

Significance
Voltage-induced calcium release (VICR) is a mechanism whereby depolarization-induced conformational changes in plasma membrane calcium channels (CaV) are allosterically coupled to the activation of calcium channels residing in the endoplasmic reticulum (ER), thereby triggering ER calcium release. Thus far, VICR has only been observed in vertebrate skeletal muscle. Here, we show that VICR also occurs in Caenorhabditis elegans body muscles, suggesting that VICR is more ancient than is currently appreciated.
Keywords: CaV1, EGL-19, ryanodine receptor, UNC-68, C. elegans
Abstract
Type 1 voltage-activated calcium channels (CaV1) in the plasma membrane trigger calcium release from the sarcoplasmic reticulum (SR) by two mechanisms. In voltage-induced calcium release (VICR), CaV1 voltage sensing domains are directly coupled to ryanodine receptors (RYRs), an SR calcium channel. In calcium-induced calcium release (CICR), calcium ions flowing through activated CaV1 channels bind and activate RYR channels. VICR is thought to occur exclusively in vertebrate skeletal muscle while CICR occurs in all other muscles (including all invertebrate muscles). Here, we use calcium-activated SLO-2 potassium channels to analyze CaV1-SR coupling in Caenorhabditis elegans body muscles. SLO-2 channels were activated by both VICR and external calcium. VICR-mediated SLO-2 activation requires two SR calcium channels (RYRs and IP3 Receptors), JPH-1/Junctophilin, a PDZ (PSD95, Dlg1, ZO-1 domain) binding domain (PBD) at EGL-19/CaV1’s carboxy-terminus, and SHN-1/Shank (a scaffolding protein that binds EGL-19’s PBD). Thus, VICR occurs in invertebrate muscles.
Action potentials (APs) trigger muscle contraction through excitation–contraction (EC) coupling. Muscle depolarization during an AP activates type 1 voltage-activated calcium channels (CaV1) in the plasma membrane, which subsequently activate calcium release channels (e.g., ryanodine receptors, RYRs) in the sarcoplasmic reticulum (SR). In vertebrate skeletal muscle, EC coupling is mediated by a direct physical coupling of CaV1 voltage-sensing domains to RYR activation, which is termed voltage-induced calcium release (VICR) (1, 2). In all other muscles, EC coupling is thought to be mediated by calcium-induced calcium release (CICR), where calcium ions entering through CaV1 channels bind and activate RYRs in the SR (3–6).
CaV1–RYR coupling occurs at specialized junctions between the plasma membrane (PM) and SR membranes. These junctions form along PM invaginations, which are termed T-tubules. Freeze fracture EM analysis of PM-SR contacts in vertebrate skeletal muscles show that CaV1.1 channels form tetrads while RYR channels form matching tetrameric foot structures (7, 8). The intermembrane spacing at PM-SR junctions is ~12 nm in both skeletal muscles and cardiac muscles (9), which is likely spanned by the large cytoplasmic domain of RYR channels (10). For these reasons, it is widely assumed that CaV1 and RYR channels are closely apposed at muscle PM-SR junctions and could be physically coupled (either directly or through intermediary proteins). These results also suggest that tight PM-SR junctions are likely required for both VICR (in skeletal muscles) and CICR (in cardiac muscles).
Several proteins are required for VICR in vertebrates. Ectopic expression of CaV1.1, CaVβ1a, RYR1, STAC3, and JPH2 is sufficient to reconstitute VICR in epithelial cells (11). STAC3 contains tandem SH3 domains, which bind CaV1.1’s II-III cytoplasmic loop (12). JPH2/Junctophilin is an SR membrane protein that binds the CaV1.1 cytoplasmic tail, thereby linking PM and SR membranes (13). Mutations that disrupt binding interactions between STAC3, JPH2, and CaV1.1 disrupt VICR and are associated with various human hereditary myopathies (12, 13).
Many decades of work suggests that VICR only occurs in vertebrate skeletal muscles (14). This phylogenetic restriction is proposed to result from several factors. First, all vertebrate genomes possess multiple CaV1, RYR, and JPH paralogs, a subset of which are expressed exclusively in skeletal muscles. By contrast, all invertebrate genomes possess single genes encoding these proteins, which are expressed in both neurons and muscles. Second, VICR is strongly disrupted in mutant mice lacking skeletal muscle isoforms (CaV1.1 and RYR1) and this defect is not rescued by ectopically expressing the cardiac isoforms (CaV1.2 and RYR2) in skeletal muscle (8, 15). Although JPH genes also underwent expansion in vertebrates, VICR can be successfully reconstituted by both skeletal muscle (JPH1 and 2) and brain-specific (JPH3) isoforms (16). Third, CaV1 channels in skeletal muscles (which undergo VICR) form tetrads (in freeze-fracture images), whereas invertebrate and cardiac muscle CaV1 channels form random arrays (14). The tetrad morphology is thought to reflect the direct physical coupling of CaV1.1 with RYR1 tetramers at SR-PM contacts in skeletal muscles. The simplest interpretation of these results is that VICR emerged in vertebrates as a consequence of gene duplications that produced skeletal muscle-specific isoforms of CaV1 and RYR (17). While VICR has primarily been studied in skeletal muscles, a few studies suggest that vertebrate neurons also exhibit VICR (18–20), suggesting that VICR is not exclusively found in muscles.
Here, we analyze CaV1-SR coupling in Caenorhabditis elegans body muscles. In C. elegans, muscle contraction is reduced but not eliminated in unc-68 RYR mutants, implying that external calcium entry can directly activate contraction (21). It remains unclear whether SR calcium release elicits contraction via CICR or VICR in C. elegans. In the nematode Ascaris Suum, muscle contraction requires external calcium and is inhibited by ryanodine (an RYR antagonist) (22), implying that EC coupling is mediated directly by external calcium and also by CICR. Like all invertebrates, C. elegans has single CaV1 (EGL-19), RYR (UNC-68), and JPH (JPH-1) orthologs (21, 23, 24). EGL-19 and UNC-68 are tightly colocalized at PM-SR junctions in body muscles and their colocalization is disrupted by mutations inactivating JPH-1/junctophilin (25). Mutations decreasing EGL-19, UNC-68, and JPH-1 function strongly impair locomotion, indicating that EC coupling has been disrupted (21, 23, 24). Surprisingly, we find that VICR also occurs in C. elegans muscles.
Results
Using SLO-2 Channels to Assay CaV1–RYR Coupling.
CaV1–RYR coupling is typically analyzed by measuring depolarization-induced changes in cytoplasmic calcium, using either fluorescent calcium dyes or muscle contraction as proxies for cytoplasmic calcium. Because cytoplasmic calcium is controlled by many mechanisms (including external calcium entry, CICR, and VICR), these assays may miss a minor contribution by VICR. To circumvent this problem, we utilized an assay that is selective for calcium in microdomains surrounding CaV1 channels. Microdomain selectivity should increase sensitivity because VICR is mediated by direct physical coupling of CaV1 and RYR channels. For these reasons, we used SLO-2 channels as an electrophysiological assay for CaV1–RYR coupling.
SLO-2 is a calcium-activated potassium channel (homologous to mammalian BK channels) (Fig. 1A) (26). SLO-2 function can be assessed in recordings utilizing internal solutions with high chloride levels (hereafter IkhiCl), which activates SLO-2 channels (26). IkhiCl recorded from body muscles is reduced in single mutants lacking either SLO-2/BK or SHK-1/KCNA channels and is eliminated in slo-2; shk-1 double mutants (27). Thus, IkhiCl comprises both SLO-2/BK and SHK-1/KCNA currents. By contrast, potassium currents recorded with low chloride internal solutions (IkloCl) are eliminated in shk-1 single mutants and are unaltered in slo-2 mutants, indicating that muscle IkloCl is solely mediated by SHK-1/KCNA channels (27).
Fig. 1.
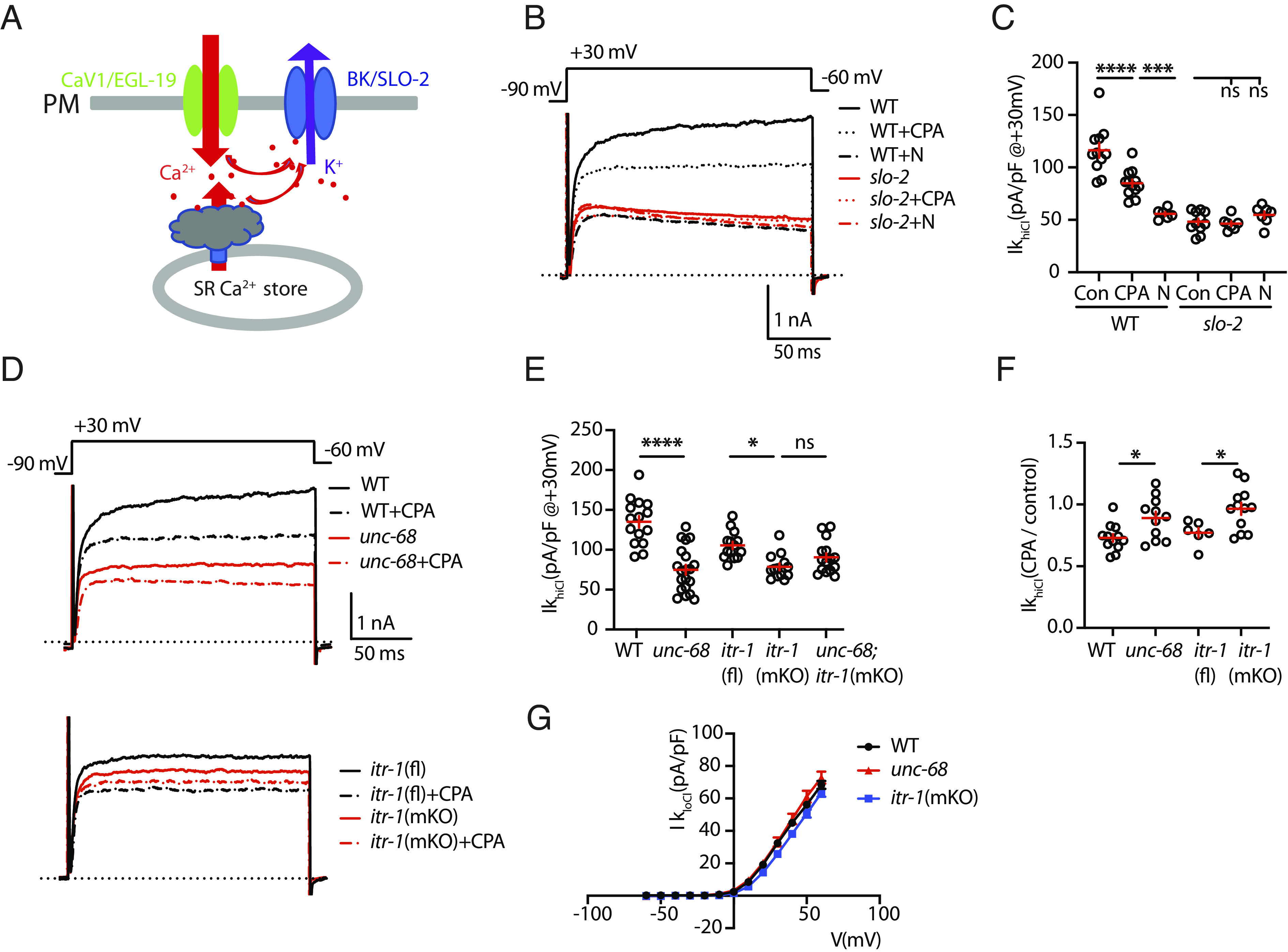
SLO-2 is activated by EGL-19/CaV1 and by SR calcium release. (A) A schematic model is shown illustrating how EGL-19 and SR calcium release contribute to SLO-2 activation. (B and C) IkhiCl was significantly decreased by Nemadipine (N, an EGL-19 antagonist) and by cyclopiazonic acid (CPA, a SERCA inhibitor). The inhibitory effects of Nemadipine and CPA are eliminated in slo-2 mutants, demonstrating that SLO-2 mediates the K current inhibited by these drugs. Representative traces (B) and mean current density (C) at +30 mV are shown. These results suggest that EGL-19 and SR calcium release are both required for SLO-2 activation. (D–F) IkhiCl was significantly decreased by mutations inactivating UNC-68/RYR and ITR-1/IP3R receptors. Representative traces (D), mean current density (E) at +30 mV, and mean current density ratio recorded with and without CPA treatment (F) are shown. For ITR-1 muscle-specific knockouts [itr-1(mKO)], we utilized a CRE inactivated allele, itr-1(fl), together with a transgene expressing CRE in body muscles. Additive IkhiCl defects were not observed in unc-68;itr-1(mKO) double mutants (E), suggesting that RYR and IP3R receptors function together to promote SLO-2 activation. CPA’s inhibitory effect on IkhiCl was significantly reduced in unc-68 and itr-1(mKO) mutants (F), suggesting that CPA inhibits SLO-2 activation by blocking SR calcium release. (G) SHK-1 (IkloCl) currents were unaffected in unc-68 and itr-1(mKO) mutants. Mean IkloCl current density is plotted as a function of membrane potential. Controls assessing ITR-1 function in the nonexcised itr-1(fl) strain and the effect of muscle CRE expression on IkhiCl currents are shown in SI Appendix, Fig. S1. Values that differ significantly are indicated (ns, not significant; *P < 0.05; ***P < 0.001; ****P < 0.0001). Error bars indicate SEM.
In principle, SLO-2 could be activated directly by external calcium (flowing through CaV channels in the PM) or by calcium released from internal stores (e.g., the SR). Following treatment with nemadipine (an EGL-19/CaV1 antagonist), IkhiCl was reduced to levels observed in slo-2 mutants (Fig. 1 B and C), as previously reported (27). Nemadipine’s inhibitory effect on IkhiCl is eliminated in slo-2 mutants, suggesting that SLO-2 mediates the nemadipine sensitive potassium current (Fig. 1 B and C). These results demonstrate that SLO-2 activation is functionally coupled to EGL-19/CaV1 channels.
Several results suggest that SLO-2 activation can occur in CaV1 microdomains (27). To prevent muscle contraction, SLO-2 currents are recorded using an intracellular solution that contains EGTA. EGTA limits free calcium levels outside of CaV1 microdomains. When intracellular solutions containing BAPTA (a fast calcium chelator) are used, IkhiCl is reduced but remains significantly higher than in slo-2 mutants (27). Collectively, these results show that SLO-2 currents can be used to assay calcium in CaV1 microdomains.
SR Calcium Release Contributes to SLO-2 Activation.
Next, we asked whether SR calcium stores are required for SLO-2 activation. Consistent with this idea, IkhiCl was significantly reduced by cyclopiazonic acid (CPA), a SERCA inhibitor (Fig. 1 B and C). CPA’s inhibitory effect on IkhiCl was eliminated in slo-2 mutants, indicating that the CPA-sensitive current was SLO-2 (Fig. 1 B and C). The CPA-resistant IkhiCl was significantly larger than that recorded from slo-2 mutants, suggesting that blocking SR calcium release reduces but does not eliminate SLO-2 activation (Fig. 1 B and C). To determine which SR calcium channel mediates SLO-2 activation, we analyzed IkhiCl in mutants lacking UNC-68/RYR and ITR-1/IP3 receptors (IP3Rs). Because itr-1 null mutants are infertile, we used CRISPR to construct a CRE-inactivated itr-1(nu774 FLOX) allele, allowing muscle-specific itr-1 knockouts. To assess the integrity of ITR-1 function in itr-1(nu774 fl) mutants, we analyzed the defecation cycle duration, which is prolonged in itr-1 mutants (28). Defecation cycle duration was unaltered in itr-1(nu774 fl) mutants (SI Appendix, Fig. S1A), suggesting that ITR-1 function was not impaired by the insertion of the LoxP sites. IkhiCl was significantly reduced in both unc-68 and itr-1(muscle KO) single mutants and was not further reduced in unc-68;itr-1 double mutants (Fig. 1 D and E). Since additive effects were not observed in the double mutants, these results suggest that UNC-68 and ITR-1 function together to activate SLO-2. CRE expression in WT body muscles had no effect on IkhiCl (SI Appendix, Fig. S1B), suggesting that the itr-1(muscle KO) mutant defects are unlikely to result from toxicity associated with CRE expression (29). CPA’s inhibitory effect on IkhiCl was significantly reduced in both unc-68 and itr-1(muscle KO) single mutants (Fig. 1F), consistent with CPA altering IkhiCl by inhibiting SR calcium release. Although SHK-1/KCNA and SLO-2/BK both contribute to IkhiCl, SHK-1/KCNA current (assessed by IkloCl) was unaltered in both unc-68 and itr-1(muscle KO) mutants (Fig. 1G); consequently, the IkhiCl defects in these mutants result from decreased SLO-2/BK currents. Collectively, these results suggest that UNC-68/RYR and ITR-1/IP3R function together to mediate voltage-activated SR calcium release thereby promoting SLO-2 activation.
SLO-2 Activation Is Mediated by Both External Calcium and VICR.
Next, we did several additional experiments to determine whether VICR promotes SR calcium release. A defining characteristic of VICR is that SR calcium release persists when external calcium entry is blocked. To determine whether SLO-2 is activated by VICR, we analyzed IkhiCl after blocking external calcium entry. IkhiCl was reduced by ~25% in recordings containing a general CaV pore blocker (Cd2+) (Fig. 2 A and B). The impact of Cd2+ on IkhiCl was eliminated in slo-2 mutants (Fig. 2 A and B), indicating that SLO-2 mediates the Cd2+-sensitive potassium current. The Cd2+-resistant IkhiCl was significantly reduced in slo-2 mutants (Fig. 2 A and B), suggesting that SLO-2 activation was reduced but not eliminated when external calcium entry is blocked. To confirm this result, we analyzed IkhiCl in recordings lacking external calcium. Compared to control recordings (with 5 mM Ca2+ext), IkhiCl was again reduced by ~25% in recordings lacking external calcium (Fig. 2 A and D). In slo-2 mutants, IkhiCl recorded in 0 mM and 5 mM Ca2+ext were not significantly different, suggesting that SLO-2 mediates the potassium current activated by external calcium (Fig. 2D). IkhiCl recorded in 0 mM Ca2+ext was significantly reduced in slo-2 mutants (Fig. 2 A and D), once again suggesting that preventing external calcium entry reduces but does not eliminate SLO-2 activation. By contrast, nemadipine (an EGL-19 antagonist) completely blocked SLO-2 activation (Fig. 1 B and C) (27). Thus, even when external calcium entry is blocked, EGL-19 activation elicits cytoplasmic calcium increases sufficient to activate SLO-2. These results also suggest that nemadipine eliminates calcium entry by blocking voltage-dependent movement of EGL-19’s voltage-sensing domains, as was previously shown for nifedipine (a structurally related dihydropyridine calcium channel antagonist) (30). Finally, CPA reduced the IkhiCl recorded in 0 mM Ca2+ext to levels found in slo-2 mutants (Fig. 2 C and D), confirming that the residual SLO-2 activation observed when external calcium entry is blocked was mediated by VICR from the SR. Taken together, these results suggest that SLO-2 channels are activated by two mechanisms, direct activation by external calcium and activation by VICR. Similar decreases in IkhiCl were observed when external calcium entry and SR calcium release were blocked (compare Figs. 1C and 2D), suggesting that these two mechanisms contribute equally to SLO-2 activation. Because VICR has not been reported in invertebrates, we further investigated the mechanism mediating VICR activation of SLO-2 channels.
Fig. 2.
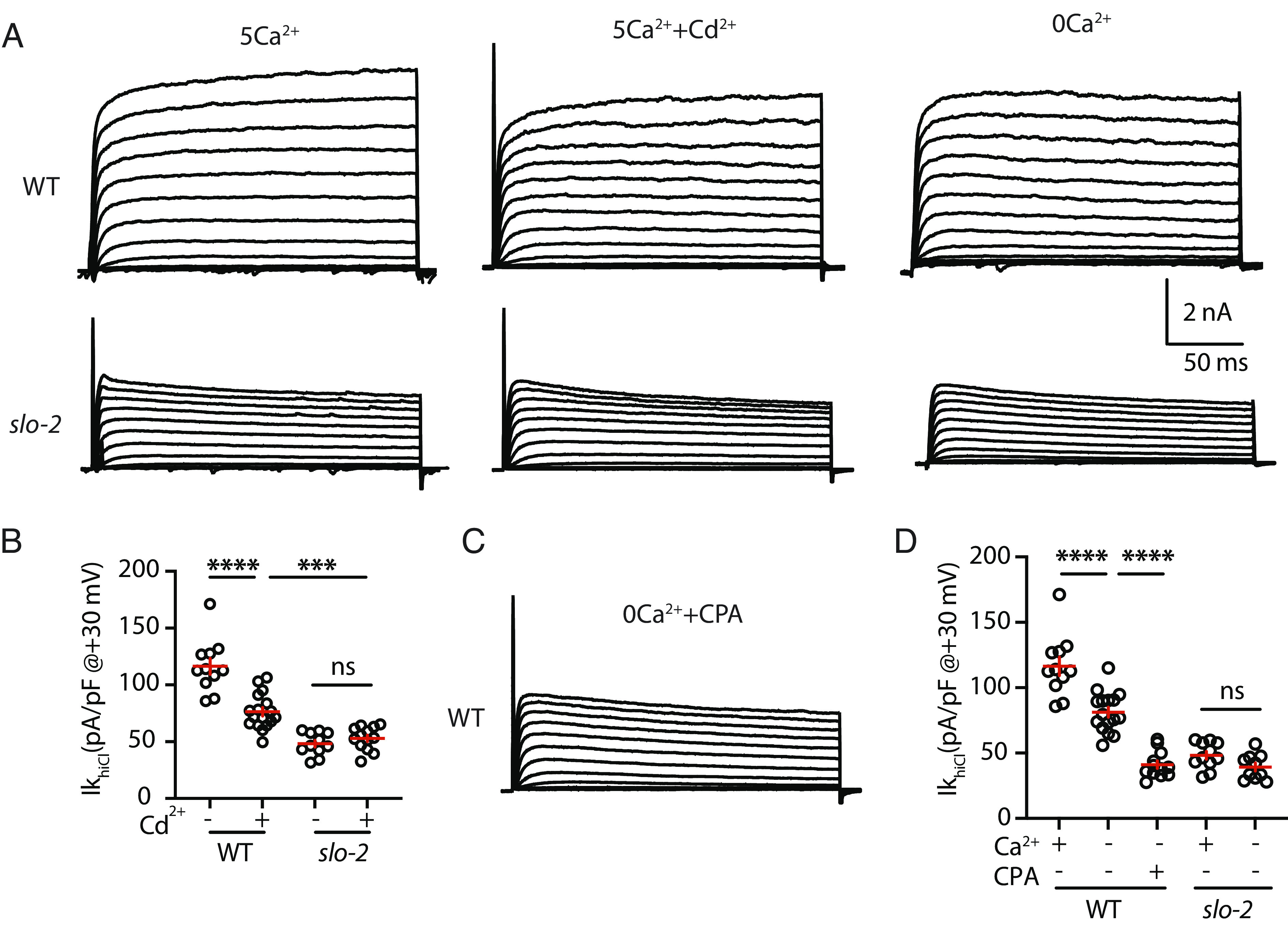
SLO-2 is activated by external calcium and by VICR. (A–D) IkhiCl was significantly decreased by a general CaV pore blocker (Cd2+) and in recordings lacking external calcium. IkhiCl currents were recorded from adult body wall muscles at holding potentials of −60 to +60 mV. Representative IkhiCl traces (A) and mean current density at +30 mV (B and D) are shown. The inhibitory effect of Cd2+ on IkhiCl was eliminated in slo-2 mutants (B), suggesting that SLO-2 mediates the Cd2+ sensitive K current. IkhiCl recorded in 5 mM and 0 external Ca2+ were not significantly different from slo-2 mutants (D), demonstrating that SLO-2 mediates the K current activated by external calcium. (C and D) CPA treatment reduced the IkhiCl recorded in 0 Ca2 to levels found in slo-2 mutants. These results suggest that the residual SLO-2 activation observed when external calcium entry is blocked is mediated by VICR. Representative traces (C) and mean current density at +30 mV (D) are shown. Values that differ significantly are indicated (ns, not significant; ***P < 0.001; ****P < 0.0001). Error bars indicate SEM.
UNC-68/RYR and ITR-1/IP3R Are Both Required for VICR Mediated SLO-2 Activation.
In vertebrates, CaV1.1 directly activates RYR1 to mediate VICR in skeletal muscle. To determine which SR calcium channel mediates VICR in C. elegans, we analyzed SLO-2 activation in unc-68 RYR and itr-1IP3R (muscle KO) mutants. Changes in SLO-2 current in these mutants could reflect impaired VICR or changes in the baseline expression or function of SLO-2, EGL-19, or UNC-68 channels. To distinguish between these possibilities, we measured the ratio of IkhiCl recorded in 0 mM and 5 mM Ca2+ext in each genotype. The IkhiCl (0/5 Ca2+ext) ratio should detect VICR defects but should be insensitive to baseline defects, as the latter would contribute equally to SLO-2 activation in both conditions. The IkhiCl (0/5 Ca2+ext) ratio was significantly reduced in both unc-68 and itr-1(muscle KO) single mutants (Fig. 3 A and B), suggesting that RYR and ITR-1 receptors are both required for VICR-mediated SLO-2 activation. The IkhiCl (0/5 Ca2+ext) ratio in unc-68; itr-1(muscle KO) double mutants was indistinguishable from that found in the corresponding single mutants (Fig. 3B). Because additive effects were not observed in the double mutants, these results suggest that UNC-68 and ITR-1 function together for VICR mediated SLO-2 activation.
Fig. 3.
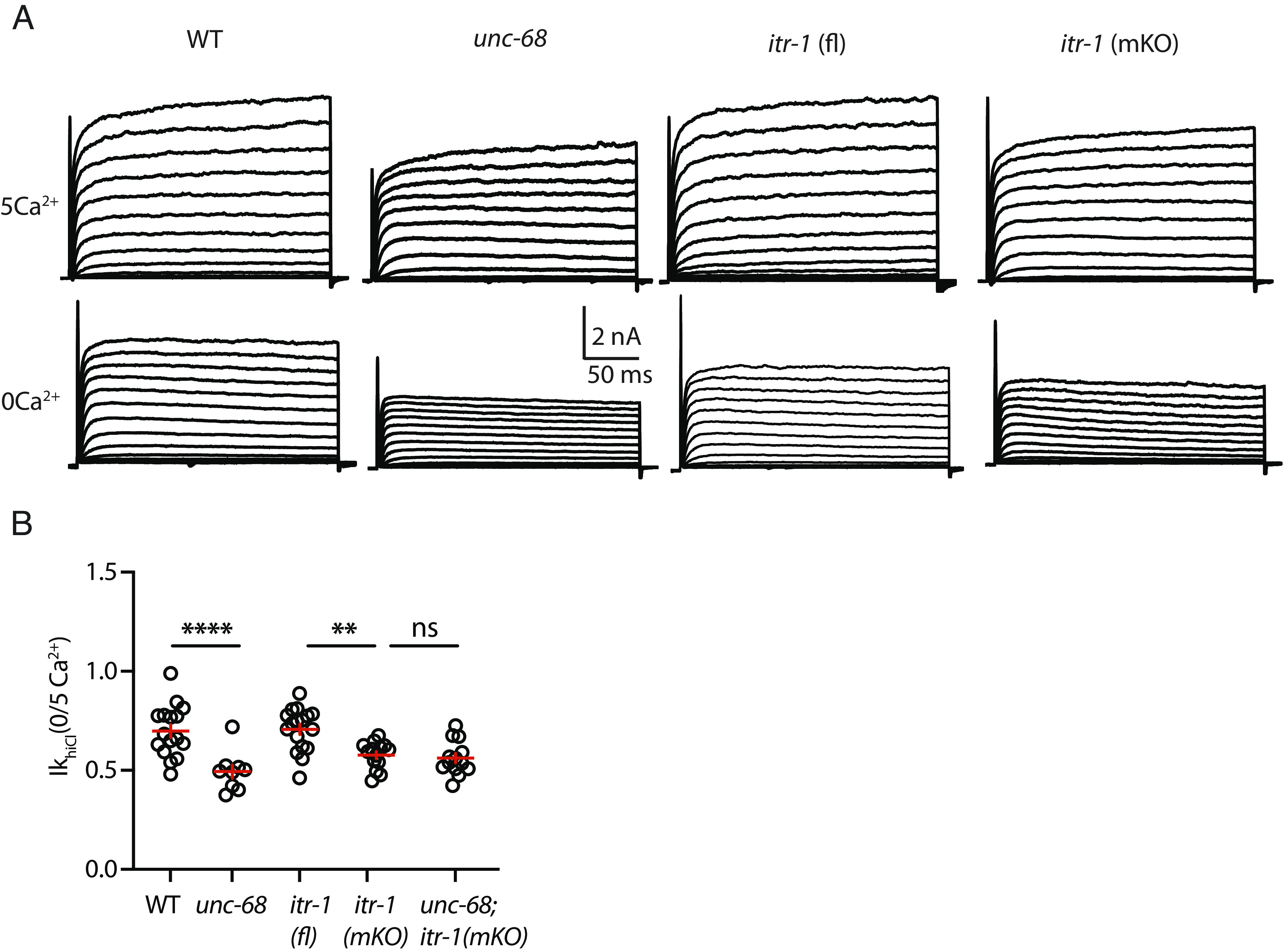
UNC-68/RYR and ITR-1/IP3R are both required for VICR-mediated SLO-2 activation. IkhiCl currents were recorded from adult body wall muscles at holding potentials of −60 to +60 mV in the presence and absence of external calcium. Representative IkhiCl traces are shown (A). The ratio of IkhiCl current density at +30 mV recorded in the absence of external calcium to the mean current density recorded in 5 mM Ca2+ [IkhiCl (0/5Ca2+)] is plotted for each genotype (B). The IkhiCl (0/5Ca2+) ratio was significantly decreased in unc-68 and itr-1(mKO) single mutants and was not further reduced in the corresponding double mutant. These results suggest that UNC-68 and ITR-1 function together to promote SLO-2 activation. Values that differ significantly are indicated (ns, not significant; **P < 0.01; ****P < 0.0001). Error bars indicate SEM.
JPH-1 Is Required for VICR.
Junctophilin is required for VICR in vertebrates (9). We previously reported that jph-1 mutations strongly disrupt EGL-19/CaV1 colocalization with UNC-68/RYR in muscles (25), implying that VICR is likely disrupted in these mutants. To confirm JPH-1’s role in VICR, we assayed SLO-2 activation in jph-1 mutants (Fig. 4 A and B). For this analysis, we used a CRE inactivated allele jph-1(nu733 FLEX) to construct muscle-specific knockouts, jph-1(muscle KO). The IkhiCl (0/5 Ca2+ext) ratio was dramatically reduced in jph-1(muscle KO) mutants compared to jph-1(nu733 FLEX) controls (Fig. 4 A and B). IkloCl was unaltered in jph-1(muscle KO) mutants (SI Appendix, Fig. S2D), suggesting that SHK-1/KCNA current was unaffected by this mutation. Collectively, these results suggest that JPH-1/Junctophilin (like its vertebrate counterpart) promotes VICR.
Fig. 4.
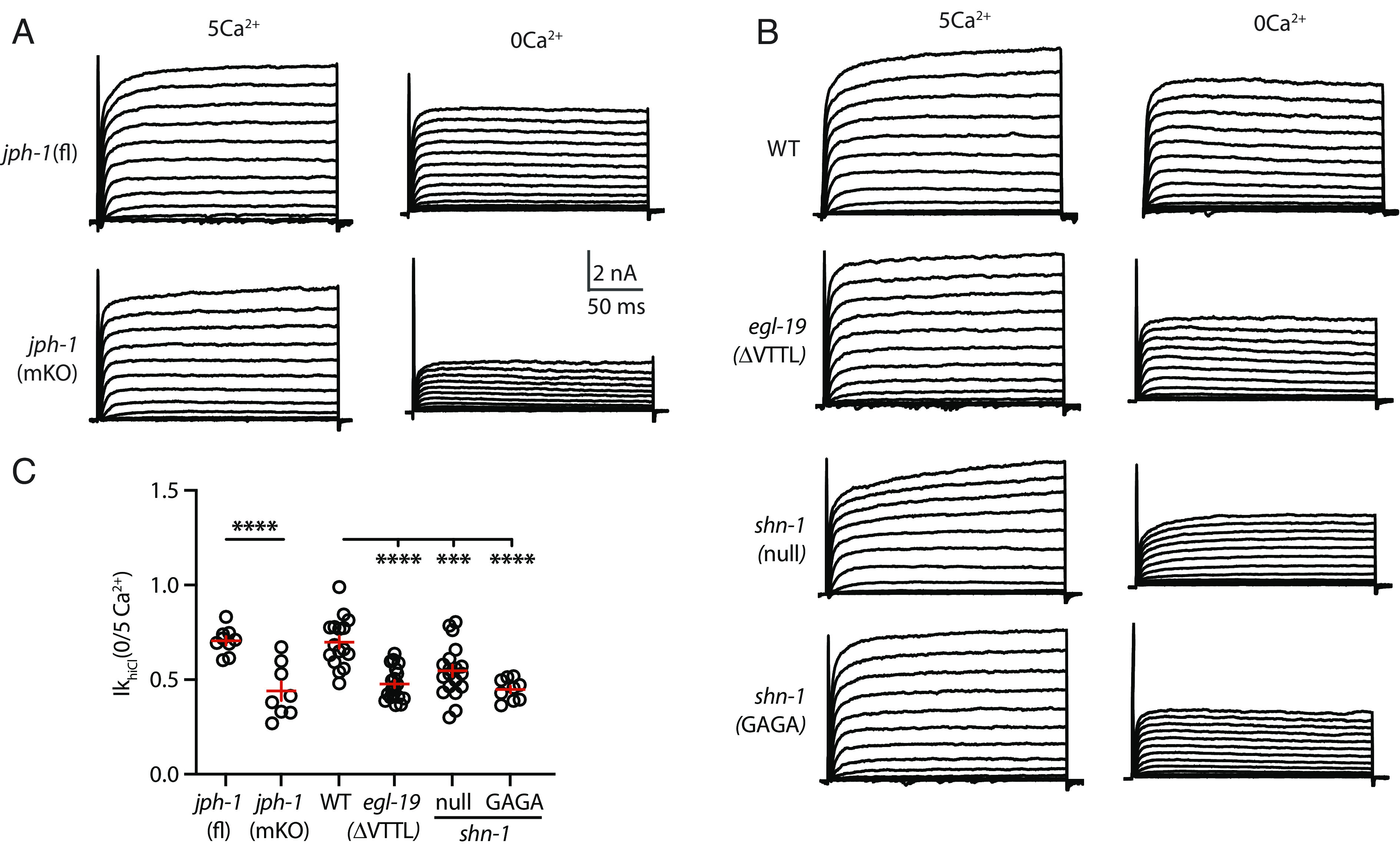
JPH-1 and SHN-1 binding to EGL-19/CaV1’s C-terminal PBD are required for VICR mediated SLO-2 activation. IkhiCl currents were recorded from adult body wall muscles at holding potentials of −60 to +60 mV in the presence and absence of external calcium. Representative IkhiCl traces are shown (A and B). The ratio of IkhiCl current density at +30 mV recorded in the absence of external calcium to the mean current density recorded in 5 mM Ca2+ [IkhiCl (0/5Ca2+)] is plotted for each genotype (C). The IkhiCl (0/5Ca2+) ratio was significantly decreased in jph-1(muscle KO), egl-19(ΔVTTL), shn-1(null), and shn-1(GAGA) mutants. These results suggest that VICR-mediated SLO-2 activation is decreased in these mutants. Nonnormalized IkhiCl IV curves are shown in SI Appendix, Fig. S2. Values that differ significantly are indicated (***P < 0.001; ****P < 0.0001). Error bars indicate SEM.
EGL-19/CaV1’s Carboxy-Terminus Is Required for VICR.
The EGL-19/CaV1 c-terminus (-VTTLCOOH) is a PDZ binding domain (PBD) (31). Muscle APs are prolonged in egl-19(nu496 ΔVTTL) mutants, which lack the PBD, and AP durations were not further increased in egl-19(ΔVTTL); slo-2 double mutants (27), implying that decreased SLO-2 activation accounts for the prolonged AP durations. Prompted by these results, we asked whether the egl-19(ΔVTTL) mutation alters SLO-2 currents. The IkhiCl (0/5 Ca2+ext) ratio was significantly reduced in egl-19(ΔVTTL) mutants (Fig. 4 A and B). IkloCl was unaltered in egl-19(ΔVTTL) mutants, suggesting that SHK-1/KCNA current was unaffected by this mutation (SI Appendix, Fig. S2D). Collectively, these results suggest that deleting EGL-19’s c-terminal PBD disrupts VICR-mediated SLO-2 activation.
SHN-1 Is Required for VICR.
EGL-19’s PBD binds the PDZ domain of SHN-1, the C. elegans ortholog of the scaffold protein Shank (31). We previously showed that SHN-1 promotes SLO-2 activation by two mechanisms (27): promoting SLO-2 clustering on the cell surface and promoting SLO-2 activation by nearby EGL-19/CaV1 channels. Next, we asked whether SHN-1 is also required for VICR-mediated SLO-2 activation. Consistent with this idea, the IkhiCl (0/5 Ca2+ext) ratio was significantly reduced in shn-1(null) mutants (Fig. 4 A and B). By contrast, IkloCl was unaltered in shn-1(null) mutants (SI Appendix, Fig. S2D), suggesting that SHK-1/KCNA current was unaffected. To confirm SHN-1’s role in VICR, we used CRISPR to mutate the PDZ domain GFGF motif to GAGA, thereby disrupting the binding pocket for c-terminal ligands (32). The shn-1(nu793 GAGA) mutation was introduced into a shn-1 allele containing a GFP11 tag. The shn-1(nu793 GAGA) mutation did not disrupt the expression of the endogenous SHN-1 protein, visualized by expressing GFP1-10 with a ubiquitous promoter (SI Appendix, Fig. S2E) nor did it alter IkloCl (SI Appendix, Fig. S2D). Similar to shn-1 null mutants, the IkhiCl (0/5 Ca2+ext) ratio was significantly reduced in shn-1(nu793 GAGA) mutants (Fig. 4 A and B), consistent with decreased VICR. Taken together, these results suggest that SHN-1 binding to EGL-19’s PBD promotes VICR-mediated SLO-2 activation.
Bidirectional Signaling between EGL-19/CaV1 and UNC-68/RYR.
VICR coupling in vertebrates is accompanied by bidirectional signaling between CaV1.1 and RYR1. VICR mediated SLO-2 activation provides evidence for anterograde (CaV1 to RYR) signaling (Fig. 2). Retrograde (RYR1 to CaV1.1) signaling is indicated by decreased EGL-19/CaV1 current in multiple VICR-deficient mutants, including egl-19(ΔVTTL) CaV1, shn-1(nu793 GAGA), unc-68(null) RYR, itr-1(muscle KO), and jph-1(muscle KO) (Fig. 5 A and B). Similar decreases in CaV1.1 current are observed in mouse mutants lacking RYR1 and JPH1 (33–35).
Fig. 5.

Retrograde regulation of EGL-19/CaV1 function and subcellular localization. EGL-19/CaV1 currents were recorded from adult body wall muscles at holding potentials of −60 to +60 mV in the indicated genotypes. Representative traces (at +20 mV) (A) and mean current density as a function of membrane potential (B) are shown. EGL-19 current was significantly decreased in VICR defective mutants. Controls showing that EGL-19 current was unaltered in strains containing the CRE inactivated itr-1 and jph-1 alleles are shown in SI Appendix, Fig. S3. (C) EGL-19(nu674 GFP11) expression is analyzed in adult body muscles. GFP11 fluorescence was reconstituted by expressing GFP1-10 in body muscles. EGL-19 puncta intensity was reduced in unc-68 mutants. Representative images of EGL-19 colocalization with UNC-68 in egl-19(ΔVTTL) and shn-1(null) mutants are shown in SI Appendix, Fig. S3. Values that differ significantly are indicated (****P < 0.0001). Error bars indicate SEM. (Scale bar indicates 4 μm.)
Bidirectional CaV1–RYR signaling is thought to result from physical coupling of these channels at PM-SR contacts. To further test this idea, we asked whether UNC-68/RYR is required for proper EGL-19/CaV1 subcellular localization. Consistent with this idea, EGL-19/CaV1 puncta intensity was 60% reduced in unc-68 RYR null mutants (Fig. 5C), which mirrors the ~40% decrease in EGL-19 current observed in these mutants (Fig. 5B). Taken together, these results suggest that the function and subcellular localization of EGL-19 is regulated in a retrograde manner by UNC-68, consistent with these channels being physically coupled at PM-SR junctions.
If VICR is mediated by direct physical contact between EGL-19/CaV1 and UNC-68/RYR at PM-SR contacts, mutations that disrupt VICR might prevent colocalization of these channels. Consistent with this idea, we previously reported that EGL-19 colocalization with UNC-68 was dramatically reduced in jph-1 mutants (25). By contrast, EGL-19 colocalization with UNC-68 was largely unaffected in two other VICR-deficient mutants, egl-19(ΔVTTL) and shn-1(null) mutants (SI Appendix, Fig. S3C). These results suggest that these mutations disrupt VICR by distinct mechanisms.
Locomotion Is Not Dramatically Altered in VICR-Deficient Mutants.
To determine whether VICR contributes to EC coupling, we analyzed egl-19(ΔVTTL) and shn-1 mutants for defects in locomotion. Because VICR-mediated SLO-2 activation is decreased in egl-19(ΔVTTL) and shn-1 mutants, we reasoned that these mutations should also prevent VICR-mediated muscle contraction. We found that locomotion rate was not significantly reduced in egl-19(ΔVTTL) and itr-1(muscle KO) mutants and was modestly reduced in shn-1(GAGA) mutants (SI Appendix, Fig. S3D). Because SLO-2 activation is impaired in all of these mutants, we also analyzed slo-2 mutants and found that rates were not significantly altered (SI Appendix, Fig. S3D). These results suggest that blocking VICR has relatively modest effects on EC coupling.
Discussion
Our results lead to six principal conclusions. First, SLO-2 activation is mediated by both external calcium entry and VICR. Second, UNC-68 and ITR-1 function together to promote VICR. Third, VICR-mediated SLO-2 activation is significantly reduced by mutations disrupting SHN-1 binding to EGL-19’s c-terminus. Fourth, EGL-19 and UNC-68 reciprocally control each other’s function. Fifth, EC coupling is not dramatically altered when VICR is blocked. And sixth, VICR is not restricted to vertebrates. Below we discuss the significance of these findings.
A Revised Phylogeny for VICR.
Decades of prior work suggested that VICR is restricted to vertebrates. Contrary to expectation, we find that VICR also occurs in C. elegans body muscles. VICR coupling in C. elegans bears several similarities to that found in vertebrates. First, VICR coupling requires a junctophilin protein (JPH-1). Second, EGL-19/CaV1 and UNC-68/RYR reciprocally control each other’s function, similar to bidirectional CaV1.1-RYR1 signaling in vertebrates. Unlike vertebrates, VICR in C. elegans is unlikely to require a STAC protein because C. elegans lacks an obvious STAC ortholog and the EGL-19/CaV1 II-III cytoplasmic loop lacks homology to the STAC3 binding site in vertebrate CaV1.1 (12).
VICR-mediated SLO-2 activation was reduced by mutations inactivating UNC-68/RYR, ITR-1/IP3R, SHN-1/Shank, and EGL-19’s c-terminal PBD. These mutations likely disrupt VICR via several mechanisms. UNC-68 and ITR-1 are likely to form SR calcium release channels, which could be activated by VICR. The UNC-68/RYR requirement for VICR was expected because RYR channels are required for VICR in vertebrates. By contrast, the ITR-1 requirement was surprising as IP3Rs have not been implicated in VICR and because itr-1 is expressed at very low levels in body muscles (36). One potential explanation for these findings is that a small subpopulation of UNC-68 subunits coassemble with ITR-1 to form heteromeric calcium release channels. Further experiments will be required to test this possibility. The defects resulting from ITR-1 inactivation were smaller than those in other VICR defective mutants. The relatively modest itr-1(mKO) phenotypes could reflect defects in the nonexcised itr-1(fl) strain, inefficient CRE-mediated itr-1 excision, or a comparatively smaller functional role for ITR-1. PM-SR contacts are grossly disrupted in unc-68 null mutants (37), which likely accounts for the dramatic change in EGL-19 abundance and subcellular localization observed in these mutants. EGL-19 and UNC-68 abundance and colocalization were also dramatically reduced in jph-1 mutants (25), consistent with a disruption of SR-PM contacts as seen in mouse JPH knockouts (9). By contrast, EGL-19 colocalization with UNC-68 was largely unaffected in shn-1 and egl-19(ΔVTTL) mutants, suggesting that PM-SR contacts were not dramatically disrupted in these mutants. Instead, we propose that SHN-1/Shank binding to EGL-19/CaV1’s PBD promotes SLO-2 clustering near PM-SR contacts (27), thereby promoting VICR-mediated SLO-2 activation.
VICR-deficient mutants exhibit relatively normal locomotion behavior, implying that EC coupling is not significantly impaired in these mutants. By contrast, we previously reported that two mutants lacking VICR, egl-19(ΔVTTL) and shn-1(null), exhibit prolonged muscle action potentials (due to decreased SLO-2 activation) (27). Thus, our results suggest that VICR in C. elegans promotes excitation–repolarization coupling while having little impact on EC coupling. Our results are also consistent with prior studies suggesting that EC coupling is mediated by external calcium entry and CICR in C. elegans and A. suum (21, 22).
A surprising aspect of our results is that we failed to detect a contribution of CICR for SLO-2 activation. In principle, external calcium entry could activate SLO-2 by two mechanisms, direct binding to SLO-2 and indirect activation by CICR. Thus, if CICR occurs in our recordings, CPA should inhibit SLO-2 activation by both external calcium entry and by VICR. Contrary to this expectation, the IkhiCl decrease (~25%) caused by blocking SR calcium release (with CPA) was very similar to the VICR-activated SLO-2 current. This result suggests that CPA’s effect on IkhiCl was limited to SLO-2 activation by VICR. One potential explanation for these results is that CICR’s contribution is diminished in our recordings due to the presence of EGTA in the intracellular solution. EGTA is included in the pipette solution to inhibit muscle contractions, which would disrupt the patch clamp recording.
A Different Physiological Function for Shank.
Shank is a synaptic scaffolding protein (containing SH3, PDZ, proline-rich, and SAM domains) (38). In humans, Shank mutations and copy number variations are linked to Autism Spectrum Disorders (ASD), schizophrenia, and mania (39, 40). Haploinsufficiency for 22q13 (which spans the Shank3 locus) occurs in Phelan-McDermid syndrome (PMS), a syndromic form of ASD (41). Shank regulates the subcellular localization of several ion channels including NMDA and AMPA type glutamate receptors (40, 42); HCN channels (43, 44); TRPV channels (45); CaV1 voltage-activated calcium channels (31, 46); and BK potassium channels (27).
We previously reported that SHN-1 promotes EGL-19/CaV1 coupling to SLO-2 channels (27). In this prior study, we showed that SHN-1 plays two roles in EGL-19 to SLO-2 coupling. First, SHN-1 promotes SLO-2 clustering on the muscle surface, as indicated by decreased SLO-2 puncta intensity in shn-1 null mutants. In addition, SHN-1 promotes EGL-19/CaV1-mediated activation of nearby SLO-2 channels, as indicated by the phenotype of SHN-1 deletion mutants lacking the PDZ domain (which exhibit decreased SLO-2 currents and unaffected SLO-2 puncta intensities). Here, we report that SHN-1 also promotes VICR-mediated SLO-2 activation. Collectively, our results suggest that SHN-1 plays multiple roles in coupling EGL-19/CaV1 to SLO-2 channels.
Shank’s role in CaV1–RYR coupling has not been tested in other organisms; however, Homer (a Shank binding protein) promotes CaV1–RYR coupling in mammalian skeletal and smooth muscles (47, 48). These results suggest that Shank protein complexes could play a conserved role in CaV1–RYR coupling. Because VICR has been reported in vertebrate neurons (18–20), it is also possible that Shank protein complexes could regulate neuronal function by promoting VICR.
Prospects for Finding VICR in Other Unexpected Tissues and Organisms.
We use an electrophysiological assay (SLO-2 activation) to detect VICR. This assay has several advantages. First, the SLO-2 assay provides a quantitative assessment of VICR’s contribution to calcium signaling even when other mechanisms (e.g., direct activation by external calcium) co-occur. Second, this assay selectively detects calcium changes in microdomains surrounding the CaV1 and RYR calcium channels due to the presence of EGTA in the recording pipet. This microdomain selectivity simplifies our results by minimizing CICR’s role. By contrast, VICR’s contribution could be missed by assays reflecting bulk cytoplasmic calcium levels. Third, this assay detects VICR independently from muscle contraction; consequently, this strategy could detect VICR in neurons or in muscles where contraction does not rely on VICR (as in C. elegans body muscles). Consequently, we speculate that similar strategies could allow VICR detection in other unexpected tissues.
Materials and Methods
Animals.
C. elegans strains were cultivated at room temperature (~22 °C) on agar nematode growth media seeded with OP50 bacteria. Unless otherwise stated, the wild-type animal refers to the Bristol N2 strain. Alleles and strains used in this study are listed in SI Appendix, Table S1. Transgenic animals were prepared by microinjection, as described (49). Single-copy transgenes were isolated by the MoSCI and miniMoS techniques (50, 51).
CRISPR Alleles.
CRISPR alleles were isolated as described (52). Briefly, Cas9 protein and guide RNAs were ordered from IDT. Repair templates shorter than 200 bp consisted of ss ULTRAMER oligos from IDT. Longer repair templates were PCR amplified from a plasmid and melted before adding to the injection mix. The injection mix included the pRF4 rol-6(gf) plasmid. Injected animals were singled and 96 F1 progeny were singled from plates that contained rollers, allowed to starve out the plate, and were then screened by PCR for the expected change. All CRISPR alleles were outcrossed at least two times. Split GFP and Cherry constructs are described in ref. 53. MiniMOS transgenes coexpressing GFP1–10 and Cherry1–10 in body muscles (nuSi456, pat-10 promoter) or GFP1-10 in all tissues (nuSi205, eft-3 promoter) were used to visualize GFP11 and Cherry11 tagged proteins.
Tissue-specific knockouts were performed using CRE inactivated alleles. CRE recombinase was expressed in muscles (myo-3 promoter) from a single copy miniMOS transgene (nuSi572). The itr-1(nu774 FLOX) allele contains LoxP sites in intron 8 and the 3’UTR of the endogenous itr-1a locus. The jph-1(nu733 FLEX) allele contains an inverted stop cassette in intron 1 of the endogenous jph-1 locus. The stop cassette consists of a synthetic exon (containing stop codons in all reading frames), a transcriptional terminator, and flanking FLEX sites (which mediate CRE induced inversions) (27, 54).
Fluorescence Imaging.
Worms were immobilized on 10% agarose pads with 3 µL of 0.1-µm-diameter polystyrene microspheres (Polysciences 00876-15, 2.5% w/v suspension). Body muscles in the head were imaged to avoid gut autofluorescence. Images were taken with a Nikon A1R confocal, using a 60×/1.49 NA oil objective, with Nyquist sampling. Image volumes spanning the muscle surface were collected (~10 planes/volume, 0.15 μm between planes, and 0.1 μm/pixel). Maximum intensity projections for each volume were autothresholded, and puncta were identified as round fluorescent objects (area > 0.1 mm2), using analysis of particles. Mean fluorescent intensity in each punctum was analyzed in deconvoluted images. All image analysis was done using FIJI.
Electrophysiology.
Whole-cell patch-clamp measurements were performed using an Axopatch 200B amplifier with pClamp 10 software (Molecular Devices). The data were sampled at 10 kHz and filtered at 5 kHz. All recordings were performed at room temperature (~19 to 21 °C).
Muscle CaV recordings.
The bath solution contained (in mM) TEA-Cl 140, CaCl2 5, MgCl2 1, 4AP 3, glucose 10, sucrose 5, and HEPES 15 (pH 7.4, 330 mOsm). The pipette solution contained (in mM) CsCl 140, TEA-Cl 10, MgCl2 5, EGTA 5 and HEPES 5 (pH 7.2, 320 mOsm). The voltage-clamp protocol consisted of −60 mV for 100 ms, −90 mV for 100 ms, test voltage (from −60 mV to +60 mV) for 200 ms.
Muscle K+ current recordings.
The bath solution contained (in mM) NaCl 140, KCl 5, CaCl2 5, MgCl2 5, dextrose 11, and HEPES 5 (pH 7.2, 320 mOsm). For IkhiCl recordings, the pipette solution contained (in mM) KCl 120, KOH 20, Tris 5, CaCl2 0.25, MgCl2 4, sucrose 36, EGTA 5 (or BAPTA 5), and Na2ATP 4 (pH 7.2, 323 mOsm). For IkloCl recordings, the pipette solution contained (in mM) Kgluconate 120, KOH 20, Tris 5, CaCl2 0.25, MgCl2 4, sucrose 36, EGTA 5, and Na2ATP 4 (pH 7.2, 323 mOsm). The voltage-clamp protocol consisted of −60 mV for 50 ms, −90 mV for 50 ms, test voltage (from −60 mV to +60 mV) for 150 ms. In figures, we show outward currents evoked at +30 mV, which corresponds to the peak amplitude of muscle APs. To block external calcium entry, bath solutions were used containing EGL-19 antagonists (5 μM nemadipine or 1 mM CdCl2) or those lacking CaCl2 (which was replaced by 5 mM EGTA). To block SR calcium release, a SERCA pump inhibitor (10 μM CPA) was added to the pipette solution.
Behavior analysis.
Locomotion.
Adult worms (~10/plate) were imaged on a fresh bacterial lawn for 200 s (5 Hz frame rate) under brightfield illumination (Artograph LightPad A920) using a FLIR-Blackfly camera (Edmund Optics BFS-U3-120S4M-CS) with a 50 mm f/2.8 lens (ThorLabs MVL50TM23, 5.25 μm/pixel). Behavioral recordings were analyzed using TIERPSY tracking software (55). The locomotion rate was assessed using the speed_midbody parameter.
Defecation.
The duration of the defecation cycle was analyzed in individual adults by measuring the time between consecutive posterior body contractions.
Quantitation and Statistical Analysis.
For normally distributed data, significant differences were assessed with unpaired t tests (for two groups) or one-way ANOVA with post hoc Tukey’s multiple comparisons test (for >2 groups). For nonnormal data, differences were assessed by Mann–Whitney U test (two groups) or Kruskal–Wallis test with post hoc Dunn’s multiple comparisons test (>2 groups). Data graphing and statistics were performed in GraphPad Prism 9. No statistical method was used to select sample sizes. Data shown in each figure represent contemporaneous measurements from mutant and control animals over a period of 1 to 2 wk. For electrophysiology, data points represent mean values for individual neuron or muscle recordings (which were considered biological replicates). For imaging studies, data points represent mean puncta fluorescence values in individual animals (which were considered biological replicates). All data obtained in each experiment were analyzed, without any exclusions.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Acknowledgments
We thank the following for strains, advice, reagents, and comments on the manuscript: Bruce Bean, C. elegans genetics stock center (CGC), Shohei Mitani, and members of the Kaplan lab. This work was supported by an NIH research grant to J.M.K. (NS32196). The CGC is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Author contributions
L.G., E.A., S.N., and J.M.K. designed research; L.G. and S.N. performed research; L.G. and E.A. analyzed data; and L.G., E.A., S.N., and J.M.K. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the SI appendix, Dataset S1.
Supporting Information
References
- 1.Rios E., Ma J. J., Gonzalez A., The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J. Muscle Res. Cell Motil. 12, 127–135 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Schneider M. F., Control of calcium release in functioning skeletal muscle fibers. Annu. Rev. Physiol. 56, 463–484 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Fabiato A., Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245, C1–14 (1983). [DOI] [PubMed] [Google Scholar]
- 4.Gyorke S., Palade P., Calcium-induced calcium release in crayfish skeletal muscle. J. Physiol. 457, 195–210 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takekura H., Franzini-Armstrong C., The structure of Ca(2+) release units in arthropod body muscle indicates an indirect mechanism for excitation-contraction coupling. Biophys. J. 83, 2742–2753 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ormerod K. G., Scibelli A. E., Littleton J. T., Regulation of excitation-contraction coupling at the Drosophila neuromuscular junction. J. Physiol. 600, 349–372 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Block B. A., Imagawa T., Campbell K. P., Franzini-Armstrong C., Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107, 2587–2600 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Protasi F., Franzini-Armstrong C., Flucher B. E., Coordinated incorporation of skeletal muscle dihydropyridine receptors and ryanodine receptors in peripheral couplings of BC3H1 cells. J. Cell Biol. 137, 859–870 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeshima H., Komazaki S., Nishi M., Iino M., Kangawa K., Junctophilins: A novel family of junctional membrane complex proteins. Mol. Cell 6, 11–22 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Yan Z., et al. , Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature 517, 50–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perni S., Lavorato M., Beam K. G., De novo reconstitution reveals the proteins required for skeletal muscle voltage-induced Ca(2+) release. Proc. Natl. Acad. Sci. U.S.A. 114, 13822–13827 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong King Yuen S. M., Campiglio M., Tung C. C., Flucher B. E., Van Petegem F., Structural insights into binding of STAC proteins to voltage-gated calcium channels. Proc. Natl. Acad. Sci. U.S.A. 114, E9520–E9528 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z. F., et al. , Structures of the junctophilin/voltage-gated calcium channel interface reveal hot spot for cardiomyopathy mutations. Proc. Natl. Acad. Sci. U.S.A. 119, e2120416119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Biase V., Franzini-Armstrong C., Evolution of skeletal type e-c coupling: A novel means of controlling calcium delivery. J. Cell Biol. 171, 695–704 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanabe T., Mikami A., Numa S., Beam K. G., Cardiac-type excitation-contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA. Nature 344, 451–453 (1990). [DOI] [PubMed] [Google Scholar]
- 16.Perni S., Beam K., Junctophilins 1, 2, and 3 all support voltage-induced Ca2+ release despite considerable divergence. J. Gen. Physiol. 154, e202113024 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mackrill J. J., Shiels H. A., Evolution of excitation-contraction coupling. Adv. Exp. Med. Biol. 1131, 281–320 (2020). [DOI] [PubMed] [Google Scholar]
- 18.De Crescenzo V., et al. , Dihydropyridine receptors and type 1 ryanodine receptors constitute the molecular machinery for voltage-induced Ca2+ release in nerve terminals. J. Neurosci. 26, 7565–7574 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wheeler D. G., Barrett C. F., Groth R. D., Safa P., Tsien R. W., CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol. 183, 849–863 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun H. L., et al. , Voltage-induced Ca(2)(+) release in postganglionic sympathetic neurons in adult mice. PLoS One 11, e0148962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maryon E. B., Coronado R., Anderson P.string-name>, unc-68 encodes a ryanodine receptor involved in regulating C. elegans body-wall muscle contraction. J. Cell Biol. 134, 885–893 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson A. P., Clark C. L., Martin R. J., Levamisole and ryanodine receptors. I: A contraction study in Ascaris suum. Mol. Biochem. Parasitol. 171, 1–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee R., Lobel L., Hengartner M., Horvitz H., Avery L., Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J. 16, 6066–6076 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida M., Sugimoto A., Ohshima Y., Takeshima H., Important role of junctophilin in nematode motor function. Biochem. Biophys. Res. Commun. 289, 234–239 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Piggott C. A., et al. , Caenorhabditis elegans Junctophilin has tissue-specific functions and regulates neurotransmission with extended-synaptotagmin. Genetics 218, iyab063 (2021), 10.1093/genetics/iyab063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan A., et al. , SLO-2, a K+ channel with an unusual Cl- dependence. Nat. Neurosci. 3, 771–779 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Gao L., et al. , Shank promotes action potential repolarization by recruiting BK channels to calcium microdomains. Elife 11, e75140 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dal Santo P., Logan M. A., Chisholm A. D., Jorgensen E. M., The inositol trisphosphate receptor regulates a 50-second behavioral rhythm in C. elegans. Cell 98, 757–767 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Speed H. E., et al. , Apparent genetic rescue of adult Shank3 exon 21 insertion mutation mice tempered by appropriate control experiments. eNeuro 6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rios E., Brum G., Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature 325, 717–720 (1987). [DOI] [PubMed] [Google Scholar]
- 31.Pym E., et al. , Shank is a dose-dependent regulator of Cav1 calcium current and CREB target expression. Elife 6, e18931 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doyle D., et al. , Crystal structures of a complexed and peptide-free membrane protein-binding domain: Molecular basis of peptide recognition by PDZ. Cell 85, 1067–1076 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Nakai J., et al. , Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380, 72–75 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Avila G., Dirksen R. T., Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca(2+) channel. J. Gen. Physiol. 115, 467–480 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakada T., et al. , Physical interaction of junctophilin and the CaV1.1 C terminus is crucial for skeletal muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 115, 4507–4512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Packer J. S., et al. , A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution. Science 365, eaax1971 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maryon E. B., Saari B., Anderson P., Muscle-specific functions of ryanodine receptor channels in Caenorhabditis elegans. J. Cell Sci. 111, 2885–2895 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Grabrucker A. M., Schmeisser M. J., Schoen M., Boeckers T. M., Postsynaptic ProSAP/Shank scaffolds in the cross-hair of synaptopathies. Trends Cell Biol. 21, 594–603 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Durand C. M., et al. , Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peca J., et al. , Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phelan K., McDermid H. E., The 22q13.3 deletion syndrome (Phelan-McDermid Syndrome). Mol. Syndromol. 2, 186–201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Won H., et al. , Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Yi F., et al. , Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu M., et al. , Shank3-deficient thalamocortical neurons show HCN channelopathy and alterations in intrinsic electrical properties. J. Physiol. 596, 1259–1276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han Q., et al. , SHANK3 deficiency impairs heat hyperalgesia and TRPV1 signaling in primary sensory neurons. Neuron 92, 1279–1293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W., et al. , Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Invest. 127, 1978–1990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng W., et al. , Homer regulates gain of ryanodine receptor type 1 channel complex. J. Biol. Chem. 277, 44722–44730 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Huang G., et al. , Ca2+ signaling in microdomains: Homer1 mediates the interaction between RyR2 and Cav1.2 to regulate excitation-contraction coupling. J. Biol. Chem. 282, 14283–14290 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Dittman J. S., Kaplan J. M., Factors regulating the abundance and localization of synaptobrevin in the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 103, 11399–11404 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frokjaer-Jensen C., et al. , Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 40, 1375–1383 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frokjaer-Jensen C., et al. , Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat. Methods 11, 529–534 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghanta K. S., Mello C. C., Melting dsDNA donor molecules greatly improves precision genome editing in Caenorhabditis elegans. Genetics 216, 643–650 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng S., et al. , Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun. 8, 370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li L., et al. , CASK and FARP localize two classes of post-synaptic ACh receptors thereby promoting cholinergic transmission. PLoS Genet. 18, e1010211 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Javer A., et al. , An open-source platform for analyzing and sharing worm-behavior data. Nat. Methods 15, 645–646 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Data Availability Statement
All study data are included in the SI appendix, Dataset S1.