

Abstract
Chronic antigen stimulation is thought to generate dysfunctional CD8 T cells. Here we identify a CD8 T cell subset in the bone marrow tumor microenvironment that, despite an apparent terminally exhausted phenotype (TPHEX), expressed granzymes, perforin, and IFNγ. Concurrent gene expression and DNA accessibility revealed that genes encoding these functional proteins correlated with BATF expression and motif accessibility. IFNγ+ TPHEX effectively killed myeloma with comparable efficacy to transitory effectors, and disease progression correlated with numerical deficits in IFNγ+ TPHEX. We also observed IFNγ+ TPHEX within CD19-targeted chimeric antigen receptor T cells, which killed CD19+ leukemia cells. Importantly, an IFNγ+ TPHEX gene signature was recapitulated in TEX cells from human cancers, including myeloma and lymphoma. Here we characterize a TEX subset in hematological malignancies that paradoxically retains function and is distinct from dysfunctional TEX found in chronic viral infections. Thus, IFNγ+ TPHEX represent a potential target for immunotherapy of blood cancers.
One sentence summary:
A subset of CD8 T cells with a terminally exhausted epigenetic profile expressed IFNγ, granzymes and perforin in bone marrow.
Introduction:
The characterization and understanding of CD8 T cell exhaustion is constantly evolving due to the discovery of several transcription factors associated with exhausted T cell (TEX) phenotypes. The gain of expression of TOX and NR4A family transcription factors and the loss of TCF-1 expression have recently been associated with a TEX phenotype in mice and humans.1–5 Maintenance of TCF-1 expression marks a precursor exhausted population (TPEX) with stem-like properties and the ability for self-renewal, which is crucial for sustaining an immune response to chronic infection and tumors.6–8 Despite debate surrounding the overall relevance of exhaustion signatures driven by chronic viral infection to the tumor microenvironment (TME), there is consensus that terminally exhausted CD8 T cells are dysfunctional in both settings.1–3,9 However, there is increasing complexity of transcriptional networks (e.g. BATF, IRF4, NFAT)10–14 that, dependent on the context and timing of expression, have been shown to drive either a cytotoxic effector or exhausted phenotype. This suggests that broad use of the terminology ‘dysfunctional’ to describe CD8 T cells with a surface marker and transcriptional profile classically associated with terminal exhaustion may not capture functional nuances. Additionally, there is considerable variation in the composition of TMEs associated with cancer subtype and anatomical site, particularly between solid and hematologic malignancies,15,16 that may further impact the functionality of TEX.
The recent description of ‘intermediate’ stages of exhaustion in CD8 T cells has added further complexity to the field.17 Firstly, CD8 T cells with a ‘restrained’ phenotype that exhibit features of both T cell exhaustion and effector function have been described in autoimmunity18 and PD-1+ T cells expressing granzyme B have been described in a murine model of lung cancer.19 Secondly, ‘transitory’ CD8 T cells, that express exhaustion markers, including TIM-3 and TOX, while maintaining expression of effector cell markers such as CX3CR1 and T-BET, have been described in cancer and chronic infection models and in response to immune checkpoint blockade in humans.17,20–22 Thirdly, TOX-expressing effector memory T cells have been described in the context of circulating viral-specific T cells in humans.23 It is important to note, that these described subsets likely represent intermediate stages of exhaustion as they retain expression of several markers for effector T cell differentiation and do not display hallmarks of terminal exhaustion. Together, these data highlight the heterogeneity that exists within CD8 T cells that are committed to the exhaustion pathway.
In this study, we sought to characterize terminal exhaustion trajectories within CD8 T cells in hematological malignancies using murine models of myeloma relapsing after autologous stem cell transplantation (ASCT) and leukemia relapsing after CD19-directed chimeric antigen receptor (CAR) T cell therapy. These immunocompetent, orthotopic murine models provide critical insight into tumor-immune cell interactions in the bone marrow (BM) TME after clinically relevant therapies. We used single cell multi-omic sequencing with confirmatory flow cytometry and identified distinct terminal TEX subsets in the BM of mice with relapsed myeloma. One TEX subset was analogous to dysfunctional cells described in solid tumors and chronic viral infection with reduced expression of effector molecules, while the other was characterized by co-expression of Havcr2 and Cd28 and had high expression of IFNγ, perforin and granzymes in mice with progressive disease. The TEX subset that expressed interferon gamma despite having a phenotype consistent with terminal exhaustion (IFNγ+ TPHEX), exhibited increased Batf, Cd28 and Ly6a expression with enhanced accessibility in the Batf motif binding domain. In mice, loss of tumor control was associated with an outgrowth of myeloma cells relative to IFNγ+ TPHEX, rather than T cell dysfunction per se, since IFNγ+ TPHEX effectively killed myeloma targets ex vivo. IFNγ+ TPHEX also differentiated within CD19 CAR T cells in response to B cell acute lymphoblastic leukemia (B-ALL) and killed B-ALL cells ex vivo, highlighting the capacity of these T cells with a terminally exhausted phenotype to retain functional capacity. An analogous IFNγ+ TPHEX signature was associated with BATF expression in tumor-infiltrating lymphocytes in patients with myeloma and analogous TIM-3+CD28+CX3CR1− CD8 T cells were expanded after ASCT.
Results:
Myeloma generates distinct TEX signatures in the bone marrow tumor microenvironment
The majority of studies exploring mechanisms of T cell exhaustion in cancer have been performed in solid tumor murine models, in which tumors often universally relapse or regress in response to interventions. Here we describe a tumor model in which some mice relapse whereas other mice maintain long-term tumor control after autologous stem cell transplantation (SCT). SCT is the clinical standard-of-care for patients with myeloma and, as faithfully recapitulated in our murine model, only a small subset of patients achieve long-term progression-free survival. SCT promotes a polyclonal myeloma-specific effector T cell response that is largely mediated by CD8 T cells.24 We used single cell RNA sequencing to capture key stages of CD8 T cell differentiation in the BM of mice with relapsed myeloma 7 weeks after SCT (Fig. 1A). Briefly, we sorted CD8 T cells into three populations based on CD38 and CD101 expression (CD38−CD101− = naïve; CD38+CD101− = activated; CD38+CD101+ = exhausted)25 and re-pooled these cells at an adjusted ratio (Fig. 1A). In this dataset, we identified 4 populations of exhausted T cells (TEX) characterized by expression of Tox and loss of Tcf7, which were distinguished by expression of Mki67, Maf, Ly6a, Havcr2, Cd28 and/or distinct cytotoxic profiles (Fig. 1A and Fig. S1A). RNA velocity analysis26 suggests a common trajectory from a precursor exhausted (TPEX) cluster towards the TEX clusters in our dataset (Fig. 1A), which also resembles described pathways of TEX differentiation in chronic infection and solid tumors.6–8 Interestingly, we observed three clusters of cells with hallmarks of terminal exhaustion in our dataset that could be differentially distinguished by expression of Ly6a, Cd28 and Havcr2 (Fig. S1A). The Cd28-expressing cluster had very high expression of genes encoding co-inhibitory molecules, IFNγ, perforin, granzyme B, in addition to exhaustion-associated markers Tox, Nr4a2, Eomes, Ctla4, and Entpd1 (Fig. 1A) and are referred to as IFNγ+ TPHEX. A second cluster expressed intermediate levels of Tox, Nr4a2, Eomes, Ifng and high levels of Jun and are referred to as Jun+ TEX. Notably, in the third cluster, loss of Ly6a and Cd28 was associated with reduced Ifng and Gzmb expression and increased expression of Gzma. This cluster is referred to as GzmA+ TEX. By using the UMAP transform function, we found that these cells were transcriptionally similar to a previously described terminally exhausted, dysfunctional population of SV40-specific T cells that recognize solid tumors expressing SV40 large T antigen (Fig. 1B, Fig. S1A).1 To draw additional parallels to published datasets of exhausted T cells, we used CD8 T cells from a melanoma model (Yummer1.7)27 as a reference dataset and found that a cluster of cells (cluster 1) were transcriptionally similar to IFNγ+ TPHEX cells using multinomial logistic regression (Fig. 1C). We next co-embedded our data with CD8 T cell data from mice infected with LCMV, either Armstrong (acute infection) or clone 13 (chronic infection) (Fig. 1D and Fig. S1B).17 The effector, cell-cycling, TPEX, and naïve T cell clusters readily co-localized across datasets, indicating similarity in gene expression across similarly labelled cells in these two datasets. Lending further validation to these findings, TPEX, identified by co-expression of Ly108 and PD-1, were TOX+ and TCF-1+ in our model, in line with published data (Fig. S1C).5,28 Several transitional and cytotoxic lymphocyte populations were specific to the LCMV dataset, however these clusters were largely found in mice infected with acute but not chronic LCMV and were absent from our myeloma dataset (Fig. S1B).17 Strikingly however, the IFNγ+ TPHEX cluster from our dataset did not imbricate with any T cell clusters from LCMV-infected mice despite the other terminally exhausted T cell subsets showing close co-localization (Fig. 1D). To determine which genes were uniquely expressed in IFNγ+ TPHEX, we compared differentially expressed genes in this subset to the other TEX clusters in our dataset and generated a IFNγ+ TPHEX signature (Fig. S2A and Data File S1). Genes associated with natural killer cells (Klrd1, Klrk1, Klre1) were differentially expressed in other TEX whereas Ccl5, Malat1, Ier3, Maf, Cd28, Id2, Gzmb and Havcr2 were differentially enriched in IFNγ+ TPHEX. The latter genes have been associated with T cell persistence, recruitment of lymphocytes to the TME, and memory differentiation.29–32
Figure 1: Myeloma generates distinct TEX signatures in the bone marrow.
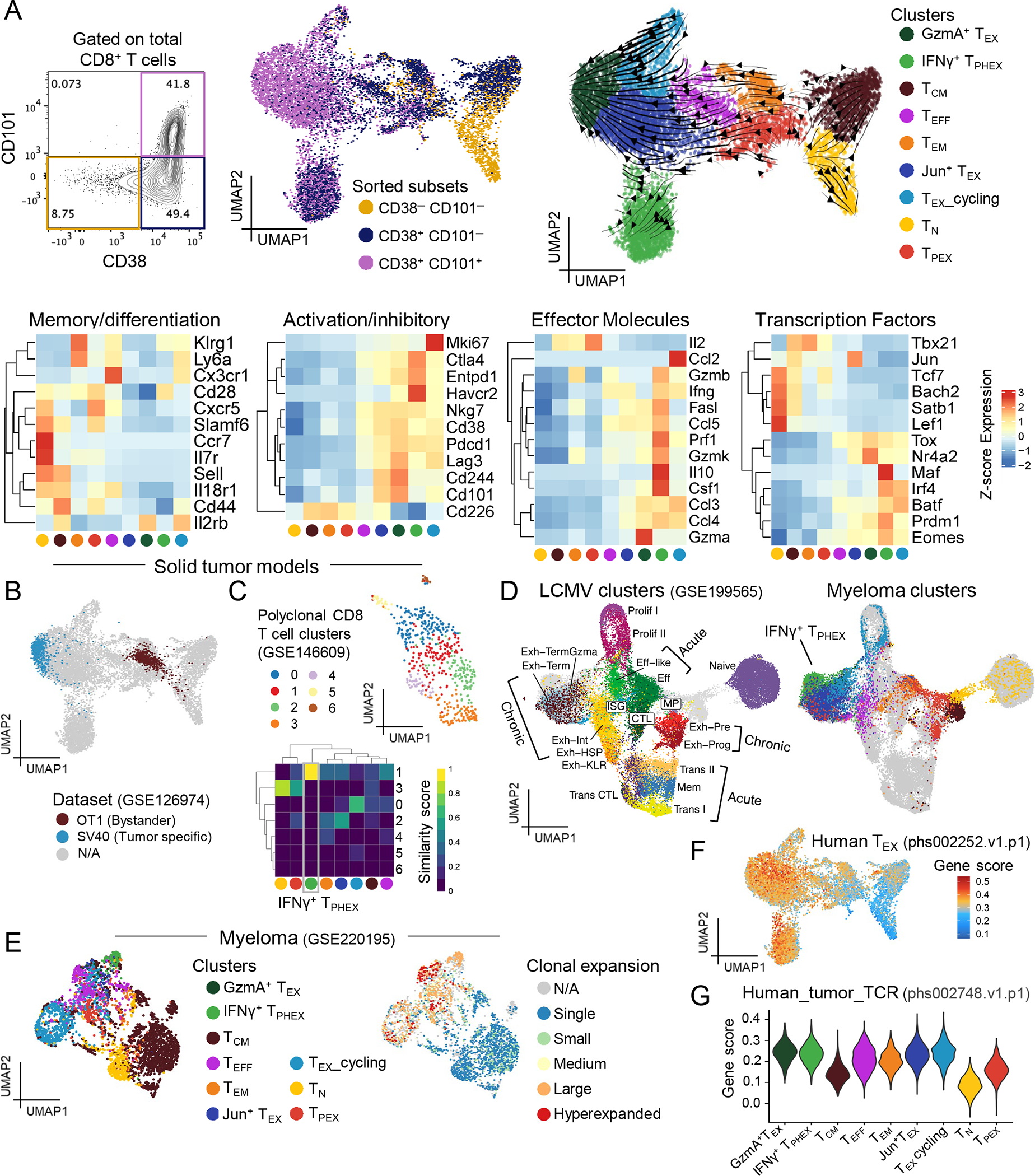
(A) UMAP embedding of RNA data from CD8 T cells isolated from BM of mice with relapsed myeloma (n = 4 mice pooled) at 7 weeks post-transplant. Cells are colored by cluster with RNA velocity vectors depicting differentiation trajectory. Heatmaps of average gene expression across identified clusters. (B) Pseudo single cell projection of bulk RNA expression data from SV40 (exhausted phenotype) and OT1 (effector phenotype) T cells. (C) Embedding of polyclonal CD8 T cells from Yummer1.7 melanoma model colored by clusters with ViewmastR heatmap identifying cluster 1 as being analogous to IFNγ+ TPHEX. (D) Co-embedding of single cell RNA expression data from CD8 T cell subsets in (A) and from LCMV infection colored by clusters. (E) Embedding of CD8 T cells from BM of MM-bearing mice at 4 weeks post-SCT showing clusters analogous to those identified in (A) using ViewmastR (left) and colored by TCR clone size (right). Small = 1–5, medium = 6–20, large = 21–100, hyperexpanded = 101–500. (F) Embedding colored by gene score from terminally exhausted human T cells from renal carcinoma dataset. (G) Violin plot of gene score from human neoantigen-specific CD8 T cells within each cluster from (A).
We next used a reference of our previously published gene-expression and TCR-sequencing data from CD8 T cells isolated from the BM of Vk*MYC MM-bearing mice at 4 weeks post-SCT to explore clonality and draw parallels to the TEX cells in our data.33 Using multinomial linear regression, we were able to identify a clearly defined IFNγ+ TPHEX cluster in the reference (Fig. 1E). Given the degree to which these cells and other ‘exhausted’ cells were clonally expanded in the reference, and our prior work showing that SCT generates myeloma-specific T cell memory in this model,24 we hypothesized that IFNγ+ TPHEX were tumor specific. In further support of tumor-reactivity, our IFNγ+ TPHEX cells displayed enrichment for gene signatures from terminally exhausted and tumor-specific T cells in humans (Fig. 1F–G).34,35
TIM-3+ CD8 T cells with a terminally exhausted phenotype produce cytotoxic molecules and IFNγ in vivo
Based on the high expression of Havcr2 within the IFNγ+ TPHEX cluster, we next used surface expression of PD-1 and TIM-3, measured by CITE-seq,36 to broadly subset TEX cells. We observed enhanced expression of Prf1, Gzmb and Ifng in TIM-3+PD-1+ compared to PD-1+TIM-3− cells (Fig. 2A–B). We confirmed these findings at the protein level with flow cytometry and found that all PD-1+ CD8 T cells expressed TOX but that cytotoxic molecules were largely found in the TIM-3+ subset (Fig. 2C). TIM-3+ cells identified in our model lacked Cx3cr1, Ki67, and Tbx21 (T-BET) expression and therefore are distinct from already described proliferating transitory cells and other intermediate exhaustion subsets (Fig. 1A and Fig. S1A).20,37 The TEFF cluster in our dataset likely include these transitory cells based on the overlap between the “Exh-int”17 subset from chronic LCMV infection and the TEFF cluster identified here (Fig. 1D).
Figure 2: CX3CR1−TIM-3+ TOX+ CD8 T cells produce cytolytic molecules and IFNγ in vivo.
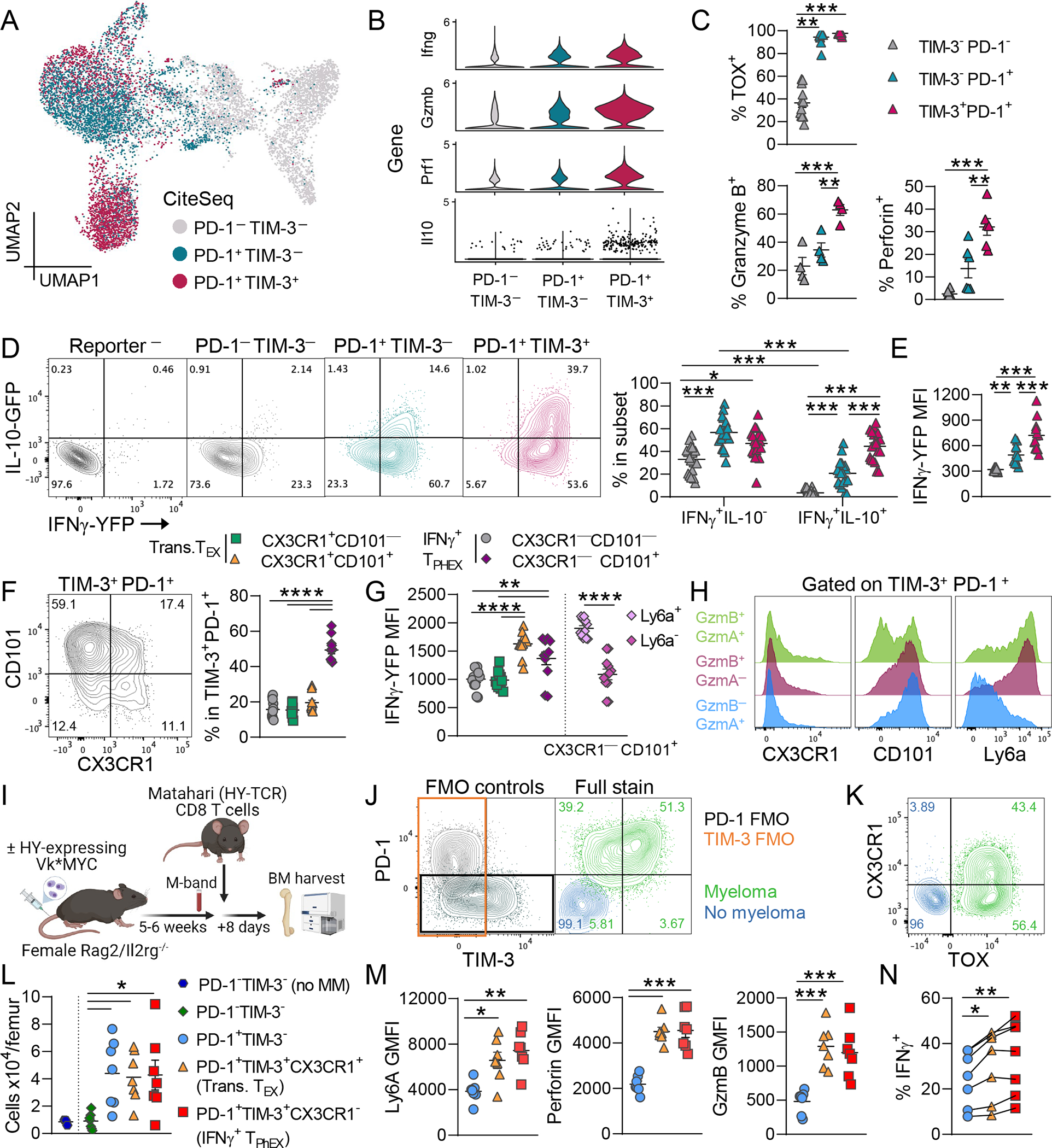
(A) UMAP embedding of RNA data from Fig. 1. Here cells are colored by subsets based on cell surface markers expression (CITE-seq) of TIM-3 and PD-1. (B) Violin plot depicting gene expression in TIM-3 and PD-1 subsets. (C) Expression of TOX (n = 12; Kruskal-Wallis test with Dunn’s test), granzyme B (n = 4), and perforin (n = 6) in TIM-3 and PD-1 subsets from BM at 7 weeks post-transplant. (D) IFNγ and IL-10 reporter protein expression in subsets with + representative plots (n = 19; Two-Way ANOVA with Sidak’s test). (E) MFI of IFNγ in IFNγ+ cells in each subset (n = 12). (F) Representative flow cytometry plot and quantification of CD101 and CX3CR1 expression on TIM-3+ PD-1+ CD8 T cells (n = 11). (G) IFNγ MFI in CD101 and CX3CR1 subsets. CX3CR1−CD101+ cells were additionally split based on expression of Ly6a (n = 11). IFNγ+ TPHEX are TIM-3+PD-1+CX3CR1−. (H) Histograms depicting CX3CR1, CD101 and Ly6a expression on TIM-3+PD-1+ T cells expressing granzyme B and granzyme A (GzmB+ GzmA+), GzmB only, and GzmA only (concatenated from n = 11). (I-N) Female Rag2/IL2rg−\− mice were naïve (no myeloma) or were injected with male HY-antigen-expressing Vk28158 (myeloma). Once M-bands were detectable, 5000 MataHari (HY-antigen specific TCR transgenic) CD8 T cells were injected, and BM was harvested 8 days later. (I) Experimental design. (J) Representative contour plots showing PD-1 and TIM-3 expression (left) FMO controls and (right) full stain and (K) CX3CR1 and TOX expression on MataHari CD8 T cells from myeloma and no myeloma mice. (L) Total number of MataHari CD8 T cells subsets based on TIM-3 and PD-1 expression. (M) MFI of Ly6a, perforin and granzyme B in subsets. (N) Frequency of IFNγ+ cells within MataHari CD8 T cell subsets after ex vivo stimulation with PMA/ionomycin (RM One-Way ANOVA). Data is mean ± SEM. Each symbol represents an individual mouse. One-Way ANOVA with Tukey’s test unless otherwise stated. * p< 0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
To validate the transcriptome data for cytokine production, we next utilized a triple-reporter mouse (IL-10-GFP x IFNγ-YFP x FoxP3-RFP) to measure cytokines without the use of ex vivo restimulation. The IFNγ reporter mice38 use a bicistronic mRNA such that the fluorescent protein is only produced when the IFNγ mRNA is translated into protein. Briefly, the enhanced-YFP reporter cassette was inserted between the translational stop codon and 3’ untranslated region (UTR)/polyA tail using an internal ribosome entry sites (IRES)-based vector. The use of IRES to generate bicistronic mRNA does not impact post-transcriptional regulation of upstream genes through the 3’UTR.39 This IFNγ reporter mouse has been validated by Reinhardt et al. and reporter protein closely correlated with IFNγ protein production in CD4 T cells.40 To confirm these findings, we show that memory CD8 T cells from spleen and BM exhibit comparable expression levels of IFNγ-YFP and IFNγ protein in response to PMA/ionomycin stimulation (Fig. S3A–B).
In our myeloma model, the majority of PD-1+ T cells expressed IFNγ-YFP with the greatest amount of in vivo IFNγ being produced by TIM-3+ cells (Fig. 2D–E and Fig. S4A). Interestingly, PMA/ionomycin stimulation did not recapitulate this effect as all subsets similarly produced IFNγ, consistent with rapid responses of memory T cells to broad stimulation in a polyclonal T cell pool (Fig. S4B). Half of the TIM-3+ cells also produced IL-10-GFP in vivo (Fig. 2D). We then measured expression of CD101 and CX3CR1 on TIM-3+PD-1+ T cells to determine whether these cells expressed previously described exhaustion (CD101) or effector (CX3CR1) markers at the protein level (Fig. 2F).20,25 The majority of TIM-3+PD-1+ cells were CD101+CX3CR1−, confirming that these cells had a terminally exhausted rather than an effector phenotype. Importantly, IFNγ production was similarly high in IFNγ+ TPHEX compared to CX3CR1+ transitory TEX, highlighting that the amount of IFNγ produced by IFNγ+ TPHEX is physiologically relevant (Fig. 2G). Additionally, IFNγ secretion was expected in CX3CR1+ cells,20,21 which provided a robust internal control for the IFNγ–YFP signal in our model. The expression of Ly6A at the protein level only, identified the most functional TIM-3+PD-1+ cells that had high expression of IFNγ and granzyme B but not granzyme A (Fig. 2G–H). To interrogate these cell subsets in a myeloma model with an antigen-specific T cell, we transferred MataHari CD8 T cells (HY-specific TCR)41,42 into female Rag2/Il2rg−/− mice that were naïve or injected with HY-antigen-expressing Vk*MYC myeloma (Vk58) (Fig. 2I). MataHari T cells expressed TIM-3, PD-1, TOX and CX3CR1 in Vk58-bearing mice but not naïve mice (Fig. 2J–K). TEX cells were expanded in tumor-bearing mice, including IFNγ+ TPHEX (PD-1+TIM-3+CX3CR1−) and transitory TEX (PD-1+TIM-3+CX3CR1+), which had higher expression of Ly6A, perforin, granzyme B and IFNγ compared to PD-1+TIM-3− cells (Fig. 2L–N). We also injected MataHari or WT T cells into recipients bearing female VK*MYC to demonstrate that the myeloma TME itself did not drive the expression of PD-1 or TIM-3 independently of tumor antigen (Fig. S5A). Notably, PD-1 and TIM-3 expression were completely absent on MataHari CD8 T cells in the absence of HY-antigen expression on myeloma (Fig. S5B). In sum, these data identify a PD-1+TIM-3+CX3CR1− T cell subset, IFNγ+ TPHEX, that produces both IFNγ and cytotoxic molecules with a terminally exhausted phenotype in the TME.
IFNγ+ TPHEX cells are marked by increased accessibility at the BATF motif
To interrogate epigenetic signatures associated with T cell control of myeloma, we performed concurrent RNA and ATAC single cell sequencing (multi-ome) on CD8 T cells from the BM of an expanded cohort of transplanted mice who either (1) were never injected with myeloma (MM-free), (2) controlled myeloma post-transplant (MM-controlled), and (3) had progressive disease (MM-relapsed) at 6 weeks post-transplant (Fig. S6A). Firstly, we broadly identified cells across stages of T cell differentiation to measure changes in expression of functional genes within the three groups (Fig. 3A). TEX cells had increased expression of Ifng, Ly6a, Il10, and Eomes with reduced expression of Tbx21 (Fig. S6B). High expression of the gene encoding the IL-10R (Il10ra; Fig. S6B) in TEX was notable given the recently described role of IL-10 signaling in maintaining TEX cell function in the TME.43,44 Next we linked gene expression to changes in chromatin accessibility using an algorithm capable of correlating these events in single cells.45 We found that TEX had greater accessibility in Ifng and Il10 linked peaks with an associated increase in gene expression (Fig. S6C). Interestingly, previously identified loci25 that showed reduced accessibility across the Ifng locus in TOX+ cells did not exhibit high levels of correlation with Ifng gene expression in our datasets (Fig.S6C). Notably, expression of Ly6a once again correlated with Ifng expression (Fig. S6B). We next identified a pseudotime trajectory from TN to TEX cells to identify transcriptomic and epigenetic differences across these transitioning cell states (Fig. 3B and Fig. S7A). Naïve and stem-like genes were highly expressed and accessible at early points in the trajectory (Bach2, Lef1). Thereafter, we observed a loss of stemness and gain of chromatin accessibility within terminal/exhaustion markers and transcription factors (Tox, Pdcd1, Havcr2, Nr4a2, Eomes) (Fig. S7A–B). Motif analysis across the psuedotime trajectory also identified NFATC1 as a regulator of exhaustion in our model (Fig. S7B). Together, these data highlight that TEX in our dataset have a canonically exhausted epigenetic signature. Yet, we again observed high expression of and accessibility within genes encoding cytotoxic molecules (Prf1 and Gzmb) and Ifng in the TEX cells found in MM-relapsed mice, which corresponded to ~75 on the pseudotime scale (Fig. 3A–C).
Figure 3: Expression of functional genes in IFNγ+ TPHEX correlates with BATF motif accessibility.
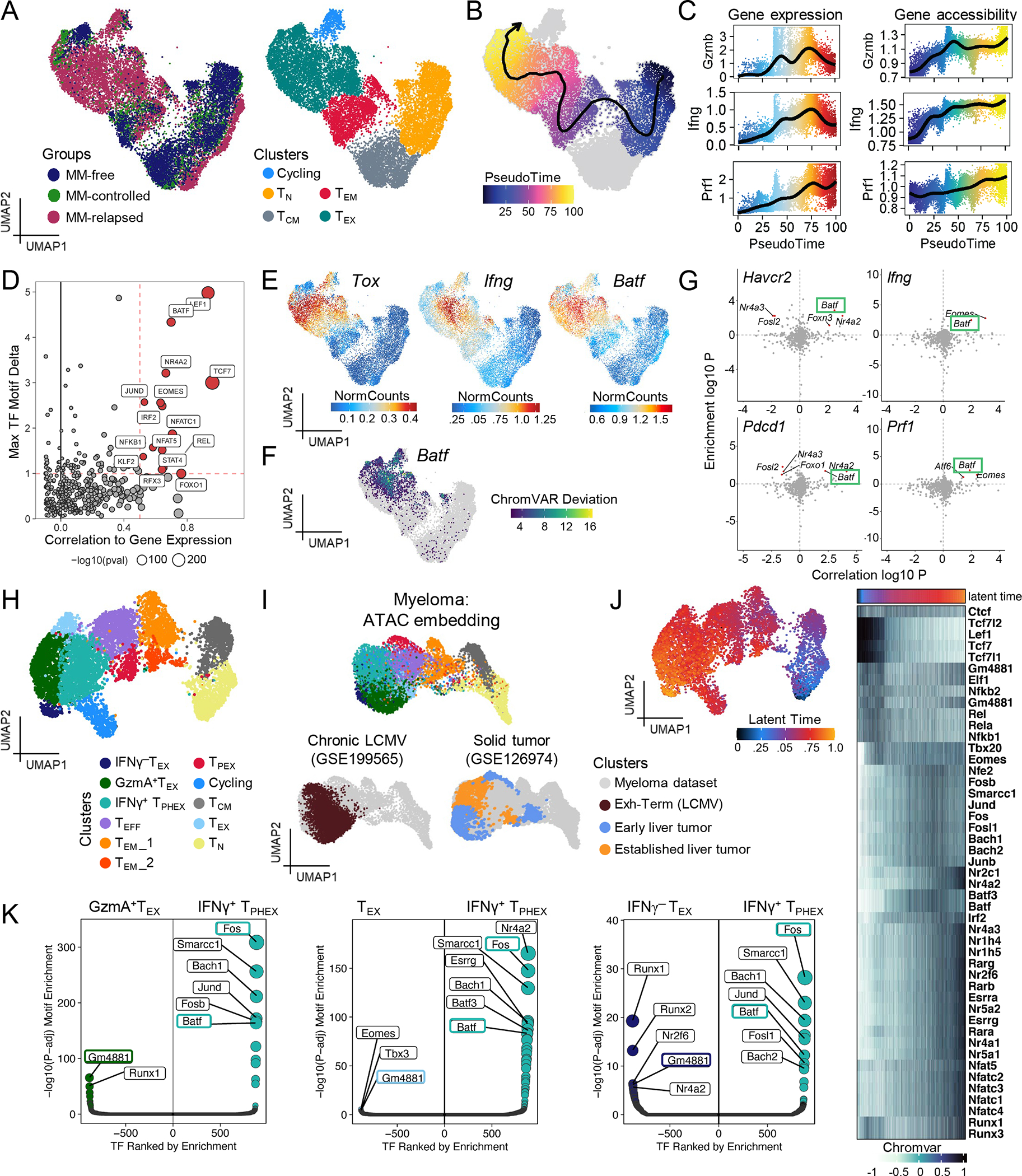
Mice were transplanted with BM and T cells from a genetically matched donor. Recipients were either never injected with tumor (MM-free) or had controlled (MM-controlled) or progressive myeloma (MM-relapsed) at 6 weeks post-transplant. BM was harvested for analysis of CD8 T cells by concurrent single cell RNA and ATAC sequencing. (A) WNN embedding colored by group (left) and identified CD8 T cell clusters (right). (B) WNN embedding with Pseudotime analysis from naïve (TN) to TEX clusters. (C) Gene expression (left) and chromatin accessibility (right) of functional proteins over pseudotime. (D) Experiment wide gene regulation score of transcription factor (TFs) motifs. (E) Gene expression of Batf, Tox and Ifng. (F) Single cell accessibility of Batf binding domain measured by ChromVAR. (G) Inferred activating (top right) and repressing (top left) TFs of Havcr2, Pdcd1, Ifng, and Prf1. (H) WNN embedding of T cells from MM-relapsed mice in (A) colored by clusters analogous to subsets identified in Fig. 1. (I) ATAC embedding colored by myeloma dataset clusters (top) and terminally exhausted clusters from chronic LCMV (bottom left) and solid tumors (bottom right). (J) WNN embedding colored by latent time with heatmap showing chromatin accessibility across latent time. (K) Differential motif accessibility of TFs in IFNγ+ TPHEX versus other TEX clusters. TFs of interest are highlighted in colored boxes.
We then correlated transcription factor (TF) expression with its motif enrichment in accessible DNA to infer highly utilized TFs across the dataset. As expected, previously described regulators of T cell exhaustion and stemness pathways had high correlation scores across the dataset: Nr4a2, Eomes, Tcf7 (Fig. 3D). This analysis also highlighted Batf as an important transcription factor, which exhibited co-expression with Tox and Ifng (Fig. 3E). Furthermore, TEX cells with the highest level of Ifng expression exhibited increased accessibility at the Batf motif (Fig. 3F). Interestingly, terminally differentiated cells from MM-free and MM-controlled mice were found within the broad TEX cluster, however these cells had lower levels of Tox expression (Fig. 3E) and likely differentiated in response to inflammatory signals during stem cell transplantation.46 Notably, there was accessibility in, but not active transcription of functional genes (Ifng, Prf1, Gzmb) in these cells (Fig. 3C). Using a method capable of linking motif enrichment with target gene expression in single cells,47 we identified target genes of Batf, which included those highly expressed in IFNγ+ TPHEX cells, such as Pdcd1, Havcr2, Prf1 and Ifng (Fig. 3G). Together, these data demonstrated that TEX cells in myeloma exhibit hallmarks of an exhausted phenotype and express BATF, a transcription factor that can be associated with effector function.11 High accessibility within the BATF motif may thus contribute to the retained production of IFNγ and granzyme B within IFNγ+ TPHEX.
Next, to specifically identify IFNγ+ TPHEX cells in the multi-ome data, we focused on a re-embedding of just the MM-relapsed dataset (Fig. 3H). We then used the IFNγ+ TPHEX signature from our scRNA seq data from the initial relapsed cohort (Fig. 1) to identify an analogous cluster (Fig. S8A). The IFNγ+ TPHEX cluster identified in this dataset also had high expression of Havcr2, Ifng, Gzmb, Prf1, Ly6a and Batf (Fig. S8B). To again draw parallels to other datasets of exhausted T cells, we projected ATAC data from chronic LCMV and solid tumor models onto our multiomic dataset. As expected, we found colocalization between our TEX and IFNγ+ TPHEX clusters with the terminally exhausted clusters identified in these models (Fig. 3I). We next performed unsupervised latent time analysis and observed a similar differentiation of CD8 T cells from TPEX and TEFF clusters towards TEX subsets (Fig. 3J), which validated our supervised pseudotime trajectory (Fig. 3B). This was also consistent with the RNA velocity analysis in Fig. 1A. Accessibility was similarly reduced within stemness genes and increased within exhaustion-associated genes over latent time (Fig. 3J). We next compared differentially accessible motifs between IFNγ+ TPHEX and the other TEX clusters in our relapsed dataset (Fig. 3K).47 Batf and Fos were significantly more accessible within IFNγ+ TPHEX while Gm4881 was more accessible in other TEX clusters. Together, these data suggest that although IFNγ+ TPHEX exhibit a chromatin accessibility profile that is largely similar to TEX previously described in solid tumors and chronic LCMV infection, they express functional genes which are strongly linked to changes in chromatin accessibility at Batf binding sites and to the expression of BATF itself.
Myeloma progression is associated with a reduced ratio of IFNγ+ TPHEX and TPEX to tumor burden
To determine why tumor control was lost, despite the presence of highly functional T cell subsets, we delineated the relationship between IFNγ+ TPHEX and tumor burden by phenotyping CD8 T cells at 6–7 weeks post-transplant in MM-free, MM-controlled, and MM-relapsed mice. We have previously shown increased expression of single inhibitory receptors on CD8 T cells at myeloma relapse48 and here we confirm that T cells co-expressing PD-1 and TIM-3 were significantly expanded in MM-relapsed mice (Fig. 4A). All PD-1+ T cells expressed CD39, a marker for tumor-specificity in humans (Fig. S9A).49 We observed increased frequency of TPEX in MM-relapsed mice compared to MM-controlled and MM-free mice (Fig. 4B). In line with the described functionality of TIM-3+ T cells in our model, IFNγ, perforin and granzyme B production was highest in MM-relapsed mice although MM-controlled mice produced more IFNγ and perforin compared to MM-free mice (Fig. 4C–E). Non-terminal bone marrow aspirates (BMA) at 3 weeks post-transplant revealed the presence of TIM-3+ cells in mice prior to frank relapse (Fig. S9B). The cytokine profile of TIM-3+ T cells in MM-controlled mice was comparable to MM-relapsed mice (Fig. S9C vs Fig. 4C). Next, to confirm that the generation of IFNγ+ TPHEX was not a transplant-specific phenomenon, we analyzed CD8 T cells from the BM of our triple-reporter mice with advanced myeloma that had not been transplanted (Fig. S9D). We observed a similar frequency of TIM-3+ T cells within CD8 T cells compared to mice that relapsed after SCT, which also produced both IFNγ and IL-10 in vivo (Fig. S9E–F). These data demonstrate that IFNγ+ TPHEX cells differentiated in response to tumor and that the presence of these cells in the BM preceded frank disease relapse.
Figure 4: Myeloma progression is associated with a reduced ratio of IFNγ+ TPHEX and TPEX to myeloma cells.
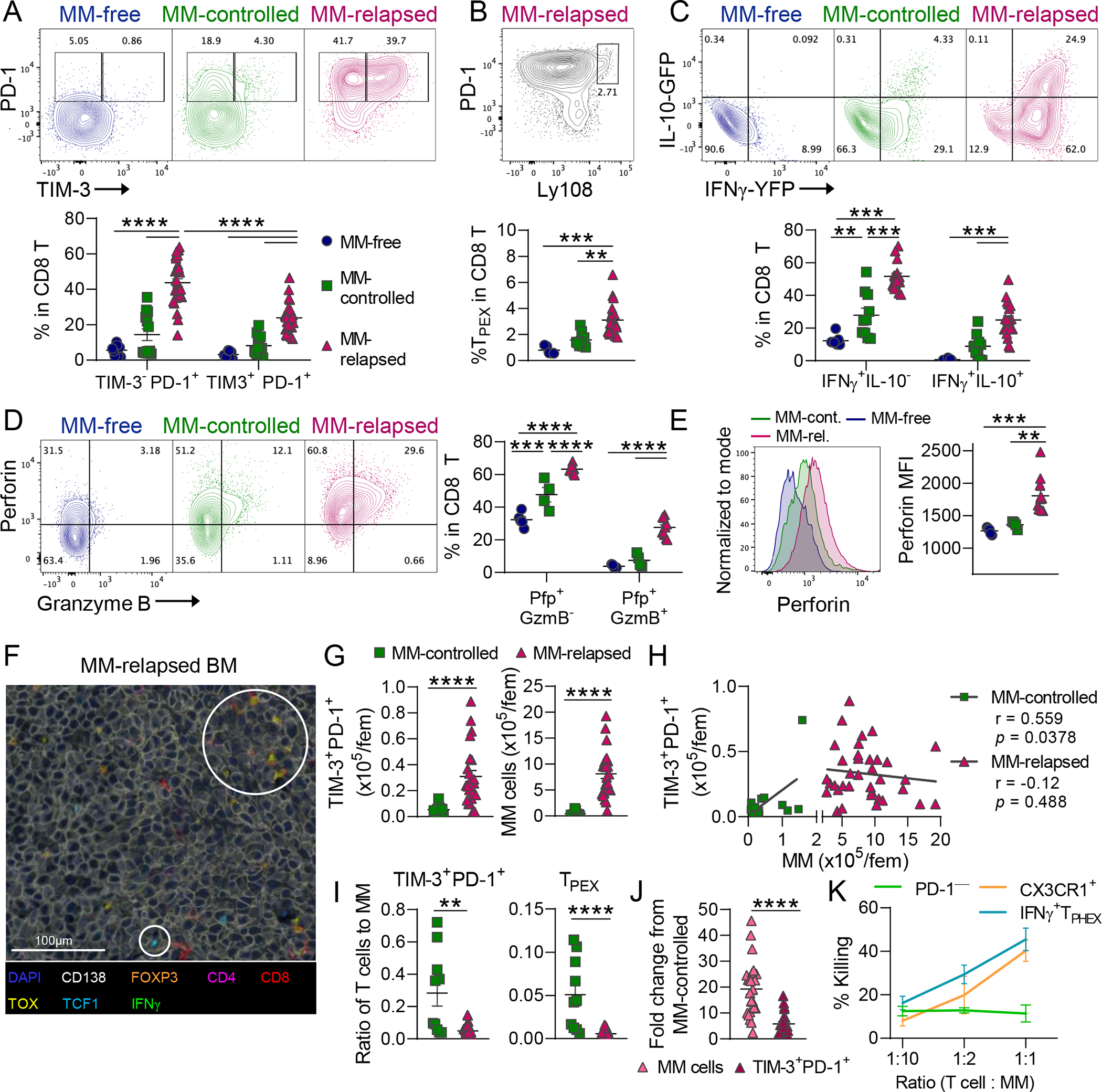
Recipient C57Bl/6 × PTPRCA (CD45.1/CD45.2) mice were transplanted with HULK donor (IFNγ-YFP × IL-10-GFP × FoxP3-RFP; CD45.2) grafts. Recipients were either never injected with tumor (MM-free) or had controlled (MM-controlled) or progressive myeloma (MM-relapsed) at 6–7 weeks post-transplant. BM was harvested for analysis of CD8 T cells using flow cytometry. (A) Representative contour plots and frequency of PD-1+TIM-3− and PD-1+TIM-3+ cells (MM-free n = 8; MM-controlled n = 10; MM-relapsed n = 19). Kruskal-Wallis with Dunn’s Test. (B) Representative contour plot and frequency of TPEX (Ly108hi PD-1+) cells. (MM-free n = 5; MM-controlled n = 9; MM-relapsed n = 24). Kruskal-Wallis with Dunn’s Test. (C) Representative contour plots and frequency of IFNγ+IL-10− and IFNγ+IL-10+ cells (MM-free n = 8; MM-controlled n = 10; MM-relapsed n = 19). One-way ANOVA with Tukey’s test. (D) Representative contour plots and frequency of Pfp+GzmB− and Pfp+ GzmB+ cells and (E) histograms and MFI of perforin in all Pfp+ cells (MM-free n = 5; MM-controlled n = 4; MM-relapsed n = 11). One-way ANOVA with Tukey’s test. (F) Sternum was harvested at MM relapse for VECTRA multispectral imaging of myeloma lesions in BM. White circles highlight populations of interest including TOX+ and TCF1+ CD8 T cells. (G) Quantification and (H) correlation of TIM-3+PD-1+ CD8 T (left) and MM (right) cell numbers in controlled and relapsed recipients. (I) ratio of T cells to myeloma for TIM-3+PD-1+ cells and TPEX cells (MM-controlled n = 10; MM-relapsed n = 24). Mann-Whitney t test and Pearson r correlation. (J) Fold change in myeloma cell and TIM-3+ T cell number in MM-relapsed mice relative to MM-controlled mice. Mann-Whitney t test. (K) Sort purified PD-1+TIM-3+CX3CR1− (IFNγ+ TPHEX), CX3CR1+, or PD-1− T cells were cultured with myeloma cells for 17 hours followed by Annexin V and 7AAD staining. % Killing (7AAD+) was calculated using a viability baseline from myeloma only wells. Error bars are from 3 biological replicates (PD-1− from 2 replicates) with 5 mice pooled per biological replicate. Data is mean ± SEM. Each symbol represents an individual mouse. ** p<0.01, *** p<0.001, **** p<0.0001.
We next performed multispectral imaging to confirm that T cells infiltrated myeloma lesions in the BM. Both TOX+ and TCF1+ CD8 T cells were sparsely seen within myeloma lesions in relapsed mice post-SCT (Fig. 4F). Moreover, myeloma cells greatly outnumbered T cells within myeloma lesions. Due to technical limitations of nuclei detection in BM sections, we used flow cytometry to quantify the total number of myeloma cells and TIM-3+ T cells in MM-controlled and MM-relapsed mice (Fig. 4G). We next correlated the number of TIM-3+ T cells with MM cell number and found a positive correlation only in MM-controlled mice (Fig. 4H). Therefore, although the total number of TIM-3+ T cells was increased at relapse, mice with controlled myeloma had a significantly higher ratio of both TIM-3+ and TPEX cells to myeloma cells (Fig. 4I). Indeed, while the number of MM cells increased ~20 fold, the number of TIM-3+ TEX only increased ~5 fold in mice with relapsed MM compared to those with controlled disease (Fig. 4J). Furthermore, freshly isolated IFNγ+ TPHEX from MM-relapsed mice killed Vk*MYC myeloma cells ex vivo with comparable efficacy to CX3CR1-expressing effector cells (Fig. 4K and Fig. S10A–B). In contrast, PD-1− cells did not have anti-myeloma activity. These data highlight that loss of immune control in myeloma is not due to dysfunction of terminally differentiated T cells but rather, progression is associated with numerical deficits in IFNγ+ TPHEX, which may be due in part to the lack of accessible chromatin within stem-like genes in these cells.
CD11c+ cells and IL-6 signaling promoted IFNγ+ TPHEX differentiation
We next sought to determine which signals contributed to IFNγ+ TPHEX differentiation and/or function in myeloma. Firstly, the role of dendritic cells (DCs) was assessed using CD11cDOG mice, whereby CD11c-expressing cells are deleted after exposure to diphtheria toxin (DT). DT treatment began at 4 weeks post-SCT and BM was harvested after 2 weeks of treatment. Using non-lethal bone marrow aspirates, IFNγ+ TPHEX frequency and function was recorded prior to DT injection to control for possible differences in tumor burden across mice (Fig. 5A). In PBS-treated mice, the frequency of IFNγ+ TPHEX within CD8 T cells increased between timepoints (Fig. 5B). In DT-treated mice, further IFNγ+ TPHEX differentiation was inhibited, while IFNγ production within IFNγ+ TPHEX was only marginally inhibited (Fig. 5B). At the end of treatment, there was no difference in myeloma burden between PBS and DT treated mice, likely due to the initiation of treatment late in the disease course (Fig. 5C). There were fewer CD8 T cells in the BM of DT-treated mice, and this was due to a selective reduction in all TEX subsets downstream from TPEX (Fig. 5D–E). Production of granzymes and perforin within IFNγ+ TPHEX was not impaired by depletion of CD11c+ cells (Fig. 5F). These data are consistent with CD11c-expressing cells broadly promoting TEX differentiation within the myeloma TME.
Figure 5: CD11c+ cells and IL-6 signaling promoted IFNγ+ TPHEX differentiation in the myeloma microenvironment.
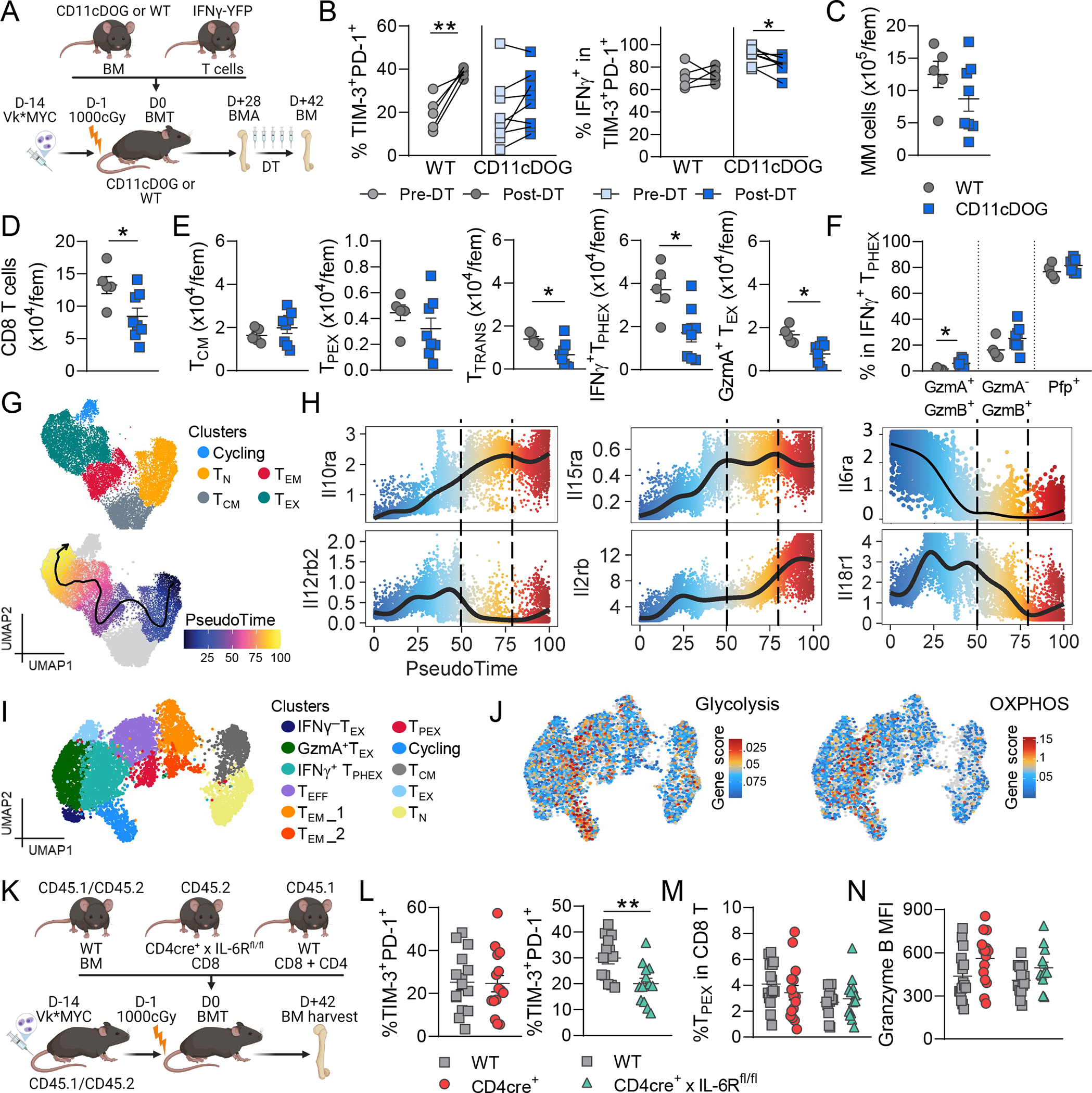
(A-F) Recipient C57Bl/6 or CD11cDOG mice were transplanted with B6. or CD11cDOG donor BM and IFNγ-YFP donor T cell grafts. BM aspirates (BMA) were taken prior to treatment with diphtheria toxin (DT) and BM was harvested two weeks later (n = 5 – 8 per group). (A) Experimental design. (B) Frequency of PD-1+TIM-3+cells within CD8 T cells and IFNγ-YFP+ cells within PD-1+ TIM-3+ cells in BMAs pre-DT and in BM post-DT. Wilcoxon Test. (C) Total number of myeloma cells (CD138+CD155+) and (D) CD8 T cells per femur post-DT. (E) Total number per femur of TCM (CD44+CD62L+), TPEX (LY108hiPD-1+), transitory TEX (PD-1+TIM-3+CX3CR1+), IFNγ+ TPHEX (PD-1+TIM-3+CX3CR1−) and GzmA+ TEX (PD-1+TIM-3−GzmA+) post-DT. (F) Frequency of granzyme B, granzyme A and perforin (Pfp) expressing cells in IFNγ+ TPHEX. (G) Repeated embeddings of multiome data from Fig. 3A and B colored by cluster and pseudotime trajectory. (H) Expression of genes encoding cytokine receptors over pseudotime. Dotted lines indicate pseudotime where TEX are found in MM-relapsed mice. (I) Repeated embedding from Fig. 3H colored by cluster and (J) by gene set enrichment for genes associated with glycolysis and OXPHOS pathways from Guo et al. Nat Immunol, 2021. (K-N) MM-bearing C57Bl/6 × Ptprca (CD45.1/CD45.2) recipients were transplanted with CD45.1/CD45.2 BM and CD8 T cells from WT (CD45.1) and transgenic (CD45.2) mice. Transgenic mice were CD4cre+ or CD4cre+ x IL-6Rfl/fl. CD4 T cells were from WT mice. Mice were sacrificed and BM was harvested at 7 weeks post-transplant (n = 13–15 from 4 experiments). (L) Frequency of PD-1+TIM-3+ cells within WT and Tg T cells in the same mice. Left graph: Tg = CD4cre+ and right graph: Tg = CD4cre+ × IL-6Rfl/fl. (M) Frequency of TPEX within WT and Tg T cells. (N) Granzyme B MFI in PD-1+TIM-3+ cells. Student’s t test. Data is mean ± SEM. Each symbol represents an individual mouse.*p<0.05, **p<0.01.
We next explored the potential role of cytokines in promoting either the differentiation or function of IFNγ+ TPHEX. We first employed our multi-ome dataset to assess changes in expression of IFNγ-related cytokine receptors throughout TEX differentiation using pseudotime (Fig. 5G–H). Il10ra, Il15ra and Il2rb increased while Il12rb, Il6ra and Il18r1 decreased during TEX differentiation. We focused on IL-6 and IL-10, which are known to be dysregulated in the myeloma microenvironment.50,51 In this model, we have previously shown that expression of the IL-10R on CD8 T cells was not required for TIM-3+ T cell differentiation.48 Given the recently described role of IL-10 signalling in augmenting metabolism in exhausted CD8 T cells in the TME44, we measured expression of gene signatures associated with glycolysis and the OXPHOS pathway in our relapsed dataset. We found enrichment for both signatures in the IFNγ+ TPHEX and Cycling clusters, consistent with high Il10r expression in TEX cells (Fig. 5H–J). Therefore, although IL-10 signalling is not required for the differentiation of IFNγ+ TPHEX cells, IL-10 may impact metabolism within these cells. On the other hand, IL-6R deletion using CD4cre+ x IL-6Rfl/fl mice resulted in reduced frequency of IFNγ+ TPHEX cells within CD8 T cells specifically within IL-6R KO transgenic T cells and not within the paired WT T cells in the same animal (Fig. 5K–L). TPEX frequency and granzyme B production within IFNγ+ TPHEX were not impacted (Fig. 5M–N). Together these data provide additional insights into the cytokines driving differentiation of IFNγ+ TPHEX in the myeloma microenvironment.
CD19-targeted CAR T cells with an IFNγ+ TPHEX phenotype are functional in the bone marrow.
To confirm that terminally differentiated cells can display features of both exhaustion and effector function in an additional model, we analyzed CD19 CAR T cells from mice with relapsing B-ALL. At 9 days post-CAR T cell transfer, the majority of the CD8 CAR T cells were CX3CR1+CD39+PD-1+ however, a subset of CAR T cells had a TEX phenotype and were CX3CR1−CD39+PD-1+TIM-3+ (Fig. S11A). These TEX cells did not express Ly108, expressed high levels of Ly6a, and produced the same amount of IFNγ as CX3CR1+ counterparts (Fig. S11B–C). Furthermore, analogous to polyclonal T cells from myeloma-bearing mice, higher expression of IFNγ, perforin and granzyme B was seen in TIM-3+ CD19 CAR T cells (Fig. S11C–F). All CAR T cells were TOX+ by 9 days post-transfer (Fig. S11E). To increase the frequency of terminally exhausted CAR T cells within the CAR T cell pool, we performed a secondary adoptive transfer of CD19 CAR T cells isolated from BM of mice with controlled B-ALL 25 days after initial CAR T cell transfer (Fig. S12A). CAR T cells isolated from BM of primary recipients were largely CD44+CD62L− and had higher CD39 expression compared to spleen yet maintained IFNγ secretion (Fig. S12B–C). After secondary transfer, the TIM-3+CX3CR1− subset now made up >35% of CD19 CAR T cells in the BM at 19 days post-transfer (Fig. 6A). TIM-3+CX3CR1− T cells produced more IFNγ and had increased CD101, CD39 and PD-1 MFI relative to TIM-3− subsets; all subsets had high expression of Ly6a (Fig. 6B–C and Fig. S12D). Interestingly, CD101 expression did not impact IFNγ secretion in TIM-3+ cells that were either CX3CR1− or CX3CR1+ likely due to the maintenance of high Ly6a expression across all subsets in this model (Fig. 6D). Single cell RNA sequencing revealed 6 clusters within CD19 CAR T cells, including a IFNγ+ TPHEX cluster with expression of Havcr2, Prf1, Ifng, Tox and Batf (Fig. 6E–F). We used 5’ TCR sequencing to assess overlap of clonotypes across clusters and observed shared clonotypes between TPEX and all TEX clusters, including IFNγ+ TPHEX (Fig. 6G). Indeed, there was an overlap of clonotypes across all clusters in this dataset, consistent with CD19 CAR T cells differentiating through multiple stages of T cell exhaustion in response to signaling via the CD19 CAR. Importantly, sort purified TIM-3+CX3CR1− (IFNγ+ TPHEX) CAR T cells (Fig. S12E) effectively killed B-ALL target cells in an ex vivo killing assay (Fig. 6H). Furthermore, IFNγ+ TPHEX CAR T cells killed a higher fraction of B-ALL cells compared to unfractionated CAR T cells at all tested effector:target ratios (Fig. 6H). Together, these data demonstrate that IFNγ+ TPHEX is a functional subset of CD19 CAR T cells able to effectively kill leukemia cells.
Figure 6: CD19 CAR T cells with a IFNγ + TPHEX phenotype effectively kill leukemia cells.
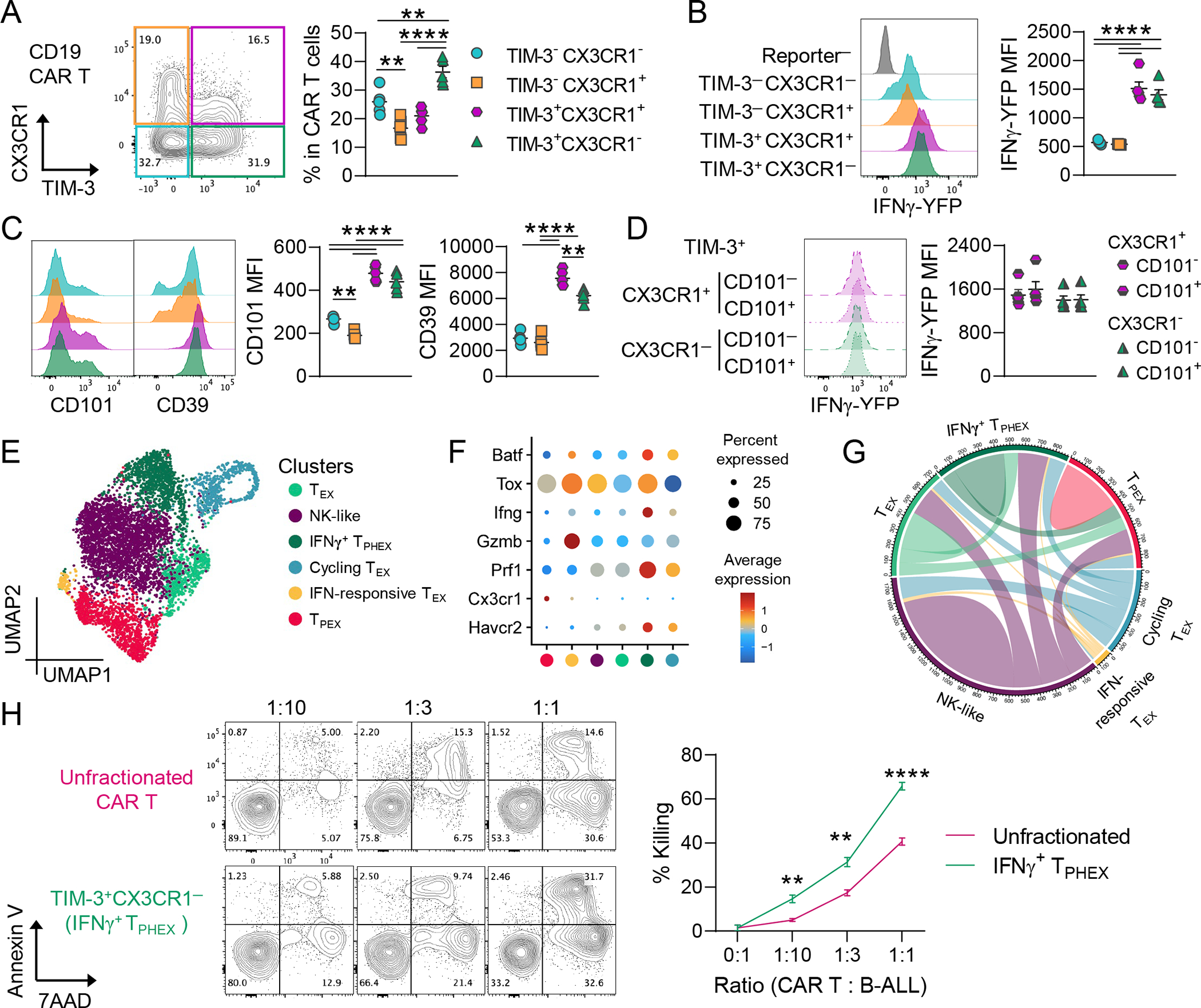
B6 mice bearing CD19+ B cell acute lymphoblastic leukemia were injected with murine CD19 CAR T cells (HULK; B6 background). CAR T cells from BM were harvested 25 days after primary transfer and were adoptively transferred to secondary B-ALL-bearing recipients. BM CAR T cells (human eGFR+) were harvested 19 days later and analyzed. (A) Representative plot of CX3CR1 and TIM-3 expression and quantification of subsets within CAR T cells. (B) Histogram of IFNγ-YFP expression (with reporter negative endogenous cells as a control) and quantification of MFI within CAR T cell subsets. (C) Histograms depicting CD101 and CD39 expression with quantification of MFI within CAR T cell subsets. (D) Histogram and quantification of IFNγ-YFP expression in TIM-3+ cells split by expression of CX3CR1 with/without CD101 co-expression. (A-D) Data is mean ± SEM. Each symbol represents an individual mouse (n = 5). One-Way ANOVA with Tukey’s test. (E-G) 5’ RNA sequencing was performed on CD8 CAR T cells. (E) UMAP embedding colored by clusters. (F) Dot plot showing gene expression within each cluster. (G) Plot depicts clonal overlap between clusters. (H) Sort purified TIM-3+ CX3CR1− CAR T cells, or unfractionated CAR T cells, were pooled from 5 biological replicates and cultured with B-ALL cells for 18 hours followed by Annexin V and 7AAD staining. Representative plots of Annexin V and 7AAD staining across effector:target ratios. The % killing was calculated using a viability baseline from B-ALL only wells (>90%). Data is mean ± SEM and error bars are from technical replicates (n = 3–5 per condition). Student’s T-test. ** p<0.01, **** p<0.0001.
IFNγ+ TPHEX are found in patients and are expanded following autologous stem cell transplantation for multiple myeloma
To determine whether IFNγ+ TPHEX were found in patients, we analysed BM samples from patients with myeloma across two centers at the following timepoints: (1) prior to ASCT (n = 23), (2) after ASCT (n = 17) and (3) at progressive disease (n = 20) (Fig. 7A). Using flow cytometry and FlowSOM to perform unbiased clustering of cells52, a population analogous to the mouse IFNγ+ TPHEX phenotype was identified, where TIM-3+ CD8 T cells were seen that expressed PD-1, TOX, CD28 and intermediate levels of granzyme B but were negative for CX3CR1 (Fig. 7A–B). In the absence of a reporter or known myeloma antigens to restimulate with, we were unable to verify IFNγ production in this subset by flow cytometry. Notably, this subset was expanded following ASCT and at myeloma relapse compared to pre-ASCT samples in patients (Fig. 7C). Focusing on patients who had paired samples at each timepoint (n =8), the frequency of TIM-3+GzmB+ cells increased after ASCT and did not further increase at disease relapse (Fig. 7D). Similar to our mouse model, the frequency of TIM-3+GzmB+ cells did not correlate with myeloma burden at relapse (Fig. 7E).
Figure 7: IFNγ+ TPHEX are found in patients with multiple myeloma and are expanded following autologous stem cell transplantation.
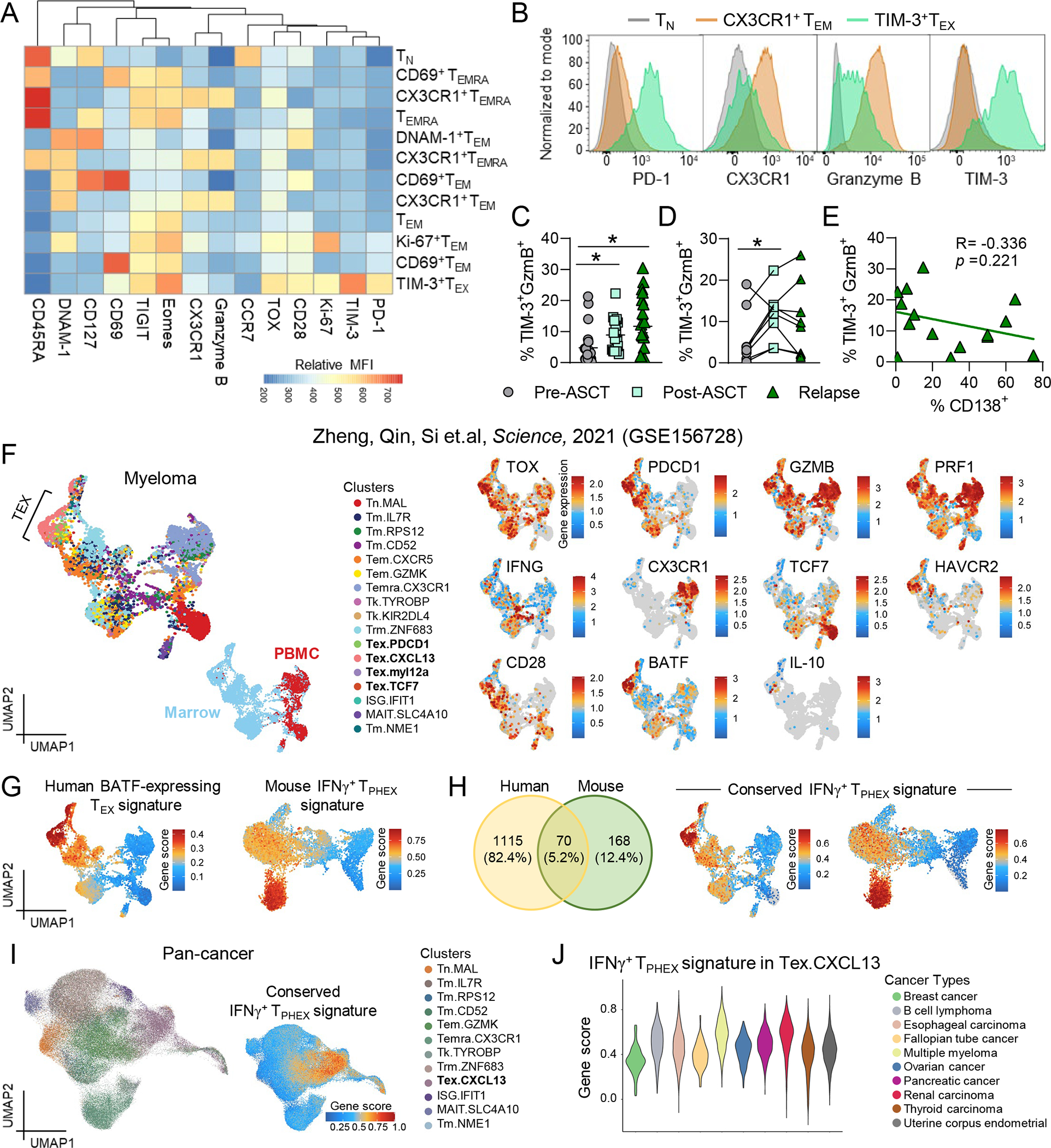
(A-E) Bone marrow samples from patients prior to autologous stem cell transplantation (ASCT; pre-ASCT), after ASCT (post-ASCT) and at disease relapse post-ASCT were thawed and stained for analysis via flow cytometry (n = 17–23). Data represent mean ± SEM. *p<0.05. (A) Heatmap of maker expression (MFI) across FlowSOM CD8 T cells populations. (B) Representative histograms of PD-1, CX3CR1, granzyme B and TIM-3 expression on naïve T cells (grey), CX3CR1+ effector cells (orange) and TIM-3+TEX cells (green). (C) Frequency of TIM-3+GzmB+ cells within CD8 T cells across the three cohorts (Kruskal-Wallis test with Dunn’s multiple comparisons test) and (D) in patients from (C) with paired samples across all timepoints (n = 8; Friedman test with Dunn’s multiple comparisons test). (E) Correlation of frequency of TIM-3+ GzmB+ cells within CD8 T cells and % CD138+ cells by morphology in patients from (C) with relapsed myeloma (n = 15). (F) Left: UMAP embedding of CD8 T cells from patients with multiple myeloma from Zheng et al. Science, 2021. Top embedding colored by cluster, bottom embedding colored by T cell location (tumor = blue, peripheral blood = red). Right: Embeddings colored by gene expression. (G) UMAP embedding colored by gene signature associated with BATF-expressing TEX in humans (left) and mouse IFNγ+ TPHEX signature from our dataset in Fig. 1 (right). (H) Venn diagram depicting the number of shared genes between the human and mouse gene signatures, which formed a ‘conserved IFNγ+ TPHEX signature’ and embeddings of human (left) and mouse (right) myeloma datasets colored by this signature. (I) Embedding of CD8 T cells from pan-cancer dataset from Zheng et al. Science, 2021 colored by cluster (left) and by expression of the conserved IFNγ+ TPHEX signature (right). (J) Violin plot showing gene score for the IFNγ+ TPHEX signature within the Tex.CXCL13 cluster stratified by cancer type. Data represent mean ± SEM. *p<0.05.
To bring additional clinical relevance to our findings, we interrogated scRNA seq data from a publicly available pan-cancer dataset, which included patients with myeloma.53 First, in the marrow of myeloma patients, we found that expression of TOX, HAVCR2, GZMB, IFNG, PRF1 and CD28 was associated with high expression of BATF in the Tex.CXCL13 cluster in this dataset (Fig. 7F). Next, to generate a human gene signature, we looked at differentially expressed genes in BATF-expressing TEX cells (Fig. 7G and Data File S2). We then compared this signature to the list of differentially expressed genes in our mouse IFNγ+ TPHEX cluster and generated a ‘conserved IFNγ+ TPHEX signature’ consisting of 70 genes across mouse and human myeloma (Fig. 7H and Data File S3). Finally, we applied our conserved IFNγ+ TPHEX signature to the pan cancer dataset from Zheng et al. and cells with the highest gene score were again largely found within the Tex.CXCL13 cluster (Fig. 7I and Fig. S13). When we quantified expression of the conserved IFNγ+ TPHEX signature in this cluster across cancer types, we found that myeloma, renal carcinoma, and B cell lymphoma had the highest expression of IFNγ+ TPHEX associated genes (Fig. 7J). Together, these data highlight that IFNγ+ TPHEX is a clinically relevant TEX phenotype that is found across a broad range of human cancers and is expanded following ASCT for myeloma.
Discussion:
Here we describe highly functional polyclonal CD8 T cells and CD19 CAR T cells in the bone marrow TME that displayed hallmarks of terminal T cell exhaustion, yet effectively killed myeloma and B-ALL target cells, respectively. Spatial imaging and flow cytometry analysis of the BM TME demonstrated that loss of T cell-mediated control in our model was underpinned by a reduced ratio of functional T cells to tumor cells rather than dysfunction of tumor-infiltrating T cells per se. It is important to highlight that while these cells can mediate anti-tumor activity, they have reduced chromatin accessibility within self-renewability genes and are therefore unlikely to generate sustained anti-tumor immunity without maintenance of TPEX populations.7,8 These data further emphasize the importance of targeting TPEX populations and suggest that identifying immunotherapies that can expand TPEX and/or promote T cell differentiation down the IFNγ+ TPHEX trajectory identified here, rather than a dysfunctional exhaustion trajectory, could greatly improve treatment of hematological malignancies. We demonstrate that IFNγ+ TPHEX differentiation was reduced after depletion of CD11c-expressing DCs, although this effect was partial. Notably, the requirement of DCs for tumor entry in myeloma has recently been described in humans.54 High expression of Cd28 in the IFNγ+ TPHEX subset relative to the dysfunctional TEX subset in our model, and in IFNγ+ TPHEX-like cells in humans, suggests that T cell interactions with other antigen presenting cells, including perhaps myeloma itself, likely also plays a role in maintaining this highly functional subset and represents another avenue for further investigation.55
The dogma in the T cell exhaustion field, that effector cytokine production is severely limited in terminally exhausted T cells, is largely built on data generated through ex vivo re-stimulation with TEX cells from solid tumor and chronic infection models.1–3,56 This discrepancy between high cytokine secretion in vivo, reported here, versus low cytokine secretion ex vivo following restimulation is possibly driven by differences in microenvironments and combinations of transcription factors regulating heterogenous TEX subsets. Differences in cytokine production could also be driven by defects in PMA/ionomycin response elements, that are bypassed in vivo where physiological IFNγ responses are primarily determined by IL-2/IL-12/IL-18 signaling.57 Specifically, we observed Nfatc motif accessibility in TEX and it is known that Nfatc2-driven effector function is negatively regulated by Dapl1 in an ionomycin-dependent manner in TEX.58 This regulatory network may underpin the lack of responsiveness of TIM-3-expressing cells to broad ex vivo restimulation. Furthermore, although TIM-3 signaling is said to inhibit effector function, in our model, NFAT1 was likely undergoing competent activation in IFNγ+ TPHEX as concurrent ATAC and RNA sequencing showed subsequent transcription of NFAT1-regulated effector genes including Prf1, Gzmb and Ifng in cells with increased Nfatc1 motif accessibility.59 Others have identified CD8 T cells with features of both exhausted (TOX, TIM-3, PD-1) and effector cells (CX3CR1, T-bet, KLRG1) in chronic infection and autoimmunity that represent intermediate stages of exhaustion.17,18,23 Together with our data, these studies highlight the heterogeneity of functional capacity within T cells at intermediate and now terminal stages of exhaustion.
Together with expression of TIM-3, CD28, and TOX, the transcription factors Maf and BATF corresponded with polyfunctionality within IFNγ+ TPHEX in mice with relapsed myeloma. In a mouse model of lung cancer, T-follicular helper cells produced IL-21 and supported PD-1+ granzyme B+ CD8 T cells in the tumor microenvironment.19 A role for IL-21 in promoting the function of effector T cells and transitory TEX cells in chronic LCMV infection has been described37,60,61 and interestingly, through STAT3, IL-21 signaling induced expression of BATF.62 BATF has been described as a driver of either effector function or exhaustion in different biological contexts.11,12,63 Our data suggest that these phenotypes are not mutually exclusive and that rather, Batf expression correlates with enhanced functionality in T cells with an otherwise phenotypically exhausted protein, transcriptomic, and epigenetic profile in mice and humans. In murine models of melanoma, Maf expression was associated with a TEX phenotype and T cell dysfunction.64,65 Notably, these studies largely utilized bulk RNA sequencing approaches to characterize the role of Maf expression in TEX, which does not allow for interrogation of discrete TEX subsets. In melanoma-specific T cells, IL-6 and TGFβ, which are highly expressed in the myeloma TME,66 induced Maf expression in antigen-stimulated CD8 T cells in vitro.64 IL-6 and TGFβ inhibited effector function in these in vitro experiments,64 however IL-6 signaling to TIM-3+ TEX did not impact granzyme B expression in our model, likely due to the reduction in Il6ra expression during TEX differentiation. A wide range of cytokines and molecules can modulate STAT3, MAF, and BATF and determining which pathways are involved in maintaining the function of IFNγ+ TPHEX in the TME is an important avenue for further investigation.
Expression of some markers observed in our mouse myeloma model (i.e. Ly6A, Maf) may be impacted by differences in protein versus gene expression and species-specific genes (e.g. Ly6A is not expressed in humans). To aid in the broad identification of IFNγ+ TPHEX in clinical datasets, we generated a conserved IFNγ+ TPHEX signature across mouse and human myeloma. We applied this signature to a pan-cancer dataset and identified several human cancers with CD8 T cells that were enriched for this phenotype. We recognize that the lack of known tumor antigen is a limitation of the Vk*MYC model, however, the induction of a naturally occurring, polyclonal T cell response to tumors is likely more representative of the heterogeneity within T cell responses generated against human cancers. Furthermore, using this same model, we observed substantial clonal expansion within polyclonal CD8 T cells with a TEX phenotype and have demonstrated that SCT generates myeloma-specific T cell responses.24,33 Here, we provided evidence for the existence of a IFNγ+ TPHEX phenotype within antigen-specific cells using two model systems including a Hy-antigen-expressing myeloma with MataHari HY-specific TCR transgenic T cells and a CD19-expressing B-ALL with CD19 CAR T cells. Subsequent studies are needed to determine the differentiation stage at which this functional vs dysfunctional trajectory is programmed (i.e., TPEX vs transitory/intermediate TEX).
In conclusion, we have identified TIM-3+CX3CR1− CD8 T cells with hallmarks of exhaustion that remain highly functional (termed IFNγ+ TPHEX) in the TME. In patients with myeloma, ASCT expanded TIM-3+ T cells and incorporation of immunotherapy early-posttransplant may augment this effect. Specifically, identification of the signaling pathways that drive differentiation of this functional, rather than dysfunctional, TEX phenotype may inform approaches to broadly optimize immunotherapy. Furthermore, the function of T cells, or lack thereof, in the TME cannot be inferred by measuring expression of inhibitory receptors or even exhaustion-associated transcription factors. These data have implications for studies investigating CD8 T cell phenotypes in tumor settings, particularly as this phenotype was observed in human TILs from diverse cancers. Finally, our data are consistent with the importance of targeting TPEX, to expand downstream TEX subsets with retained anti-tumor function, as a means of generating sustained responses to immunotherapies.
Methods:
Study Design
This study was designed to characterize T cell differentiation in the bone marrow tumor microenvironment. We interrogated CD8 T cell phenotypes in the bone marrow of polyclonal and antigen-specific murine models using flow cytometry and multi-ome single cell RNA/ATAC sequencing approaches. We then performed confirmatory analysis of BM T cells in a myeloma patient cohort and a publicly available human pan-cancer dataset. Mice were randomly assigned to groups in all experiments without investigator blinding. All n values reflect biological replicates unless otherwise stated and numbers of mice per group are included, with the statistical test performed, in the legend for each figure.
Mice
C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and Rag2xIl2rg−/− mice were purchased from Taconic Biosciences (La Jolla, CA, USA). PTPxC57 (CD45.1/CD45.2), HULK (IFNγ-YFP x IL-10-GFP x FoxP3-RFP) reporter, CD4cre x IL-6Rfl/fl, and MataHari41 mice were bred in house at Fred Hutchinson Cancer Research Center (Seattle, WA, USA). All individual reporter strains in the HULK line were either generated on a B6 background or were backcrossed more than 10 generations (IFNγ-YFP40; IL-10-GFP JAX #:008379; FoxP3-RFP JAX #:008374). Mice were housed in sterile microisolator cages, fed normal chow, and given autoclaved pH 2.5 water. All animal procedures were performed in accordance with protocols approved by the institutional animal ethics committee. All recipient mice were female at 8–12 weeks of age at the initiation of experiments.
Human Samples
The use of human bone marrow samples for research in the USA was approved by the Fred Hutchinson Cancer Research Center IRB. Samples from Melbourne Australia were approved by the Alfred Hospital Human Research Ethics Committee. All samples were from respective biorepositories of myeloma patients who provided written informed consent. Samples were taken prior to autologous stem cell transplantation, at 3 months post-transplantation, and at disease progression.
Antibodies
The following antibodies for flow cytometry were purchased from: eBiosciences; CD101 (Moushi101), c-MAF (sym0F1), TIM3 (CD366; RMT3–23), FOXP3 (FJK-16s), TOX (TXRX10) and from Biolegend; Perforin (S16009A), CD319 (SLAMF7; 4G2), PD-1 (CD279; RMPI-30, 29F.1A12), CD38 (T10), CD39 (Duha59), CD90.2 (53–2.1), CD49d (R1–2), Granzyme B (QA16A02), CD69 (H1.2F3), CD226 (DNAM-1; TX42.1), CD3 (145–2C11) and from BD Bioscience; CD122 (TM-β1), Granzyme B (GB11), CD44 (IM7), TIGIT (1G9), CD62L (MEL-14), CD45 (30-F11), CD45.1 (A20), CD4 (GK1.5), LY108 (13G3), PD-1 (CD279; J43), CD8 (53–6.7).
Myeloma model and stem cell transplantation
Mice were injected intravenously with Vk12653, Vk12598, or Vk28158 (1×106 cells; MM-bearing mice), which originated from Vk*MYC transgenic mice.67,68 Vk28158 expresses the male Hy-antigen and was generously provided by Marta Chesi. For transplantation experiments, recipients were injected with Vk*MYC two weeks, or were naïve (MM-free), prior to lethal irradiation split across two doses 3 hours apart (1000cGy, 137Cs source). Recipients were transplanted the following day with 10 × 106 BM and 5 × 106 T cells via tail vein injection as described previously.48,69 PTPxC57 recipients were utilized in all experiments with HULK reporter donors to allow for the exclusion of reporter negative recipient T cells from downstream analysis. Rag2/Il2rg−/− mice were used for all Vk28158 experiments. Serum samples were collected every two weeks from MM-bearing recipients and M-band was quantified using serum protein electrophoresis (HYDRASYS 2 Scan) as previously described.67 Recipients were monitored daily and sacrificed when clinical scores reached ≥6 or hind limb paralysis was observed. In experiments using CD11cDOG mice, all groups were treated with diphtheria toxin three times per week, for two weeks, at 160ng per dose I.P.
CD19 CAR T cell model
Construct of the CD19 CAR (m1928E) has been described previously which encodes a mouse CD19 CAR with CD28 co-stimulation domain and an EGFRt selectable marker.70 Retrovirus was produced by transfection of Platinum-E packaging cells with CD19 CAR expressing plasmid using Calcium Phosphate Transfection kit (Takara) per manufacturer’s protocol. Retroviral supernatant was harvested, filtered through 0.45μM filters (Millipore) and frozen down at −80°C for future use. Mouse CAR T cells were generated as previously described with modifications.70 Briefly, CD4+ and CD8+ T cells were isolated from spleens of B6 donors (H2Db, CD45.2+) with magnetic activated cell sorting or MACS (Miltenyi) and stimulated with plate-bound anti-CD3 (clone 2C11, 1μg/ml) and anti-CD28 (clone N37.51, 1μg/ml) for 20 – 30 hours in IMDM complete medium.71 Retrovirus was thawed, loaded onto Retronectin (Takara) coated non-tissue culture treated plates, spun at 3000g at 32°C for 2 hours and then removed. Stimulated T cells were mixed with CD3/CD28 Dynabeads (Giboco) at 1:1 ratio, loaded onto the retrovirus-coated plates, incubated overnight and subsequently transferred to fresh medium. The cells were cultured in the presence of rhIL-2 (Miltenyi, 100U/ml) for the first 3 days, washed out and cultured in rmIL-15 (PeproTech, 50ng/mL) for another 4 days. After 7 days of culture, transduced T cells were harvested, cleared of Dynabeads with magnet, labelled with anti-human EGFR PE (clone AY13, Biolegend), followed by MACS selection with anti-PE microbeads (Miltenyi) resulting in highly pure products (>98% EGFR+). CD4+ and CD8+ cells were mixed at 1:1 ratio before adoptive transfer. PTP X C57 (H2Db, CD45.1+, CD45.2+) recipients were transplanted with TCD BM (H2Db, CD45.1+) and acute lymphocyte leukemia cells (H2Db, CD45.2+) which harbor the transgenes Arf−/− MSCV-BCR-ABL1-ires-luciferase and MSCV-IK6-ires-GFP.72 CD4+ and CD8+ CAR T cells were adoptively transferred on day +3 after transplant. Relapse is this model was defined by >50% GFP+ tumor cells in bone marrow or blood and/or a white blood cell count about 50 ×106/ml.
For the secondary transfer experiment, EGFR+ CD19 CAR T cells were isolated from BM 25 days after initial adoptive transfer. CAR T cells were pooled from 10 primary CAR T cell recipients and a total of 1 × 106 CAR T cells were injected into each secondary B-ALL-bearing recipient. After 19 days, CAR T cells were harvested from BM of secondary recipients for flow cytometry phenotyping and an ex vivo killing assay. Briefly, isolated CAR T cells were sorted based on CX3CR1 and TIM-3 expression (CX3CR1− TIM-3+) or were left unsorted prior to being cultured at 37 °C with B-ALL cells for 18 hours at CAR T: B-ALL ratios of 1:1, 1:3 and 1:10. Target lysis was measured using Annexin V and 7AAD staining according to the manufacturer’s protocol (BD Biosciences).
Flow Cytometry
Mice were sacrificed at time points indicated in Figure Legends and BM was flushed with media (RPMI + 1% FCS) to harvest T cells. For bone marrow aspirates, mice were anesthetized and treated with a local analgesic (0.5% lidocaine) followed by injection of 30 μL of PBS into the femur to allow up to 10 μL of marrow to be aspirated. For analysis of human BM, samples were thawed and up to 5 × 106 total cells were stained for surface markers prior to fixation for intracellular staining. For all FACS analysis, whole BM was incubated with Fc-block prior to staining with fluorescently tagged antibodies on ice for 30 minutes. For intracellular staining, cells were permeabilized after surface staining (Foxp3 Staining Buffer Kit; eBioscience) prior to incubation with intracellular antibodies for 60 minutes at room temperature. To measure restimulated IFNγ production, cells were stimulated with PMA (500 ng/mL) and ionomycin (50 ng/mL) (Sigma-Aldrich) with Brefeldin A (BioLegend) for 4 hours at 37°C. The ex vivo killing assay in the myeloma model was performed as described above in the CAR T cell model, with additional sort purification of CX3CR1+PD-1+ and PD-1− subsets. All samples were acquired on a BD FACSymphony A3 (BD Biosciences) and analyzed using FlowJo software (v10). FlowSOM, which used a Self-Organizing Map method of data visualization, analysis was performed on concatenated samples, which included 5000 CD8 T cells per patient sample.52
Single-cell RNA sequencing
For all sequencing experiments, BM was harvested from femurs and T cell populations were FACS sorted prior to sample preparation according to 10x Genomics protocols. In some experiments, individual mice or purified T cell populations were stained with BioLegend Hashtag TotalSeqB/TotalSeqC reagents, and cell surface markers (including PD-1, TIM-3, CD4 and CD8), according to manufacturer’s protocol to allow pooling of samples for capture. T cells (gene expression) or nuclei (multiome, also herein referred to as cells) were captured and libraries were generated according to manufacturer’s specifications. Libraries were sequenced using an Illumina NovaSeq 6000 or NextSeq 2000 targeting a depth of 25,000 reads per cell per library and 5,000 reads per cell for TCR libraries.
Single cell RNA sequencing of MM-relapsed CD8 cells
Illumina BCL reads were demultiplexed with cellranger count. We log-normalized antibody features and removed cells with reads not meeting the positive threshold criteria HTO1 > 5, HTO2 > 5.2, HTO3 > 5 in more than one hashtag oligo were removed. Cells with over 5% mitochondrial RNA were considered likely dead and removed. Satellite cell clusters expressing Cd4a, Itgam, Cd19, or low Cd8a were identified as contamination and eliminated. Iterative LSI reduced RNA counts. Cell types were identified through Louvain and expert annotation of top marker genes. We categorized cells into CD38−CD101−, CD38+CD101−, and CD38+CD101+ gates based on HTO1, HTO2, and HTO3 expressions. Bimodal PD1 and TIM3 surface expression thresholds identified double positive, negative, and single positive cells.
Loom files were created with velocyto and analyzed with scvelo.26,73 We calculated bulk RNA projections using a modified LSI projection method for RNA counts.74 IFNγ+ TPHEX signatures in polyclonal and myeloma infiltrating CD8s were measured with softmax regression using the viewmastR package (https://github.com/furlan-lab/viewmastR). We compared relapsed CD8s to a previously published LCMV model through Seurat integration. Single cell TCR data was analyzed with scRepertoire.75 We computed average expression scores for published gene-sets with AddModuleScore(). The IFNγ+ TPHEX gene signature was derived by comparing IFNγ+ TPHEX to other cell types with FindMarkers().
Single cell RNA and ATAC sequencing of MM-free, MM-controlled, and MM-relapsed CD8 cells
Illumina BCL reads were demultiplexed using cellranger ARC. MACS276 peak calls and filtered feature matrices were used to create a Seurat object. We retained cells based on specific criteria: percent.mt <= 15, 3 <= log_ATAC <= 5, 3 <= log_RNA <= 4.5, and Frip >= 0.4. RNA counts were normalized with SCT transform. ATAC counts were normalized using term-frequency inverse-document-frequency, singular value decomposition, and Harmony.77 An integrated RNA and ATAC UMAP was generated using FindMultiModalNeighbors(). Cell types were identified via top marker analysis with expert annotation. A complementary ArchR object was created for further analysis.78 ATAC fragments and RNA matrices were processed through the ArchR workflow, and UMAP coordinates were transferred from Seurat. We calculated supervised pseudotime trajectories with addTrajectory(). Single cell motif enrichment was computed using addDeviationsMatrix() with the “cisbp” motif set. Transcription factor expression and motif accessibility were evaluated using correlateMatrices(). Target genes of transcription factors were inferred with FigR.47
Relapsed cells were re-embedded and analyzed as above. Single cell and bulk ATAC datasets were projected onto ATAC UMAP coordinates using projectBulkATAC(). Unsupervised latent time was calculated with multivelo. Cell cluster annotation overlap among single cell RNA signatures of CD38/101 sorted CD8 cells was determined with viewmastR. The IFNγ+ TPHEX signature was assessed using AddModuleScore().
Single cell sequencing analysis of CD19 CAR-T CD8 cells
BCL Illumina reads were demultiplexed and filtered with cellranger multi. We retained cells meeting these thresholds: rna_Cd4 <= 0, rna_Cd8a > 0, adt_CD8 > 1.5, HTO_classification.global == Singlet, 3.25 < log_RNA < 4.25, percent.mt < 5. RNA counts were normalized with SCT transform, and ADT counts underwent CLR normalization. Cell types were visualized and identified using the standard Seurat workflow with expert annotation. TCR data was analyzed with scRepertoire.75
Single cell RNA of human tumor infiltrating CD8 cells
The human neoantigen TCR dataset was from Lowery et.al34 and was incorporated into our data as described above. The human terminal exhaustion dataset was from Braun et.al35 and was incorporated into our data as described. Single cell RNA counts of human myeloma specific CD8 cells were obtained from GSE156728, specifically the GSE156728_MM_10X.CD8.counts.txt file.53 RNA counts were normalized and visualized using the standard Seurat workflow. BATF-specific cells were identified through gene expression and cluster analysis. This signature was intersected with the previously created IFNγ+ TPHEX signature to form a conserved human-mouse IFNγ+ TPHEX gene set. Single cell RNA analysis of pan-cancer CD8 cells was downloaded from Zenodo using the CD8.thisStudy_10X.seu.rds file. Additional analysis was performed with Seurat.
Statistical analysis
Survival curves were plotted using Kaplan-Meier estimates and compared by Log-rank (Mantel-Cox) test. M-bands were modeled as described previously 24,48 and the M-band relapse threshold (G/A above 0.282) has been reported.24,48 Comparisons between two groups were performed with Mann-Whitney U test or t-test and those between three or more groups were performed with Kruskal-Wallis and Dunn’s multiple comparisons test or one-way ANOVA and Tukey’s multiple comparisons tests for nonparametric data and normally distributed data respectively. All data presented as mean ± SEM and p<0.05 was considered significant.
Supplementary Material
Acknowledgements:
SAM is supported by a Klorfine Fellowship, ASTCT New Investigator Award, and SITC-BMS Translational Research Fellowship. JRB is supported by an NIH NCATS KL2TR002317 and NIH NCI T32CA009351. MC is funded by NCI CA272426 and CA186781, and MC receives royalties from the licensing of Vk*MYC, hCRBN and derivative mice. This work was supported by a research grant from the National Cancer Institute of the NIH (U01 CA244291) and a Specialized Center of Research Award from the Leukemia and Lymphoma Society. Scientific Computing Infrastructure at Fred Hutch was supported by ORIP grant S10OD028685. This research was supported in part through funding from Seattle Translation Tumor Research (STTR).
Footnotes
Competing Interests:
SRR is a founder and shareholder of Lyell Immunopharma and Juno Therapeutics, a Bristol Myers Squibb company. SRR serves as an advisor to Lyell Immunopharma and Adaptive Biotechnologies, has intellectual property licensed to Lyell Immunopharma and Juno/BMS, and receives research support from Lyell Immunopharma and Bristol Myers Squibb. DJG has received research funding, has served as an advisor and has received royalties from Juno Therapeutics, a Bristol-Myers Squibb company; has served as an advisor and received research funding from Seattle Genetics; has served as an advisor for GlaxoSmithKline, Celgene, Janssen Biotech, Ensoma and Legend Biotech; and has received research funding from SpringWorks Therapeutics, Sanofi, and Cellectar Biosciences. GRH has consulted for Generon Corporation, NapaJen Pharma, iTeos Therapeutics, Neoleukin Therapeutics, CSL Behring, Cynata Therapeutics and has received research funding from Compass Therapeutics, Syndax Pharmaceuticals, Applied Molecular Transport, Serplus Technology, Heat Biologics, Laevoroc Oncology and iTeos Therapeutics. All other authors declare that they have no competing interests.
Data and materials availability:
Normalized counts and metadata have been deposited in the GEO repository and are available under GSE262306. The code for generating single cell figures is available at https://github.com/furlan-lab/TIM3_CD8_myeloma. All other data are present in the paper or Supplementary Materials, including IFNγ+ TPHEX gene signatures (Data file S1-3) and the raw data used to generate figures (Data file S4).
References:
- 1.Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, Zamarin D, Walther T, Snyder A, Femia MR, Comen EA, Wen HY, Hellmann MD, Anandasabapathy N, Liu Y, Altorki NK, Lauer P, Levy O, Glickman MS, Kaye J, Betel D, Philip M, Schietinger A. TOX is a critical regulator of tumour-specific T cell differentiation. Nature. 2019;571(7764):270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, Wherry EJ. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571(7764):211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seo H, Chen J, González-Avalos E, Samaniego-Castruita D, Das A, Wang YH, López-Moyado IF, Georges RO, Zhang W, Onodera A, Wu CJ, Lu LF, Hogan PG, Bhandoola A, Rao A. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proceedings of the National Academy of Sciences of the United States of America. 2019;116(25):12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao C, Sun H-W, Lacey NE, Ji Y, Moseman EA, Shih H-Y, Heuston EF, Kirby M, Anderson S, Cheng J, Khan O, Handon R, Reilley J, Fioravanti J, Hu J, Gossa S, Wherry EJ, Gattinoni L, McGavern DB, O’Shea JJ, Schwartzberg PL, Wu T. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nature immunology. 2019;20(7):890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, Yan P, Nzingha K, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Huang AC, Wherry EJ. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity. 2020;52(5):825–841.e828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity. 2019;50(1):195–211.e110. [DOI] [PubMed] [Google Scholar]
- 7.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue HH, Ahmed R. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537(7620):417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Utzschneider DT, Gabriel SS, Chisanga D, Gloury R, Gubser PM, Vasanthakumar A, Shi W, Kallies A. Early precursor T cells establish and propagate T cell exhaustion in chronic infection. Nature immunology. 2020;21(10):1256–1266. [DOI] [PubMed] [Google Scholar]
- 9.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. [DOI] [PubMed] [Google Scholar]
- 10.Man K, Gabriel SS, Liao Y, Gloury R, Preston S, Henstridge DC, Pellegrini M, Zehn D, Berberich-Siebelt F, Febbraio MA, Shi W, Kallies A. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity. 2017;47(6):1129–1141.e1125. [DOI] [PubMed] [Google Scholar]
- 11.Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio CJ, Rao A, Hogan PG. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nature immunology. 2021;22(8):983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, Zander RA, Wu X, Schauder DM, Kasmani MY, Shen J, Zheng S, Burns R, Taparowsky EJ, Cui W. BATF regulates progenitor to cytolytic effector CD8(+) T cell transition during chronic viral infection. Nature immunology. 2021;22(8):996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, DiIorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, Wherry EJ, Haining WN. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nature immunology. 2014;15(4):373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, Lamperti ED, Ansel KM, Mempel TR, Lähdesmäki H, Hogan PG, Rao A. The transcription factor NFAT promotes exhaustion of activated CD8⁺ T cells. Immunity. 2015;42(2):265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Qi T, Milner JJ, Lu Y, Cao Y. Speed and Location Both Matter: Antigen Stimulus Dynamics Controls CAR-T Cell Response. 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montironi C, Muñoz-Pinedo C, Eldering E. Hematopoietic versus Solid Cancers and T Cell Dysfunction: Looking for Similarities and Distinctions. Cancers. 2021;13(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giles JR, Ngiow SF, Manne S, Baxter AE, Khan O, Wang P, Staupe R, Abdel-Hakeem MS, Huang H, Mathew D, Painter MM, Wu JE, Huang YJ, Goel RR, Yan PK, Karakousis GC, Xu X, Mitchell TC, Huang AC, Wherry EJ. Shared and distinct biological circuits in effector, memory and exhausted CD8(+) T cells revealed by temporal single-cell transcriptomics and epigenetics. Nature immunology. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grebinoski S, Zhang Q, Cillo AR, Manne S, Xiao H, Brunazzi EA, Tabib T, Cardello C, Lian CG, Murphy GF, Lafyatis R, Wherry EJ, Das J, Workman CJ, Vignali DAA. Autoreactive CD8(+) T cells are restrained by an exhaustion-like program that is maintained by LAG3. Nature immunology. 2022;23(6):868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M, Cheung JF, Mao T, Askari AS, Chen S, Fitzgerald B, Foster GG, Eisenbarth SC, Zhao H, Craft J, Joshi NS. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. 2021;184(25):6101–6118.e6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin J-X, Konieczny BT, Im SJ, Freeman GJ, Leonard WJ, Kissick HT, Ahmed R. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity. 2019;51(6):1043–1058.e1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen J, Blank CU, Horlings HM, David E, Baran Y, Bercovich A, Lifshitz A, Schumacher TN, Tanay A, Amit I. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 2019;176(4):775–789.e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, Kuchroo VK, Regev A, Anderson AC. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1−CD8+ Tumor-Infiltrating T Cells. Immunity. 2019;50(1):181–194.e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sekine T, Perez-Potti A, Nguyen S, Gorin J-B, Wu Vincent H, Gostick E, Llewellyn-Lacey S, Hammer Q, Falck-Jones S, Vangeti S, Yu M, Smed-Sörensen A, Gaballa A, Uhlin M, Sandberg Johan K, Brander C, Nowak P, Goepfert Paul A, Price David A, Betts Michael R, Buggert M. TOX is expressed by exhausted and polyfunctional human effector memory CD8+ T cells. Science immunology. 2020;5(49):eaba7918. [DOI] [PubMed] [Google Scholar]
- 24.Vuckovic S, Minnie SA, Smith D, Gartlan KH, Watkins TS, Markey KA, Mukhopadhyay P, Guillerey C, Kuns RD, Locke KR, Pritchard AL, Johansson PA, Varelias A, Zhang P, Huntington ND, Waddell N, Chesi M, Miles JJ, Smyth MJ, Hill GR. Bone marrow transplantation generates T cell-dependent control of myeloma in mice. J Clin Invest. 2019;129(1):106–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, Schietinger A. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545(7655):452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, Fan J, Borm LE, Liu Z, van Bruggen D, Guo J, He X, Barker R, Sundström E, Castelo-Branco G, Cramer P, Adameyko I, Linnarsson S, Kharchenko PV. RNA velocity of single cells. Nature. 2018;560(7719):494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, Fischer S, Jackson R, Flavell RA, Wang J, Sanmamed MF, Bosenberg MW, Ring AM. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583(7817):609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, Yu S, Kurachi M, Herati RS, Vella LA, Baxter AE, Wu JE, Khan O, Beltra JC, Giles JR, Stelekati E, McLane LM, Lau CW, Yang X, Berger SL, Vahedi G, Ji H, Wherry EJ. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Schlossman SF, Edwards RA, Ou CN, Gu J, Wu MX. Impaired apoptosis, extended duration of immune responses, and a lupus-like autoimmune disease in IEX-1-transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(2):878–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanbar JN, Ma S, Kim ES, Kurd NS, Tsai MS, Tysl T, Widjaja CE, Limary AE, Yee B, He Z, Hao Y, Fu XD, Yeo GW, Huang WJ, Chang JT. The long noncoding RNA Malat1 regulates CD8+ T cell differentiation by mediating epigenetic repression. The Journal of experimental medicine. 2022;219(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cannarile MA, Lind NA, Rivera R, Sheridan AD, Camfield KA, Wu BB, Cheung KP, Ding Z, Goldrath AW. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nature immunology. 2006;7(12):1317–1325. [DOI] [PubMed] [Google Scholar]
- 32.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347(6294):669–671. [DOI] [PubMed] [Google Scholar]
- 33.Minnie SA, Waltner OG, Ensbey KS, Olver SD, Collinge AD, Sester DP, Schmidt CR, Legg SRW, Takahashi S, Nemychenkov NS, Sekiguchi T, Driessens G, Zhang P, Koyama M, Spencer A, Holmberg LA, Furlan SN, Varelias A, Hill GR. TIGIT inhibition and lenalidomide synergistically promote antimyeloma immune responses after stem cell transplantation in mice. The Journal of Clinical Investigation. 2023;133(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowery Frank J, Krishna S, Yossef R, Parikh Neilesh B, Chatani Praveen D, Zacharakis N, Parkhurst Maria R, Levin N, Sindiri S, Sachs A, Hitscherich Kyle J, Yu Z, Vale Nolan R, Lu Y-C, Zheng Z, Jia L, Gartner Jared J, Hill Victoria K, Copeland Amy R, Nah Shirley K, Masi Robert V, Gasmi B, Kivitz S, Paria Biman C, Florentin M, Kim Sanghyun P, Hanada K-i, Li Yong F, Ngo Lien T, Ray S, Shindorf Mackenzie L, Levi Shoshana T, Shepherd R, Toy C, Parikh Anup Y, Prickett Todd D, Kelly Michael C, Beyer R, Goff Stephanie L, Yang James C, Robbins Paul F, Rosenberg Steven A. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science (New York, NY).0(0):eabl5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB, Gohil SH, Schindler N, Pomerance L, Hirsch L, Bakouny Z, Hou Y, Forman J, Huang T, Li S, Cui A, Keskin DB, Steinharter J, Bouchard G, Sun M, Pimenta EM, Xu W, Mahoney KM, McGregor BA, Hirsch MS, Chang SL, Livak KJ, McDermott DF, Shukla SA, Olsen LR, Signoretti S, Sharpe AH, Irizarry RA, Choueiri TK, Wu CJ. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer cell. 2021;39(5):632–648.e638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods. 2017;14(9):865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, Cui W. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity. 2019;51(6):1028–1042.e1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reinhardt RL, Liang H-E, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nature immunology. 2009;10(4):385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouabe H, Fässler R, Heesemann J. Improvement of reporter activity by IRES-mediated polycistronic reporter system. Nucleic acids research. 2008;36(5):e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reinhardt RL, Liang H-E, Bao K, Price AE, Mohrs M, Kelly BL, Locksley RM. A novel model for IFN-γ-mediated autoinflammatory syndromes. Journal of immunology (Baltimore, Md : 1950). 2015;194(5):2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szyska M, Herda S, Althoff S, Heimann A, Russ J, D’Abundo D, Dang TM, Durieux I, Dörken B, Blankenstein T, Na IK. A Transgenic Dual-Luciferase Reporter Mouse for Longitudinal and Functional Monitoring of T Cells In Vivo. Cancer Immunol Res. 2018;6(1):110–120. [DOI] [PubMed] [Google Scholar]
- 42.Valujskikh A, Lantz O, Celli S, Matzinger P, Heeger PS. Cross-primed CD8+ T cells mediate graft rejection via a distinct effector pathway. Nature immunology. 2002;3(9):844–851. [DOI] [PubMed] [Google Scholar]
- 43.Emmerich J, Mumm JB, Chan IH, LaFace D, Truong H, McClanahan T, Gorman DM, Oft M. IL-10 directly activates and expands tumor-resident CD8(+) T cells without de novo infiltration from secondary lymphoid organs. Cancer Res. 2012;72(14):3570–3581. [DOI] [PubMed] [Google Scholar]
- 44.Guo Y, Xie Y-Q, Gao M, Zhao Y, Franco F, Wenes M, Siddiqui I, Bevilacqua A, Wang H, Yang H, Feng B, Xie X, Sabatel CM, Tschumi B, Chaiboonchoe A, Wang Y, Li W, Xiao W, Held W, Romero P, Ho P-C, Tang L. Metabolic reprogramming of terminally exhausted CD8+ T cells by IL-10 enhances anti-tumor immunity. Nature immunology. 2021;22(6):746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stuart T, Srivastava A, Madad S, Lareau CA, Satija R. Single-cell chromatin state analysis with Signac. Nature Methods. 2021;18(11):1333–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maurice NJ, Berner J, Taber AK, Zehn D, Prlic M. Inflammatory signals are sufficient to elicit TOX expression in mouse and human CD8+ T cells. JCI Insight. 2021;6(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kartha VK, Duarte FM, Hu Y, Ma S, Chew JG, Lareau CA, Earl A, Burkett ZD, Kohlway AS, Lebofsky R, Buenrostro JD. Functional inference of gene regulation using single-cell multi-omics. Cell Genomics. 2022;2(9):100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Minnie SA, Kuns RD, Gartlan KH, Zhang P, Wilkinson AN, Samson L, Guillerey C, Engwerda C, MacDonald KPA, Smyth MJ, Markey KA, Vuckovic S, Hill GR. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood. 2018;132(16):1675–1688. [DOI] [PubMed] [Google Scholar]
- 49.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, Duan K, Ang N, Poidinger M, Lee YY, Larbi A, Khng AJ, Tan E, Fu C, Mathew R, Teo M, Lim WT, Toh CK, Ong BH, Koh T, Hillmer AM, Takano A, Lim TKH, Tan EH, Zhai W, Tan DSW, Tan IB, Newell EW. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–579. [DOI] [PubMed] [Google Scholar]
- 50.Treon SP, Anderson KC. Interleukin-6 in multiple myeloma and related plasma cell dyscrasias. Current opinion in hematology. 1998;5(1):42–48. [PubMed] [Google Scholar]
- 51.Alexandrakis MG, Goulidaki N, Pappa CA, Boula A, Psarakis F, Neonakis I, Tsirakis G. Interleukin-10 Induces Both Plasma Cell Proliferation and Angiogenesis in Multiple Myeloma. Pathology & Oncology Research. 2015;21(4):929–934. [DOI] [PubMed] [Google Scholar]
- 52.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, Saeys Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry Part A : the journal of the International Society for Analytical Cytology. 2015;87(7):636–645. [DOI] [PubMed] [Google Scholar]
- 53.Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, Ren X, Wang L, Wu X, Zhang J, Wu N, Zhang N, Zheng H, Ouyang H, Chen K, Bu Z, Hu X, Ji J, Zhang Z. Pan-cancer single-cell landscape of tumor-infiltrating T cells. 2021;374(6574):abe6474. [DOI] [PubMed] [Google Scholar]
- 54.Robinson MH, Villa NY, Jaye DL, Nooka AK, Duffy A, McCachren SS, Manalo J, Switchenko JM, Barnes S, Potdar S, Azeem MI, Horvat AA, Parihar VC, Gong J, Liang Y, Smith GH, Gupta VA, Boise LH, Kaufman JL, Hofmeister CC, Joseph NS, Lonial S, Dhodapkar KM, Dhodapkar MV. Regulation of antigen-specific T cell infiltration and spatial architecture in multiple myeloma and premalignancy. J Clin Invest. 2023;133(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duraiswamy J, Turrini R, Minasyan A, Barras D, Crespo I, Grimm AJ, Casado J, Genolet R, Benedetti F, Wicky A, Ioannidou K, Castro W, Neal C, Moriot A, Renaud-Tissot S, Anstett V, Fahr N, Tanyi JL, Eiva MA, Jacobson CA, Montone KT, Westergaard MCW, Svane IM, Kandalaft LE, Delorenzi M, Sorger PK, Färkkilä A, Michielin O, Zoete V, Carmona SJ, Foukas PG, Powell DJ, Rusakiewicz S, Doucey M-A, Dangaj Laniti D, Coukos G. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer cell. 2021;39(12):1623–1642.e1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. Regulation of interferon-gamma production by IL-12 and IL-18. Curr Opin Immunol. 1998;10(3):259–264. [DOI] [PubMed] [Google Scholar]
- 58.Zhu L, Zhou X, Gu M, Kim J, Li Y, Ko C-J, Xie X, Gao T, Cheng X, Sun S-C. Dapl1 controls NFATc2 activation to regulate CD8+ T cell exhaustion and responses in chronic infection and cancer. Nature Cell Biology. 2022;24(7):1165–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nature Reviews Immunology. 2004;4(11):900–911. [DOI] [PubMed] [Google Scholar]
- 60.Fröhlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, Marsland BJ, Oxenius A, Kopf M. IL-21R on T Cells Is Critical for Sustained Functionality and Control of Chronic Viral Infection. 2009;324(5934):1576–1580. [DOI] [PubMed] [Google Scholar]
- 61.Yi JS, Du M, Zajac AJ. A Vital Role for Interleukin-21 in the Control of a Chronic Viral Infection. Science (New York, NY). 2009;324(5934):1572–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xin G, Schauder DM, Lainez B, Weinstein JS, Dai Z, Chen Y, Esplugues E, Wen R, Wang D, Parish IA, Zajac AJ, Craft J, Cui W. A Critical Role of IL-21-Induced BATF in Sustaining CD8-T-Cell-Mediated Chronic Viral Control. Cell Reports. 2015;13(6):1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsao H-W, Kaminski J, Kurachi M, Barnitz RA, DiIorio Michael A, LaFleur Martin W, Ise W, Kurosaki T, Wherry EJ, Haining WN, Yosef N. Batf-mediated epigenetic control of effector CD8+ T cell differentiation. Science immunology.7(68):eabi4919. [DOI] [PubMed] [Google Scholar]
- 64.Giordano M, Henin C, Maurizio J, Imbratta C, Bourdely P, Buferne M, Baitsch L, Vanhille L, Sieweke MH, Speiser DE, Auphan-Anezin N, Schmitt-Verhulst AM, Verdeil G. Molecular profiling of CD8 T cells in autochthonous melanoma identifies Maf as driver of exhaustion. The EMBO journal. 2015;34(15):2042–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, Kurtulus S, Law T, Etminan Y, Nevin J, Buckley CD, Burkett PR, Buenrostro JD, Rozenblatt-Rosen O, Anderson AC, Regev A, Kuchroo VK. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. 2018;558(7710):454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prabhala RH, Pelluru D, Fulciniti M, Prabhala HK, Nanjappa P, Song W, Pai C, Amin S, Tai YT, Richardson PG, Ghobrial IM, Treon SP, Daley JF, Anderson KC, Kutok JL, Munshi NC. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood. 2010;115(26):5385–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK, Fonseca R, Kremer R, Cattoretti G, Bergsagel PL. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer cell. 2008;13(2):167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, Stewart AK, Johnstone RW, Bergsagel PL. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood. 2012;120(2):376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Markey KA, Burman AC, Banovic T, Kuns RD, Raffelt NC, Rowe V, Olver SD, Don AL, Morris ES, Pettit AR, Wilson YA, Robb RJ, Randall LM, Korner H, Engwerda CR, Clouston AD, Macdonald KP, Hill GR. Soluble lymphotoxin is an important effector molecule in GVHD and GVL. Blood. 2010;115(1):122–132. [DOI] [PubMed] [Google Scholar]
- 70.Paszkiewicz PJ, Frassle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, Sadelain M, Liu L, Jensen MC, Riddell SR, Busch DH. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. 2016;126(11):4262–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang P, Lee JS, Gartlan KH, Schuster IS, Comerford I, Varelias A, Ullah MA, Vuckovic S, Koyama M, Kuns RD, Locke KR, Beckett KJ, Olver SD, Samson LD, Montes de Oca M, de Labastida Rivera F, Clouston AD, Belz GT, Blazar BR, MacDonald KP, McColl SR, Thomas R, Engwerda CR, Degli-Esposti MA, Kallies A, Tey SK, Hill GR. Eomesodermin promotes the development of type 1 regulatory T (TR1) cells. Science immunology. 2017;2(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, Payne-Turner D, Althoff MJ, Song G, Chen SC, Ma J, Rusch M, McGoldrick D, Edmonson M, Gupta P, Wang YD, Caufield W, Freeman B, Li L, Panetta JC, Baker S, Yang YL, Roberts KG, McCastlain K, Iacobucci I, Peters JL, Centonze VE, Notta F, Dobson SM, Zandi S, Dick JE, Janke L, Peng J, Kodali K, Pagala V, Min J, Mayasundari A, Williams RT, Willman CL, Rowe J, Luger S, Dickins RA, Guy RK, Chen T, Mullighan CG. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer cell. 2015;28(3):343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nature biotechnology. 2020;38(12):1408–1414. [DOI] [PubMed] [Google Scholar]
- 74.Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, Parks B, Gars E, Liedtke M, Zheng GXY, Chang HY, Majeti R, Greenleaf WJ. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nature biotechnology. 2019;37(12):1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Borcherding N, Bormann NL, Kraus G. scRepertoire: An R-based toolkit for single-cell immune receptor analysis. F1000Research. 2020;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7(9):1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, Greenleaf WJ. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nature genetics. 2021;53(3):403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.