

Abstract
Objectives
Seasonal influenza viruses cause roughly 650 000 deaths annually despite available vaccines. CD8+ T cells typically recognise influenza‐derived peptides from internal structural and non‐structural influenza proteins and are an attractive avenue for future vaccine design as they could reduce the severity of disease following infection with diverse influenza strains. CD8+ T cells recognise peptides presented by the highly polymorphic Human Leukocyte Antigens class I molecules (HLA‐I). Each HLA‐I variant has distinct peptide binding preferences, representing a significant obstacle for designing vaccines that elicit CD8+ T cell responses across broad populations. Consequently, the rational design of a CD8+ T cell‐mediated vaccine would require the identification of highly immunogenic peptides restricted to a range of different HLA molecules.
Methods
Here, we assessed the immunogenicity of six recently published novel influenza‐derived peptides identified by mass‐spectrometry and predicted to bind to the prevalent HLA‐B*18:01 molecule.
Results
Using CD8+ T cell activation assays and protein biochemistry, we showed that 3/6 of the novel peptides were immunogenic in several HLA‐B*18:01+ individuals and confirmed their HLA‐B*18:01 restriction. We subsequently compared CD8+ T cell responses towards the previously identified highly immunogenic HLA‐B*18:01‐restricted NP219 peptide. Using X‐ray crystallography, we solved the first crystal structures of HLA‐B*18:01 presenting immunogenic influenza‐derived peptides. Finally, we dissected the first TCR repertoires specific for HLA‐B*18:01 restricted pathogen‐derived peptides, identifying private and restricted repertoires against each of the four peptides.
Conclusion
Overall the characterisation of these novel immunogenic peptides provides additional HLA‐B*18:01‐restricted vaccine targets derived from the Matrix protein 1 and potentially the non‐structural protein and the RNA polymerase catalytic subunit of influenza viruses.
Keywords: CD8+ T cells, HLA‐B*18:01, immunodominance hierarchy, immunogenic, influenza
In this study, we have investigated 6 novel influenza‐derived peptides predicted to be restricted to the understudied HLA‐B*18:01 molecule. We identified that 3/6 of the novel peptides were immunogenic in samples from several donors and restricted to HLA‐B*18:01 as predicted and were further characterised.
Introduction
The seasonal strains of influenza A (A/H1N1 & A/H3N2) virus circulate annually and cause mild to severe disease in humans. This typically results in roughly 650 000 mortalities each year 1 despite vaccines being available to many worldwide. Influenza‐like infections also result in economic consequences. Indeed, it is estimated that the total cost of influenza‐like illnesses equated to more than $8 billion USD in the United States alone. Widely approved influenza vaccines are typically designed to induce a strong neutralising antibody response against the viral surface glycoprotein haemagglutinin (HA), which is highly susceptible to antigenic drift that results in reduced vaccine efficacy over time. 2 , 3 Furthermore, these yearly vaccines are immensely reliant on World Health Organisation (WHO) predictions of the prevalent circulating strains up to 6 months in advance to enable timely generation. 1 This has caused variable vaccine efficacies in the past, such as in the 2014–2015 season, where an overall 6% vaccine efficacy against the A/H3N2 strain was reported. 4 While high‐end estimates of 78% vaccine efficacy were identified in young adolescents. 5 Nevertheless, in the event of a spillover from an animal reservoir, current vaccines may not be adequate for protection, as a mismatched influenza strain typically leads to severely lowered vaccine efficacy. 2 , 6 As such, there is a need for updated vaccines that can protect broad populations against emerging influenza virus strains.
Unlike antibodies, CD8+ T cells typically recognise more conserved internal proteins of influenza viruses. They are also long‐lived, together meaning they have the potential to protect against different influenza virus strains across multiple years and are an attractive target for future influenza virus vaccines. 3 , 7 , 8 Undoubtedly, CD8+ T cells are important against influenza virus infections, as they have also been associated with improved disease outcomes. 3 , 9 , 10 , 11 CD8+ T cells recognise peptides presented by HLA class I molecules (HLA‐I). 12 , 13 These HLA‐I molecules are highly polymorphic, with over 25 000 HLA‐I molecules described thus far. 14 , 15 Moreover, different HLA‐I molecules also favor different peptide sequence motifs for binding, meaning that they typically present unique peptide repertoires, causing a significant challenge for rational vaccine design. 16 Adding further to this complexity, HLA‐I profiles often differ between different ethnicities and geographical locations. Thus, for rational vaccine design, it is critical that we identify and characterise the best, most immunogenic peptides for the range of prevalent HLA molecules that are expressed worldwide.
HLA‐B*18:01 is the 15th most prevalent HLA‐B molecule, estimated to be expressed by ~2.3% of the world's population, spanning the Middle East, Africa, Southeast Asia and Europe. 17 According to the immune epitope database (IEDB), 18 only six influenza‐derived immunogenic HLA‐B*18:01‐restricted peptides have so far been published, with only 2 confirmed using specific HLA‐B*18:01 antigen‐presenting cells in an intracellular cytokine staining (ICS) assay. This includes the highly conserved immunogenic NP219‐226 peptide identified by our group, 11 and the slightly longer NP219‐228. 19 Together, this makes HLA‐B*18:01 severely understudied compared to other HLA molecules. 18 In 2022, a paper published by Nicholas et al., 20 identified six novel influenza‐derived, HLA‐I‐bound peptides by mass spectrometry. Given the HLA type of the donor cells used, these peptides were predicted to bind to HLA‐B*18:01. 20
In this study, we tested the potential immunogenicity of these six novel peptides, 20 characterising the CD8+ T cell responses in cells derived from HLA‐B*18:01+ individuals. We determined that 3/6 peptides were indeed immunogenic in several HLA‐B*18:01+ individuals and we confirmed the expected HLA‐B*18:01 restriction by tetramer staining. We then compared the immune response towards these novel HLA‐B*18:01‐restricted peptides with the HLA‐B*18:01‐restricted NP219‐226 peptide (herein referred to as NP219) previously identified. 11 A clear immunodominance hierarchy was observed, with CD8+ T cell responses strongest towards NP219 followed by M15, PB1177 and finally NS2111. Further, we solved the first crystal structures of HLA‐B*18:01 presenting immunogenic influenza‐derived peptides. Three of the four peptides were presented by HLA‐B*18:01 in a similar configuration while the longer PB1177 was different. Moreover, we identified that peptide stability did not influence this immunodominance hierarchy. Finally, we report the first T cell receptor (TCR) repertoires of CD8+ T cells recognising HLA‐B*18:01+ restricted pathogen‐derived antigens. The TCR repertoire towards each of the four epitopes was restricted and private, unique to each individual. Overall, our study has characterised novel conserved immunogenic peptides from a range of proteins representing novel potential vaccine targets for HLA‐B*18:01+ individuals.
Results
Novel influenza‐derived peptides are highly conserved
Several novel influenza‐derived peptides were identified by Nicholas et al. 20 in 2022 who infected human lung‐derived cells from several donors with a laboratory‐adapted A/X‐31/H3N2 virus or the human‐derived A/H3N2/Wisconsin/67/2005 influenza virus, before subjecting them to mass spectrometry. Given the HLA types of the donors, 6 of these peptides were predicted to be restricted by HLA‐B*18:01. To investigate these six peptides as potential vaccine candidates that cover HLA‐B*18:01+ individuals, we assessed whether these peptide sequences were conserved in either the H1N1 (A/Victoria/2570/2019(H1N1)) and the H3N2 (A/Darwin/9/2021(H3N2)) influenza virus strains from the 2022 WHO recommended Southern Hemisphere influenza vaccine strains (Table 1). For 5/6 peptides, the published sequences were conserved in at least one of the vaccine strains and were selected for further investigation. Conversely, the PB1177 peptide sequence was the same in both vaccine strains (EEIEITTHF), differing from the published sequence by two amino acids (EEMGITTHF). Consequently, the EEIEITTHF; PB1177 peptide was selected peptide for immunogenicity screening and characterisation.
Table 1.
Conservation of the novel peptides in recommended vaccine strains
| Source | Strain | Peptide | |||||
|---|---|---|---|---|---|---|---|
| M15 | NS2109 | NS2111 | NS290 | NP45 | PB1177 | ||
| Nicholas et al. | A/X‐31/H3N2 | TEVETYVL | VEQEIRTF | QEIRTFSF | TENSFEQITF | TELKLSDY | EEMGITTHF |
| Nicholas et al. | A/H3N2/Wisconsin/67/2005 (H3N2) | TEVETYVL | VEQEIRTF | QEIRTFSF | N/A | N/A | N/A |
| 2022 Vaccine H3N2 strain | A/Darwin/9/2021 (H3N2) | TEVETYVL | VEQEIRTF | QEIRTFSF | TENSFEQITF | TELKLSDH | EEIEITTHF |
| 2022 Vaccine H1N1 strain | A/Victoria/2570/2019 (H1N1) | TEVETYVL | VEQEIRAF | QEIRAFSF | TENSFEQITF | TELKLSDY | EEIEITTHF |
| Sequence selected for this study | TEVETYVL | VEQEIRTF | QEIRTFSF | TENSFEQITF | TELKLSDY | EEIEITTHF | |
Sequences of identified peptides from Nicholas et al. 20 were aligned with the WHO recommended 2022 Southern Hemisphere Influenza virus vaccine strains A/Darwin/9/2021 (H3N2) and A/Victoria/2570/2019 (H1N1). 45 Protein sequences of Matrix Protein 1, Nuclear Export Protein, Nucleoprotein and RNA‐directed RNA polymerase catalytic subunit were obtained from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/). 46 Sequences were aligned using Clustal Omega Multiple Sequence Alignment (https://www.ebi.ac.uk/Tools/msa/clustalo/) 47 and amino acid variations are shown in red.
Three of six novel peptides are immunogenic in multiple HLA‐B*18:01+ samples
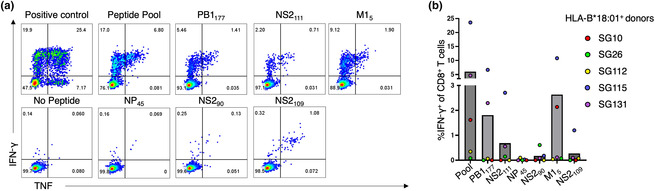
To determine the immunogenicity of these six influenza‐derived peptides, 20 CD8+ T cell lines were generated by stimulating PBMCs from HLA‐B*18:01+ donors (n = 5) against the pool of peptides for 10+ days. CD8+ T cell lines were subsequently restimulated with the peptide pool, or individual peptides, and immunogenicity was assessed in an ICS assay. The NS2109 and NS290 were immunogenic in samples from 1 out of 5 donors (SG115) and we did not observe CD8+ T cell activation for the NP45 peptide in any of the samples tested (Supplementary figure 2). Three of the six peptides induced a CD8+ T cell response in multiple donors (Figure 1, Supplementary figure 2). Of these, the M15 peptide induced the largest CD8+ T cell response (average; 2.6% IFN‐γ+ CD8+ T cells) followed by PB1177 (average; 1.8% IFN‐γ+ CD8+ T cells) and NS2111 (average; 0.68% IFN‐γ CD8+ T cells). For donor SG10, only M15 introduced a positive IFN‐γ+ CD8+ T cell response.
Figure 1.
Identification of immunogenic HLA‐B*18:01 predicted peptides. PBMCs from HLA‐B*18:01+ individuals (n = 5) were stimulated with a pool of peptides (2 μm per peptide) for 10+ days to generate pooled CD8+ T cell lines. CD8+ T cells were stimulated with the peptide pool (2 μm) and each peptide individually (10 μm), along with negative (no peptide) and positive (×500) controls, and responses were assessed using an ICS assay. (a) Representative FACS plots from donor SG115. (b) A bar graph outlining the average proportion of IFN‐γ+ of CD8+ T cells, minus the no peptide control, in five HLA‐B*18:01+ donors, with each coloured dot representing a donor's response (red, SG10; green, SG26; yellow, SG112; blue, SG115; purple, SG131).
NP219 induces an immunodominant CD8+ T cell response in HLA‐B*18:01+ samples
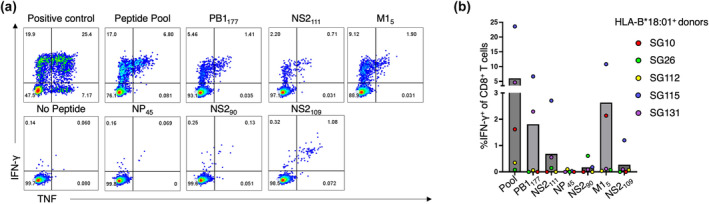
To confirm the expected HLA‐B*18:01 restriction of the novel influenza‐derived peptides, individual peptide‐specific CD8+ T cell lines were generated against PB1177, NS2111 and M15 peptides individually using PBMCs of HLA‐B*18:01+ individuals (Figure 2, n = 3). The magnitude of epitope‐specific CD8+ T cell populations and the HLA‐B*18:01 restriction of PB1177, NS2111 and M15 was confirmed using tetramer staining (Figure 2a and c), where clear tetramer‐specific populations could be seen in all individuals.
Figure 2.
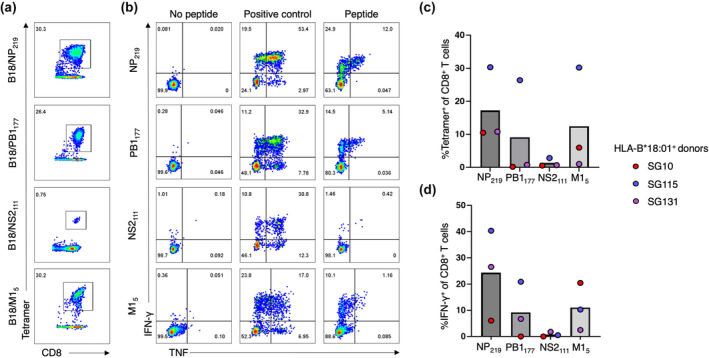
Novel immunogenic peptides are all HLA‐B*18:01 restricted. Peptide‐specific CD8+ T cell lines (n = 3 HLA‐B*18:01+ individuals) were generated against 10 μm of the PB1177, NS2111, M15 and NP219 peptides individually and cultured for 10+ days. CD8+ T cell line specificity was assessed using tetramer staining and an ICS assay. (a) Representative FACS plots of peptide‐specific CD8+ T cell lines recognising tetramers of their cognate peptide. (b) Representative FACS plots of peptide‐specific CD8+ T cell lines producing cytokines in response towards their cognate peptide or controls. (c) Summary bar graph outlining the average percentage of tetramer+ of CD8+ T cells specific for B18/PB1177, B18/NS2111, B18/M15 and B18/NP219. Each dot represents the percentage of tetramer+ CD8+ T cells for each donor (red: SG10, blue: SG115 and purple: SG131). (d) A bar graph outlining the average proportion of IFN‐γ+ of CD8+ T cells, minus the no peptide control, in three HLA‐B*18:01+ donors, with each coloured dot representing a donor's response (red, SG10; blue, SG115; purple, SG131).
The NP219 influenza‐derived epitope is another HLA‐B*18:01‐restricted peptide known to induce a strong CD8+ T cell response in HLA‐B*18:01+ individuals. 11 , 21 Therefore, we wanted to compare the CD8+ T cell responses towards the three novel immunogenic peptides with the response towards the NP219 peptide (Figure 2). To achieve this, individual peptide‐specific CD8+ T cell lines were also generated against the NP219 peptide using PBMCs of HLA‐B*18:01+ individuals (Figure 2, n = 3). The magnitude of CD8+ T cell populations specific to all four peptides was determined using tetramer staining (Figure 2a and c) and the functional CD8+ T cell responses were assessed using an ICS assay (Figure 2b and d).
We noticed a clear immunodominance hierarchy across the donors, with the largest CD8+ T cell populations expanded against NP219 (average; 17.2% tetramer+ CD8+ T cells) followed by M15 (average; 12.4% tetramer+ CD8+ T cells), PB1177 (average; 9.1% tetramer+ CD8+ T cells) and NS2111 (average; 1.3% tetramer+ CD8+ T cells). The same immunodominance hierarchy was observed with the ICS assay (Figure 2b and d), with the NP219 peptide inducing the largest functional CD8+ T cell responses (average; 24.3% IFN‐γ+ CD8+ T cells) followed by M15 (average; 11.1% IFN‐γ+ CD8+ T cells), PB1177 (average; 9.2% IFN‐γ+ CD8+ T cells) and NS2111 (average; 0.76% IFN‐γ+ CD8+ T cells) as measured by IFN‐γ production (Figure 2). TNF, MIP‐1β, CD107a and IL2 production were also individually recorded with the same immunodominance hierarchy observed (Supplementary figure 3). The polyfunctionality of the CD8+ T cell responses towards the four HLA‐B*18:01‐restricted epitopes was assessed (Figure 3a and b). We interpret a polyfunctional response as the ability of CD8+ T cells to produce more than one effector function upon peptide stimulation. All four peptides induced a polyfunctional CD8+ T cell response in cells from all 3 donors. Interestingly, the three most immunogenic peptides, NP219, M15 and PB1177, induced a similar proportion of polyfunctionality by CD8+ T cells, with up to four effector functions by CD8+ T cells across a range of cytokine permutations. Conversely, the least immunogenic peptide, NS2111, induced at most three functions, across a range of effector function combinations.
Figure 3.
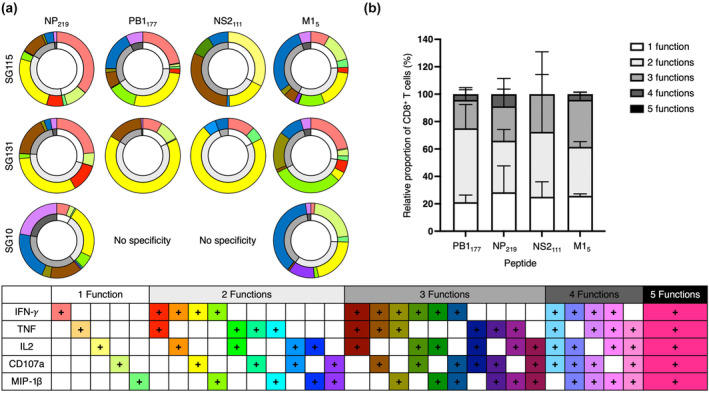
Polyfunctional CD8+ T cell analysis of immunogenic HLA‐B*18:01 specific peptides. PB1177, NS2111, M15 and NP219 peptide‐specific T cell lines from three HLA‐B*18:01+ donors, SG10, SG115 and SG131 (n = 3) were generated as per Figure 2 and CD8+ T cell production of IFN‐γ, TNF, CD107a, IL2 and MIP‐1β were measured in an ICS assay. (a) Summary of cytokine production, minus the no peptide control, represented as a double ring graph where the inner ring is representative of the number of functions, while the outer shell denotes the different cytokine combinations. SG10 CD8+ T cell lines grown against PB1177 and NS2111 displayed no specificity (as per Figure 2) and were not analysed for polyfunctionality. (b) A summary bar graph representing the proportion of CD8+ T cells expressing an effector function. All results were normalised to 100% and the error bars represent the mean with standard deviation (SD).
Next, we assessed whether these immunogenic peptides have been conserved over time, or whether mutations in the amino‐acid sequence have occurred in past influenza virus strains. We aligned the peptides against the ‘vaccine only’ strains from the influenza virus database, 22 as representative of the major circulating strains, to determine the level conservation of these peptide sequences (Table 2). Three of the four immunogenic peptides were conserved in > 94% of the virus strains analysed, including the two most immunogenic peptides, NP219 and M15, which were both conserved in 100% of the aligned sequences. Conversely, the PB1177 peptide was conserved in only 23% of the vaccine virus strains.
Table 2.
Conservation and stability of the immunogenic HLA‐B*18:01‐restricted peptides
| Peptide | Sequence | Conservation % | Tm (°C) SEM |
|---|---|---|---|
| M15 | TEVETYVL | 100% (n = 35) | 62.51 ± 0.04 |
| NS2111 | QEIRTFSF | 94% (n = 31) | 61.14 ± 0.72 |
| PB1177 | EEIEITTHF | 23% (n = 30) | 64.45 ± 0.99 |
| NP219 | YERMCNIL | 100% (n = 36) | 63.00 ± 0.08 |
Immunogenic peptides were aligned with recent vaccine strains of the influenza virus. Full length M1, NS2, PB1 and NP sequences from vaccine‐only strains were obtained from the NCBI Influenza Research Database https://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph‐select.cgi?go=database 22 on the 2nd of February 2024. Sequences were aligned using Clustal Omega Multiple Sequence Alignment (https://www.ebi.ac.uk/Tools/msa/clustalo/), 47 and conservation was determined. Conservation is listed as a proportion of total sequences aligned that had an identical amino acid sequence as the peptide under investigation. Tm represents the midpoint thermal melt temperature calculated at two concentrations, in duplicate with two independent samples, with the error as standard error from the mean value (SEM).
Immunodominance hierarchy of the HLA‐B*18:01‐restricted peptides is unrelated to peptide‐HLA stability
To understand the immunodominance hierarchy observed towards the immunogenic HLA‐B*18:01‐restricted epitopes, we refolded and purified HLA‐B*18:01 with NP219, M15, PB1177 and NS2111 to measure the thermal stability of each pHLA complexes (Table 2). All four peptide‐HLA‐B*18:01 complexes were exhibiting the same stability, within 1–2 °C difference between each of the complexes (Table 2), with a Tm value of about 63 °C in line with previously reported values for pHLA‐B*18:01. 23 Therefore, the differences in immunogenicity of the four epitopes was not driven by differences in the pHLA complexes stability.
We next assessed the presentation of each peptide by HLA‐B*18:01 by determining their crystal structure at high resolution (1.60–1.15 Å, Supplementary table 1, Supplementary figure 4). We previously reported the structure of an influenza‐derived peptide bound to HLA‐B*18:01 (PDB code: 6MT3), 24 but this peptide was not immunogenic in the tested HLA‐B*18:01+ samples, rather in the HLA‐B*37:01+ samples. Therefore, here, we present the first structures of immunogenic influenza‐derived peptides in complex with HLA‐B*18:01. Overall, the four peptides bound canonically in the cleft of HLA‐B*18:01, with the preferred P2‐Glu in the B pocket and a large hydrophobic residue at PΩ residue (L/F/Y/M) 25 (Figure 4). The three 8mer peptides (NP219, NS2111 and M15) adopt a similar conformation with an average root mean square deviation (r.m.s.d.) between 0.33 and 0.44 Å for their Cα atoms (Figure 4a, d and g). In addition, the antigen binding cleft (residues 1–180) also shared a similar conformation across the four complexes with a r.m.s.d. between 0.14 and 0.20 Å for their Cα atoms (Figure 4a).
Figure 4.
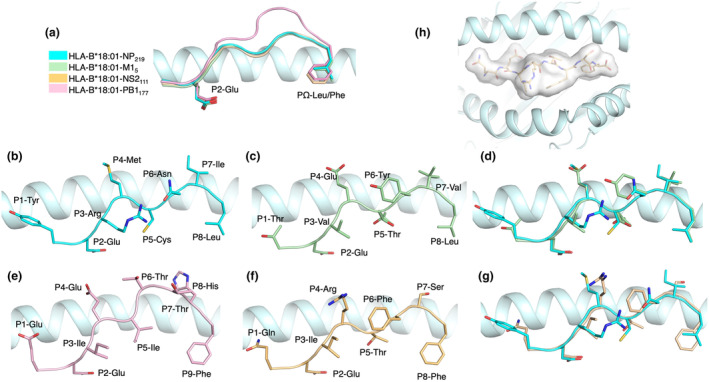
Crystal structures of the HLA‐B*18:01 binding cleft, presenting four immunogenic influenza peptides. (a) Structural overlay of the peptide backbone and anchor positions (cartoon) for NP219 (cyan), M15 (green), NS2111 (yellow) and PB1117 (pink), within the binding cleft of HLA‐B*18:01 (light cyan). Side view of the HLA‐B*18:01 binding cleft presenting NP219 (b), M15 (c) and their overlay (d). Side view of the HLA‐B*18:01 binding cleft presenting PB1117 (e), NS2111 and their overlay (f). (b–g) Peptide residue side chains are represented in stick. (h) Top‐down view of NS2111 is represented as a transparent surface (white) within the binding cleft of HLA‐B*18:01 (light cyan cartoon).
The immunogenic peptides NP219, M15 and PB1177 are more featured than the NS2111
Despite a similar overall structure, some differences were observed that might help distinguish the features driving the immunogenicity hierarchy of these epitopes. The NP219 peptide is firmly anchored into the HLA‐B*18:01 cleft, with the canonical P2‐Glu and P8‐Leu binding to the B and F pockets, respectively (Figure 4b). In addition, the P5‐Cys is acting as secondary anchor residue contacting His9, Arg97 and Tyr74 in the cleft. The peptide NP219 is constrained 26 by intra‐peptide contacts between P3‐Arg and P6‐Asn side‐chains, forming a network of interaction with the Gln155 as well (Supplementary figure 5a). The large side chains of the P1‐Tyr, P4‐Met and P7‐Ile point upwards out of the cleft for direct TCR interaction (Figure 4b).
Similarly, the M15 peptide has the canonical P2‐Glu and P8‐Leu as primary anchor residues (Figure 4c) and P5‐Thr as secondary anchor interacting with Asn70, Tyr74 and Arg97 (Supplementary figure 5b). As a result, M15 and NP219 adopt similar peptide conformations in the cleft of HLA‐B*18:01 (Figure 4d), with P1‐Thr, P4‐Glu and P7‐Val residues also exposed to the solvent and ready for TCR interaction in the M15 peptide (Figure 4c).
The longer 9mer peptide PB1177 was also binding with P2‐Glu and P9‐Phe to HLA‐B*18:01 (Figure 4e) and as canonically observed for 9mer peptide was bulging more outside the antigen cleft than the 8mer peptides described above (Figure 4a). In addition, the P5‐Ile acts as a secondary anchor as per the other peptides described above. Interestingly, despite a difference in length, the backbone from P1 to P4 residues remains overall similar because of the shared secondary anchor residue at position 5 of the peptides (Figure 4a). The PB1177 peptide exposed the side chains of the residues P1‐Glu, P4‐Glu, P6‐Thr, P8‐His and P7‐Thr is partially solvent exposed (Figure 4e).
The NS2111 peptide followed the same P2‐Glu and P8‐Phe primary anchor residues and P5‐Thr as secondary anchor (Figure 4f). As a result, the backbone of the NS2111 was overlayed well with the backbone of the NP219 peptide (Figure 4g). While the P1‐Gln, P4‐Arg, P6‐Phe, P7‐Ser residues of the NS2111 peptide were solvent exposed and the overall surface was comparable to the NP219 peptide (~240 Å2), intra‐peptide contacts limit the access of their side‐chains (Supplementary figure 6). The NS2111 peptide P4‐Arg side‐chain is forming a hydrogen bond with the Gln155 of the HLA α2‐helix and hydrophobic bond network with the peptide P3‐Ile and P6‐Phe shielding them (Supplementary figure 6e and f). This interaction network is limiting the accessibility for the TCR as there is no space between the peptide backbone and the α2‐helix for the CDR loops to engage without structural rearrangement (Supplementary figure 6f). This is in contrast with the three other peptides (Supplementary figure 6b, d and h), where P4 residue side chains are upright, closer to the HLA α1‐helix and perhaps more easily accessible for CDR loops binding.
Private and restricted TCR repertoires recognise HLA‐B*18:01‐restricted epitopes
Finally, we assessed the αβTCR repertoire of the epitope‐specific CD8+ T cells from our HLA‐B*18:01+ samples, representing the first TCR repertoire specific for HLA‐B*18:01‐restricted pathogenic peptides. To do this, peptide‐specific CD8+ T cell lines assayed above (Figures 2 and 3) were stained with the relevant pHLA tetramer and single‐cell sorted. TCR repertoire was subsequently assessed by single‐cell multiplex PCR as previously described. 27 The TCR repertoires specific for each of the HLA‐B*18:01‐restricted peptides were restricted and entirely private, with no shared αβTCR sequences identified between donors or across epitopes (Figure 5, Supplementary table 2) and few TRAV and TRBV biases were identified within epitope‐specific TCR repertoires between donors.
Figure 5.
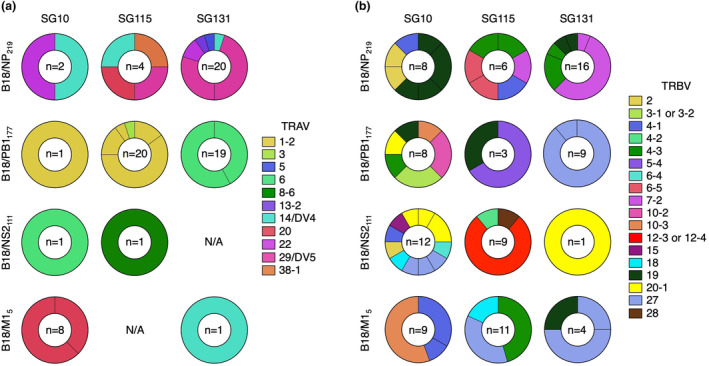
TCR repertoire of identified HLA‐B*18:01 immunogenic peptides. PB1177, NS2111, M15 and NP219 peptide‐specific T cell lines from three HLA‐B*18:01+ donors were tetramer stained with the cognate peptide, single‐cell sorted and subject to single‐cell multiplex PCR. Pie charts representing (a) TRAV and (b) TRBV gene usage for CD8+ T cell recognition of B18/NP219, B18/PB1177, B18/NS2111 and B18/M15 tetramers. A total of 22–26 cells were sequenced, while ‘n’ denotes the number of clonotypes that were resolved. TRAV and TRBV genes are depicted in different colours, while unique clonotypes are depicted as separate segments of the pie chart.
For B18/NP219, TRAV14 usage was seen in all 3 donors, while TRAV29 and TRAV22 were seen in samples from 2/3 donors. Similarly, TRBV7‐2, TRBV4‐1, TRBV4‐3 and TRBV19 were seen in samples from 2/3 donors (Figure 5). In the few αβTCR pairs that were resolved, we found pairing of TRAV14/TRBV4‐3, TRAV38‐1/TRBV4‐3, TRAV29/TRBV7‐2 and TRAV20/TRBV4‐1 (Supplementary table 2). CDR3 lengths varied from 12 to 16 amino acids long for CDR3α and from 14 to 17 amino acids for CDR3β. No distinct motifs were identified with the small number of clonotypes available for analysis at each amino‐acid length (Supplementary figure 7).
B18/PB1177‐specific CD8+ T cells displayed a bias in the TCR repertoire towards TRAV1‐2 in 2/3 donors, while the third donor made exclusive use of TRAV6 (Figure 5). Only TRBV19 was shared across 2 donors, while a range of other TRBVs was utilised, the most common being TRBV27, TRBV5‐4, TRBV10‐2 and TRBV3 (Figure 5). In the single αβTCR pairs that were able to be resolved, we saw pairings of TRAV6/TRBV27 (Supplementary table 3). The CDR3α length varied from 13 to 16 amino acids long, while the CDR3β was between 14 and 17 amino acids, with no clear motifs identified from the few clonotypes of each length available for analysis (Supplementary figure 7).
CD8+ T cells specific for B18/NS2111 likewise used a variety of αβTCRs for recognition (Figure 5). The two αTCRs resolved displayed TRAV6 as well as TRAV8‐6, which both paired with TRBV4‐2 (Figure 5, Supplementary table 4). TRBV20‐1 was identified in 2/3 of donors, while other prevalent TRBVs included TRBV12 and TRBV27 (Figure 5). Similar to both NP219 and PB1177, the CDR3α and CDR3β length varied from 15 to 17 and 13 to 17 amino acids long, respectively, with no distinct motifs observed from the data available (Supplementary figure 7).
Similar to B18/NS2111, the αTCRs specific to B18/M15 that were resolved used TRAV20 or TRAV14 (Figure 5) paired with TRBV4‐1 or TRBV27, respectively, where αβTCR pairs were resolved (Supplementary table 5). TRBV27 was identified in 2/3 donors, while a few other TRBV genes were utilised including TRBV10‐3, TRBV4‐1/4–3, TRBV18 and TRBV19. Once again, the CDR3α length varied from 12 to 14 amino acids, while the CDR3β length varied from 14 to 17 amino acids, with no distinct motifs observed from the data available (Supplementary figure 7).
When looking at the αβTCR repertoires specific to the four distinct epitopes within an individual donor, no distinct TRAV gene usage biases were observed (Figure 5a). The only TCRα that appeared across multiple epitopes in any of the donors was TRAV14, which was observed in the B18/NP219 and B18/M15‐specific TCR repertoires of SG131, with very different CDR3α sequences of ‘CAMRPYGGSQGNLIF’ and ‘CAMREDGNKLVF’, respectively (Figure 5a, Supplementary tables 2 and 5).
In comparison, several TRBV genes were detected within multiple epitope‐specific αβTCR repertoires within each donor (Figure 5b). Of the three donors, SG10 had the most TRBV genes shared across the four peptide‐specific αβTCR repertoires. TRBV19 was identified in both the B18/NP219 and B18/PB1177‐specific TCR repertoires (Figure 5b). TRBV2 was identified in both the B18/NP219 and the B18/NS2111‐specific TCR repertoires (Figure 5b). TRBV10‐3 was detected in both the B18/PB1177 and B18/M15‐specific TCR repertoires and TRBV20‐1 was observed in the B18/PB1177 and B18/NS2111‐specific TCR repertoires (Figure 5b). Finally, TRBV4‐3 was seen across 3/4 epitope‐specific TCR repertoires in donor SG10, including B18/NP219, B18/NS2111 and B18/M15 (Figure 5b). Donor SG115 had only a single shared TRBV gene across multiple epitopes, namely TRBV4‐3, seen in both the B18/NP219 and B18/M15‐specific TCR repertoires (Figure 5b, Supplementary tables 2 and 5). Lastly, Donor SG131 had two shared TRBVs across the four epitopes. TRBV19 was identified in both the B18/NP219 and B18/M15‐specific TCR repertoires, while TRBV27 was seen in the B18/PB1177 and B18/M15‐specific TCR repertoires (Figure 5b). Importantly, despite being from the same donor, recognising peptides restricted by the same HLA molecule and sharing a TRBV bias, the CDR3β sequences were unique (Supplementary tables 2–5).
Discussion
As influenza viruses continue to cause significant morbidity and mortality annually, there is an urgent need for the design and production of a new generation of influenza vaccines that provide long‐lasting protection against different influenza virus strains. One potential answer to this may be the activation of long‐lived CD8+ T cells. 28 CD8+ T cells typically recognise internal influenza‐derived peptides that are less likely to be mutated and hence more conserved throughout different strains. However, one significant challenge towards designing a CD8+ T cell‐mediated vaccine is the polymorphism of HLA‐I, each with their own peptide‐binding preferences. 25 As such, there is a need to identify and characterise novel conserved and highly immunogenic peptides restricted to a range of prevalent HLA‐I molecules to expand the pool of epitopes that could be selected for future vaccine design.
Our study addresses this by characterising novel influenza A virus‐derived peptides that were predicted to bind to the HLA‐B*18:01 molecule. The HLA‐B*18:01 allele is ranked 15th in the world in terms of HLA‐B frequency and accounts for roughly 2.3% of the global population. 17 This extends to roughly 183 million individuals and is prevalent across Europe, Africa, the Middle East and Southeast Asia. 17 Therefore, given its prevalence it would represent a potential target, in combination with other HLA molecules, towards the development of broadly applicable T‐cell‐based vaccine for the global population. 21 , 29
A recent study identified six novel influenza A virus‐derived peptides predicted to be restricted to HLA‐B*18:01. 20 Our results identified three of the six peptides, namely PB1177, NS2111 and M15 to be immunogenic in multiple of the samples tested, with different donors responding to different levels. Tetramer staining then confirmed the HLA‐B*18:01 restriction of these three peptides. We then compared CD8+ T cell responses towards these peptides versus the known highly immunogenic HLA‐B*18:01 NP219 peptide. 11 , 21 A striking immunogenicity hierarchy was observed across all donors, with CD8+ T cell responses strongest towards NP219, followed by M15, PB1177 and finally NS2111. The immunodominance hierarchy can be influenced by many factors 30 , 31 including naïve precursor frequency of responding CD8+ T cells, 32 , 33 other HLA‐I expression within an individual, 34 antigen dose, 32 the cell processing and presenting the peptide to naïve CD8+ T cells, 35 TCR affinity. 36 We assessed whether the stability of the pHLA complexes correlated with the immunodominance hierarchy and found that the four peptides were all able to stabilise the HLA‐B*18:01 molecule similarly, with a Tm value averaging 63 °C and as such, the pHLA complexes stability had no influence on the immunodominance hierarchy.
CD8+ T cells capable of exhibiting multiple effector functions have long been considered to provide superior protection against viral infections. 37 Indeed, a study has shown that vaccination against the vaccinia virus induces high levels of polyfunctional T cell responses and has been associated with an increase of CD8+ T cells producing IFN‐γ. 38 All four of the peptides, we tested induced polyfunctional CD8+ T cell responses across a range of permutations. IFN‐γ was consistently the most expressed cytokine across all the peptides and donors. Interestingly, the three most immunogenic peptides, namely NP219, M15 and PB1177 displayed up to 4 effector functions, while NS2111 with the lowest immunogenicity, displayed at most 3 functions. This is consistent with studies that have shown that more immunogenic peptides induce increased polyfunctionality and may suggest an increase in antiviral protection. 34 , 39 Thus, it is important to consider not only the level of immunogenicity, but also the level of polyfunctionality when picking targets for future vaccine design.
We characterised the presentation of the four epitopes by HLA‐B*18:01 by X‐ray crystallography. The crystal structures revealed that all four epitopes shared the HLA‐B*18:01‐restricted peptides canonical primary anchor residues and a secondary anchor residue at position 5 of the peptide, 25 which might provide a basis for the similar stability observed. αβTCR docking occurs mainly ‘on the top’ of the pHLA and that canonical interaction between TCR and peptide occurs by primary contact from the CDR3 loops wrapped around a few key peptide residues. 16 Therefore, the conformation of the NS2111 P4‐Arg, shielding a portion of the peptide and the HLA α2‐helix, may generate an unfavorable surface for TCR interaction. Compared with the three other more immunogenic peptides (NP219, M15 and PB1177), the central part of the NS2111 would limit the ability of the CDR loop to wrap around the residues side chains without structural rearrangement. This might explain the lower immunogenicity of the NS2111 peptide observed in the samples tested here.
Aside from a two TCRs recognising melanoma‐associated antigens, 40 , 41 there is no information of the TCR repertoire of CD8+ T cells recognising pathogen‐derived peptides presented by HLA‐B*18:01. The TCR repertoire was entirely private, with no shared clonotypes between donors. This is unsurprising as TCRs can exhibit a huge diversity, with some estimating that αβTCR diversity ranges from 2.5 × 107 to 3.8 × 108 in a single individual. 42 , 43 Within each peptide, few TCR biases were seen between donors, with at most 2/3 of donors utilising the same TRAV or TRBV in the recognition of the same peptide. CDR3 lengths ranged from 12 to 18 amino acids long in line with other TCR repertoires published across a range of HLA‐I molecules 24 , 27 , 44 and no distinct CDR3 motifs were evident from the available data. Interestingly, although these 4 peptides are all presented by HLA‐B*18:01, there were no TCR biases evident within donors recognising each of the 4 peptides.
In summary, we screened six newly reported influenza A‐derived peptides and identified three new immunogenic peptides restricted to the prevalent HLA‐B*18:01 molecule in samples from several donors. When compared to the known highly immunogenic NP219 peptide, a clear immunodominance hierarchy was observed comprising the strongest CD8+ T cell responses towards NP219, followed by M15, PB1117 and NS2111, and this immunodominance hierarchy was not dependent on pHLA stability. We then present the first structures of HLA‐B*18:01 presenting immunogenic pathogen‐derived epitopes showing a conserved conformation and the presence of a secondary anchor residue at position 5 of the peptide that provide the basis for the similar pHLA stability observed. Finally, we report the first TCR repertoire of CD8+ T cells recognising pathogen‐derived peptides presented by HLA‐B*18:01, showing that private TCR repertoires were used with little evidence of TCR biases. Overall, this study provides new knowledge for the field and suggests that both NP219 and M15, which are well conserved and induce immunodominant, polyfunctional CD8+ T cell, may be of interest for future influenza vaccine design.
Methods
Donors, ethics and peripheral blood mononuclear cells
Whole peripheral blood from volunteers and Buffy coats that were obtained from the Australian Red Cross Lifeblood were the source of peripheral blood mononuclear cells (PBMCs) used in all cellular experiments. PBMCs were isolated using the Ficoll density gradient centrifugation method as previously described. 24 , 27 PBMCs were cryogenically stored until required. HLA typing was undertaken by CareDx Pty Ltd, Fremantle, Western Australia (AlloSeq Tx). All research was approved by the La Trobe University Human Ethics Committee (Approval No: HEC21097) and undertaken in accordance with the declaration of Helsinki. Donor age and sex are listed in Table 3.
Table 3.
Donor details
| Donor | Age | Sex |
|---|---|---|
| SG10 | 35 | Female |
| SG26 | 29 | Male |
| SG112 | 43 | Female |
| SG115 | 41 | Male |
| SG131 | 53 | Male |
HLA‐B*18:01+ Donors whose samples were used in this study.
Peptide sequence alignment and conservation analysis
The WHO recommended 2022–2023 Southern Hemisphere Influenza virus vaccine strains A/Darwin/9/2021 (H3N2) and A/Victoria/2570/2019 (H1N1) 45 proteins were obtained from the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/). 46 Sequences were aligned to the peptides of interest using Clustal Omega Multiple Sequence Alignment (https://www.ebi.ac.uk/Tools/msa/clustalo/) 47 and are reported in Table 1. Full‐length ‘Vaccine only’ influenza A virus strains of M1, NS2, PB1 and NP were obtained from the NCBI Influenza Research Database https://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph‐select.cgi?go=database 22 on the 2nd of February 2024. Sequences were aligned to the peptides of interest using Clustal Omega Multiple Sequence Alignment (https://www.ebi.ac.uk/Tools/msa/clustalo/) 47 and conservation, as a proportion of strains with an identical amino acid sequence, is reported in Table 2.
Peptides and the generation of pool and peptide‐specific CD8+ T cell lines
Pure (> 80%) peptides TEVETYVL (M15), VEQEIRTF (NS2109), QEIRTFSF (NS2111), TENSFEQITF (NS290), TELKLSDY (NP45), EEIEITTHF (PB1177) and YERMCNIL (NP219) were ordered from GenScript Biotech (Piscataway, USA). CD8+ T cell lines were generated as previously described. 48 Briefly, PBMCs from healthy HLA‐B*18:01+ individuals were stimulated with either a pool of peptides (peptide pool) or were stimulated with individual peptides to generate peptide‐specific T cell lines. One‐third of PBMCs were peptide‐pulsed with either 2 μm/peptide for pooled CD8+ T cell lines, or 10 μm of peptide for peptide‐specific CD8+ T cell lines. Peptide‐pulsed PBMCs were then incubated for 90 min at 37 °C with 5% CO2. The peptide‐pulsed PBMCs were then washed twice by centrifugation and added to the remaining two‐thirds of responder PBMCs. Cell lines were then cultured for 10+ days in RPMI‐1640 (Gibco, New York, USA) supplemented with 1× Penicillin Streptomycin Glutamine (Gibco), 1× non‐essential amino acids (100× NEAA; Gibco), 2 mM l‐glutamine (Sigma‐Aldrich, St. Louis, USA), 5 mM HEPES (Sigma‐Aldrich), 50 μm β‐mercaptoethanol (Sigma‐Aldrich) and foetal calf serum (FCS; Sigma‐Aldrich). On day 4, 10 IU mL−1 of recombinant human IL2 (Peprotech, Rocky Hill, USA) was added, then twice weekly or as necessary.
Protein production and purification
The sequence encoding HLA‐B*18:01 heavy chain was obtained from the IPD‐IMGT/HLA database. 14 The cDNA construct for the soluble protein insert, excluding the transmembrane domain, was sub‐cloned into the pET30 plasmid vector using NedI/HindIII restriction enzymes (GenScript, Piscataway, USA). The recombinant protein and human β2‐microglobulin (β2m) were then individually expressed in BL21‐RIL Escherichia coli cells, resulting in inclusion bodies that were extracted and purified through centrifugation. 49 The peptides described in this study were chemically synthesised (GenScript; Tables 1 and 2). Refolding was performed using 5 mg of peptide, 30 mg of HLA‐B*18:01 heavy chain and 10 mg of β2m in refolding buffer [3 m Urea (ThermoFisher, Scoresby, VIC, Australia), 0.4 m l‐Arginine, (Merck, Darmstadt, Germany) 0.1 m Tris–HCl pH 8 (ThermoFisher), 2 mm Na‐EDTA pH 8 (Merck), 0.16% w/v reduced Glutathione (Goldbio, St Louis, USA) and 0.03% w/v oxidised Glutathione (Goldbio)]. This solution was dialyzed in 10 mM Tris–HCl pH 8 (ThermoFisher) and the peptide‐HLA‐B*18:01 complexes were purified using a two‐stage anion exchange chromatography (Cytiva, Marlborough, Massachusetts, USA).
Generation of monomers and tetramers
The HLA‐B*18:01 heavy chain with a BirA enzyme tag was obtained as described above (GenScript). Subsequently, after anion exchange, the protein complex solution was desalted using a HiTrap column (Cytiva). The monomeric protein was combined with 10% v/v 0.5 m bicine pH 8.3 (Avidity, Aurora, USA), 10% v/v 100 mM ATP, 100 mM magnesium acetate, 500 μm D‐biotin (Avidity) and 5 μg of biotin protein ligase per mg of protein. After overnight incubation at 4 °C, the solution was loaded onto a size exclusion (Superdex S200 10/300, Marlborough, USA) column. The pure protein fraction was pooled and the amount of biotinylated protein monomer was measured via native gel electrophoresis. Each peptide‐HLA‐B*18:01 monomer was conjugated at an 8:1 peptide‐HLA to Streptavidin‐PE molar ratio (BD Biosciences, Franklin Lakes, USA) to form tetramers of HLA‐B*18:01 presenting M15, NS2111, PB1177 or NP219 (hereafter, tetramers and tetramer+ cells are referred to as B18/M15, B18/NS2111, B18/PB1177 or B18/NP219, respectively).
Tetramer staining and single‐cell sorting of peptide‐specific CD8+ T cell lines
CD8+ T cell lines were stained with 1 μL of tetramer (1:100) and incubated at room temperature for 1 h in the dark. Cells were then washed and surface stained with CD3‐BV480 (1:100; BD Biosciences), anti‐human CD19‐APCH7 (1:100; BD Biosciences) anti‐human CD8‐PerCPCy5.5 (1:50; BD Biosciences), CD4‐BV680 (1:50; BD Biosciences) anti‐human CD14‐APCH7 (1:200; BD Biosciences), Live/Dead‐NIR (1:1000; Molecular Probes, Eugene, USA), CCR7‐PeCy7 (1:50; BD Biosciences), CD27‐APC (1:100; BD Biosciences), CD45RA‐FITC (1:100; BD Biosciences), CD95‐BV421 (1:50; BD Biosciences) and PD1‐BV605 (1:50; BD Biosciences). Cells were then washed and either resuspended with 100 μL per well of 1% PFA (Thermoscientific) and acquired using the Beckman Coulter CytoflexS (Beckman Coulter) or resuspended in PBS and single‐cell sorted into 96‐well PCR plates (Eppendorf, Hamburg, Germany) on the BD FACSAria Fusion (BD Biosciences). Plates were centrifuged at 1200 g and then stored at −80 °C until use. All data were gated as per Supplementary figure 1 using FlowJo version 10.8.1.
Single‐cell multiplex PCR
Multiplex polymerase chain reaction (PCR) of tetramer+ CD8+ T cells single‐cell sorted above was undertaken as previously described. 24 First, a reverse transcription cDNA synthesis was completed using a cDNA synthesis kit (Invitrogen, Waltham, USA) at 1/20 of the manufacturer's recommendation. Briefly, 2.5 μL of 5× VILO (Invitrogen, Waltham, USA), 10× superscript (Ingen, Waltham, USA), 1% Triton X (G‐Biosciences, St Louis, USA) and HPLC (Sigma‐Aldrich) water was added per well. Plates were cycled at 25 °C for 10 min, 42 °C for 120 min, 85 °C for 5 min and 4 °C hold. Next, an external TCR amplification was completed by adding 22.5 μL/well of 10× PCR buffer (Qiagen, Hilden, Germany), dNTPs (Thermoscientific), TRAV, TRAC, TRBV and TRBC external primers (Sigma‐Aldrich), Taq polymerase (Qiagen) and HPLC. Following this, an internal TCR amplification was then prepared by transferring 2.5 μL of the external PCR product into a new PCR plate and adding 22.5 μL of 10× PCR buffer, dNTPs, either TRAV and TRAC or TRBV and TRBC internal primers (Sigma‐Aldrich), Taq polymerase and HPLC. Plates were cycles as per the internal PCR round. PCR products were confirmed on a 1–2% DNA agarose gel. Lastly, 5 μL of all PCR products were added to a new PCR plate with 1 μL of ExoSap‐IT (Applied Biosystems, Waltham, USA). Products were then purified at 37 °C for 15 min and 80 °C for 15 min. An aliquot of 2 μL of purified PCR product was added to 10 μL of TRAC or TRBC internal primers with HPLC and were sent to the Australian Genome Research Facility (AGRF) for sequencing.
Intracellular cytokine staining assay
Intracellular cytokine staining was undertaken as previously described. 27 Peptide‐expanded CD8+ T cell lines were counted using the trypan blue exclusion methods and 1–2 × 105 cells/well were utilised for all conditions. For CD8+ T cell lines raised against peptide pools or individual peptides, 2 μm per peptide or 10 μm of the cognate peptide were added, respectively, as well as no peptide as a negative control and cell stimulation cocktail 500× (eBiosciences, San Diego, USA) as a positive control. Cells were then incubated at 37 °C with 5% CO2 for 5 h with the presence of 1:1000 Golgi‐Plug (BD Biosciences), 1:1400 Golgi‐stop (BD Biosciences) and 1:200 dilution of anti‐human CD107a‐AF488 (eBiosciences). Following incubation, cells were surface stained for 30 min at 4 °C with anti‐human CD4‐BV650 (1:100; BD Biosciences), anti‐human CD3‐BV480 (1:100; BD Biosciences), anti‐human CD8‐PerCPCy5.5 (1:50; BD Biosciences), anti‐human CD19‐APCH7 (1:100; BD Biosciences), anti‐human CD14‐APCH7 (1:100; BD Biosciences) and Live/Dead‐NIR (1:1000; Molecular Probes, Eugene, USA). Cells were washed in PBS, then fixed and permeabilised using BD‐Fix Perm buffer (BD Biosciences) for 20 min at 4 °C. Lastly, cells were intracellularly stained with anti‐human IFNγ‐BV421 (1:100; BD Biosciences), anti‐human TNF‐PeCy7 (1:100; BD Biosciences), anti‐human MIP‐1b‐APC (1:100; BD Biosciences) and anti‐human IL2‐PE (1:50; BD PharMingen). All samples were acquired on the BD FACSymphony A3 analyser (BD Biosciences) or the Beckman Coulter CytoflexS (Beckman Coulter, Brea, USA) and were examined via FlowJo (BD Biosciences) version 10.8.1. All samples were gated as per Supplementary figure 1.
Crystallisation and crystal structure determination
The purified peptide‐HLA‐B*18:01 complexes in 10 mM Tris–HCl pH 8 (ThermoFisher) and 150 mM NaCl (ThermoFisher) were used to grow crystals via hanging drop vapour diffusion at 20 °C, with a reservoir‐to‐drop ratio of 1:1. Crystals of HLA‐B*18:01‐NP219 and ‐M15 were grown in a solution containing 0.2 m Potassium Nitrate, 20% w/v PEG 3350 at pH 6.5 (Hampton Research, Aliso Viejo, USA). Crystals of HLA‐B*18:01‐NS2111 and ‐PB1177 grew in a solution containing 0.2 m Lithium Acetate, 20% w/v PEG 3350 (Hampton Research) and 10 mM MgCl2 (Merck). The crystals were subsequently soaked in a solution containing the mother liquor with the PEG 3350 (ThermoFisher) concentration increased to 30% for cryoprotection prior to being flash frozen in liquid nitrogen. Diffraction data were collected using the MX2 beamline as the Australian Synchrotron, Melbourne, Australia. 50 The data were processed using XDS 51 and the CCP4 suite 52 PHASER program 53 was utilised for molecular replacement, using HLA‐B*18:01 without the peptide as model (PDB code: 6MT3). 24 Manual model building was carried out with COOT, 54 followed by further refinement using PHENIX (version 1–9). 55 All molecular representations were created using PyMOL (Schrodinger v2.5). The final models were validated and deposited using the PDB OneDep System with the following PDB accession codes: 8ROO for HLA‐B*18:01‐NP219; 8RNG for HLA‐B*18:01‐M15; 8ROP for HLA‐B*18:01‐NS2111; and 8RNH for HLA‐B*18:01‐PB1177.
Thermal melting assay using differential scanning fluorimetry
The thermal stability of each peptide‐HLA‐B*18:01 complex was assessed using the ViiA 7 real‐time PCR system (ThermoFisher, Scoresby, Australia), with excitation and emission channels set to the TAMRA reporter (x3−m3 filter) at an excitation wavelength of approximately 550 nm and detection at about 587 nm. Protein complexes were diluted to concentrations of 0.5 and 1 mg mL−1 using 10 mM Tris–HCl pH 8 and 150 mM NaCl with duplicate samples for each concentration. SYPRO Orange dye (ThermoFisher) was subsequently added to reach a final concentration of 10×. Each sample underwent heating from 25 °C to 95 °C at a rate of 1 °C min−1. Normalisation of the obtained fluorescence data was performed using GraphPad Prism 9 (v9.3), with the thermal melting point (Tm) value for each complex determined at 50% of the maximum fluorescence intensity. This value corresponds to the approximate temperature at which 50% of the protein has unfolded. The thermal melting assay was performed with 2 independent samples in duplicate at two concentrations.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Samuel Liwei Leong: Data curation; formal analysis; funding acquisition; investigation; methodology; project administration; validation; writing – original draft; writing – review and editing. Lawton Murdolo: Data curation; formal analysis; funding acquisition; investigation; methodology; project administration; validation; writing – original draft; writing – review and editing. Janesha C Maddumage: Data curation; formal analysis; investigation; methodology; project administration; supervision; validation; writing – original draft; writing – review and editing. Marios Koutsakos: Funding acquisition; resources; writing – review and editing. Katherine Kedzierska: Funding acquisition; resources; writing – review and editing. Anthony W Purcell: Funding acquisition; methodology; writing – review and editing. Stephanie Gras: Conceptualization; data curation; formal analysis; funding acquisition; project administration; supervision; validation; writing – original draft; writing – review and editing. Emma J Grant: Conceptualization; data curation; formal analysis; funding acquisition; project administration; supervision; validation; writing – original draft; writing – review and editing.
Supporting information
Supporting Information
Acknowledgments
We thank the Australian Red Cross Lifeblood and all our volunteer donors for the supply of samples used in this study. We acknowledge the Bioimaging Platform, La Trobe University for flow cytometry undertaken in this study. We also acknowledge the Australian Synchrotron for all X‐ray crystallography experiments. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. We also thank Dr Nicole Mifsud and Dr Patricia Illing for discussions and advice related to this project. SLL is supported by the La Trobe University RTP Scholarship and an AINSE Ltd. Honours Scholarship. LM is supported by the La Trobe University RTP Scholarship and an AINSE Ltd Top Up Scholarship. MK is supported by the NHMRC Emerging Leader Investigator Grant (#1195698). KK is supported by the NHMRC Leadership Investigator Grant (#1173871). AWP is supported by the NHMRC Investigator Grant (#2016596). SG is supported by the NHMRC Senior Research Fellowship (#1159272) and EJG is supported by the Australian Research Council DECRA Fellowship (DE210101479) and the AINSE Early Career Research Grant. Open access publishing facilitated by La Trobe University, as part of the Wiley ‐ La Trobe University agreement via the Council of Australian University Librarians.
Contributor Information
Stephanie Gras, Email: s.gras@latrobe.edu.au.
Emma J Grant, Email: e.grant@latrobe.edu.au.
Data availability statement
The crystal structure data that support the findings of this study are openly available in the Protein Data Bank (https://www.rcsb.org/) and PDB code accession number are summarised in Supplementary table 1. The data that support the findings of this study are available from the corresponding author, EJG, upon reasonable request.
References
- 1. Hay AJ, McCauley JW. The WHO global influenza surveillance and response system (GISRS)‐a future perspective. Influenza Other Respir Viruses 2018; 12: 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guthmiller JJ, Utset HA, Wilson PC. B cell responses against influenza viruses: Short‐lived humoral immunity against a life‐long threat. Viruses 2021; 13: 965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grant EJ, Quiñones‐Parra SM, Clemens EB, Kedzierska K. Human influenza viruses and CD8+ T cell responses. Curr Opin Virol 2016; 16: 132–142. [DOI] [PubMed] [Google Scholar]
- 4. Eiden J, Volckaert B, Rudenko O et al. M2‐deficient single‐replication influenza vaccine–induced immune responses associated with protection against human challenge with highly drifted H3N2 influenza strain. J Infect Dis 2022; 226: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orrico‐Sánchez A, Valls‐Arévalo Á, Garcés‐Sánchez M, Álvarez Aldeán J, Ortiz de Lejarazu Leonardo R. Efficacy and effectiveness of influenza vaccination in healthy children. A review of current evidence. Enferm Infecc Microbiol Clin (Engl Ed) 2023; 41: 396–406. [DOI] [PubMed] [Google Scholar]
- 6. Agor JK, Özaltın OY. Models for predicting the evolution of influenza to inform vaccine strain selection. Hum Vaccin Immunother 2018; 14: 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McMichael AJ, Gotch FM, Noble GR, Beare PAS. Cytotoxic T‐cell immunity to influenza. N Engl J Med 1983; 309: 13–17. [DOI] [PubMed] [Google Scholar]
- 8. Leong SL, Gras S, Grant EJ. Fighting flu: Novel CD8+ T‐cell targets are required for future influenza vaccines. Clin Transl Immunology 2024; 13: e1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sridhar S, Begom S, Bermingham A et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med 2013; 19: 1305–1312. [DOI] [PubMed] [Google Scholar]
- 10. Clemens EB, Grant EJ, Wang Z et al. Towards identification of immune and genetic correlates of severe influenza disease in indigenous Australians. Immunol Cell Biol 2016; 94: 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grant E, Wu C, Chan KF et al. Nucleoprotein of influenza A virus is a major target of immunodominant CD8+ T‐cell responses. Immunol Cell Biol 2013; 91: 184–194. [DOI] [PubMed] [Google Scholar]
- 12. Wieczorek M, Abualrous ET, Sticht J et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front Immunol 2017; 8: 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kreijtz JHCM, De Mutsert G, Van Baalen CA, Fouchier RAM, Osterhaus ADME, Rimmelzwaan GF. Cross‐recognition of avian H5N1 influenza virus by human cytotoxic T‐lymphocyte populations directed to human influenza A virus. J Virol 2008; 82: 5161–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barker DJ, Maccari G, Georgiou X et al. The IPD‐IMGT/HLA database. Nucleic Acids Res 2023; 51: D1053–D1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gonzalez‐Galarza FF, McCabe A, Santos E et al. Allele frequency net database (AFND) 2020 update: Gold‐standard data classification, open access genotype data and new query tools. Nucleic Acids Res 2020; 48: D783–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szeto C, Lobos CA, Nguyen AT, Gras S. TCR recognition of peptide‐MHC‐I: Rule makers and breakers. Int J Mol Sci 2020; 22: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Solberg OD, Mack SJ, Lancaster AK et al. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: A meta‐analytic review of 497 population studies. Hum Immunol 2008; 69: 443–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vita R, Mahajan S, Overton JA et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 2019; 47: D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu C, Zanker D, Valkenburg S et al. Systematic identification of immunodominant CD8+ T‐cell responses to influenza A virus in HLA‐A2 individuals. Proc Natl Acad Sci USA 2011; 108: 9178–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nicholas B, Bailey A, Staples KJ, Wilkinson T, Elliott T, Skipp P. Immunopeptidomic analysis of influenza A virus infected human tissues identifies internal proteins as a rich source of HLA ligands. PLoS Pathog 2022; 18: e1009894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quiñones‐Parra S, Grant E, Loh L et al. Preexisting CD8+ T‐cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc Natl Acad Sci USA 2014; 111: 1049–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bao Y, Bolotov P, Dernovoy D et al. The influenza virus resource at the National Center for Biotechnology Information. J Virol 2008; 82: 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rist MJ, Theodossis A, Croft NP et al. HLA peptide length preferences control CD8+ T cell responses. J Immunol 2013; 191: 561–571. [DOI] [PubMed] [Google Scholar]
- 24. Grant EJ, Josephs TM, Loh L et al. Broad CD8+ T cell cross‐recognition of distinct influenza A strains in humans. Nat Commun 2018; 9: 5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen AT, Szeto C, Gras S. The pockets guide to HLA class I molecules. Biochem Soc Trans 2021; 49: 2319–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Theodossis A, Guillonneau C, Welland A et al. Constraints within major histocompatibility complex class I restricted peptides: Presentation and consequences for T‐cell recognition. Proc Natl Acad Sci USA 2010; 107: 5534–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nguyen AT, Lau HMP, Sloane H et al. Homologous peptides derived from influenza A, B and C viruses induce variable CD8+ T cell responses with cross‐reactive potential. Clin Transl Immunology 2022; 11: e1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klebanoff CA, Chandran SS, Baker BM, Quezada SA, Ribas A. T cell receptor therapeutics: Immunological targeting of the intracellular cancer proteome. Nat Rev Drug Discov 2023; 22: 996–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koutsakos M, Illing PT, Nguyen THO et al. Human CD8+ T cell cross‐reactivity across influenza A, B and C viruses. Nat Immunol 2019; 20: 613–625. [DOI] [PubMed] [Google Scholar]
- 30. Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I‐restricted T lymphocyte responses. Annu Rev Immunol 1999; 17: 51–88. [DOI] [PubMed] [Google Scholar]
- 31. Kedl RM, Kappler JW, Marrack P. Epitope dominance, competition and T cell affinity maturation. Curr Opin Immunol 2003; 15: 120–127. [DOI] [PubMed] [Google Scholar]
- 32. La Gruta NL, Kedzierska K, Pang K et al. A virus‐specific CD8+ T cell immunodominance hierarchy determined by antigen dose and precursor frequencies. Proc Natl Acad Sci USA 2006; 103: 994–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi EY, Christianson GJ, Yoshimura Y et al. Immunodominance of H60 is caused by an abnormally high precursor T cell pool directed against its unique minor histocompatibility antigen peptide. Immunity 2002; 17: 593–603. [DOI] [PubMed] [Google Scholar]
- 34. Sant S, Quiñones‐Parra SM, Koutsakos M et al. HLA‐B*27:05 alters immunodominance hierarchy of universal influenza‐specific CD8+ T cells. PLoS Pathog 2020; 16: e1008714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crowe SR, Turner SJ, Miller SC et al. Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J Exp Med 2003; 198: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sette A, Vitiello A, Reherman B et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol 1994; 153: 5586–5592. [PubMed] [Google Scholar]
- 37. Almeida JR, Price DA, Papagno L et al. Superior control of HIV‐1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 2007; 204: 2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Precopio ML, Betts MR, Parrino J et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8+ T cell responses. J Exp Med 2007; 204: 1405–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Streeck H, Brumme ZL, Anastario M et al. Antigen load and viral sequence diversification determine the functional profile of HIV‐1–specific CD8+ T cells. PLoS Med 2008; 5: e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goncharov M, Bagaev D, Shcherbinin D et al. VDJdb in the pandemic era: A compendium of T cell receptors specific for SARS‐CoV‐2. Nat Methods 2022; 19: 1017–1019. [DOI] [PubMed] [Google Scholar]
- 41. Murata K, Nakatsugawa M, Rahman MA et al. Landscape mapping of shared antigenic epitopes and their cognate TCRs of tumor‐infiltrating T lymphocytes in melanoma. elife 2020; 9: e53244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science 1999; 286: 958–961. [DOI] [PubMed] [Google Scholar]
- 43. Sun X, Nguyen T, Achour A et al. Longitudinal analysis reveals age‐related changes in the T cell receptor repertoire of human T cell subsets. J Clin Invest 2022; 132: e158122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lineburg KE, Grant EJ, Swaminathan S et al. CD8+ T cells specific for an immunodominant SARS‐CoV‐2 nucleocapsid epitope cross‐react with selective seasonal coronaviruses. Immunity 2021; 54: 1055–1065.e1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. WHO . Recommended composition of influenza virus vaccines for use in the 2022–2023 northern hemisphere influenza season. Available from: https://www.who.int/publications/m/item/recommended-composition-of-influenza-virus-vaccines-for-use-in-the-2022-2023-northern-hemisphere-influenza-season.
- 46. Sayers EW, Bolton EE, Brister JR et al. Database resources of the national center for biotechnology information. Nucleic Acids Res 2022; 50: D20–D26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Madeira F, Pearce M, Tivey ARN et al. Search and sequence analysis tools services from EMBL‐EBI in 2022. Nucleic Acids Res 2022; 50: W276–W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grant EJ, Gras S. Protocol for generation of human peptide‐specific primary CD8+ T cell lines. STAR Protoc 2022; 3: 101590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chatzileontiadou DSM, Szeto C, Jayasinghe D, Gras S. Protein purification and crystallization of HLA‐A*02:01 in complex with SARS‐CoV‐2 peptides. STAR Protoc 2021; 2: 100635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McPhillips TM, McPhillips SE, Chiu HJ et al. Blu‐ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 2002; 9: 401–406. [DOI] [PubMed] [Google Scholar]
- 51. Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr 2010; 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Agirre J, Atanasova M, Bagdonas H et al. The CCP4 suite: Integrative software for macromolecular crystallography. Acta Crystallogr D Struct Biol 2023; 79: 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010; 66: 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Afonine PV, Grosse‐Kunstleve RW, Echols N et al. Towards automated crystallographic structure refinement with phenix.Refine. Acta Crystallogr D Biol Crystallogr 2012; 68: 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The crystal structure data that support the findings of this study are openly available in the Protein Data Bank (https://www.rcsb.org/) and PDB code accession number are summarised in Supplementary table 1. The data that support the findings of this study are available from the corresponding author, EJG, upon reasonable request.