Biol. 2022, 434, 167453. 10.1016/j.jmb.2022.167453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W.; Zhao H.; Chen Y.; Zhang B.; Yang Y.; Zang J.; Wu J.; Lin S. Chimeric Design of Pyrrolysyl-tRNA Synthetase/tRNA Pairs and Canonical Synthetase/tRNA Pairs for Genetic Code Expansion. Nat. Commun. 2020, 11, 3154. 10.1038/s41467-020-16898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis J. C. W.; Chin J. W. Mutually Orthogonal Pyrrolysyl-tRNA Synthetase/tRNA Pairs. Nat. Chem. 2018, 10, 831–837. 10.1038/s41557-018-0052-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J. T.; Söll D.; Tharp J. M.. Directed Evolution of Methanomethylophilus Alvus Pyrrolysyl-tRNA Synthetase Generates a Hyperactive and Highly Selective Variant. Front. Mol. Biosci. 2022, 9. 10.3389/fmolb.2022.850613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke R.; Swenson C. V.; Roe L. T.; Hamlish N. X.; Shah B.; Zhang Z.; Ficaretta E.; Ad O.; Smaga S.; Gee C. L.; et al. Expanding the Substrate Scope of Pyrrolysyl-Transfer RNA Synthetase Enzymes to Include Non-α-Amino Acids in Vitro and in Vivo. Nat. Chem. 2023, 15, 960–971. 10.1038/s41557-023-01224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkelmann D. L.; Piedrafita C.; Dickson A.; Liu K. C.; Elliott T. S.; Fiedler M.; Bellini D.; Zhou A.; Cervettini D.; Chin J. W. Adding α,α-Disubstituted and β-Linked Monomers to the Genetic Code of an Organism. Nature 2024, 625, 603–610. 10.1038/s41586-023-06897-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W.; Huang Y.; Wang Z.; Russell W. K.; Pai P.-J.; Russell D. H.; Liu W. R. A Facile System for Genetic Incorporation of Two Different Noncanonical Amino Acids into One Protein in Escherichia Coli. Angew. Chem., Int. Ed. 2010, 49, 3211–3214. 10.1002/anie.201000465. [DOI] [PubMed] [Google Scholar]
- Neumann H.; Wang K.; Davis L.; Garcia-Alai M.; Chin J. W. Encoding Multiple Unnatural Amino Acids via Evolution of a Quadruplet-Decoding Ribosome. Nature 2010, 464, 441–444. 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- Oller-Salvia B.; Chin J. W. Efficient Phage Display with Multiple Distinct Non-Canonical Amino Acids Using Orthogonal Ribosome-Mediated Genetic Code Expansion. Angew. Chem., Int. Ed. 2019, 58, 10844–10848. 10.1002/anie.201902658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkelmann D. L.; Willis J. C. W.; Beattie A. T.; Chin J. W. Engineered Triply Orthogonal Pyrrolysyl-tRNA Synthetase/tRNA Pairs Enable the Genetic Encoding of Three Distinct Non-Canonical Amino Acids. Nat. Chem. 2020, 12, 535–544. 10.1038/s41557-020-0472-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinck M.; Piedrafita C.; Robertson W. E.; Elliott T. S.; Cervettini D.; de la Torre D.; Chin J. W. Genetically Programmed Cell-Based Synthesis of Non-Natural Peptide and Depsipeptide Macrocycles. Nat. Chem. 2023, 15, 61–69. 10.1038/s41557-022-01082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredens J.; Wang K.; de la Torre D.; Funke L. F. H.; Robertson W. E.; Christova Y.; Chia T.; Schmied W. H.; Dunkelmann D. L.; Beránek V.; Uttamapinant C.; Llamazares A. G.; Elliott T. S.; Chin J. W. Total Synthesis of Escherichia Coli with a Recoded Genome. Nature 2019, 569, 514–518. 10.1038/s41586-019-1192-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaye D. L.; Geigerman C. M.; Fuller R. E.; Akyildiz A.; Parkos C. A. Direct Fluorochrome Labeling of Phage Display Library Clones for Studying Binding Specificities: Applications in Flow Cytometry and Fluorescence Microscopy. J. Immunol. Methods 2004, 295, 119–127. 10.1016/j.jim.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Sojitra M.; Sarkar S.; Maghera J.; Rodrigues E.; Carpenter E. J.; Seth S.; Ferrer Vinals D.; Bennett N. J.; Reddy R.; Khalil A.; Xue X.; et al. Genetically Encoded Multivalent Liquid Glycan Array Displayed on M13 Bacteriophage. Nat. Chem. Biol. 2021, 17, 806–816. 10.1038/s41589-021-00788-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-L.; Sojitra M.; Carpenter E. J.; Hayhoe E. S.; Sarkar S.; Volker E. A.; Wang C.; Bui D. T.; Yang L.; Klassen J. S.; Wu P.; Macauley M. S.; Lowary T. L.; Derda R. Chemoenzymatic Synthesis of Genetically-Encoded Multivalent Liquid N-Glycan Arrays. Nat. Commun. 2023, 14, 5237. 10.1038/s41467-023-40900-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer M. A.; Lu W.; Dwyer J. J.; Kossiakoff A. A. Biosynthetic Phage Display: A Novel Protein Engineering Tool Combining Chemical and Genetic Diversity. Chem. Biol. 2000, 7, 263–274. 10.1016/S1074-5521(00)00102-2. [DOI] [PubMed] [Google Scholar]
- Carrico Z. M.; Farkas M. E.; Zhou Y.; Hsiao S. C.; Marks J. D.; Chokhawala H.; Clark D. S.; Francis M. B. N-Terminal Labeling of Filamentous Phage To Create Cancer Marker Imaging Agents. ACS Nano 2012, 6, 6675–6680. 10.1021/nn301134z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan K. F.; Stroh J. G. Site-Directed Conjugation of Nonpeptide Groups to Peptides and Proteins via Periodate Oxidation of a 2-Amino Alcohol. Application to Modification at N-Terminal Serine. Bioconjugate Chem. 1992, 3, 138–146. 10.1021/bc00014a008. [DOI] [PubMed] [Google Scholar]
- Ng S.; Jafari M. R.; Matochko W. L.; Derda R. Quantitative Synthesis of Genetically Encoded Glycopeptide Libraries Displayed on M13 Phage. ACS Chem. Biol. 2012, 7, 1482–1487. 10.1021/cb300187t. [DOI] [PubMed] [Google Scholar]
- Hudak J. E.; Yu H. H.; Bertozzi C. R. Protein Glycoengineering Enabled by the Versatile Synthesis of Aminooxy Glycans and the Genetically Encoded Aldehyde Tag. J. Am. Chem. Soc. 2011, 133, 16127–16135. 10.1021/ja206023e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjhung K. F.; Kitov P. I.; Ng S.; Kitova E. N.; Deng L.; Klassen J. S.; Derda R. Silent Encoding of Chemical Post-Translational Modifications in Phage-Displayed Libraries. J. Am. Chem. Soc. 2016, 138, 32–35. 10.1021/jacs.5b10390. [DOI] [PubMed] [Google Scholar]
- Chou Y.; Kitova E. N.; Joe M.; Brunton R.; Lowary T. L.; Klassen J. S.; Derda R. Genetically-Encoded Fragment-Based Discovery (GE-FBD) of Glycopeptide Ligands with Differential Selectivity for Antibodies Related to Mycobacterial Infections. Org. Biomol. Chem. 2018, 16, 223–227. 10.1039/C7OB02783D. [DOI] [PubMed] [Google Scholar]
- Ng S.; Bennett N. J.; Schulze J.; Gao N.; Rademacher C.; Derda R. Genetically-Encoded Fragment-Based Discovery of Glycopeptide Ligands for DC-SIGN. Bioorg. Med. Chem. 2018, 26, 5368–5377. 10.1016/j.bmc.2018.08.036. [DOI] [PubMed] [Google Scholar]
- Vinals D. F.; Kitov P. I.; Tu Z.; Zou C.; Cairo C. W.; Lin H. C.-H.; Derda R. Selection of Galectin-3 Ligands Derived from Genetically Encoded Glycopeptide Libraries. Pept. Sci. 2019, 111, e24097 10.1002/pep2.24097. [DOI] [Google Scholar]
- Yan K.; Triana V.; Kalmady S. V.; Aku-Dominguez K.; Memon S.; Brown A.; Greiner R.; Derda R. Learning the Structure-Activity Relationship (SAR) of the Wittig Reaction from Genetically-Encoded Substrates. Chem. Sci. 2021, 12, 14301–14308. 10.1039/D1SC04146K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triana V.; Derda R. Tandem Wittig/Diels-Alder Diversification of Genetically Encoded Peptide Libraries. Org. Biomol. Chem. 2017, 15, 7869–7877. 10.1039/C7OB01635B. [DOI] [PubMed] [Google Scholar]
- Kitov P. I.; Vinals D. F.; Ng S.; Tjhung K. F.; Derda R. Rapid, Hydrolytically Stable Modification of Aldehyde-Terminated Proteins and Phage Libraries. J. Am. Chem. Soc. 2014, 136, 8149–8152. 10.1021/ja5023909. [DOI] [PubMed] [Google Scholar]
- Kather I.; Bippes C. A.; Schmid F. X. A Stable Disulfide-Free Gene-3-Protein of Phage Fd Generated by In Vitro Evolution. J. Mol. Biol. 2005, 354, 666–678. 10.1016/j.jmb.2005.09.086. [DOI] [PubMed] [Google Scholar]
- Jafari M. R.; Deng L.; Kitov P. I.; Ng S.; Matochko W. L.; Tjhung K. F.; Zeberoff A.; Elias A.; Klassen J. S.; Derda R. Discovery of Light-Responsive Ligands through Screening of a Light-Responsive Genetically Encoded Library. ACS Chem. Biol. 2014, 9, 443–450. 10.1021/cb4006722. [DOI] [PubMed] [Google Scholar]
- McCarthy K. A.; Kelly M. A.; Li K.; Cambray S.; Hosseini A. S.; van Opijnen T.; Gao J. Phage Display of Dynamic Covalent Binding Motifs Enables Facile Development of Targeted Antibiotics. J. Am. Chem. Soc. 2018, 140, 6137–6145. 10.1021/jacs.8b02461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay A.; McCarthy K. A.; Kelly M. A.; Gao J. Targeting Bacteria via Iminoboronate Chemistry of Amine-Presenting Lipids. Nat. Commun. 2015, 6, 6561. 10.1038/ncomms7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M.; Cambray S.; McCarthy K. A.; Wang W.; Geisinger E.; Ortiz-Marquez J.; van Opijnen T.; Gao J. Peptide Probes of Colistin Resistance Discovered via Chemically Enhanced Phage Display. ACS Infect. Dis. 2020, 6, 2410–2418. 10.1021/acsinfecdis.0c00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H.; Xiao F.; Zhan K.; Kim Y.-P.; Xie H.; Xia Z.; Rao J. A Biocompatible Condensation Reaction for the Labeling of Terminal Cysteine Residues on Proteins. Angew. Chem., Int. Ed. 2009, 48, 9658–9662. 10.1002/anie.200903627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton J. T.; Lalonde T. J.; Tharp J. M.; Kurra Y.; Alugubelli Y. R.; Roundy C. M.; Hamer G. L.; Xu S.; Liu W. R. Novel Regioselective Approach to Cyclize Phage-Displayed Peptides in Combination with Epitope-Directed Selection to Identify a Potent Neutralizing Macrocyclic Peptide for SARS-CoV-2. ACS Chem. Biol. 2022, 17, 2911–2922. 10.1021/acschembio.2c00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M.; Haeffner F.; Gao J. N-Terminal Cysteine Mediated Backbone-Side Chain Cyclization for Chemically Enhanced Phage Display. Chem. Sci. 2022, 13, 8349–8354. 10.1039/D2SC03241D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Li Z.; Gao W.; Meng X.; Li X.; Luk L. Y. P.; Zhao Y.; Tsai Y.-H.; Wu C. Condensation of 2-((Alkylthio)(Aryl)Methylene)Malononitrile with 1,2-Aminothiol as a Novel Bioorthogonal Reaction for Site-Specific Protein Modification and Peptide Cyclization. J. Am. Chem. Soc. 2020, 142, 5097–5103. 10.1021/jacs.9b11875. [DOI] [PubMed] [Google Scholar]
- Zheng M.; Gao J. Phage Display of Two Distinct Warheads to Inhibit Challenging Proteins. ACS Chem. Biol. 2023, 18, 2259–2266. 10.1021/acschembio.3c00297. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Chen S. Cyclic Peptide Drugs Approved in the Last Two Decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. 10.1039/D1CB00154J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucana M. C.; Arruga Y.; Petrachi E.; Roig A.; Lucchi R.; Oller-Salvia B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. 10.3390/pharmaceutics13122065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeur E.; Guéret S. M.; Adihou H.; Gopalakrishnan R.; Lemurell M.; Waldmann H.; Grossmann T. N.; Plowright A. T. New Modalities for Challenging Targets in Drug Discovery. Angew. Chem., Int. Ed. 2017, 56, 10294–10323. 10.1002/anie.201611914. [DOI] [PubMed] [Google Scholar]
- McConnell S. J.; Kendall M. L.; Reilly T. M.; Hoess R. H. Constrained Peptide Libraries as a Tool for Finding Mimotopes. Gene 1994, 151, 115–118. 10.1016/0378-1119(94)90640-8. [DOI] [PubMed] [Google Scholar]
- O’Neil K. T.; Hoess R. H.; Jackson S. A.; Ramachandran N. S.; Mousa S. A.; DeGrado W. F. Identification of Novel Peptide Antagonists for GPIIb/IIIa from a Conformationally Constrained Phage Peptide Library. Proteins Struct. Funct. Bioinforma. 1992, 14, 509–515. 10.1002/prot.340140411. [DOI] [PubMed] [Google Scholar]
- Assem N.; Ferreira D. J.; Wolan D. W.; Dawson P. E. Acetone-Linked Peptides: A Convergent Approach for Peptide Macrocyclization and Labeling. Angew. Chem., Int. Ed. 2015, 54, 8665–8668. 10.1002/anie.201502607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokoyny A. M.; Zou Y.; Ling J. J.; Yu H.; Lin Y.-S.; Pentelute B. L. A Perfluoroaryl-Cysteine SNAr Chemistry Approach to Unprotected Peptide Stapling. J. Am. Chem. Soc. 2013, 135, 5946–5949. 10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo H.; Meinhardt N.; Wu Y.; Kulkarni S.; Hu X.; Low K. E.; Davies P. L.; DeGrado W. F.; Greenbaum D. C. Development of α-Helical Calpain Probes by Mimicking a Natural Protein-Protein Interaction. J. Am. Chem. Soc. 2012, 134, 17704–17713. 10.1021/ja307599z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diderich P.; Bertoldo D.; Dessen P.; Khan M. M.; Pizzitola I.; Held W.; Huelsken J.; Heinis C. Phage Selection of Chemically Stabilized α-Helical Peptide Ligands. ACS Chem. Biol. 2016, 11, 1422–1427. 10.1021/acschembio.5b00963. [DOI] [PubMed] [Google Scholar]
- Kale S. S.; Villequey C.; Kong X.-D.; Zorzi A.; Deyle K.; Heinis C. Cyclization of Peptides with Two Chemical Bridges Affords Large Scaffold Diversities. Nat. Chem. 2018, 10, 715–723. 10.1038/s41557-018-0042-7. [DOI] [PubMed] [Google Scholar]
- Eshwar V.; Kamath A.; Shastry R.; Shenoy A. K.; Kamath P. A Review of the Safety of Interleukin-17A Inhibitor Secukinumab. Pharmaceuticals 2022, 15, 1365. 10.3390/ph15111365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonschorek P.; Zorzi A.; Maric T.; Le Jeune M.; Schüttel M.; Montagnon M.; Gómez-Ojea R.; Vollmar D. P.; Whitfield C.; Reymond L.; et al. Phage Display Selected Cyclic Peptide Inhibitors of Kallikrein-Related Peptidases 5 and 7 and Their In Vivo Delivery to the Skin. J. Med. Chem. 2022, 65, 9735–9749. 10.1021/acs.jmedchem.2c00306. [DOI] [PubMed] [Google Scholar]
- Kalhor-Monfared S.; Jafari M. R.; Patterson J. T.; Kitov P. I.; Dwyer J. J.; Nuss J. M.; Derda R. Rapid Biocompatible Macrocyclization of Peptides with Decafluoro-Diphenylsulfone. Chem. Sci. 2016, 7, 3785–3790. 10.1039/C5SC03856A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J. Y. K.; Ekanayake A. I.; Kharchenko S.; Kirberger S. E.; Qiu R.; Kelich P.; Sarkar S.; Li J.; Fernandez K. X.; Alvizo-Paez E. R.; Miao J.; et al. Genetically Encoded Discovery of Perfluoroaryl Macrocycles That Bind to Albumin and Exhibit Extended Circulation in Vivo. Nat. Commun. 2023, 14, 5654. 10.1038/s41467-023-41427-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Liu W.; Liu Z.; Zhao Y.; Wu C. Biocompatible and Rapid Cyclization of Peptides with 2,4-Difluoro-6-Hydroxy-1,3,5-Benzenetricarbonitrile for the Development of Cyclic Peptide Libraries. Bioconjugate Chem. 2020, 31, 2085–2091. 10.1021/acs.bioconjchem.0c00363. [DOI] [PubMed] [Google Scholar]
- Bellotto S.; Chen S.; Rentero Rebollo I.; Wegner H. A.; Heinis C. Phage Selection of Photoswitchable Peptide Ligands. J. Am. Chem. Soc. 2014, 136, 5880–5883. 10.1021/ja501861m. [DOI] [PubMed] [Google Scholar]
- Jafari M. R.; Lakusta J.; Lundgren R. J.; Derda R. Allene Functionalized Azobenzene Linker Enables Rapid and Light-Responsive Peptide Macrocyclization. Bioconjugate Chem. 2016, 27, 509–514. 10.1021/acs.bioconjchem.6b00026. [DOI] [PubMed] [Google Scholar]
- Ng S.; Derda R. Phage-Displayed Macrocyclic Glycopeptide Libraries. Org. Biomol. Chem. 2016, 14, 5539–5545. 10.1039/C5OB02646F. [DOI] [PubMed] [Google Scholar]
- Ekanayake A. I.; Sobze L.; Kelich P.; Youk J.; Bennett N. J.; Mukherjee R.; Bhardwaj A.; Wuest F.; Vukovic L.; Derda R. Genetically Encoded Fragment-Based Discovery from Phage-Displayed Macrocyclic Libraries with Genetically Encoded Unnatural Pharmacophores. J. Am. Chem. Soc. 2021, 143, 5497–5507. 10.1021/jacs.1c01186. [DOI] [PubMed] [Google Scholar]
- Chen S.; Lovell S.; Lee S.; Fellner M.; Mace P. D.; Bogyo M. Identification of Highly Selective Covalent Inhibitors by Phage Display. Nat. Biotechnol. 2021, 39, 490–498. 10.1038/s41587-020-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M.; Chen F.-J.; Li K.; Reja R. M.; Haeffner F.; Gao J. Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display. J. Am. Chem. Soc. 2022, 144, 15885–15893. 10.1021/jacs.2c07375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppewal T. R.; Jansen I. D.; Hekelaar J.; Mayer C. A Strategy to Select Macrocyclic Peptides Featuring Asymmetric Molecular Scaffolds as Cyclization Units by Phage Display. J. Am. Chem. Soc. 2022, 144, 3644–3652. 10.1021/jacs.1c12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinis C.; Rutherford T.; Freund S.; Winter G. Phage-Encoded Combinatorial Chemical Libraries Based on Bicyclic Peptides. Nat. Chem. Biol. 2009, 5, 502–507. 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- Chen S.; Rentero Rebollo I.; Buth S. A.; Morales-Sanfrutos J.; Touati J.; Leiman P. G.; Heinis C. Bicyclic Peptide Ligands Pulled out of Cysteine-Rich Peptide Libraries. J. Am. Chem. Soc. 2013, 135, 6562–6569. 10.1021/ja400461h. [DOI] [PubMed] [Google Scholar]
- Kong X.-D.; Moriya J.; Carle V.; Pojer F.; Abriata L. A.; Deyle K.; Heinis C. De Novo Development of Proteolytically Resistant Therapeutic Peptides for Oral Administration. Nat. Biomed. Eng. 2020, 4, 560–571. 10.1038/s41551-020-0556-3. [DOI] [PubMed] [Google Scholar]
- Wong J. Y.-K.; Mukherjee R.; Miao J.; Bilyk O.; Triana V.; Miskolzie M.; Henninot A.; Dwyer J. J.; Kharchenko S.; Iampolska A.; et al. Genetically-Encoded Discovery of Proteolytically Stable Bicyclic Inhibitors for Morphogen NODAL. Chem. Sci. 2021, 12, 9694–9703. 10.1039/D1SC01916C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeriswyl V.; Rapley H.; Pollaro L.; Stace C.; Teufel D.; Walker E.; Chen S.; Winter G.; Tite J.; Heinis C. Bicyclic Peptides with Optimized Ring Size Inhibit Human Plasma Kallikrein and Its Orthologues While Sparing Paralogous Proteases. ChemMedChem. 2012, 7, 1173–1176. 10.1002/cmdc.201200071. [DOI] [PubMed] [Google Scholar]
- Angelini A.; Diderich P.; Morales-Sanfrutos J.; Thurnheer S.; Hacker D.; Menin L.; Heinis C. Chemical Macrocyclization of Peptides Fused to Antibody Fc Fragments. Bioconjugate Chem. 2012, 23, 1856–1863. 10.1021/bc300184m. [DOI] [PubMed] [Google Scholar]
- Bertoldo D.; Khan M. M. G.; Dessen P.; Held W.; Huelsken J.; Heinis C. Phage Selection of Peptide Macrocycles against β-Catenin To Interfere with Wnt Signaling. ChemMedChem. 2016, 11, 834–839. 10.1002/cmdc.201500557. [DOI] [PubMed] [Google Scholar]
- Baeriswyl V.; Calzavarini S.; Gerschheimer C.; Diderich P.; Angelillo-Scherrer A.; Heinis C. Development of a Selective Peptide Macrocycle Inhibitor of Coagulation Factor XII toward the Generation of a Safe Antithrombotic Therapy. J. Med. Chem. 2013, 56, 3742–3746. 10.1021/jm400236j. [DOI] [PubMed] [Google Scholar]
- Rentero Rebollo I.; McCallin S.; Bertoldo D.; Entenza J. M.; Moreillon P.; Heinis C. Development of Potent and Selective S. Aureus Sortase A Inhibitors Based on Peptide Macrocycles. ACS Med. Chem. Lett. 2016, 7, 606–611. 10.1021/acsmedchemlett.6b00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Bertoldo D.; Angelini A.; Pojer F.; Heinis C. Peptide Ligands Stabilized by Small Molecules. Angew. Chem., Int. Ed. 2014, 53, 1602–1606. 10.1002/anie.201309459. [DOI] [PubMed] [Google Scholar]
- Diderich P.; Heinis C. Phage Selection of Bicyclic Peptides Binding Her2. Tetrahedron 2014, 70, 7733–7739. 10.1016/j.tet.2014.05.106. [DOI] [Google Scholar]
- Chen S.; Morales-Sanfrutos J.; Angelini A.; Cutting B.; Heinis C. Structurally Diverse Cyclisation Linkers Impose Different Backbone Conformations in Bicyclic Peptides. ChemBioChem. 2012, 13, 1032–1038. 10.1002/cbic.201200049. [DOI] [PubMed] [Google Scholar]
- Lee Y.-J.; Kurra Y.; Liu W. R. Phospha-Michael Addition as a New Click Reaction for Protein Functionalization. Chembiochem Eur. J. Chem. Biol. 2016, 17, 456–461. 10.1002/cbic.201500697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd G. E.; Brown A.; Chen L.; van Rietschoten K.; Watcham S.; Teufel D. P.; Pavan S.; Lani R.; Huxley P.; Bennett G. S. Identification and Optimization of EphA2-Selective Bicycles for the Delivery of Cytotoxic Payloads. J. Med. Chem. 2020, 63, 4107–4116. 10.1021/acs.jmedchem.9b02129. [DOI] [PubMed] [Google Scholar]
- Mudd G. E.; Scott H.; Chen L.; van Rietschoten K.; Ivanova-Berndt G.; Dzionek K.; Brown A.; Watcham S.; White L.; Park P. U.; et al. Discovery of BT8009: A Nectin-4 Targeting Bicycle Toxin Conjugate for the Treatment of Cancer. J. Med. Chem. 2022, 65, 14337–14347. 10.1021/acs.jmedchem.2c00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G.; Lutz R.; Park P.; Harrison H.; Lee K. Abstract 1167: Development of BT1718, a Novel Bicycle Drug Conjugate for the Treatment of Lung Cancer. Cancer Res. 2017, 77, 1167. 10.1158/1538-7445.AM2017-1167. [DOI] [Google Scholar]
- Luzi S.; Kondo Y.; Bernard E.; Stadler L. K. J.; Vaysburd M.; Winter G.; Holliger P. Subunit Disassembly and Inhibition of TNFα by a Semi-Synthetic Bicyclic Peptide. Protein Eng. Des. Sel. 2015, 28, 45–52. 10.1093/protein/gzu055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari M. R.; Yu H.; Wickware J. M.; Lin Y.-S.; Derda R. Light-Responsive Bicyclic Peptides. Org. Biomol. Chem. 2018, 16, 7588–7594. 10.1039/C7OB03178E. [DOI] [PubMed] [Google Scholar]
- Chen F.-J.; Pinnette N.; Yang F.; Gao J. A Cysteine-Directed Proximity-Driven Crosslinking Method for Native Peptide Bicyclization. Angew. Chem., Int. Ed. 2023, 62, e202306813 10.1002/anie.202306813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushottam L.; Adusumalli S. R.; Singh U.; Unnikrishnan V. B.; Rawale D. G.; Gujrati M.; Mishra R. K.; Rai V. Single-Site Glycine-Specific Labeling of Proteins. Nat. Commun. 2019, 10, 2539. 10.1038/s41467-019-10503-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle V.; Kong X.-D.; Comberlato A.; Edwards C.; Díaz-Perlas C.; Heinis C. Generation of a 100-Billion Cyclic Peptide Phage Display Library Having a High Skeletal Diversity. Protein Eng. Des. Sel. 2021, 34, gzab018. 10.1093/protein/gzab018. [DOI] [PubMed] [Google Scholar]
- Esvelt K. M.; Carlson J. C.; Liu D. R. A System for the Continuous Directed Evolution of Biomolecules. Nature 2011, 472, 499–503. 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André A. S.; Moutinho I.; Dias J. N. R.; Aires-da-Silva F.. In Vivo Phage Display: A Promising Selection Strategy for the Improvement of Antibody Targeting and Drug Delivery Properties. Front. Microbiol. 2022, 13. 10.3389/fmicb.2022.962124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J.; Dickerson M. T.; Owen N. K.; Landon L. A.; Deutscher S. L. Biodistribution of Filamentous Phage Peptide Libraries in Mice. Mol. Biol. Rep. 2004, 31, 121–129. 10.1023/B:MOLE.0000031459.14448.af. [DOI] [PubMed] [Google Scholar]
- Weber T.; Pscherer S.; Gamerdinger U.; Teigler-Schlegel A.; Rutz N.; Blau W.; Rummel M.; Gattenlöhner S.; Tur M. K. Parallel Evaluation of Cell-Based Phage Display Panning Strategies: Optimized Selection and Depletion Steps Result in AML Blast-Binding Consensus Antibodies. Mol. Med. Rep. 2021, 24, 767. 10.3892/mmr.2021.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters J. M.; Telleman P.; Junghans R. P. An Optimized Method for Cell-Based Phage Display Panning. Immunotechnology 1997, 3, 21–29. 10.1016/S1380-2933(96)00056-5. [DOI] [PubMed] [Google Scholar]
- Aviner R. The Science of Puromycin: From Studies of Ribosome Function to Applications in Biotechnology. Comput. Struct. Biotechnol. J. 2020, 18, 1074–1083. 10.1016/j.csbj.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.