

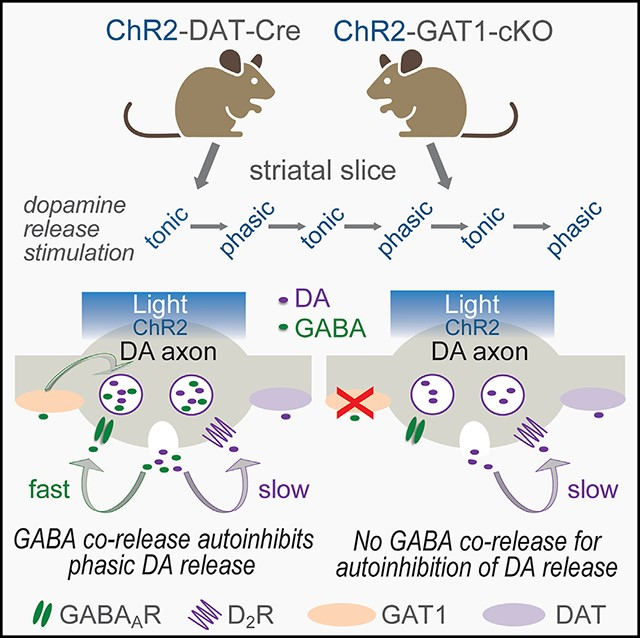
SUMMARY
Striatal dopamine axons co-release dopamine and gamma-aminobutyric acid (GABA), using GABA provided by uptake via GABA transporter-1 (GAT1). Functions of GABA co-release are poorly understood. We asked whether co-released GABA autoinhibits dopamine release via axonal GABA type A receptors (GABAARs), complementing established inhibition by dopamine acting at axonal D2 autoreceptors. We show that dopamine axons express α3-GABAAR subunits in mouse striatum. Enhanced dopamine release evoked by single-pulse optical stimulation in striatal slices with GABAAR antagonism confirms that an endogenous GABA tone limits dopamine release. Strikingly, an additional inhibitory component is seen when multiple pulses are used to mimic phasic axonal activity, revealing the role of GABAAR-mediated autoinhibition of dopamine release. This autoregulation is lost in conditional GAT1-knockout mice lacking GABA co-release. Given the faster kinetics of ionotropic GABAARs than G-protein-coupled D2 autoreceptors, our data reveal a mechanism whereby co-released GABA acts as a first responder to dampen phasic-to-tonic dopamine signaling.
Graphical Abstract
In brief
Striatal dopamine (DA) axons co-release GABA as well as DA. The actions and consequences of GABA co-release are not fully understood. Patel et al. show co-released GABA is an autoregulatory messenger that curtails burst-evoked DA release via GABAARs on DA axons thereby rapidly suppressing the contrast in phasic-to-tonic DA signaling.
INTRODUCTION
Dopamine (DA) is a crucial regulator of striatal neuronal output, thereby facilitating movement, motivation, and reward-based learning.1–7 Striatal axonal DA release is governed in part by the somatic activity of DA neurons,8,9 which transitions between single spikes (tonic pacemaker activity) and phasic bursts.10–12 Additionally, axonal DA release can be triggered locally via sparse but powerfully effective cholinergic interneurons (ChIs) via synaptic-like axo-axonal contacts with DA axons.13–15 Acetylcholine (ACh) release activates nicotinic ACh receptors (nAChRs) on DA axons, resulting in locally generated action potentials and DA release.13–16 The magnitude, spatial precision, and sphere of influence of DA is sculpted further by other striatal transmitters and modulators, including GABA and glutamate.17–21 Thus, regulation of striatal DA signaling and its impact on behavior is multivalent.1–6,22
In addition to extrinsic regulators, axonal DA release is modulated by cell-autonomous factors, most notably by DA itself through activation of inhibitory DA D2 autoreceptors.23–31 This enables DA axons to respond to and regulate DA signaling in the short term by decreasing further release and in the longer term by inhibiting DA synthesis.30,32
Additionally, DA axons release gamma-aminobutyric acid (GABA) as well as DA.33–40 GABA co-released from optically activated DA axons elicits GABAA receptor (GABAAR)-mediated inhibitory postsynaptic currents (oIPSCs) in striatal spiny projection neurons (SPNs),33,34,37 even though DA axons lack conventional molecular machinery for GABA synthesis and storage, e.g., glutamic acid decarboxylase (GAD) for conversion of glutamate to GABA and the vesicular GABA transporter (vGAT) for uptake into vesicles.34,37 An alternative mechanism for de novo GABA synthesis by aldehyde dehydrogenase 1a1 (ALDH1a1) has been proposed,37 but increasing evidence indicates that DA axons do not utilize ALDH1a1 for GABA synthesis; rather, they acquire GABA from the extracellular compartment, using the plasma membrane GABA transporter-1 (GAT1).34,38 Packaging of GABA into vesicles in DA axons then occurs via the vesicular monoamine transporter 2 (vMAT2),33,38 suggesting that GABA and DA cohabit the same vesicles (although see Zych and Ford39).
We hypothesized that co-released GABA might autoregulate DA release via GABAARs on DA axons, whatever the intracellular storage site. To test this, we first identified the presence of GABAARs on striatal DA axons. Then, using a combination of pharmacology, optogenetics, and genetic manipulations, we evaluated the functional impact of endogenous GABA on DA release evoked by single optical pulses (1 p) that represent the single action potentials of tonic pacemaker firing by DA neurons, or by brief pulse trains (10 p/10 Hz) that mimic phasic bursts of action potentials. Our studies identify a mechanism by which DA axons autoregulate DA release through co-released GABA that dampens the contrast between phasic and tonic DA signals.
RESULTS
Anatomical studies reveal α3-GABAAR subunits on DA axons throughout the striatum
Although the presence of GABAARs on striatal DA axons has been shown functionally,41,42 direct anatomical evidence is lacking. We used fluorescence immunohistochemistry with confocal microscopy and immunoelectron microscopy (i-EM) to explore whether striatal DA axons express GABAARs. The GABAAR is an ionotropic receptor with five pore-forming subunits: two α, two β, plus one γ or δ subunit (Figure S1A).43–47 However, at least 19 genes encode the subunit subtypes in humans. We used the RNA sequencing (RNA-seq) analysis resource DropViz (DropViz online analysis portal: http://dropviz.org)48 and found that midbrain tyrosine hydroxylase (TH)-containing cells in the substantia nigra pars compacta (SNc) and ventral tegmental area (VTA) express mRNA from many GABAAR-encoding genes, with relatively high mRNA levels for some genes, and undetectable mRNA levels for others (Figure S1A). Single-cell RT-PCR data show that, although mRNA transcripts for several GABAAR subunits are expressed in SNc and VTA DA neurons, α3 and γ2 subunits are present in a majority of these cells.49
The γ2 subunit mRNA is also present in striatal SPNs, however, which could obscure immuno-labeling in DA axons, whereas α3-GABAAR subunit mRNA (DropViz, data not shown) and protein expression50,51 are virtually absent in SPNs. Therefore, for our anatomical studies, we selected the α3-GABAAR subunit and confirmed with DropViz the relatively homogeneous expression of α3 in midbrain TH cell subclusters (Figure S1B).
We performed immunohistochemistry using an antibody targeted to α3 subunits assessed in previous studies, including in the reticular thalamic nucleus (RTN) and in VTA DA neurons.52,53 We first ensured that the general brain-wide expression pattern of α3 immunoreactivity was in line with that reported previously for the same or another α3-subunit-selective antibody. We found strong α3 immunolabeling in the RTN (Figure S2A), the islands of Calleja (iCj) (Figure S2B), deeper layers of the cortex (not shown), and in midbrain DA neurons (not shown) as seen by others.50,52–56 We also discovered that immunolabeling for α3 was present throughout the striatal DA axon network, as delineated by immunolabeling for TH (Figures S2A–S2C). Immunostaining in the RTN, iCj, and striatum was absent when the α3 antibody was preincubated with its immunogenic blocking peptide (n = 4 mice) (Figure S2). This verifies that the antibody recognizes the specified peptide sequence with high affinity, albeit this alone is not proof that the antibody binds to the target molecule in the tissue. Importantly, we obtained evidence for co-localization of α3 subunits in striatal TH-labeled DA axons and DA axonal varicosities in z stacks taken at high magnification in both the dorsal striatum (dStr) and the nucleus accumbens (NAc) core (Figures 1A and S3; Video S1).
Figure 1. Anatomical and functional localization of GABAAR subunits on DA axons.
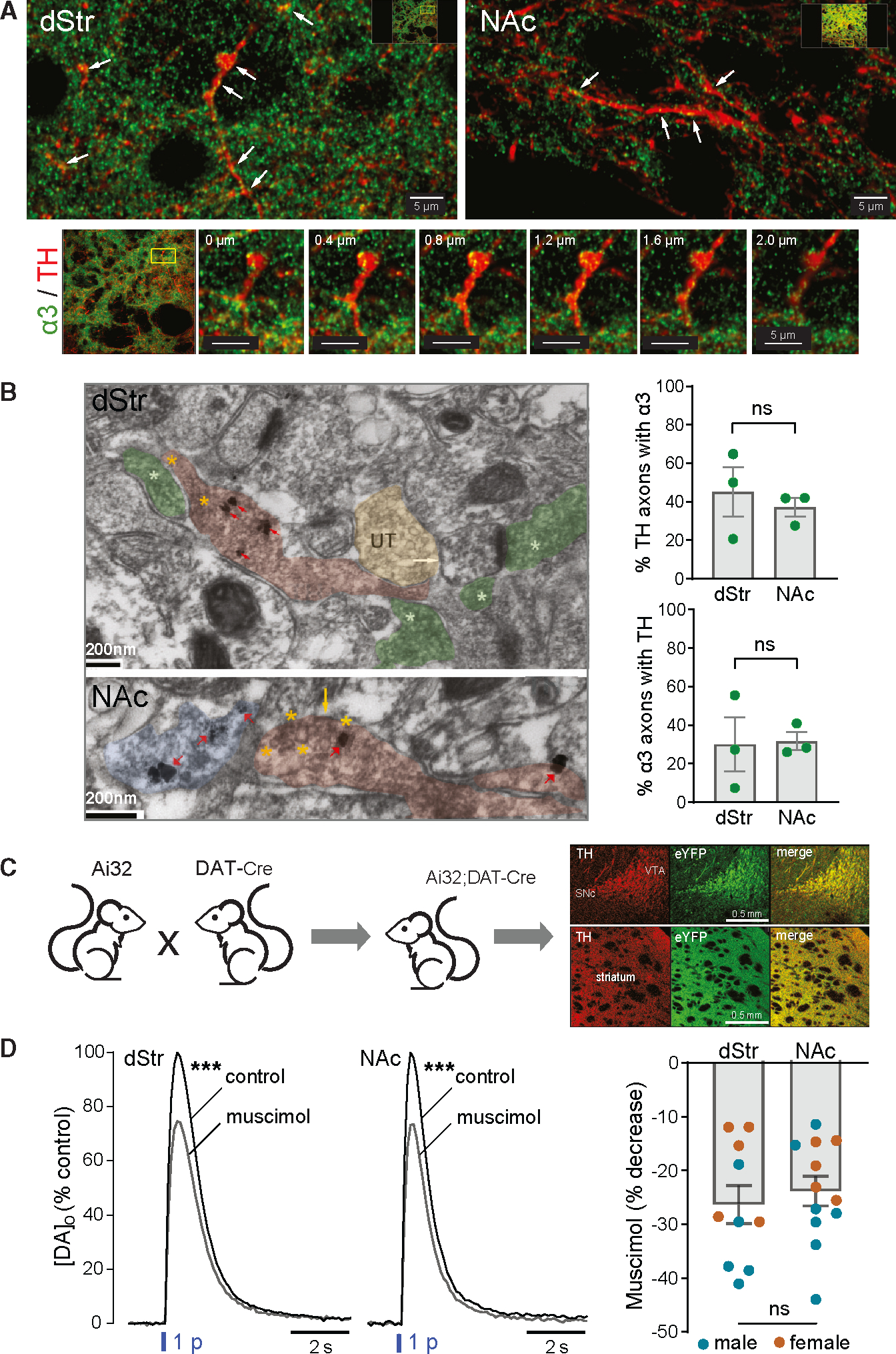
(A) Top: immunofluorescent images obtained with confocal microscopy showing co-localization of α3-GABAAR subunits with TH-labeled DA axons in dStr (left) and NAc core (right). White arrows indicate where co-localization is seen. Images are from a single plane of a z stack captured with a 60× Nikon oil-immersion objective (1.4 NA). Scale bar, 5 μm. Bottom: expanded views of a region of interest in the dStr at different levels in the z stack, demonstrating localization of α3-puncta in DA axonal profiles. Scale bar, 5 μm.
(B) Left: electron micrographs showing α3-GABAAR subunits co-localized with TH-labeled DA axons in dStr and NAc core. Top: TH axons in dStr were identified by the presence of more than one SIG particle within an axonal profile packed with vesicles (red arrows), while presence of α3-GABAAR subunits was identified by electron-dense diffuse DAB-horseradish peroxidase (HRP) labeling of vesicular and plasma membranes (asterisks). α3 subunits within TH axons (red overlay) are highlighted by yellow asterisks, whereas α3-GABAAR subunits in non-TH axons (green overlay) are highlighted by white asterisks; UT marks an unlabeled axon terminal (yellow overlay). The white arrow points to the plasma membrane of a non-TH profile, identifiable as a dendritic spine receiving excitatory synaptic input from a non-TH axon terminal (rightmost asterisk), presumably glutamatergic and α3 positive. Scale bar, 200 nm. Bottom: TH axons and α3-GABAAR subunits in NAc core were identified as above and shown at higher magnification to highlight immunolabeling of the plasma membrane of a dually labeled axon (yellow arrow) and of vesicle membranes (encircled by yellow asterisks). Blue overlay shows a TH axon without α3. Scale bar, 200 nm. Right: average data, quantifying the percentage of TH axons expressing α3 (upper graph) and α3 axons expressing TH (lower graph) in dStr and NAc (n = 3 mice; mean ± SEM; ns is not signficant).
(C) Cartoon showing generation of Ai32;DAT-Cre mice with genetic ChR2 expression in DAT-containing neurons and images of fixed coronal brain sections confirming selective fluorescent labeling of eYFP-ChR2 expression in midbrain DA (TH) soma and striatal axons. Scale bars, 0.5 mm.
(D) Right: average optically evoked single-pulse (1 p) [DA]o transients recorded in dStr and NAc in the absence or presence of the GABAAR agonist muscimol (Mus, 10 μM); error bars omitted for clarity. Muscimol significantly decreased 1 p-evoked [DA]o in both the dStr (n = 10 mice) and NAc (n = 12 from 11 mice). ***p < 0.01 versus respective control (ratio paired t test). Left: graph showing individual data (mean ± SEM) for the percentage decrease of peak [DA]o by muscimol in males (blue circles) plus females (orange circles) in dStr versus NAc (p > 0.05, unpaired t test; n is not significant).
We then utilized i-EM to identify and quantify α3 subunits on striatal TH-labeled DA axons at the subcellular level. TH was visualized with silver-intensified gold (SIG) and α3 visualized with 3,3′-diaminobenzidine (DAB) (Figures 1B and S4). Of the TH-labeled axons examined in the dStr (n = 101 from 3 mice), approximately half (45% ± 13%) were also labeled with α3. In the NAc core, the proportion of TH-positive axons (n = 121 from 3 mice) dually expressing α3-GABAAR subunits was slightly, but not significantly, lower (37% ± 5%) (p = 0.7000 versus dStr; Mann-Whitney test) (Figure 1B). In general, α3 subunits were expressed in a patchy fashion along a DA axon. Most of the DAB signal was cytosolic likely reflecting membrane trafficking/turnover of α3 subunits. Notably, α3-subunit expression was visible on the plasma membrane of vesicle-containing varicosities in DA axons (Figures 1B and S4) and thus well-positioned for detecting GABA release from the same axonal release site.
In the same sections, approximately 30% of α3-expressing axons in the dStr and NAc core contained TH labeling (30% ± 14% in dStr, n = 123 axons from 3 mice versus 32% ± 5% in NAc, n = 140 from 3 mice; p > 0.9999, Mann-Whitney test) indicating that α3 subunits are also present in non-DA axons (Figure 1B). Labeling for this subunit was also seen in dendrites (not quantified) but typically not in somata, consistent with the absence of α3-mRNA (DropViz) or protein50,51 in SPNs. Because DA neurons lack mRNA for d-containing GABAAR subunits (Figure S1A), we processed striatal sections with a δ-subunit selective antibody as a control experiment for our i-EM procedure. None of the TH axons spanning the dStr and NAc core (n = 92 axons from 3 mice) exhibited multiple SIG particles used to immunolabel δ subunits, with two exhibiting just one SIG particle each, likely reflecting background labeling (not shown). This same antibody has been used by the Aoki lab to identify δ subunits in hippocampus.57,58
Functional consequences of GABAARs on striatal DA axons
In the absence of selective pharmacological ligands for α3-GABAAR subunits, we tested whether GABAARs on DA axons are functional using a non-subunit selective agonist, muscimol. Single-pulse (1 p) optically evoked DA release was detected by fast-scan cyclic voltammetry (FSCV) in striatal slices from mice that gentically express channelrhodopsin2 (ChR2) in dopamine transporter (DAT) containing DA neurons in a Cre-dependent manner (Ai32+/−;DAT-Cre+/− mice) (Figure 1C). Activation of GABAARs by muscimol (10 μM) attenuated 1 p-evoked increases in extracellular DA concentration ([DA]o) in the dStr (p < 0.0001, n = 10 slices from 10 mice; ratio paired t test) and NAc (p < 0.0001, n = 12 slices from 11 mice; ratio paired t test) of male and female mice (Figure 1D). Moreover, the extent of inhibition by muscimol was similar (~25%) between striatal subregions (p = 0.5773; unpaired t test) (Figure 1D). These data verify the functionality of inhibitory GABAARs on DA axons throughout the striatal complex, as shown by others.41,42
Endogenous GABA tone at GABAARs inhibits striatal DA release
In striatal slices, levels of basal DA release are insufficient to provide a DA tone at D2 autoreceptors.23–27,29,31 However, ambient levels of GABA are sufficient to provide a GABA tone at GABAARs on SPNs.34,59 Moreover, DA release is suppressed by ambient GABA acting predominantly at GABABRs.41 Evidence for this was shown by an enhancing effect of GABA-receptor antagonists on DA release evoked by 1 p electrical stimulation when nAChRs were blocked or by targeted optical stimulation of DA axons following adeno-associated virus (AAV)-ChR2 midbrain injections. We tested whether endogenous GABA tone also suppresses DA release via GABAARs specifically using 1 p optical stimulation in slices from Ai32;DAT-Cre mice. Note: 1 p optically evoked DA release in striatal slices is independent of regulation by nAChRs.14,60 Application of picrotoxin (PTX, 100 μM), which blocks GABAAR chloride channels (Figure S1A),61 increased 1-p optically evoked [DA]o in both dStr and NAc of male and female mice, revealing inhibition of DA release by ambient GABA throughout the striatum (Figures 2A and 2B). Within each subregion, the effect of PTX at a given recording site differed across slices, with increases ranging from 4% to 77% in dStr and 6% to 46% in NAc. The variable efficacy did not correlate with initial control evoked [DA]o in either region (Figure 2C), indicating that GABA tone is not a primary determinant of DA release heterogeneity. The average effect of PTX was slightly greater in dStr (32% ± 6% increase, n = 13 from 11 mice) than in NAc (19% ± 4% increase, n = 10 from 9 mice), although the difference was not significant (p = 0.1068; unpaired t test).
Figure 2. Endogenous ambient GABA tone suppresses 1 p-evoked DA release via GABAARs on DA axons.
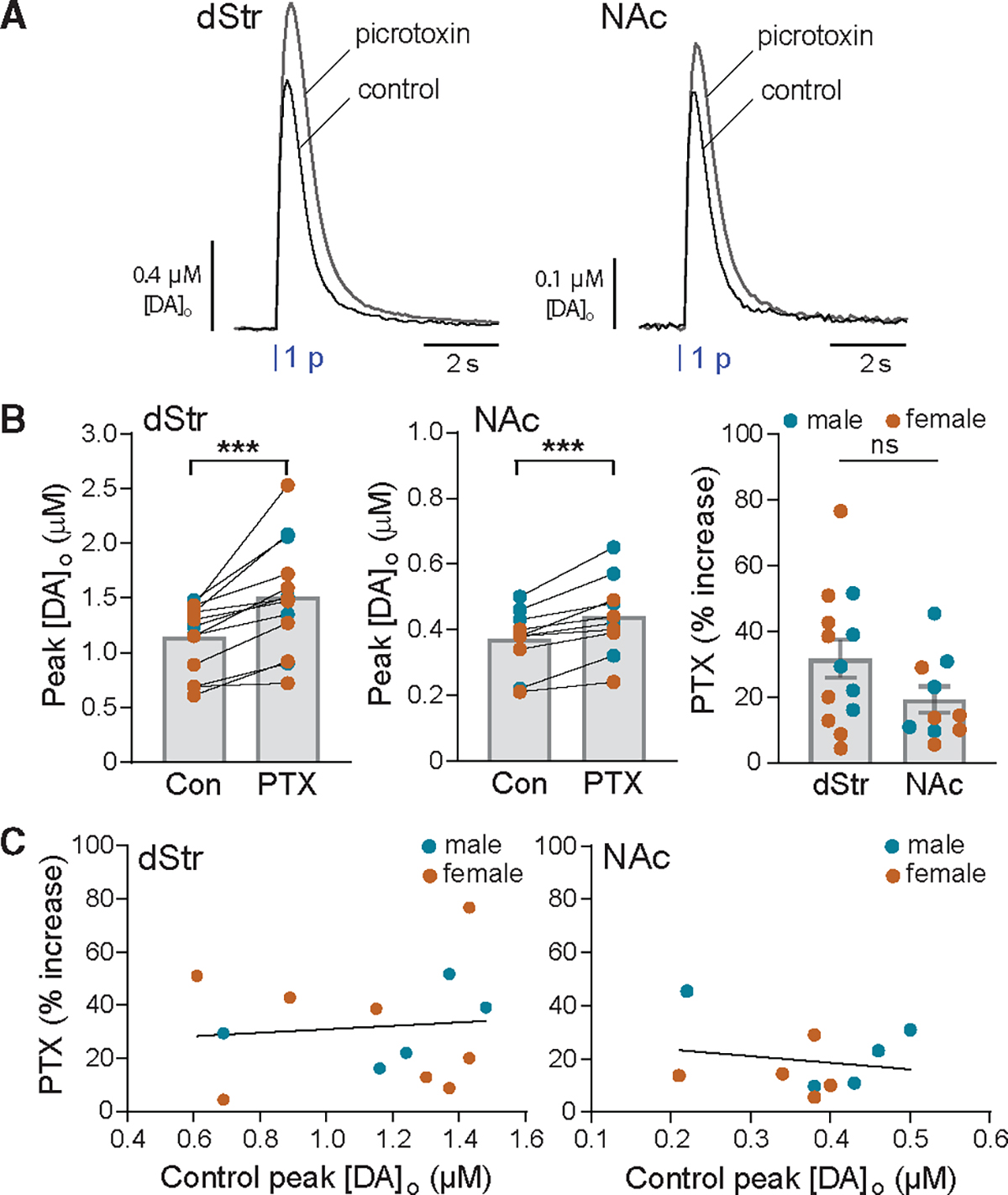
(A) Average optically evoked single-pulse (1 p) [DA]o transients recorded in dStr and NAc in the absence or presence of the GABAAR blocker picrotoxin (PTX, 100 μM); error bars omitted for clarity.
(B) PTX significantly increased peak [DA]o evoked by 1 p in males (blue circles) plus females (orange circles) in both the dStr (n = 13 from 11 mice) and NAc (n = 10 from 9 mice). ***p < 0.001 versus respective control (ratio paired t test), thereby revealing inhibition of DA release by ambient GABA acting at GABAARs. The effect of PTX did not differ between dStr and NAc (p > 0.05, unpaired t test). Data are mean ± SEM; ns is not significant.
(C) Efficacy of PTX-induced enhancement of DA release was variable but not related to initial evoked [DA]o under control conditions in either dStr (simple linear regression with Pearson correlation: slope = 6.494, R2 = 0.001, F = 0.1075(1,11); p = 0.7492, n = 13) or NAc (simple linear regression with Pearson correlation: slope = −24.85, R2 = 0.034, F = 0.2835(1,8); p = 0.6089, n = 10).
Endogenous GABA released from DA axons during phasic stimulation inhibits DA release via GABAARs
In previous FSCV studies, application of a D2 receptor (D2R) antagonist, e.g., sulpiride, unmasked the autoinhibitory effect of DA released by the first pulse of a burst on DA release during subsequent pulses.24–27,31 By analogy, we assessed autoregulation by GABA with multiple-pulse optical stimulation of DA axons in the presence of PTX to reveal the effect of GABA co-released during the first few pulses on DA released by subsequent pulses. Inhibition of axonal DA release by D2 autoreceptors is maximal using 10-Hz stimulation.25,26,31 Here we used 10-pulse trains at 10 Hz, which also mimics phasic firing patterns of DA neurons.11,12 We found that PTX (100 μM) amplified 10 p optically evoked [DA]o in both dStr and NAc core (Figures 3A and 3B). The effect of PTX on pulse-train evoked [DA]o was similar (p = 0.5911, unpaired t test) between regions (dStr, 47% ± 4% increase, n = 13 from 13 mice; NAc, 44% ± 5% increase, n = 12 from 11 mice). The efficacy of PTX within each region varied (29%–75% increase in dStr and 20%–68% increase in NAc) but, again, with no correlation between initial evoked [DA]o and the amplifying effect of PTX in either region (Figure 3C).
Figure 3. Evoked co-released GABA from DA axons suppresses pulse-train evoked DA release via GABAARs.
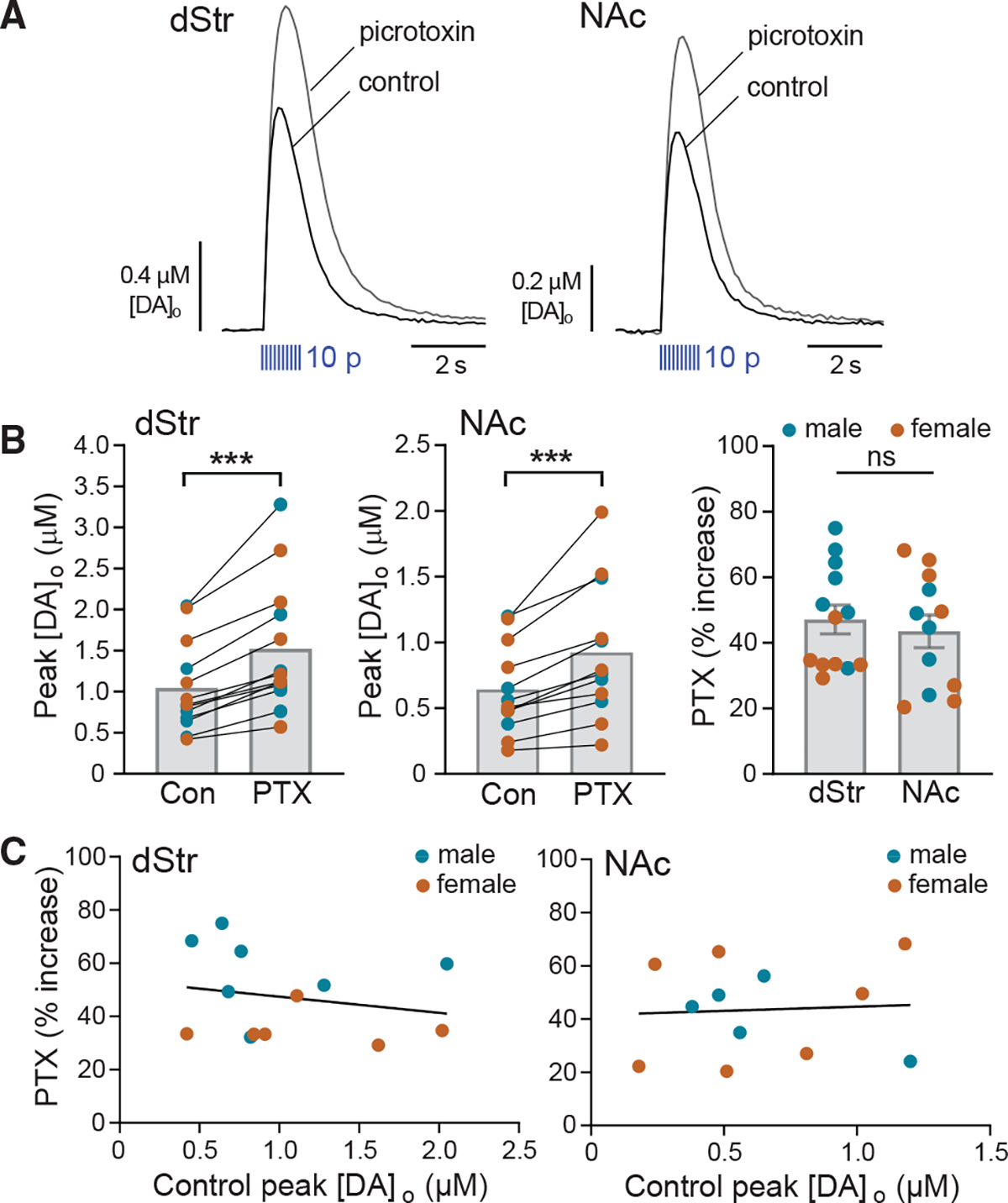
(A) Average multiple-pulse (10 p at 10 Hz) evoked [DA]o transients recorded in dStr and NAc in the absence and presence of PTX (100 μM); error bars omitted for clarity.
(B) PTX significantly increased peak [DA]o evoked by 10 p in males (blue circles) plus females (orange circles) in both the dStr (n = 13 mice) and NAc (n = 12 from 11 mice). ***p < 0.001 versus respective control (ratio paired t test), thereby revealing inhibition of DA release by endogenous GABA acting at GABAARs. The effect of PTX was similar in dStr versus NAc (p > 0.05, unpaired t test). Data are mean ± SEM; ns is not significant.
(C) Efficacy of PTX-induced enhancement of DA release did not correlate with initial evoked [DA]o under control conditions in either the dStr (simple linear regression with Pearson correlation: slope = −6.065, R2 = 0.04425, F = 0.5093(1,11); p = 0.4903, n = 13) or NAc (simple linear regression with Pearson correlation: slope = 3.155, R2 = 0.003883, F = 0.03898(1,10); p = 0.8474, n = 12).
To evaluate whether co-released GABA might autoregulate DA release, we compared PTX amplification of [DA]o evoked by 10 p versus 1 p optical stimulation, recorded in separate slices (Figures 2 and 3). We documented a significantly larger enhancement with 10 p versus 1 p in both dStr (p = 0.0433, unpaired t test) and NAc (p = 0.0015, unpaired t test) (Figure 4A). A greater effect of PTX on 10 p-evoked [DA]o implies an additional component to DA release inhibition, during phasic activity, beyond that from ambient GABA alone.
Figure 4. Greater amplification of DA release by PTX on 10 p versus 1 p implies autoinhibition by co-released GABA during phasic DA axon activity.
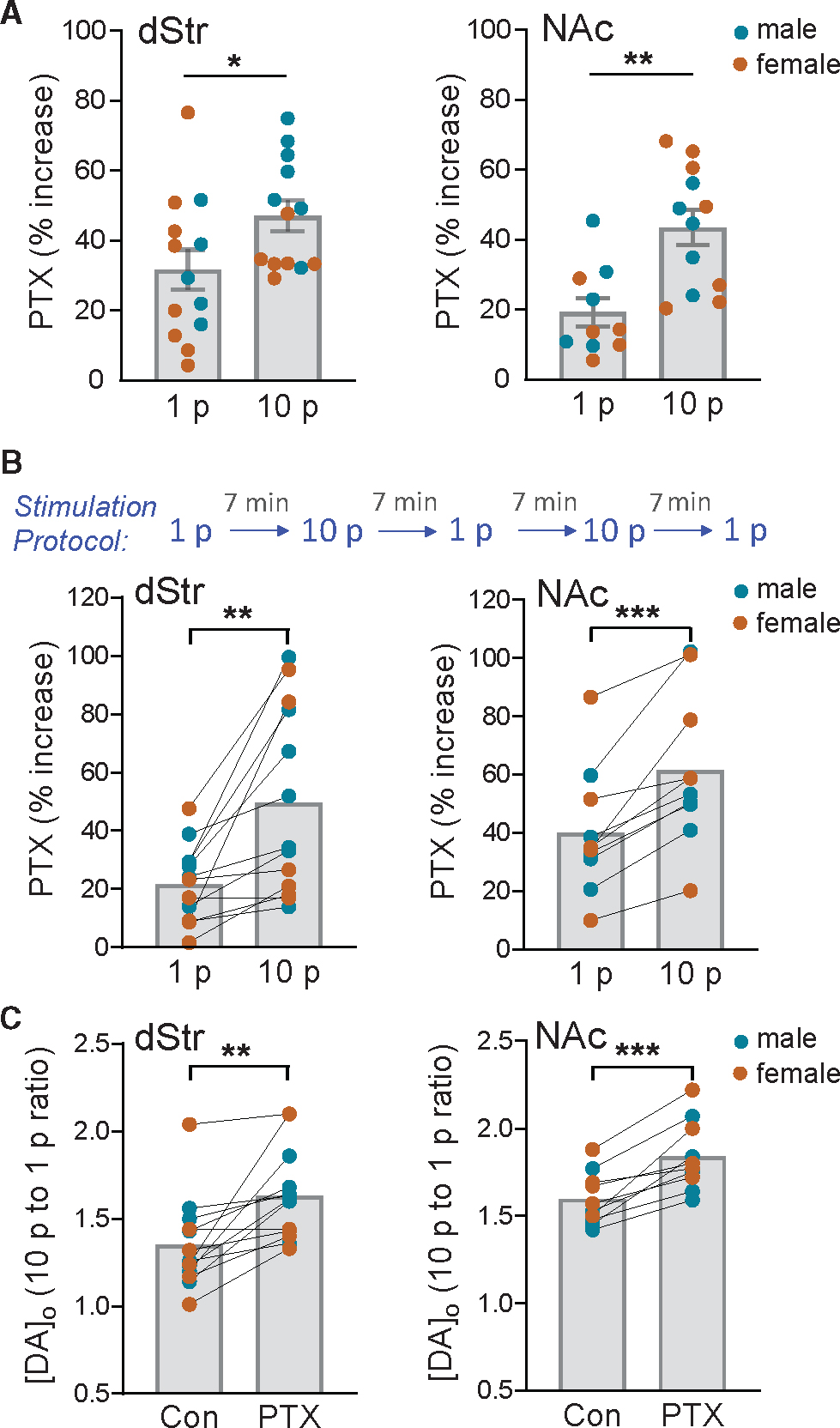
The effect of PTX (100 μM) on increasing peak optically evoked [DA]o was significantly greater when release was evoked by multiple-pulses (10 p at 10 Hz) than with a single pulse (1 p) in dStr and NAc when recorded in (A) separate experiments (unpaired t test) or in (B) the same recording site using an alternating stimulation protocol (paired t test). Consequently, the ratio of phasic (10 p) to tonic (1 p) DA release was enhanced by PTX in both the dStr (n = 13) and NAc (n = 10) (C), indicating that an additional source of GABA inhibits DA release during phasic stimulation. *p < 0.05; **p < 0.01; ***p < 0.001 versus respective 1 p stimulation or control in males (blue circles) plus females (orange circles). Data are mean ± SEM.
Because the effects of PTX on 1 p- and 10 p-evoked DA release were conducted at different recording sites in different slices and the effect of PTX was variable (Figures 2 and 3), we tested the hypothesis of DA-release regulation by co-released GABA by comparing the PTX-induced enhancement of 10 p- versus 1 p-evoked [DA]o at a single recording site in a given slice. For these experiments, we alternated 1 p and 10 p simulation at 7-min intervals at a single site in dStr or NAc (Figure 4B). We found that in dStr, PTX caused a 22% ± 4% increase of 1 p-evoked [DA]o but a significantly greater increase of 50% ± 9% with 10 p stimulation (p = 0.0022, n = 13; paired t test) (Figure 4B). Using the same alternating stimulation paradigm, we also found a greater amplification of 10 p- versus 1 p-evoked [DA]o with PTX in the NAc, with an increase of 40% ± 7% with 1 p but 61% ± 8% with 10 p (p = 0.0005, n = 10; paired t test) (Figure 4B). Significant increases in evoked [DA]o with PTX were seen in both striatal subregions from males and females (Figure S5), although the responses in females were highly variable. Consequently, the greater effect of PTX on 10 p- versus 1 p-evoked [DA]o was less statistically robust in the dStr from females (p = 0.0870, n = 6, paired t test) than males (p = 0.0184, n = 7; paired t test). In NAc, however, a significant increase was seen in both sexes (females, p = 0.0411, n = 5; males, p = 0.0110, n = 5; paired t tests) (Figure S5). Overall, these data support our hypothesis that co-released GABA autoinhibits DA release from DA axons.
By comparing the effects of PTX on 1 p- and 10 p-evoked [DA]o at a given recording site, we obtained a direct index of the influence of endogenous GABA on phasic versus tonic DA signaling. The greater influence of GABA on phasic DA release is documented by the higher 10 p-to-1 p ratio for evoked [DA]o in PTX versus control in both dStr (PTX, 1.63 ± 0.07 versus control, 1.35 ± 0.07; p = 0.0019, n = 13; paired t test) and NAc core (PTX, 1.84 ± 0.06 versus control,1.60 ± 0.05; p = 0.0003, n = 10; paired t test) (Figure 4C). These data not only support autoinhibition of axonal DA release by co-released GABA but also indicate that co-released GABA acts to dampen the contrast in phasic versus tonic DA signaling.
It should be noted that these same-site experiments were conducted at room temperature to minimize the rundown of optically evoked [DA]o seen at higher temperatures.60 Lower temperatures would be expected to decrease GABA transporter activity, which might artificially amplify GABA tone and spillover of co-released GABA. We therefore verified key findings in male dStr in slices maintained at 30°C–32°C. To avoid rundown, we used multi-site recording in a given brain slice, with 1 p- and 10 p-evoked increases in [DA]o monitored at several different sites in the absence and presence of PTX (100 μM). Under these conditions, PTX still caused significant increases in [DA]o evoked with either 1 p (p = 0.0018, n = 5 mice; paired t test) or 10 p stimulation (p = 0.0003; n = 5 mice, paired t test) (Figure S6). Moreover, the greater effect of PTX on 10 p- versus 1 p-evoked [DA]o persisted at the higher temperature (1 p, 27% ± 4% increase; 10 p, 47% ± 6% increase; p = 0.0303). These data confirm that ambient GABA tone at GABAARs on DA axons limits DA release at near-physiological temperatures and that autoregulation of phasic DA release by co-released GABA is a physiological process.
Why was only PTX used for these experiments? In preliminary tests, we found that PTX was the only agent we tested that did not interfere with quantification of evoked [DA]o using FSCV and carbon-fiber microelectrodes (Figure S7A). By contrast, two other commonly used GABAAR antagonists, (+)bicuculline and gabazine,41,42 did interfere with DA detection (Figures S7B and S7C). We found that bicuculline is electroactive, producing oxidation currents that obscure DA detection, and that gabazine is not only electroactive but also decreases electrode sensitivity to DA by 50% (sensitivity: F5,12 = 13.3; p < 0.0001, one-way ANOVA with p > 0.9999 for control versus PTX; p > 0.9999 for control versus bicuculline; p < 0.0001 for control versus gabazine, Bonferroni’s multiple comparison test). Thus, PTX is the best available drug to assess GABAergic regulation of DA release with our methods. That said, a potential caveat for the use of PTX is that it not only blocks GABAAR chloride channels but also antagonizes other Cys-loop receptors, including nAChRs and glycine receptors,62 as do bicuculline and gabazine.62,63 Previous work, however, has shown that nAChR antagonism decreases 1 p DA release evoked with electrical stimulation in striatal slices64–67 and has no effect on 1 p DA release evoked by selective optical stimulation of DA axons.14,60 Both of these findings contrast with the amplification of evoked DA release induced by PTX. We tested whether the effects of PTX might involve antagonism of glycine receptors by monitoring the effect of a glycine receptor antagonist, strychnine (10 μM), on DA release. Strychnine decreased [DA]o evoked by either 1 p or 10 p optical stimulation at a given recording site (1 p, 31% ± 6% decrease; 10 p, 34% ± 5% decrease, p = 0.165, paired t test, n = 4 male mice) with no effect on phasic-to-tonic ratio (10 p to 1 p ratio: control, 1.17 ± 0.05; strychnine, 1.12 ± 0.03; p = 0.2032, paired t test). Together, these data argue against off-target effects of PTX at nAChRs or glycine receptors in the amplification of DA release.
Inhibiting striatal GABA synthesis attenuates regulation of phasic DA release by GABA
What is the source of co-released GABA involved in DA-release regulation by GABAARs? Although DA neurons lack GAD,34,37 GAD-dependent GABA synthesis does take place in striatal cells and contributes to ambient GABA that inhibits DA release via GABAARs and GABABRs.20,68 To determine whether regulatory GABA released during phasic DA axon stimulation is also dependent on GAD, we preincubated striatal slices for >90 min in the GAD inhibitor, 3-mercaptopropionic acid (3-MPA) (500 μM), then assessed the effect of PTX on 10 p-evoked [DA]o in the continued presence of 3-MPA. We found that compromising GABA availability by limiting synthesis via GAD attenuated the increase induced by PTX in the dStr (47% ± 4% in PTX alone, n = 13 mice versus 31% ± 4% in 3-MPA+PTX, n = 10 mice; p = 0.0160, unpaired t test) and in the NAc core (44% ± 5% in PTX alone, n = 12 from 11 mice versus 28% ± 3% in 3-MPA+PTX, n = 13 from 12 mice; p = 0.0233, unpaired t test). Thus, inhibition of phasic DA release by co-released GABA relies on GAD-dependent synthesis.
Regulation of phasic DA release is lost in GAT1 conditional knockout mice
Because GABA synthesis does not occur in DA neurons, GABA used for co-release is instead taken up by DA axons via plasma membrane GATs.34,38 Initial evidence for this mechanism was the loss of GABAAR currents in SPNs evoked by optical stimulation of DA axons in a cocktail of GAT1 and GAT4 inhibitors.34 Given that DA neurons exhibit robust mRNA expression for GAT1, but weaker expression for GAT4, GAT1 was proposed to be the primary carrier supplying GABA to DA axons.34 An essential role for GAT1 in GABA co-release was demonstrated by the complete loss of GABAAR oIPSCs in dStr SPNs from mice in which GAT1 was selectively deleted in a DAT-Cre-dependent manner (GAT1 conditional knockout [cKO] mice).38 Thus, as a final test of our hypothesis that co-released GABA autoinhibits DA release, we examined the effect of PTX on [DA]o evoked by alternating tonic 1 p and phasic 10 p optical stimulation in the dStr of male GAT1-cKO mice, with ChR2 targeted to DA neurons by Cre-dependent AAV vector-mediated SNc injections (Figure 5A).
Figure 5. Autoregulation of DA release by co-released GABA is lost with GAT1 deletion from DA axons.
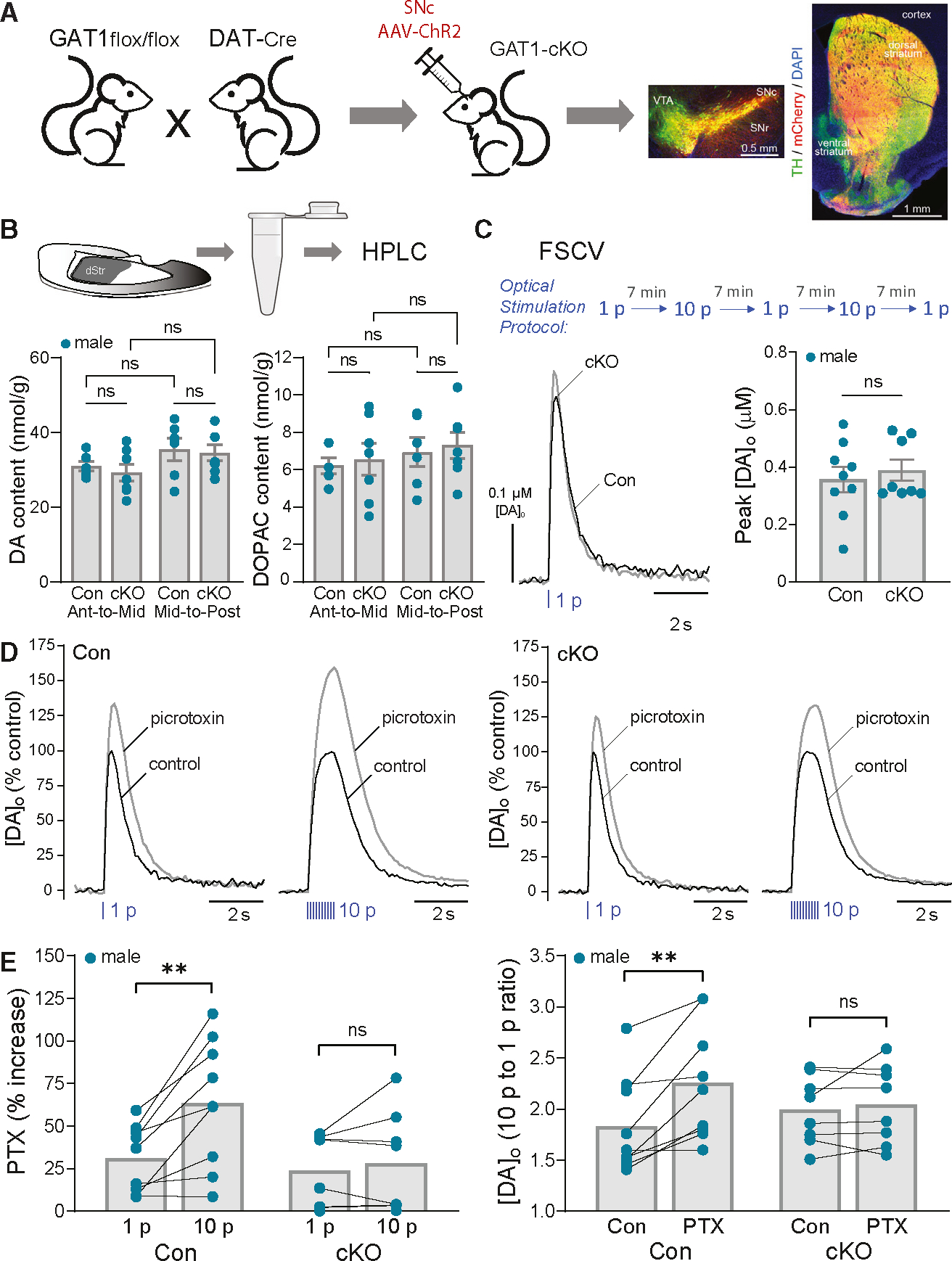
(A) Cartoon showing generation of GAT1;DAT-Cre cKO mice (GAT1-cKO) with SNc AAV-ChR2-mCherry injection and images of fixed coronal brain sections showing fluorescent labeling of ChR2 expression (mCherry) in midbrain DA (TH) somata and striatal axons; DAPI was used to label all cells. Scale bar, 0.5 mm.
(B) Tissue collection of dStr for HPLC-EC analysis. Tissue content of DA and the DA metabolite DOPAC did not differ in anterior-to-mid or mid-to-posterior dStr (p > 0.05, two-way ANOVA) between GAT1;DAT-Cre cKOs (n = 7 samples for each subregion from 7 mice) versus DAT-Cre-Het controls (n = 6 samples for each subregion from 6 mice).
(C) Same-site recording with alternating single-pulse (1 p) and multiple-pulse (10 p at 10 Hz) optical stimulation. Average 1 p evoked [DA]o transients recorded in control and GAT1-cKO mice under control conditions; error bars omitted for clarity. Average DA release for 1 p did not differ significantly between genotypes (unpaired t test, n = 8 GAT1-cKO mice versus n = 9 control mice).
(D) Average normalized 1 p and 10 p optically evoked [DA]o transients in control and GAT1-cKO mice in the absence and presence of PTX (100 μM).
(E) PTX had a significantly greater effect of increasing peak [DA]o evoked by 10 p versus 1 p in control mice (paired t test, n = 9) but not in GAT1-cKO mice (paired t test, n = 8). Consequently, the enhanced ratio of phasic (10 p) totonic (1 p) DA release in control mice was lost in cKOs.
**p < 0.01 versus respective 1 p or control. Data are mean ± SEM; ns is not significant.
We first assessed tissue DA content using high-performance liquid chromatography with electrochemical detection (HPLC-EC) to ascertain whether preventing GABA uptake into DA axons, and consequently into DA vesicles, might also alter vesicular storage of DA. We found no difference in DA content in anterior-to-mid or mid-to-posterior regions of the dStr of GAT1-cKO mice versus control DAT-Cre+/− mice (F1,22 = 0.03224; p = 0.8591, two-way ANOVA; n = 7 samples for each dStr region from 7 cKO mice; n = 6 samples for each dStr region from 6 control mice), or in tissue content of the DA metabolite, 3,4-dihydroxyphenylacetic acid (DOPAC) (F1,22 = 0.0004340; p = 0.9836, two-way ANOVA) (Figure 5B). We then used FSCV to test whether GABAARs on DA axons are functional in cKO mice by applying muscimol (10 μM) to striatal slices from ChR2-injected GAT1-cKO mice. Muscimol decreased 1 p optically evoked [DA]o by 37% ± 4% (n = 5 mice) in the dStr of cKO mice, confirming that GABAARs on DA axons remain operational.
We next examined the effect of PTX on alternating 1 p versus 10 p optically evoked DA release. Under control conditions, peak amplitude of 1 p-evoked [DA]o in the dStr did not differ between cKO and control mice (cKO, 0.39 ± 0.04 μM DA, n = 8 mice; controls, 0.36 ± 0.04 μM DA, n = 9 mice; p = 0.5931, unpaired t test) (Figure 5C). Unsurprisingly, given that GABA tone is intact in GAT1-cKO mice,38 PTX-induced amplification of 1 p-evoked [DA]o was not altered in GAT1-cKO mice compared to controls (cKO, 24% ± 8% increase, n = 8 mice; controls, 31% ± 7% increase, n = 9 mice; p = 0.4736, unpaired t test). Strikingly, however, the usual greater effect of PTX on 10 p versus 1 p was absent in cKO mice lacking GAT1 in DA axons (1 p, 24.0% ± 7.5% versus 10 p, 28% ± 10% increase, n = 8 cKO mice; p = 0.4114, paired t test) (Figures 5D and 5E), whereas it persisted in controls (10 p, 63.6% ± 12.5% increase versus 1 p, 31.3% ± 6.5%, n = 9 control mice; p = 0.004, paired t test). Consequently, the typical increases in the ratio of 10 p-to-1 p DA release seen in control mice in PTX (2.25 ± 0.19 versus 1.83 ± 0.16; p = 0.0044, n = 9; paired t test) were absent in GAT1-cKOs (2.04 ± 0.14 versus 1.99 ± 0.12; p = 0.4609, n = 8; paired t test) (Figures 5D and 5E). These data from mice that lack GABA co-release from DA axons38 confirm that co-released GABA provides an autoregulatory signal via GABAARs for rapid inhibition of DA release to constrain phasic-to-tonic DA signaling.
Does co-released glutamate play a role in DA-release regulation by co-released GABA?
Striatal DA axons also release glutamate,40,69–75 albeit primarily from distinct vesicles76 in different varicosities and axonal branches77,78 and with regional72 as well as subregional79,80 heterogeneity. Key targets for co-released glutamate in lateral dStr and NAc are ChIs.79–83
Given that ChIs directly trigger DA release by activating nAChRs on DA axons,13,14,16 a potential factor in our experiments using phasic optical stimulation of DA axons is that co-released glutamate might drive ChIs to activate DA axons and the co-release of DA plus GABA.36 This would provide an additional catalyst for GABA autoregulation of DA release. Also, because ~50% of ChIs in the lateral dStr express GAD65, with a subset of those able to co-release GABA,84,85 glutamate-activated ChIs could provide an alternative source of evoked GABA. We tested this possibility by examining the effect of PTX on alternating 1 p and 10 p optically evoked DA release in the presence of regionally appropriate glutamate receptor antagonists.
In lateral dStr, phasic optical stimulation of DA axons induces a short pause of firing in ChIs, followed by a delayed burst of activity mediated by activation of group 1 metabotropic glutamate receptors (mGluR1s) by co-released glutamate.79,80 We tested a possible role for this dStr microcircuit by applying PTX in the presence of an mGluR1 antagonist, JNJ-16259685 (10 μM).80 PTX enhanced 1 p and 10 p optically evoked [DA]o in the dStr in the presence of the antagonist, with the usual greater amplification of 10 p versus 1 p (p = 0.0121, paired t test, n = 6 mice) indicating a lack of involvement of mGluR1 on DA-release regulation by co-released GABA (Figure 6A). Similarly, in the NAc and olfactory tubercle, glutamate co-released from DA axons can drive ChI burst firing via ionotropic glutamate receptor activation: AMPA receptors (AMPARs) in NAc81 and NMDA receptors (NMDARs) in the olfactory tubercle.82 We found that, in the NAc core, PTX continued to augment 1 p- and 10 p-evoked increases in [DA]o in the presence of a cocktail of an AMPAR antagonist (DNQX, 10 μM) and an NMDAR antagonist (D-AP5, 50 μM),73 again with significantly greater PTX-induced enhancement of [DA]o evoked by 10 p versus 1 p (p = 0.0098, paired t test, n = 6 mice) (Figure 6B).
Figure 6. GABAAR autoinhibition of phasic DA release persists when effects of glutamate co-release or nAChR activation are prevented, confirming direct action by co-released GABA.
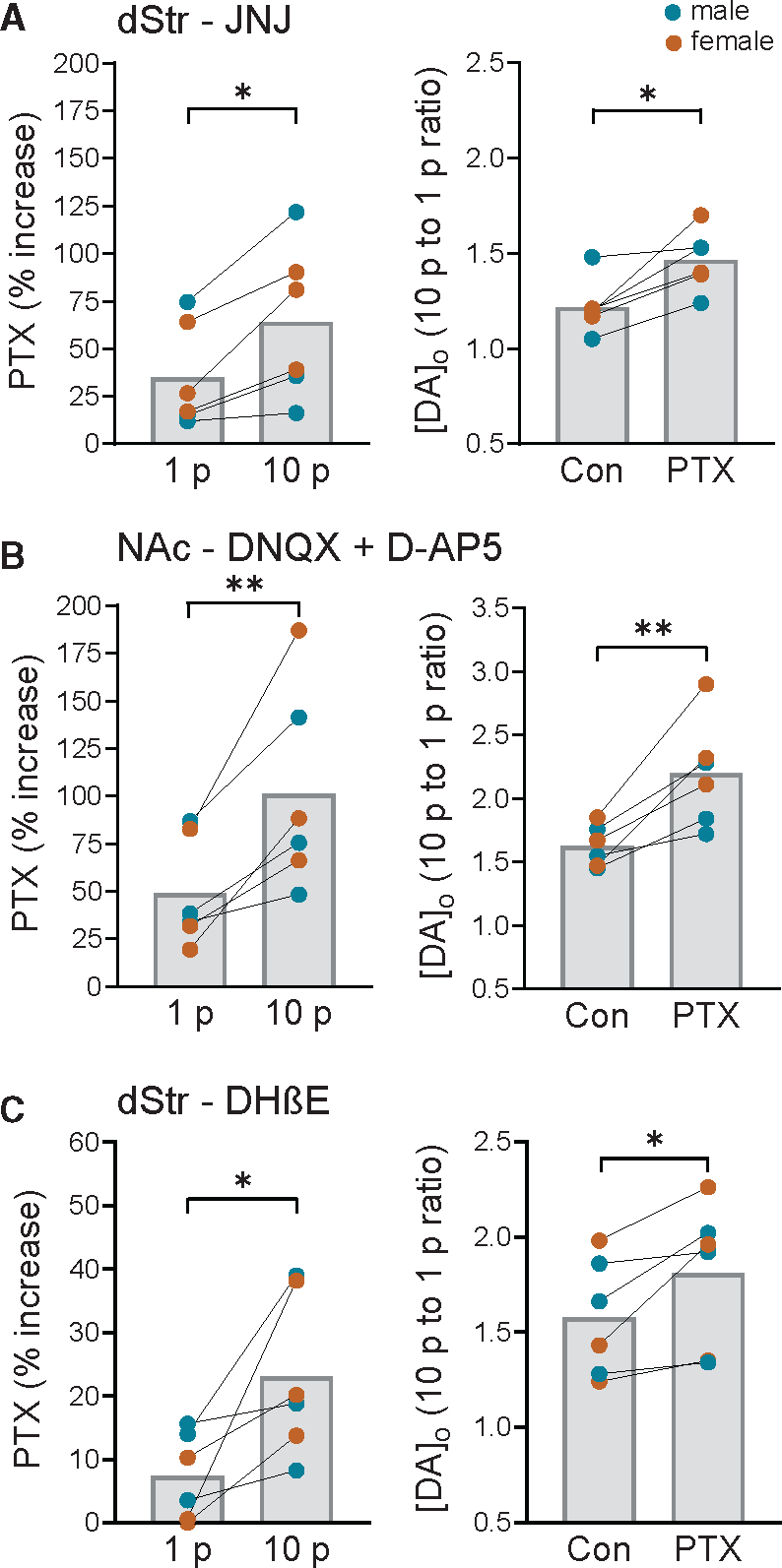
(A) In the dStr, PTX (100 μM) continued to produce a greater enhancement of 10 p- versus 1 p-evoked [DA]o in the presence of a mGluR1 antagonist (JNJ-16259685, 10 μM) and significantly increased the ratio of phasic (10 p) to tonic (1 p) DA signaling (n = 6 mice, paired t test).
(B) In the NAc core, greater enhancement of 10 p- versus 1 p-evoked [DA]o with PTX persisted in a cocktail of antagonists for AMPARs (DNQX, 10 μM) and NMDARs (D-AP5, 50 μM), as did the increased ratio of phasic-to-tonic DA release (n = 6 mice, paired t test).
(C) In the dStr, PTX continued to produce a greater enhancement of 10 p- versus 1 p-evoked [DA]o in the presence of a nAChR antagonist (DHβE, 1 μM) and significantly increased the ratio of phasic (10 p) to tonic (1 p) DA signaling (n = 6 mice, paired t test).
**p < 0.01, ***p < 0.001 versus respective 1 p stimulation or control (paired t tests) in males (blue circles) plus females (orange circles). Data are mean ± SEM.
We then examined the effect of PTX on 1 p versus 10 p optically evoked [DA]o in the dStr in the presence of dihydro-β-erythroidine (DHβE, 1 μM), a selective antagonist of nAChRs. The differential increase in phasic versus tonic DA release with PTX application persisted in DHβE (p = 0.0339, paired t test, n = 6 mice), excluding a role for ChIs in DA-release regulation by co-released GABA (Figure 6C). Thus, the inhibitory influence of GABA co-release on phasic DA signaling is unaffected by additional effects of glutamate co-release from DA axons on ChIs with subsequent activation of nAChRs, or GABA co-release from ChIs.
DISCUSSION
The concept that fast, classical transmitters such as GABA and glutamate are co-released with neuromodulators such as DA is relatively new, and the functional consequences of co-release are still under study. The phenomenon of co-released GABA and DA from striatal DA axons is a case in point. Apart from the initial finding that co-released GABA produces IPSCs in striatal SPNs,33 little is known. An unusual aspect of GABA and DA co-release is that striatal DA axons lack vGAT, which is normally required to transport GABA into synaptic vesicles. Instead, GABA storage requires vMAT2,33,38 indicating that GABA and DA may reside in the same vesicles, i.e., mediating true co-release (albeit subject to differential regulation).39 This possible co-packaging led us to propose that GABA, as well as DA, might provide inhibitory feedback modulation of ongoing axonal DA signaling. Here, we confirm that ambient GABA acting at ionotropic GABAARs establishes a tone that limits axonal DA release. We also identify an anatomical substrate for DA regulation by GABA by demonstrating the presence of α3-GABAAR subunits near striatal DA axon varicosities. Moreover, we show that GABAARs on DA axons mediate inhibition of DA release by co-released GABA. Thus, an important function of co-released GABA is to provide a rapid, GABAAR-mediated, autoinhibitory signal that curtails DA release probability during phasic DA activity, thereby dampening the contrast between phasic versus tonic DA signaling.
GABAARs on DA axons
The question of whether GABAARs are anatomically expressed by striatal DA axons is not trivial. Almost all striatal neurons are GABAergic, including the SPNs that represent 96% of striatal neurons,86 as well as most striatal interneurons.87,88 The striatum also receives GABAergic inputs from other brain regions, including the globus pallidus for input to dStr89,90 and the ventral pallidum and VTA for input to NAc.91,92 Unsurprisingly, therefore, GABAARs are widely expressed throughout the striatum, with additional complexity from diversity in GABAAR subunit composition (e.g., Figure S1).
Our dual-labeling immunohistochemistry and i-EM data provide anatomical evidence for GABAARs on striatal DA axons, complementing existing functional evidence for GABAAR-dependent regulation of DA release.20,41,42 At the electron microscopy (EM) level, we found that almost half of DA axons were associated with the α3-subunit protein, with mRNA for α3 found in almost all DA neurons49 (Figures 1, S1, and S4). Because midbrain DA neurons also express other GABAAR subunits49 (Figure S1), this seeming mismatch in mRNA and protein expression might suggest dynamic recruitment of α3-subnunits into GABAAR assemblies or simply incomplete labeling in i-EM studies because of low GABAAR expression on DA axons. Indeed, α3-labeling is patchy along segments of a given DA axon but includes localization near vesicle-containing varicosities, suggesting that GABAARs are located near DA release sites and therefore poised to respond to co-released GABA at those sites. Given that only ~20% of DA axon varicosities have exocytotic proteins and release DA,93–95 it would be interesting to determine whether α3-GABAARs are located selectively at active varicosities. Our studies do not exclude involvement of GABAARs containing other subunits, or of GABABRs. Indeed, evidence suggests that ambient GABA in other brain areas acts at extrasynaptic α4-containing, α5-containing, or δ-containing GABAARs96; we note, however, that RNA-seq data (DropViz) indicate DA neurons lack mRNA for α5 and δ subunits (Figure S1).
Given that GABAergic SPNs and most GABAergic striatal interneurons are not spontaneously active in slices,87,88 the question of the source of ambient GABA remains unanswered. It is possible that activity from sparse spontaneously active GABAergic interneurons, including low-threshold spiking interneurons that inhibit DA release via GABABRs but not GABAARs,97 is sufficient to provide a GABA tone in slices. However, a GABA tone at GABAARs persists in the presence of tetrodotoxin (TTX),68,98,99 implying at least some contribution from spontaneous miniature events arising from SPNs and GABAergic interneurons. Recent evidence suggests that extracellular GABA pools in the dStr, though not NAc, are regulated by uptake through GAT1 and GAT3 transporters on local astrocytes that act as a GABA sink.68 Whether astrocytes are also a source of GABA, as reported in some other brain regions,100–102 has yet to be determined.
GABA co-release from DA axons activates GABAARs to dynamically autoinhibit DA release
Although ambient GABA would be expected to contribute to the dampening of DA release when multiple pulses are used to stimulate DA axons, the greater enhancement of [DA]o evoked by 10 p versus 1 p in both dStr and NAc when GABAARs are blocked by PTX unmasks an additional component to DA-release regulation by GABAARs. The most parsimonious explanation is the involvement of an autoinhibitory signal by co-released GABA that curtails DA release and, presumably, further GABA release as well. With evidence for stronger GABA co-release in dStr than in NAc,33,34,37,38 we anticipated that DA-release regulation by co-released GABA might be minimal in the NAc. However, this was not the case. Robust PTX-dependent increases in phasically evoked [DA]o were seen in the NAc as well as in dStr (Figure 4), indicating that co-released GABA autoinhibits DA release and dampens phasic-to-tonic DA signaling throughout the striatum.
In direct patch-clamp recording of striatal DA axons, activation of GABAARs with muscimol causes depolarization rather than hyperpolarization, because the resting membrane potential of DA axons is more hyperpolarized than the reversal potential for GABAARs.42 Furthermore, these studies found that the mechanism by which muscimol inhibits DA release is through a combination of inactivating voltage-gated sodium channels and current shunting, presumably through the chloride channel pore.42 It is likely that similar mechanisms become engaged from activation of GABAARs on DA axons by co-released GABA, raising the possibility that co-released GABA may not only autoinhibit DA release but also limit propagation of subsequent action potentials throughout the vast axonal network of a DA neuron.
Because DA axons co-release glutamate, as well as GABA,72,73,103,104 it was important to test possible involvement of co-released glutamate. We found that greater PTX-induced enhancement of 10 p- versus 1 p-evoked [DA]o persisted in the presence of a group 1 mGluR antagonist in the dStr and in ionotropic AMPAR and NMDAR antagonists in the NAc, supporting the independence of DA-release regulation by indirect consequences from co-released glutamate acting via local microcircuits. Notably, although GABA co-release is abolished in GAT1-cKO mice, the amplitude of excitatory postsynaptic currents in SPNs induced by glutamate co-released from DA axons is unchanged.38 Nevertheless, we observed that differential amplification of phasic versus tonic DA release with PTX was lost in GAT1-cKO mice, consistent with the absence of glutamate involvement shown in our pharmacological studies. Together, these observations establish that autoregulation of DA release by co-released GABA from DA axons is direct and does not involve glutamate co-release, activation of ChIs, DA release facilitation by ACh acting at nAChRs,13–16,64–67 GABA co-release from ChIs,84,85 or indirect modulation of DA release through activation of postsynaptic GABAARs on SPNs by GABA co-released from DA axons.
GABA co-release and autoregulation of DA release by co-released GABA requires GAT1 in DA axons
Midbrain DA neurons lack conventional GAD enzymes used for GABA synthesis34,37 in contrast to other GABA-releasing cells like including retinal amacrine cells that co-release GABA and DA but express GAD.105 Two possible explanations for this condundrum have emerged. One is that DA neurons synthesize GABA via a non-conventional pathway that involves conversion of putrescine to γ-aminobutyraldehyde by the enzyme diamine oxidase, with formation of GABA by ALDH1a1,37 which is expressed in subsets of DA neurons located in the ventral tier of SNc and ventromedial VTA.106–110 Indeed, co-released GABA-driven oIPSCs in striatal SPNs are decreased by pharmacological inhibition of ALDH or global ALDH1a1 deletion.37,68 However, recent evidence shows that selective overexpression of ALDH1a1 in DA neurons after global ALDH1a1 deletion does not enhance oIPSCs in SPNs from co-released GABA.38 The second and currently stronger explanation is that GABA from the interstitial compartment is sequestered in DA axons by GAT1 transporters on the plasma membrane.34,38 Convincing evidence for this comes from the complete loss of oIPSCs in SPNs from optical activation of DA axons in the dStr of GAT1-cKO mice.38 Our finding that PTX-induced enhancement of 10 p- versus 1 p-evoked [DA]o is attenuated when GAD activity is compromised aligns with an external GAD-dependent source of modulatory GABA for DA-release regulation.
In a final test of our autoregulatory hypothesis, we took advantage of the availability of GAT1-cKO mice in which GABA co-release is absent.38 Viral transfection of ChR2 in SNc DA neurons allowed selective optical stimulation of nigrostriatal axons in cKOs, and robust axonal DA release in slices (Figure 5). Our finding that PTX amplifies 1 p-evoked [DA]o to a similar extent in cKO and control mice is consistent with the persistence of an inhibitory GABAAR tone in SPNs in cKO mice38 and demonstrates that GABAAR functionality and ambient GABA tone are preserved in these animals.
Previous work shows that vGluT2-dependent glutamate uptake in DA axons synergistically promotes DA storage in vesicular pools111–113 and increases DA release,114 even though current evidence suggests that most glutamate-releasing vesicles are distinct from those containing DA76 and are localized to separate varicosities or axonal branches.77,78 In this light, given that DA and GABA may cohabit the same vesicles, loss of GABA uptake in DA axons and vesicles might be expected to alter vesicular DA uptake, DA content, and consequent DA release. Analysis of DA tissue content and 1 p DA release suggests that this is not the case, however: DA and DOPAC tissue levels in dStr were indistinguishable between GAT1-cKO mice and controls, as was 1 p-evoked [DA]o. In contrast, the differential enhancement by PTX on alternating 10 p versus 1 p optically evoked [DA]o was attenuated in GAT1-cKO mice and the increased 10 p-to-1 p ratio lost (Figure 5), confirming autoinhibition of DA release by co-released GABA.
The value of autoregulation of DA release by co-released GABA
It is well established that axonal DA release is potently inhibited by D2 autoreceptors.23–31 In striatal slices, application of D2R agonists like quinpirole can completely suppress 1 p-evoked DA release,115,116 whereas D2R antagonists enhance pulse-train-evoked release in a pulse-dependent and frequency-dependent manner, unmasking inhibition by endogenous DA.25–27 Marked suppression of DA release and inability to respond to quinpirole in mice with selective deletion of D2Rs in DA neurons confirms DA autoinhibitory feedback.30 Notably, D2Rs are metabotropic receptors that couple to G-protein second-messenger cascades and typically operate with slower kinetics than ionotropic receptors. With paired-pulse stimulation in striatal slices, D2R-mediated inhibition of DA release begins at ~100 ms with a maximum effect at 500–700 ms and termination within 5 s,24,29,31 with slightly faster kinetics in vivo.28 On the other hand, GABAAR-mediated oIPSCs in SPNs from co-released GABA begin within 2 ms and last for ~100 ms. Of course, the extent of autoinhibition will depend not only on the time for autoreceptors to be activated, but also on the pattern of firing that initiates transmitter release, the concentration of transmitter released by the first pulse, and the extent of rundown by subsequent pulses. Nevertheless, we predict that GABA released within the first 200 ms after activation of a DA axon (i.e., by the first two pulses of a 10-Hz train) would inhibit DA release without significant D2 autoreceptor activation, with a diminishing contribution from GABA as D2 autoreceptor inhibition increases. Thus, by utilizing their own ionotropic GABAARs, DA axons can deploy GABA as a first responder during a burst of neuronal activity to limit subsequent DA release before slower DA autoinhibition by D2Rs can occur.
Together, our data introduce a physiological mechanism of striatal DA autoregulation by co-released GABA that rapidly dampens phasic-to-tonic DA signaling via GABAARs on DA axons. Beyond revealing a functional role for co-released GABA, these results contribute to a new concept in neurobiology: autoregulation of transmitter release by a co-released transmitter. This regulation is likely to extend to other transmitters in other brain circuits and regions.
Limitations of the study
One limitation of this study, as in many that involve immunolabeling, is the confirmation of specificity of the antibody used to label α3-GABAAR subunits.117–119 In our i-EM images, most of the labeling visualized with DAB was cytosolic, even though the antibody used recognizes an extracellular epitope of the α3 subunit. Detection of DAB on extracellular membrane surfaces for other receptor subunits has been reported.120,121 Our pattern of staining may reflect the presence of intracellular protein, along with limited accumulation of DAB on the extracellular side of axonal surfaces because of diffusion of the label with the methods used here. In support of selective detection of α3 subunits in TH+ axons, we find strong immunoreactivity in brain areas reported to contain high levels of α3, including the RTN and the iCj. Within striatum, immunolabeling was clearly associated with DA axons but absent over profiles associated with SPNs known to lack α3 subunits.50,51 Moreover, immunoreactivity was absent when the antibody was preadsorbed with its immunogenic peptide. This alone does not confirm specificity, but, together with our other evidence, supports α3-subunit detection. Definitive proof would require showing absence of striatal staining in an appropriate α3-knockout mouse.
As mentioned earlier, there are limitations in the reliance on PTX as a GABAAR antagonist. Another point worth noting is that we found greater variability in the effect of PTX on DA release in female striatum. A possible contributing factor to this observation is that some GABAAR subunits, including α3, are regulated by ovarian hormones,122 which were not assessed here. Future studies powered to assess the influence of sex and estrous cycle on DA-release regulation by ambient GABA, as well as DA autoregulation by GABA co-release, could provide insight into known sex- and sex-hormone-dependent differences in DA transmission123 and responsiveness to psychostimulants.124,125
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Margaret E. Rice (margaret.rice@nyu.edu).
Materials availability
These studies did not generate unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
All animal procedures were in accordance with NIH guidelines “Principles of Laboratory Animal Care” (NIH publication number 85–23) and were approved by the NYU Langone Health Animal Care and Use Committee. Efforts were made to minimize the number of animals used. Mice were kept on a 12 h light:dark cycle, with lights on from 06:30 to 18:30 local time, and were housed in an AAALAC-accredited facility in groups of ≤5 with ad libitum access to food and water. Temperature and humidity were maintained and monitored by the vivarium staff. Adult (>2 months old) male and female mice were used unless indicated otherwise; in some control experiments and when GAT1 cKO mice were examined, males only were used given the lower variability of PTX-induced increases in this sex. Transgenic lines were maintained on a C57BL/6J background.
Ai32; DAT-cre mice
Mice expressing a blue light sensitive ChR2 (H134R variant) selectively in DA neurons were generated as previously.60 Breeder knock-in mouse lines were originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA). We crossed B6; 129S-Gt(ROSA)26Sortm32(CAG—COP4*H134R/EYFP)Hze (Ai32; JAX stock #024109) homozygous mice126 with B6.SJL-Slc6a3tm1.1(cre)Bkmn/J (DAT-IRES-Cre; JAX stock No 006660) heterozygous (Het) mice.127 The progeny were self-crossed to produce a line of breeders used to generate Ai32+/−;DAT-Cre+/− (Het/Het) mice with restricted expression of the ChR2 ion channel fused to enhanced-yellow fluorescent protein (eYFP) in DAT-containing DA neurons for experiments. It should be noted that DAT-Cre Het mice have a small but significantly lower maximum DA uptake velocity, Vmax, and significantly higher peak extracellular DA concentration ([DA]o) evoked by single pulse electrical stimulation versus littermate control mice in both dStr and NAc core.60
GAT1-DAT-cre-ChR2 mice
Mice lacking the plasma membrane GAT1, encoded by Slc6a1, in DA neurons were derived by crossing a floxed GAT1 mouse line (GAT1flox/flox) generated by geneOway38 with DAT-IRES-Cre mice (see above). These mice have no gross abnormalities or disruption to DA neuronal development.38 Incorporation of ChR2 into DA neurons was achieved by intracranial microinjection of a Cre-dependent adeno-associated virus (AAV) construct containing ChR2 fused to mCherry (AAV8-EF1a-double floxed-hChR2(H134R)-mCherry-WPRE-HGHpA, which was s gift from Karl Deisseroth obtained from Addgene, MA, USA (Addgene viral prep No 20297-AAV8) into the right substantia nigra pars compacta (SNc) of male control (DAT-Cre-Het) mice and cKO (GAT1flox/flox;DAT-Cre-Het) mice anesthetized with isoflurane (Covetrus, Portland, ME, USA).38 Coordinates for SNc injections were (from bregma): AP −3.12 mm, ML +1.6 mm and DV −4.2 mm (from pia).128 A volume of 1 μL of 1 × 1012 GC/mL AAV was injected at a rate of 100 nL/sec as previously.38 Optically evoked DA recording in striatal slices was conducted at least three weeks after AAV microinjection to enable ChR2 expression throughout striatal DA axons in cKO and age-matched control mice. All mice in the GAT1 cKO cohort were homozygous except for one that was heterozygous. In some mice the dStr from slices of the left hemisphere were dissected, weighed and frozen on dry-ice for HPLC analysis (see below). After recording experiments slices were fixed in 4% paraformaldehyde and processed to verify expression and location of mCherry-fused viral infection by fluorescence imaging.
Genotyping.
Ai32;DAT-Cre mice were genotyped by obtaining toe clippings that were analyzed by standard PCR procedures at the NYU Langone Genotyping Core Facility using protocols published by Jackson Labs. Genotyping of GAT1; DAT-Cre mice were from tail samples using GAT1 primers and protocols designed by genOway and performed in either the Tritsch Lab or at the NYU Langone Genotyping Core Facility.
METHOD DETAILS
Fluorescence immunohistochemistry (i-HC) with confocal microscopy
Immunohistochemical procedures were carried out based on modifications of our previous methods.129,130 Mice (C57BL6/J) were anesthetized with either sodium pentobarbital (Euthasol 60 mg/kg, i.p.) or isoflurane inhalation, then perfused transcardially with 20 mL phosphate buffered saline (PBS) followed by 20 mL of freshly prepared 4% paraformaldehyde (PFA) in 0.1 M PBS. After extraction, brains were kept in PFA overnight at 4°C, cut into blocks containing relevant brain region(s) and cryoprotected in 20% sucrose at room temperature for 2 to 3 h and then in 30% sucrose overnight at 4°C. Brain blocks were embedded in Tissue-Tek O.C.T. compound (Electron Microscopy Sciences, Hatfield, PA, USA), cooled to −18°C to −20°C and frozen coronal sections (50 μm thick) cut through the striatum or midbrain using a Cryocut 1800 cryostat (Belair Instrument Company, Springfield, NJ, USA). Sections were kept free-floating in PBS at 4°C before being processed for i-HC.
All i-HC incubations took place at room temperature on a shaking platform. Brain sections were washed for 20 min in PBS followed by 2 × 20 min (in PBS with 0.1% Triton X-100), then incubated in a blocking solution (PBS with 0.3% Triton X-100, 20% normal donkey serum (NDS)) for 1 h. Thereafter, sections were incubated in primary antibodies (in PBS with 0.3% Triton X-100, 2% NDS) for 18–24 h. Following 2 × 20 min washes in PBS (containing 0.3% Triton X-100), sections were incubated in fluorescent secondary antibodies (diluted 1:200 in PBS with 0.3% Triton X-100) for 2 h. After 2 × 20 min final washes in PBS alone, sections were mounted on slides, dehydrated in graded alcohols (50%, 70%, 95%, 100%), air dried, and coverslipped with Vectashield (Vector Laboratories, Newark, California, USA).
The primary antibodies used were sheep-anti-tyrosine hydroxylase (TH) polyclonal (catalog #ab113; Abcam Inc., Cambridge, MA, USA) at 1:1000 for labeling of DA neurons and a rabbit anti-α3-GABAAR subunit (extracellular) polyclonal (catalog #AGA-003; Alomone Labs, Jerusalem, Israel) at 1:200 for labeling GABAARs. It should be noted that the α3-subunit antibody used has been knockout (KO) validated.131 Moreover, the supplier states that each lot is verified using Western blot analysis, which provides evidence that the antibody can recognize the denatured target protein, and that the peptide sequence is confirmed by amino acid analysis and mass spectrometry. Secondary antibodies were donkey anti-sheep Cy5 and donkey anti-rabbit Cy3 (Jackson ImmunoResearch Laboratories Inc, West Grove, Pennsylvania, USA). To test the specificity of the α3-GABAAR primary antibody to the target epitope, preadsorbtion with its corresponding antigenic blocking peptide (catalog #BLP-GA003; Alomone Labs) at 1x (w/w) was conducted at room temperature for 1 h before applying to sections. Primary and secondary antibodies used are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Biotinylated donkey anti-rabbit | Jackson ImmunoResearch Labs | Cat No: 711-065-152; RRID: AB_2340593 |
| Biotinylated goat anti-mouse | Vector Laboratories | Cat No: BA-9200; RRID: AB_2336171 |
| Chicken anti-Green Fluorescent Protein | Aves Labs, Inc | Cat No: GFP-1020; RRID: AB_10000240 |
| Donkey anti-mouse IgG conjugated to ultrasmall (0.8 nm) colloidal gold | Electron Microscopy Sciences | Cat No: 25810 |
| Donkey anti-rabbit IgG conjugated to ultrasmall (0.8 nm) colloidal gold | Electron Microscopy Sciences | Cat No: 25701 |
| Donkey anti-chicken Cy3 | Jackson ImmunoResearch Labs | Cat No: 703-165-155; RRID: AB_2340363 |
| Donkey anti-rabbit Cy3 | Jackson ImmunoResearch Labs | Cat No: 711-165-152; RRID: AB_2307443 |
| Donkey anti-rabbit Cy2 | Jackson ImmunoResearch Labs | Cat No: 711-225-152; RRID: AB_2340612 |
| Donkey anti-sheep Cy5 | Jackson ImmunoResearch Labs | Cat No: 713-175-147; RRID: AB_2340730 |
| Mouse anti-Tyrosine hydroxylase monoclonal | Boehringer Mannheim | Cat No: 1017-381 |
| Rabbit anti-GABAAR α3 (extracellular) polyclonal | Alomone Labs | Cat No: AGA-003; RRID: AB_2039866 |
| Rabbit anti-GABAAR α3 blocking peptide | Alomone Labs | Cat No: BLP-GA-003 |
| Rabbit ant-GABAAR δ (extracellular) | Dr. W. Sieghart | N/A |
| Rabbit anti-Tyrosine hydroxylase polyclonal | Millipore | Cat No: AB152; RRID: AB_390204 |
| Sheep anti-Tyrosine hydroxylase polyclonal | Abcam | Cat No: ab113; RRID: AB_297905 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAV8-EF1a-double floxed-hChR2(H134R)-mCherry-WPRE-HGHpA | pAAV-EF-1a-double floxed-hChR2(H134R)-mCherry-WPRE-HGHpA was a gift from Karl Deisseroth | Addgene viral prep Cat No: 20297-AAV8 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| D-AP5 | Hello Bio | Cat No: HB0225 |
| (+)Bicuculline | Sigma-Aldrich | Cat No: 14340 |
| Calcium chloride dihydrate (CaCl2.2H2O) | Sigma-Aldrich | Cat No: C5080 |
| DAB (3,3’-Diaminobenzidine tetrahydrochloride) | Sigma-Aldrich | Cat No: D5905 |
| DHβE hydrobromide | Tocris Bioscience | Cat No: 2349 |
| DMSO (dimethyl sulfoxide) | Sigma-Aldrich | Cat No: 276855 |
| DNQX | Hello Bio | Cat No: HB0261 |
| DOPAC (3,4-Dihydroxyphenylacetic acid) | Sigma-Aldrich | Cat No: D-9128 |
| Dopamine hydrochloride (3-hydroxytryramine) | Sigma-Aldrich | Cat No: H-8502 |
| EDTA (Ethylenediaminetetraacetic acid disodium salt dihydrate) | Sigma-Aldrich | Cat No: E1644 |
| Ethanol, 200 proof | Fisher Scientific | Cat No: A962P-4 |
| Euthasol | Covetrus | SKU No: 009444 |
| Gabazine (SR 95531) | Hello Bio | Cat No: HB0901 |
| D-(+)-Glucose | Sigma-Aldrich | Cat No: G-5767 |
| HEPES (Acid) | Sigma-Aldrich | Cat No: H-3375 |
| HEPES (Sodium Salt) | Sigma-Aldrich | Cat No: H-7006 |
| Isoflurane | Covetrus (NYULH-DCM) | SKU No: 029405 |
| JNJ-16259685 | Tocris Bioscience | Cat No: 2333 |
| 3-mercaptopropionic acid (3-MPA) | Sigma-Aldrich | Cat No: M5801 |
| Magnesium sulfate heptahydrate (MgSO4.7H2O) | Sigma-Aldrich | Cat No: M-1880 |
| Methanol | Sigma-Aldrich | Cat No: 34860 |
| Muscimol | Tocris Bioscience | Cat No: 0289 |
| Muscimol | Hello Bio | Cat No: HB0887 |
| Normal Donkey Serum (NDS) | Sigma-Aldrich | Cat No: S30-M |
| Paraformaldehyde | Fisher | Cat No: T353 |
| Paraformaldehyde for EM (4% in 0.1M Phosphate Buffer, PH 7.4 | Electron Microscopy Sciences | Cat No: 15735-20S |
| Phosphate Buffered Saline (PBS) | Sigma-Aldrich | Cat No: P7059 |
| Picrotoxin | Sigma-Aldrich | Cat No: P1675 |
| Picrotoxin | Hello Bio | Cat No: HB0506 |
| Potassium chloride (KCl) | Sigma-Aldrich | Cat No: P-3911 |
| Potassium phosphate monobasic (KH2PO4) | Sigma-Aldrich | Cat No: P-0662 |
| Sodium bicarbonate (NaHC03) | Sigma-Aldrich | Cat No: S-6014 |
| Sodium-1-heptanesulfanate (C7H15O3SNa) | Sigma-Aldrich | Cat No: H2766 |
| Sodium chloride (NaCl) | Sigma-Aldrich | Cat No: S-7653 |
| Sodium octyl sulfate | Sigma-Aldrich | Cat No: O-4003 |
| Sodium phosphate monobasic monohydrate (NaH2PO4.H2O) | Sigma-Aldrich | Cat No: S9638 |
| Sucrose | Sigma-Aldrich | Cat No: S-7903 |
| Strychnine hydrochloride | Sigma-Aldrich | Cat No: S8759 |
| Triton X-100 | Sigma-Aldrich | Cat No: T8787 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Silver Enhancer Kit for Microscopy | Kirkegaard & Perry Laboratories (KPL), Inc | Cat No: 50-22-01 |
| Vectastain Elite ABC-HRP Kit | Vector Labs | Cat No: PK6100 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Ai32 | Jackson Laboratory | Stock No: 024109 |
| Mouse: C57BL/6J | Jackson Laboratory | Stock No: 000664 |
| Mouse: DAT-IRES-Cre | Jackson Laboratory | Stock No: 006660 |
| Mouse: GAT1flox/flox | Tritsch Lab, NYULH | geneOway JAX stock No: 037219 |
|
| ||
| Software and algorithms | ||
|
| ||
| DropViz RNA-Seq Data | Saunders et al.48 | https://DropViz.org |
| Fiji ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Fusion Benchtop Image Acquisition Software (ver. 1.101) | Andor Technology Ltd. Oxford Instruments | https://andor.oxinst.com/ |
| Imaris Microscope Imaging Software (ver. 10.0) | Oxford Instruments | https://imaris.oxinst.com |
| Graphpad Prism 9 & 10 | Graphpad Software | https://www.graphpad.com/ |
| Adobe Photoshop CC2015 | Adobe Systems | https://www.adobe.com/ |
|
| ||
| Other | ||
|
| ||
| DAPI Fluoromount-G | SouthernBiotech | Cat No: 0100-20 |
| Tissue-Tek® O.C.T. Compound | Electron Microscopy Sciences | Cat No: 62550 |
| Vectashield® Antifade Mounting Media | Vector Labs | Cat No:H1000-10 |
| Epoxy resin, grids, and most other electron microscopic supplies | Electron Microscopy Sciences | |
Images of immunostained tissue were obtained with a Bench Top BC43 spinning disk confocal microscope controlled with Fusion image acquisition software (Benchtop version 1.101, Andor Technology Ltd. Oxford Instruments, Belfast, Ireland) and processed with Imaris microscope imaging software (version 10.0, Oxford Instruments). Images were captured with either epifluorescence using a 2x objective (0.06 NA) or with confocal imaging using a 60x oil-immersion objective (1.4 NA, 1 Airy unit). Any changes in brightness and/or contrast were made on the entire image. Comparisons of immunostained control and preabsorbed tissue were on adjacent sections from the same brain, using identical intensity and exposure settings. Each primary antibody was tested on multiple tissue sections from at least two mice. Additional controls including processing some sections without addition of primary antibodies to test for the specificity of the secondary antibodies for their respective primary.
Immunoelectron microscopy (i-EM)
The presence of α3 containing GABAAR subunits on tyrosine hydroxylase (TH) expressing axons was examined using double immuno-EM as previously.132,133 Striatal vibratome sections (50 μm thick) were cut from paraformaldehyde-perfused adult C57BL/6J mice (~8 weeks postnatal), then incubated simultaneously in rabbit anti-GABAAR α3 (extracellular) antibody (1:200) (catalog #AGA-003, Lot #AGA003AN0502; Alomone Labs, Jerusalem, Israel) and mouse anti-TH monoclonal antibody (1:5) (catalog #1017-381, Lot #10968624-01, clone 9B9; Boehringer Mannheim, Mannheim, Germany). The secondary antibody for α3 was biotinylated donkey anti-rabbit (1:200) (catalog #711-065-152, Jackson ImmunoResearch Laboratories) and for TH it was donkey anti-mouse IgG conjugated to ultrasmall (0.8 nm) colloidal gold (1:40) (catalog #25810, temporary RRID: AB_2631210; Electron Microscopy Sciences). Sections were treated with 3,3′-Diaminobenzidine (DAB, Sigma Aldrich, St. Louis, MO, USA) plus avidinbiotin-horseradish-peroxidase (HRP) complex (Vectastain Elite ABC-HRP Kit, catalog #PK6100, Vector Laboratories, Burlingame, CA, USA) to visualize α3 and silver intensified using a Silver Enhancer Kit for Microscopy (catalog #50-22-01; Kirkegaard & Perry Laboratories (KPL), Inc., Gaithersburg, MD, USA) for 17 min to enlarge the colloidal gold particles (silver-intensified gold, SIG) for visualizing TH, and processed for EM as previously.132,133 Ultra-thin sections were cut from sections embedded in resin and collected onto EM grids. Digital images to quantify co-localization were captured by a 1.2-megapixel Hamamatsu CCD camera (AMT, Boston, MA, USA) attached to a JEOL 1200XL electron microscope and running AMT’s software. Images from both dStr and NAc core were evaluated. DA axonal profiles with multiple SIG particles were considered to be TH-positive. The proportion of TH axons expressing α3-GABAARs in dStr and NAc was quantified, as was the proportion of α3-GABAAR containing axonal profiles that were TH-positive. We avoided bias in sampling by taking digital images strictly in the order of encounter along the surface of vibratome sections, where immunolabeling would be optimal. The vibratome section surfaces were created randomly, with the only rule being that the plane of section be coronal. Ultrathin sections were tangential to the vibratome sections, with no pre-selection of the plane of section except to be as tangential as possible, so as to maximize the sampling of vibratome surfaces while remaining within striatum, based on light microscopic inspection of the vibratome section.
As controls, other sections were processed in parallel, using intentionally unmatched secondary antibodies, i.e., gold-conjugated anti-mouse IgG following incubation of sections with rabbit anti-α3 (1:200) and biotinylated anti-rabbit IgG following incubation of other sections with mouse anti-TH (1:5). These sections were verified by light and EM to show little labeling, indicating negligible cross-reactivity of the two secondary antibodies used to detect the primary antibodies. The low level of labeling provided our basis for judging the extent of background immunolabeling, which was assessed to be no more than a single SIG particle per profile.
We also used similar dual i-EM for detecting δ subunits of GABAARs that are absent in DA neurons49 (Figure 1A). For this evaluation, we used the same lot of a rabbit anti-d subunit antibody (1:80, gift from Dr. W. Sieghard, Medical University, Vienna, Austria), characterized for its specificity previously50,134,135 and detected using the SIG immunolabel as previously.132,133 This was combined with TH, detected by DAB-HRP using the same primary antibody (1:5) as above. Secondary antibodies for the δ subunit TH dual i-EM were ultrasmall gold-conjugated donkey anti-rabbit IgG (1:40) (catalog #25701; Electron Microscopy Sciences) and biotinylated goat antimouse IgG (1:200) (catalog #BA-9200; Vector Laboratories). The DAB-HRP reaction time was 15 min, and silver-intensification of ultrasmall gold was 28 min.
Voltammetric detection of DA release
Slice preparation.
Procedures for preparing striatal slices were as described previously.60,67,136 Adult mice were deeply anesthetized by isoflurane inhalation before decapitation and brain removal; these procedures were typically initiated between 12:00 and 14:30 each experimental day. Coronal striatal slices (300 μm thick) were prepared using a vibrating microtome (VT1200S; Leica Microsystems, Bannockburn, IL, USA). Slices were allowed to recover at room temperature in a HEPES-buffered artificial cerebrospinal fluid (aCSF) containing (in mM): NaCl (120); NaHCO3 (20); HEPES acid (6.7); KCl (5); HEPES sodium salt (3.3); MgSO4 (2); glucose (10); CaCl2 (2); equilibrated with 95% O2/5% CO2 for at least 90 min. In some experiments the recovery HEPES-buffered aCSF also contained the GAD enzyme inhibitor, 3-mercaptopropionic acid (3-MPA, 500 μM), for slice preincubation. After the recovery period a slice was transferred to a submersion recording chamber (Warner Instruments, Harvard Bioscience, Holliston, MA, USA) superfused at 1.3 to 1.5 mL/min with bicarbonate-buffered aCSF containing (in mM): NaCl (124); KCl (3.7); NaHCO3 (26); MgSO4 (1.3); KH2PO4 (1.3); glucose (10); CaCl2 (2.4); equilibrated with 95% O2/5% CO2. Dopamine recordings. Given that optically-evoked DA release evoked by pulse-train stimulation in the same recording site in Ai32+/−;DAT-Cre+/− mice is subject to rundown at our usual recording temperature of 30°C–32°C,60 we used slightly lower temperatures (23°C–27°C) here that enabled stable evoked DA release for the duration of a given experiment. However, key findings were verified using a multi-site recording protocol at 30°C–32°C. Most experiments examined one slice per mouse; when two slices per mouse were tested simultaneously in separate slice chambers, care was taken to ensure that conditions between chambers were similar including aCSF flow rate, temperature, and slice orientation.
In each slice, DA release was evoked and recorded in a single site in either the central portion of the dStr or in the NAc core, within 100 μm dorsal of the anterior commissure.128 Optical stimulation was achieved using a TTL-driven 473 nm blue laser light source (Laserglow Technologies, North York, Ontario, Canada) coupled to a 200 μm diameter optical fiber with a ceramic ferrule (Part #M80L01; Thorlabs, Newton, NJ, USA) positioned just above the desired recording site in the slice.60 Light pulse duration (1 ms) and timing were controlled by a Master-8 (A.M.P.I., Jerusalem, Israel). Power intensity was determined to be 400–600 μW using a power meter equipped with a photodiode sensor (Part #PM100USB; Thorlabs). Optically evoked DA release using these parameters is TTX sensitive.60 Increases in [DA]o were monitored at carbon fiber microelectrodes (CFMs: 7 μm in diameter and 50 to 70 μm in length, positioned in the slice ~100 μm away from the optical fiber) and FSCV using a Millar Voltammeter (Julian Millar, Barts and the London School of Medicine and Dentistry, UK) scanning from −700 mV to +1300 mV to −700 mV at a rate of 800 V/s every 100 ms.136,137
Stimulation protocols used included 1 p stimulation or 10 p stimulation in a given recording site in separate slices or alternating 1 p and 10 p in the same recording site in a given slice. Stimulus interval was 5 min for 1 p, 10 min for 10 p and 7 min when these were interleaved. Drugs were applied after 3 to 4 consistent baseline evoked [DA]o records were obtained; these records were averaged to provide control peak amplitude for the stimulus used. Subsequently, 3–4 responses in the drug (after 30–45 min) were averaged. In some experiments a multi-site recording protocol was used in which 1 p stimulations and 10 p stimulations were applied only once in 3–4 recording sites in the absence and then presence of drug in a given slice. Data for 1 p or 10 p evoked [DA]o obtained in these multiple sites under control conditions were averaged and compared with the average evoked [DA]o obtained in drug.
Sex differences have been reported in electrically-evoked [DA]o in rodent striatal slices with generally higher evoked [DA]o in females and greater uptake.132,138 Notably, differential DAT expression between the sexes is lost in heterozygous DAT-Cre mice.139 Here, all DA recordings were performed in mice with heterozygous DAT-Cre. With the exception of experiments conducted at a higher recording temperature (Figure S6) and experiments using GAT1-cKO mice in which data was obtained from male mice only (Figure 5), we used balanced numbers of each sex per group, when possible. We found no significant difference in 1 p or 10 p optically-evoked DA release between males and females, allowing us to combine data from both sexes for statistical analyses. Individual data points obtained from males or females are identified on graphs by color.
At the end of each slice experiment, evoked [DA]o was quantified by calibration of the carbon-fiber recording electrode in the recording chamber at the same temperature as slice recordings using 1 μM DA in control aCSF and in any drug-containing solution used during the preceding experiment. No change in electrode sensitivity to DA or evidence of inherent electroactivity was seen in with any drug used in DA release recordings, including PTX, although gabazine and (+)bicuculine were excluded for these reasons (Figure S7).
HPLC-EC analysis of tissue DA and DOPAC content
Striatal tissue collected from the left hemisphere of age-matched DAT-Cre-Het control mice and GAT1flox/flox;DAT-Cre-Het cKO mice were analyzed for DA and DOPAC content using HPLC-EC, as previously.140,141 Immediately after slicing, two tissue samples (3–7 mg) containing the dStr were dissected in ice-cold aCSF from two anterior-to-mid and two mid-to-posterior striatal slices; care was taken not to include the overlying cortex or corpus callosum; samples were weighed in Eppendorf tubes, frozen on dry-ice and stored at −80°C.
The HPLC mobile phase (6.90 g/L NaH2PO4.H2O, 232 mg/L C7H15O3SNa, 80 mg/L EDTA, 30 mg/L sodium octyl sulfate, 10% methanol, pH 3.1) was deoxygenated with argon, filtered and maintained under argon during sample analysis. Tissue samples were diluted 100-fold in ice-cold, deoxygenated eluent, sonicated, then centrifuged for 2 min at 13,000 g. The supernatant was injected directly onto a BAS HPLC-EC system (West Lafayette, Indiana, USA) with a C18 reverse phase column, and amperometric detection at a glassy carbon electrode set at 600 mV versus Ag/AgCl. Peak heights were quantified by comparison with those of DA and DOPAC calibration standards and expressed as nmol/g tissue wet weight.
General and pharmacological reagents
Salts for all buffers, as well as DA and DOPAC were purchased from Sigma Aldrich (St. Louis, MO, USA). The source of PTX was either Sigma Aldrich or Hello Bio Inc. (Princeton, NJ, USA), muscimol was from Tocris Bioscience (R&D Systems Inc., Minneapolis, MN, USA) or Hello Bio, D-AP5, DNQX and gabazine (SR 95531) were from Hello Bio, JNJ-16259685 and Dihydro-β-erythroidine hydrobromide (DHβE) were from Tocris Bioscience, and 3-mercaptopropionic acid (3-MPA), (+)bicuculline, strychnine hydrochloride were from Sigma Aldrich. Experimental solutions of PTX were prepared by vigorous stirring in aCSF for ~10 min immediately before use. 3-MPA was added to the recovery HEPES buffer for preincubation of slices and to aCSF during slice recordings. DNQX, JNJ-16259685 and (+)bicuculline were dissolved in dimethyl sulfoxide (DMSO, Sigma Aldrich) as stock solutions. Working solutions with the final experimental concentrations were prepared in aCSF immediately before use, with a maximal final DMSO concentration of <0.1%. All other drugs were prepared and stored as aqueous stocks in deionized water before dilution in aCSF.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism versions 9 and 10 for Windows (GraphPad Software, La Jolla California USA). Data are expressed as means ± SEM where n denotes the number of mice or recordings sites, as indicated. Data sets were tested for normality using a Shapiro-Wilk test and/or Kolmogorov-Smirnov test. Significance of differences between two groups was assessed by non-parametric Mann-Whitney tests for data sets that were not normally-distributed. For normally distributed data, parametric unpaired, paired or ratio paired Student’s t-tests were used, with or without Welch correction for unequal variance, as appropriate. two-way ANOVA was used to analyze DA and DOPAC tissue content between genotypes and striatal subregion. Significance was considered to be p < 0.05 for all comparisons.
Supplementary Material
Highlights.
Dopamine axons express GABAARs that inhibit optically evoked dopamine (DA) release
GABA inhibition of DA release is greater with phasic burst vs. tonic single-pulse stimulation
Enhanced inhibition of phasic DA release is absent in mice lacking GABA co-release
GABA co-released from DA axons acts as a physiological autoregulator of DA release
ACKNOWLEDGMENTS
Funding was provided by NIH grants R01DA038616 (M.E.R.), R01DA050165 (M.E.R.), and DP2NS105553 (N.X.T.); the Attilio and Olympia Ricciardi Research Fund (M.E.R. and J.C.P.); and a Marlene and Paolo Fresco Postdoctoral Fellowship (R.M.). We thank the NYU Langone Genotyping Core and the Division of Comparative Medicine for their services. We also thank Megan Fernandes and Vanessa Khachaturyan for assistance with immunohistochemistry and Ryan Feeley for assistance with mouse colony maintenance. Some components of the graphical abstract were from BioRender.
Footnotes
DECLARATION OF INTERESTS
B.O. is currently employed at Addgene but was not when contributing to this study.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113834.
REFERENCES
- 1.Gerfen CR, and Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tritsch NX, and Sabatini BL (2012). Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76, 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, and Janak PH (2013). A causal link between prediction errors, dopamine neurons and learning. Nat. Neurosci. 16, 966–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamid AA, Pettibone JR, Mabrouk OS, Hetrick VL, Schmidt R, Vander Weele CM, Kennedy RT, Aragona BJ, and Berke JD (2016). Mesolimbic dopamine signals the value of work. Nat. Neurosci. 19, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox J, and Witten IB (2019). Striatal circuits for reward learning and decision-making. Nat. Rev. Neurosci. 20, 482–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohebi A, Pettibone JR, Hamid AA, Wong JMT, Vinson LT, Patriarchi T, Tian L, Kennedy RT, and Berke JD (2019). Dissociable dopamine dynamics for learning and motivation. Nature 570, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sippy T, and Tritsch NX (2023). Unraveling the dynamics of dopamine release and its actions on target cells. Trends Neurosci. 46, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chergui K, Suaud-Chagny MF, and Gonon F (1994). Nonlinear relationship between impulse flow, dopamine release and dopamine elimination in the rat brain in vivo. Neuroscience 62, 641–645. [DOI] [PubMed] [Google Scholar]
- 9.Venton BJ, Zhang H, Garris PA, Phillips PEM, Sulzer D, and Wightman RM (2003). Real-time decoding of dopamine concentration changes in the caudate-putamen during tonic and phasic firing. J. Neurochem. 87, 1284–1295. [DOI] [PubMed] [Google Scholar]
- 10.Grace AA, and Bunney BS (1984). The control of firing pattern in nigral dopamine neurons: single spike firing. J. Neurosci. 4, 2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grace AA, and Bunney BS (1984). The control of firing pattern in nigral dopamine neurons: burst firing. J. Neurosci. 4, 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman AS, Meltzer LT, and Bunney BS (1985). Firing properties of substantia nigra dopaminergic neurons in freely moving rats. Life Sci. 36, 1983–1994. [DOI] [PubMed] [Google Scholar]
- 13.Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, and Cheer JF (2012). Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, and Cragg SJ (2012). Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75, 58–64. [DOI] [PubMed] [Google Scholar]
- 15.Kramer PF, Brill-Weil SG, Cummins AC, Zhang R, Camacho-Hernandez GA, Newman AH, Eldridge MAG, Averbeck BB, and Khaliq ZM (2022). Synaptic-like axo-axonal transmission from striatal cholinergic interneurons onto dopaminergic fibers. Neuron 110, 2949–2960.e4. 10.1016/j.neuron.2022.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu C, Cai X, Ritzau-Jost A, Kramer PF, Li Y, Khaliq ZM, Hallermann S, and Kaeser PS (2022). An action potential initiation mechanism in distal axons for the control of dopamine release. Science 375, 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice ME, Patel JC, and Cragg SJ (2011). Dopamine release in the basal ganglia. Neuroscience 198, 112–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sulzer D, Cragg SJ, and Rice ME (2016). Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia 6, 123–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nolan SO, Zachry JE, Johnson AR, Brady LJ, Siciliano CA, and Calipari ES (2020). Direct dopamine terminal regulation by local striatal microcircuitry. J. Neurochem. 155, 475–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts BM, Lopes EF, and Cragg SJ (2021). Axonal modulation of striatal dopamine release by local γ-aminobutric acid (GABA) signaling. Cells 10, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C, Goel P, and Kaeser PS (2021). Spatial and temporal scales of dopamine transmission. Nat. Rev. Neurosci. 22, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohebi A, and Berke JD (2020). Dopamine release drives motivation, independently from dopamine cell firing. Neuropsychopharmacology 45, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Limberger N, Trout SJ, Kruk ZL, and Starke K (1991). “Real time” measurement of endogenous dopamine release during short trains of pulses in slices of rat neostriatum and nucleus accumbens: role of autoinhibition. Naunyn-Schmiedeberg’s Arch. Pharmacol. 344, 623–629. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy RT, Jones SR, and Wightman RM (1992). Dynamic observation of dopamine autoreceptor effects in rat striatal slices. J. Neurochem. 59, 449–455. [DOI] [PubMed] [Google Scholar]
- 25.Patel J, Trout SJ, and Kruk ZL (1992). Regional differences in evoked dopamine efflux in brain slices of rat anterior and posterior caudate putamen. Naunyn-Schmiedeberg’s Arch. Pharmacol. 346, 267–276. [DOI] [PubMed] [Google Scholar]
- 26.Trout SJ, and Kruk ZL (1992). Differences in evoked dopamine efflux in rat caudate putamen, nucleus accumbens and tuberculum olfactorium in the absence of uptake inhibition: influence of autoreceptors. Br. J. Pharmacol. 106, 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cragg SJ, and Greenfield SA (1997). Differential autoreceptor control of somatodendritic and axon terminal dopamine release in substantia nigra, ventral tegmental area, and striatum. J. Neurosci. 17, 5738–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benoit-Marand M, Borrelli E, and Gonon F (2001). Inhibition of dopamine re-lease via presynaptic D2 receptors: time course and functional characteristics in vivo. J. Neurosci. 21, 9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips PEM, Hancock PJ, and Stamford JA (2002). Time window of autoreceptor-mediated inhibition of limbic and striatal dopamine release. Synapse 44, 15–22. [DOI] [PubMed] [Google Scholar]
- 30.Anzalone A, Lizardi-Ortiz JE, Ramos M, De Mei C, Hopf FW, Iaccarino C, Halbout B, Jacobsen J, Kinoshita C, Welter M, et al. (2012). Dual control of dopamine synthesis and release by presynaptic and postsynaptic dopamine D2 receptors. J. Neurosci. 32, 9023–9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin JH, Adrover MF, and Alvarez VA (2017). Distinctive Modulation of Dopamine Release in the Nucleus Accumbens Shell Mediated by Dopamine and Acetylcholine Receptors. J. Neurosci. 37, 11166–11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolf ME, and Roth RH (1990). Autoreceptor regulation of dopamine synthesis. Ann. N. Y. Acad. Sci. 604, 323–343. [DOI] [PubMed] [Google Scholar]
- 33.Tritsch NX, Ding JB, and Sabatini BL (2012). Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature 490, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tritsch NX, Oh WJ, Gu C, and Sabatini BL (2014). Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. Elife 3, e01936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tritsch NX, Granger AJ, and Sabatini BL (2016). Mechanisms and functions of GABA co-release. Nat. Rev. Neurosci. 17, 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson AB, Hammack N, Yang CF, Shah NM, Seal RP, and Kreitzer AC (2014). Striatal cholinergic interneurons drive GABA release from dopamine terminals. Neuron 82, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JI, Ganesan S, Luo SX, Wu YW, Park E, Huang EJ, Chen L, and Ding JB (2015). Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 350, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melani R, and Tritsch NX (2022). Inhibitory co-transmission from midbrain dopamine neurons relies on presynaptic GABA uptake. Cell Rep. 39, 110716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zych SM, and Ford CP (2022). Divergent properties and independent regulation of striatal dopamine and GABA co-transmission. Cell Rep. 39, 110823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace ML, and Sabatini BL (2023). Synaptic and circuit functions of multitransmitter neurons in the mammalian brain. Neuron 111, 2969–2983. 10.1016/j.neuron.2023.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopes EF, Roberts BM, Siddorn RE, Clements MA, and Cragg SJ (2019). Inhibition of nigrostriatal dopamine release by striatal GABAA and GABAB receptors. J. Neurosci. 39, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kramer PF, Twedell EL, Shin JH, Zhang R, and Khaliq ZM (2020). Axonal mechanisms mediating γ-aminobutyric acid receptor type A (GABA-A) inhibition of striatal dopamine release. Elife 9, e55729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barnard EA, Skolnick P, Olsen RW, Mdhler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, and Langer SZ (1998). International Union of Pharmacology. XV. Subtypes of GABA receptors: Classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 50, 291–313. [PubMed] [Google Scholar]
- 44.Bocquet N, Nury H, Baaden M, LePoupon C, Changeux JP, Delarue M, and Corringer PJ (2009). X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 457, 111–114. [DOI] [PubMed] [Google Scholar]
- 45.Olsen RW, and Sieghart W (2008). International Union of Pharmacology. LXX. Subtypes of GABAA receptors: classification on the basis of subunit composition, pharmacology, and function. Pharmacol. Rev. 60, 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olsen RW (2018). GABAA Receptor: Positive and Negative Allosteric Modulators. Neuropharmacology 136, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu S, Noviello CM, Teng J, Walsh RM Jr., Kim JJ, and Hibbs RE (2018). Structure of a human synaptic GABAA receptor. Nature 559, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okada H, Matsushita N, Kobayashi K, and Kobayashi K (2004). Identification of GABAA receptor subunit variants in midbrain dopaminergic neurons. J. Neurochem. 89, 7–14. [DOI] [PubMed] [Google Scholar]
- 50.Fritschy JM, and Mohler H (1995). GABAA-receptor heterogeneity in the adult rat brain: Differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 359, 154–194. [DOI] [PubMed] [Google Scholar]
- 51.Boccalaro IL, Cristiá-Lara L, Schwerdel C, Fritschy JM, and Rubi L (2019). Cell type-specific distribution of GABAA receptor subtypes in the mouse dorsal striatum. J. Comp. Neurol. 527, 2030–2046. [DOI] [PubMed] [Google Scholar]
- 52.Seo S, and Leitch B (2014). Altered thalamic GABAA-receptor subunit expression in the stargazer mouse model of absence epilepsy. Epilepsia 55, 224–232. [DOI] [PubMed] [Google Scholar]
- 53.Tossell K, Dodhia RA, Galet B, Tkachuk O, and Ungless MA (2021). Tonic GABAergic inhibition, via GABAA receptors containing αβƐ subunits, regulates excitability of ventral tegmental area dopamine neurons. Eur. J. Neurosci. 53, 1722–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, and Sperk G (2000). GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101, 815–850. [DOI] [PubMed] [Google Scholar]
- 55.Schwarzer C, Berresheim U, Pirker S, Wieselthaler A, Fuchs K, Sieghart W, and Sperk G (2001). Distribution of the major γ-Aminobutyric acidA receptor subunits in the basal ganglia and associated limbic brain areas of the adult rat. J. Comp. Neurol. 433, 526–549. [DOI] [PubMed] [Google Scholar]
- 56.Studer R, von Boehmer L, Haenggi T, Schweizer C, Benke D, Rudolph U, and Fritschy JM (2006). Alteration of GABAergic synapses and gephyrin clusters in the thalamic reticular nucleus of GABAA receptor alpha3 subunit-null mice. Eur. J. Neurosci. 24, 1307–1315. [DOI] [PubMed] [Google Scholar]
- 57.Shen H, Sabaliauskas N, Sherpa A, Fenton AA, Stelzer A, Aoki C, and Smith SS (2010). A critical role for alpha4betadelta GABAA receptors in shaping learning deficits at puberty in mice. Science 327, 1515–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sabaliauskas N, Shen H, Homanics GE, Smith SS, and Aoki C (2012). Knockout of the γ-aminobutyric acid receptor subunit α4 reduces functional d-containing extrasynaptic receptors in hippocampal pyramidal cells at the onset of puberty. Brain Res. 1450, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ade KK, Janssen MJ, Ortinski PI, and Vicini S (2008). Differential Tonic GABA Conductances in Striatal Medium Spiny Neurons. J. Neurosci. 28, 1185–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Neill B, Patel JC, and Rice ME (2017). Characterization of optically and electrically evoked dopamine release in striatal slices from digenic knock-in mice with DAT-driven expression of channelrhodopsin. ACS Chem. Neurosci. 8, 310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olsen RW (2006). Picrotoxin-like channel blockers of GABAA receptors. Proc. Natl. Acad. Sci. USA 103, 6081–6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Juárez EH, Ochoa-Cortés F, Miranda-Morales M, Espinosa-Luna R, Montaño LM, Barajas-López C, and Barajas-López C (2014). Selectivity of antagonists for the Cys-loop native receptors for Ach, 5-HT and GABA in guinea-pig myenteric neurons. Auton. Autacoid Pharmacol. 34, 1–8. [DOI] [PubMed] [Google Scholar]
- 63.Jackson C, Bermudez I, and Beadle DJ (2002). Pharmacological properties of nicotinic acetylcholine receptors in isolated locusta migratoria neurons. Microsc. Res. Tech. 56, 249–255. [DOI] [PubMed] [Google Scholar]
- 64.Zhou FM, Liang Y, and Dani JA (2001). Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat. Neurosci. 4, 1224–1229. [DOI] [PubMed] [Google Scholar]
- 65.Rice ME, and Cragg SJ (2004). Nicotine amplifies reward-related dopamine signals in striatum. Nat. Neurosci. 7, 583–584. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H, and Sulzer D (2004). Frequency-dependent modulation of dopamine release by nicotine. Nat. Neurosci. 7, 581–582. [DOI] [PubMed] [Google Scholar]
- 67.Patel JC, Rossignol E, Rice ME, and Machold RP (2012). Opposing regulation of striatal dopamine release and exploratory motor behavior by forebrain and brainstem cholinergic inputs. Nat. Commun. 3, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roberts BM, Doig NM, Brimblecombe KR, Lopes EF, Siddorn RE, Threlfell S, Connor-Robson N, Bengoa-Vergniory N, Pasternack N, Wade-Martins R, et al. (2020). GABA uptake transporters support dopamine release in dorsal striatum with maladaptive downregulation in a parkinsonism model. Nat. Commun. 11, 4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sulzer D, Joyce MP, Lin L, Geldwert D, Haber SN, Hattori T, and Rayport S (1998). Dopamine neurons make glutamatergic synapses in vitro. J. Neurosci. 18, 4588–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, and Rayport S (2004). Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J. Neurosci. 24, 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chuhma N, Choi WY, Mingote S, and Rayport S (2009). Dopamine neuron glutamate cotransmission: frequency-dependent modulation in the mesoventromedial projection. Neuroscience 164, 1068–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stuber GD, Hnasko TS, Britt JP, Edwards RH, and Bonci A (2010). Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J. Neurosci. 30, 8229–8233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Koós T, and Koos T (2010). Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J. Neurosci. 30, 7105–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trudeau LE, Hnasko TS, Wallén-Mackenzie A, Morales M, Rayport S, and Sulzer D (2014). The multilingual nature of dopamine neurons. Prog. Brain Res. 211, 141–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eskenazi D, Malave L, Mingote S, Yetnikoff L, Ztaou S, Velicu V, Rayport S, and Chuhma N (2021). Dopamine neurons that cotransmit glutamate, from synapses to circuits to behavior. Front. Neural Circ. 15, 665386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silm K, Yang J, Marcott PF, Asensio CS, Eriksen J, Guthrie DA, Newman AH, Ford CP, and Edwards RH (2019). Synaptic Vesicle Recycling Pathway Determines Neurotransmitter Content and Release Properties. Neuron 102, 786–800.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang S, Qi J, Li X, Wang HL, Britt JP, Hoffman AF, Bonci A, Lupica CR, and Morales M (2015). Dopaminergic and glutamatergic microdomains in a subset of rodent mesoaccumbens axons. Nat. Neurosci. 18, 386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fortin GM, Ducrot C, Giguère N, Kouwenhoven WM, Bourque M-J, Pacelli C, Varaschin RK, Brill M, Singh S, Wiseman PW, and Trudeau LÉ (2019). Segregation of dopamine and glutamate release sites in dopamine neuron axons: regulation by striatal target cells. Faseb. J. 33, 400–417. [DOI] [PubMed] [Google Scholar]
- 79.Cai Y, and Ford CP (2018). Dopamine Cells Differentially Regulate Striatal Cholinergic Transmission across Regions through Corelease of Dopamine and Glutamate. Cell Rep. 25, 3148–3157.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chuhma N, Mingote S, Yetnikoff L, Kalmbach A, Ma T, Ztaou S, Sienna A-C, Tepler S, Poulin J-F, Ansorge M, et al. (2018). Dopamine neuron glutamate cotransmission evokes a delayed excitation in lateral dorsal striatal cholinergic interneurons. Elife 7, e39786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chuhma N, Mingote S, Moore H, and Rayport S (2014). Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron 81, 901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wieland S, Du D, Oswald MJ, Parlato R, Köhr G, and Kelsch W (2014). Phasic dopaminergic activity exerts fast control of cholinergic interneuron firing via sequential NMDA, D2, and D1 receptor activation. J. Neurosci. 34, 11549–11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai Y, Nielsen BE, Boxer EE, Aoto J, and Ford CP (2021). Loss of nigral excitation of cholinergic interneurons contributes to parkinsonian motor impairments. Neuron 109, 1137–1149.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lozovaya N, Eftekhari S, Cloarec R, Gouty-Colomer LA, Dufour A, Riffault B, Billon-Grand M, Pons-Bennaceur A, Oumar N, Burnashev N, et al. (2018a). GABAergic inhibition in dual-transmission cholinergic and GABAergic striatal interneurons is abolished in Parkinson’s disease. Nat. Commun. 9, 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lozovaya N, Ben-Ari Y, and Hammond C (2018b). Striatal dual cholinergic/GABAergic transmission in Parkinson disease: friends or foes? Cell Stress 2, 147–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith AD, and Bolam JP (1990). The neural network of the basal ganglia as revealed by the study of synaptic connections of identified neurones. Trends Neurosci. 13, 259–265. [DOI] [PubMed] [Google Scholar]
- 87.Assous M, Faust TW, Assini R, Shah F, Sidibe Y, and Tepper JM (2018). Identification and characterization of a novel spontaneously active bursty GABAergic interneuron in the mouse striatum. J. Neurosci. 38, 5688–5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tepper JM, Koós T, Ibanez-Sandoval O, Tecuapetla F, Faust TW, and Assous M (2018). Heterogeneity and diversity of striatal GABAergic interneurons: Update 2018. Front. Neuroanat. 12, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bevan MD, Booth PA, Eaton SA, and Bolam JP (1998). Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. J. Neurosci. 18, 9438–9452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Smith Y, Bevan MD, Shink E, and Bolam JP (1998). Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 86, 353–387. [DOI] [PubMed] [Google Scholar]
- 91.Breton JM, Charbit AR, Snyder BJ, Fong PTK, Dias EV, Himmels P, Lock H, and Margolis EB (2019). Relative contributions and mapping of ventral tegmental area dopamine and GABA neurons by projection target in the rat. J. Comp. Neurol. 527, 916–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vachez YM, Tooley JR, Abiraman K, Matikainen-Ankney B, Casey E, Earnest T, Ramos LM, Silberberg H, Godynyuk E, Uddin O, et al. (2021). Ventral arkypallidal neurons inhibit accumbal firing to promote reward consumption. Nat. Neurosci. 24, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pereira DB, Schmitz Y, Mészáros J, Merchant P, Hu G, Li S, Henke A, Lizardi-Ortiz JE, Karpowicz RJ Jr., Morgenstern TJ, et al. (2016). Fluorescent false neurotransmitter reveals functionally silent dopamine vesicle clusters in the striatum. Nat. Neurosci. 19, 578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu C, Kershberg L, Wang J, Schneeberger S, and Kaeser PS (2018). Dopamine Secretion Is Mediated by Sparse Active Zone-like Release Sites. Cell 172, 706–718.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Banerjee A, Imig C, Balakrishnan K, Kershberg L, Lipstein N, Uronen RL, Wang J, Cai X, Benseler F, Rhee JS, et al. (2022). Molecular and functional architecture of striatal dopamine release sites. Neuron 110, 248–265.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hannan S, Minere M, Harris J, Izquierdo P, Thomas P, Tench B, and Smart TG (2020). GABA A R isoform and subunit structural motifs determine synaptic and extrasynaptic receptor localization. Neuropharmacology 169, 107540. [DOI] [PubMed] [Google Scholar]
- 97.Holly EN, Davatolhagh MF, España RA, and Fuccillo MV (2021). Striatal low-threshold spiking interneurons locally gate dopamine. Curr. Biol. 31, 4139–4147.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kirmse K, Dvorzhak A, Kirischuk S, and Grantyn R (2008). GABA transporter 1 tunes GABAergic synaptic transmission at output neurons of the mouse neostriatum. J. Physiol. 586, 5665–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wójtowicz AM, Dvorzhak A, Semtner M, and Grantyn R (2013). Reduced tonic inhibition in striatal output neurons from Huntington mice due to loss of astrocytic GABA release through GAT-3. Front. Neural Circ. 7, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, and Lee CJ (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796. [DOI] [PubMed] [Google Scholar]
- 101.Lee M, McGeer EG, and McGeer PL (2011). Mechanisms of GABA release from human astrocytes. Glia 59, 1600–1611. [DOI] [PubMed] [Google Scholar]
- 102.Yoon B-E, and Lee CJ (2014). GABA as a rising gliotransmitter. Front. Neural Circ. 8, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mingote S, Chuhma N, Kusnoor SV, Field B, Deutch AY, and Rayport S (2015). Functional Connectome Analysis of Dopamine Neuron Glutamatergic Connections in Forebrain Regions. J. Neurosci. 35, 16259–16271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Papathanou M, Creed M, Dorst MC, Bimpisidis Z, Dumas S, Pettersson H, Bellone C, Silberberg G, Lüscher C, and Wallén-Mackenzie Å (2018). Targeting VGLUT2 in mature dopamine neurons decreases mesoaccumbal glutamatergic transmission and identifies a role for glutamate co-release in synaptic plasticity by increasing baseline AMPA/NMDA ratio. Front. Neural Circ. 12, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hirasawa H, Betensky RA, and Raviola E (2012). Co-release of dopamine and GABA from a retinal dopaminergic neuron. J. Neurosci. 38, 13281–13291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sgobio C, Wu J, Zheng W, Chen X, Pan J, Salinas AG, Davis MI, Lovinger DM, and Cai H (2017). Aldehyde dehydrogenase 1-positive nigrostriatal dopaminergic fibers exhibit distinct projection pattern and dopamine release dynamics at mouse dorsal striatum. Sci. Rep. 7, 5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, Chan CS, Dombeck DA, Deisseroth K, and Awatramani R (2018). Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat. Neurosci. 21, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu J, Kung J, Dong J, Chang L, Xie C, Habib A, Hawes S, Yang N, Chen V, Liu Z, et al. (2019). Distinct Connectivity and Functionality of Aldehyde Dehydrogenase 1a1-Positive Nigrostriatal Dopaminergic Neurons in Motor Learning. Cell Rep. 28, 1167–1181.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Carmichael K, Sullivan B, Lopez E, Sun L, and Cai H (2021). Diverse midbrain dopaminergic neuron subtypes and implications for complex clinical symptoms of Parkinson’s disease. Ageing Neurodegener. Dis. 1. 10.20517/and.2021.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pereira Luppi M, Azcorra M, Caronia-Brown G, Poulin JF, Gaertner Z, Gatica S, Moreno-Ramos OA, Nouri N, Dubois M, Ma YC, et al. (2021). Sox6 expression distinguishes dorsally and ventrally biased dopamine neurons in the substantia nigra with distinctive properties and embryonic origins. Cell Rep. 37, 109975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Birgner C, Nordenankar K, Lundblad M, Mendez JA, Smith C, le Grevès M, Galter D, Olson L, Fredriksson A, Trudeau LE, et al. (2010). VGLUT2 in dopamine neurons is required for psychostimulant-induced behavioral activation. Proc. Natl. Acad. Sci. USA 107, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, and Edwards RH (2010). Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron 65, 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hnasko TS, and Edwards RH (2012). Neurotransmitter corelease: mechanism and physiological role. Annu. Rev. Physiol. 74, 225–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Aguilar JI, Dunn M, Mingote S, Karam CS, Farino ZJ, Sonders MS, Choi SJ, Grygoruk A, Zhang Y, Cela C, et al. (2017). Neuronal Depolarization Drives Increased Dopamine Synaptic Vesicle Loading via VGLUT. Neuron 95, 1074–1088.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Palij P, Bull DR, Sheehan MJ, Millar J, Stamford J, Kruk ZL, and Humphrey PP (1990). Presynaptic regulation of dopamine release in corpus striatum monitored in vitro in real time by fast cyclic voltammetry. Brain Res. 509, 172–174. [DOI] [PubMed] [Google Scholar]
- 116.Patel J, Mooslehner KA, Chan PM, Emson PC, and Stamford JA (2003). Presynaptic control of striatal dopamine neurotransmission in adult vesicular monoamine transporter 2 (VMAT2) mutant mice. J. Neurochem. 85, 898–910. [DOI] [PubMed] [Google Scholar]
- 117.Rhodes KJ, and Trimmer JS (2006). Antibodies as valuable neuroscience research tools versus reagents of mass distraction. J. Neurosci. 26, 8017–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fritschy JM (2008). Is my antibody-staining specific? How to deal with pitfalls of immunohistochemistry. Eur. J. Neurosci. 28, 2365–2370. [DOI] [PubMed] [Google Scholar]
- 119.Frevert CW, Johnson B, and Stahl WL (2014). Pathobiology of Human Disease. In Immunohistochemistry: Antibody Specificity (Academic Press; ), pp. 3807–3816. [Google Scholar]
- 120.Baude A, Molnár E, Latawiec D, McIlhinney RA, and Somogyi P (1994). Synaptic and nonsynaptic localization of the GluRl subunit of the AMPA-type excitatory amino acid receptor in the rat cerebellum. J. Neurosci. 14, 2830–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nusser Z, Roberts JD, Baude A, Richards JG, Sieghart W, and Somogyi P (1995). Immunocytochemical localization of the alpha 1 and beta 2/3 subunits of the GABAA receptor in relation to specific GABAergic synapses in the dentate gyrus. Eur. J. Neurosci. 7, 630–646. [DOI] [PubMed] [Google Scholar]
- 122.Berggaard N, Witter MP, and van der Want JJL (2019). GABAA Receptor Subunit α3 in Network Dynamics in the Medial Entorhinal Cortex. Front. Syst. Neurosci. 13, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zachry JE, Nolan SO, Brady LJ, Kelly SJ, Siciliano CA, and Calipari ES (2021). Sex differences in dopamine release regulation in the striatum. Neuropsychopharmacology 46, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Becker JB, and Koob GF (2016). Sex differences in animal models: focus on addiction. Pharmacol. Rev. 68, 242–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Calipari ES, Juarez B, Morel C, Walker DM, Cahill ME, Ribeiro E, Roman-Ortiz C, Ramakrishnan C, Deisseroth K, Han M-H, and Nestler EJ (2017). Dopaminergic dynamics underlying sex-specific cocaine reward. Nat. Commun. 8, 13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Madisen L, Mao T, Koch H, Zhuo JM, Berenyi A, Fujisawa S, Hsu YWA, Garcia AJ 3rd, Gu X, Zanella S, et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 15, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, and Tomac AC (2006). Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis 44, 383–390. [DOI] [PubMed] [Google Scholar]
- 128.Paxinos G, and Franklin KBJ (2008). The Mouse Brain in Stereotaxic Coordinates (Academic Press; ). [Google Scholar]
- 129.Hikima T, Lee CR, Witkovsky P, Chesler J, Ichtchenko K, and Rice ME (2021). Activity-dependent somatodendritic dopamine release in the substantia nigra autoinhibits the releasing neuron. Cell Rep. 35, 108951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mancini M, Patel JC, Affinati AH, Witkovsky P, and Rice ME (2022). Leptin promotes striatal dopamine release via cholinergic interneurons and regionally distinct signaling pathways. J. Neurosci. 42, 6668–6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ordaz RP, Garay E, Limon A, Pérez-Samartín A, Sánchez-Gómez MV, Robles-Martínez L, Cisneros-Mejorado A, Matute C, and Arellano RO (2021). GABAA receptors expressed in oligodendrocytes cultured from the neonatal rat contain α3 and γ1 subunits and present differential functional and pharmacological properties. Mol. Pharm. 99, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Aoki C, Sabaliauskas N, Chowdhury T, Min J-Y, Colacino AR, Laurino K, and Barbarich-Marsteller NC (2012). Adolescent female rats exhibiting activity-based anorexia express elevated levels of GA-BA(A) receptor α4 and δ subunits at the plasma membrane of hippocampal CA1 spines. Synapse 66, 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Aoki C, Chen YW, Chowdhury TG, and Piper W (2018). α4βδ-GABAA receptors in dorsal hippocampal CA1 of adolescent female rats traffic to the plasma membrane of dendritic spines following voluntary exercise and contribute to protection of animals from activity-based anorexia through localization at excitatory synapses. J. Neurosci. Res. 96, 1450–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jechlinger M, Pelz R, Tretter V, Klausberger T, and Sieghart W (1998). Subunit composition and quantitative importance of hetero-oligomeric receptors: GABAA receptors containing alpha6 subunits. J. Neurosci. 18, 2449–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Peng Z, Hauer B, Mihalek RM, Homanics GE, Sieghart W, Olsen RW, and Houser CR (2002). GABA(A) receptor changes in delta subunit-deficient mice: Altered expression of alpha4 and gamma2 subunits in the forebrain. J. Comp. Neurol. 446, 179–197. [DOI] [PubMed] [Google Scholar]
- 136.Patel JC, and Rice ME (2013). Monitoring axonal and somatodendritic dopamine release using fast-scan cyclic voltammetry in brain slices. Methods Mol. Biol. 964, 243–273. [DOI] [PubMed] [Google Scholar]
- 137.Patel JC (2016). Voltammetry: Electrochemical detection of neurotransmitters in the brain. In Encyclopedia of Life Sciences (eLS) (John Wiley & Sons, Ltd; ). [Google Scholar]
- 138.Walker QD, Rooney MB, Wightman RM, and Kuhn CM (2000). Dopamine release and uptake are greater in female than male rat striatum as measured by fast cyclic voltammetry. Neuroscience 95, 1061–1070. [DOI] [PubMed] [Google Scholar]
- 139.Costa KM, Schenkel D, and Roeper J (2021). Sex-dependent alterations in behavior, drug responses and dopamine transporter expression in heterozygous DAT-cre mice. Sci. Rep. 11, 3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen BT, Avshalumov MV, and Rice ME (2001). H2O2 is a novel, endogenous modulator of synaptic dopamine release. J. Neurophysiol. 85, 2468–2476. [DOI] [PubMed] [Google Scholar]
- 141.Longo F, Mancini M, Ibraheem PL, Aryal S, Mesini C, Patel JC, Penhos E, Rahman N, Mamcarz M, Santini E, et al. (2021). Cell-type-specific disruption of PERK-eIF2α signaling in dopaminergic neurons alters motor and cognitive function. Mol. Psychiatr. 26, 6427–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.