

Abstract
Carbohydrate analogs are an important, well-established class of clinically useful medicinal agents that exhibit potent antimicrobial activity. Thus, we explored the various therapeutic potential of methyl α-D-mannopyranoside (MαDM) analogs, including their ability to synthesize and assess their antibacterial, antifungal, and anticancer properties; additionally, molecular docking, molecular dynamics simulation, and ADMET analysis were performed. The structure of the synthesized MαDM analogs was ascertained by spectroscopic techniques and physicochemical and elemental analysis. In vitro antimicrobial activity was assessed and revealed significant inhibitory effects, particularly against gram-negative bacteria along with the prediction of activity spectra for substances (PASS). Concurrently, MαDM analogs showed good results against antifungal pathogens and exhibited promising anticancer effects in vitro, demonstrating dose-dependent cytotoxicity against Ehrlich ascites carcinoma (EAC) cancer cells while sparing normal cells from compound 5, with an IC50 of 4511.65 µg/mL according to the MTT colorimetric assay. A structure–activity relationship (SAR) study revealed that hexose combined with the acyl chains of decanoyl (C-10) and benzenesulfonyl (C6H5SO2-) had synergistic effects on the bacteria and fungi that were examined. Molecular docking was performed against the Escherichia coli (6KZV) and Candida albicans (1EAG) proteins to acquire insights into the molecular interactions underlying the observed biological activities. The docking results were further supported by 100 ns molecular dynamics simulations, which provided a dynamic view of the stability and flexibility of complexes involving MαDM and its targets. In addition, ADMET analysis was used to evaluate the toxicological and pharmacokinetic profiles. Owing to their promising drug-like properties, these MαDM analogs exhibit potential as prospective therapeutic candidates for future development.
Keywords: Mannopyranoside (MαDM), Antimicrobial, Anticancer, Molecular docking, MD, ADMET
1. Introduction
The therapeutic potential of small molecules and natural compounds has been increasing in recent years, as they offer promising opportunities to develop novel drugs for various medical conditions (Newman and Cragg, 2020, Thomford et al., 2018). The most common biological material on earth is carbohydrates, which have a variety of physiological and physical characteristics and various health advantages (Cummings et al., 2010; J. Wang et al., 2021). Numerous studies have been conducted on the use of carbohydrates and their derivatives as therapeutic agents for bacterial and viral infections, cancer, diabetes, cardiovascular disease, neurological system abnormalities, and many other conditions (Cao et al., 2022, Oppenheimer et al., 2008). The vast array of structural variations, including differences in functional groups, linkages, and numbers of rings, provides these molecules with great resources for designing and developing biologically active glycoconjugates (Kabir et al., 2004, Matsumoto et al., 2012, Metallo and Heiden, 2013, Mirajul et al., 2019). Carbohydrates are crucial for metabolism and enable interactions between cells by supplying the necessary energy for different cellular activities. These complex molecules are a fundamental fuel source for our bodies, enabling metabolic pathways that power basic cellular processes and more intricate functions in multicellular organisms (Jequier, 1994; Matsumoto et al., 2022). Most of the time, aromatic compounds and their derivatives in which halogen, sulfur, or nitrogen atoms are replaced have shown promise as antibacterial agents (Hosen et al., 2022, Kabir et al., 2008). Regioselective acylation enhances the antibacterial activity of carbohydrate compounds by adding heterocyclic aromatic rings with electron-donating or electron-attracting groups and introduces novel possibilities for medicinal use against microbial infections (Kawsar et al., 2022, Rana et al., 2021). Methyl α-D-mannopyranoside, a naturally occurring monosaccharide derivative, has shown promise as a therapeutic option in the present study because of its unique structural properties and range of biological activities (Farhana et al., 2021, Maowa et al., 2021, Bhat et al., 2021). Because it is involved in many biological processes, such as cell–cell recognition, signal transduction, and immune response modulation, its numerous derivatives have been researched for their therapeutic potential in great detail (Actor et al., 2009; Kawsar et al., 2011). The development of effective antiviral and antibacterial candidates has resulted from changing the hydroxyl group in nucleosides and monosaccharides by adding aliphatic and aromatic groups (Mahmoud et al., 2018). Carbohydrate-based drugs, e.g., dapagliflozin, tofogliflozin, plazomicin and azvudine may be regarded as potential therapeutic drugs for treating both previously treated and untreated patients with antidiabetic, antibacterial and antiviral agents (Fig. 1).
Fig. 1.

Marketed drugs with carbohydrate moieties.
In this research, we observed new analogs (2–7) derived from methyl α-D-mannopyranoside that bind rarely used aliphatic and aromatic groups. Density functional theory is used for accurately predicting electronic and nuclear structures in molecular systems. Moreover, HOMO and LUMO analyses are particularly valuable for assessing chemical stability and reactivity (van Mourik et al., 2014). Molecular docking, an essential aspect of drug discovery, provides insights into drug–protein binding, energy changes, and binding site identification (Meng et al., 2011). Lipophilicity is crucial in assessing both aquatic and nonaquatic toxicity profiles during drug discovery (Lobo, 2020). The ADME and toxicity profiles of monosaccharides and their derivatives were examined in this work (Pires et al., 2015). The PASS web-based tool was used to anticipate a wide range of biological effects, including anti-inflammatory, anticancer, antifungal, antiviral, antidiabetic, and antibacterial effects (Filimonov et al., 2014). In this study, acyl substituents were added to methyl α-D-mannopyranoside, which was subsequently optimized using quantum mechanical techniques, after which its thermal and electrical stability and biochemical characteristics were assessed. This study employed a number of computational tools, including density functional theory (DFT) analysis, molecular docking, molecular dynamics simulation, and MM-GBSA, to evaluate the compounds' stability, antibacterial properties, and pharmacokinetics.
2. Materials and methods
2.1. Equipment and resources
For evaporation of the solvents, the mixture was subjected to reduced pressure using a VV–1 type vacuum rotatory evaporator from Germany and a BUCHI-461 water bath from Switzerland operating at temperatures less than 40 °C. Low pressure was maintained using a 0.25H. A P vacuum pump from England was used. We used an electrothermal melting point device made in England to determine the melting points in the laboratory. The spectroscopic measurements included infrared (IR) spectra recorded at the Chemistry Department, University of Chittagong, Bangladesh, using an IR Affinity Fourier Transform Infrared Spectrophotometer (SHIMADZU, Japan) with CHCl3 or KBr discs and films. Nuclear magnetic resonance (NMR) spectra, specifically 1H NMR (400 MHz), were recorded with a CDCl3 spectrometer at the Wazed Miah Science Research Center, Jahangirnagar University, Savar, Dhaka, Bangladesh, using tetramethylsilane (TMS) as an internal standard and a Bruker DPX-400 spectrometer (400 MHz). These instruments and techniques were used for detailed investigations and structural analysis of the compounds included in the present study.
2.2. Synthesis
2.2.1. Methyl 6-O-p-toluoyl-α-D-mannopyranoside (2)
A methyl α-D-mannopyranoside (1) solution containing 200 mg (1.03 mmol) of 3 ml of dry dimethyl formamide (DMF)/triethyl amine (Et3N) was cooled to 0 °C, after which 0.15 mg (1.1 M eq.) of p-toluoyl chloride was added. After being stirred constantly for 6 h at 0 °C, the reaction mixture was left to stand overnight at room temperature. The reaction was subsequently performed using T.L.C. (methanol-chloroform, 1:5), which showed that the starting material was completely converted into a single product (Rf = 0.52). The resulting mixture was purified by silica gel column chromatography (with MeOH-CHCl3, 1:5 as the eluant) to afford the p-toluoyl derivative (2, 134.80 mg) as a crystalline solid, followed by recrystallization (with EtOAc–C6H14), m.p. 103–104 °C.
Yield: 71.40 %, Rf = 0.51; FTIR (KBr): 1680, 1711, (C = O), 3300–3500 (br., –OH) cm−1. 1H NMR (400 MHz, CDCl3) δH ppm 8.01 (2H, d, J = 8.3 Hz, Ar-H), 7.33 (2H, d, J = 8.6 Hz, Ar-H), 5.09 (1H, d, J = 3.3 Hz, H-1), 4.61 (1H, dd, J = 4.2 and 12.0 Hz, H-6a), 4.56 (1H, dd, J = 2.0 and 12.1 Hz, H-6b), 4.28 (1H, t, J = 9.0 Hz, H-4), 3.91 (1H, t, J = 9.2 Hz, H-3), 3.76 (1H, dd, J = 3.5 and 10.0 Hz, H-2), 3.73 (1H, ddd, J = 2.2, 9.4 and 12.1 Hz, H-5), 3.40 (3H, s, 1-OCH3), 2.43 (3H, s, 4-CH3. C6H4CO-, p-toluoyl). 13C NMR (100 MHz, CDCl3): δC 167.90 (4-CH3. C6H4CO-), 145.01, 131.35, 131.37, 130.22, 129.31, 129.30 (4-CH3. C6H4CO-), 97.09 (C-1), 72.92 (C-2), 71.32 (C-4), 70.62 (C-3), 69.38 (C-5), 63.05 (C-6), 55.16 (1-OCH3), 21.41 (4-CH3.C6H4CO-). LC–MS [M + 1]+ 313.12. Calcd. for C15H20O7: C = 57.59, H = 6.35, O = 35.86; found: C = 57.58, H = 6.34, O = 35.88 %.
General procedure for the synthesis of p-toluoyl derivatives of mannopyranoside (3–7).
2.2.2. Methyl 2,3,4-tri-O-heptanoyl-6-O-p-toluoyl-α-D-mannopyranoside (3)
A solution of the p-toluoyl derivative (2, 232 mg, 0.745 mmol) in anhyd. DMF/Et3N was treated with heptanoyl chloride (0.5 ml, 5.0 mmol). The mixture was stirred at −5°C for ∼6 hrs and then left overnight at room temperature. The reaction was monitored using T.L.C., which is a mixture of methanol and chloroform at a ratio of 1:5. The results showed that the starting material was completely converted into a single product with an Rf value of 0.52. The residue was purified by passing through a silica gel column with MeOH–CHCl3 (1:5) as the eluent, which provided the 2,3,4-tri-O-heptanoyl derivative (3, 155 mg) as a crystalline solid, m.p. 103–104 °C (EtOAc–C6H14).
A similar reaction and purification procedure was used to prepare compound 4 (61 mg) as needles, m.p. 107–108 °C (EtOAc–C6H14), compound 5 (77 mg) as needles, m.p. 112–113 °C (EtOAc–C6H14), compound 6 (164 mg) as needles, 115–117 °C (EtOAc–C6H14) and compound 7 (260 mg) as needles, 85–86 °C (EtOAc–C6H14).
Yield: 66.81 %, Rf = 0.53, FTIR (KBr): 1708 (C = O) cm−1. 1H NMR (400 MHz, CDCl3) δH ppm 8.0 (2H, d, J = 8.3 Hz, Ar-H), 7.28 (2H, d, J = 8.6 Hz, Ar-H), 5.10 (1H, d, J = 3.3 Hz, H-1), 5.0 (1H, dd, J = 3.5 and 10.1 Hz, H-2), 4.77 (1H, t, J = 9.4 Hz, H-3), 4.67 (1H, t, J = 9.4 Hz, H-4), 4.01 (1H, dd, J = 11.0 and 6.4 Hz, H-6a), 3.98 (1H, t, J = 10.1 Hz, H-6b), 3.96 (1H, m, H-5), 3.50 (3H, s, 1-OCH3), 2.43 (3H, s, 4-CH3. C6H4CO-, p-toluoyl), 2.38 {6H, m, 3 × CH3(CH2)4CH2CO-}, 1.65 {6H, m, 3 × CH3(CH2)3CH2CH2CO–}, 1.28 {18H, m, 3 × CH3(CH2)3CH2CH2CO–}, 0.90 {9H, m, 3 × CH3(CH2)5CO-}. 13C NMR (100 MHz, CDCl3): δC 174.50, 174.73, 171.88 {3 × CH3(CH2)5CO-}, 163.87 (4-CH3. C6H4CO-), 145.05, 131.37, 131.35, 130.20, 129.33, 129.31 (4-CH3. C6H4CO-), 97.06 (C-1), 72.91 (C-2), 71.34 (C-4), 70.65 (C-3), 69.32 (C-5), 63.03 (C-6), 55.11 (1-OCH3), 34.09, 34.05, 34.02, 33.82, 31.21(×3), 24.77, 24.53, 24.44, 24.41, 22.27 (×3), 21.87 {3 × CH3(CH2)5CO-}, 21.40 (4-CH3. C6H4CO-), 13.83, 13.81, 13.80 {3 × CH3(CH2)5CO-}. LC–MS [M + 1]+ 649.39. Calcd. for C36H56O10: C = 66.64, H = 8.70, O = 24.66; found: C = 66.63, H = 8.71, O = 24.65 %.
2.2.3. Methyl 2,3,4-tri-O-octanoyl-6-O-p-toluoyl-α-D-mannopyranoside (4)
Yield: 90.0 %, Rf = 0.52; FTIR (KBr): 1708 (C = O) cm−1. 1H NMR (400 MHz, CDCl3) δH ppm 8.0 (2H, d, J = 8.2 Hz, Ar-H), 7.26 (2H, d, J = 8.4 Hz, Ar-H), 5.22 (1H, d, J = 3.2 Hz, H-1), 4.90 (1H, dd, J = 3.3 and 10.2 Hz, H-2), 4.72 (1H, t, J = 9.3 Hz, H-3), 4.58 (1H, t, J = 9.2 Hz, H-4), 3.97 (1H, dd, J = 11.3 and 6.3 Hz, H-6a), 3.88 (1H, t, J = 10.2 Hz, H-6b), 3.87 (1H, m, H-5), 3.49 (3H, s, 1-OCH3), 2.41 (3H, s, 4-CH3. C6H4CO-, p-toluoyl), 2.40 {6H, m, 3 × CH3(CH2)5CH2CO-}, 1.64 {6H, m, 3 × CH3(CH2)4CH2CH2CO-}, 1.30 {24H, m, 3 × CH3(CH2)4CH2CH2CO-}, 0.90 {9H, m, 3 × CH3(CH2)6CO-}. 13C NMR (100 MHz, CDCl3): δC 174.0, 171.89, 171.04 {3 × CH3(CH2)6CO-}, 163.86 (4-CH3. C6H4CO-), 145.69, 135.44, 135.35, 132.67, 129.34, 129.32 (4-CH3. C6H4CO-), 97.05 (C-1), 72.97 (C-2), 71.30 (C-4), 70.61 (C-3), 69.33 (C-5), 63.01 (C-6), 55.17 (1-OCH3), 34.21, 34.11 (×3), 32.0 (×3), 31.05 (×3), 25.13, 22.61 (×2), 21.45 (×2), 20.07 (×3) {3 × CH3(CH2)6CO-}, 21.43 (4-CH3. C6H4CO-), 14.15, 14.09, 14.01 {3 × CH3(CH2)6CO-}. LC–MS [M + 1]+ 691.43. Calcd. for C39H62O10: C = 67.80, H = 9.01, O = 23.16; found: C = 67.79, H = 9.02, O = 23.15 %.
2.2.4. Methyl 2,3,4-tri-O-nonanoyl-6-O-p-toluoyl-α-D-mannopyranoside (5)
Yield: 71.0 %, Rf = 0.55, FTIR (KBr): 1717 (C = O) cm−1. 1H NMR (400 MHz, CDCl3) δH ppm 8.0 (2H, d, J = 8.0 Hz, Ar-H), 7.27 (2H, d, J = 8.2 Hz, Ar-H), 4.97 (1H, d, J = 3.1 Hz, H-1), 4.95 (1H, dd, J = 3.1 and 10.0 Hz, H-2), 4.70 (1H, t, J = 9.2 Hz, H-3), 4.55 (1H, t, J = 9.0 Hz, H-4), 4.17 (1H, dd, J = 11.1 and 6.2 Hz, H-6a), 4.0 (1H, t, J = 10.1 Hz, H-6b), 3.88 (1H, m, H-5), 3.48 (3H, s, 1-OCH3), 2.41 (3H, s, 4-CH3. C6H4CO-, p-toluoyl), 2.38 {6H, m, 3 × CH3(CH2)6CH2CO-}, 1.65 {6H, m, 3 × CH3(CH2)5CH2CH2CO-}, 1.28 {30H, m, 3 × CH3(CH2)5CH2CH2CO-}, 0.89 {9H, m, 3 × CH3(CH2)7CO-}. 13C NMR (100 MHz, CDCl3): δC 171.11, 170.23, 169.55 {3 × CH3(CH2)7CO-}, 169.52 (4-CH3. C6H4CO-), 145.69, 135.70, 135.32, 132.20, 129.33, 129.37 (4-CH3. C6H4CO-), 97.11 (C-1), 72.87 (C-2), 71.44 (C-4), 70.59 (C-3), 69.35 (C-5), 63.06 (C-6), 55.11 (1-OCH3), 34.20, 34.22, 34.24 (×3), 32.02 (×3), 31.08 (×3), 25.21, 22.55, 22.45 (×2), 21.32 (×2), 21.30, 20.09 (×3) {3 × CH3(CH2)7CO-}, 21.42 (4-CH3. C6H4CO-), 14.09, 14.04, 13.97 {3 × CH3(CH2)7CO-}. LC–MS [M + 1]+ 733.48. Calcd. for C42H68O10: C = 68.82, H = 9.35, O = 21.83; found: C = 68.80, H = 9.36, O = 21.84 %.
2.2.5. Methyl 2,3,4-tri-O-decanoyl-6-O-p-toluoyl-α-D-mannopyranoside (6)
Yield: 83.17 %, Rf = 0.53; FTIR (KBr): 1709 (C = O) cm−1. 1H NMR (400 MHz, CDCl3) δH ppm 8.01 (2H, d, J = 8.2 Hz, Ar-H), 7.28 (2H, d, J = 8.0 Hz, Ar-H), 5.01 (1H, d, J = 3.0 Hz, H-1), 4.90 (1H, dd, J = 3.0 and 10.1 Hz, H-2), 4.75 (1H, t, J = 9.0 Hz, H-3), 4.57 (1H, t, J = 9.1 Hz, H-4), 4.23 (1H, dd, J = 11.2 and 6.0 Hz, H-6a), 4.10 (1H, t, J = 10.0 Hz, H-6b), 3.78 (1H, m, H-5), 3.50 (3H, s, 1-OCH3), 2.44 (3H, s, 4-CH3. C6H4CO-, p-toluoyl), 2.36 {6H, m, 3 × CH3(CH2)7CH2CO-}, 1.65 {6H, m, 3 × CH3(CH2)6CH2CH2CO-}, 1.29 {36H, m, 3 × CH3(CH2)6CH2CH2CO-}, 0.90 {9H, m, 3 × CH3(CH2)8CO-}. 13C NMR (100 MHz, CDCl3): δC 172.43, 172.23, 172.20 {3 × CH3(CH2)8CO-}, 167.85 (4-CH3. C6H4CO-), 145.69, 132.67, 131.28, 130.20, 129.27, 129.26 (4-CH3. C6H4CO-), 97.04 (C-1), 72.86 (C-2), 71.33 (C-4), 70.64 (C-3), 69.34 (C-5), 63.11 (C-6), 55.13 (1-OCH3), 34.39 (×2), 34.26, 34.02 (×3), 31.70 (×2), 31.68 (×3), 31.63 (×3), 29.55 (×3), 25.07, 22.67, 22.63, 22.60 (×2), 22.56, 20.05 {3 × CH3(CH2)8CO-}, 21.39 (4-CH3. C6H4CO-), 13.51, 13.47, 13.44 {3 × CH3(CH2)8CO-}. LC–MS [M + 1]+ 773.55. Calcd. for C46H76O9: C = 71.47, H = 9.91, O = 18.63; found: C = 71.46, H = 9.93, O = 18.64 %.
2.2.6. Methyl 2,3,4-tri-O-benzenesulfonyl-6-O-p-toluoyl-α-D-mannopyranoside (7)
Yield: 80.32 %, Rf = 0.52; FTIR (KBr): 1709 (C = O) cm−1. 1H NMR (400 MHz, CDCl3) δH ppm 8.09 (2H, d, J = 8.3 Hz, Ar-H), 7.86 (3 × 2H, m, 3 × Ar-H), 7.64 (3 × 1H, m, 3 × Ar-H), 7.27 (3 × 2H, m, 3 × Ar-H), 7.24 (2H, d, J = 8.4 Hz, Ar-H), 4.96 (1H, d, J = 3.5 Hz, H-1), 4.94 (1H, dd, J = 3.1 and 10.2 Hz, H-2), 4.65 (1H, t, J = 9.2 Hz, H-3), 4.64 (1H, t, J = 9.0 Hz, H-4), 4.0 (1H, dd, J = 11.1 and 6.1 Hz, H-6a), 3.95 (1H, t, 10.2 Hz, H-6b), 3.76 (1H, m, H-5), 3.50 (3H, s, C6H4CO-, p-toluoyl). 13C NMR (100 MHz, CDCl3): δC 174.32 (4-CH3. C6H4CO-), 145.06, 133.71, 133.26, 130.19, 129.74, 129.26 (4-CH3. C6H4CO-), 144.35, 144.28, 144.07, 135.40, 135.17, 135.02, 129.79 (×3), 129.11 (×3),126.90 (×3), 126.45 (×3) {3 × C6H5SO2-}, 97.02 (C-1), 77.37 (C-2), 76.73 (C-4), 70.61 (C-3), 69.55 (C-5), 63.27 (C-6), 55.11 (1-OCH3), 21.81 (4-CH3.C6H4CO-). LC–MS [M + 1]+ 733.10. Calcd. for C33H32O13S3: C = 54.09, H = 4.40, O = 28.38; found: C = 54.11, H = 4.41, O = 28.39 %.
2.3. In vitro antimicrobial test and microorganisms
The antibacterial activities of the synthesized compounds (1–7) were tested in vitro according to the following procedures. Four types of human pathogenic bacteria were employed (viz. Escherichia coli ATCC-8739, Bacillus subtilis ATCC-6633, Staphylococcus aureus ATCC 6538, and Salmonella typhi AE 14612) were used to evaluate the antibacterial effects of the test compounds via the disc diffusion method (Bauer et al., 1959). Dimethyl sulfoxide (DMSO) was utilized as a solvent for the test compounds, and a 5 mg/ml solution of the substance was used in the experiment. DMSO was used as a negative control, while the known antibiotic azithromycin was used as a positive control. The MIC and MBC were used to determine the effectiveness of antibiotics on organisms. The MIC refers to the concentration of a substance that can effectively hinder the development of microorganisms following an extended period of incubation. On the other hand, minimum bactericidal concentrations (MBCs) are concentrations that completely prevent the growth of microorganisms after they have been transferred to culture media without any antibiotics. Microbial susceptibility testing was employed to validate the resistance and ascertain the novel antibacterial activity of the strains. The MIC of each substance was determined using the broth microdilution method, a basic antimicrobial susceptibility testing method that was performed according to CLSI instructions (CLSI, 2012, Wiegand et al., 2008). This involves preparing twofold dilutions (e.g., 1, 2, 4, 8, 16 and 32 µg/mL) of the agent in a liquid growth medium using 96-well plates. Mueller Hinton broth (MHB) media was poured into the wells, and each well was inoculated with a microbial inoculum. The plates were incubated at 37 °C for 20–22 h, with azithromycin serving as a positive control and DMSO serving as a negative control. After incubation, the plates were assayed with tetrazolium salt, and the MIC was calculated visually. The MIC was estimated as the first dilution that completely inhibited bacterial growth in MH broth media. The method can be easily converted to determine the MBC.
2.4. Analyzing test compounds in relation to fungi
Fungal cultures, including Aspergillus niger (ATCC 16404) and Candida albicans (ATCC 18804), were collected from the Microbiology Laboratory at the University of Chittagong. Throughout the investigation, standard potato dextrose agar (PDA) media, which included potato, dextrose, agar, and distilled water, were utilized. Sliced potatoes were boiled in distilled water supplemented with dextrose and agar to generate PDA media, which were subsequently autoclaved. Test tube slants of PDA medium were utilized for culture maintenance, with small mycelial portions transferred from old cultures to these tubes. Stock cultures were prepared by placing mycelial portions of each fungus in sterilized Petri dishes filled with test compounds and melted PDA media. The antifungal activity of the synthesized chemicals was evaluated using the mycelial growth test based on the “food poisoning” technique (Bhuyan et al., 2015, Islam et al., 2022). Sterilized medium, test chemicals (dissolved in DMSO at 20 µg/µL), and fungal inoculums were used for the test.
2.5. In vitro anticancer assessment
The MTT colorimetric assay was used in anticancer research to assess cell viability and cytotoxicity (Ahmed et al., 2017). MTT, or 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, is a yellow tetrazolium salt that, when reduced by metabolically active cells, results in the formation of a purple formazan product. This reduction is performed by cellular oxidoreductase enzymes that depend on NAD(P)H, indicating the quantity of living cells. The reduction process relies on the metabolic activity of cells, leading to the formation of formazan, which is purple in color. The metabolic activity and reduction of tetrazolium dye can be influenced by the conditions during the assay, but they do not have an impact on the viability of the cells. In the anticancer test, adult Swiss albino mice were used, and Ehrlich's ascites carcinoma (EAC) cells were collected and checked for viability. The in vitro proliferation of EAC cells was then assessed using the MTT colorimetric assay (Ahmed et al., 2017). Viable EAC cells were plated in culture plates supplemented with different concentrations of compound no. 1–7 and incubated. The absorbance at 570 nm was measured after incubation, and the cell proliferation inhibition ratio was calculated.
2.6. Structure-activity relationship (SAR)
To comprehend how a molecule's chemical structure and biological activity connect to one another, the structure–activity relationship (SAR) concept is fundamental to the drug development process. Systematic modification of specific structural features can reveal the impact of these changes on the compound's activity, selectivity, and safety. SAR studies investigate elements such as functional groups, steric effects, electron density, and conformational flexibility to elucidate the key factors influencing a molecule's interaction with its biological target. In our investigation, we specifically examined the SAR in line with the concept of membrane permeation, as described by Hunt (Hunt, 1975) and Kim (Kim et al., 2007). This knowledge is instrumental in optimizing lead compounds and guiding the development of new drugs with improved therapeutic properties.
2.7. Enhancing drug discovery: Bioactivity with PASS
The biological activity spectrum refers to the scope of biological activities that occur as a result of a compound's interaction with different biological entities. These activities are evaluated qualitatively, indicating whether they are present (“yes”) or absent (“none”). The Prediction of Activity Spectra for Substances (PASS) is a software program that uses the structures of organic drug-like chemicals to predict their total biological potential. This can be achieved by simultaneously predicting a range of diverse biological activities. (Kawsar et al., 2022). The results are shown as the probabilities Pa (for the active chemical) and Pi (for the inactive molecule). The Pa and Pi values range from 0.00 to 1.00. Typically, Pa + Pi does not equal 1, as these potentialities are anticipated independently. The results of the PASS computation are interpreted and applied in a versatile manner, with a high level of analytical probability when Pa exceeds 0.7 and a low level of analytical probability when Pa is less than 0.5.
2.8. Structural optimization strategies
In this work, Gaussian 09 software was used to analyze the various mannopyranoside derivatives, and GaussView 6 was used to construct the compounds (Frisch et al., 2009). Initially, density functional theory (DFT) and the B3LYP approach with a 6-31G(d,p) basis set were used to optimize the molecular structures (Sim et al., 1992). The optimization aimed to achieve configurations with minimal free energy and predict thermal and molecular orbital properties. Various parameters, including dipole moment, enthalpy, free energy, and electrical energy, were calculated. After optimization, the structures were utilized for molecular docking, dynamics and prediction of ADMET characteristics and molecular reactivity descriptors. We used known techniques to calculate chemical reactivity values and related descriptors (Becke, 1988, Lee et al., 1988). Parameters such as the HOMO-LUMO energy gap, hardness (η), and softness (S) were calculated for each mannopyranoside analog (Pearson, 1986).
2.9. Molecular docking
With antibiotic resistance becoming a growing concern, the identification of new antibacterial agents has become an absolute necessity (Islam et al., 2024). The molecular docking technique has been used to explore the interactions between various molecules and the active sites of specific proteins, with the aim of discovering the essential structural features that influence their binding efficiency (Sultana et al., 2024). The 3D crystallographic structures of these target proteins, such as those of Escherichia coli (PDB ID: 6KZV) (Ushiyama et al., 2020) and Candida albicans (PDB ID: 1EAG) (Cutfield et al., 1995), were extracted from the Protein Data Bank (PDB) and prepared by excluding water molecules, ligands, and nonprotein components. Subsequently, specialized software, including Discovery Studio, PyRx, and AutoDock Vina (Trott and Olson, 2010) was used to examine the interactions between compounds and protein targets and to assess the energy of these interactions using a 3D grid. The dimensions of the central grid box were fine-tuned to (0.493, 11.964, and 2.528) for 6KZV and (47.965, 21.534, and 8.561) for 1EAG to precisely position the ligands within the complex. The results were then examined using 2D and 3D visualizations to provide more in-depth information on these binding interactions.
2.10. In silico pharmacokinetics ADMET
Advances in computer technology have had a profound impact on the field of drug discovery, enabling more efficient and precise development of new drug candidates (Hadni et al., 2023). In silico studies provide a better understanding of the absorption, distribution, metabolism, excretion and toxicity (ADMET) of compounds. To conduct preliminary evaluations during drug discovery, this approach uses pharmacokinetic parameters and drug similarities. Using the pkCSM online tool (Ahmed et al., 2023), it is possible to evaluate a compound's capacity for absorption in the human small intestine, distribution throughout the body, elimination routes of metabolic changes, and toxicity levels. Hence, computer technology is crucial for assessing ADMET pharmacokinetic parameters in pharmaceutical research.
2.11. Molecular dynamic simulation
In this research, we conducted molecular dynamics simulations utilizing the NAMD software (Phillips et al., 2005) and the CHARMM36 force field (Jo et al., 2008). To set up the simulation environment, we placed the system in a cubic box with dimensions of 10 Å filled with TIP3P water molecules and neutralized it by introducing NaCl ions at a concentration of 0.154 M, employing the Monte Carlo method (Im et al., 2000). We initiated the simulations with an energy minimization phase, employing a gradient descent approach of more than 10,000 steps for each system. Subsequently, we conducted a 100 ns equilibration phase in the NVT (number of particles, volume, temperature) ensemble, maintaining a temperature of 310 K. Following this, an unconstrained 100 ns molecular dynamics simulation was performed in the NPT (number of atoms, pressure, temperature) ensemble for each system. To assess the stability of the systems, we analyzed the molecular dynamics trajectories using visual molecular dynamics (VMD) software (Humphrey et al., 1996).
2.12. Free energy binding calculations
To determine the binding energy of the docked poses, we used the Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) approach with AmberTools 22 software (Kollman et al., 2000). This approach represents a crucial step in assessing the structural stability and function of docked complexes through molecular dynamics (MD) simulations (Hadni and Elhallaoui, 2022). The binding free energies (ΔEbind) were calculated using the MM/GBSA approach as follows:
where Gcomplex, Greceptor, and Gligand denote the energies of the complex, protein and unbound ligand, respectively. ΔGgas represents changes in molecular mechanical energies in the gas phase, including bound and unbound energies from electrostatic (ΔGele) and van der Waals (ΔEvdW) interactions. ΔGsol represents the free energy of solvation, ΔGPB represents the polar free energy of solvation, and ΔGSA represents the nonpolar free energy of solvation. Finally, several studies (Adasme-Carreño et al., 2014, Genheden and Ryde, 2010, Kollman et al., 2000) have confirmed that entropy (ΔS) has no influence on the MM-GBSA calculation of binding free energies for complexes. As a result, we neglected the entropy contribution in all the analyzed complexes.
3. Results
3.1. Structure and chemistry
The major goal of this work was to use a direct acylation approach to perform targeted p-toluoylation of methyl α-D-mannopyranoside (1). Concurrently, diverse acylating agents were used to synthesize derivatives of the resultant p-toluoylation products. This strategy aims to provide evidence for structural elucidation while yielding new derivatives with synthetic and biological significance. Fig. 2 provides a detailed flow diagram of the research process, outlining sequential steps, methodologies, and key elements. This visual representation enhances comprehension of the study, highlighting each crucial aspect of the investigation. This study aimed to achieve its objectives while providing a clear narrative for readers to understand the selective p-toluoylation and derivative synthesis process (Scheme 1). The identities of the synthesized compounds were confirmed by multiple spectroscopic methods (Fig. 3, Fig. 4).
Fig. 2.

Comprehensive study workflow diagram: sequential steps and key elements.
Scheme 1.

Synthesis of (MαDM) analogs (2–7).
Fig. 3.

The FTIR spectra of the (MαDM) analogs (2–7).
Fig. 4.

HMBC correlations of derivative 2.
The antimicrobial activity and bioactivity of the resulting MαDM analogs (Table 1) were analyzed using PASS and bioactivity spectra. The synthesized analogs showed significant bactericidal and fungicidal effects against the tested strains in vitro. Physicochemical properties were studied through DFT optimization, molecular docking, dynamic simulations, and in silico pharmacokinetic and drug-likeness predictions to assess their potential applicability.
Table 1.
Optimized structure of the synthesized (MαDM) analogs (2–7).
| Entry | Chemical structure | Optimized 3D structure |
|---|---|---|
| 1 | 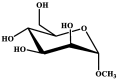 |
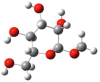 |
| 2 | 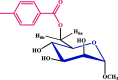 |
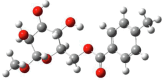 |
| 3 |  |
 |
| 4 |  |
 |
| 5 | 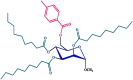 |
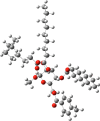 |
| 6 | 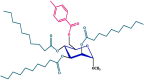 |
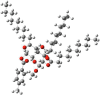 |
| 7 |  |
 |
3.2. In vitro antimicrobial results analysis
In this work, several acylated MαDM analogs (2–7) were selected and screened for their antibacterial activity against four human pathogenic bacteria (Fig. 5). Incorporating various acyl groups into carbohydrates increases antimicrobial efficacy (Amin et al., 2021a, Alam et al., 2021). Table S1 displays the inhibition zone diameter measurements resulting from the impact of the test substances.
Fig. 5.

The antibacterial efficacy of MαDM analogs.
The results of an antimicrobial susceptibility test that specifically measures the diameter of the zone of inhibition in millimeters for various bacterial strains exposed to 50 μg/μL/disc of azithromycin. Both gram-positive (S. aureus, B. subtilis) and gram-negative (S. typhi, E. coli) species are present in the bacterial strains. Analysis of the data revealed that various bacterial strains had various zones of inhibition. The synthesized MαDM analogs (2–7) exhibited varying degrees of inhibition of bacterial growth across the different strains (Fig. 5).
3.3. MIC and MBC determination
Compounds 5 and 6 exhibited significantly greater effectiveness against the tested organisms, and we further investigated them by estimating their minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) (Tables S2 and S3 and Fig. 6, Fig. 7). Compound 5 consists of a p-toluoyl group attached to the 6-O-position of mannopyranoside, along with a nonanoyl acylating group attached to the 2, 3, and 4-O-positions.
Fig. 6.

MIC test results for compounds 5 and 6.
Fig. 7.

MBC test results for compounds 5 and 6.
3.4. Impact of test substances on fungal organisms
The antifungal activity of the synthesized MαDM analogs is demonstrated in this section, as indicated in Table 2. The results are expressed as the percentage of fungal mycelial growth inhibited in millimeters (mm) at a concentration of 20 μg/μL. The table presents the inhibitory effects of each analog on two separate strains of fungi, Aspergillus niger and Candida albicans, together with comparison data for the positive control DMSO and the standard antibiotic nystatin. Among the tested analogs, compound 1, which is a methyl-α-D-mannopyranoside, did not inhibit fungal growth. In contrast, analog 2 effectively inhibited the growth of Aspergillus niger and Candida albicans, with substantial decreases of 70.67 ± 0.43 and 73.34 ± 0.61, respectively. Analog 3 showed moderate antifungal activity against Aspergillus niger, inhibiting growth by approximately 50.65 ± 2.9 %. Analog 5 stood out for its strong antifungal effectiveness against Aspergillus niger and Candida albicans, achieving high inhibition rates of 78.67 ± 0.69 and 80.41 ± 0.7, respectively. Analogs 4, 6, and 7 had good effects on the growth of Aspergillus niger and Candida albicans. In comparison, the positive control, nystatin, inhibited 65.51 ± 0.6 % of the bacteria from Aspergillus niger and 71.60 ± 0.65 % of the bacteria from Candida albicans (Figs S13 and S14).
Table 2.
Antifungal activities of the (MαDM) analogs.
| Entry | % inhibition in cm (20 μg/μl) |
|
|---|---|---|
|
Aspergillus niger (48 h) |
Candida albicans (48 h) |
|
| 1 | NI | NI |
| 2 | *70.67 ± 0.43 | *73.34 ± 0.61 |
| 3 | 50.65 ± 2.9 | *77.45 ± .32 |
| 4 | *70.51 ± 0.57 | 62.55 ± 1.60 |
| 5 | *78.67 ± 0.69 | *80.41 ± 0.7 |
| 6 | *68.52 ± 0.38 | *78.52 ± 0.63 |
| 7 | *69.87 ± 0.67 | *75.73 ± 0.76 |
| **Nystatin | **65.51 ± 0.6 | **71.60 ± 0.65 |
= good inhibition, Nystatin = standard antibiotic, NI = no inhibition.
3.5. In vitro studies investigating the potential of the anticancer properties
This study aimed to evaluate the anticancer activity of our synthesized MαDM analogs against EAC cells using an MTT calorimetric assay (Ahmed et al., 2017). The substances were evaluated at concentrations ranging from 31.25 to 500 µg/mL. Among compounds 1–7, only compound 5 [methyl 2,3,4-tri-O-nonanoyl-6-O-p-toluoyl-α-D-mannopyranoside] has shown potential activity. Compound 5 was found to be inhibitory at doses of 500, 250, 125, 62.5, and 31.25 µg/mL, with inhibition rates of 8.25 %, 6.82 %, 5.11 %, 4.46 %, and 2.96 %, respectively (Table S4). The inhibitory effect diminished as the concentration of the extract gradually decreased (Fig. 8). The IC50 of compound 5 was determined to be 4511.65 µg/mL.
Fig. 8.

MTT assay of compound 5 in EAC cells (n = 5, mean ± SD).
3.6. SAR studies
The structure–activity relationship (SAR) is vital in medicinal chemistry and drug design. The chemical structure of a compound affects its biological activity. SAR studies examine the function of certain groups of molecules to help synthesize new chemicals. The structure–activity relationship (SAR) helps enhance the molecular structure to increase the desired biological activity while minimizing undesired consequences. The atlas identifies the locations where few structural alterations result in significant modifications in cellular function. Antimicrobials are mostly heterocyclic compounds with five or six carbon atoms that are essential for cellular metabolism (Amin et al., 2021b). Because nucleosides are involved in practically every basic metabolic process in cells, it should be no surprise that carbohydrate analogs can target enzymes involved in protein synthesis, chitin biosynthesis in fungi, and bacterial peptidoglycan biosynthesis, among many other processes. Furthermore, condensed ring structures have proven to be highly effective pharmaceutical platforms and have attracted considerable interest due to their wide range of physiological activities. According to the antimicrobial activity data provided in Tables S1-S3, modifications to the mannopyranoside skeleton considerably impacted the antibacterial activity (Fig. 9).
Fig. 9.

SAR analysis of synthesized MαDM analogs.
Interestingly, native mannopyranoside did not demonstrate any activity against pathogenic bacteria. These findings highlight the significant impact that structural alterations have on the effectiveness of antibacterial agents. The parent compound's activity is improved by the addition of benzene-substituted acyl groups, especially when the groups are attached to a benzenesulfonyl group. As the acyl chain length increases, straight-chain acyl groups (nonanoyl, decanoyl) become more active. The nonanoyl (5) and decanoyl (6) derivatives worked better against most of the bacteria that were tested than did the other derivatives. Higher concentrations of synthesized compounds (128 µg/L) are required to kill gram-negative bacteria than gram-positive bacteria. The structural differences between the cell walls of gram-positive and gram-negative bacteria are the reason behind their varied behavior (Fig. 10).
Fig. 10.

Interactions of different chains of the compound 6.
Lipopolysaccharides (LPS) cover the peptidoglycan layer of gram-negative bacteria and are wrapped within the outer membrane. This outer membrane strictly regulates the diffusion of chemicals by acting as an impenetrable barrier. The LPS layer is an important component of selective permeability and functions as a barrier for preventing different chemicals from passing through rapidly. Hydrophobicity plays a crucial role in determining bioactivity, as it can influence the integrity and permeability of cellular membranes (Lien et al., 1982). Research suggests that the effectiveness of aliphatic alcohols is linked to their lipid solubility, which is primarily driven by hydrophobic interactions with lipid membrane regions (Hunt, 1975, Judge et al., 2013). Similarly, the lipid-like components and acyl chains of the methyl α-D-mannopyranoside derivatives can form hydrophobic interactions in gram-negative bacteria (Fig. 11). In the case of gram-positive bacteria, interactions involve sugar moieties. Sugars are carbohydrate sources that bacteria may use for metabolism.
Fig. 11.

Proposed mechanism of action of various MαDM analogs for studying the antibacterial effects of SAR.
3.7. Results of the PASS prediction
PASS, a computational tool, predicts the biological activities of compounds based on their chemical structure, aiding researchers in prioritizing experimental testing but requires caution and experimental validation. Unknown harmful side effects and toxicity have prevented many research initiatives from reaching the final stage. On the other hand, PASS, an internet server, makes it feasible to forecast more than 3,500 pharmacological effects and the biological characteristics of chemicals. Table 3 displays the PASS findings in two different forms: Pa (to be active) and Pi (to be inactive). The antibacterial, antifungal, anticarcinogenic and anti-inflammatory PASS predictions for compounds 1–7 were 0.472 < Pa < 0.561, 0.498 < Pa < 0.662, 0.314 < Pa < 0.731, and 0.543 < Pa < 0.710, respectively.
Table 3.
Prediction of the antibacterial, antifungal, anticarcinogenic and anti-inflammatory activities of the MαDM analogs.
| Entry |
Antibacterial |
Antifungal |
Anti carcinogenic |
Anti-inflammatory |
||||
|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| 1 | 0.561 | 0.012 | 0.628 | 0.016 | 0.731 | 0.008 | 0.645 | 0.021 |
| 2 | 0.523 | 0.016 | 0.629 | 0.016 | 0.635 | 0.011 | 0.628 | 0.026 |
| 3 | 0.531 | 0.018 | 0.662 | 0.012 | 0.496 | 0.020 | 0.621 | 0.028 |
| 4 | 0.481 | 0.031 | 0.542 | 0.024 | 0.314 | 0.053 | 0.710 | 0.012 |
| 5 | 0.534 | 0.014 | 0.662 | 0.012 | 0.496 | 0.020 | 0.634 | 0.025 |
| 6 | 0.534 | 0.011 | 0.662 | 0.012 | 0.496 | 0.020 | 0.612 | 0.028 |
| 7 | 0.482 | 0.029 | 0.498 | 0.034 | 0.449 | 0.007 | 0.543 | 0.041 |
3.8. Analysis of thermodynamics
Minor alterations in the molecular structure significantly influence characteristics such as thermal properties and molecular orbital parameters. The free energy and enthalpy data can be utilized to determine a reaction's spontaneity and stability (Lien et al., 1982). During the synthesis of drugs, the dipole moment affects the formation of hydrogen bonds and noncovalent interactions. The free energy (G) is crucial to how binding members interact. A negative number for (G) means that binding and interaction occur spontaneously. Our results suggest that, compared with the original mannopyranoside, the mannopyranoside analogs had stronger negative values for electronic energy (E), enthalpy (H), Gibbs free energy (G) and polarizability; these findings may provide evidence that the interactions and binding of these molecules could be enhanced by the attachment of an acyl group (Table S5). The mannopyranoside analog (7) has the highest enthalpy (−3427.8419 Hatree), electronic energy (−3428.5682 Hatree), Gibbs free energy (−3427.9550 Hatree) and dipole moment (10.2465 Debye).
3.9. Frontier molecular orbital (FMO) analysis
Frontier molecular orbitals (FMOs) are crucial concepts in theoretical chemistry that help researchers understand the reactivity and properties of molecules. These orbitals are associated with the highest and lowest energy molecular orbitals involved in chemical reactions. The two main types of FMOs are the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). (HOMO) is the outermost electron shell of a molecule and often interacts with the LUMO to facilitate electron transport. (LUMO) is crucial for understanding the electrophilic nature of a molecule. FMO theory is used to predict chemical reactions, with favorable reactions occurring when the HOMO of one reactant interacts with the LUMO of another, leading to the bond formation or bond breaking. Since more energy is needed to remove electrons from the ground-state HOMO to the excited-state LUMO, a larger energy gap is indicative of chemical stability and less chemical reactivity in molecules (Pearson, 1986). On the other hand, decreased chemical stability is indicated by a decreased energy gap. FMO theory is also used in organic chemistry to predict regioselectivity and stereoselectivity, aiding in the optimization of reaction conditions. The FMO data for all the analogs are shown in Table 4.
Table 4.
FMO data of the MαDM analogs.
| Parameters | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| εHOMO | −6.5132 |
−6.8734 |
−6.5982 |
−6.6256 |
−6.5598 |
−6.8352 |
| εLUMO | −1.5651 |
−1.3071 |
−0.9341 |
−0.9154 |
−1.0133 |
−1.6221 |
| Gap (Δε) = |
4.9481 |
5.5663 |
5.6641 |
5.7102 |
5.5465 |
5.2131 |
| Hardness η = |
2.4741 |
2.7832 |
2.8321 |
2.8551 |
2.7733 |
2.6066 |
| Softness S = |
0.4042 |
0.3593 |
0.3531 |
0.3503 |
0.3606 |
0.3836 |
| Chemical potential µ = |
−4.0392 |
−4.0903 |
−3.7662 |
−3.7705 |
−3.7866 |
−4.2287 |
| Electronegativity χ = − |
4.0392 |
4.0903 |
3.7662 |
3.7705 |
3.7866 |
4.2287 |
| Electrophilicity ω = |
3.2972 |
3.0056 |
2.5042 |
2.4897 |
2.5850 |
3.4301 |
Table 4 reveals that, compared to the other MαDM analogs, analog 2 had a smaller energy gap of 4.9481 eV (i.e., most reactive, less stable), while analog 5 had a greater energy gap of 5.7102 eV (i.e., less reactive, more stable) (Fig. 12). In quantum chemistry, the terms “chemical hardness” or “chemical softness” refers to the degree to which an atom or molecule resists changes in electron density. These concepts are often used to understand and predict chemical species reactivity, stability, and other properties. With a chemical hardness of 2.8551 eV and a softness of 0.3503 eV, analog (5) was the hardest and softest of the synthesized compounds.
Fig. 12.



Graphs showing the HOMO and LUMO positions of the MαDM analogs at DFT/B3LYP/6-31G(d,p).
3.10. Molecular docking
The molecular docking method is used to discover how drugs bind to the active site of a protein and the specific interactions that take place. In this context, we carried out molecular docking simulations involving synthetic ligands and proteins from Escherichia coli (6KZV) and Candida albicans (1EAG) (Fig. 13). This enabled us to understand how these ligands bind and interact with these bacterial and fungal proteins. The results, including binding affinities and hydrogen bonding details such as residues, angles and distances, are presented in Table 5. 2D visualization of the interaction types is shown in Fig. 14, Fig. 15.
Fig. 13.

3D positions of all the compounds in the active site of the Escherichia coli (a) and Candida albicans (b) proteins.
Table 5.
Molecular docking scores (binding affinities) and hydrogen bonding interactions of Escherichia coli and Candida albicans with all the ligands.
| Entry | Binding Energy of Escherichia coli (6KZV) (kcal/mol) | Ligand groups – Distances (Å) – Residue groups | Compound no. | Binding Energy of Candida albicans (1EAG) (kcal/mol) | Ligand groups – Distances (Å) – Residue groups |
|---|---|---|---|---|---|
| 1 | −5.3 | C-O – 2.89 – ARG76 | 1 | −5.4 | O–H – 2.55 – ASP32 O–H – 2.54 – ASP32 O–H – 2.10 – GLY34 C-O – 1.97 – GLY85 C-O – 2.71 – ASP86 O–H – 2.82 – GLY220 |
| 2 | −7.8 | O–H – 2.53 – GLY77 | 2 | −7.1 | O–H – 2.29 – ASP32 O–H – 2.58 – ASP32 O–H – 2.43 – ASP220 O–H – 2.79 – GLY220 |
| 3 | −5.5 | C-O – 3.08 – ARG76 C-O – 2.79 – ARG76 |
3 | −6.8 | C = O – 3.02 – GLY85 C = O – 2.32 – ASP86 C = O – 2.17 – SER88 C-O – 2.11 – THR222 |
| 4 | −5.3 | C-O – 2.76 – ARG76 | 4 | −6.7 | C-O – 1.86 – GLY85 C-O – 2.67 – THR221 |
| 5 | −6.1 | C-O – 2.85 – ARG76 C = O – 2.80 – ARG76 |
5 | −6.3 | C = O – 2.93 – THR221 C-O – 2.12 – THR222 C = O – 2.01 –THR222 |
| 6 | −5.3 | C-O – 2.21 – ARG76 C-O – 3.05 – ARG76 C = O – 2.15– ARG76 |
6 | −5.6 | C = O – 2.31 – THR222 C = O – 2.66 –THR222 |
| 7 | −7.2 | S = O – 2.06 – ARG76 S = O – 3.09 – ARG76 S = O – 2.58– ARG76 |
7 | −9.0 | C-O – 2.41 – GLY85 S = O – 2.44 – THR222 |
| Azithromycin | −6.6 | O–H – 2.24 – ASP49 O–H – 3.60 – GLU50 C-O – 2.50– ARG76 C-O – 2.32 – ARG76 |
Nystatin | −9.2 | O–H – 2.94 – ASP53 O–H – 2.32 – ASP218 |
Fig. 14.

2D visualization of the interaction types between the ligands and Escherichia coli (6KZV).
Fig. 15.

2D visualization of the interaction types between the best ligands and Candida albicans (1EAG).
3.11. According to the in silico ADMET prediction
The ability of bioactive compounds to function as drugs against bacterial and antifungal proteins was assessed via the prediction of ADMET pharmacokinetic parameters. The results of the ADMET prediction are presented in Table 6.
Table 6.
In silico prediction of ADMET for all ligands (MαDM analogs).
| Entry | Absorption |
Distribution |
Metabolism |
Excretion |
Toxicity |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intestinal absorption (human) |
VDss (human) |
BBB permeability |
CNS permeability |
Substrate |
Inhibitor |
Total Clearance |
AMES toxicity |
||||||
| CYP | |||||||||||||
| 2D6 |
3A4 |
1A2 |
2C19 |
2C9 |
2D6 |
3A4 |
|||||||
| Numeric (% Absorbed) |
Numeric(LogL/kg) | Numeric (LogBB) | Numeric (LogPS) |
Categorical (Yes/No) | Numeric (Log ml/min/kg) |
Categorical (Yes/No) | |||||||
| 1 | 33.429 | −0.331 | −0.992 | −3.622 | No | No | No | No | No | No | No | 0.671 | No |
| 2 | 53.662 | −0.552 | −0.889 | −3.473 | No | No | No | No | No | No | No | 0.408 | No |
| 3 | 100 | −0.332 | −1.86 | −2.629 | No | Yes | No | No | No | No | Yes | 1.473 | No |
| 4 | 89.852 | 0.332 | −1.968 | −2.438 | No | Yes | No | No | No | No | Yes | 1.546 | No |
| 5 | 100 | 0.085 | −2.003 | −2.155 | No | Yes | No | No | No | No | Yes | 1.616 | No |
| 6 | 100 | −0.221 | −2.069 | −2.091 | No | Yes | No | No | No | No | Yes | 1.665 | No |
| 7 | 100 | −0.404 | −2.784 | −4.127 | No | Yes | No | No | No | No | Yes | 0.353 | No |
3.12. Molecular dynamic simulation
Following the evaluation of the molecular docking results and in silico ADMET predictions for the synthesized compounds, we carried out MD simulation studies on compounds 3 and 5, which exhibited the best antibacterial and antifungal activities, respectively. These studies were carried out by comparing them with the drugs azithromycin and nystatin. The RMSD (root mean square deviation) and RMSF (root mean square fluctuation) of the Escherichia coli 6KZV and Candida albicans 1EAG protein complexes with the best synthesized compounds are presented in Fig. 16, Fig. 17, respectively.
Fig. 16.

a) The RMSD values of Escherichia coli in complex with the best ligand and the drug. b) The RMSD values of Candida albicans in complex with the best ligand and the drug.
Fig. 17.

a) The RMSF values of Escherichia coli residues in complex with the best ligand and the drug. b) The RMSF values of Candida albicans residues in complex with the best ligand and the drug.
3.13. MM-GBSA analysis
Binding free energy calculations for the simulated trajectories of all the complexes were performed using the MM-GBSA method, and the results are presented in Table 7.
Table 7.
The binding free energy of the complexes was calculated by the MM-GBSA method.
| Energy Parameter (kcal/mol) |
Escherichia coli (6KZV) |
Candida albicans (1EAG) |
||
|---|---|---|---|---|
| 3 | Azithromycin | 5 | Nystatin | |
| ΔGvdw | −52.946 | −48.373 | −47.860 | −35.697 |
| ΔGele | 7.591 | −1.866 | −4.040 | −15.135 |
| ΔGGB | 13.065 | 28.873 | 30.135 | 51.071 |
| ΔGSA | −5.946 | −5.145 | −6.394 | −4.9426 |
| ΔGgas | −48.568 | −53.832 | −60.622 | −59.461 |
| ΔGsol | 7.118 | 23.728 | 23.741 | 46.128 |
| ΔGbind | −41.449 | −30.104 | −36.881 | −13.333 |
Based on the analysis presented in Table 7, negative ΔGgas values suggest a favorable energy contribution in all the complexes. Conversely, negative ΔGsol values indicate an unfavorable energy contribution to the overall binding free energy. Additionally, ΔGvdw has a more significant contribution to the binding free energy than does ΔGele.
4. Discussion
The primary objective of this research was to carry out a specific p-toluoylation (Scheme 1) of methyl a-D-mannopyranoside (1) utilizing a direct acylation technique (Shagir et al., 2016) and synthesize various derivatives of acylated mannopyranoside (Akter et al., 2023). The primary acylation products and their derivatives were characterized through analysis of their FTIR and 1H NMR spectra. First, methyl α-D-mannopyranoside (1) was treated with p-toluoyl chloride as the acylating agent in anhydrous DMF/Et3N at −5 °C. Following standard procedures, compound (1) was obtained in high yield as a crystalline solid at a m.p. of 103–104 °C. This compound exhibited absorption bands in its FTIR spectrum (Fig. 3) at 1680 cm−1 and 1711 cm−1 for –CO stretching and 3300–3500 cm−1 (br.) for –OH stretching. The 1H NMR spectra (Fig. S1) provided evidence for the formation of a mono-substituted product. Two-proton doublets were observed at δ 8.01 (J = 8.4 Hz, Ar-H) and 7.33 (J = 8.8 Hz, Ar-H), as was a three-proton singlet at δ 2.43 (4-CH3). C6H4CO-), which can be attributed to the presence of a p-toluoyl group in the compound. The C-6 proton of the compound underwent a downfield shift to δ 4.61 (dd, J = 4.2 and 12.0 Hz, H-6a) and δ 4.56 (dd, J = 2.0 and 12.1 Hz, H-6b) compared to the standard mannopyranoside (1) values (∼4.00 ppm) (Hosen et al., 2022). The resonances of other protons were observed at their expected positions, indicating that the attachment of the p-toluoyl group occurred at a less hindered and more reactive position, specifically at position 6. The molecular ion peak at m/z [M + 1]+ 313.12, corresponding to the molecular formula C15H20O7, and the 13C NMR spectrum (Fig. S2) confirmed the structure of compound 2. On the basis of a comprehensive examination of FTIR and 1H NMR spectra and other characteristics, we determined that the structure of this molecule is methyl 6-O-p-toluoyl-α-D-mannopyranoside (2). In addition, assignments of the signals were made by analyzing their 2D spectral data with the 13C NMR spectrum (Fig. 4), which was confirmed as methyl 6-O-p-toluoyl-α-D-mannopyranoside (2).
The structure of compound 2 was further confirmed by converting it to compound 3, known as the heptanoyl derivative, and identifying it. The FTIR spectrum of the compound (Fig. 3) exhibited an absorption peak at 1708 cm−1, indicating carbonyl (–CO) stretching, while hydroxyl stretching was not observed. The presence of three heptanoyl groups in the molecule was demonstrated by the presence of two multiplets, each consisting of six protons at δ 2.38 {3 × CH3(CH2)4CH2CO-}, δ 1.65 {3 × CH3(CH2)3CH2CH2CO–}, eighteen-proton multiplets at δ 1.28 {3 × CH3(CH2)3CH2CH2CO–} and nine-proton multiplets at δ 0.90 {3 × CH3(CH2)5CO-} in its 1H NMR spectrum, suggesting the presence of three heptanoyl groups on the triol molecule. The remaining FTIR, 1H NMR and 13C NMR signals were as expected, suggesting that the compound's structure was methyl 2,3,4-tri-O-heptanoyl-6-O-p-toluoyl-α-D-mannopyranoside (3) (Figs. S3 and S4).
The same p-toluoyl derivative (2) was then transformed to compound 4, and its FTIR spectrum (Fig. 3) displayed an absorption band at 1708 cm1 corresponding to –CO stretching. In its 1H NMR spectrum, two six-proton multiplets, δ 2.40 {3 × CH3(CH2)5CH2CO-} and 1.64 {3 × CH3(CH2)4CH2CH2CO-}, twenty-four-proton multiplets at δ 1.30 {3 × CH3(CH2)4CH2CH2CO-} and a nine-proton multiplet at δ 0.90 {3 × CH3(CH2)6CO-}, were observed due to the presence of three octanoyl groups on the molecule. Like the structure of the heptanoyl derivative (3), which was established by analyzing FTIR, 1H NMR, and 13C NMR data and elemental data, we propose that its structure is methyl 2,3,4-tri-O-octanoyl-6-O-p-toluoyl-α-D-mannopyranoside (4) (Figs. S5 and S6). We repeated the process using nonanoyl chloride to derivatize compound 2. According to the results of FTIR and 1H NMR and 13C NMR analyses, compound 5 exhibited characteristic peaks at δ 2.38, δ 1.65, δ 1.28, and δ 0.89 based on previous descriptions of comparable compounds, and the structures of these compounds were positively identified as methyl 2,3,4-tri-O-nonanoyl-6-O-p-toluoyl-α-D-mannopyranoside (5) (Figs. S7 and S8). Based on the positive outcomes achieved from the previous acylation of the triol (2), we proceeded to utilize decanoyl chloride as the subsequent acylating agent. By detailed analysis of the FTIR, 1H NMR, and 13C NMR spectra (Figs. S9 and S10), its structure was confidently assigned as methyl 2,3,4-tri-O-decanoyl-6-O-p-toluoyl-α-D-mannopyranoside (6). Based on the results obtained thus far, we then used benzenesulfonyl chloride and its 1H NMR spectrum, and the peaks at δ 7.86, δ 7.64 and δ 7.27 corresponded to the protons of the three phenyl groups. The downfield shifts of C-2 to δ 4.94, C-3 to δ 4.65 and C-4 to δ 4.64 from their usual values, led us to propose this compound as methyl 2,3,4-tri-O-benzenesulfonyl-6-O-p-toluoyl-α-D-mannopyranoside (7) (Figs. S11 and S12).
From the antibacterial results compound 1, the starting material methyl-α-D-mannopyranoside, had no inhibitory effect on any of the bacterial strains. Compound 2 has a strong inhibitory effect on B. subtilis, with a zone of inhibition measuring 7.0 ± 1.6 mm. Compound 3 showed the highest inhibition zone against E. coli, at approximately 7.0 ± 0.6 mm. Compound 4 did not cause any inhibition. Compound 5 has moderate activity against both gram-positive and gram-negative strains, with B. subtilis showing the highest zone of inhibition (8.3 ± 0.6 mm). Compound 6 had consistent inhibitory effects on all the tested strains, with the most significant effect on B. subtilis (8.3 ± 0.23 mm). It is essential to consider the standard deviations associated with the mean values, as they provide insights into the precision of the measurements. Compound 6 is characterized by the presence of a p-toluoyl group attached to the 6-O-position of mannopyranoside, as well as a decanoyl acylating group attached to the 2, 3, and 4-O-positions. Compound 5 had a minimum inhibitory concentration (MIC) of 16 μg/L against S. typhi, while compound 6 had an MIC of 16 μg/L against S. aureus (Fig. 6) (Misbah et al., 2020). Additionally, the MBC for compound 5 was determined to be 128 μg/L against all three bacteria except for S. typhi, for which the MBC was approximately 64 mg/L. Similarly, compound 6 displayed an MBC of 128 μg/L against four microorganisms (Fig. 7). Based on the MIC and MBC values, these substances had better bacteriostatic and bactericidal activities, in the order of 6 > 5. Therefore, after studying their side effects and performing any other necessary tests, these two chemicals can be used as antibacterial medicines to treat a number of diseases that are spread by these test organisms (Amin et al., 2021a, Farhana et al., 2021).
Antifungal findings suggest that analogs 2 (methyl 6-O-p-toluoyl-α-D-mannopyranoside) and 5 (methyl 2,3,4-tri-O-nonanoyl-6-O-p-toluoyl-α-D-mannopyranoside) exhibit the most favorable antifungal efficacy, particularly against C. albicans (Kawsar et al., 2023, Mandloi et al., 2020). Therefore, further investigations should be conducted to explore the potential of these materials as antifungal medications. Notably, the zone of inhibition for these analogs was greater than that of the standard antibiotic nystatin (Figs. S13 and S14) (de Castro et al., 2015, Kayes et al., 2023). The acylation of mannopyranoside was shown to enhance antibacterial activity (Islam et al., 2024). The results indicate that the inclusion of various acyl groups, such as p-toluoyl, heptanoyl, octanoyl, nonanoyl, decanoyl, and benzenesulfonyl groups, significantly increased the antibacterial effectiveness of the mannopyranoside analogs.
Additionally, in terms of anticancer activity, compound 5 had an IC50 value of 4511.65 µg/mL. Noticeable decreases in cell viability were observed at higher concentrations compared to those in the control group. The IC50 values for each chemical against the EAC cells were computed based on the dose–response curves derived from the MTT assay results. The IC50 values ranged from 4000 to 4500 µg/mL, revealing the ability of the investigated drugs to suppress the growth of cancer cells (Ahmed et al., 2017, Islam et al., 2022).
However, a SAR study revealed that synthesized compounds that contain long-chain hydrocarbons, disrupt membrane permeability through hydrophobic interactions, ultimately leading to bacterial death (Guha, 2013, Kim et al., 2007, Lien et al., 1982).
A PASS study showed that starting compound (1) has strong antifungal, anticarcinogenic and anti-inflammatory properties. However, the antifungal properties of analogs 2–7 were enhanced when different acylating agents were added to them. Compounds 5 and 6 have nonanoyl and decanoyl long-chain hydrocarbon groups, which may increase antifungal activity. Our wet laboratory test results confirmed that compounds 5 and 6 also have better antifungal activity. Compound 7, which has a bulky benzenesulfonyl group in its structure, may decrease the antifungal activity, but wet laboratory tests have shown different results (Kawsar et al., 2022). Hence, according to the PASS results, the relative antifungal activity of the MαDM analogs was greater than that of the other antimicrobial agents (0.314 < Pa < 0.731) (Amin et al., 2021b).
Frontier molecular orbital (FMO) data offer valuable insights into the electronic characteristics and reactivity of MαDM analogs. It provides information on the (HOMO) and (LUMO) energies, indicating a molecule's electron-donating and electron-accepting capabilities, respectively. The energy gap (ΔE) signifies the molecule's reactivity, with a smaller gap indicating higher reactivity and susceptibility to electronic transitions (Kawsar and Hosen, 2020). Compound 5 exhibited the highest energy gap, approximately −5.7102 eV (Table 4), suggesting greater stability. The hardness and softness parameters further elucidate a molecule's stability and susceptibility to electronic changes. A higher hardness indicates enhanced stability, while a lower softness implies susceptibility to electronic transitions. Compound 5, noted for its significant biological activity, possesses a structural arrangement comprising long-chain hydrocarbons and a benzene ring, attributed to its bioactive properties (Hunt., 1975; Judge et al., 2013). Thermodynamic analysis confirmed its stability and structural suitability for biological activities.
In thermodynamic studies, the number of intermolecular interactions in a molecule has been determined by its dipole moment (Lien et al., 1982), which is essential for defining its electrical properties. A more polar character is indicated by a larger dipole moment (Heinz and Suter, 2004). The presence of a bulky acylating group suggested a possible improvement in polarizability; derivative 6 showed the highest polarizability, 492.8468 a.u. As more carbon atoms were added to the substituents, the scores for all the factors steadily increased (2–7). Altering the hydroxyl (–OH) groups of mannopyranoside will likely significantly improve the thermal properties of these products (Sultana et al., 2024).
The docking simulation data are presented in Table 5. The binding affinities of all the ligands of the E. coli protein receptor (6KZV) ranged from −5.3 to −7.2 kcal/mol. Furthermore, it is important to note that ligand 7 exhibits a particularly high binding affinity, surpassing even that obtained with the drug azithromycin (Paul et al., 2017). In contrast, the binding affinities of the other ligands were slightly lower but very close to those of azithromycin, suggesting strong competition in the protein's active site, as shown by the 3D visualization of the E. coli active site in Fig. 14. Previous studies have indicated that the 6KZV active site pocket is mainly formed by key residues, namely, GLU50, ASP73, ARG76, and ARG136 (Ushiyama et al., 2020). As shown in Fig. 14, all the ligands established one or more hydrogen bonding interactions with these key residues in the E. coli active site, with the exception of ligand 2. Modelling of the contact zones between all ligands, with the exception of ligand 2, and the E. coli receptor showed that these interactions are mainly stabilized by hydrogen bonds involving acceptor groups linked to the key residue ARG76, which plays a very important role in antibacterial activity (Chtita et al., 2022, Desai et al., 2020).
For the binding site of the C. albicans receptor (1EAG) to all ligands, the binding affinities vary between −5.4 and −9.2 kcal/mol, and it is noteworthy that the drug nystatin has the highest binding affinity among all the ligands. 3D visualization of the C. albicans active site is shown in Fig. 15, which reveals that all the ligands efficiently occupy the active site pocket, even though the affinity of the complexes varies. This finding theoretically supports the antifungal efficacy observed experimentally. Furthermore, important active sites for the 6H2L receptor include residues ASP32, GLY34, GLY85, ASP86, ASP218, GLY220, THR221 and THR222 (Cutfield et al., 1995). Remarkably, all the ligands established several hydrogen bonding interactions involving hydroxide and carbonyl groups, as shown in Fig. 15. In these interactions, the ligands act as both acceptors and donors in relation to the key residue, stabilizing the complexes that formed. This stability justifies the high antifungal activities of these ligands, even when compared to those of the drug nystatin (de Castro et al., 2015).
According to the in silico ADMET analyses presented in Table 6, all the compounds had favorable absorption rates in the human intestine, with absorption values above 30 %. The volume of distribution (VDss) is a pharmacokinetic parameter used to gauge how much volume would be needed to evenly distribute a drug dose to achieve the same concentration as in the blood plasma. When the VDss value falls below −0.15, it is considered low, and when it surpasses 0.45, it is deemed high. The VDss in this study ranged from −0.552 to 0.332 logL/kg, reflecting how the compounds are distributed throughout the body. The blood–brain barrier (BBB) is a specialized, partially permeable boundary separating the blood circulation from the brain's interstitial fluid. It enables the regulation of molecule and ion passage between the bloodstream and the brain, a LogBB value below −1 indicates limited distribution in the brain, while a LogBB value above 0.3 suggests the possibility of BBB penetration. In addition, a LogPS value greater than −2 indicates penetration of the central nervous system (CNS), while values less than −3 indicate difficulty accessing the CNS. The results obtained suggest that all the compounds, with the exception of 1, 2 and 7, demonstrate the ability to overcome biological barriers (Bouamrane et al., 2022, Daoui et al., 2022, Daoui et al., 2023). Drug metabolism is a fundamental process involving the biochemical transformation of pharmaceutical compounds in the human body. This metabolic process gives rise to a variety of metabolites, each with distinct pharmacological, pharmacokinetic and physicochemical properties. Understanding drug metabolism is essential in the field of pharmacology, as it has a direct impact on drug efficacy and potential interactions. The inhibition of cytochrome P450, particularly CYP1A2, 2C19, 2C9, 2D6 and 3A4, plays an important role in drug metabolism and may lead to drug interactions. These cytochrome P450 enzymes can also be therapeutic targets (Cheng et al., 2011). CYP3A4 was the inhibitor that played a very important role in this study. All the compounds, with the exception of compounds 1 and 3, were found to be substrates or inhibitors of CYP3A4. Furthermore, a reduced total clearance value implies a prolonged duration of drug presence in the body, a potential advantage for certain pharmaceutical products. Finally, the results of this research confirmed the nontoxic nature of all the compounds, a critical step in the drug development process. The results of the in-silico predictions of ADMET are particularly promising, as the synthesized molecules exhibit favorable kinetic properties, meet the essential criteria for drug similarity and exhibit significant biological activity.
In a molecular dynamics study of the E. coli protein (Fig. 16a), analysis of the RMSD trajectories revealed a rapid increase in fluctuations, from 0.7 to 1.2 Å in the space of 12 ns region. This increase may be mainly due to the initial kinetic impact experienced by all the complexes during the transition period (Decherchi and Cavalli, 2020). Subsequently, all the complexes stabilize and reach equilibrium. The mean RMSD for the E. coli protein complexed with ligand 3 was 1.425 Å, while the mean RMSD for the E. coli protein complexed with the drug azithromycin, used as the reference ligand in this study, was 1.833 Å. An average RMSD value of less than 2 Å is an indicator of the conformational stability of the complexes (Perola and Charifson, 2004, Ouassaf et al., 2021). This finding suggested that ligand 3 remains stable in the active site of the protein, even compared to the reference ligand. Analysis of the RMSD trajectories of the C. albicans protein (Fig. 16b) revealed a rapid increase in fluctuations, ranging from 0.8 to 2 Å within 25 ns. Following this initial phase, both complexes fluctuated within a similar distance range of 2 to 2.6 Å, implying that both complexes were stable. The average RMSD for the C. albicans protein complexed with ligand 5 was 2.342 Å, while the average RMSD for the C. albicans protein complexed with the drug nystatin, which served as the reference ligand in this study, was 2.363 Å. The RMSD plot for the two complexes showed similar fluctuations, and we also observed that the mean RMSD values for the two complexes were very close, with a slight increase in stability for ligand 5. This finding suggested that ligand 5 forms a more stable complex with the C. albicans protein than does the drug nystatin (Bouamrane et al., 2022). These results have significant implications for the understanding of ligand–protein interactions and can potentially inform experimental results in vitro.
The RMSF trajectories are an important way of assessing how ligand binding affects the flexibility of a receptor throughout an MD simulation. This information plays an essential role in assessing the stability of ligand–receptor binding throughout the simulation (Mortier et al., 2015). A high RMSF value indicates a high degree of residue flexibility, while a low RMSF value indicates a higher level of residue stability. For the E. coli protein (as illustrated in Fig. 17a), the majority of residues had comparable RMSF values, with fluctuations of greater amplitude evident within various regions, including ASP6 (3.532 Å), THR80 (2.864 Å), ALA100 (2.014 Å), GLY200 (1.858 Å) and GLY220 (4.226 Å). These fluctuations, which are located in inactive areas of the E. coli protein, are of less important. In contrast, key residues in the active site, such as GLU50, ASP73, ARG76 and ARG136, exhibited small RMSF variations (less than 0.7 Å). This observation suggested that the hydrogen bonds established between these residues play a substantial role in stabilizing the ligand–protein complexes with the E. coli protein (Hubbard and Kamran, 2010). For the C. albicans protein (as shown in Fig. 17b), analysis of the RMSF values revealed that certain residues, such as ASP53 (2.474 Å), SER89 (2.486 Å), ASN247 (5.095 Å) and ASP287 (2.911 Å), exhibited more pronounced fluctuations. These residues are not directly involved in the active site of the protein and do not contribute significantly to receptor–ligand interactions. However, the residues crucial for the most important interactions, namely, GLY34, ASP218, THR221 and THR222, had RMSF values less than 0.5 Å. These findings suggest that these residues establish hydrogen bonds that help stabilize the complexes formed by the C. albicans protein with its ligands. These results confirm the information obtained from the RMSD analysis, demonstrating that interactions between ligands and bacterial and fungal proteins are characterized by increased conformational stability, largely due to the formation of strong hydrogen bonding interactions.
MM-GBSA analysis of the E. coli protein revealed that the ΔGbind value for the complex with ligand 3 was −41.449, suggesting a stronger positive interaction with the target protein (Genheden and Ryde, 2010). In addition, this complex exhibited greater stability than the complex of the E. coli protein with azithromycin, where the ΔGbind value was −30.104. For the C. albicans protein, the ΔGbind value of the C. albicans protein complex with ligand 5 was −41.449, suggesting a better energetic affinity for the target protein. In addition, the complex of the C. albicans protein with nystatin had a ΔGbind value of −13.333, indicating that the more energetically favorable ligand 5 formed a more stable complex than the drug nystatin. The binding free energy results obtained using the MM-GBSA method were in perfect agreement with the molecular dynamics data. Indeed, ligands with more favorable binding free energies also displayed greater complex stability than the reference drug (Amin et al., 2021a, Islam et al., 2024).
5. Conclusions and future perspective
This work investigated various MαDM analogs in great detail by testing them for antibacterial, thermodynamic, molecular docking, drug-like, and molecular dynamics effects both in vitro and in silico. One of the most important factors affecting the biological activity of these products is the addition of various aromatic and aliphatic groups to the mannopyranoside structure. Notably, compounds 5 and 6, which are nonanoyl- and decanoyl-substituted, exhibited enhanced pharmacokinetic and biological profiles, respectively, and their effectiveness against bacterial species also increased. Further evidence was provided by molecular docking experiments, which showed that these MαDM analogs had promising antibacterial and antifungal effects on the receptors E. coli (6KZV) and C. albicans (1EAG). Compound 7 showed promising in silico activity against the 6KZV and 1EAG receptors, with favorable binding interactions and energies. After reviewing the molecular docking data and in silico predictions of the synthesized compounds, compounds 3 and 5 were subjected to MD simulation experiments to prove their superior antibacterial and antifungal properties. The reference ligand in this investigation, azithromycin, demonstrated an average RMSD of 1.833 Å, suggesting that ligand 3 is stable inside the active site of the E. coli protein complex. These compounds have favorable kinetic properties, adhere to essential drug similarity requirements, and exhibit substantial biological activity. The results of in silico ADMET predictions are particularly intriguing, and these factors together suggest that these MαDM analogs could be attractive therapeutic candidates. To validate the effectiveness of MαDM analogs as possible therapeutics against antibacterial and antifungal infections, further validation through wet-laboratory tests, which include both in vivo and in vitro examinations, is needed. Finally, the extensive results highlight the possibility of using MαDM analogs as viable agents for drug development, which should be subjected to additional experimental confirmation.
CRediT authorship contribution statement
Md. Ahad Hossain: Methodology, Investigation, Formal analysis. Shahin Sultana: Formal analysis, Data curation. Mohammed M. Alanazi: Validation, Resources, Funding acquisition. Hanine Hadni: Software, Investigation, Formal analysis. Ajmal R. Bhat: Writing – review & editing, Formal analysis. Imtiaj Hasan: Investigation, Formal analysis. Sarkar M.A. Kawsar: Writing – review & editing, Writing – original draft, Supervision, Project administration, Methodology, Conceptualization.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R628), King Saud University, Riyadh, Saudi Arabia for funding this work. This work was also partially supported by a research grant from the Research and Publication Office (2023-2024), CU, Bangladesh. The authors are very thankful to the Director, WMSRC, JU, Savar, Dhaka, Bangladesh for taking the spectra.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jsps.2024.102093.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Actor J.K., Hwang S.A., Kruzel M.L. Lactoferrin as a natural immune modulator. Curr. Pharm. Des. 2009;15:1956–1973. doi: 10.2174/138161209788453202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adasme-Carreño F., Muñoz-Gutierrez C., Caballero J., Alzate-Morales J.H. Performance of the MM/GBSA scoring using a binding site hydrogen bond network-based frame selection: the protein kinase case. Phys. Chem. Chem. Phys. 2014;16:14047–14058. doi: 10.1039/c4cp01378f. [DOI] [PubMed] [Google Scholar]
- Ahmed F.R.S., Amin R., Hasan I., Asaduzzaman A.K.M., Kabir S.R. Antitumor properties of a methyl-β-D-galactopyranoside specific lectin from Kaempferia rotunda against Ehrlich ascites carcinoma cells. Int. J. Biol. Macromol. 2017;102:952–959. doi: 10.1016/j.ijbiomac.2017.04.109. [DOI] [PubMed] [Google Scholar]
- Ahmed S., Mahendiran D., Bhat A.R., Rahiman A.K. Theoretical, in vitro anti-proliferative, and in silico molecular docking and pharmacokinetics studies of heteroleptic nickel(II) and copper(II) complexes of thiosemicarbazone-based ligands and pefloxacin. Chem. Biodivers. 2023;20:e202300702. doi: 10.1002/cbdv.202300702. [DOI] [PubMed] [Google Scholar]
- Akter S., Alhatlani B.Y., Abdallah E.M., Saha S., Ferdous J., Hossain M.E., Ali F., Kawsar S.M.A. Exploring cinnamoyl-substituted mannopyranosides: synthesis, evaluation of antimicrobial properties, and molecular docking studies targeting H5N1 influenza a virus. Molecules. 2023;28:8001. doi: 10.3390/molecules28248001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A., Hosen M.A., Islam M., Ferdous J., Fujii Y., Ozeki Y., Kawsar S.M.A. Synthesis, antibacterial and cytotoxicity assessment of modified uridine molecules. Curr. Adv. Chem. Biochem. 2021;6:114–129. [Google Scholar]
- Amin M.R., Yasmin F., Dey S., Mahmud S., Saleh M.A., Emran T.B., Hasan I., Rajia S., Ogawa Y., Fujii Y., Yamada M., Ozeki Y., Kawsar S.M.A. Methyl β-D-galactopyranoside esters as potential inhibitors for SARS-CoV-2 protease enzyme: synthesis, antimicrobial, PASS, molecular docking, molecular dynamics simulations and quantum computations. Glycoconj. J. 2021;39:261–290. doi: 10.1007/s10719-021-10039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin M.R., Yasmin F., Hosen M.A., Dey S., Mahmud S., Saleh M.A., Emran T.B., Hasan I., Fujii Y., Yamada M., Ozeki Y., Kawsar S.M.A. Synthesis, antimicrobial, anticancer, PASS, molecular docking, molecular dynamic simulations, and pharmacokinetic predictions of some methyl β-D-galactopyranoside analogs. Molecules. 2021;26:7016. doi: 10.3390/molecules26227016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A.W., Perry D.M., Kirby W.M.M. Single-disk antibiotic-sensitivity testing of staphylococci: an analysis of technique and results. AMA Arch. Intern. Med. 1959;104:208–216. doi: 10.1001/archinte.1959.00270080034004. [DOI] [PubMed] [Google Scholar]
- Becke A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Bhat A.R., Dongre R.S., Almalki F.A., Berredjem M., Aissaoui M., Touzani R., Hadda T.B., Akhter M.S. Synthesis, biological activity and POM/DFT/docking analyses of annulated pyrano[2,3-d]pyrimidine derivatives: identification of antibacterial and antitumor pharmacophore sites. Bioorg. Chem. 2021;106 doi: 10.1016/j.bioorg.2020.104480. [DOI] [PubMed] [Google Scholar]
- Bhuyan P.D., Tamuli P., Boruah P. In-vitro efficacy of certain essential oils and plant extracts against three major pathogens of Jatropha curcas L. Am. J. Plant Sci. 2015;6:362–365. [Google Scholar]
- Bouamrane S., Khaldan A., Hajji H., El-mernissi R., Maghat H., Ajana M.A., Sbai A., Bouachrine M., Lakhlifi T. 3D-QSAR, molecular docking, molecular dynamic simulation, and ADMET study of bioactive compounds against candida albicans. Mor. J. Chem. 2022;10:523–541. [Google Scholar]
- Cao X., Du X., Jiao H., An Q., Chen R., Fang P., Wang J., Yu B. Carbohydrate-based drugs launched during 2000–2021. Acta Pharm. Sin. B. 2022;12:3783–3821. doi: 10.1016/j.apsb.2022.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Yu Y., Shen J., Yang L., Li W., Liu G., Lee P.W., Tang Y. Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J. Chem. Inf. Model. 2011;51:996–1011. doi: 10.1021/ci200028n. [DOI] [PubMed] [Google Scholar]
- Chtita S., Belaidi S., Qais F.A., Ouassaf M., Almogren M.M., Al-Zahrani A.A., Bakhouch M., Belhassan A., Zaki H., Bouachrine M., Lakhlifi T. Unsymmetrical aromatic disulfides as SARS-CoV-2 Mpro inhibitors: Molecular docking, molecular dynamics, and ADME scoring investigations. J. King Saud Univ. Sci. 2022;34 doi: 10.1016/j.jksus.2022.102226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI (Clinical Laboratory Standards Institute). 2012. M07-A9: Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-ninth edition. Available at: www.clsi.org.
- Cummings J.H., Roberfroid M.B., Andersson H., Barth C., Ferro-Luzzi A., Ghoos Y., Gibney M., Hermansen K., James W.P.T., Korver O., Lairon D., Pascal G., Voragen A.G.S. Erratum: a new look at dietary carbohydrate: chemistry, physiology and health. Eur. J. Clin. Nutr. 2010;64:334. doi: 10.1038/sj.ejcn.1600427. [DOI] [PubMed] [Google Scholar]
- Cutfield S.M., Dodson E.J., Anderson B.F., Moody P., Marshall C.J., Sullivan P.A., Cutfield J.F. The crystal structure of a major secreted aspartic proteinase from candida albicans in complexes with two inhibitors. Structure. 1995;3:1261–1271. doi: 10.1016/s0969-2126(01)00261-1. [DOI] [PubMed] [Google Scholar]
- Daoui O., Elkhattabi S., Chtita S. Rational identification of small molecules derived from 9,10-dihydrophenanthrene as potential inhibitors of 3CLpro enzyme for COVID-19 therapy: a computer-aided drug design approach. Struct. Chem. 2022;33:1667–1690. doi: 10.1007/s11224-022-02004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoui O., Nour H., Abchir O., Elkhattabi S., Bakhouch M., Chtita S. A computer-aided drug design approach to explore novel type II inhibitors of c-Met receptor tyrosine kinase for cancer therapy: QSAR, molecular docking, ADMET and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2023;41:7768–7785. doi: 10.1080/07391102.2022.2124456. [DOI] [PubMed] [Google Scholar]
- de Castro R.D., de Souza T.M.P.A., Bezerra L.M.D., Ferreira G.L.S., de Brito Costa E.M.M., Cavalcanti A.L. Antifungal activity and mode of action of thymol and its synergism with nystatin against candida species involved with infections in the oral cavity: an in vitro study. BMC Complement. Altern. Med. 2015;15:417. doi: 10.1186/s12906-015-0947-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decherchi S., Cavalli A. Thermodynamics and kinetics of drug-target binding by molecular simulation. Chem. Rev. 2020;120:12788–12833. doi: 10.1021/acs.chemrev.0c00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai N.C., Vaja D.V., Jadeja K.A., Joshi S.B., Khedkar V.M. Synthesis, biological evaluation and molecular docking study of pyrazole, pyrazoline clubbed pyridine as potential antimicrobial agents. Anti-Infect. Agent. 2020;18:306–314. [Google Scholar]
- Farhana Y., Amin M.R., Hosen M.A., Bulbul M.Z.H., Dey S., Kawsar S.M.A. Monosaccharide derivatives: synthesis, antimicrobial, PASS, antiviral, and molecular docking studies against SARS-CoV-2 mpro inhibitors. J. Cellulose Chem. Technol. 2021;55:477–499. [Google Scholar]
- Filimonov D.A., Lagunin A.A., Gloriozova T.A., Rudik A.V., Druzhilovskii D.S., Pogodin P.V., Poroikov V.V. Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem. Heterocycl. Compd. 2014;50:444–457. [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., et al., 2009. Gaussian 09.
- Genheden S., Ryde U.L.F. How to obtain statistically converged MM/GBSA results. J. Comput. Chem. 2010;31:837–846. doi: 10.1002/jcc.21366. [DOI] [PubMed] [Google Scholar]
- Guha R. In: Kortagere S., editor. vol 993. Humana Press; Totowa, NJ. Page: 2013. On Exploring Structure-Activity Relationships; pp. 81–94. (Silico Models for Drug Discovery. Methods in Molecular Biology). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadni H., Benjelloun A.T., Benzakour M., Mcharfi M., Benbrahim M. Identification of terpenoids as potential inhibitors of SARS-CoV-2 (main protease) and spike (RBD) via computer-aided drug design. J. Biomol. Struct. Dyn. 2023;7:1–14. doi: 10.1080/07391102.2023.2245051. [DOI] [PubMed] [Google Scholar]
- Hadni H., Elhallaoui M. Discovery of anti-colon cancer agents targeting wild-type and mutant p53 using computer-aided drug design. J. Biomol. Struct. Dyn. 2022;41:10171–10189. doi: 10.1080/07391102.2022.2153919. [DOI] [PubMed] [Google Scholar]
- Heinz H., Suter U.W. Atomic charges for classical simulations of polar systems. The J. Phys. Chem. b. 2004;108:18341–18352. [Google Scholar]
- Hosen M.A., Munia N.S., Al-Ghorbani M., Faisal M.B., Almalki A., Hadda T.B., Ali F., Mahmud S., Saleh M.A., Laaroussi H., Kawsar S.M.A. Synthesis, antimicrobial, molecular docking and molecular dynamics studies of lauroyl thymidine analogs against SARS-CoV-2: POM study and identification of the pharmacophore sites. Bioorg. Chem. 2022;125 doi: 10.1016/j.bioorg.2022.105850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard R.E., Kamran H.M. Wiley; In Encyclopedia of Life Sciences: 2010. Hydrogen Bonds in Proteins: Role and Strength. [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hunt W.A. The effects of aliphatic alcohols on membrane function's biophysical and biochemical correlates. Adv. Exp. Med. Biol. 1975;56:195–210. doi: 10.1007/978-1-4684-7529-6_9. [DOI] [PubMed] [Google Scholar]
- Im W., Seefeld S., Roux B. A grand canonical monte Carlo-Brownian dynamics algorithm for simulating ion channels. Biophys. J. 2000;79:788–801. doi: 10.1016/S0006-3495(00)76336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam A.U., Hadni H., Ali F., Abuzreda A., Kawsar S.M.A. Synthesis, antimicrobial activity, molecular docking, molecular dynamics simulation, and ADMET properties of the mannopyranoside derivatives as antimicrobial agents. J. Taibah Univ. Sci. 2024;18:2327101. [Google Scholar]
- Islam S., Hosen M.A., Ahmad S., ul Qamar M.T., Dey S., Hasan I., Fujii Y., Ozeki Y., Kawsar S.M.A. Synthesis, antimicrobial, anticancer activities, PASS prediction, molecular docking, molecular dynamics and pharmacokinetic studies of designed methyl α-D-glucopyranoside esters. J. Mol. Struct. 2022;1260 [Google Scholar]
- Jequier E. Carbohydrates as a source of energy. Am. J. Clin. Nutr. 1994;59 doi: 10.1093/ajcn/59.3.682S. (682S-685S) [DOI] [PubMed] [Google Scholar]
- Jo S., Kim T., Iyer V.G., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Judge V., Narasimhan B., Ahuja M., Sriram D., Yogeeswari P., Clercq E., Pannecouque C., Balzarini J. Synthesis, antimycobacterial, antiviral, antimicrobial activity and QSAR studies of N2-acyl isonicotinic acid hydrazide derivatives. Med. Chem. 2013;9:53–76. doi: 10.2174/157340613804488404. [DOI] [PubMed] [Google Scholar]
- Kabir A.K.M.S., Kawsar S.M.A., Bhuiyan M.M.R., Islam M.R., Rahman M.S. Biological evaluation of some mannopyranoside derivatives. Bull. Pure Appl. Sci. 2004;23:83–91. [Google Scholar]
- Kabir A.K.M.S., Kawsar S.M.A., Bhuiyan M.M.R., Rahman M.S., Banu B. Biological evaluation of some octanoyl derivatives of methyl 4,6-O-cyclohexylidene-α-D-glucopyranoside. Chittagong Univ. J. Biol. Sci. 2008;3:53–64. [Google Scholar]
- Kawsar S.M.A., Matsumoto R., Fujii Y., Matsuoka H., Masuda N., Iwahara C., Yasumitsu H., Kanaly R.A., Sugawara S., Hosono M., Nitta K., Ishizaki N., Dogasaki C., Hamako J., Matsui T., Ozeki Y. Cytotoxicity and glycan-binding profile of α-D-galactose-binding lectin from the eggs of a Japanese sea hare (Aplysia kurodai) Protein J. 2011;30:509–519. doi: 10.1007/s10930-011-9356-7. [DOI] [PubMed] [Google Scholar]
- Kawsar S.M.A., Kumer A., Munia N.S., Hosen M.A., Chakma U., Akash S. Chemical descriptors, PASS, molecular docking, molecular dynamics and ADMET predictions of glucopyranoside derivatives as inhibitors to bacteria and fungi growth. Org. Commun. 2022;15:1–20. [Google Scholar]
- Kawsar S.M.A., Almalki F.A., Hadda T.B., Laaroussi H., Khan M.A.R., Hosen M.A., Mahmud S., Aounti A., Maideen N.M.P., Heidarizadeh F., Soliman S.S.M. Potential antifungal activity of novel carbohydrate derivatives validated by POM, molecular docking and molecular dynamic simulations analyses. Mol. Simul. 2023;49:60–75. [Google Scholar]
- Kawsar S.M.A., Hosen M.A. An optimization and pharmacokinetic studies of some thymidine derivatives. Turkish Comp. Theo. Chem. 2020;4:59–66. [Google Scholar]
- Kayes M.R., Saha S., Alanazi M.M., Ozeki Y., Hadda T.B., Legssyer A., Kawsar S.M.A. Macromolecules: Synthesis, antimicrobial, POM analysis and computational approaches of some glucoside derivatives bearing acyl moieties. Saudi Pharm. J. 2023;31 doi: 10.1016/j.jsps.2023.101804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Farrah S., Baney R.H. Structure–antimicrobial activity relationship for silanols, a new class of disinfectants, compared with alcohols and phenols. Int. J. Antimicrob. Agents. 2007;29:217–222. doi: 10.1016/j.ijantimicag.2006.08.036. [DOI] [PubMed] [Google Scholar]
- Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A., Cheatham T.E. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. b. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Lien E.J., Guo Z.-R., Li R.-L., Su C.-T. Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J. Pharm. Sci. 1982;71:641–655. doi: 10.1002/jps.2600710611. [DOI] [PubMed] [Google Scholar]
- Lobo S. Is there enough focus on lipophilicity in drug discovery? Expert Opin. Drug Discov. 2020;15:261–263. doi: 10.1080/17460441.2020.1691995. [DOI] [PubMed] [Google Scholar]
- Mahmoud S., Hasabelnaby S., Hammad S.F., Sakr T.M. Antiviral nucleoside and nucleotide analogs; a review. J. Adv. Pharm. Res. 2018;2:73–88. [Google Scholar]
- Mandloi D., Dabade S.J., Bajaj A.V., Atre H. Molecular docking and QSAR studies for modeling antifungal activity of triazine analogues against therapeutic target NMT of candida albicans. Int. J. Pharm. Sci. Drug Res. 2020;13:140–146. [Google Scholar]
- Maowa J., Alam A., Rana K.M., Dey S., Hosen A., Fujii Y., Hasan I., Ozeki Y., Kawsar S.M.A. Synthesis, characterization, synergistic antimicrobial properties, and molecular docking of sugar-modified uridine derivatives. Ovidius Univ. Ann. Chem. 2021;32:6–21. [Google Scholar]
- Matsumoto R., Fujii Y., Kawsar S.M.A., Kanaly R., Yasumitsu H., Koide Y., Hasan I., Iwahara C., Ogawa Y., Chang H., Sugawara S., Hosono M., Nitta K., Hamako J., Matsui T., Ozeki Y. Cytotoxicity and glycan-binding properties of an 18 kDa lectin isolated from the marine sponge Halichondria okadai. Toxins. 2012;4:323–338. doi: 10.3390/toxins4050323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X.-Y., Zhang H.-X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo C.M., Heiden M.G.V. Understanding metabolic regulation and its influence on cell physiology. Mol. Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirajul M.I., Arifuzzaman M., Monjur M.R., Rahman A., Kawsar S.M.A. Novel methyl 4,6-O-benzylidene-α-D-glucopyranoside derivatives: synthesis, structural characterization and evaluation of antibacterial activities. Hacettepe J. Biol. Chem. 2019;47:153–164. [Google Scholar]
- Misbah M.M., Ferdous J., Dey S., Bulbul M.Z., Chowdhury T.S., Hasan I., Kawsar S.M.A. Evaluation of MIC, MBC, MFC and anticancer activities of acylated methyl β-D-galactopyranoside esters. Int. J. Biosci. 2020;16:299–309. [Google Scholar]
- Mortier J., Rakers C., Bermudez M., Murgueitio M.S., Riniker S., Wolber G. The impact of molecular dynamics on drug design: applications for the characterization of ligand–macromolecule complexes. Drug Discov. Today. 2015;20:686–702. doi: 10.1016/j.drudis.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Newman D.J., Cragg G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Oppenheimer S.B., Alvarez M., Nnoli J. Carbohydrate-based experimental therapeutics for cancer. HIV/AIDS and Other Diseases. Acta Histochem. 2008;110:6–13. doi: 10.1016/j.acthis.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouassaf M., Belaidi S., Khamouli S., Belaidi H., Chtita S. Combined 3D-QSAR and molecular docking analysis of thienopyrimidine derivatives as staphylococcus aureus inhibitors. Acta Chim. Slov. 2021;68:289–303. [PubMed] [Google Scholar]
- Paul S., Banerjee S., Vogel H.J. Ligand binding specificity of the Escherichia coli periplasmic histidine binding protein. HisJ. Protein Sci. 2017;26:268–279. doi: 10.1002/pro.3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. u.s.a. 1986;83:8440–8441. doi: 10.1073/pnas.83.22.8440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola E., Charifson P.S. Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding. J. Med. Chem. 2004;47:2499–2510. doi: 10.1021/jm030563w. [DOI] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana K.M., Maowa J., Alam A., Dey S., Hosen A., Hasan I., Fujii Y., Ozeki Y., Kawsar S.M.A. In silico DFT study, molecular docking, and ADMET predictions of cytidine analogs with antimicrobial and anticancer properties. In Silico Pharmacol. 2021;9:42. doi: 10.1007/s40203-021-00102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shagir A.C., Bhuiyan M.M.R., Ozek Y., Kawsar S.M.A. Simple and rapid synthesis of some nucleoside derivatives: structural and spectral characterization. Current Chem. Lett. 2016;5:83–92. [Google Scholar]
- Sim, F., St. Amant, A., Papai, I., Salahub, D.R., 1992. Gaussian density functional calculations on hydrogen-bonded systems. J. Am. Chem. Soc. 114, 4391–4400.
- Sultana S., Hossain A., Islam M.M., Kawsar S.M.A. Antifungal potential of mannopyranoside derivatives through computational and molecular docking studies against candida albicans 1IYL and 1AI9 proteins. Curr. Chem. Lett. 2024;13:1–14. [Google Scholar]
- Thomford N.E., Senthebane D.A., Rowe A., Munro D., Seele P., Maroyi A., Dzobo K. Natural products for drug discovery in the 21st century: innovations for novel drug discovery. Int. J. Mol. Sci. 2018;19:1578. doi: 10.3390/ijms19061578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushiyama F., Amada H., Takeuchi T., Tanaka-Yamamoto N., Kanazawa H., Nakano K., Mima M., Masuko A., Takata I., Hitaka K., Iwamoto K., Sugiyama H., Ohtake N. Lead identification of 8-(Methylamino)-2-oxo-1,2-dihydroquinoline derivatives as DNA gyrase inhibitors: hit-to-lead generation involving thermodynamic evaluation. ACS Omega. 2020;5:10145–10159. doi: 10.1021/acsomega.0c00865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mourik T., Bühl M., Gaigeot M.-P. Density functional theory across chemistry, physics and biology. Philos. Trans. A Math. Phys. Eng. Sci. 2014;372:20120488. doi: 10.1098/rsta.2012.0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhang Y., Lu Q., Xing D., Zhang R. Exploring carbohydrates for therapeutics: a review on future directions. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.756724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand I., Hilpert K., Hancock R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008;3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.