

Abstract
Cancer cells rewire metabolism to favour the generation of specialized metabolites that support tumour growth and reshape the tumour microenvironment1,2. Lysine functions as a biosynthetic molecule, energy source, and antioxidant3–5, but little is known about its pathological role in cancer. Here, we show that glioblastoma stem cells (GSCs) reprogram lysine catabolism through the upregulation of lysine transporter SLC7A2 and crotonyl-CoA-producing enzyme GCDH (glutaryl-CoA dehydrogenase) with downregulation of crotonyl-CoA hydratase ECHS1 (enoyl-CoA hydratase, short chain 1), leading to accumulation of intracellular crotonyl-CoA and histone H4 lysine crotonylation (Kcr). Reduction of histone Kcr by either genetic manipulation or lysine restriction impaired tumour growth. In the nucleus, GCDH interacts with crotonyltransferase CBP to promote histone Kcr. Loss of histone Kcr promotes immunogenic cytosolic dsRNA and dsDNA generation through enhanced H3K27ac, which stimulates RNA sensor MDA5 and DNA sensor cGAS to boost type I interferon (IFN) signalling, leading to compromised GSC tumourigenic potential and elevated CD8+ T cell infiltration. A lysine-restricted (LR) diet synergized with MYC inhibition or anti-PD-1 therapy to slow tumour growth. Collectively, GSCs co-opt lysine uptake and degradation to shunt production of crotonyl-CoA, remodelling the chromatin landscape to evade IFN-induced intrinsic effects on GSC maintenance and extrinsic effects on immune response.
INTRODUCTION
Glioblastoma (GBM) displays an immune suppressive microenvironment that dampens T cell infiltration, activation, and function, also limiting the efficacy of immunotherapy6. Within GBMs, GSCs self-renew, differentiate, and contribute to therapeutic resistance1,7,8. Amino acid metabolism modulates T cell activation and function, and dietary amino acid interventions limit tumour growth in vivo2,9,10. Lysine catabolism produces bioactive intermediate metabolites, including crotonyl-CoA3–5. Crotonyl-CoA is the precursor for histone Kcr to regulate the fates of embryonic/neural stem cells by remodelling the local chromatin environment11,12, but its role in cancer remains unclear. Although lysine is an essential amino acid, LR diets are established therapy for multiple inborn errors of metabolism, including glutaric acidemia type I and pyridoxine-dependent epilepsy13–16.
RESULTS
GSCs avidly uptake lysine through SLC7A2
We interrogated a public dataset17 with a comparative metabolomic analysis of 40 gene sets18, which revealed enrichment of pathways related to lysine, folate, pyrimidine, purine, arginine metabolism in GSCs compared to matched differentiated GBM cells (DGCs) (Fig. 1a, Extended Data Fig. 1a). Folate, pyrimidine and purine metabolism maintain GSCs and promote GBM growth19–21. Arginine deprivation alters the immune microenvironment of a non-arginine-auxotrophic cellular background and improves GBM treatment22. Lysine catabolic activity was increased in classical GBM relative to non-tumour brain tissue or other subtypes, and inversely correlated with patient survival (Extended Data Fig. 1b, c). Next, we used mass spectrometry (MS) to compare the levels of free amino acids in patient-derived GSCs, matched DGCs, and neural stem cells (NSCs) (Fig. 1b, Supplementary Table 1). Lysine was consistently increased in GSCs (Supplementary Table 2). The half-maximal effective concentration (EC50) of lysine for GSC growth was 0.1–0.2 mM (Fig. 1c). Thus, we controlled L-lysine culture concentrations (0.2, 0.8, and 2 mM) of cells and observed a concentration-dependent increase of cell proliferation specifically in GSCs, but not in DGCs or NSCs (Extended Data Fig. 1d–f). Impaired growth of GSCs cultured in media containing 0.2 mM lysine was independent of protein synthesis, as shown by inactivated GCN2-eIF2α axis, a protein synthesis inhibitory checkpoint that senses amino acid deficiency, and unchanged puromycin incorporation rates (Extended Data Fig. 1g).
Fig. 1. GSCs upregulate lysine catabolism through SLC7A2.
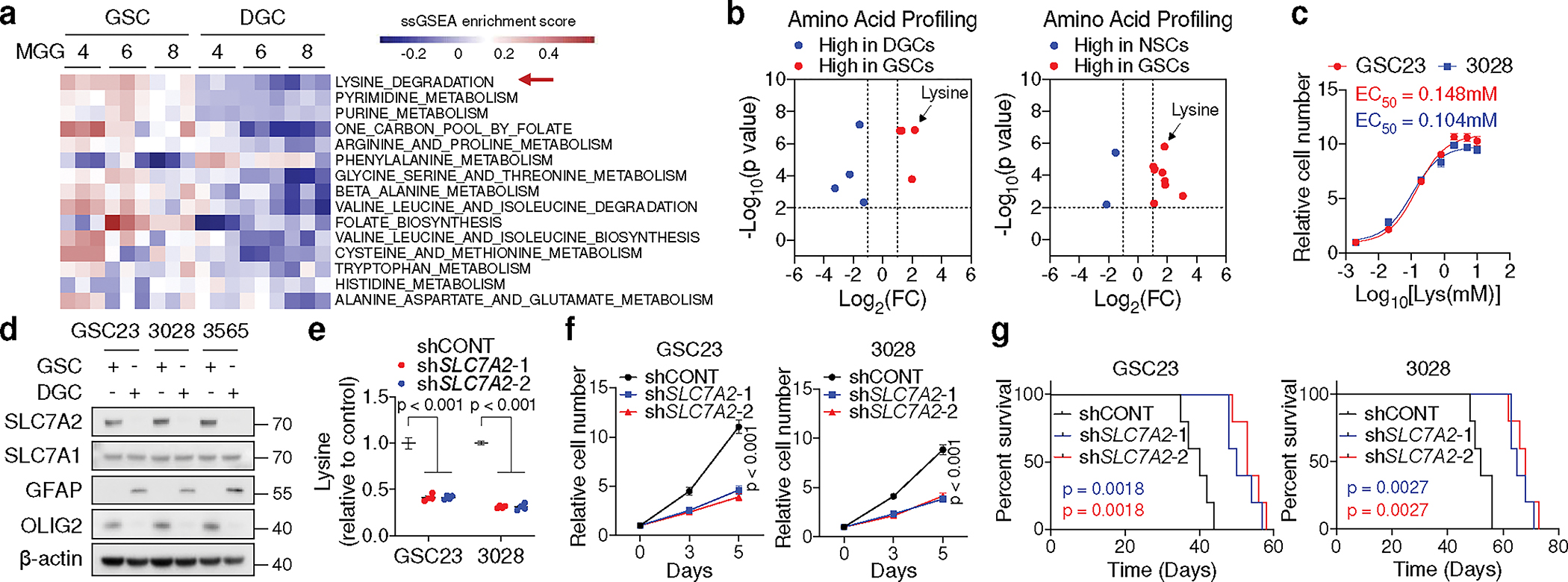
a, ssGSEA of metabolic gene sets from KEGG database in three GSCs (MGG4, MGG6, MGG8) and matched DGCs. Each cell contains 3 replicates from GSE54791.
b, Volcano plots summarizing intracellular amino acid levels across GSCs, DGCs, and NSCs from 5 biological replicates. Fold changes in amino acid levels relative to the average of GSCs are displayed.
c, Relative cell number of two GSCs cultured in media with different doses (0.002, 0.02, 0.2, 0.8, 2, 5, 10 mM) of L-lysine for 5 days.
d, IB analysis of matched GSCs and DGCs.
e, f, Relative intracellular lysine levels (n = 4 biologically independent samples, e) and cell proliferation (f) of GSCs with or without SLC7A2 KD.
g, Kaplan-Meier plots showing survival of immunocompromised mice bearing intracranial tumours from GSCs with or without SLC7A2 KD (n = 5).
Data are presented from three independent experiments in c and f. Representative of two independent experiments in d. In c, e and f, data are presented as mean ± SEM. Two-tailed unpaired t test for b, one-way ANOVA followed by multiple comparisons with adjusted p values for e, two-way ANOVA followed by multiple comparisons with adjusted p values for f, log-rank test for g.
Extracellular lysine and arginine are transported via SLC7A1, SLC7A2, and SLC7A32. SLC7A3 was undetectable in GBMs across two transcriptomic datasets17,23. In cancer, SLC7A1 preferentially transports arginine3. Increased expression and an active promoter of SLC7A2, but not SLC7A1, were observed in GSCs compared to both DGCs and NSCs (Fig. 1d, Extended Data Fig. 1h–j). SLC7A2 protein was exclusively accumulated on GSC membrane, as shown by immunofluorescence (IF) (Extended Data Fig. 1k). MS-based analysis identified that SLC7A2 knockdown (KD) in two GSCs dramatically reduced the intracellular levels of lysine, rather than other amino acids (Fig. 1e, Extended Data Fig. 1l–n, Supplementary Table 3). SLC7A2 KD impaired GSC growth and tumourigenesis (Fig. 1f, g). Thus, GSCs upregulate SLC7A2 to increase lysine uptake and tumour growth.
Lysine catabolism generates crotonyl-CoA
The saccharopine pathway is considered the major pathway of lysine catabolism that ultimately yields two acetyl-CoA units (Fig. 2a). In line with GSC-specific expression of SLC7A2, 13C6-lysine tracing demonstrated an increased conversion of lysine to acetyl-CoA in GSCs compared to DGCs (Extended Data Fig. 2a). Interrogation of TCGA GBM dataset24 revealed that SLC7A2, AASS, and GCDH were amplified or highly expressed in approximately 30% of GBM cases (Extended Data Fig. 2b). GCDH mRNA and lysine catabolic activity were increased in GBM samples compared to cerebral cortex tissues (Extended Data Fig. 2c). Further, GCDH was highly expressed in classical GBM, although its expression was not associated with EGFR amplification (Extended Data Fig. 2d–g). We next analysed single-cell RNA sequencing (scRNA-seq) data25 of 65,655 early-passage GSCs from 24 patients and 14,207 malignant tumour cells from 7 GBM tumours (Extended Data Fig. 2h). A logistic regression classifier was trained to find GSC-like tumour cells and differentiated tumour cells within GBM tumours (Extended Data Fig. 2i, j). The GSC-like tumour pool was enriched for a gene signature related to lysine degradation (Extended Data Fig. 2k). The differentiated tumour cells expressed differentiation markers, such as GFAP (Extended Data Fig. 2l). GCDH expression was increased in GSCs compared to DGCs and NSCs (Fig. 2b, c, Extended Data Fig. 2m). In contrast, ECHS1 was deleted in approximately 6% of GBM cases, and exhibited lower expression in classical GBM samples (Extended Data Fig. 2b–d). ECHS1 displayed decreased expression in GSCs (Fig. 2b, c, Extended Data Fig. 2m).
Fig. 2. GSCs reprogram lysine catabolism to promote Kcr and tumourigenesis.
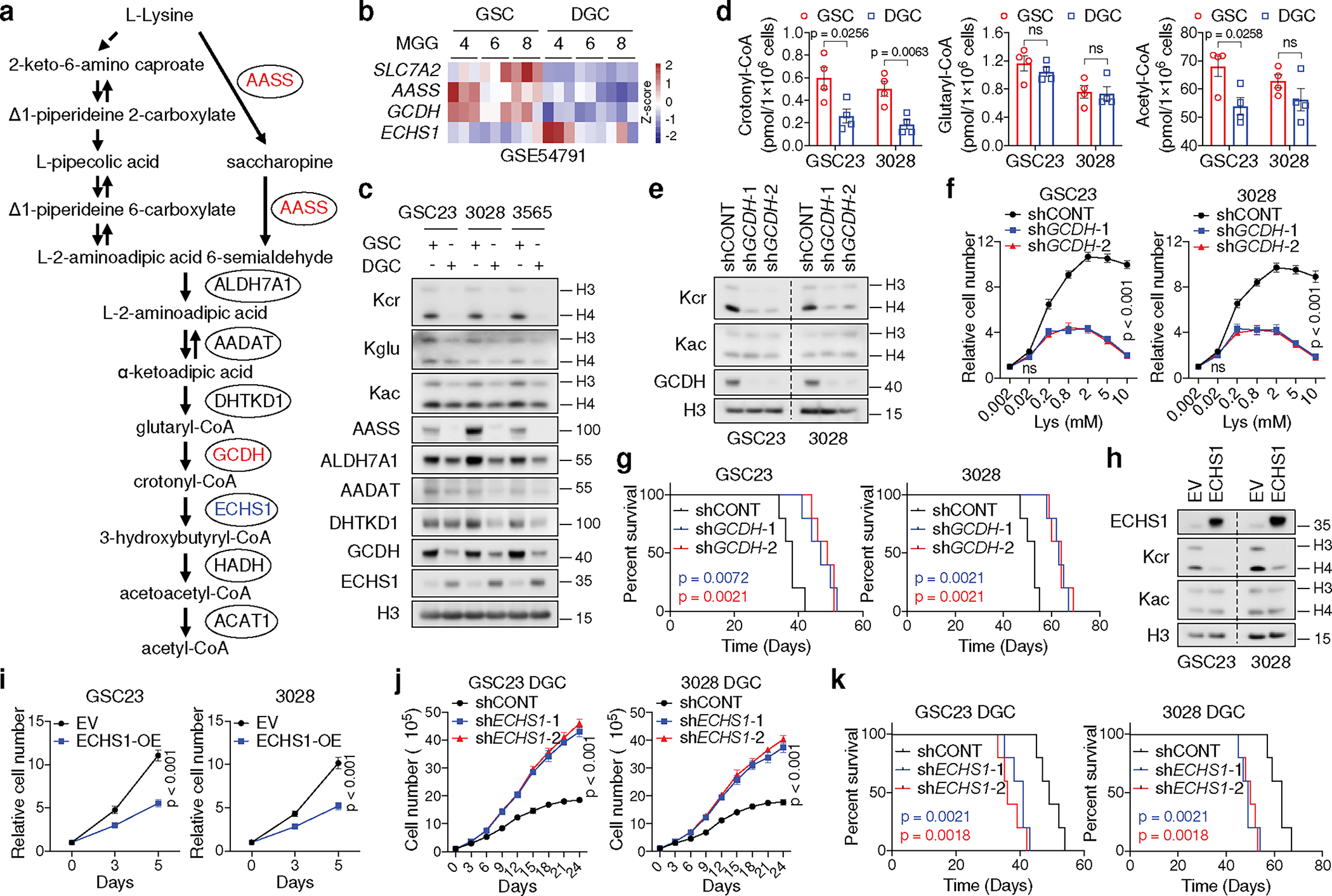
a, Diagram showing lysine catabolism pathway.
b, Heatmap summarizing gene expression in matched GSCs and DGCs.
c, IB analysis of matched GSCs and DGCs.
d, Intracellular acyl-CoAs (n = 4 biologically independent samples) in GSCs and DGCs.
e, IB analysis of GSCs cultured in typical media.
f, Relative cell number of two GSCs with or without GCDH KD cultured in media with increasing doses of L-lysine for 5 days.
g, Kaplan-Meier survival plots of immunocompromised mice as indicated (n = 5).
h, i, IB (h) and cell proliferation (i) of two GSCs with or without ECHS1 overexpression.
j, Cumulative cell number of two DGCs with or without ECHS1 KD.
k, Kaplan-Meier survival plots of immunocompromised mice as indicated (n = 5). Representative of two independent experiments in c, e and h. Data are presented from three independent experiments in f, i and j. Data are presented as mean ± SEM in d, f, i and j. Two-tailed unpaired t test for d, two-way ANOVA followed by multiple comparisons with adjusted p values for f, i and j, log-rank test for g and k. ns, not significant.
We hypothesized that GSCs reprogram lysine catabolism to accumulate crotonyl-CoA through upregulation of GCDH and downregulation of ECHS1. Crotonyl-CoA acts as the precursor for histone Kcr catalysed by P300/CBP and MOF in mammalian cells26,27. ECHS1 hydrolyses crotonyl-CoA and negatively regulates Kcr28. We compared levels of histone Kcr, histone glutarylation (Kglu), and histone acetylation (Kac), as well as the enzymes related to lysine catabolism in GSCs and matched DGCs (Fig. 2c). Kcr, but not Kglu or Kac, was enhanced in GSCs, accompanied by increased intracellular crotonyl-CoA, but not glutaryl-CoA or short-chain fatty acids (SCFAs) based on MS (Fig. 2c, d, Extended Data Fig. 2n). Intracellular levels of acetyl-CoA were slightly elevated in GSCs compared to DGCs, likely due to different growth rates (Fig. 2d). Notably, Kcr was mainly enriched in histone H4 compared to H3 and located in the nucleus, as detected by a pan-anti-Kcr antibody, PTM-50111,12,26,27,29 (Fig. 2c, Extended Data Fig. 2o). The GCDH promoter region displayed enhanced H3K27ac occupancy (Extended Data Fig. 2p), and targeting GCDH impaired H4 Kcr to a greater degree than H3 Kcr, without affecting Kac (Fig. 2e). GCDH depletion abolished the enhanced cell growth induced by L-lysine supplementation (Fig. 2f). Targeting GCDH further restricted cell growth and promoted histone Kglu under culture media with more than fivefold increase of lysine compared to normal media (Fig. 2f, Extended Data Fig. 2q), consistent with the findings that GCDH-deficient mice exhibited impaired glutaryl-CoA breakdown and neuropathologic features when exposed to fivefold elevated dietary lysine15. Mice bearing intracranial xenografts of shGCDH-transduced GSCs survived longer than those transduced with a control shRNA (shCONT) (Fig. 2g). As expected, ECHS1 overexpression attenuated histone Kcr and proliferation in GSCs (Fig. 2h, i). In contrast, ECHS1 KD in GSCs only exhibited minor effects on cell growth (Extended Data Fig. 2r, s). To mimic the reduced levels of ECHS1 found in GSCs, we depleted ECHS1 in DGCs, as they have high basal ECHS1 expression. DGCs deficient for ECHS1 exhibited accelerated growth in vitro and in vivo (Fig. 2j, k). Collectively, GSCs reprogram lysine catabolism to promote histone Kcr and tumour growth through enhanced GCDH and reduced ECHS1 expression.
Kcr interventions affect IFN signalling
Catabolism of lysine or tryptophan generates crotonyl-CoA, catalysed by GCDH. Circulating crotonate can be converted into crotonyl-CoA by ACSS2. In addition, fatty acid β-oxidation produces crotonyl-CoA upon oxidation of butyryl-CoA, catalysed by ACADS, ACOX1 or ACOX311 (Fig. 3a). Only GCDH, but not other enzymes, was highly expressed in GSCs compared to DGCs and NSCs (Extended Data Fig. 3a, b). The expression of tryptophan transporter, SLC7A5, was either decreased or unchanged in GSCs compared to DGCs or NSCs, supporting a specific role for lysine instead of tryptophan as a metabolite source for GCDH (Extended Data Fig. 3c). Targeting GCDH only minimally impacted cell growth of DGCs and NSCs (Extended Data Fig. 3d–g). To further assess the source of crotonyl-CoA in GSCs, we examined the intracellular acyl-CoA levels by MS after knocking down key transporters or enzymes for crotonyl-CoA generation (Extended Data Fig. 3h). We observed a trend of nearly 50% reduction of crotonyl-CoA and slight decrease of acetyl-CoA after disruption of lysine catabolism (Extended Data Fig. 3i). GCDH depletion abolished increased Kcr induced by L-lysine supplementation (Fig. 3b). L-lysine supplementation enhanced intracellular crotonyl-CoA levels measured by MS, which was abrogated by GCDH KD (Fig. 3c). Disrupting lysine catabolism did not substantially affect butyryl-CoA production and overall cellular fatty acid metabolism despite minor variations (Fig. 3c, Extended Data Fig. 3j). Therefore, increased Kcr levels in GSCs can be attributed to accumulated intracellular crotonyl-CoA, generated from reprogrammed lysine catabolism, but not other tested acyl-CoAs or SCFAs.
Fig. 3. Perturbation of lysine catabolism affects type I IFN signalling.
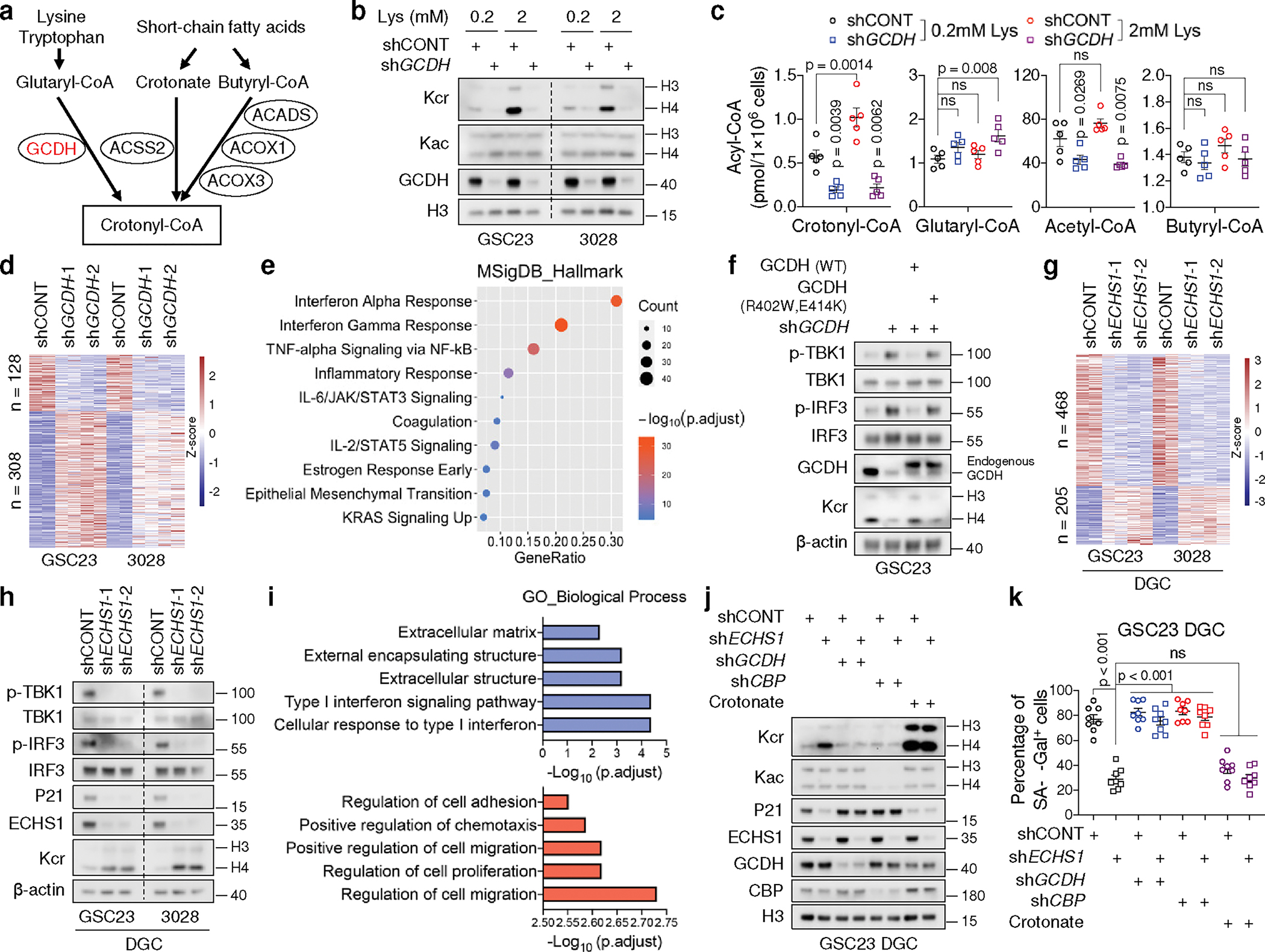
a, Key metabolic pathways that produce crotonyl-CoA. Red, upregulation.
b, c, IB (b) and intracellular acyl-CoAs (n = 5 biologically independent samples from GSC23, c) of control and GCDH KD GSCs cultured in media with indicated concentrations of L-lysine.
d, Heatmap summarizing DEGs upon GCDH KD in two GSCs.
e, Dot plot summarizing top 10 signalling pathways enriched in the upregulated genes upon GCDH KD in GSCs, ranked by adjusted p values.
f, IB analysis of control and 3’UTR-targeted shGCDH GSC23 with or without ectopic expression of WT, GCDH enzymatic mutant (R402W, E414K).
g, Heatmap summarizing DEGs upon ECHS1 KD in two early-passage DGCs.
h, IB analysis of two DGCs with or without ECHS1 KD.
i, GO enrichment analysis of downregulated (blue) and upregulated (red) genes upon ECHS1 KD in two DGCs, ranked by adjusted p values.
j, k, IB (j) and SA-β-Gal+ percentage (k) of DGCs with indicated gene depletion or 2mM crotonate supplementation in culture media for 1 week.
Representative of two independent experiments in b, f, h and j. Data are presented from three independent experiments in k. In c and k, data are presented as mean ± SEM. One-way ANOVA followed by multiple comparisons with adjusted p values for c and k. ns, not significant.
RNA-seq revealed that 308 genes were induced by GCDH depletion in two GSCs (Fig. 3d, Supplementary Table 4). GSEA and Gene Ontology (GO) analyses indicated enrichment of type I IFN signalling in the upregulated genes, which represent more than 70% of the differentially expressed genes (DEGs) upon GCDH loss (Fig. 3e, Extended Data Fig. 4a). As activation of type I IFN promotes cellular proliferation arrest30, quantifying the proliferation rate by 5-ethynyl-2’-deoxyuridine (EdU) incorporation confirmed the restrained growth of GCDH-depleted GSCs (Extended Data Fig. 4b). Increased IFN-stimulated genes (ISGs) upon GCDH KD are displayed in a heatmap (Extended Data Fig. 4c). IFN signalling can be initiated by RIG-I/MDA5 (encoded by DDX58/IFIH1) and cyclic GMP-AMP synthase (cGAS), which detect exogenous pathogens as well as endogenous dsRNA and cytosolic DNA31,32. Activation of either pathway stimulates TBK1-drived phosphorylation and activation of IFN regulatory factor (IRF) 3/7, which translocate to the nucleus and functions as a transcription factor (TF) to drive IFN expression30. DDX58/IFIH1 increased upon GCDH depletion (Extended Data Fig. 4c). Robust activation of cGAS-STING can trigger the production of senescence-associated secretory phenotype (SASP) factors, thereby promoting paracrine senescence and growth inhibition30. Concordantly, GCDH depletion raised the mRNA levels of multiple SASP factors (Extended Data Fig. 4c). Reintroduction of WT GCDH but not catalytically compromised GCDH (bearing mutations R402W and E414K)33 into GCDH KD GSC23 abolished the activation of the IFN pathway (Fig. 3f), suggesting the demand of GCDH catalytic activity for IFN signalling repression.
Targeting ECHS1 in DGCs accelerated proliferation and reduced senescence (Extended Data Fig. 4d, e). RNA-seq of two early-passage DGCs revealed that 468 genes were repressed and 205 genes were induced by ECHS1 loss (Fig. 3g, Supplementary Table 5). Contrary to the functions of GCDH in GSCs, silencing ECHS1 in DGCs eliminated the activation of IFN signalling, decreased the protein levels of senescence marker p21CIP1/WAF1, and increased Kcr (Fig. 3h), which was validated by GO and GSEA analyses of downregulated genes (Fig. 3i, Extended Data Fig. 4f). Cell migration and proliferation signatures were highly enriched in upregulated genes (Fig. 3i). To further determine the functional importance of Kcr, we investigated whether crotonate, a crotonyl-CoA precursor11,26, directly affected DGC senescence. Crotonate promoted histone Kcr but repressed senescence without altering Kac (Fig. 3j, k). In contrast, targeting GCDH or CBP abolished Kcr elevation and senescence evasion induced by ECHS1 depletion (Fig. 3j, k).
To examine the connection between lysine catabolism and IFN signalling, we augmented L-lysine concentrations in GSC culture media, revealing enrichment of IFN signalling in the downregulated genes, representing around 70% DEGs in two GSCs (Extended Data Fig. 4g–i, Supplementary Table 6). Among 278 downregulated genes induced by higher L-lysine, 181 genes designated as “Lysine catabolism-repressed genes” were inversely upregulated upon GCDH depletion in GSCs and enriched for cytokine-mediated IFN signalling pathways (Extended Data Fig. 4j, k, Supplementary Table 7). Collectively, GSCs reprogram lysine catabolism to modulate type I IFN signalling.
IFN signalling suppresses tumour growth by inducing senescence and inhibiting proliferation and by regulation of immune response30,31. Two days after GCDH loss, the expression of cyclin-dependent kinase inhibitor, p21CIP1/WAF1, did not change, but IFN signalling was activated (Extended Data Fig. 5a), indicating the enhanced IFN signalling appeared to be the cause of cell-cycle arrest and senescence. Enhanced secretion of IFNα and IFNβ were detected in the culture supernatant of GSCs with GCDH KD (Extended Data Fig. 5b). Persistent treatment of either IFNβ or the supernatant from GSCs with GCDH KD for 6 days inhibited GSC maintenance (Extended Data Fig. 5c, d). Blocking IFNα/β receptor subunit 2 (IFNAR2) using a neutralizing antibody largely abolished the effects induced by supernatant stimulation in recipient cells (Extended Data Fig. 5d). In support of these data, GCDH loss induced the accumulation of IFNα and repression of Kcr in GSC-derived intracranial tumours without affecting Kac and Kglu, accompanied by increased expression of human ISGs (Extended Data Fig. 5e, f). We compared the abundance of SOX2+ CD133+ GSCs in CD147+ human tumour cells in vivo (Extended Data Fig. 5g). Decreased GSC proportion was observed upon GCDH loss in GSC-derived intracranial tumours, which was largely restored by human IFNAR blocking antibody treatment (Extended Data Fig. 5h, i). Thus, lysine catabolism disruption-induced IFN signalling mediates GSC fate.
Nuclear-GCDH binds to CBP and boosts Kcr
GCDH most frequently co-localized with the mitochondrial marker COX IV, but >20% of total GCDH localized to nuclei in GSCs (Fig. 4a, b). Likewise, GCDH and Kcr were localized to cellular nuclei to various degrees in GBM surgical specimens, with enrichment in SOX2-positive GSC-like tumour cells (Extended Data Fig. 6a). Other than ECHS1, other lysine degrading enzymes partially localized to the nucleus (Extended Data Fig. 6b, c), suggesting a nuclear role of lysine catabolism. Next, we sought to determine whether nuclear-localized GCDH directly linked histone crotonyltransferases. 58 proteins were specifically identified by MS in the Flag-GCDH protein complex from nuclear fraction of 293T cells (Supplementary Table 8). CBP was detected in GCDH immunoprecipitates (Extended Data Fig. 6d). Two highly selective inhibitors of the CBP/P300 complex attenuated global Kcr levels in GSCs at low concentrations to a greater degree than Kac (Extended Data Fig. 6e). Although both CBP and P300 possess histone acetyltransferase activity predominantly towards H3, only CBP overexpression preferentially enhanced Kcr on H4 (Fig. 4c). GCDH interacted with CBP more strongly than P300 (Fig. 4c). Endogenous nuclear-GCDH interacted with CBP in GSCs, which was verified by in vitro interaction (Extended Data Fig. 6f, g). Crotonyl-CoA produced from glutaryl-CoA by GCDH was utilized by CBP for histone Kcr (Fig. 4d). L-lysine supplementation induced Kcr and cell growth were abolished by CBP depletion (Extended Data Fig. 6h, i). In contrast, targeting P300 in GSCs induced only a minor reduction in H4 Kcr compared to H3 (Extended Data Fig. 6j). To identified the amino acid sequences in GCDH that govern its nuclear translocation and interaction with CBP, we truncated the full-length GCDH (GCDH-FL) to generate vectors encoding amino acids:1–400, 1–380, and 1–360, which were designated fragments F1, F2, and F3, respectively. GCDH-FL and GCDH-F1, but not other fragments, associated with CBP and translocated into nuclei (Extended Data Fig. 6k, l). GCDH bearing a nuclear export signal (GCDH-NES) or nuclear localization signal (GCDH-NLS) was used as a negative or positive control (Extended Data Fig. 6k, l). Therefore, amino acids 380–400 of GCDH are required for its nuclear localization and direct interaction with CBP. Only nuclear-localized WT GCDH, but not the mutant, coupled with CBP promoted Kcr (Extended Data Fig. 6m). NLS-tagged GCDH more efficiently generated Kcr and promoted cell growth than WT GCDH or NES-tagged GCDH (Fig. 4e, f), suggesting crotonyl-CoA generated by nuclear GCDH complexes serves as a local source for CBP-dependent Kcr. MS and IB analyses indicated that H4K5cr, H4K8cr, and H4K12cr were enriched in GSCs and modulated in response to lysine catabolism interventions (Fig. 4g, h, Extended Data Fig. 6n, o, Supplementary Table 9). Thus, nuclear GCDH coupled with CBP is crucial for Kcr regulation.
Fig. 4. GCDH interacts with CBP in nucleus to modulate Kcr and confer MYC-dependence.
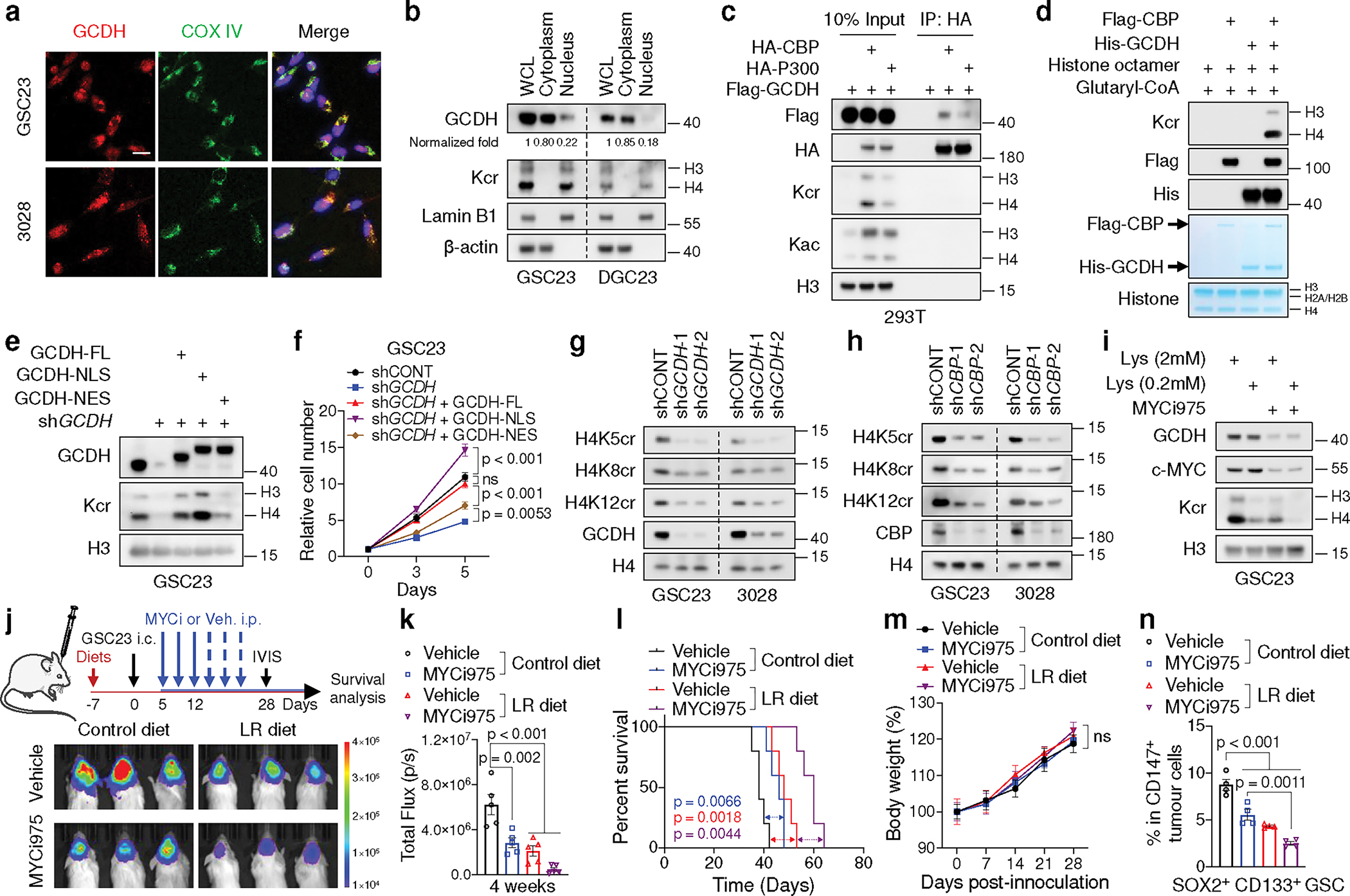
a, IF staining of GCDH in GSCs. Scale bar, 20 μm.
b, Nuclear localization of GCDH in GSCs and DGCs. Band intensity of GCDH was measured and normalized to β-actin or Lamin B1 based on the intensity of each lane after background subtraction, and the ratios are shown. WCL, whole cell lysate.
c, IB analysis of WCLs and IP of 293T cells transfected with indicated plasmids.
d, IB analysis of Kcr by in vitro crotonylation assay.
e, f, IB (e) and cell proliferation (f) of control and 3’UTR-targeted shGCDH GSC23 with or without ectopic expression of WT, NLS-tagged and NES-tagged GCDH.
g, h, IB analysis of GSCs with or without GCDH (g) or CBP (h) KD.
i, IB analysis of GSCs cultured in media with indicated concentrations of L-lysine and treated with or without 0.2 μM MYCi975 for 5 days.
j-n, In vivo bioluminescence imaging at 4 weeks post-transplantation (j), quantification of total flux (n = 5 biologically independent mice, k), Kaplan-Meier survival curves (n = 5, l), weekly mice weight (n = 5 biologically independent mice, m) and percentages of SOX2+ CD133+ GSCs (n = 4 biologically independent mice, n) from immunodeficient mice bearing GSC23-derived intracranial tumours with MYCi975, LR diet or the combination treatment.
Representative of two independent experiments for a-e and g-i. Data are presented from three independent experiments in f. In f, k, m and n, data are presented as mean ± SEM. Two-way ANOVA followed by multiple comparisons with adjusted p values for f and m, one-way ANOVA followed by multiple comparisons with adjusted p values for k and n, log-rank test for l. ns, not significant.
Specific inhibitors of GCDH are not currently available. Based on a computational biology framework “Lisa”34, the top 20 enriched TFs potentially responsible for the perturbation of DEGs between GSCs and DGCs/NSCs were used for analysis, including 12 common TFs (Extended Data Fig. 7a, b, Supplementary Table 10). To determine the upstream TFs that potentially bind the promoter of and regulate GCDH, a regulatory potential score was used to estimate the regulatory strength of TFs on GCDH (Extended Data Fig. 7c). Among the top 20 regulators for GCDH transcription, MYC was the only candidate predicted to regulate GCDH expression and confer selective dependencies in GSCs (Extended Data Fig. 7d). Indeed, MYC directly bound to the promoter region of GCDH harbouring a conserved MYC-binding element (Extended Data Fig. 7e, f). MYC mRNA and MYC-driven gene signatures positively correlated with the levels of GCDH (Extended Data Fig. 7g, h). MYC KD in GSCs reduced the steady-state mRNA levels of GCDH and GCDH nascent transcripts measured by nuclear run-on (NRO) RT-qPCR35, together with decreased GCDH and histone Kcr protein levels (Extended Data Fig. 7i, j). Targeting MYC by MYCi97536 inhibited the steady-state and nascent transcription of GCDH, as well as its protein levels in GSCs (Fig. 4i, Extended Data Fig. 7k). Combined lysine restriction with MYC inhibitor (MYCi) treatment synergistically impaired Kcr and GSC growth in vitro (Fig. 4i, Extended Data Fig. 7l).
To assess the effect of LR diet in vivo, healthy or tumour-bearing NOD.SCID γc-deficient (NSG) mice were randomized to a control diet (1.44% lysine, w/w) or a LR diet (0.144% lysine, w/w). The LR diet reproducibly caused about 55% decrease of serum lysine in healthy and tumour-bearing mice, along with more than 60% reduction of free lysine in tumour tissue (Extended Data Fig. 7m), similar to intracellular lysine changes induced by lysine restriction or SLC7A2 depletion in vitro (Extended Data Fig. 7n). The LR diet did not alter protein synthesis measured by O-Propargyl-puromycin (OP-puro) incorporation rates in vivo (Extended Data Fig. 7o). MYCi975 and LR diet displayed combinatorial benefit against tumour growth without causing additional discernible toxicity (Fig. 4j–m). Flow cytometric analysis and IF staining of tumour-bearing brains revealed that the LR diet additionally inhibited GSC maintenance and Kcr levels in tumour tissues without affecting GCDH and MYC expression in mice treated with MYCi975 (Fig. 4n, Extended Data Fig. 7p).
LR diet potentiates anti-tumour immunity
We hypothesized that crotonyl-CoA competes with acetyl-CoA as a CBP substrate. Indeed, crotonyl-CoA concentration-dependently repressed histone Kac and H3K27ac in vitro (Extended Data Fig. 8a). Histone Kcr signals exhibited a genome-wide decrease upon GCDH depletion in GSC23, together with enhanced H3K27ac signals from ChIP-seq (Fig. 5a). However, lysine catabolism-repressed genes (Extended Data Fig. 4j) did not appear to be direct targets of GCDH-mediated Kcr (Extended Data Fig. 8b), suggesting IFN signalling was an indirect effect of Kcr restriction. Increased H3K27ac was observed at their promoter regions (Extended Data Fig. 8b). 26774 GCDH-dependent Kcr peaks were lost after GCDH KD, displaying enhanced H3K27ac and decreased H3K9me3 (Fig. 5b). Specifically, 86.8% of GCDH-dependent Kcr peaks exhibited increased H3K27ac modifications (Extended Data Fig. 8c), mainly located at promoters (39.1%) and repetitive DNA or heterochromatin elements (25.8%) (Fig. 5c).
Fig. 5. Lysine catabolism disruption stimulates anti-tumour immunity via immunogenic retrotransposons.
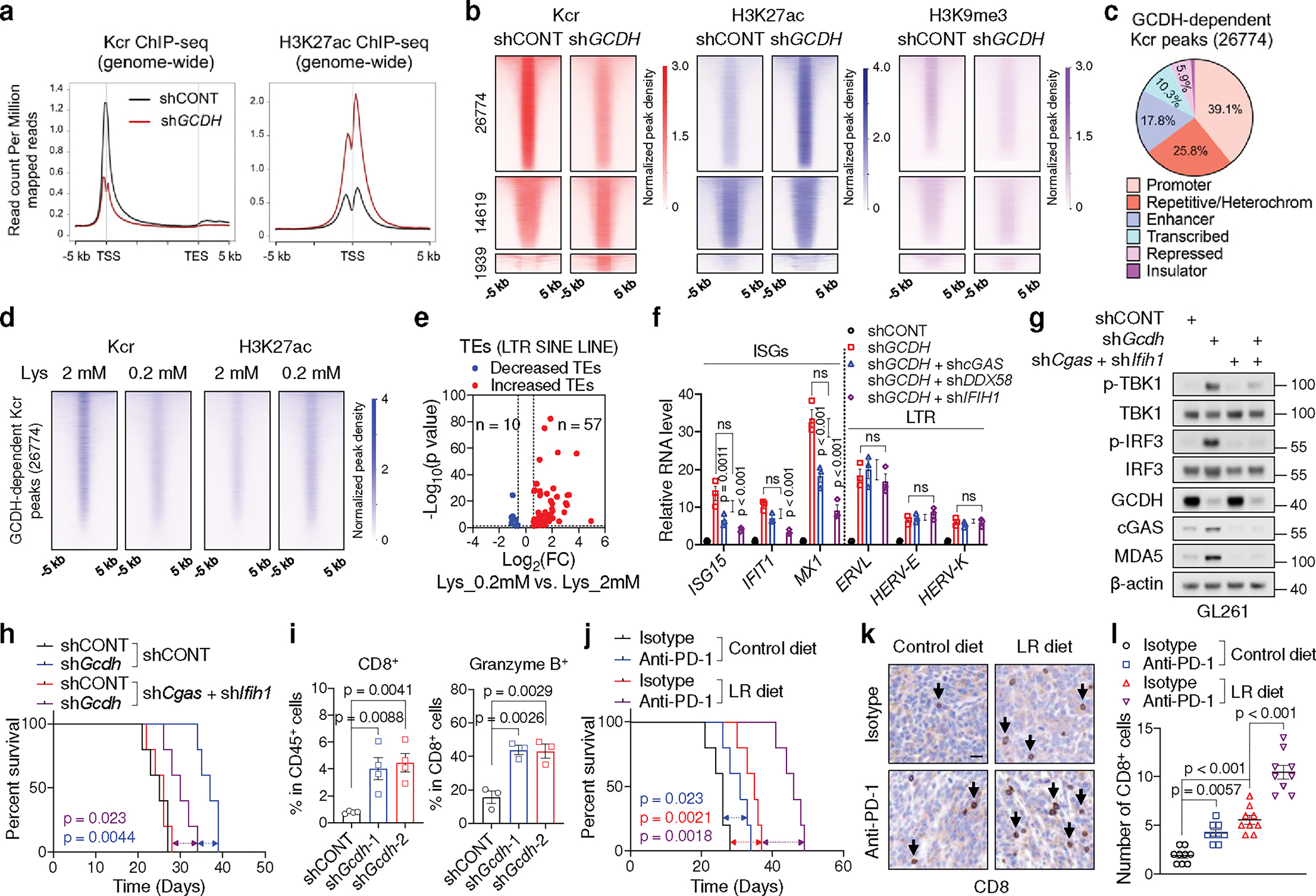
a, Genome-wide profiles of Kcr and H3K27ac in control and GCDH KD GSC23.
b, Heatmaps of Kcr, H3K27ac and H3K9me3 ChIP-seq signals sorted by differential Kcr peaks between control and GCDH KD GSC23. 26774 peaks exhibited reduced Kcr signals upon GCDH loss were named as GCDH-dependent Kcr peaks.
c, Genomic annotations of GCDH-dependent Kcr peaks by chromosome features.
d, Heatmaps of Kcr and H3K27ac ChIP-seq signals within GCDH-dependent Kcr peaks in GSC23 cultured in media with indicated concentrations of L-lysine.
e, Volcano plot showing transcript levels of TEs in GSCs cultured with 0.2 mM L-lysine versus 2 mM L-lysine, defined by adjusted p < 0.05 and fold change > 1.5.
f, RT-qPCR analysis of representative ISGs and LTR-containing ERVs in GSC23.
g, IB analysis in control and Gcdh KD GL261 with or without Cgas and Ifih1 KD.
h, Kaplan-Meier survival curves of immunocompetent mice bearing GL261-derived intracranial tumours (n = 5).
i, Flow cytometric analysis of CD8+ T cells (n = 4 biologically independent mice) and Granzyme B+ CD8+ T cells (n = 3 biologically independent mice) in GL261-derived intracranial tumours at day 14 post-injection.
j, Kaplan-Meier survival curves of immunocompetent mice bearing GL261-derived intracranial tumours (n = 5).
k, l, Representative IHC staining (k) and quantification (n = 3 biologically independent mice, l) of CD8+ T cells from indicated tumours at day 21 post-transplantation. Scale bar, 20 μm.
Data are presented from three independent experiments in f. Representative of two independent experiments for g. In f, i and l, data are presented as mean ± SEM. One-way ANOVA followed by multiple comparisons with adjusted p values for f, i and l, log-rank test for h and j. ns, not significant.
Transposable elements (TEs) are major repetitive DNA that account for about 50% of the human genome31,32. Apart from DNA transposons (which account for ~3% of the human genome), retrotransposons are the majority class of TEs, consisting of long terminal repeat (LTR)-based endogenous retroviruses (ERVs), non-LTR-based long/short interspersed nuclear elements (LINEs/SINEs)31,32. We found that attenuated Kcr binding sites induced by GCDH loss were localized to retrotransposons (Extended Data Fig. 8d). The signalling pathways related to RNA metabolic process and viral infections were enriched in the GCDH-dependent Kcr modified genes (Extended Data Fig. 8e). GCDH-dependent Kcr peaks showed enriched binding for immune regulators and zinc-finger proteins (Extended Data Fig. 8f). TEs are decorated by repressive H3K9me3 mark31. Enhanced H3K27ac and decreased H3K9me3 enrichment at repetitive DNA might compromise its integrity to generate TEs in GCDH-depleted GSCs. Upon GCDH loss, ChIP-seq confirmed the attenuation of Kcr in TEs along with enhanced H3K27ac modification (Extended Data Fig. 8g). Lysine restriction in GSCs also abolished histone Kcr and induced H3K27ac enrichment within GCDH-dependent Kcr loci, accompanied by increased transcription of retroelements from RNA-seq (Fig. 5d, e, Supplementary Table 11). GCDH loss or lysine restriction in GSCs triggered the transcription of TEs that displayed diminished histone Kcr and H4K12cr modification, increased H3K27ac, and decreased H3K9me3 occupancy (Extended Data Fig. 8h–o, Supplementary Table 12). Together, Kcr derived from reprogrammed lysine catabolism serves as competitive epigenetic machinery to limit immunogenic TEs through affecting H3K27ac and H3K9me3.
Consistent with the notion that retrotransposon transcription contributes to immunogenic cytosolic dsRNA and DNA generation and consequently IFN activation31,32, targeting GCDH induced the accumulation of cytosolic dsRNA and DNA in GSCs, in line with the enrichment of signalling related to dsRNA binding in the upregulated genes upon GCDH loss (Extended Data Fig. 9a, b). IP of isolated cytosolic dsRNA confirmed enhanced dsRNA generation from TE subtypes in GCDH KD cells (Extended Data Fig. 9c). Re-transfection of cytosolic dsRNA or DNA isolated from GCDH KD cells activated IFN signalling in recipient cells (Extended Data Fig. 9d). Experimentally increasing histone Kcr largely compromised the induction of TEs and IFN signalling, while restored cell proliferation in GCDH KD GSCs (Extended Data Fig. 9e–h). Abrogation of DNA sensor cGAS or RNA sensor MDA5, but not RIG-I, diminished ISG induction without altering LTR-containing ERV expression in GCDH KD GSCs (Fig. 5f). To directly assess whether GCDH loss-induced dsRNA was recognized by MDA5, we performed an MDA5-protection assay37, followed by RT-qPCR analysis of TEs. Crotonate supplementation reversed the promotion of GCDH silencing on MDA5-protected TEs (Extended Data Fig. 9i). We hypothesized that cytosolic DNA can be generated through the reverse transcription of TEs and recognized by cGAS32,38. As expected, DNA-containing dsDNA budding off the nucleus co-stained with cGAS clusters in GCDH KD GSC23 (Extended Data Fig. 9j). Treatment with reverse transcriptase inhibitor (RTi) zidovudine abrogated the accumulation of cytosolic dsDNA and retroelement cDNA, and partially repressed the activation of IFN signalling in GCDH KD GSC23 (Extended Data Fig. 9j–l). Simultaneous depletion of both cGAS and MDA5 in mouse glioma cells GL261 blocked IFN pathway activation induced by GCDH inhibition, and largely restored the in vivo tumour growth ability of Gcdh KD cells (Fig. 5g, h, Extended Data Fig. 10a, b). Thus, local loss of histone Kcr at retrotransposons promotes cytosolic dsRNA and DNA generation, which trigger MDA5/cGAS-dependent IFN signalling in GSCs.
Accumulated self-derived dsRNA or dsDNA upon dysregulated epigenetic modification enhance therapeutic benefits of immunotherapy31,32. The lysine catabolism-repressed signature was negatively associated with GCDH mRNA levels and positively correlated with CD4 T cell, CD8 T cell, and NK cell signatures39 in a GBM patient dataset40 (Extended Data Fig. 10c). GCDH and ECHS1 mRNA levels were negatively or positively correlated, respectively, with immune scores in TCGA GBM dataset (Extended Data Fig. 10d). Enhanced infiltration and activity of CD8+ T cells were observed in Gcdh KD GL261 tumours from immunocompetent hosts, consistent with the negative correlation between GCDH expression and CD8+ T cell infiltration scores (Fig. 5i, Extended Data Fig. 10e–g). The LR diet more efficiently slowed tumour growth and improved survival in immunocompetent hosts compared to that in NSG mice (Fig. 5j, Extended Data Fig. 10h–j). In line with the enhanced IFN signalling and functional CD8+ T cell infiltration upon GCDH loss, the LR diet augmented the efficacy of anti-PD-1 therapy to restrain tumour growth and promoted IFNγ+ CD8+ T cells infiltration (Fig. 5j–l, Extended Data Fig. 10i–k).
DISCUSSION
Here we show that GSCs reprogram lysine catabolism to propagate an immunosuppressive state. Lysine restriction renders GSC population vulnerable to synergistic therapy, setting the stage for translational studies in GBM (Extended Data Fig. 10l). Nuclear-GCDH bound CBP, which transferred crotonyl-CoA to target histones immediately upon its generation, although leaked crotonyl-CoA from mitochondria might also contribute to Kcr. Due to the extremely low intracellular levels and inherent instability of crotonyl-CoA, the dynamic balance between Kac and Kcr is more sensitive to the changes of crotonyl-CoA than acetyl-CoA.
Mechanisms that repress TEs are cell type- and developmental stage-specific, commonly involving DNA or histone modification, as well as post-transcriptional control through RNA editing, methylation and RNA interference31,38,41–43. Inhibitions of epigenetic regulators DNMTs, KDM5B, LSD1, and SETDB1 enhance type I IFN signalling and immunotherapy response via the activation of TE-derived immunogenic elements38,41–43. Although Kcr has generally been associated with increased gene transcription, enrichment of H4 Kcr from reprogrammed lysine catabolism in GSCs, rather than the well-characterized H3 Kcr12,26 might lead to the different functions of histone Kcr.
METHODS
GSC and NSC derivation and maintenance
GBM tissues were obtained from excess surgical resection samples from patients at the Case Western Reserve University (Cleveland, OH) after reviewed by neuropathology with written informed consent from the patients and in accordance with an institutional review board-approved protocol (090401). All specimens were examined by neuropathologists. All patient studies were carried out in accordance with the Declaration of Helsinki. To minimize in vitro cell culture-based artifacts, patient-derived xenografts were propagated as a renewable source of GSCs. GSC23 was derived from a recurrent GBM biopsy specimen from a 63-year-old male patient and was provided as a generous gift by Dr. Erik Sulman (NYU Langone Health). GSC3028 was derived from a recurrent GBM from a 65-year-old female patient. GSC3565 was derived by our laboratory and transferred via a material transfer agreement from Duke University (Durham, NC). HNP1 Human Neural Progenitors (ArunA Biomedical, Inc., Athens, GA) are fully differentiated and derived as adherent cells from the hESC WA09 line. Human NSC line NSC11 (ALSTEM, Richmond, CA, USA) was derived from human IPS cells. ENSA (ENStem-A, Millipore, Billerica, MA) are human embryonic stem derived neural progenitors. All GSC and NSC lines were typically cultured in Neurobasal media containing 0.8 mM L-lysine unless otherwise noted (cat: 12349015; Gibco) supplemented with B27 without vitamin A (cat: 12587010; Gibco), 20 ng/ml EGF (cat: 236-EG-01M; R&D Systems), and 20 ng/ml recombinant human bFGF (cat: 4114-TC-01M; R&D Systems), sodium pyruvate (cat: 11360070; Gibco), GlutaMAX (cat: 35050061; Gibco) and streptomycin/penicillin (cat: 15140122; Gibco) at 37°C with 5% CO2. Matched DGCs were maintained in Dulbecco’s modified Eagle’s media (DMEM) supplemented with 10% fetal bovine serum (cat: 26140079; Gibco) to maintain differentiation status. The custom Neurobasal media without L-lysine were purchased from Boca Scientific. Apart from removing L-lysine in the custom media, all the other components are the same as the Neurobasal media (cat: 12349015; Gibco). All experiments conform to relevant regulatory standards.
Cell Culture and lentiviral transduction
293T and GL261 cells were purchased from American Type Culture Collection, and cultured in DMEM supplemented with 10% fetal bovine serum (cat: 26140079; Gibco) and streptomycin/penicillin (cat: 15140122; Gibco) at 37°C with 5% CO2. Lentiviral clones expressing two non-overlapping shRNAs directed against SLC7A2, GCDH, ECHS1, ACSS2, CBP, P300, c-MYC, cGAS, DDX58, IFIH1 or a non-targeting control shRNA encoding a sequence not present in the mammalian genome (shCONT) were obtained from Sigma-Aldrich. Lentiviral overexpression plasmid for GCDH or ECHS1 and empty vector were ordered from VectorBuilder Inc. 293T cells were used to generate lentiviral particles through co-transfection of the packaging vectors pCMV-dR8.2 dvpr and pCI-VSVG. Cells were transfected with lentivirus for 24 h, followed by recovery for 24 h before selection. Infected cells were selected with 2 mg/ml puromycin until uninfected control cells were dead. For siRNA transfection, cells were transfected with siRNA duplexes (60–100 nM) using Lipofectamine 3000 (cat: L3000008; Thermo Fisher Scientific) according to manufacturer’s instructions. All cell lines in this study were tested to confirm lack of mycoplasma contamination. The genomic identity of each cell line has been authenticated by Duke University DAN Analysis Facility. The siRNA and shRNA sequences are listed in Supplementary Table 13.
Tumour dissociation and GSC culture
GSCs were isolated immediately following dissociation or following transient xenograft passage in immunocompromised NSG mice using prospective sorting, followed by assays to confirm stem cell marker expression, sphere formation, and secondary tumour initiation. Briefly, dissociation of xenografted tumours was performed using a papain dissociation system according to the manufacturer’s instructions. Cells were then cultured in Neurobasal media supplemented with 2% B27, 1% GlutaMAX, 1% sodium pyruvate, 1% penicillin/streptomycin, 20 ng/ml bFGF, and 20 ng/ml EGF for at least 6 h to recover expression of surface antigens. AC133-positive populations were isolated using CD133/1 antibody-conjugated magnetic beads. Cells were then labeled with a CD133/2(293C)-APC antibody, and CD133+ cells were sorted and analyzed by flow cytometry. The sorted CD133+ cells were cultured in complete Neurobasal media. Expression of stem cell markers (SOX2 and OLIG2), functional assays of self-renewal (serial neurosphere passage) and tumour propagation using in vivo limiting dilution assays were used to validate GSC phenotypes.
Xenograft models
All mouse experiments were performed in accordance with the Institutional Animal Care and Use Committee of the University of Pittsburgh (protocol 21049014). All mice were housed in specific-pathogen-free conditions at an ambient temperature of 20–26 °C and humidity of 30–70% with a 12 h:12 h light:dark cycle before use. A maximal tumour size of 15 mm in any direction was not exceeded in any experiment. Both male and female mice were used in studies. For intracranial xenografts of human-derived GSCs or DGCs, healthy NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, strain: 005557; The Jackson Laboratory), 5–6 weeks old, were randomly selected for intracranial injection by implanting 10,000 GSCs or 100,000 DGCs into the right cerebral cortex at a depth of 3.5 mm following standard procedures. Intracranial xenografts of mouse glioma cells were created by implanting 50,000 mouse glioma cells (GL261) into the right cerebral cortex of 5–6-week-old C57BL/6J mice (The Jackson Laboratory) at a depth of 3.5 mm. Housing conditions and animal status were supervised by a veterinarian. Animals were monitored until neurologic signs were observed, at which point they were sacrificed. Neurologic signs included hunched posture, gait changes, lethargy, and weight loss. Brains were harvested and fixed in 4% paraformaldehyde (PFA), cryopreserved in 30% sucrose, and then cryosectioned. Hematoxylin and eosin (H&E) staining was performed on sections for histological analysis. Mouse survival was analyzed in GraphPad Prism software, and the statistical significance was tested by log-rank test. Mouse brains implanted with GSCs or GL261 that were labeled with firefly luciferase were monitored by the bioluminescence imaging. Animals were treated with D-Luciferin (50 mg/kg, cat: P1042; Promega) intraperitoneally and anesthetized with isoflurane for the imaging analysis. The bioluminescence images were captured by an IVIS imaging system (PerkinElmer).
Lysine-restricted diet
Custom diets containing purified amino acids rather than whole protein as a source of amino acids were purchased from Envigo. The control diet (TD.01084, Envigo) contained 1.44% lysine is a modification of diet TD.99366 where vitamins have been increased to compensate for losses during irradiation. The lysine-restricted diet (TD.210714, Envigo) with defined lysine levels was modified from the control diet by reducing lysine to 0.144%. Mice of NSG or C57BL/6J background, 5–6 weeks old, were subjected to the control or lysine-restricted diet from 1 week before GSCs or GL261 inoculation until the end point, and mice weight was monitored weekly after the dietary treatments.
Lysine quantification in vitro and in vivo
Healthy NSG mice or NSG mice harboring GSC23-derived tumour were randomized subjected to control or lysine-restricted diet for 4 weeks prior to serum and brain tissues harvest. Mice were deeply anesthetized with isoflurane and blood was collected via intracardiac sampling. Next, mice were euthanized quickly, and non-tumour brain tissue and matched tumour tissue were dissected, weighed and snap-frozen in liquid nitrogen. Lastly, serum was isolated from whole blood samples and frozen until lysine quantification. Non-tumour brain tissue and matched tumour tissue were lysed, and measured using Lysine Assay Kit (cat: MET-5130; Cell Biolabs) according to the manufacturer’s instructions. GSC23 and 3028 cultured in complete Neurobasal media with indicated concentrations of L-lysine were harvested and weighed before analysis. The Lysine Assay Kit was used to quantify free lysine in cellular extracts from GSC23 and 3028.
In vivo treatment
Mice of NSG or C57BL/6J background were randomly subjected to control diet or lysine-restricted diet from 1 week before GSC23, GSC3028 or GL261 inoculation. Animals were monitored until neurologic signs were observed, at which point they were sacrificed. For in vivo IFNAR blocking antibody treatment, the immunodeficient mice bearing GSC23-derived intracranial tumours were treated with 100 μg human IFNAR neutralizing antibody (cat: MAB4015; R&D Systems) or isotype-matched control antibody via i.p. injection every 5 days starting at 5 days post-inoculation, followed with analysis of SOX2+ and CD133+ GSCs by flow cytometry. For MYCi975 and Lysine-restricted diet treatment, mice were treated with MYCi975 (50 mg/kg, cat: HY-129601; MedChemExpress) or vehicle on day 5 post-inoculation via intraperitoneal injection 3 times per week for 4 to 6 weeks until neurological signs are observed. For anti-PD-1 and Lysine-restricted diet treatment, intracranial xenografts were created by implanting GL261 into the right cerebral cortex of 5–6-week-old C57BL/6J mice at a depth of 3.5 mm. After 5 days post-inoculation, mice were randomly selected into four groups (control, anti-PD-1 only, lysine-restricted diet only, anti-PD-1 + lysine-restricted diet), and were administered 200 μg rat IgG2a isotype control (cat: BP0089; Bio X Cell) or anti-PD-1 (cat: BP0273; Bio X Cell) antibody via i.p. injection every 5 days for the duration of experiments.
Measurement of protein synthesis rate in vitro and in vivo
To evaluate protein synthesis in vitro, GSC23 cultured in complete Neurobasal media with indicated concentrations of L-lysine were supplemented with 1 μM Puromycin 30 min prior to IB analysis. To measure protein synthesis of GSC-derived xenograft in vivo, NSG mice were subjected to the control and lysine-restricted diet from 1 week before GSC23 intracranial inoculation in the right hemisphere until the end point. 20 ul of 1.5 mM O-Propargyl-puromycin (OP-puro, cat: 1407–5; Click Chemistry Tools) was injected into the contralateral hemisphere on day 28 using the following coordinates: −0.5/1.2/−2 mm, and then mice were sacrificed 30 min later as previously described44. The brains were harvested, processed to cell digestion, antibodies labeling, click reaction (cat: C10643; Thermo Fisher Scientific) and flow cytometric analysis. In vivo protein synthesis rate was compared based on the fluorescence intensity of Alexa Fluor 647-OP-puro in human CD147+ tumour cells.
Type I IFN measurement and experiments
To quantify IFNα and IFNβ in GSC culture supernatant, GSCs with or without GCDH KD were cultured under standard conditions for 5 days before supernatants were collected and subjected to ELISA analysis. IFNα (cat: BMS216; Thermo Fisher Scientific) and IFNβ (cat: 41435; Pestka Biomedical Laboratories) ELISA were performed according to manufacturer’s instructions. For GSC in vitro treatment, GSCs were cultured in complete Neurobasal media with 5 ng/ml of human IFNβ (cat: 300–02BC; PeproTech), and IFNβ was added every 2 days for the duration of experiments. To block GCDH loss induced activation of IFN signalling, GSCs were preincubated with 10 μg/ml of human IFNAR blocking antibody (cat: MAB4015; R&D Systems) for 2 h in 37 °C, followed with stimulation of the culture supernatants from control or GCDH KD GSCs. The supernatants of each culture condition with or without IFNAR blocking antibody were replaced every 2 days.
Proliferation and neurosphere formation assay
Cell proliferation experiments were performed by seeding 2,000 cells/well in a 96-well plate with three to six replicates. CellTiter-Glo (Promega) was used to measure cell proliferation at days 0, 3, and 5. All data were normalized to day 0.
Click-iT EdU incorporation assay
The Click-iT EdU reaction was performed following the instructions of Click-iT™ EdU Cell Proliferation Kit (Cat: C10339; Thermo Fisher Scientific). GSCs cultured in complete Neurobasal media were incubated with 10 μM EdU for 2 h. DGCs cultured in DMEM supplemented with 10% FBS were incubated with 10 μM EdU for 12 h. Ten random fields per condition were acquired with a ×40 magnification objective. The percentage of EdU+ cells was determined according to the number of EdU-positive cells and DAPI-positive cells using ImageJ.
Cumulative growth curve and SA-β-Gal assay
Populations of primary DGCs with or without ECHS1 depletion (1 × 105 per dish) were counted by trypan blue exclusion every three days. SA-β-Gal staining was performed according to the manufacturer’s instructions (cat: 9860S; Cell Signaling Technology), followed by images capture and quantification.
Cytoplasm and nucleus fractionation
The cytoplasmic and nuclear fraction were obtained using Nuclear Extract Kit (cat: 40010; Active motif) according to the manufacturer’s guidelines. The whole-cell, cytoplasmic, nuclear extracts were prepared from GSCs and analyzed by IB using indicated antibodies.
Co-IP analysis
Briefly, cells were lysed in IP buffer (cat: 87788; Thermo Fisher Scientific) supplemented with 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF and protease-inhibitor cocktail (cat: A32953; Thermo Fisher Scientific). Cell debris was removed by centrifugation, and lysates were then incubated with IgG antibody of the same species as the IP antibody and protein G beads (cat: 10003D; Thermo Fisher Scientific) for 2 h at 4°C to reduce nonspecific binding. The cleared lysates were incubated with indicated primary antibody and protein G beads or anti-flag M2 beads (cat: M8823; Sigma) overnight at 4°C. The IP was washed three times with the IP buffer before boiled and analyzed by IB according to the standard methods.
IF and IHC
For IF analyses, cells were grown on matrigel-coated coverslips, and fixed with 4% PFA for 15 min followed by permeabilization with 0.1% Triton X-100 in PBS for 15 min. After washing with PBS for three times, cells were blocked in PBS buffer with 5% goat serum for 1 h. The relevant primary antibodies diluted in blocking buffer were then incubated at 4°C overnight, followed by secondary antibodies incubation for 1 h at room temperature according to manufacturer’s instructions. Cryosectioned tumour-bearing brain tissues were permeabilized and blocked in PBS buffer with 0.1% Triton X-100 and 5% goat serum. Sections were then incubated with primary antibodies at 4°C overnight and appropriate secondary antibodies for 1 h at room temperature. For IHC staining, paraffin-embedded sections were deparaffinized and underwent antigen retrieval in sodium citrate buffer in a microwave oven for 15 min. Sections were permeabilized and blocked in PBS buffer with 0.1% Triton X-100 and 5% goat serum. Sections were then incubated with CD8 antibody (cat: 14-0808-82; eBioscience; 1: 400) at 4°C overnight and appropriate secondary antibody for 1 h at room temperature and performed as manufacturer’s instructions.
In vitro interaction and histone crotonylation assay
To validated the interaction between GCDH and CBP in vitro, His-tagged recombinant GCDH (cat: NBP1-50838; Novus Biologicals) was incubated with or without recombinant Flag-tagged CBP (cat: 31590; Active motif) overnight at 4 °C in binding buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1 mM EDTA, 0.1% NP-40 and 5% glycerol, 1 mM PMSF) supplemented with protease-inhibitor cocktail (Roche), followed by incubation with anti-Flag beads for 2 h at 4 °C. Beads were subsequently harvested through centrifugation and washed three times by binding buffer before boiled and analyzed by IB using indicated antibodies. To analyze GCDH-dependent histone crotonylation, we incubated His-tagged recombinant GCDH with recombinant Flag-tagged CBP, recombinant histone octamer (cat: 31470; Active motif), 2 mM Glutaryl-CoA (cat: G9510; Sigma), and 2mM FAD (flavin adenine dinucleotide, cat: F8384; Sigma) in the presence of HAT buffer (50mM Tris-base, pH 8.0, 10% glycerol, 0.1mM EDTA and 1mM dithiothreitol, 1mM PMSF, 10mM sodium butyrate) at 30 °C for 2 h. To analyze nuclear GCDH-CBP complex-dependent histone crotonylation, the cytoplasmic and nuclear GCDH from 293T cells transfected with indicated plasmids were purified through anti-Flag beads according to the standard IP protocol. Then, the immunoprecipitated Flag-tagged GCDH complex was incubated with recombinant histone octamer, 2 mM Glutaryl-CoA, and 2mM FAD in the presence of HAT buffer at 30 °C for 2 h with shaking. These reactions were stopped by heat to 95 °C in the presence of LDS loading buffer for 10 min and then probed with the indicated antibodies.
Quantification of amino acids using MS
Mass spectrometry was used to quantify the concentrations of free amino acids in GSCs, DGCs and NSCs. The GSCs and NSCs were cultured in complete Neurobasal media, whereas DGCs were cultured in DMEM supplemented with 10% FBS for at least 1 week, and transferred to complete Neurobasal media for 24 h before harvesting. Each cell sample contains 5 replicates. Metabolic quenching and polar metabolite pool extraction were performed by adding ice cold 80% methanol (aqueous) at a ratio of 500 μl buffer per 2 million cells. Deuterated (D3)-creatinine and (D3)-alanine, (D4)-taurine and (D3)-lactate (Sigma-Aldrich) were added to the sample lysates as internal standards at a final concentration of 10 μM. Samples were homogenized using a 25 °C water bath sonicator and the supernatant was then cleared of protein by centrifugation at 16,000 ×g. 2 μl of cleared supernatant was subjected to online LC-MS analysis. Analyses were performed by untargeted LC-HRMS. Briefly, Samples were injected via a Thermo Vanquish UHPLC and separated over a reversed phase Thermo HyperCarb porous graphite column (2.1×100mm, 3μm particle size) maintained at 55 °C. For the 20 min LC gradient, the mobile phase consisted of the following: solvent A (water / 0.1% FA) and solvent B (ACN / 0.1% FA). The gradient was the following: 0–1 min 1%B, increase to 15%B over 5 min, continue increasing to 98%B over 5 min, hold at 98%B for 5 min, re-equilibrate at 1%B for 5 min. The Thermo IDX tribrid mass spectrometer was operated in both positive and ion mode, scanning in ddMS2 mode (2 μscans) from 70 to 800 m/z at 120,000 resolution with an AGC target of 2e5 for full scan, 2e4 for ms2 scans using HCD fragmentation at stepped 15,35,50 collision energies. Source ionization setting was 3.0 and 2.4kV spray voltage respectively for positive and negative mode. Source gas parameters were 35 sheath gas, 12 auxiliary gas at 320 °C, and 8 sweep gas. Calibration was performed prior to analysis using the Pierce™ FlexMix Ion Calibration Solutions (Thermo Fisher Scientific). Purified standards were purchased and compared in retention time, m/z, along with ms2 fragmentation patterns to validate the identity of Integrated peak areas extracted manually using Quan Browser (Thermo Fisher Xcalibur ver. 2.7). The ratios of MS-based peak areas of analytes to internal standards were used to represent amino acid levels. Data were normalized to cell numbers and the average of control group. The cell lysates from GSCs with or without SLC7A2 depletion were used to identify differences of amino acid abundance. Each cell sample contains 4 replicates.
Short-chain acyl-CoA measurement
GSCs were cultured in complete Neurobasal media until harvesting. DGCs were cultured in DMEM supplemented with 10% FBS for at least 1 week, and transferred to complete Neurobasal media for 24 h before harvesting by the addition of 1 ml of ice cold 10% trichloroacetic acid (TCA). Samples were spiked with SILEC acyl-CoA internal standards generated as previously described45. Calibration curves were prepared from commercially available acyl-CoAs (Sigma-Aldrich), then samples and calibrators were analyzed for the abundances of crotonyl-CoA, acetyl-CoA, and other short-chain fatty acyl-CoA using LC-quadrupole/Orbitrap high-resolution mass spectrometry as previously described46. To quantify the acyl-CoAs that are altered by lysine catabolism, control and GCDH KD GSC23 (4 × 106, n = 5) were cultured with indicated concentrations of L-lysine, then metabolism was quenched by the addition of 1 ml of ice cold 10% TCA. To compare intracellular crotonyl-CoA and acetyl-CoA levels after knocking down SLC7A2, SLC7A5, GCDH or ACSS2, GSCs with indicated gene depletion were harvested and processed using the methods described above. The quantification of intracellular acyl-CoAs was calculated by interpolation of the integrated area under the curve for each analyte/13C15N-acyl-CoA internal standard to a calibration curve prepared from increasing amounts of commercially available standards. For experiments indicated on the y-axis, pmol amounts of acyl-CoAs were divided by the cell count of individual replicates.
For tracing assays, GSC23 or DGC23 were cultured in complete Neurobasal media lacking lysine, and given 0.2 mM or 0.8 mM 13C6-lysine (cat: 89988; Thermo Fisher Scientific) for 12 h before metabolites were extracted with 10% TCA. Samples were spiked with SILEC acyl-CoA internal standards to monitor alteration of MS signals, and processed following the above methods. Intracellular acetyl-CoA was calculated by interpolation of the integrated area under the curve for acetyl-CoA/13C15N-acety-CoA internal standard to a calibration curve prepared from increasing amounts of commercially available standards. The enrichment of acetyl-CoA (m+2) derived from 13C6-lysine was calculated by a correction for the naturally occurring isotopologue distribution using linear algebra we coded into an online tool47. The correction matrix was generated by using theoretical or experimental isotopologue distribution for a chosen analyte according to a well-established algorithm in FluxFix47.
Short-chain fatty acid measurement
Cells were homogenized with 80% methanol at a ratio of 0.5ml/2million cells. 5 μg/ml Deuterated internal standards: (D2)-formate, (D4)-acetate, (D5)-butyrate, (D6)-propionate, (D2)-valerate and (D4)-hexanoate (CDN Isotopes, Quebec, Canada) were added. Samples were homogenized using a FastPrep-24 system (MP-Bio), with Matrix D at 60hz for 30 seconds, before being cleared of protein by centrifugation at 16,000 ×g. 60 μl cleared supernatants were collected and derivatized using 3-nitrophenylhydrazine. Each sample was mixed with 20 μl of 200 mM 3-nitrophenylhydrazine in 50% aqueous acetonitrile and 20 μl of 120 mM N-(3-dimethylaminopropyl)-N0-ethylcarbodiimide −6% pyridine solution in 50% aqueous acetonitrile. The mixture was incubated at 50 °C for 40 min and the reaction was stopped with 0.45 ml of 50% acetonitrile. Derivatized samples were injected (50 μl) via a Thermo Vanquish UHPLC and separated over a reversed phase Phenomenex Kinetex 150mm × 2.1mm 1.7μM particle C18 maintained at 55 °C. For the 20 min LC gradient, the mobile phase consisted of the following: solvent A (water / 0.1% FA) and solvent B (ACN / 0.1% FA). The gradient was the following: 0–2 min 15%B, increase to 60%B over 10 min, continue increasing to 100%B over 1 min, hold at 100%B for 3 min, re-equilibrate at 15%B for 4 min. The Thermo IDX tribrid mass spectrometer was operated in both positive ion mode, scanning in ddMS2 mode (2 μscans) from 75 to 1000 m/z at 120,000 resolution with an AGC target of 2e5 for full scan, 2e4 for ms2 scans using HCD fragmentation at stepped 15,35,50 collision energies. Source ionization setting was 3.0kV spray voltage respectively for positive mode. Source gas parameters were 45 sheath gas, 12 auxiliary gas at 320 °C, and 3 sweep gas. Calibration was performed prior to analysis using the PierceTM FlexMix Ion Calibration Solutions (Thermo Fisher Scientific). Integrated peak areas were then extracted manually using Quan Browser (Thermo Fisher Xcalibur ver. 2.7). The ratios of MS-based peak areas of analytes to internal standards were used to represent SCFA levels. Data were normalized to cell numbers and the average of control group.
Mass spectrometric analysis
To identify GCDH-interacting proteins in the nucleus, GCDH and its interacting proteins were immunoprecipitated from the nuclear fraction of 293T cells transfected with or without Flag-GCDH plasmid using anti-Flag beads. The protein sample was heated to 95 °C in the presence of LDS loading buffer for 10 min and then loaded into 4–12% Bis-Tris gel (cat: NP0322BOX; Thermo Fisher Scientific) via electrophoresis at 100 V for 10 min to gain a quick assessment of the major proteins in short separated gel. Each sample contains 3 replicates. Gel bands were digested with trypsin and tryptic peptides were analyzed by nano reverse phase HPLC interfaced with a mass spectrometer. The raw data files were processed using Proteome Discoverer software, and the International Protein Index database was searched for spectra by using Mascot software. The identified peptides and proteins were further statistically validated with the Scaffold software. Search results were trimmed to a 1% false discovery rate using Percolator algorithm (Matrix Science).
To identify lysine catabolism-dependent crotonylation on histone H4, core histones derived from GSCs cultured in a complete Neurobasal medium with low (0.2 mM) or high (2 mM) L-lysine were prepared using a commercial Histone Extraction Kit (cat: 40028; Active motif). The histones were immunoprecipitation with a pan-anti-Kcr antibody (cat: PTM-501; PTM Biolab). The eluted proteins were quantified with the Bradford protein assay (Bio-Rad Laboratories). The enriched Kcr proteins were then digested by trypsin (1:50; Promega) for 16.5h in 100 mM ammonium bicarbonate (Sigma-Aldrich) buffer and then concentrated by vacuum. To make sure loading amounts were consistent among different samples, equal amounts of proteins were subjected to digestion by trypsin. The resulting tryptic peptides were loaded onto a home-made column packed with 12 cm length × 3 μm ID C18 resin (Dr. Maisch GmbH). LC-MS/MS was performed on an Orbitrap Exploris™ 480 mass spectrometer (Thermo Fisher Scientific) coupled with an EASY-nLC 1000 system (Thermo Fisher Scientific) as described earlier48. Database search and label-free quantification were performed by Proteome Discoverer 2.5. The peptide intensities were normalized and scaled for label-free quantification. Normalization was performed according to the abundances of all peptides. The peptide-spectrum matches (PSMs) for all crotonylated peptides were manually verified as previously described49.
Flow cytometry
To quantify T cells and T cell cytokine expression, single-cell suspensions were prepared from fresh mouse tumour tissues on day 14 post-injection. Brain tumours were dissected, minced and digested with 5 mg/ml Collagenase Type II (cat: 17101-015; Gibco) and 0.1 mg/ml DNase I recombinant (cat: 4536282001; Sigma-Aldrich) at 37 °C for 30 min. Cells were passed through a 40 μm filter to remove clumps and maintain single cell suspensions. Brain cell suspensions were separated over 30% percoll gradients at 700g for 30 min at 4 °C to enrich tumour-infiltrating immune cells. Red blood cells were removed by ACK Lysing Buffer (Thermo Fisher Scientific). The remaining cells were suspended in a staining buffer (cat: 554656; BD Biosciences) and blocked with CD16/CD32 antibody (cat: 156604; Biolegend; 1:500) at 4 °C for 10 min. Cells were then stained with anti-CD45 (cat: 103133; Biolegend; 1:200), anti-CD3 (cat: 555275; BD Biosciences; 1:200), anti-CD8 (cat: 100705; Biolegend; 1:100) antibodies for 45 min on ice, followed by fixation and permeabilization (cat: 88-8824-00; ebioscience) for cytokine staining. After permeabilized with fixation/permeabilization buffer, cell suspension was incubated with Granzyme B (cat: 396407; Biolegend; 1:100) antibody at 4 °C for 45 min. Splenocytes were stained and examined by flow cytometry to characterize the immune phenotype of CD45+ infiltrating cells.
For GSC isolation in vivo, brain single cells from NSG mice bearing GSC23-derived tumour were generated and stained with anti-human CD147 (cat: 306204; Biolegend; 1:200) and anti-human CD133 (cat: 566594; BD Biosciences; 1:50) antibodies using the methods described above. Anti-human CD147 antibody can be used to gating GSC-derived tumour cells in brain cell suspensions. Cells were then fixed and permeabilized before staining with anti-SOX2 (cat: 656111; Biolegend; 1:200). Similar amounts of total cells (around 100,000 cells) were injected into Flow Cytometer and gated for further analysis of CD147+ cells in mouse brain tissue. SOX2+ CD133+ cells in CD147+ human tumour cells were defined as GSCs. All flow samples were run on LSR Fortessa (BD) and data were analyzed with FlowJo v.10.
Cytoplasmic dsRNA IP and analysis
Cytoplasmic fractions were extracted from GSC23 (1 × 107) with or without GCDH depletion after the nuclear fraction was removed through centrifugation using Nuclear Extract Kit (cat: 40010; Active motif). The cytoplasmic RNA was extracted and dissolved in RNase-free H2O using Direct-zol RNA Miniprep kit from cytoplasmic fractions of GSC23. IgG and J2 antibodies (cat: MABE1134; Millipore) were conjugated (2 μg per pulldown) to 10 μl protein G beads by shaking for 2 h at 4 °C in advance. 50 μg total RNA was used for each IP in 400 μl IP buffer (350 mM NaCl, 25 mM Tris-HCl pH7.5, 5 mM DTT and 0.5% NP-40), followed by the addition of 1 μl of RNase A (10 mg/ml, cat: R6513; Sigma-Aldrich) and incubation with conjugated protein G beads for 2 h at 4 °C. Beads were washed 3 times with IP buffer and incubated in 50 μl proteinase K digestion solution (1× TE, 100 mM NaCl, 1% SDS, and 1 μl of 20 mg/ml Proteinase K solution (cat: AM2546; Thermo Fisher Scientific)) for 20 min at 45 °C. dsRNA was extracted from the elution solution (50 μl) using Direct-zol RNA Miniprep kit, followed by reverse transcription using High-Capacity cDNA Reverse Transcription Kit. RT-qPCR detection for selected TEs was performed using indicated primers (Supplementary Table 13).
MDA5-protection assay
To identify histone Kcr-regulated cytosolic RNA that activates MDA5, we performed RNase A protection assay followed by RT-qPCR to detect TEs as previously described37. Briefly, GSC cytosolic fraction with RNase inhibitors and protease inhibitors was used for MDA5 IP according to standard ribonucleoprotein immunoprecipitation (RIP) protocol. Lysates containing same amounts of protein were incubated with IgG or MDA5 conjugated protein G beads for 2 h at 4°C, followed by addition of RNase A (final concentration 2 ng/μl) and DNase I (final concentration 2 U/μl), and incubation for another 5 min at room temperature after washed with RIP wash buffer for three times. RNase A digestion was inactivated by adding TRIzol reagent and the RNA was purified with Direct-zol RNA Miniprep kit. The extracted RNA was further purified using QIAquick PCR purification kit (QIAGEN) to remove small fragments resulting from RNase A digestion (less than 100 bp). The final RNA was used for the cDNA preparation through reverse transcription.
Cytoplasmic dsRNA/DNA isolation and re-transfection
The cytoplasmic total RNA from GSC23 with or without GCDH depletion was isolated using Direct-zol RNA Miniprep kit. 10 μg total RNA from each sample was dissolved in 49 μl RNase protection buffer (10 mM Tris-HCl pH7.5, 350 mM NaCl). 1 μl RNase A (10 mg/ml, cat: R6513; Sigma-Aldrich) was added and the mixture was incubated at 37 °C for 30 min. dsRNA was extracted and denatured at 95 °C for 5 min, followed by digestion with 1 μl RNase III (cat: AM2290; Thermo Fisher Scientific) or 1 μl H2O (mock digestion) in 10 μl reaction mixture for 60 min at 37 °C prior to re-transfection into GSC23 with 1.5 μl Lipofectamine 3000. For cytoplasmic DNA isolation, control or GCDH KD GSC23 (2 × 106) were harvested and cytoplasmic fractions were extracted using Nuclear Extract Kit (cat: 40010; Active motif) according to the manufacturer’s instructions. cytoplasmic fraction (400 μl solution) was combined with 25 μl 5M NaCl, and treated with 8 μl RNase A/T1 Mix (cat: EN0551; Thermo Fisher Scientific) for 30 min at 37 °C prior to DNA extraction using Gel Extraction Kit (QIAGEN no. 28704). Cytoplasmic DNA from GSC23 with GCDH depletion was digested with 1 μl H2O or 1 μl DNase I (cat: EN0521; Thermo Fisher Scientific) in 10 μl reaction mixtures for 30 min at 37 °C prior to re-transfection into GSC23. After 48 h, RNA was isolated for following RT-qPCR reactions using indicated primers.
Quantification of nascent transcription
Nuclear run-on (NRO) was used to estimate the effect of MYC KD or MYCi975 treatment on GCDH nascent transcription based on a published protocol35. Briefly, cell nuclei from GSCs with or without MYC loss were collected by gentle lysis of the plasma membrane and low-speed centrifugation, followed by in vitro NRO transcription reaction in the presence of bromouridine. Incorporation of bromouridine into nascent transcripts (NRO-RNAs) enables their specific detection against a background of unlabeled nuclear RNAs. Labeled nascent transcripts were purified by bromouridine immunocapture, and transcript levels were determined by RT-qPCR. Changes in GCDH NRO-RNA abundance are therefore a direct quantification of MYC-mediated transcriptional regulation of GCDH.
RNA isolation and RT-qPCR
Cellular total RNA was isolated using TRIzol (cat: 15596018; Life Technologies) and Direct-zol RNA Miniprep kit (cat: R2052; Zymo Research). 1 μg total RNA was used for cDNA synthesis using High-Capacity cDNA Reverse Transcription Kit (cat: 4368814; Thermo Fisher Scientific). Quantitative real-time PCR was performed using SYBR Green Master mix (cat: 4309155; Thermo Fisher Scientific) on an Applied Biosystems 7900HT Real-Time PCR System and normalized to β-actin. The primers used for gene expression analysis of human TEs and genes are shown in Supplementary Table 13.
RNA-seq
Total RNA was extracted and used for library construction using Illumina TruSeq Stranded Total RNA Library Prep Kit, which was performed by Novogene. The library was sequenced as a paired-end 150bp read. Raw FASTQ reads were trimmed using Trim Galore, followed by transcript mapping with Hisat2 to human reference genome (hg38). Samtools was used for sorting, indexing and format conversion from SAM files. Quantification and differential analysis were performed using FeatureCounts and DESeq2 with paired sample analysis. The list of significance was established by setting fold changes threshold at level of 2 and p < 0.05 using DEseq2. The differentially expressed gene lists generated were subsequently analyzed for enrichment of biological themes using ClusterProfiler or GSEA. The REACTOME_INTERFERON_SIGNALING and GOBP_INNATE_IMMUNE_RESPONSE signature genes were derived from GSEA MSigDB database. Signature genes were compared using GSEA to the list of differentially expressed genes ranked by fold change. The clinical relevance of lysine catabolism-related signature was queried using R package “GSVA” in public GBM database. For analysis of RE expression from RNA-seq data, reads were mapped to hg38 using Bowtie2 and assigned to TEs using RepEnrich2 with the recommended parameters50. The TE annotation file (hg38_repeatmasker_clean.txt) was provided by RepEnrich2. The resulting counts for TEs were analyzed by the DESeq2 package.
Chromatin-immunoprecipitation assay
ChIP assays were performed according to the ChIP kit (cat: 17-10085; Millipore) instruction manual provided by the manufacturer. Briefly, 1–2 × 107 GSC23 were cross-linked with 1% formaldehyde for 10 min, followed by adding glycine to a final concentration of 0.125 M for 5 min. Cells were harvested and washed with cold PBS for 3 times, and then re-suspended in cell lysis buffer to remove cytosol. The released nuclei were lysed and sonicated to shear chromatin to 200–500bp in nuclear lysis buffer. After centrifugation, the supernatant was diluted 1:10 by adding ChIP dilution buffer. The sheared crosslinked chromatin was incubated with indicated antibody overnight followed by magnetic Protein A/G beads incubation. After washing, elution and reverse-crosslinking, the ChIP DNA was purified and used to prepare sequencing libraries by the NEBNext Ultra DNA Library Prep Kit for Illumina sequencing. Raw FASTQ files were trimmed using Trim Galore and aligned to the hg38 human genome using Bowtie2 with default parameters. Samtools was used to sort and index the BAM files. Peak calling was performed using MACS2 with the narrow peak mode. For subsequent visualization of ChIP-seq signal, bigwig files were generated using Deeptools “bamCoverage” script. Deeptools or ngsplot was used to generate heatmaps and profile plots. Signal tracks were visualized using Integrative Genomics Viewer (IGV). Annotation of ChIP-seq peaks was performed using R package “ChIPseeker”. GO and KEGG enrichment of target genes was analyzed and visualized by ClusterProfiler. The Chromatin State Segmentation files were from ENCODE/Broad(http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHmm/wgEncodeBroadHmmHmecHMM.bed.gz). To identify Kcr and H3K27ac signals in TEs, a list of Kcr peaks associated with TEs was created by intersecting GCDH-dependent Kcr peaks with TE loci obtained from RepeatMasker annotation file in UCSC web browser, which was used for following analysis.
Single-cell RNA-seq analysis
The original data and code for the re-analysis of public scRNA-seq datasets were from published literature25. To characterize heterogeneity of GSC fraction and the cellular architecture of GBM, scRNA-seq data of 65,655 cells from 28 early-passage GSC cultures derived from 24 patients and 14,207 malignant tumour cells from 7 GBM tumours were used for further analysis. Malignant tumour cells were determined by a combination of unbiased clustering, cell-type marker expression and CNV inference in R. The differentially expressed genes between GSCs and tumour cells were called by Findmarkers (Seurat package) in R. On the training set, a logistic regression classifier was trained using the best hyperparameter determined from fivefold cross validation using Caret package in R based on top 20 differentially expressed genes. We repeated the 80/20 train test split randomly (stratified) for 30 times and tested the accuracy and stability of these models. We found that the model was robust with high accuracy and then picked the best performing model (highest test accuracy) to predict the entire dataset. The correctly classified tumour cells were reassigned as “Differentiated Tumour” and the misclassified tumour cells were reassigned as “GSC-like Tumour”. Lysine catabolic activity in single cells, except for the cell-cycle stage, was quantified using AUCell. AUCell scores were used to compare the activity of lysine catabolism between differentiated tumour cells and GSC-like tumour cells.
GSC dataset interrogation and transcription regulator analysis
RNA-seq data for three pairs of GSCs and DGCs were downloaded from GSE54791. RNA-seq-derived counts-per-million matrix was obtained and differential expression analysis was performed using R package “limma” to obtain a fold change ranking metric for GSC vs. DGC comparison. To identify the metabolic pathways that are highly active in GSCs, 40 gene sets18 related to vitamin, amino acid and nucleotide metabolism were queried using R package “GSVA” to get the gene signature score. RNA-seq data for 44 GSC and 9 NSC cell lines were downloaded from GSE119834. R package “limma” was used for differential expression analysis to obtain the fold change and p value in GSC vs. NSC. To rank the transcriptional factors (TFs), including transcription and chromatin regulators in GSC, the DEGs between GSCs and DGCs/NSCs were used to evaluate and rank TFs effect by integrative modelling of chromatin landscapes and cistromes from Cistrome Data Browser (CistromeDB) based on a computational biology framework “Lisa”34. The motivation of Lisa is to use public along with in-house chromatin profile data from a comprehensive TFs ChIP-seq, DNase-seq, H3K27ac ChIP-seq database to discover the underlying regulatory TFs that are directly responsible for the perturbation of a differentially expressed gene set. Top 20 enriched TFs ranked by p value were used for further analysis of overall 1316 TFs identified in GSC. To quantitatively determine the upstream TFs that can regulate and bind GCDH, we applied a regulatory potential (RP) score by considering the binding site’s distance to GCDH transcription start site (TSS) and the signal strength of TFs ChIP-seq peaks by integrating datasets in CistromeDB51. RP scores derived from the BETA algorithm are used to estimate how likely a factor regulates GCDH. For a ChIP-seq, DNase-seq, or ATAC-seq sample, BETA uses a distance-weighted measure to gauge the RP of all the binding sites of the factor within a certain distance to GCDH. Therefore, the RP score indicates the regulatory strength of TFs on the specific gene. ChIP-seq data of MYC from 8 cell lines were downloaded from the ENCODE database.
Public glioma patient database
Genetic alterations of lysine-catabolism related genes in glioblastoma multiforme were from cBioPortal (http://www.cbioportal.org). TCGA TARGET GTEx from UCSC Xena database (https://xenabrowser.net/datapages/) were used to extract FPKM values of TCGA_GBM and all normal brain cortex samples, followed by a query of GCDH expression, ECHS1 expression, and lysine catabolic activity. Public GBM databases from GlioVis (http://gliovis.bioinfo.cnio.es) were used to interrogate GCDH or ECHS1 expression levels in GBM and normal tissues. Z-scores calculated by mean and standard deviation of the expression level for each gene across all samples were used to convert the expression value to z-score based on the following formula: Z-score = (expression value − mean)/standard deviation. The R package “GSVA” was used to calculate a gene signature score with the “ssgsea” method. Survival analysis of the indicated gene signature was analyzed using Kaplan-Meier curve and log-rank test. Pearson correlation was done in Prism 9. ESTIMATE (https://bioinformatics.mdanderson.org/public-software/estimate/) or TIMER2.052 (http://timer.cistrome.org/) website were used to calculate the correlation between GCDH or ECHS1 expression and immune scores in TCGA GBM dataset.
Statistical analysis
For all statistical tests, p value less than 0.05 was accepted for statistical significance. Statistical tests, including two-tailed unpaired t test, one-way ANOVA, two-way ANOVA, Pearson or Spearman correlation, Log-rank test were performed using R or Prism 9 software. Unless otherwise indicated, data in the figures are presented as the mean ± SEM. For the box-and-whisker plot, boxes represent data within the 25th to 75th percentiles. Whiskers depict the range of all data points. Horizontal lines within boxes represent mean values.
Extended Data
Extended Data Fig. 1. SLC7A2 augments lysine consumption to support GSC function.
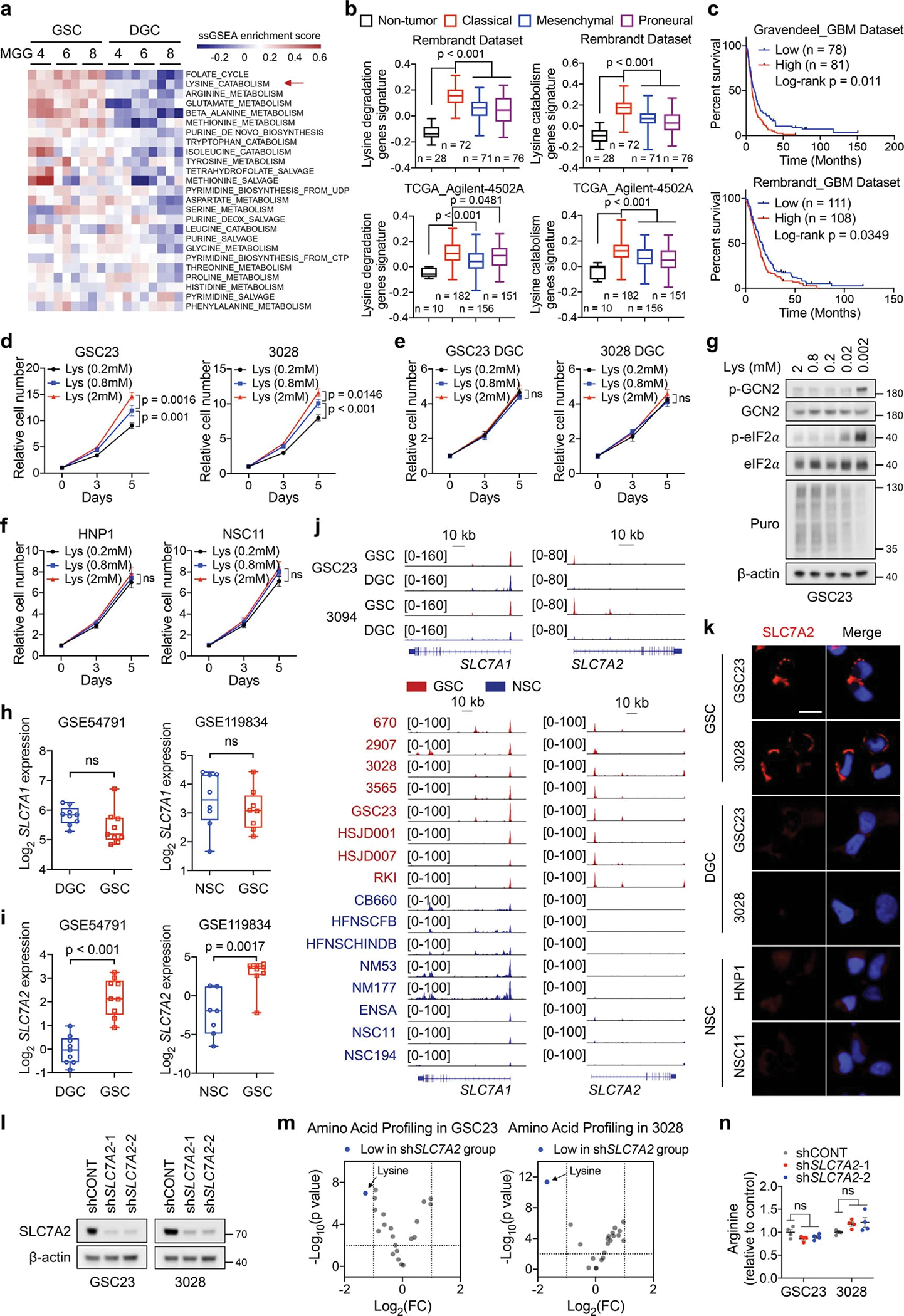
a, ssGSEA of metabolic gene sets from BioCyc database in three GSCs and matched DGCs. Each cell contains 3 replicates from GSE54791.
b, Box plots comparing lysine catabolic activity defined by corresponding gene set signatures in non-tumour brain tissue and three subtypes of GBM across two datasets from GlioVis. Boxes represent data within the 25-to-75 percentiles. Whiskers depict the range of all data points. Horizontal lines within boxes represent mean values. n indicates the number of biologically independent samples.
c, Kaplan-Meier plots of GBM patients grouped by lysine catabolism signature score.
d-f, Cell proliferation of GSCs (d), DGCs (e) and NSCs (f) cultured in media with indicated concentrations of L-lysine. Lysine-deprived DMEM supplemented with 10% dialyzed FBS and indicated concentrations of L-lysine was used to culture DGCs.
g, IB analysis of GSC23 cultured in media with indicated concentrations of L-lysine for 5 days. 1 μM Puromycin was added 30 min before harvest.
h, i, SLC7A1 (h) and SLC7A2 (i) expression levels in GSCs, DGCs, and NSCs. Boxes represent data within the 25-to-75 percentiles. Whiskers depict the range of all data points. Horizontal lines within boxes represent mean values. Three GSCs (MGG4, MGG6 and MGG8, each cell contains 3 replicates) and matched DGCs were queried in GSE54791. n = 8 biologically independent cells in NSCs and GSCs from GSE119834.
j, H3K27ac ChIP-seq tracks at SLC7A1 and SLC7A2 gene loci.
k, IF staining of SLC7A2 in GSCs, DGCs and NSCs. Scale bar, 20 μm.
l, IB analysis of GSCs with or without SLC7A2 KD.
m, Volcano plot showing altered amino acids upon SLC7A2 KD in two GSCs. Fold changes in amino acid levels relative to the average of control group are displayed.
n, Relative arginine levels (n = 4 biologically independent samples) in two GSCs with or without SLC7A2 KD by MS.
Data are presented from three independent experiments in d-f. Representative of two independent experiments in g, k and l. Data are presented as mean ± SEM in d-f and n. One-way ANOVA followed by multiple comparisons with adjusted p values for b and n, log-rank test for c, two-way ANOVA followed by multiple comparisons with adjusted p values for d-f, two-tailed unpaired t test for h, i and m. ns, not significant.
Extended Data Fig. 2. Reprogrammed lysine catabolism affects GSC growth via GCDH.
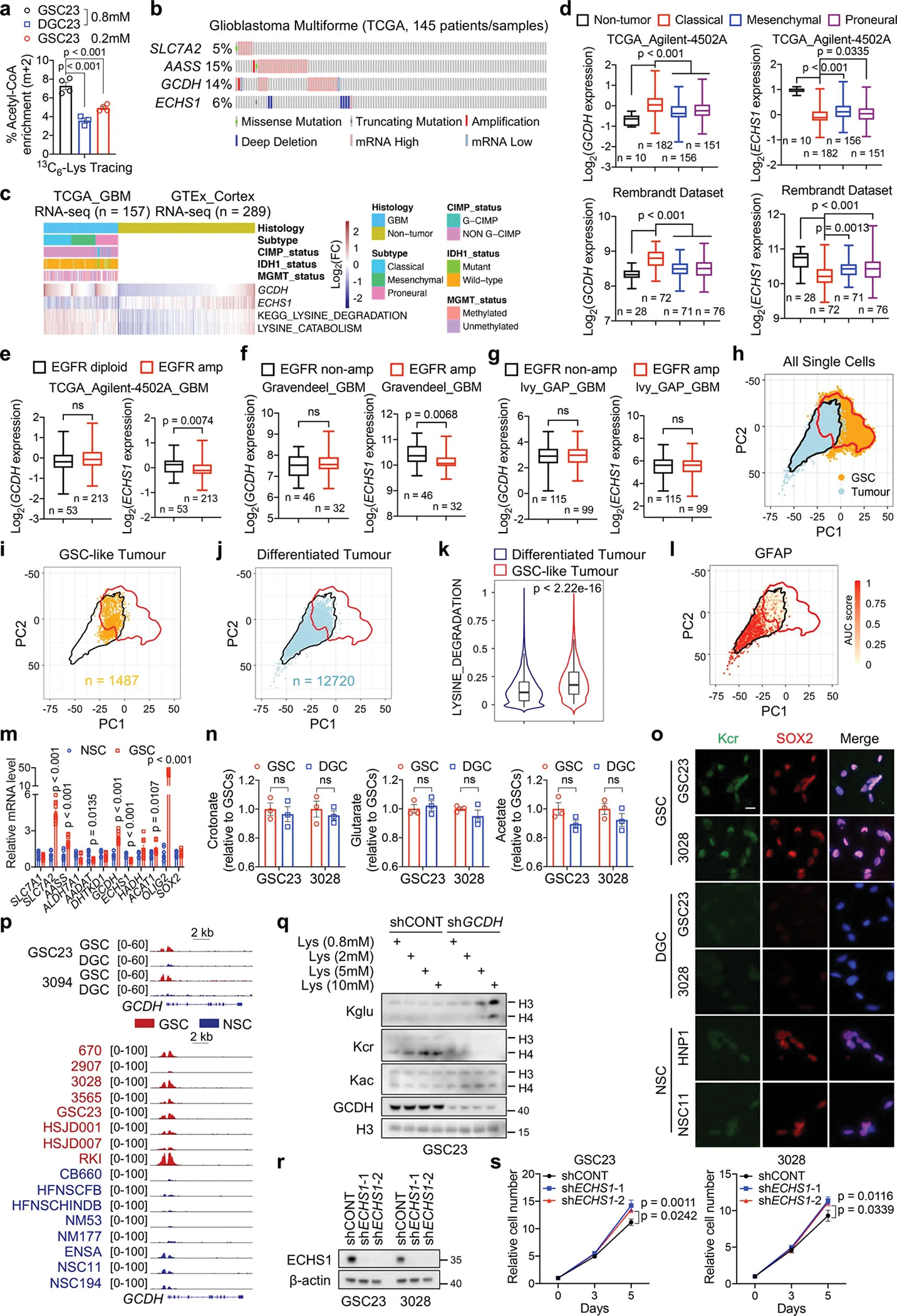
a, 13C6-lysine tracing assays in GSCs (n = 4 biologically independent samples) and DGCs (n = 3 biologically independent samples). Cells were given 13C6-lysine for 12 h.
b, Genomic alterations of SLC7A2, AASS, GCDH, and ECHS1 in TCGA GBM dataset from cBioPortal.
c, Heatmap displaying clinical information, GCDH, ECHS1 mRNA levels and lysine catabolism signature scores.
d-g, GCDH and ECHS1 expression in non-tumour brain tissue and three subtypes of GBM (d), and GBM samples with or without EGFR amplification (e-g) across datasets from GlioVis. Boxes represent data within the 25-to-75 percentiles. Whiskers depict the range of all data points. Horizontal lines within boxes represent mean values. n indicates the number of biologically independent samples.
h, PCA highlights overlap between GSCs (n = 65,655 cells from 28 GSC cultures) and malignant GBM tumour cells (n = 14,207 cells from 7 GBM tumours). Red and black lines represent contour encompassing 99% of GSCs and tumour cells, respectively.
i, j, GBM tumour cells classified as GSC-like tumour cells (i) or differentiated tumour cells (j).
k, Scoring of lysine degradation gene signature (12,720 differentiated tumour cells and 1,487 GSC-like tumour cells). Violin plots represent the overall distribution of data points. Boxes show median, upper and lower quartiles. Whiskers depict 1.5 times the interquartile range. l, Visualization of GFAP expression within GBM tumour cells.
m, RT-qPCR analysis of indicated genes in three GSCs (GSC23, 3028, and 3565) and NSCs (HNP1, NSC11, and ENSA).
n, Relative intracellular SCFAs (n = 3 biologically independent samples) in GSCs and DGCs.
o, IF staining of Kcr in GSCs, DGCs and NSCs. Scale bar, 20 μm.
p, H3K27ac ChIP-seq tracks at GCDH gene locus.
q, IB analysis of GSC23 cultured in media with indicated concentrations of L-lysine for 5 days.
r, s, IB (r) and cell proliferation (s) of two GSCs with or without ECHS1 KD.
Data are presented from three independent experiments in m and s. Representative of two independent experiments in o, q and r. Data are presented as mean ± SEM in a, m, n and s. One-way ANOVA followed by multiple comparisons with adjusted p values for a and d, two-tailed unpaired t test for e-g, k, m and n, two-way ANOVA followed by multiple comparisons with adjusted p values for s. ns, not significant.
Extended Data Fig. 3. The dependence of key crotonyl-CoA producing enzymes in GSCs.
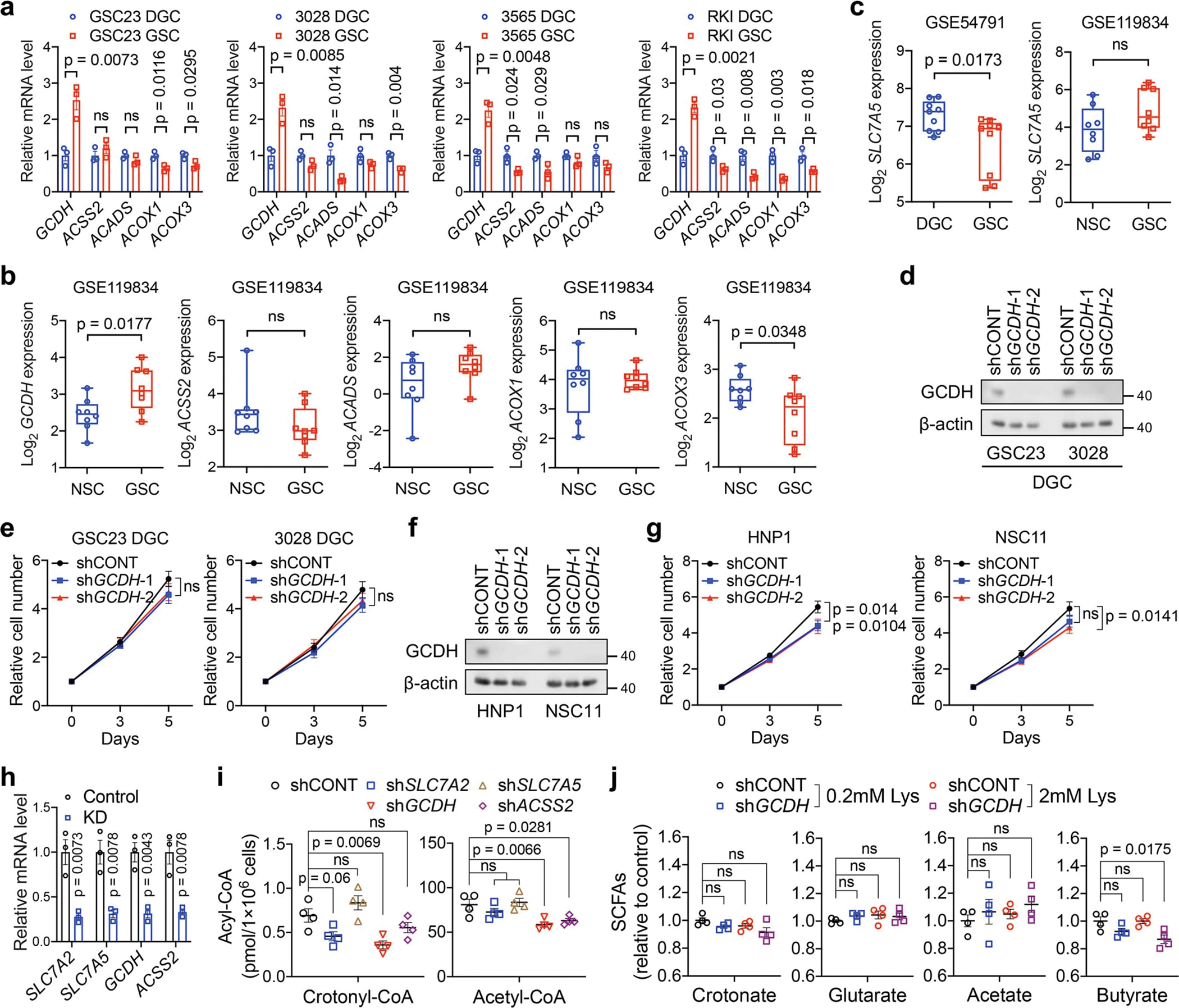
a, RT-qPCR analysis of key crotonyl-CoA-producing enzymes in paired GSCs and DGCs.
b, c, Expression levels of indicated genes from public datasets. n = 8 biologically independent cells in NSCs and GSCs from GSE119834. Three GSCs (MGG4, MGG6 and MGG8, each cell contains 3 replicates) and matched DGCs were queried in GSE54791.
d-g, IB analysis (d, f) and cell proliferation (e, g) in DGCs (d, e) or NSCs (f, g) with or without GCDH KD.
h, i, RT-qPCR analysis (h) and intracellular acyl-CoAs (i, n = 4 biologically independent samples) in GSC23 with or without indicated gene depletion.
j, Relative intracellular SCFAs (n = 4 biologically independent samples) in GSC23 with or without GCDH KD cultured in media with indicated concentrations of L-lysine.
Data are presented from three independent experiments in a, e, g and h. Representative of two independent experiments in d and f. Data are presented as mean ± SEM in a, e and g-j. Boxes represent data within the 25-to-75 percentiles in b and c. Whiskers depict the range of all data points. Horizontal lines within boxes represent mean values. Two-tailed unpaired t test for a-c and h, two-way ANOVA followed by multiple comparisons with adjusted p values for e and g, one-way ANOVA followed by multiple comparisons with adjusted p values for i and j. ns, not significant.
Extended Data Fig. 4. Lysine catabolism modulates IFN signalling and cell growth in vitro.
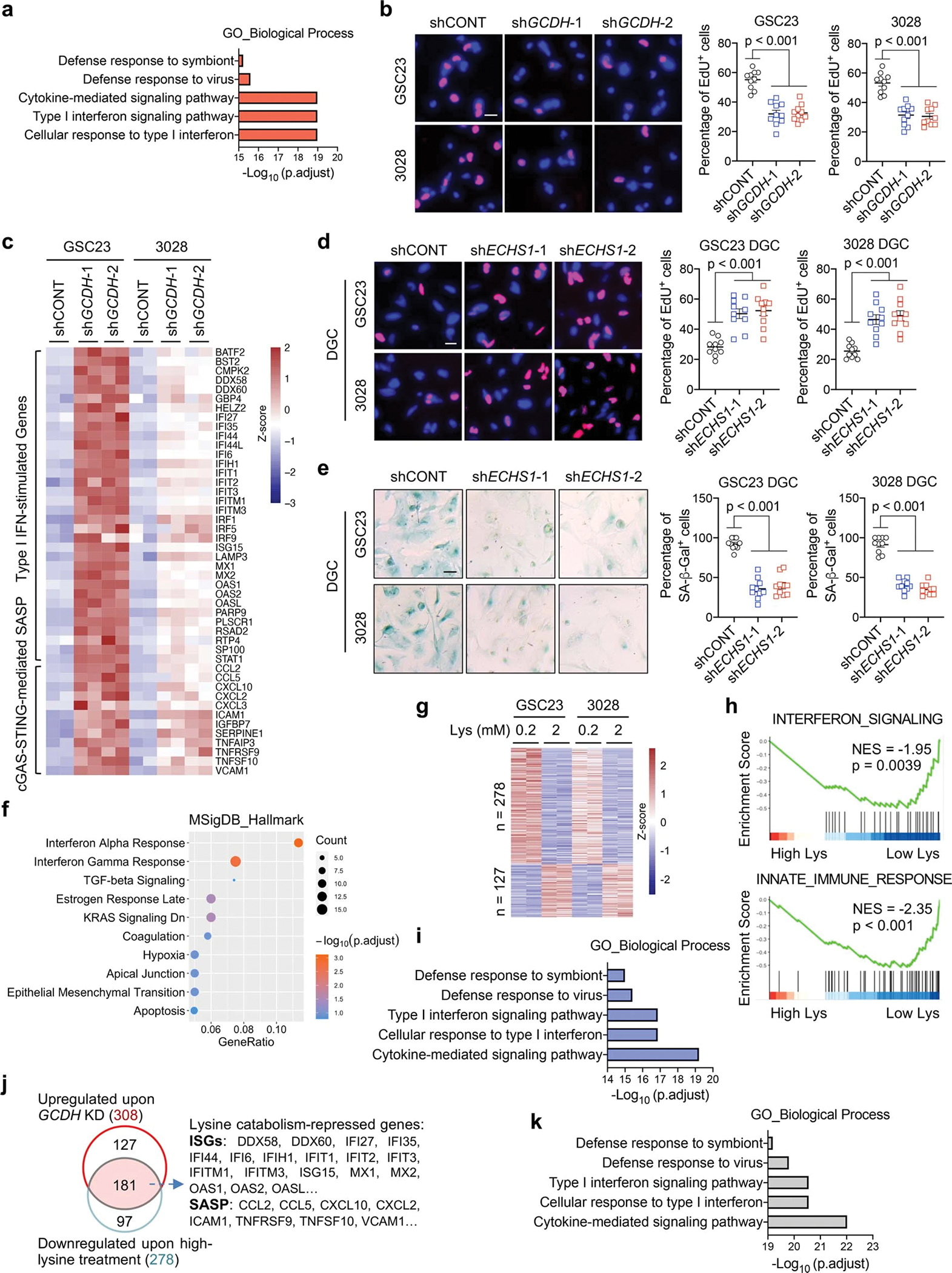
a, GO enrichment analysis of 308 upregulated genes upon GCDH KD, ranked by adjusted p values.
b, Representative images and quantification of EdU+ proportion of GSCs with or without GCDH KD. GSCs were exposed to 10 μM EdU for 2 h. Scale bar, 20 μm.
c, Heatmap showing the upregulated ISGs and SASP factors from RNA-seq upon GCDH loss in GSCs.
d, e, Proportion of EdU+ (d) and SA-β-Gal+ (e) DGCs with or without ECHS1 KD after 21 days in culture. DGCs were exposed to 10 μM EdU for 12 h. Scale bar, 20 μm.
f, Dot plot summarizing top 10 signalling pathways enriched in the 468 downregulated genes upon ECHS1 KD in two DGCs, ranked by adjusted p values.
g, Heatmap summarizing the DEGs of two GSCs cultured in media with indicated concentrations of L-lysine.
h, GSEA of two top pathways among DEGs from two GSCs cultured in media with high or low L-lysine. Normalized enrichment score (NES) and adjusted p values are shown.
i, Enrichment analysis of the 278 downregulated genes upon high L-lysine treatment in GSCs, ranked by adjusted p values.
j, Venn diagram showing the overlapping genes with increased expression upon GCDH depletion and decreased expression by high-lysine treatment in two GSCs.
k, Enrichment analysis of the 181 overlapping genes, ranked by adjusted p values. Representative of two independent experiments in b, d and e, and data are presented as mean ± SEM. One-way ANOVA followed by multiple comparisons with adjusted p values for b, d and e.
Extended Data Fig. 5. IFN signalling suppresses GSC maintenance.
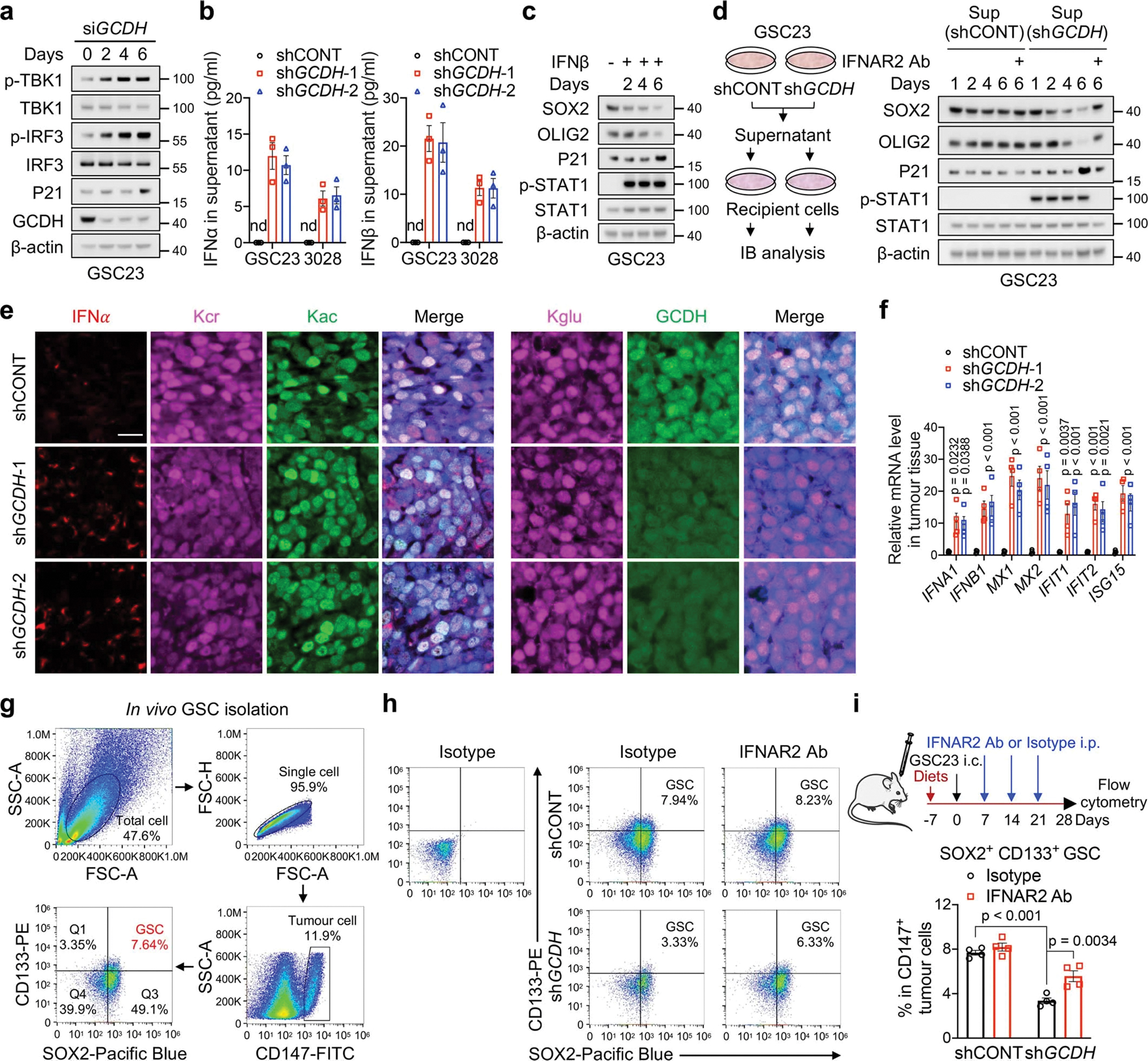
a, IB analysis of GSC23 after GCDH KD at the indicated time.
b, ELISA quantification of IFNα and IFNβ in culture supernatants from GSCs with or without GCDH KD. nd, not detected.
c, d, IB analysis of GSC23 treated with IFNβ (5 ng/ml, c) or culture supernatants from GSC23 with or without GCDH KD (d) for indicated time. IFNβ was added every 2 days for the duration of experiments. The supernatants with or without IFNAR blocking antibody were replaced every 2 days.
e, IF staining for IFNα, Kcr, Kac, Kglu and GCDH in indicated sections from GSC23-derived intracranial tumours (n = 3 biologically independent mice). Scale bar, 20 μm.
f, RT-qPCR analysis of human ISGs in GSC23-derived intracranial tumour tissues (n = 4 biologically independent mice).
g, The gating strategy of GSCs in flow cytometric analysis.
h, i, Flow cytometry plots (h) and quantification (i, n = 4 biologically independent mice) of SOX2+ CD133+ GSCs in CD147+ human tumour cells as indicated.
Representative of two independent experiments in a, c and d. Data are presented from three independent experiments in b. In b, f and i, data are presented as mean ± SEM. One-way ANOVA followed by multiple comparisons with adjusted p values for f and i.
Extended Data Fig. 6. Nuclear-localized GCDH interacts with CBP to affect histone H4 Kcr.
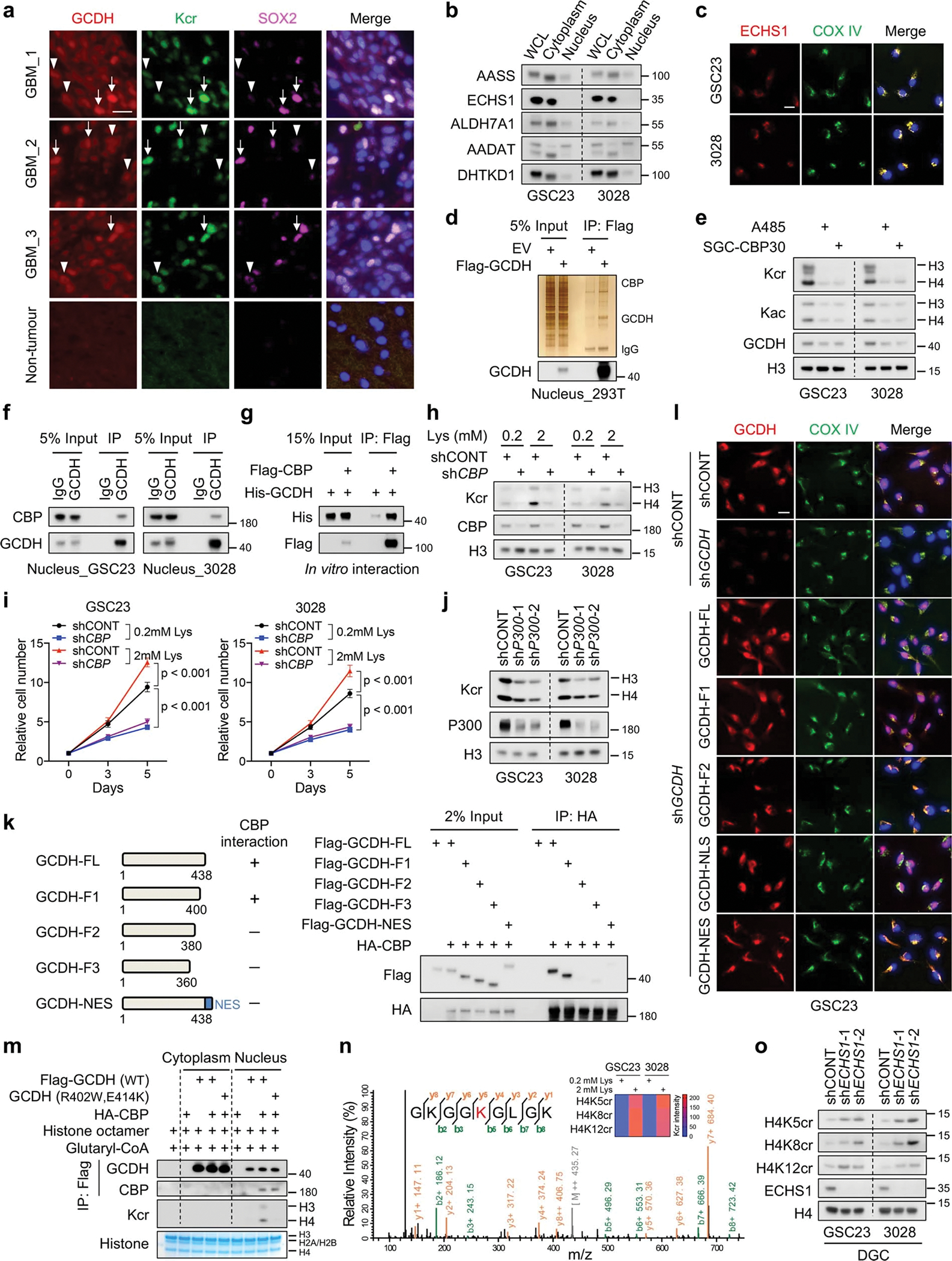
a, IF staining of GCDH, Kcr and SOX2 in tumour (n = 3 biologically independent samples) and non-tumour brain tissues of GBM surgical specimens. Arrows, GSC-like tumour cells; arrowheads, differentiated tumour cells. Scale bar, 20 μm.
b, IB analysis of cytoplasmic and nuclear fractions of indicated proteins in GSCs.
c, IF staining of ECHS1 in GSCs. Scale bar, 20 μm.
d, IP-purified Flag-GCDH protein complex from the nuclear fraction of 293T cells was analysed by silver staining and IB.
e, IB analysis of GSCs treated with 1 μM A485 or 2 μM SGC-CBP30 for 3 days.
f, IB analysis of endogenous interaction between GCDH and CBP in the nuclei of GSCs.
g, In vitro interaction assay with recombinant GCDH and CBP proteins.
h, i, IB (h) and cell proliferation (i) of GSCs with or without CBP KD cultured in media with indicated concentrations of L-lysine.
j, IB analysis of Kcr in GSCs upon P300 KD.
k, IB analysis of IP in 293T cells co-transfected with the indicated plasmids. Summary of GCDH fragments used to interact with CBP is shown on the left.
l, IF staining of GCDH fragments in GSC23. Scale bar, 20 μm.
m, Nuclear GCDH complex crotonylates histone in vitro. Purified cytoplasmic or nuclear GCDH complexes from 293T cells transfected with indicated plasmids were incubated with histone octamer and glutaryl-CoA.
n, Representative MS spectrum of histone H4K8 crotonylated peptide from GSC23 cultured in media with 2 mM L-lysine. Heatmap summarizes the quantification of crotonylation signals at H4K5, H4K8 and H4K12 in GSCs.
o, IB analysis of H4 Kcr in DGCs with or without ECHS1 KD.
Representative of two independent experiments in a-h, j-m and o. Data are presented as mean ± SEM from three independent experiments in i. Two-way ANOVA followed by multiple comparisons with adjusted p values for i.
Extended Data Fig. 7. MYC directly regulates GCDH expression.
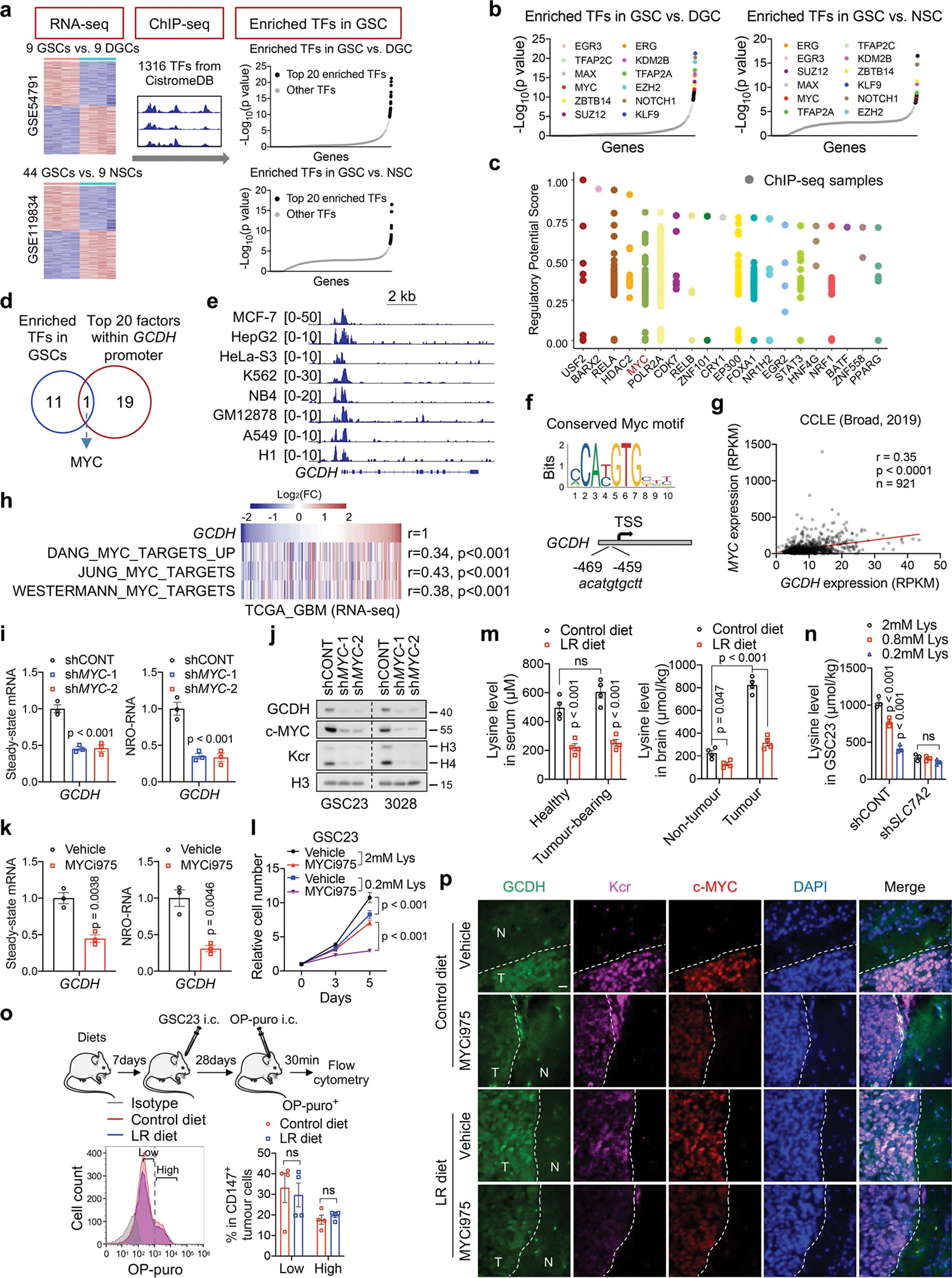
a, Diagram depicting the screening strategy to identify enriched TFs with selective dependency in GSCs.
b, 12 overlapped TFs responsible for DEGs between GSCs and DGCs/NSCs among the top 20 enriched TFs.
c, Top 20 upstream regulators for GCDH transcription, ranked by regulatory potential (RP) score from BETA algorithm. Each dot represents a ChIP-seq sample with analysed TFs labelled on the X axis. TFs with high RP scores are more likely to regulate GCDH.
d, Venn diagram showing the overlapped TFs.
e, MYC ChIP-seq tracks at GCDH gene locus in 8 human cell lines from ENCODE database.
f, The promoter of GCDH harbours a conserved MYC-binding element.
g, h, Correlation between GCDH and MYC mRNA (g) or signature (h).
i-k, RT-qPCR (i, k) and IB (j) analysis of steady-state mRNA, nascent transcripts or protein of GCDH in GSCs with or without MYC inhibition. NRO, nuclear run-on.
l, Cell proliferation of GSC23 cultured in indicated media and treated with or without 0.2 μM MYCi975 for 5 days.
m, Lysine levels in serum and brain tissues from healthy NSG mice or NSG mice bearing GSC23-derived intracranial tumours after dietary lysine restriction for 4 weeks (n = 4 biologically independent mice).
n, Intracellular lysine levels of GSC23 cultured in indicated media for 5 days.
o, Flow cytometry plots and quantification (n = 4 biologically independent mice) of protein synthesis rate in CD147+ human tumour cells as indicated.
p, IF staining of indicated sections from GSC23-derived intracranial tumours (n = 3 biologically independent mice). N, non-tumour; T, tumour. Scale bar, 20 μm.
Data are presented from three independent experiments in i, k, l and n. Representative of two independent experiments in j. Data are presented as mean ± SEM in i and k-o. Pearson’s correlation with two-tailed test for g and h, one-way ANOVA followed by multiple comparisons with adjusted p values for i, m and n, two-tailed unpaired t test for k and o, two-way ANOVA followed by multiple comparisons with adjusted p values for l. ns, not significant.
Extended Data Fig. 8. Histone Kcr governs retrotransposon expression by remodelling the chromatin landscape.
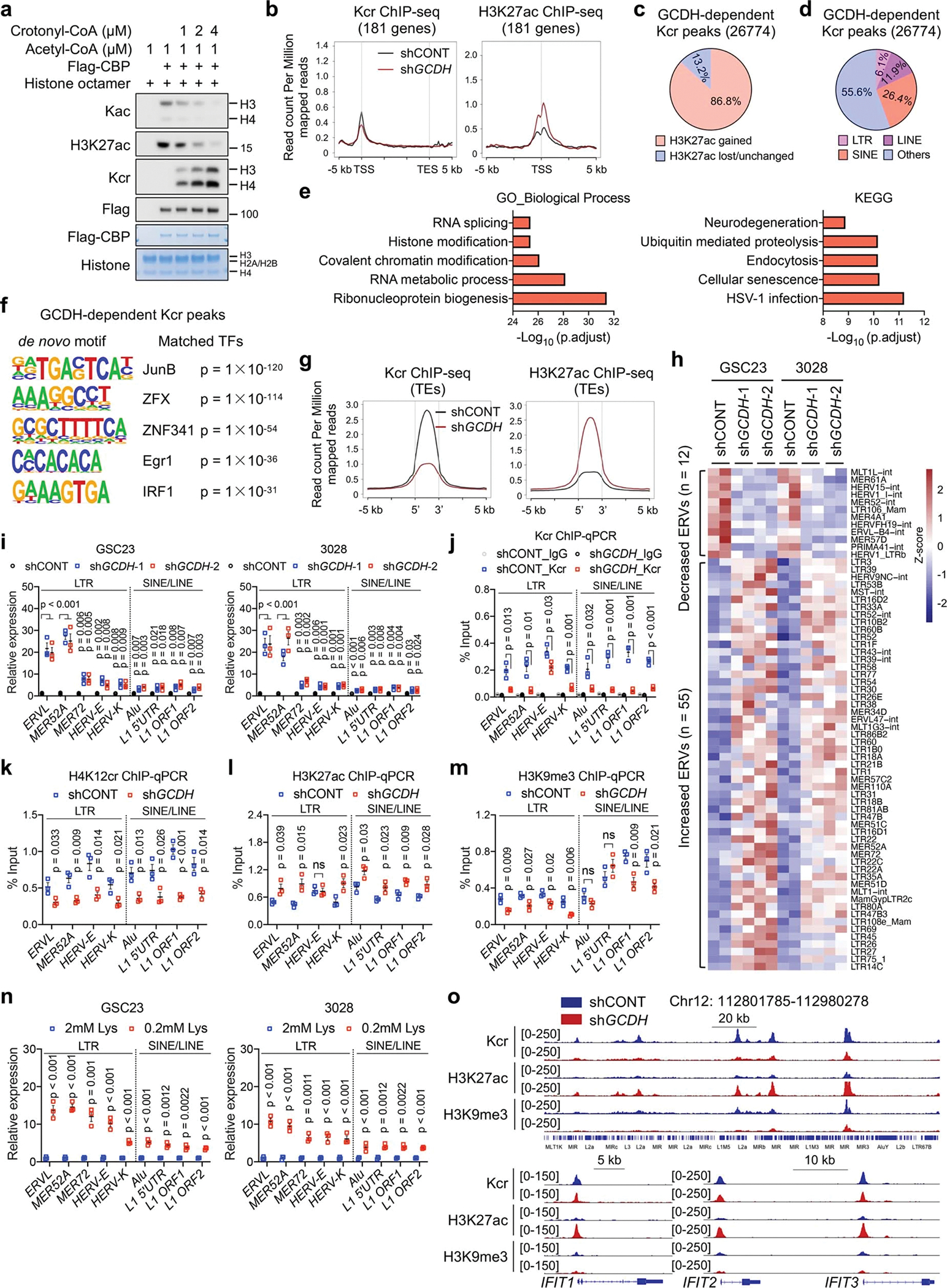
a, IB analysis of Kac and Kcr by in vitro acetylation and crotonylation assays.
b, Kcr and H3K27ac ChIP-seq signals at genomic loci of lysine catabolism-repressed genes in control and GCDH KD GSC23.
c, Pie charts showing proportions of increased and lost/unchanged H3K27ac signals within GCDH-dependent Kcr peaks after GCDH loss.
d, Percentages of TEs (LTRs, LINEs and SINEs) in GCDH-dependent Kcr peaks.
e, GO and KEGG analysis of GCDH-dependent Kcr occupied genes, ranked by adjusted p values.
f, Motif sequences (left) and 5 best matched TFs with corresponding p value (right) from de novo motif analysis of GCDH-dependent Kcr peaks.
g, Tag density of Kcr and H3K27ac profiles on TEs (LTRs, LINEs and SINEs) upon GCDH loss.
h, Heatmap summarizing the differential transcript expression of LTR-containing ERVs from multiple genomic loci between control and GCDH KD GSCs.
i-n, RT-qPCR (i, n) and ChIP-qPCR (j-m) analyses of representative TEs with primers that detected overall levels of the corresponding TE subfamilies from multiple genomic loci. Data are presented as mean ± SEM from three independent experiments. Two-tailed unpaired t test was used for statistical analysis. ns, not significant.
o, Genome browser snapshots of Kcr, H3K27ac and H3K9me3 signals at indicated genomic loci of TEs (upper panel) and ISGs (bottom panel) from control and GCDH KD GSC23.
Extended Data Fig. 9. Retrotransposons boost IFN signalling through cytosolic dsRNA and dsDNA sensing pathways.
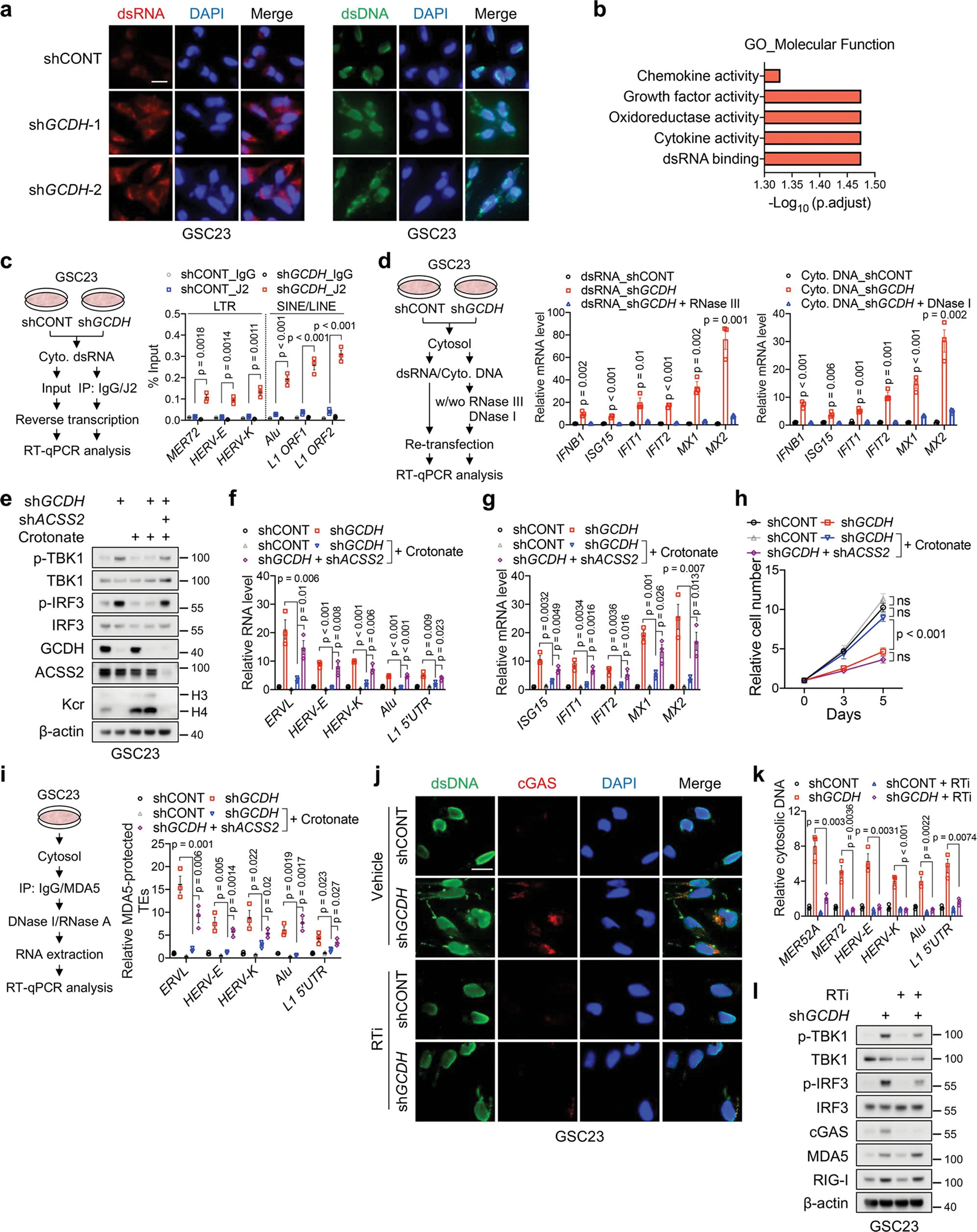
a, IF staining of dsRNA (left panels) and dsDNA (right panels) in GSC23 with or without GCDH KD. Scale bar, 20 μm.
b, GO enrichment analysis of 308 upregulated genes upon GCDH KD in two GSCs, ranked by adjusted p values.
c, RT-qPCR analysis of indicated retrotransposon transcripts captured by dsRNA-specific antibody (J2) in pulldown assay. Protocol is shown on the left.
d, RT-qPCR analysis of ISGs in GSC23 transfected with cytosolic dsRNA or DNA.
e-h, IB (e), RT-qPCR analysis of representative TEs (f) and ISGs (g), and cell proliferation (h) of GSC23 with indicated gene depletion or 2mM crotonate supplementation in culture media for 5 days.
i, RT-qPCR analysis of representative TEs in MDA5-protected fractions after indicated gene depletion or 2mM crotonate supplementation in culture media for 5 days in GSC23.
j-l, IF staining (j), qPCR analysis of TEs in cytosolic DNA (k) and IB analysis (l) of control and GCDH KD GSC23 treated with or without a reverse transcriptase inhibitor (RTi, 10 μM) for 48 h. Each TE level in cytosolic DNA was normalized to its expression in DNA isolated from nuclear lysate and control group. Scale bar, 20 μm.
Representative of two independent experiments in a, e, j and l. Data are presented as mean ± SEM from three independent experiments in c, d, f-i and k. One-way ANOVA followed by multiple comparisons with adjusted p values for c, d, f, g, i and k, two-way ANOVA followed by multiple comparisons with adjusted p values for h. ns, not significant.
Extended Data Fig. 10. Lysine catabolism disruption enhances tumour immunogenicity.
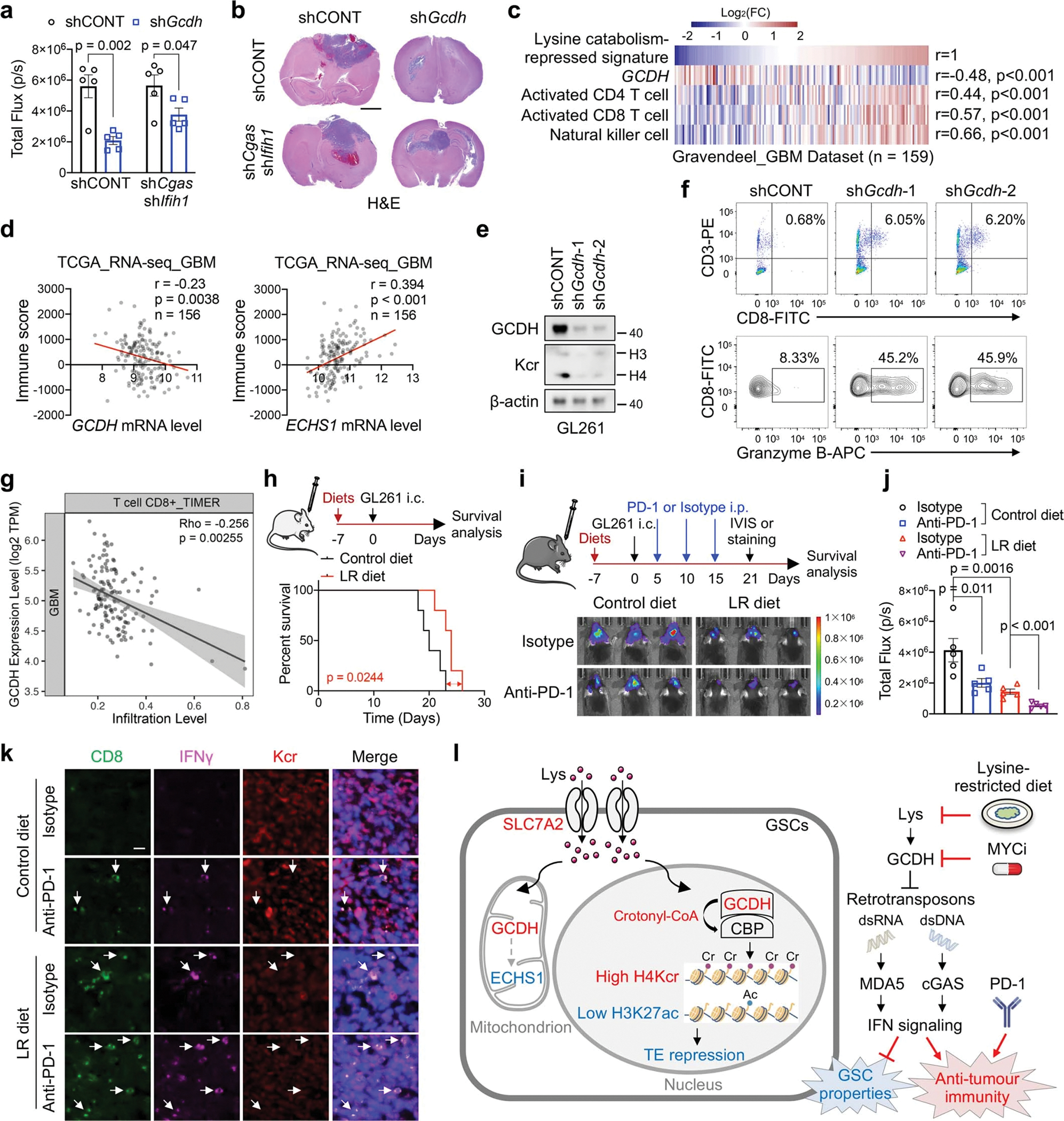
a, b, Quantification of in vivo bioluminescence imaging (n = 5 biologically independent mice, a) and H&E-stained coronal sections (b) of immunocompetent mice bearing GL261-derived intracranial tumours at 3 weeks. Scale bar, 2 mm.
c, Correlations between lysine catabolism-repressed signature, GCDH mRNA level, activated CD4 T cell, activated CD8 T cell and NK cell in GBM dataset.
d, Correlation of GCDH, ECHS1 mRNA levels and immune scores based on ESTIMATE algorithm in TCGA GBM dataset.
e, IB analysis of Kcr in GL261 upon Gcdh KD.
f, Flow cytometric analysis of CD8+ T cells (upper panel) and Granzyme B+ CD8+ T cells (bottom panel) in GL261-derived intracranial tumours at day 14 post-injection.
g, Scatter plots showing the correlation between GCDH expression levels and CD8+ T cell infiltration scores in TCGA GBM dataset from TIMER database. The gray area indicates 95 % confidence interval for the regression line.
h, Kaplan-Meier survival plots of immunodeficient mice bearing GL261-derived intracranial tumours fed control or LR diet starting at 7 days before transplantation (n = 5).
i, j, In vivo bioluminescence imaging (i) and quantification of total flux (n = 5 biologically independent mice, j) of immunocompetent mice bearing GL261-derived intracranial tumours with anti-PD-1, LR diet or the combination treatment.
k, IF staining for CD8, IFNγ and Kcr in indicated sections (n = 3 biologically independent mice). Scale bar, 20 μm.
l, A working model showing that disruption of reprogrammed lysine catabolism-derived histone Kcr suppresses GSC maintenance and induces anti-tumour immune response by promoting immunogenic retroelements and IFN signalling.
Representative of two independent experiments in e. In a and j, data are presented as mean ± SEM. Two-tailed unpaired t test for a, Pearson’s correlation with two-tailed test for c and d, Spearman’s Rho with two-tailed test was used for g, log-rank test for h, one-way ANOVA followed by multiple comparisons with adjusted p values for j.
Supplementary Material
Acknowledgements
We thank the Metabolomics and Lipidomics Core for mass spectrometry analysis, the Biospecimen Core for histology analysis and the Flow Cytometry Core Facility in University of Pittsburgh. We thank Steven J. Mullett for technical support in metabolomics. H.Y. is supported by Hillman Fellow for Innovative Cancer Research Program. This work was supported by start-up funds from the University of Pittsburgh (J.N.R.). Y.Z. is supported by Nancy and Leonard Florsheim family fund, NIH grants GM135504, AR078555, and CA251677. N.W.S. is supported by NIH grant R01GM132261. J.N.R. is supported by NIH grants R35CA197718, R01CA238662, and R01NS103434.
Footnotes
Competing interests
Y.Z. is a founder, board member, advisor to, and inventor on patents licensed to PTM Bio Inc. (Hangzhou, China and Chicago, IL) and Maponos Therapeutics Inc. (Chicago, IL). The other authors declare no competing interests.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
RNA-seq data for three pairs of GSCs and DGCs are from GSE54791. RNA-seq data for 44 GSC and 9 NSC lines are from GSE119834. Matched pairs of GSCs and DGCs H3K27ac ChIP-seq data are from GSE129438. GSCs and NSCs H3K27ac ChIP-seq are from GSE119755. scRNA-seq and snRNA-seq data are publicly available in Broad Institute Single-Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP503). Original western blots are included in the Supplementary Information. All metabolomics data are provided in the Supplementary Table and Source Data. Raw sequencing data and processed data generated in this study are available on the Gene Expression Omnibus through accession number GSE208618. Source data are provided with this paper.
REFERENCES
- 1.Gimple RC, Yang K, Halbert ME, Agnihotri S & Rich JN Brain cancer stem cells: resilience through adaptive plasticity and hierarchical heterogeneity. Nat. Rev. Cancer 22, 497–514 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Wang W & Zou W Amino Acids and Their Transporters in T Cell Immunity and Cancer Therapy. Mol. Cell 80, 384–395 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieu EL, Nguyen T, Rhyne S & Kim J Amino acids in cancer. Exp. Mol. Med. 52, 15–30 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olin-Sandoval V et al. Lysine harvesting is an antioxidant strategy and triggers underground polyamine metabolism. Nature 572, 249–253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Z et al. TPO-Induced Metabolic Reprogramming Drives Liver Metastasis of Colorectal Cancer CD110+ Tumor-Initiating Cells. Cell Stem Cell 17, 47–59 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Jackson CM, Choi J & Lim M Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat. Immunol. 20, 1100–1109 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Singh SK et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Bao S et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Maddocks ODK et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Gao X et al. Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang Y et al. Histone crotonylation promotes mesoendodermal commitment of human embryonic stem cells. Cell Stem Cell 28, 748–763 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai SK et al. Histone crotonylation regulates neural stem cell fate decisions by activating bivalent promoters. EMBO Rep 22, e52023 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minet P et al. Clinical and biochemical outcome of a patient with pyridoxine-dependent epilepsy treated by triple therapy (pyridoxine supplementation, lysine-restricted diet, and arginine supplementation). Acta Neurol. Belg. 121, 1669–1675 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Kava MP et al. Beneficial outcome of early dietary lysine restriction as an adjunct to pyridoxine therapy in a child with pyridoxine dependant epilepsy due to Antiquitin deficiency. JIMD Rep 54, 9–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zinnanti WJ et al. A diet-induced mouse model for glutaric aciduria type I. Brain 129, 899–910 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Biagosch C et al. Elevated glutaric acid levels in Dhtkd1-/Gcdh-double knockout mice challenge our current understanding of lysine metabolism. Biochim Biophys Acta Mol Basis Dis 1863, 2220–2228 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Suva ML et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 157, 580–594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su A et al. The Folate Cycle Enzyme MTHFR Is a Critical Regulator of Cell Response to MYC-Targeting Therapies. Cancer Discov. 10, 1894–1911 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okada M et al. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. Int. J. Mol. Sci. 22, 11633 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med. 11, eaau4972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 20, 661–673 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hajji N et al. Arginine deprivation alters microglial polarity and synergizes with radiation to eradicate non-arginine-auxotrophic glioblastoma tumors. J. Clin. Invest. 132, e142137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mack SC et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J. Exp. Med. 216, 1071–1090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards LM et al. Gradient of Developmental and Injury Response transcriptional states defines functional vulnerabilities underpinning glioblastoma heterogeneity. Nat Cancer 2, 157–173 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Sabari BR et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 58, 203–215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X et al. MOF as an evolutionarily conserved histone crotonyltransferase and transcriptional activation by histone acetyltransferase-deficient and crotonyltransferase-competent CBP/p300. Cell Discov 3, 1–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang X et al. Short-Chain Enoyl-CoA Hydratase Mediates Histone Crotonylation and Contributes to Cardiac Homeostasis. Circulation 143, 1066–1069 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Tan M et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanpouille-Box C, Demaria S, Formenti SC & Galluzzi L Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 34, 361–378 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Chen YG & Hur S Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 23, 286–301 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller KN et al. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184, 5506–5526 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keyser B et al. Disease-causing missense mutations affect enzymatic activity, stability and oligomerization of glutaryl-CoA dehydrogenase (GCDH). Hum. Mol. Genet. 17, 3854–3863 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Qin Q et al. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 21, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts TC et al. Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nat. Protoc. 10, 1198–1211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han H et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 36, 483–497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehdipour P et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 588, 169–173 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Zhang SM et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 598, 682–687 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Charoentong P et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 18, 248–262 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Gravendeel LA et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 69, 9065–9072 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Chiappinelli KB et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162, 974–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheng W et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffin GK et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 595, 309–314 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Llorens-Bobadilla E et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell 17, 329–340 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Snyder NW et al. Production of stable isotope-labeled acyl-coenzyme A thioesters by yeast stable isotope labeling by essential nutrients in cell culture. Anal. Biochem. 474, 59–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frey AJ et al. LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and 13C-isotopic labeling of acyl-coenzyme A thioesters. Anal. Bioanal. Chem. 408, 3651–3658 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trefely S, Ashwell P & Snyder NW FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC Bioinformatics 17, 485 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Y, Kwon SW, Kim SC & Zhao Y Integrated Approach for Manual Evaluation of Peptides Identified by Searching Protein Sequence Databases with Tandem Mass Spectra. J. Proteome Res. 4, 998–1005 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Criscione SW, Zhang Y, Thompson W, Sedivy JM & Neretti N Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15, 1–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng R et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li T et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data for three pairs of GSCs and DGCs are from GSE54791. RNA-seq data for 44 GSC and 9 NSC lines are from GSE119834. Matched pairs of GSCs and DGCs H3K27ac ChIP-seq data are from GSE129438. GSCs and NSCs H3K27ac ChIP-seq are from GSE119755. scRNA-seq and snRNA-seq data are publicly available in Broad Institute Single-Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP503). Original western blots are included in the Supplementary Information. All metabolomics data are provided in the Supplementary Table and Source Data. Raw sequencing data and processed data generated in this study are available on the Gene Expression Omnibus through accession number GSE208618. Source data are provided with this paper.