

Abstract
Purpose:
Leiomyosarcomas (LMS) are clinically and molecularly heterogeneous tumors. Despite recent large scale genomic studies, current LMS risk stratification is not informed by molecular alterations. We propose a clinically applicable genomic risk stratification model.
Experimental Design:
We performed comprehensive genomic profiling in a cohort of 195 soft tissue LMS (STLMS), 151 primary at presentation, and a control group of 238 uterine LMS (ULMS), 177 primary at presentation, with at least one-year follow up.
Results:
In STLMS, FNCLCC grade but not tumor size predicted progression-free survival (PFS) or disease-specific survival (DSS). In contrast, in ULMS, tumor size, mitotic rate and necrosis were associated with inferior PFS and DSS. In STLMS, a 3-tier genomic risk stratification performed well for DSS: high risk – co-occurrence of RB1 mutation and chr12q deletion (del12q)/ATRX mutation; intermediate risk – presence of RB1 mutation, ATRX mutation or del12q; low risk – lack of any of these three alterations. The ability of RB1 and ATRX alterations to stratify STLMS was validated in an external AACR GENIE cohort. In ULMS, a 3-tier genomic risk stratification was significant for both PFS and DSS: high risk – concurrent TP53 mutation and chr20q amplification/ATRX mutations; intermediate risk – presence of TP53 mutation, ATRX mutation or amp20q; low risk – lack of any of these three alterations. Longitudinal sequencing showed that most molecular alterations were early clonal events that persisted during disease progression.
Conclusions:
Compared to traditional clinicopathologic models, genomic risk stratification demonstrates superior prediction of clinical outcome in STLMS and is comparable in ULMS.
Keywords: leiomyosarcoma, genomic risk, nomogram, soft tissue, uterine
INTRODUCTION
Leiomyosarcoma (LMS) is one of the most common sarcoma histotypes, defined phenotypically by its consistent differentiation along smooth muscle lines. Although considered a single disease based on its uniform smooth muscle differentiation pattern, the clinical presentation and behavior of LMS are tightly linked to the anatomic location, with metastatic rates ranging from 53–71% in uterine, 31–50% in soft tissue, to 0% in cutaneous LMS (1–3). It remains undetermined if the site-specific behavior relates to intrinsic differences of the cells of origin or due to acquired, unique driver gene alterations. In keeping with this site-specific biologic behavior, the histologic criteria applied for risk of malignancy and pathologic grading are also dependent on tumor location. Tumor grade (by French Federation of Cancer Centers, FNCLCC), tumor size and deep location are deemed the most important prognostic factors in soft tissue but not for uterine and cutaneous LMS (1,4–7), while the presence of coagulative necrosis and high mitotic activity are required for assigning high risk to ULMS (2,8). Moreover, challenges in pathologic grading, such as interobserver variability, tumor sampling and site-specific criteria (uterine vs soft tissue) have limited its benefit for establishing risk. Despite the wide application of genomic testing in clinical practice, which includes a high proportion of advanced/metastatic LMS patients, none of the profiled molecular alterations are used for risk stratification or therapeutic decisions. In this study, we sought to define molecular determinants of aggressive clinical behaviors that can be used as the next-generation, genomic-based risk prediction of clinical outcomes in LMS. Moreover, we further undertook a comparative genomic risk evaluation using a control group of uterine LMS, typically evaluated by different criteria for histologic grading. The ultimate goal of this work is to improve progression-free survival (PFS) and disease-specific survival (DSS) risk assessment and facilitate therapy decisions, such as neoadjuvant or adjuvant therapy.
METHODS
Case Selection and Study Cohort
All cases were identified from the Memorial Sloan Kettering Cancer Center (MSKCC) surgical pathology archives with confirmed pathologic diagnosis between 1991 and 2022 and consented to clinical protocols approved by the MSKCC Institutional Review Board that allowed for MSK-IMPACT next generation sequencing (9). All study subjects provided written informed consent to the use of their genomic data for research. This study has been conducted in accordance with the International Ethical Guidelines for Biomedical Research Involving Human Subjects (CIOMS). There were 195 soft tissue LMS (STLMS) (excluding germline-associated cases, cutaneous and bone LMS), 151 (77%) primary at presentation, and a control group of 238 uterine LMS (ULMS), 177 (74%) primary at presentation, with at least one-year follow up. The STLMS samples used for MSK IMPACT included 104 (53%) primary tumors and 91 (47%) metastatic samples. Among the ULMS cases, 119 (50%) were primary samples, 119 (50%) were metastatic samples. Tumors across the entire grading spectrum of STLMS were included. All conventional ULMS fulfilled histologic criteria for malignancy and were definitionally high-grade. At least two of the following features were seen among ULMS: diffuse moderate to severe nuclear atypia, tumor necrosis, and at least 10 mitotic figures per 10 high power fields. The following clinicopathologic parameters were collected: sex, age, primary site, tumor size (greatest dimension in cm), tumor differentiation, mitotic rate (per 10 high power fields, HPF), tumor necrosis, FNCLCC tumor grade, date of diagnosis, stage at diagnosis, development of progression (recurrence/metastasis), date of progression, date of last follow-up or date of death.
Targeted DNA Sequencing, Copy Number and Mutational Profiling
Detailed descriptions of MSK-IMPACT workflow and data analysis, a hybridization capture-based targeted matched tumor-normal DNA NGS assay targeting 341 to 505 genes for solid tumor were previously described (9). Briefly, for somatic mutation calling, MSK-IMPACT uses genomic DNA from tumor samples with matched patient-derived normal samples from peripheral blood. For whole gene copy number calling, a set of normal FFPE samples were used for reference diploid genome comparison; coverage of targeted regions was computed using the GATK DepthOfCoverage tool (RRID: SCR_001876). Normalized coverage values from tumor samples were divided by corresponding values in normal samples, and log-transformed to yield log-ratios. Recurrent gene-level mutations and copy number alterations present in at least 3% of all samples were kept for downstream analysis. Mutations and gene-level copy number alterations were visualized and summarized using the R package “ComplexHeatmap” version 2.8.0 (RRID:SCR_017270). Germline variants were not included in this study. Mutational signature calling was described previously (10). Briefly, using the pattern and nucleotide context of all observed silent and non-silent substitutions, mutations from each sample were assigned to constituent mutational signatures from the set of 30 COSMIC single base substitution (SBS) signatures described previously (11). Dominant mutation signatures were defined by tumors where > 40% mutations were attributable to that signature.
Overlapping segments were derived using the “CNTools” package version 1.52.0 (RRID: SCR_000281). Copy number variation (CNV) was considered present if the absolute segmentation mean is greater or equal to 0.5. The average copy number signals were then calculated at 1 MB genomic windows and visualized using the R package “ComplexHeatmap” version 2.8.0. Arm-level chromosomal amplifications and deletions for each sample were derived using the ASCETS algorithm (12). Arm-level amplifications and deletions that are present in at least 15% of all samples were kept for downstream analysis.
Gene-level variants and outcome data from 317 cases of STLMS from the AACR GENIE consortium cBioPortal database were also retrieved for downstream analysis (13). The AACR GENIE cohort included predominantly tumor-only targeted panels as follows (listed are panels contributing > 90% of cases): DFCI-ONCOPANEL (v1–3), PROV-TSO500HT-v2, COLU-CCCP, UCSF-IDTV5-TO, VICC-01-D2, and YALE-OCP-V3. In contrast, the MSK-IMPACT cohort is a matched tumor-normal platform. Of the 317 cases available in the AACR GENIE cohort, segmented copy number data were available in only 10 cases, precluding the ability to derive chromosomal arm level CNV data. Moreover, depending on probe and assay design, the same genes interrogated by different NGS panels may be covered at variable breadth and depth of coverage.
Immunohistochemistry
Randomly selected STLMS cases with genetic alterations detected in TP53, RB1, ATRX and/or PTEN and with available formalin-fixed, paraffin-embedded tissue were retrieved for immunohistochemical staining using antibodies raised against TP53, RB1, ATRX and PTEN. Detailed information on antibody clone and staining protocol is found in Supplementary Table S1. The result interpretation was done independently by two sarcoma pathologists (JD, CRA), blinded from the molecular results. Null (complete absence of expression) or overexpression (diffuse nuclear or cytoplasmic expression) of TP53 was considered a mutant pattern. A wild-type TP53 pattern was defined as heterogeneous nuclear expression of variable intensity. Null expression (complete absence of nuclear ATRX and RB1 and cytoplasmic PTEN staining) was considered a mutant pattern for these proteins. Wild-type patterns were defined as positive nuclear ATRX and RB1 or cytoplasmic PTEN expression.
Survival Analysis
Survival analysis by comparison of hazard ratios using log-rank P testing and visualization of Kaplan-Meier curves were performed using R packages “survminer” version 0.4.9 (RRID: SCR_021094) and “survival” version 3.2.13 (RRID: SCR_021137). Median time (in years) to disease progression was defined as the time interval between initial diagnosis (presence of tumor seen radiographically or by pathologic confirmation) and the first instance of tumor recurrence or distant metastases after initial surgical resection. PFS was defined as time from surgery to first local recurrence or metastasis or last clinical follow-up. All but five of the disease progression events in the STLMS cohort and all disease progression events in the ULMS cohort were metastases. DSS was defined as time from diagnosis to last clinical follow-up or death caused by disease. Among STLMS patients who died, all but two cases died of LMS. Among ULMS patients who died, all died of LMS. Therefore, DSS and OS were similar in the MSKCC cohort. From the AACR GENIE cohort, among the STLMS cases with documented survival status at last contact, overall survival (OS) was calculated by subtracting “Age at Which Sequencing was Reported” from “Interval in days from DOB to date of last contact” or “Interval in days from DOB to DOD”. DSS and PFS data were not available from the AACR GENIE validation cohort. There was an insufficient number of cases of ULMS with follow-up data in the AACR GENIE cohort to validate our genomic risk stratification model.
Genomic Risk Stratification
Genomic data (single gene mutations/copy number alterations and chromosomal arm level changes) were fed into the OncoCast-MPM (14) algorithm. The OncoCast machine-learning tool repeatedly and randomly splits the entire cohort into training (two-thirds of the cohort) and test (a third of the cohort) sets 100 times to generate an ensemble of classifiers with a varying selection of genomic variables. For the OncoCast function, the elastic net (ENET) method model (recommended) with “Cox” family selection was performed over 100 runs. ENET is a penalized regression method that combines the properties of LASSO (least absolute shrinkage and selection operator) and RIDGE regression to create a model that is parsimonious, breaks multicollinearity and performs selection of predictors to maximize prediction accuracy (14). The training cohort was used to build the elastic-net model, whereas the testing cohort was used to assess the performance of the model generated. Thereafter, based on the frequency and hazard ratios of each parameter and their distribution among the OncoCast risk groups, statistically significant genomic alterations were selected and assigned to our proposed genomic risk stratification model.
Data Availability Statement
All clinical and genomic sequencing data generated by MSK-IMPACT described in this manuscript are publicly available at: https://www.cbioportal.org/study/summary?id=lms_msk_2024. The raw sequencing data are protected. Deidentified data are available under restricted access to protect patient privacy in accordance with federal and state law. These data can be requested for research use from the corresponding author via execution of a data transfer agreement with MSKCC, which will retain all title and rights to the data and results from their use.
RESULTS
Genomic Landscapes in STLMS and ULMS
Using MSK-IMPACT, we profiled gene-level mutations and CNA, as well as chromosomal arm-level gains and losses in a clinically well-characterized cohort of 195 STLMS and 238 ULMS with at least one-year follow-up. Among the STLMS cohort, median age was 58 years old (range 21–86), 124 female (64%) and 71 male (46%), with the majority located in the retroperitoneal/intraabddominal/pelvic sites (145, 74%) (Table 1). Among the ULMS cohort, median age was 54 years old (range 22–79) (Suppl. Table S2).
Table 1.
Clinicopathologic summary of soft tissue leiomyosarcoma (STLMS)
| Clinicopathologic variables | All cases N (%) | Primary at Presentation N% | Univariate P-value* | Multivariate P-value* |
|---|---|---|---|---|
| Total | 195 | 151 | ||
| Age (years) | 0.511 | |||
| ≤ 60 | 110 (56) | 86 (57) | ||
| > 60 | 85 (44) | 65 (43) | ||
| Sex | 0.489 | |||
| Female | 124 (64) | 96 (64) | ||
| Male | 71 (46) | 55 (36) | ||
| Site | 0.566 | |||
| Retroperitoneal/intraabdominal/pelvic | 145 (74) | 110 (73) | ||
| Extremity | 37 (20) | 31 (21) | ||
| Trunk | 13 (6) | 10 (7) | ||
| Size (cm) | 0.229 | |||
| ≤ 5 | 49 (26) | 46 (31) | ||
| > 5 to ≤ 10 | 77 (41) | 61 (40) | ||
| > 10 | 64 (33) | 44 (29) | ||
| Mitotic rate (per 10 HPF) | 0.0039 | 0.0045 | ||
| ≤ 10 | 96 (49) | 70 (46) | ||
| > 10 to ≤ 20 | 60 (31) | 51 (34) | ||
| > 20 | 39 (20) | 30 (20) | ||
| Necrosis | 0.0371 | 0.2572 | ||
| Present | 134 (81) | 108 (79) | ||
| Absent | 31 (19) | 29 (21) | ||
| Unknown | 30 | 14 | ||
| Progression | ||||
| Yes – Distant metastasis | 171 (88) | 127 (84) | ||
| Yes – Local recurrence | 5 (3) | 5 (3) | ||
| No | 19 (9) | 19 (13) | ||
| Median survival (months) | ||||
| Disease-specific survival | 80.1 | 109 | ||
| Progression-free survival | 26.3 |
DSS analysis among 151 STLMS primary at presentation
By genomic profiling, STLMS and ULMS were both characterized by frequent TP53 mutations (56% STLMS, 55% ULMS) and deletions (12% STLMS, 16% ULMS), RB1 mutations (26% STLMS, 13% ULMS) and deletions (24% STLMS 39% ULMS), ATRX mutations (10% STLMS, 32% ULMS), and PTEN deletions / occasional mutations (12% STLMS, 18% ULMS) (Figure 1A). As expected in tumor suppressor genes, these mutations were seen throughout the entire coding regions of TP53, RB1 and ATRX across various functional domains (Suppl. Fig. S1A). CDKN2A deletion was more common in ULMS (10%) than in STLMS (2.6%). ULMS, in addition, harbored recurrent MED12 mutations (15%), which were infrequent among STLMS (1%).
Figure 1.
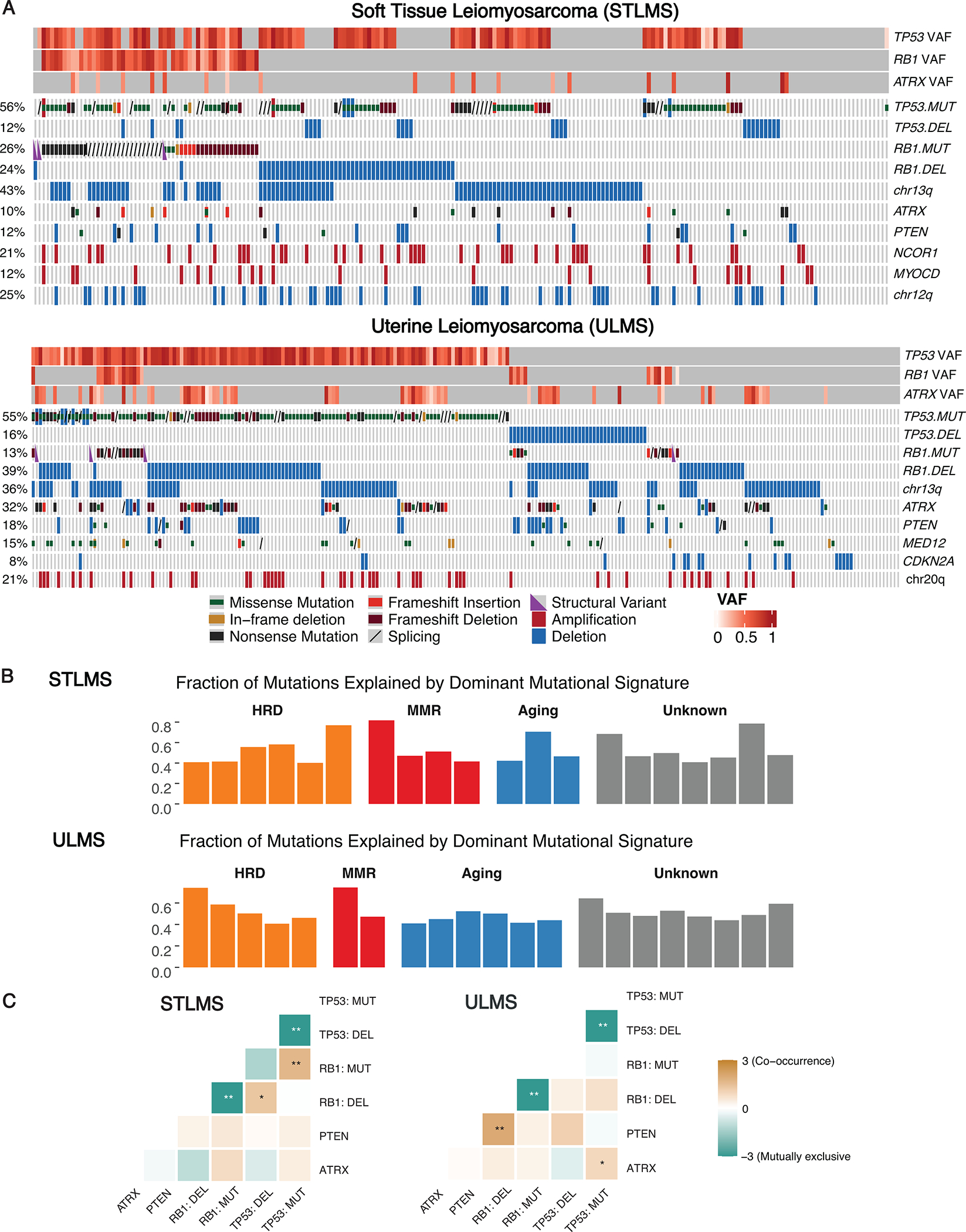
Spectrum of genomic alterations in STLMS and ULMS.
A. Genomic alterations in soft tissue (STLMS) and uterine leiomyosarcomas (ULMS). OncoPrint depicts key gene-level alterations and chromosomal arm level copy number variations. Variant allele frequency (VAF) of TP53, RB1, and ATRX mutations are represented by heatmaps. B. Individual STLMS and ULMS tumors harboring dominant mutation signatures. Bar charts display the fraction of mutations explained by the mutational signature. C. Mutual exclusivity and co-occurrence plots for the most frequent genomic alterations detected in STLMS and ULMS. Color of heatmap represents log2 odds ratio (* P < 0.05; ** P < 0.01).
Tumor mutation burden (TMB) was low: STLMS and ULMS had a median of TMB of 2 and 3 mt/mb, respectively, with 99% and 98% of STLMS and ULMS cases harboring a TMB < 10 mt/mb. From the subset of cases harboring dominant SBS mutation signatures (20 STLMS, 22 ULMS), the following recurrent signatures were identified: aging (Signature 1); homologous recombination deficiency (HRD) (Signature 3); DNA mismatch repair (MMR) (Signatures 6, 15, and 26); and unknown etiology (Signature 5,12 and 30) (Figure 1B). Among STLMS, dominant HRD, MMR and aging signatures were detected in 6, 4 and 3 cases, respectively. Among ULMS, dominant HRD, MMR and aging signatures were detected in 5, 2 and 6 cases, respectively.
In both STLMS and ULMS, RB1 mutations and RB1 deletions were mutually exclusive (P < 0.01), as were TP53 mutations and TP53 deletions (P < 0.01). On the other hand, in STLMS, RB1 mutations and TP53 mutations tended to co-occur (P < 0.01), as did RB1 deletions and TP53 deletions (P < 0.05). In ULMS, RB1 deletions often co-occurred with PTEN deletions (P < 0.01), as did TP53 mutations and ATRX mutations (P < 0.05) (Figure 1C). In terms of clonality, TP53 and RB1 alterations had higher VAFs than ATRX and PTEN, suggesting that they might be an earlier/dominant clonal events or have loss of heterozygosity (Figure 1A).
Longitudinal Sequencing in STLMS and ULMS
In a cohort of 18 patients with STLMS and 15 patients with ULMS, we interrogated sequential tumors in the same patient. The median time intervals between sequential cases were 3 years (range 1–6) for STLMS and 4 years (range 1–9) for ULMS, respectively. Longitudinal sequencing showed that most of the molecular alterations appeared to be an early clonal event that persisted during disease progression, with a subset of cases harboring acquired molecular alterations, e.g., RB1 loss or ATRX mutations, not detected in the initial tumor that was sequenced (Figure 2A, B). Additionally, when we compared the distributions of recurrent genetic alterations in STLMS and ULMS, respectively, between primary vs metastatic samples (unpaired), there were no significant differences in frequencies (Suppl. Fig. S1B,C).
Figure 2.
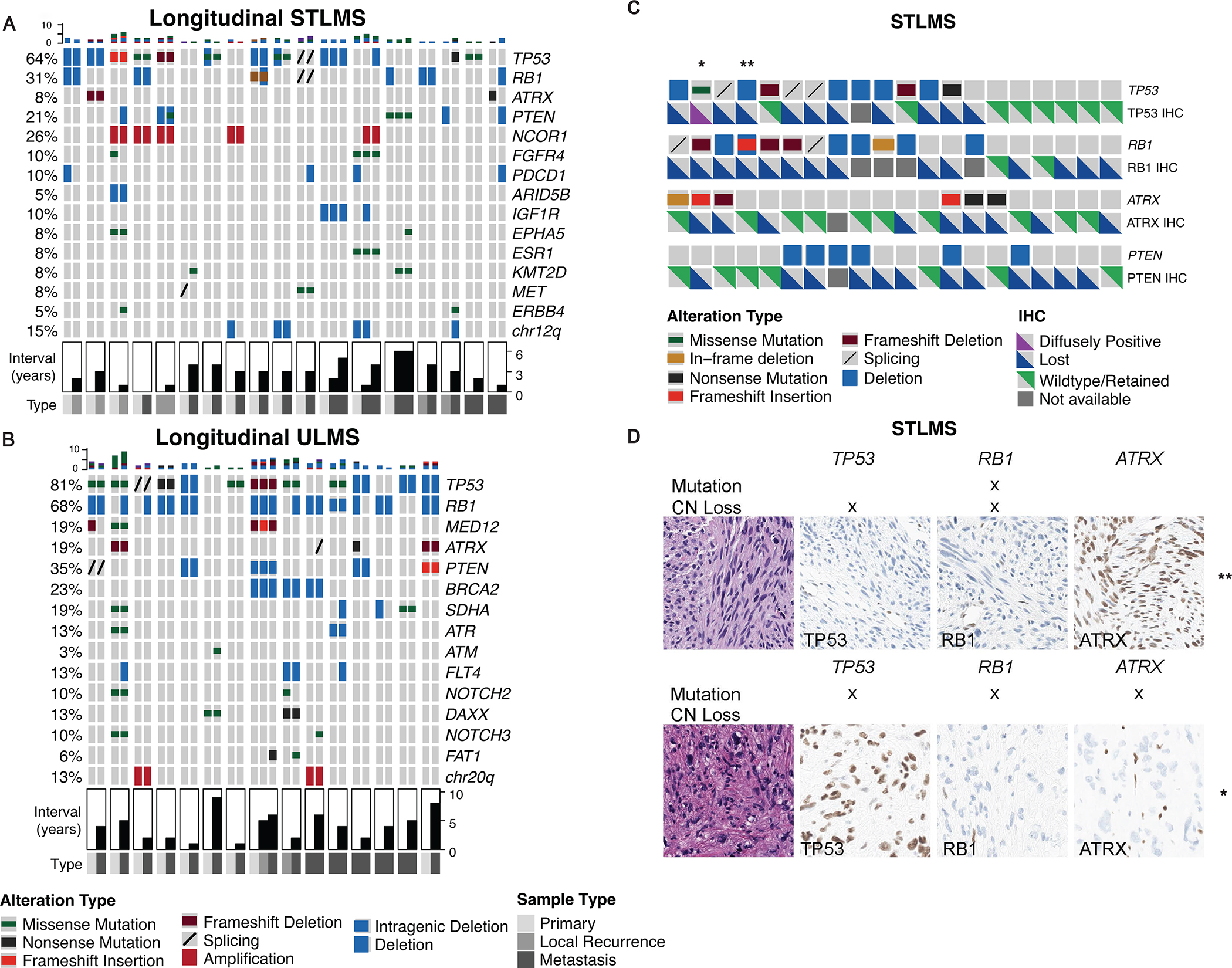
Longitudinal sequencing and molecular-based immunohistochemical (IHC) profiles.
A-B, OncoPrint depicting 18 patients with soft tissue (STLMS) (A) and 14 patients with uterine leiomyosarcomas (ULMS) (B) with more than one sample sequenced longitudinally. Samples from the same patient were grouped and arranged chronologically. Also annotated is sample type (primary versus metastatic) at the time of sequencing and time interval in years between sequential samples. C-D, Molecular-based immunohistochemical (IHC) profiles for TP53, RB1 and ATRX in STLMS. C, Oncoprint depicting the correlation of molecular alterations and IHC expression in TP53, RB1, ATRX and PTEN. D, Illustrated are two cases of STLMS (marked by asterisks in A) with either mutations or deletions in TP53, RB1 and/or ATRX, and their corresponding wild-type and mutant expression patterns by IHC.
Immunohistochemistry as Surrogate Biomarkers
Given their frequency and potential prognostic significance, we investigated the ability of immunohistochemistry (IHC) to act as a surrogate for molecular alterations in TP53, RB1, ATRX and PTEN in 20 random cases of STLMS. As shown in Figure 2C,D, we detected diffuse loss of protein expression in STLMS cases [or diffuse and strong expression (“mutant pattern”) in one case for TP53] harboring inactivating point mutations or copy number losses in TP53, RB1 and ATRX, suggesting the potential use of IHC as a surrogate prognostic biomarker. Nonetheless, we did detect loss of these proteins in some cases without detectable genetic alterations, which may imply that either some loss-of-function genetic changes were missed by our molecular assay, or that these IHC markers were relatively sensitive but not specific for the presence of genetic alterations. Moreover, PTEN IHC results were more equivocal and difficult to interpret, as the cytoplasmic staining appeared weak/”blush” even in retained cases. Overall, the sensitivity and specificity of IHC expression for molecular alterations in TP53 were: 83% (10/12) and 86% (6/7), in RB1 were: 100% (8/8) and 25% (2/8), in ATRX were: 83% (5/6) and 69% (9/13), and in PTEN were: 100% (5/5) and 50% (7/14), respectively.
Genomic vs Traditional Risk Stratification in STLMS
For multivariate survival analysis, we included gene-level genomic mutations and CNA present in > 3% of cases and arm level variations > 15% of cases in the OncoCast ML model (13). To generate a risk stratification model that is widely applicable, we selected genomic parameters with high selection frequency and a hazard ratio > 1.3 that showed strong distribution bias for the high and low risk groups using the OncoCast model, a next-generation sequencing-derived machine-learning risk-prediction model (OncoCast-MPM) (13) (Suppl. Fig. S2).
Of 195 STLMS [44 (23%) metastatic] cases from the MSKCC cohort, 151 (77%) cases were primary at presentation. Among the STLMS that were primary at presentation, the overall median DSS was 109 months (5-year DSS: 79%). FNCLCC grading (3-tier system based on a combination of tumor differentiation, mitotic count, and necrosis) (global log-rank P = 0.047) but not tumor size (global log-rank P = 0.47) predicted worse DSS (Figure 3A, Suppl. Fig. 3, Table 1). Our analysis generated a three-tier genomic risk stratification model that performed well for DSS (global log-rank P = 0.04): high risk – co-occurrence of RB1 inactivating mutations (exclusive of RB1 deletion) and chr12q deletion (del12q) or co-occurrence of RB1 mutation and ATRX mutation (n = 10; median DSS: 75 months); intermediate risk – presence of any of the following: RB1 mutation, ATRX mutation or del12q (n = 60; median DSS: 101 months); low risk – lack of any of these three alterations (n = 81; median DSS: 158 months) (Figure 3B). Among the 44 cases of STLMS metastatic at presentation, the presence of RB1 mutations (n = 14) predicted worse DSS (log-rank P = 0.044; median DSS: 38 vs. 61 months) (Figure 3C). This risk stratification model remained significant when applied to OS (Suppl. Fig. S3).
Figure 3.
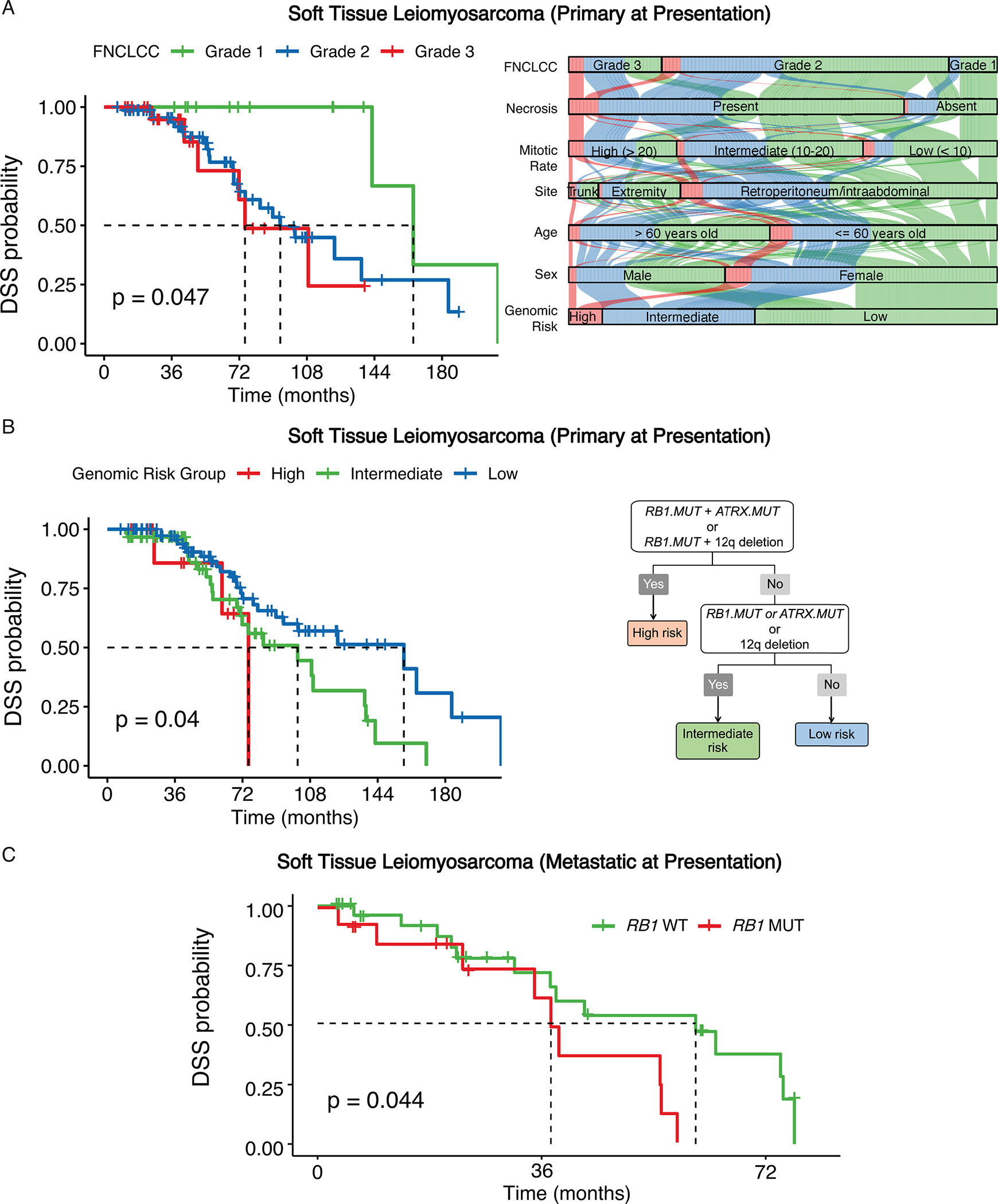
Primary and metastatic STLMS risk stratification.
A, Disease-specific survival (DSS) for patients from the MSK cohort stratified by FNCLCC grade (A) represented by Kaplan-Meier curves. Alluvial diagram comparing age (≤ 60 years old, > 60 years old), sex, primary site, histologic grade (FNCLCC, mitotic count, necrosis) (versus genomic risk groups in STLMS. B, A 3-tier genomic stratification scheme is proposed for DSS from the MSK cohort of 151 STLMS patients that were primary at presentation. C, DSS for 44 patients with metastatic STLMS from the MSK cohort stratified by RB1 mutation status is represented by Kaplan-Meier curve.
Comparing the traditional versus genomic risk categories, cases stratified as high or intermediate genomic risks were present in low, intermediate and high traditional risk tiers (based on FNCLCC and tumor size), implying a potential for traditional risk stratification to under-grade some cases (Figures 3A, Suppl. Fig. S4). On multivariate Cox regression analysis, compared with mitotic rate and necrosis, only high genomic risk group remained significant prognostically for DSS in STLMS (Suppl. Fig. S5).
Of the entire cohort of 195 STLMS cases, the overall median DSS was 80.1 months (5-year DSS: 71%). This 3-tier genomic risk stratification scheme remained significant for DSS among the entire group (global log-rank P = 0.00057), with significantly different pairwise survival differences after multiple comparison correction. Specifically, high risk (n = 18): median DSS – 55 months, 5-year DSS – 43%; intermediate risk (n = 76): median DSS – 77 months, 5-year DSS – 64%; low risk (n = 101): median DSS – 102 months, 5-year DSS – 79% (Figure 4A).
Figure 4.
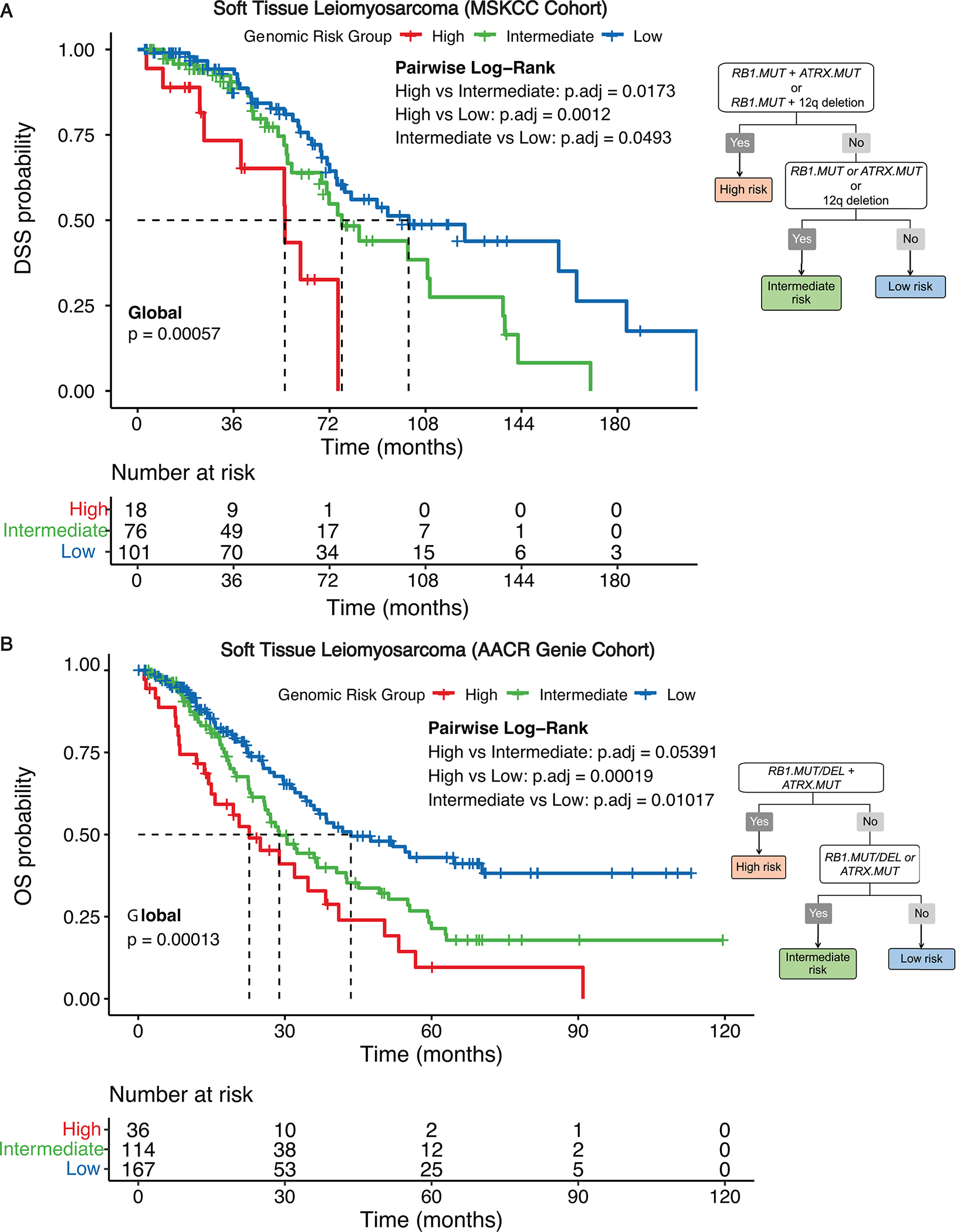
STLMS genomic risk stratification for disease-specific survival (DSS)
A 3-tier genomic risk stratification scheme is proposed for DSS. DSS for 195 STLMS patients from the MSK (A) and OS for 317 STLMS patients from AACR GENIE cohorts (B) in low-, intermediate-, and high-risk genomic groups is represented by Kaplan-Meier curves for 195 STLMS cases (global log-rank P and Benjamin-Hochberg adjusted P-values by pairwise analysis).
To validate our risk stratification model in an external cohort, we analyzed the AACR GENIE dataset, which was comprised of STLMS cases from multiple tumor-only targeted NGS panels from different institutions (excluding MSKCC cases). Since majority of these datasets only included gene-level alterations but not chromosomal arm level gains and losses, we focused on the ability of RB1 and ATRX alterations to stratify STLMS. In this cohort, RB1 mutations and deletions were mutually exclusive with each other, as were TP53 mutations and deletions, while RB1 and TP53 mutations tend to co-occur, confirming our observation in the MSKCC cohort (Suppl. Fig. S6). Of 317 STLMS cases [128 (40%) metastatic at presentation] with follow-up data (documented survival status at least one-year post diagnosis), the following simplified 3-tier genomic risk stratification model performed well for OS (global log-rank P = 0.00013), with significantly different pairwise survival differences after multiple comparison correction: high risk – co-occurrence of RB1 alterations (inactivating mutation/deletion) and ATRX mutation (n = 36; median OS: 23 months; 5-year OS: 10%); intermediate risk – presence of any of the following: RB1 alterations or ATRX mutation (n = 114; median OS: 29 months; 5-year OS: 21%); low risk – lack of RB1 or ATRX alterations (n = 167; median OS: 44 months; 5-year OS: 43%) (Figure 4B). This modified 3-tier genomic risk stratification scheme that excludes del12q remained significant for DSS in the MSK cohort (Suppl. Fig. S7). There were slight differences in the frequencies of detection of RB1 mutation and RB1 deletion: 20% and 16% of cases from the AACR GENIE cohort harbored RB1 mutation and RB1 deletion, respectively. In contrast, 26% and 24% of cases from the MSKCC cohort harbored RB1 mutation and RB1 deletion, respectively.
Focusing on PFS, of the 151 STLMS cases that were primary at presentation with at least one-year follow up, the median PFS was 26.3 months (5-year PFS: 14%). The presence of any of the following: RB1 mutations, ATRX mutations or PTEN deletions (n = 57), was predictive of worse PFS (log-rank P = 0.049; median PFS: 5-year PFS: 5.5% vs 19%) (Suppl. Figure S8A). On the other hand, only FNCLCC grade 3 but not tumor size (> 10 cm vs 5–10 cm vs < 5 cm) predicted worse PFS (Suppl. Fig. 8B, C). TP53 mutations/deletions were not found to be associated with worse OS or PFS in STLMS.
Genomic vs Traditional Risk Stratification in ULMS.
In a control group of 238 well characterized ULMS, 177 primary at presentation [61 (26%) metastatic at presentation], with at least one-year follow up, we applied the same methodology using Oncocast-MPM to evaluate for prognostically significant genomic variants (Suppl. Fig. S9). The overall median DSS among the 177 primary at presentation ULMS cases was 86.9 months (5-year DSS: 64%). Our modeling generated a three-tier genomic risk stratification model that performed well for DSS (global log-rank P = 0.00012) with significantly different pairwise survival differences after multiple comparison correction (high vs low P = 0.00014, high vs intermediate P = 0.00623): high risk – co-occurrence of TP53 mutation (exclusive of TP53 deletion) and ATRX mutation or co-occurrence of TP53 mutation and chr20q amplification (amp20q) (n = 50; median DSS: 52 months; 5-year DSS: 41%); intermediate risk – presence of any of the following: TP53 mutation, ATRX mutation or amp20q (n = 66; median DSS: 92 months; 5-year DSS: 67%); low risk – lack of any of these three alterations (n = 61; median DSS: 157 months; 5-year DSS: 79%) (Figure 5A).
Figure 5.
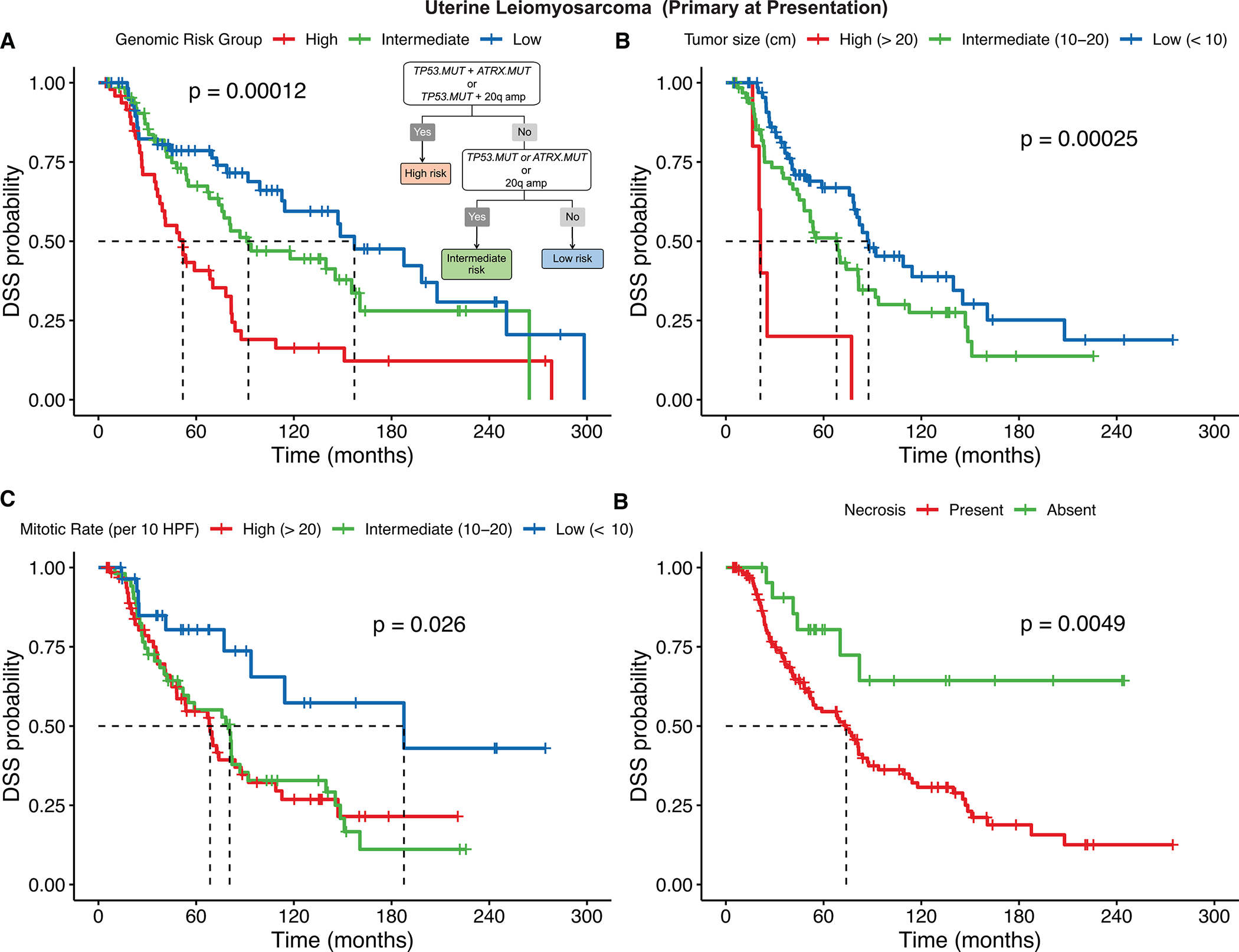
Primary ULMS genomic and traditional risk stratification for disease-specific survival (DSS)
A, A 3-tier genomic risk group is proposed for DSS. DSS for 177 ULMS patients (primary at presentation) from the MSK cohort in low, intermediate, and high-risk genomic groups is represented by Kaplan-Meier curves (global log-rank P and Benjamin-Hochberg adjusted P-values by pairwise analysis). B-D, DSS stratification by tumor size (B), mitotic rate (C) and presence of necrosis (D) are also represented.
The overall median DSS among the entire cohort of 238 ULMS cases was 75.9 months (5-year DSS: 55%). This 3-tier genomic risk stratification scheme remained significant for DSS among the 238 ULMS cases (global log-rank P = 0.00011), with significantly different pairwise survival differences after multiple comparison correction: high risk (n = 72): median DSS – 41 months, 5-year DSS – 38%; intermediate risk (n = 94): median DSS – 74 months, 5-year DSS – 54%; low risk (n = 72): median DSS – 149 months, 5-year DSS – 73% (Suppl. Figure S10).
Focusing on PFS, the median PFS of the primary at presentation ULMS cases was 24 months (3-year PFS: 33%). The same genomic risk model was also predictive of PFS in ULMS (global log-rank P = 0.014): high risk (n = 50; median PFS: 15 months; 3-year PFS: 17%); intermediate risk (n = 66; median PFS: 25 months; 3-year PFS: 36%); low risk (n = 61; median PFS: 29 months; 3-year PFS: 42%) (Suppl. Fig. S11A).
In terms of traditional clinicopathologic risk stratification in ULMS, as FNCLCC grading is not generally applied, we examined the relationship of tumor size, mitotic rate, and tumor necrosis with clinical outcome. For both DSS and PFS, tumor size (> 20 cm vs 10–20 cm vs < 10 cm), mitotic rate > 10 per 10 high power fields, and presence of necrosis were significantly associated with worse outcome (Figure 5B–D, Suppl. Fig. S11B–D, Suppl. Table S2). On multivariate Cox regression analysis, compared with tumor size, mitotic rate and necrosis, only tumor size and high genomic risk group remained significant prognostic biomarkers for DSS in ULMS (Suppl. Fig. S12).
DISCUSSION
Recent genomic efforts in leiomyosarcoma (LMS), including large integrated datasets (WES, WGS and RNAseq) from TCGA and other groups, have identified frequent alterations in TP53, RB1, PTEN and ATRX (15). In addition to LMS, RB1 and TP53 alterations drive the pathogenesis of most other genomically complex sarcomas, often co-occurring at a significantly higher prevalence compared to translocation-associated sarcomas (16). However, some recurrent amplifications of 17p11.2-p12, involving myocardin (MYOCD), MAP2K4 and NCOR1 genes, and of 15q IGF1R locus appear pathognomonic for LMS (17–19). Among the most frequent recurrent genomic alterations involving muscle genes in LMS are intragenic deletions of the dystrophin (DMD) gene (16%) and focal MYOCD amplifications (39%) (20). MYOCD is a transcriptional cofactor of serum response factor regulating smooth muscle development and differentiation (21–23). It was initially reported by Perot et al. to be highly amplified and overexpressed in a subset of retroperitoneal LMS (20), and further confirmed by other studies either by FISH (18) or by NGS (20, 24). In our study, although most of the muscle related genes (LMOD1, CALD1, MYOCD, DMD, ACTG2, DES, CFL2, SLMAP) are not covered by the MSK-IMPACT panel, using the copy number gains at their corresponding genomic locations as a surrogate marker, e.g., chr17p12 amplification as a surrogate for MYOCD gains, we did not observe association of the presence of amplifications at these loci with differences in survival.
In a large comprehensive genomic study using targeted DNA next generation sequencing (NGS), similar to other high grade genomically complex sarcomas, both STLMS and ULMS showed high entropy levels and could be divided in relatively comparable genomic subclusters based on alterations of TP53 and RB1 pathways (TP53, RB, TP53-RB, TP53-ATRX, TP53-ATRX-RB) (25). Among these, STLMS had the highest incidence of RB1 mutations (19%), while ULMS had the highest incidence of ATRX alterations (predominantly loss-of-function mutations and some deletions) (32%) (26). TP53 mutations were detected in 45–52% of STLMS and ULMS, respectively (25). While TP53 and RB1 alterations were found to co-occur in multiple sarcoma types (25), mutual exclusivity between RB1 mutations and deletions, as well as TP53 mutations and deletions, had yet to be formally reported in soft tissue sarcomas. Similar to what was previously reported in LMS and other complex karyotype sarcomas, STLMS and ULMS consistently demonstrate low TMB in our study (16, 25). We also detected HRD and MMR mutational signatures in a subset of STLMS and ULMS, which had been reported in some LMS and was proposed to be a potential therapeutic target (24).
Despite our enhanced understanding of the genomic landscape of LMS, studies focusing on the utility of clinically available targeted DNA sequencing datasets in predicting outcome or informing on targeted therapy have not been employed. In this study we sought to develop a risk assessment tool that compares novel molecular biomarkers and conventional pathologic risk metrics (e.g., tumor size, location, and mitotic account) using machine learning (ML) in a well characterized clinical and molecular cohort of soft tissue LMS compared to a uterine LMS group. In our cohort, compared to traditional clinicopathologic models based on tumor size (surrogate for staging), mitotic count and necrosis (parameters for histologic grade), genomic risk stratification demonstrates superior performance in STLMS and is comparable in ULMS. As the vast majority of STLMS was classified as “conventional LMS” and received a tumor differentiation score of 2 based on the FNCLCC scheme, we did not examine tumor differentiation specifically as a prognostic factor. Compared to the study from Gladdy et al. (7), which investigated 353 primary STLMS patients diagnosed between 1982–2006, our cohort comprised only of cases that underwent MSK-IMPACT genomic profiling from 2013–2022. In a head-to-head comparison with the older cohort, our group of 151 STLMS cases that were primary at presentation contained a significantly higher percentage of patients harboring tumors that were retroperitoneal or intraabdominal (73% vs 41%, chi-square P < 0.0001) and that went on to develop distant metastasis (84% vs 31%, chi-square P < 0.0001). This bias towards more aggressive STLMS cases in our study is likely explained by the preferential patient selection with higher grade tumors for NGS analysis, which may account for the reason why tumor size based on a cutoff of 5 and 10 cm was a significant prognostic predictor in the older cohort, but not in the present study.
One limitation of this study is the inherent variability of the genomic NGS platforms used for our validation cohort, as different institutions participating to the AACR GENIE cohort include tumor-only, versus the MSK-IMPACT cohort, which is a matched tumor-normal platform. The AACR GENIE dataset also did not contain chromosomal arm level CNV data, limiting our ability to compare arm level CNV as prognostic variables across datasets. Further, the calculation of OS for the AACR GENIE cohort from the time of sequencing rather than diagnosis (data not available) is potentially problematic. Moreover, different NGS panels are associated with a variable breadth of probe coverage. These may account for the differences in sensitivity in detecting RB1 mutations and RB1 deletions, which may explain why RB1 mutation, exclusive of RB1 deletion, was a significant prognostic factor for the MSKCC STLMS cohort, while RB1 alterations (mutations + deletions) were significant for the AACR GENIE cohort. The AACR GENIE STLMS cohort also had a significantly higher percentage of cases that were metastatic at presentation (40%) compared to the MSKCC cohort (23%), which likely accounted for the lower median and 5-year OS in the former. Despite these limitations, our results showed a good concordance of the overall frequencies of TP53, RB1, ATRX, and PTEN alterations and their respective prognostic significance in DSS/OS in STLMS. These findings suggest that a simplified version of the genomic risk stratification, incorporating mostly mutations and gene-level CNV, may be feasible to implement across different institutions using tumor tissue-only input and slightly different targeted NGS panels.
With the advent of whole-genome sequencing (WGS) and transcriptome profiling, nearly universal TP53 and RB1 inactivation has emerged as a unifying feature of LMS development (22). In the TCGA study, the types of RB1 alterations included 14% deep deletions, 78% shallow deletions and 15% mutations (15). Moreover, based on these integrated genomic approaches, a wider spectrum of biallelic disruption mechanisms was uncovered, including less frequent non-recurrent structural rearrangements (fusions), microdeletions, inversions, exon skipping, etc. (26). The variant allele frequencies (VAF) of SNVs and indels affecting TP53 and RB1 were found to be congruent with tumor purity, highlighting TP53 and RB1 inactivation as truncal events in LMS development (28). Furthermore, phylogenetic reconstruction by WGS demonstrated that in addition to somatic TP53 substitutions, which are likely the first initiating alteration, arm-level or whole-chromosome deletions of tumor suppressor genes, i.e. RB1, TP53, PTEN, are early events, preceding DNA amplifications and whole genome duplication (WGD) (20).
Our results of RB1 mutations but not RB1 deletions having a negative effect on survival in STLMS at least in the MSKCC cohort raises questions on the functional impact of different mechanisms of tumor suppressor inactivation. Notably, the presence of 13q14/RB1 deletion is the hallmark alteration of many benign mesenchymal lesions (e.g., spindle cell lipoma, cellular angiofibroma, mammary-type myofibroblastoma, etc.), which typically do not acquire additional genetic alterations in their disease course (27). One hypothesis is that RB1 deletion alone may be insufficient to induce malignant transformation, and this finding requires further functional analyses. Similarly, the presence of ATRX and TP53 mutations but not deletions were found to have prognostic significance in ULMS. This is in keeping with the observation that most TP53 abnormalities in human tumors are point mutations, resulting in the expression of an aberrant, mutant form of the protein, which has an enhanced oncogenic role, either as a dominant negative or gain-of-function effect, compared to a simple loss of the wild-type function (28). On a similar note, the introduction of TP53 hotspot mutations was more efficient in triggering sarcomagenesis in murine models compared to TP53 loss alone and led to spontaneous metastasis in 13% of cases (28).
Few genomic studies have evaluated low grade LMS or histologic progression to a high grade LMS, with one study showing 13q14 loss as an early genetic alteration in the progression of the disease, with 3 of the 4 low grade LMS tested showed this abnormality (19). Similarly, two of the 3 low grade LMS in the MSK cohort harbored chr13q deletion without concurrent RB1/TP53/ATRX variants. The remaining case harbored PTEN deletion. At the opposite end, late events occurring in LMS progression includes WGD, which occurs in 44% of cases, as well as kataegis and chromothripsis, which generate widespread genetic diversity within a single primary or relapsed LMS tumor (20).
Molecular risk stratification using clinically validated targeted NGS panels recently has been applied in other sarcomas, including gastrointestinal stromal tumor (GIST) (29). In this recent study, GIST was divided in a 3-tier genomic risk groups, with different sets of genetic alterations found to be prognostic for the two main clinical subsets, gastric and small bowel. For example, small bowel GISTs were classified as high risk if MAX/MGA/MYC, CDKN2A, or RB1 alterations were present, and intermediate risk if chr1p deletion or chr5q amplification was present. Moreover, artificial Intelligence-based models, such as the Optimal Classification Trees, are emerging as superior predictors of the probability of recurrence after surgery, compared to traditional pathologic criteria (Miettinen system, etc.) or sarcoma-specific nomograms, by capturing non-linear relationships among predictors of recurrence (30, 31). Historically, pathologic factors that predicted metastatic spread in STLMS included tumor size, necrosis, histologic grade and disruption, however, site, depth or mitotic activity were not significant (7, 32).
In summary, no prior study has investigated directly the impact of the various mechanisms of inactivation on survival of LMS patients. Of special interest is the finding that different sets of mutations were associated with inferior survival in our two LMS cohorts, specifically, RB1 and ATRX in STLMS, and TP53 and ATRX in ULMS. Compared to traditional clinicopathologic models, genomic risk stratification demonstrates superior performance in STLMS and is comparable in ULMS. Further refinements of this model may include integration of existing clinicopathologic factors to the genomic risk model into a combined nomogram.
Supplementary Material
Translational Relevance.
Despite recent large scale genomic studies, current risk stratification schemes in leiomyosarcoma (LMS) rely solely on clinicopathologic metrics: tumor size and tumor grade. These risk models have performed inconsistently in differentiating potentially aggressive from indolent clinical behavior, offering limited guidance on systemic therapy. We sought to better define molecular determinants of survival that can be used as the next-generation, genomic-based risk prediction in soft tissue and uterine LMS. By integrating molecular markers and utilizing multivariate statistical models and machine learning methods, we developed a novel genomic risk stratification model for soft tissue and uterine LMS.
Acknowledgement
Supported in part by: P50 CA217694 (CRA), P30 CA008748 (CRA, SC, MLH, WT, SM, RGM), Cycle for Survival (CRA, MLH), Kristin Ann Carr Foundation (CRA), Suzie Wolf Coffland Fund (MLH), Leiomyosarcoma SPORE Career Enhancement Program Award (JKD).
We gratefully acknowledge the members of the Molecular Diagnostics Service in the Department of Pathology and would like to acknowledge the Center Core grant (P30 CA008748) and the Marie-Josee and Henry R. Kravis Center for Molecular Oncology for use of MSK-IMPACT data. All authors report no conflict of interests related to this study.
REFERENCES
- 1.Coindre JM, Terrier P, Guillou L, Le Doussal V, Collin F, Ranchère D, et al. Predictive value of grade for metastasis development in the main histologic types of adult soft tissue sarcomas: a study of 1240 patients from the French Federation of Cancer Centers Sarcoma Group. Cancer 2001;91:1914–26. [DOI] [PubMed] [Google Scholar]
- 2.Tirumani SH, Deaver P, Shinagare AB, Tirumani H, Hornick JL, George S, Ramaiya NH. Metastatic pattern of uterine leiomyosarcoma: retrospective analysis of the predictors and outcome in 113 patients. J Gynecol Oncol 2014;25:306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kraft S, Fletcher CD. Atypical intradermal smooth muscle neoplasms: clinicopathologic analysis of 84 cases and a reappraisal of cutaneous “leiomyosarcoma”. Am J Surg Pathol 2011;35:599–607. [DOI] [PubMed] [Google Scholar]
- 4.Pisters PW, Leung DH, Woodruff J, Shi W, Brennan MF. Analysis of prognostic factors in 1,041 patients with localized soft tissue sarcomas of the extremities. J Clin Oncol 1996;14:1679–89. [DOI] [PubMed] [Google Scholar]
- 5.Farshid G, Pradhan M, Goldblum J, Weiss SW. Leiomyosarcoma of somatic soft tissues: a tumor of vascular origin with multivariate analysis of outcome in 42 cases. Am J Surg Pathol 2002;26:14–24. [DOI] [PubMed] [Google Scholar]
- 6.Miyajima K, Oda Y, Oshiro Y, Tamiya S, Kinukawa N, Masuda K, Tsuneyoshi M. Clinicopathological prognostic factors in soft tissue leiomyosarcoma: a multivariate analysis. Histopathology. 2002;40:353–9. [DOI] [PubMed] [Google Scholar]
- 7.Gladdy RA, Qin LX, Moraco N, Agaram NP, Brennan MF, Singer S. Predictors of survival and recurrence in primary leiomyosarcoma. Ann Surg Oncol 2013;20:1851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prayson RA, Goldblum JR, Hart WR. Epithelioid smooth-muscle tumors of the uterus: a clinicopathologic study of 18 patients. Am J Surg Pathol 1997;21:383–91. [DOI] [PubMed] [Google Scholar]
- 9.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spurr LF, Touat M, Taylor AM, Dubuc AM, Shih J, Meredith DM, et al. Quantification of aneuploidy in targeted sequencing data using ASCETS. Bioinformatics 2021;37:2461–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7:818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zauderer MG, Martin A, Egger J, Rizvi H, Offin M, Rimner A, et al. The use of a next-generation sequencing-derived machine-learning risk-prediction model (OncoCast-MPM) for malignant pleural mesothelioma: a retrospective study. Lancet Digit Health 2021;3:e565–e576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network. Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017;171:950–965.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gounder MM, Agaram NP, Trabucco SE, Robinson V, Ferraro RA, Millis SZ, et al. Clinical genomic profiling in the management of patients with soft tissue and bone sarcoma. Nat Commun 2022;13:3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428(6979):185–9. [DOI] [PubMed] [Google Scholar]
- 18.Pérot G, Derré J, Coindre JM, Tirode F, Lucchesi C, Mariani O, et al. Strong smooth muscle differentiation is dependent on myocardin gene amplification in most human retroperitoneal leiomyosarcomas. Cancer Res 2009;69:2269–78. [DOI] [PubMed] [Google Scholar]
- 19.Agaram NP, Zhang L, LeLoarer F, Silk T, Sung YS, Scott SN, et al. Targeted exome sequencing profiles genetic alterations in leiomyosarcoma. Genes Chromosomes Cancer 2016;55:124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson ND, Babichev Y, Fuligni F, Comitani F, Layeghifard M, Venier RE, et al. Lineage-defined leiomyosarcoma subtypes emerge years before diagnosis and determine patient survival. Nat Commun 2021;12:4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du KL, Ip HS, Li J, Chen M, Dandre F, Yu W, Lu MM, Owens GK, Parmacek MS. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol 2003;23:2425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, Wang DZ, Pipes GC, Olson EN. Myocardin is a master regulator of smooth muscle gene expression. Proc Natl Acad Sci U S A 2003;100:7129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev 2006;20:1545–56. [DOI] [PubMed] [Google Scholar]
- 24.Chudasama P, Mughal SS, Sanders MA, Hübschmann D, Chung I, Deeg KI, et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat Commun 2018;9:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nacev BA, Sanchez-Vega F, Smith SA, Antonescu CR, Rosenbaum E, Shi H, et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun 2022;13:3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mäkinen N, Aavikko M, Heikkinen T, Taipale M, Taipale J, Koivisto-Korander R, Bützow R, Vahteristo P. Exome Sequencing of Uterine Leiomyosarcomas Identifies Frequent Mutations in TP53, ATRX, and MED12. PLoS Genet. 2016;12:e1005850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahlén A, Debiec-Rychter M, Pedeutour F, Domanski HA, Höglund M, Bauer HC, et al. Clustering of deletions on chromosome 13 in benign and low-malignant lipomatous tumors. Int J Cancer 2003;103:616–23. [DOI] [PubMed] [Google Scholar]
- 28.Doyle B, Morton JP, Delaney DW, Ridgway RA, Wilkins JA, Sansom OJ. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J Pathol 2010;222:129–37. [DOI] [PubMed] [Google Scholar]
- 29.Dermawan JK, Kelly C, Gao Z, Smith S, Jadeja B, Singer S, et al. Novel Genomic Risk Stratification Model for Primary Gastrointestinal Stromal Tumors (GIST) in the Adjuvant Therapy Era. Clin Cancer Res 2023;29:3974–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bertsimas D, Margonis GA, Tang S, Koulouras A, Antonescu CR, et al. An interpretable AI model for recurrence prediction after surgery in gastrointestinal stromal tumour: an observational cohort study. EClinicalMedicine 2023;64:102200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gold JS, Gönen M, Gutiérrez A, Broto JM, García-del-Muro X, Smyrk TC, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localised primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol 2009;10:1045–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farshid G, Pradhan M, Goldblum J, Weiss SW. Leiomyosarcoma of somatic soft tissues: a tumor of vascular origin with multivariate analysis of outcome in 42 cases. Am J Surg Pathol 2002;26:14–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All clinical and genomic sequencing data generated by MSK-IMPACT described in this manuscript are publicly available at: https://www.cbioportal.org/study/summary?id=lms_msk_2024. The raw sequencing data are protected. Deidentified data are available under restricted access to protect patient privacy in accordance with federal and state law. These data can be requested for research use from the corresponding author via execution of a data transfer agreement with MSKCC, which will retain all title and rights to the data and results from their use.