

Abstract
Effective control of epidemics, individualized medicine, and new drugs with virologic response-dependent dose and timing require, among other things, simple, inexpensive, multiplexed molecular detection platforms suitable for point of care and home use. Herein, we describe our progress towards developing such a platform that includes sample lysis, nucleic acid isolation, concentration, purification, and amplification. Our diagnostic device comprises a sliding component that houses the nucleic acid isolation membrane and a housing containing three amplification reaction chambers with dry stored reagents, blisters with buffers and wash solutions, and absorption pads to facilitate capillarity pull and waste storage. After sample introduction, the user slides the slider within the housing from one station to another to carry out various unit operations. The slider motion induces blisters to discharge their contents, effectuating washes, and eventual elution of captured nucleic acids into reaction chambers. The slider cassette mates with a processor that incubates isothermal amplification but can also be made to operate instrumentation-free. We demonstrate our cassette’s utility for the co-detection of the human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV). These three blood-borne pathogens co-infect many people worldwide with severe personal and public health consequences.
Keywords: Molecular Diagnostics, Multiplexed, Sample to answer, Hepatitis B virus (HBV), Hepatitis C virus (HCV), Human immunodeficiency virus (HIV), Microfluidics, Loop-mediated isothermal amplification (LAMP)
1. Introduction
Viral infections, ranging from seasonal influenza and other respiratory illnesses such as COVID-19 and SARS, to mosquito-borne infections such as zika and dengue, to sexually-transmitted diseases such as HIV (human immunodeficiency virus) and HPV (human papilloma virus) burden global health [1–5]. Over 400 million people worldwide are infected with the three blood-borne viruses: hepatitis B (HBV), hepatitis C (HCV), and HIV [6, 7]. HBV causes liver cirrhosis, hepatitis, and hepatocellular carcinoma, infecting nearly 4 million people annually. Over 350 million people carry the HBV virus [8], of whom 90% are unaware of their predicament [4]. Mortality from viral hepatitis is increasing because of poor access to diagnostics and treatment [9]. The hepatitis virus HCV infects over 71 million people [5] and cause chronic liver diseases and cancer [10]. In 2019, AIDS related deaths numbered over 770,000 worldwide [6]. Only 60 % of HIV patients take antiviral drugs continuously [11] and over 20% of HIV infected individuals are unaware of their condition. It is estimated that 2.3 million of those infected with HIV are co-infected with HCV, while 2.6 million of HIV infected individuals are co-infected with HBV [12]. In view of the availability and affordability of treatments, there is a need to scale-up testing. Furthermore, there is a need to co-test for HIV, HBV and HCV due to their common path of transmission, the impact of co-infections on therapy, and to enable public health officials to enact informed polices and control measures [13].
Current diagnostics for HBV, HCV, and HIV are PCR-based and require stringent sample preparation, skilled personnel, cold chain, and comparatively expensive equipment such as precise (± 1 °C), rapid (~ 10 °C/s), and costly thermal cyclers [14]. Thus, PCR-based tests are inappropriate for the point of care and there is an insufficient PCR-based testing capacity in resource-limited settings.
The advent of isothermal enzymatic nucleic acid amplification assays provides opportunities for simple molecular tests [15]. These methods use proteins with strand displacement activity or helicase instead of temperature to denature dsDNA and, therefore, can be carried out at a constant temperature, eliminating the need for expensive temperature cycling equipment. Examples of isothermal amplification methods are: recombinase polymerase amplification (RPA) [16, 17], helicase-dependent amplification (HDA) [18], loop-mediated isothermal amplification (LAMP) [19–21], and rolling circle amplification (RCA) [11]. Furthermore, isothermal amplification assays are often more tolerant of inhibitors than PCR; can process small untreated volumes of blood [19, 20, 22], and produce abundance of amplicons, enabling colorimetric detection of amplification products.
A few molecular diagnostic systems for use at the point of care have been previously described, ranging from paper-based microfluidics [2] to test tubes [23, 24]. These devices require the user to carry out a few unit operations with auxiliary equipment outside the device and interact with the device during various processing steps, and may not be suitable for inexperienced users. Also, many of the devices reported in the literature are designed for single plex assays. Fully automated devices are bulky and unaffordable for resource poor settings and home use [25]. The development of a cost-effective, reliable, user-friendly, and multiplexed system for use at the point of need by minimally-trained individuals is still a challenge.
Herein, we address this challenge with a manually operated, slider cassette for multiplexed molecular detection of HBV, HCV, HIV at the point-of-need. All reagents (in dry form) and buffers are pre-stored in the cassette for refrigeration-free operation. Once the user introduces the raw sample into the cassette, the user pushes a sliding bar that contains a nucleic acid isolation membrane, comprised of chitosan coated glass fiber, through a sequence of unit operations. The slider motion actuates blisters that discharge, in sequence, wash and elution solutions. At the end of this process, the nucleic acids are subjected to LAMP amplification amenable to real time fluorescent and/or end point colorimetric detection. We demonstrate the entire molecular diagnostic process from sample to answer. Table S3 of the electronic supplement compares our slider cassette with other platforms described in the literature. Among devices in its class, ours has the advantages of more complete integration, including on-cassette storage of reagents and buffers and minimal need for user intervention.
2. Materials and methods
2.1. Cassette
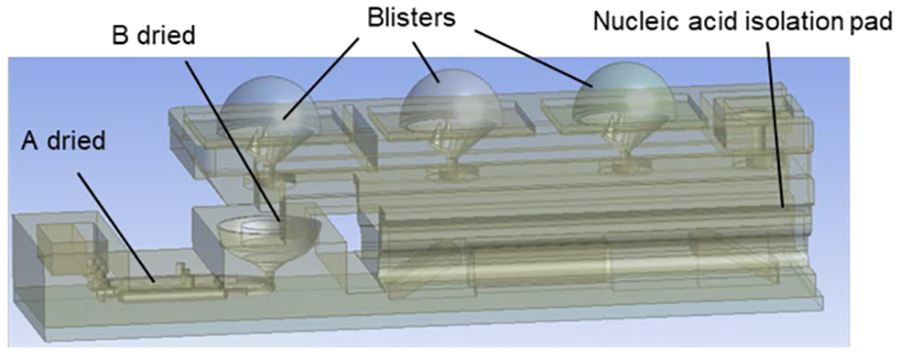
Our 3D-printed cassette (Figures 1 and S1) comprises a slider (130 mm (L) × 16 mm (W) × 4 mm (H)), a housing (90 mm (L) × 25 mm (W) × 20 mm (H)), and a blister-actuating sheath (128 mm (L) × 34 mm (W) × 16 mm (H)). The housing (Figure 1a) accommodates three reaction chambers (⑥), a blister pack (②), blister seats with lances to pierce through the blisters’ bottoms (Figure S1), and absorption pads mounted on an elastic support (⑤). The absorption pads imbibe wash solutions and store waste. Dried LAMP reaction mixes are pre-stored in the reaction chambers and liquids in the blisters.
Figure 1.
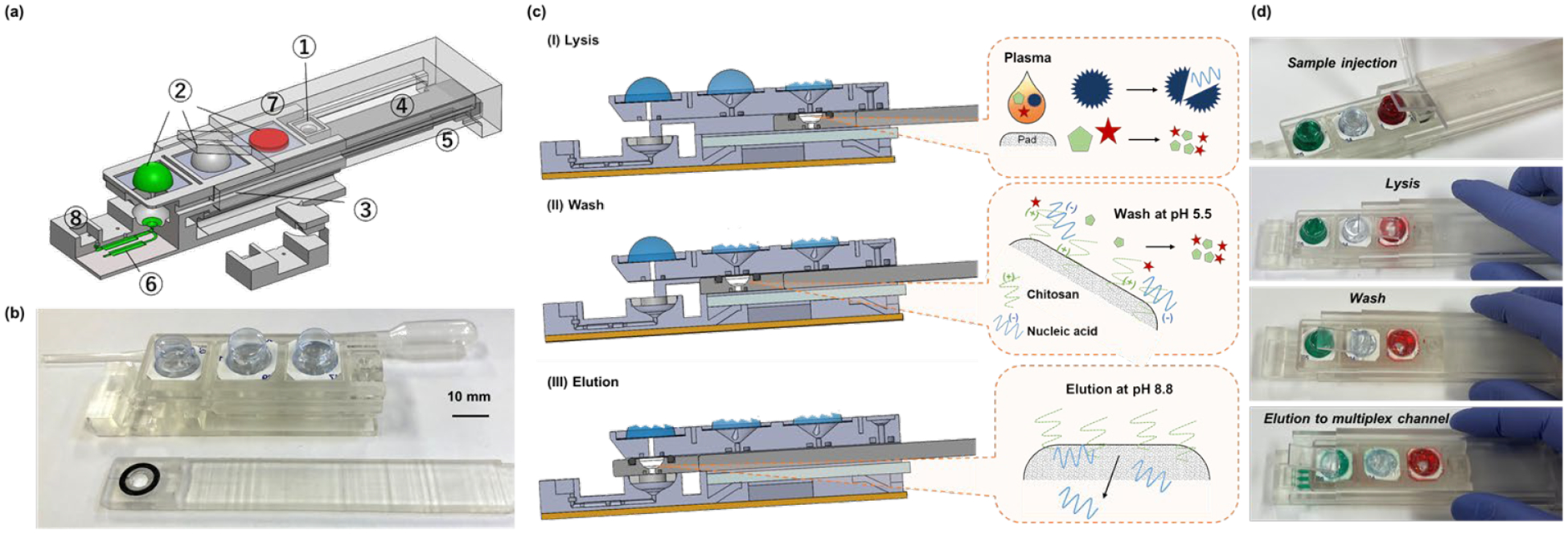
Our handheld slider cassette. (a) An exploded view of the cassette housing, slider, and sheath displaying embedded features. ① sample injection port; ② blisters storing lysis buffer, wash solution, and elution buffer; ③ chitosan-coated, nucleic acid isolation membrane located in the slider; ④ slider; ⑤ spring-supported absorption pad for capillary imbibition and waste storage; ⑥ three reaction chambers; ⑦ sheath with a wedge to compress blisters; and ⑧ phase change material for the sealing of the reaction chambers. (b) A photograph of the 3D-printed cassette-housing (top) and slider with nucleic acid isolation membrane (bottom). (c) Cross-section and (d) photographs of the cassette during (I) lysis; (II) wash; and (III) elution.
The slider (④) houses a fiber glass nucleic acid separation membrane treated with chitosan (③) (section 2.2). The sheath includes a blister actuation wedge that moves in tandem with the slider. As the slider travels in its housing, the sheath compresses one blister at a time to discharge the wash solutions contained in the blisters through the nucleic acid isolation membrane into the absorption pad; and elution buffer into the elution well (⑧) located upstream of the reaction chamber array. Alternatively, the blisters can be finger-actuated.
The cassette was designed with SolidWorks™ (Dassualt Systems). The SolidWorks file was then converted to a computer-aided design (CAD) software and exported as an STL file to the Form 3, low-force stereolithographic, desktop 3D printer (Formlabs, Sommerville, MA). We manufactured our cassettes with clear photopolymer resin (Formlabs™, FLGPCL04, specific mass 1.1 g cm−3 thermal conductivity of 0.11 W m−1 K−1) at a resolution of ± 0.05 mm. Following manufacturer’s recommendations, after printing and removing printing supports, we washed the various components with isopropyl alcohol for 15 minutes; and post cured them for 15-minute with 405-nm UV light at 60 °C in the Form Cure unit.
2.2. Nucleic acid binding membrane
The slider houses a 3-mm diameter nucleic acid capture membrane punched out from Whatman GF/F glass fiber filter paper (Milipore, Billerica, MA, USA). A 10 μL of chitosan (0.1% w/v) mixed with 10 mM MES buffer (2-(N-morpholino)ethanesulfonic acid, pH 5.5) was pipetted on the GF/F membrane and dried at 60 °C for 10 minutes.
2.3. Dry-stored LAMP reagents
Dried compounds A (Table 1) comprised of LAMP reagents, enzymes, and primers were pre-stored in the various reaction chambers, specializing each reaction chamber to a specific target. The nontarget-specific compound B (Table 1) needed to reconstitute the reaction mixes and the detection dye was stored in the common elution receiving well located at the head of the manifold that distributes the eluent to the various reaction chambers. In operation, the elution buffer (Tris-HCl) hydrates the dried reagents and reconstitutes the reaction mixes.
Table 1.
Chemical compositions of (a) solutions dried in our system and (b) liquid solutions in the blisters.
|
||||||
|---|---|---|---|---|---|---|
| (a) | Nucleic acid isolation pad | Dried compound A | Dried compound B | |||
| Chemical | Concentration | Chemical | Concentration | Chemical | Concentration | |
| MES (pH 5.5) | 10 mM | 0.4 μM F3/B3 | Tris-HCl (pH 8.8) | 30 mM | ||
| LAMP Primer mix | 3.2 μM FIP/BIP | |||||
| Chitosan | 0.1 % | 1.6 μM LF/LB | KCl | 30 mM | ||
| dNTPs | 2.8 mM | Betaine | 2.4 M | |||
| MgSO4 | 16 mM | Tween 20 | 0.3 % | |||
| (NH4)2SO4 | 20 mM | HNB | 360 μM | |||
| Bst polymerase | 0.64 U/uL | EvaGreen | 6X (7.5 μM) | |||
| (b) | Solution 1: Lysis buffer | Solution 2: Wash solution | Solution 3: Elution buffer | |||
| Chemical | Concentration | Chemical | Concentration | Chemical | Concentration | |
| Triton X-100 | 5% (v/v) | Molecular water | - | Tris-HCl (pH 8.8) | 10 mM | |
KCl, MgSO4, (NH4)2SO4, Betaine, Tween 20, Triton X-100, Chitosan was purchased from Sigma (St. Louis, MO, USA). Tris-HCl and MES buffer were purchased from Boston Bioproducts (Ashland, MA, USA). dNTPs were purchased from Denville Scientific (South Plainfield, NJ, USA). 20X Evagreen dye was purchased from Biotium Inc. (Hayward, CA, USA). Warmstart Bst 2.0 polymerase was purchased from New England Biolab (Ipswich, MA, USA).
To dry-store compound A, we introduced 20 μL of aqueous solution containing, among other things, bst polymerase and LAMP primer sets for either HIV, HBV, or HCV (Table S1) through 1-mm diameter injection ports into the respective reaction chambers. Next, we introduced compound B (30 μL) into the elution receiving well. The compositions of compounds A and B were previously optimized [2]. We used these compounds at higher concentrations than previously reported [2] to reduce the liquid volumes injected into the cassette prior to drying, accounting for the minute sizes of our reaction chambers. Compound A and B were then dried at room conditions for 5 hours. After drying, the injection ports were sealed with a PCR tape (Bio-Rad Microseal® B). The cassettes with dry-stored reagents were stored under room conditions and were viable for a few weeks (Table S4).
2.4. Blisters for liquids storage and delivery
Plastic blisters (one-piece book-fold style medication cards; Type MA-1090BA, cylindrical clear PVC, 12 mm diameter × 8 mm tall, Medi-Aid®, Drug Package, Inc, O’Fallon, MO) stored the lysis buffer (30 μl), wash solution (350 μl), and elution buffer (250 μl) (Figure S4). These liquid volumes exceed manufacturers’ recommendations by about 50% to compensate for dead volumes in our system. Once filled, the blisters were placed in a soft cork tray (Drug Package, Inc. MA-0631), covered with paper-backed foil card (included with the blister packs), and sealed with a hydraulic press (~104 Pa static pressure, Carver, Inc, Wabash, IN) with its platens heated to 140°C for 15 seconds. The individual blisters were then trimmed, leaving a 2 mm edge, and attached to the blister seats in the cassette housing with double-sided adhesive tape (3M Scotch tape).
In operation, the wedge mounted on the sheath compresses one blister at a time. When compressed, the blister’s bottom contacted the piercing lances that pierced the seal. The discharged buffer drains through the nucleic acid isolation membrane, and then imbibes into the absorption pad that doubles as waste storage.
2.5. Wax-based, Spontaneous Seals
During incubation, it is necessary to seal the reaction chambers to prevent liquid evaporation. Furthermore, it is desirable to maintain the reaction chambers permanently sealed after the amplification process to prevent amplicons from discharging to the ambient and contaminating the test site. To achieve inexpensive sealing without a need for active devices or manual operation, we use a phase change material. A 50 μl of molten docosane wax (Sigma-Aldrich, St. Louis, MO, USA) was deposited in the well located at the distal end of the LAMP reactor array and in the elution receiving well and allowed to solidify. During this process, the cassette was slightly tilted to prevent the wax from blocking the reaction chambers’ exits and the entrance into the inlet manifold. When the cassette was heated to the LAMP incubation temperature (65 °C), the docosane (melting temperature: 42 – 45 °C) melted and sealed the reaction chambers’ common inlet and outlets. We selected docasane for this purpose because of its compatibility with LAMP amplification and its melting temperature is above the typical room temperatures but below the incubation temperature of the LAMP process.
2.6. Processor
The cassette mates with a portable, custom-made processor that provides power, thermal control, and optical detection of fluorescence (Figure S5). Once the reactors have been filled with eluent from the membrane, the cassette’s part containing the reaction chambers is inserted in a Styrofoam insulated, light-tight box, contacting the reaction chambers with a 1-mm thick copper plate located atop a flexible polyimide heater (20 mm × 110 mm, Minco Minneapolis, MN, part HK6911). A type K thermocouple (Omega TT-K-30-SLE), bonded to the copper plate, provides input to the proportional-integral-derivative (PID) closed loop microcontroller (Raspberry Pi 4 mode B). A USB fluorescent camera (DinoLite AM4113T-GFBW Dino-Lite Premier, AnMo Electronics, Taipei, Taiwan) with blue LEDs (465 nm wavelength) and a CCD camera with a 510–545 nm wavelength band pass filter is mounted over the cassette reaction chambers to excite and monitor fluorescence emission during incubation.
2.7. Samples
HBV plasma from patients (No.9231137, 30,277,330 IU/mL) was purchased from Seracare life science (Milford, MA, USA). HCV genotype 1 plasma sample (CV900106678051314DD, 69,000,000 IU/mL) for clinical research was purchased from Discovery life science Inc. (Huntsville, AL, USA). HIV-1 control (964003, 3.1 × 107 IU/mL) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Samples containing multiple targets were formed by blending two or more of the above samples. Samples were serially diluted in PBS buffer (Thermo Fisher Scientific, pH 7.2) for sensitivity test, and pure PBS was used for a negative control.
2.8. Workflow
At the start of the test, the slider was positioned to center the nucleic acid isolation membrane under the sample loading port. The sample (5 – 100 μl) was then pipetted onto the membrane through the sample loading port. The entire sample was absorbed into or remained above the nucleic acid isolation membrane. The slider was then propelled to place the membrane at the lysis station. During this process, the sheath’s wedge compresses the first blister; and the blister’s seal (bottom) pushes against the lances. The lances pierced through the foil’s seal; and the lysis buffer discharged through the membrane and into the absorption pad. Similar process was repeated with the wash solution. Then, the sheath actuated the elution buffer blister. Th elution buffer filtered through the membrane, desorbing nucleic acids. The eluent discharged into the eluent well and continued through the manifold into the reaction chambers. As an alternative to the sheath, the blisters can be finger-actuated [26].
After the reaction chambers had been filled, the slider and sheath were removed, and the cassette was placed in the processor for incubation. After turning on the power, the USB camera located in the processor captured an image once a minute. These images were exported to a laptop computer and analyzed with a custom-written MATLAB® program. The imaging program averaged three time-wise readings of each pixel intensity to reduce noise. Then the signal was averaged over selected areas of each reaction chamber to produce intensity as a function of time plots for each reaction chamber. Zero time is selected at 5-minutes after the heater was turned on – reflecting the ramp up time needed for the reaction chambers to reach their desired incubation temperature.
3. Results and discussion
3.1. The handheld slider cassette and its operation
Our disposable, single-use cassette is comprised of a housing, slider, and sheath (Figures 1 and S1). The housing includes sample introduction port (①), liquid-filled blisters (②), spring-supported absorption pads (⑤), and three reaction chambers (⑥). The sheath is equipped with a flared flap (wedge) at its leading edge (Figure S2a). The sheath and the slider are linked. As the slider (④) travels, the sheath slides along the length of the housing in a grooved track and the flap compresses one blister at a time, in the desired succession. Alternatively, the blisters can be compressed by finger actuation [26].
A plasma sample, ranging in volume from 5 μL to 100 μL is introduced through the 1 mm diameter sample introduction port (①) located in the housing, saturating, and pooling above the nucleic acid isolation membrane (③) (Figure S2b). The nucleic acid isolation membrane is confined within a bio-compatible O-ring to prevent liquid leakage into the gap between the slider and its housing.
After sample introduction, the slider is translocated to bring the nucleic acid isolation membrane to the lysis station, wherein the blister laden with lysis buffer is compressed. As a result, the blister’s bottom is pierced (Figure S1), discharging low pH lysis solution through the nucleic acid isolation membrane and into the absorption pad (Figure 1c). The lysis buffer lyses viruses and bacteria and denatures proteins. At lysis buffer’s low pH (5.5), the chitosan-coated nucleic acid isolation membrane retains the negatively charged nucleic acids.
After lysis, the slider is translocated to its wash station, wherein molecular water is filtered through the membrane to remove contaminants, proteins, and residues of the lysis buffer, purifying the captured nucleic acids. Next, the slider is propelled to its elution station, wherein high pH (8.8) elution buffer filters through the nucleic acid isolation membrane, desorbing nucleic acids from the membrane [2, 27]. The eluent flows into the elution well, hydrates part B of the reaction mix, dispenses through the manifold into three reaction chambers, wherein the eluent hydrates and reconstitutes part A of the reaction mix. The eluent flow halts at the capillary valves located at the distal ends of the reaction chambers.
After the elution step, the cassette is inserted into our custom-made processor (Figure S5). The processor houses a heater, thermal controller, and a USB camera equipped with LED to excite fluorescence. During the temperature ramp up, docosane pre-stored in the elution well and in the exit well melts and seals the reaction chamber array’s inlet and exits to block evaporation. Once the test is completed and the cassette has cooled to the room temperature, the docosane solidifies, entombing the amplification products. During incubation, the USB camera collects fluorescent images - one per minute. These captured images are analyzed with a custom MATLAB® program to produce normalized fluorescence intensity plots for each chamber after censoring the first 5-minute data to accommodate the temperature ramp up time.
3.2. Manifold and reaction chamber array
Three conduits bifurcate from the elution well and connect to the three reaction chambers (Figure 2a). When the elution blister is actuated, the discharged buffer laden with desorbed nucleic acids hydrates component B (Table 1) that was pre-dried at the head of the manifold. The solution then aliquots into the three conduits and discharges into the three reaction chambers, where it hydrates component A that contains LAMP primers specific to each reaction chamber. The solution is halted at the distal ends of the reaction chambers with capillary valves.
Figure 2.
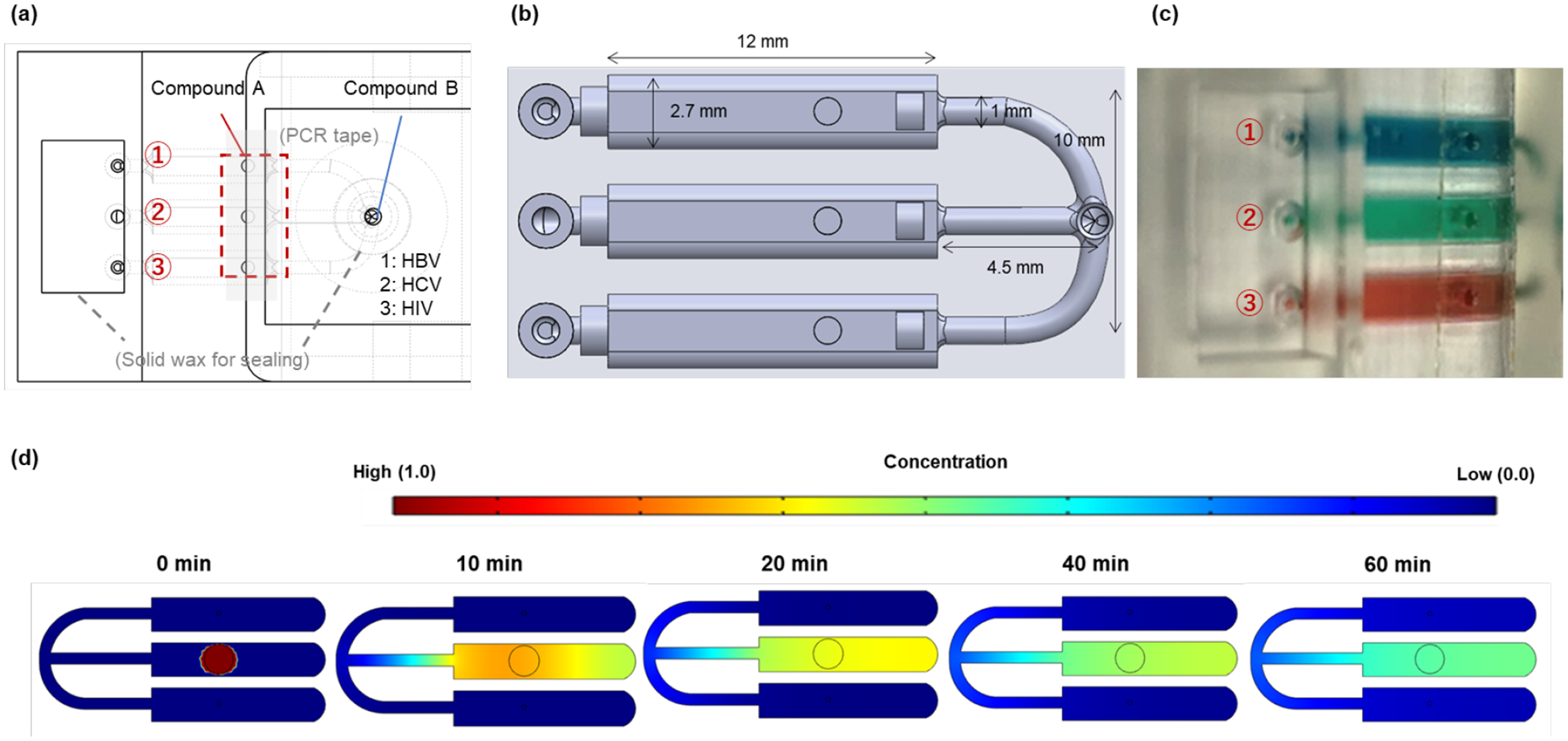
Reactor array and distribution manifold. (a) Top view of the reactor array, indicating the locations of the dried reagents. Enzymes and target-specific primers (Component A) are stored in the individual reaction chambers. Dried salts and detergent common to all reactions (Component B) are stored in the receiving well, upstream of the distribution manifold. (b) The reaction chamber and manifold. (c) To test for possible cross-talk among the reaction chambers, red, green, and blue food coloring dyes were dried in the individual reaction chambers; hydrated by the addition of water through the head of the manifold. (d) Two-dimensional finite element (COMSOL) simulation tracking the diffusion of the red blob (initial, dimensionless concentration one) positioned in the central reactor as a function of time.
Migration of primers among the reaction chambers is of concern as it may result in false positives. Such migration may take place either during the drying process of specific compounds A in the individual reaction chambers and/or during incubation. The manifold legs’ lengths and cross-sections were designed to minimize such eventualities (Figure 2a, 2b). Migration of primers from one reaction chamber to another during the drying of compounds A was prevented by maintaining an air gap among the various reaction chambers during the drying process. To examine the diffusion of species among the reaction chambers, we dried blue, green, and food colorings in the various reaction chambers, hydrated the chambers through the elution well and monitored the spreads of the dyes as functions of time while the cassette was incubated at 65°C. No significant dye blending was observed within 60 min of incubation (Figure 2c). Furthermore, we carried out two-dimensional, finite element simulations of the diffusion of a blob of dye from the central reaction chamber to the other two reactors with similar results (Figure 2d). The proof is, of course, in the testing. None of our experiments with various targets compositions exhibited false positives.
3.3. Cassette Heating
The cassette was heated with a custom thermal controller (Figure S5). The cassette’s reaction chamber array contacted copper plate that, in turn, was in contact with a resistance heater controlled in a feedback mode. The input temperature was measured at the interface between the copper plate and the cassette. The controller adjusted the power input in proportion to the deviation of the measured temperature from the set temperature.
Since the polymerase enzyme (Bst) is temperature-sensitive; too high a temperature denatures the enzyme, too low a temperature yields suboptimal performance. Hence, it is necessary to operate the cassette in the temperature range between 60°C and 67°C. To examine various cassette designs and operating conditions, we carried out finite element numerical simulations (Ansys Fluent; Figure S3). Our model accounts for the presence of aqueous solutions in the reaction chambers and convective boundary conditions at the cassette’s surface. Our theoretical predictions agreed with temperature measurements carried out with a thermocouple inserted in one of the reaction chambers. The ramp up time lasted less than 5 minutes (Figure S3c).
3.4. Wash solutions and Nucleic acid extraction
To assess our cassette’s performance, we used the LAMP threshold time as a figure of merit. For a given primer design, the LAMP threshold time depends on the quantity of eluted template available for amplification, presence of inhibitors, and the incubation temperature. In our experiments reported herein, unless stated otherwise, sample introduction, lysis, nucleic acid isolation, and elution were carried out in our cassette. To control the volumes of the lysis and wash solutions, we pipetted these solutions directly onto the nucleic acid isolation membrane and eluted into a test tube. We then amplified the eluent with a benchtop thermal cycler operating at the LAMP incubation temperature.
We placed 10 μL of plasma laden with HBV (3.0 × 107 IU/mL) on the nucleic acid isolation membrane, applied various volumes of lysis solution, washed the membrane with molecular water, and eluted the captured nucleic acids into a vial. As the lysis buffer volume increased, the threshold time decreased, achieving a minimum at ~20 μL, and then increased slightly as the volume of the lysis buffer further increased (Figure S4b). The assay depended only weakly on the lysis buffer volume in the range from 20 μL to 200 μL.
We evaluated various wash solutions (200μL): molecular water (DI water), 10 mM MES buffer (2-(N-morpholino)ethanesulfonic acid, pH 5.5), 15% IPOH (isopropyl alcohol), and 80% ethanol. Molecular water provided the best results among the tested solutions (Figure S4c). As the molecular water volume increased, the threshold time decreased, attained a minimum at 200 μl and then increased slightly again (Figure S4d) with little sensitivity to the water volume in the range from 150 to 250 μL. In all subsequent experiments, we used 20 μL lysis buffer and 200 μL of molecular water as the wash solution (Table 1b).
To assess the various sample preparation steps, we processed 10 μL of plasma laden with HBV (30,277,330 IU/mL) omitting one or more of the sample-preparation steps (Figure 3b). In the absence of lysis (1) and in the presence (2) of 20 μL lysis but without wash, the eluant from the membrane failed to amplify and LAMP did not produce any detectable signal, likely due to the inhibitory effect of the lysis buffer.
Figure 3.
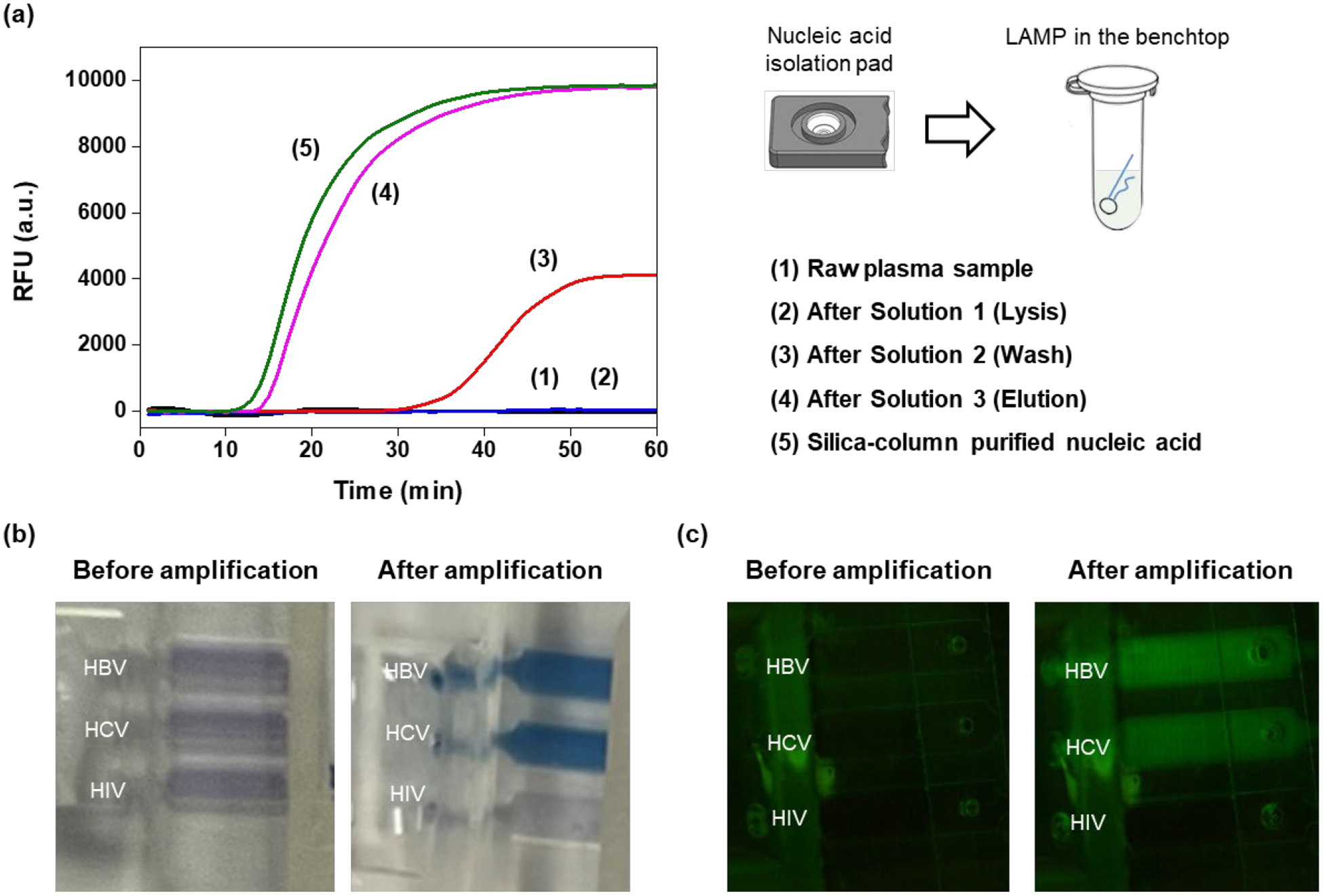
Assessment of individual sample preparation steps. (a) 10 μL of plasma containing HBV were placed on the nucleic acid isolation membrane. LAMP amplification curves of eluant from the nucleic acid isolation membrane in the absence (1) and the presence (2) of lysis. (3) Amplification curve from wash solution filtered through the nucleic acid isolation membrane. (4) Amplification curve of a sample fully processed with our cassette. (5) Amplification curve of the same sample as in (4) processed with standard laboratory procedures and benchtop equipment. (b) End point colorimetric detection of amplicons with HNB dye of a sample containing HBV and HCV. (c) Fluorescence emission from intercalating dye (EvaGreen) before (left) and after (right) amplification.
Then, following lysis, we washed the membrane with 200 μL water and subjected the filtrate to LAMP. LAMP produced a weak, delayed signal (3), indicating that only a small fraction of the membrane-adsorbed nucleic acids desorbed in water. Amplification curve (3) should be contrasted with the sample that has undergone proper processing steps comprised of lysis, wash, and elution (4). In case (4), the entire process was carried out with our cassette. The results obtained with our cassette are comparable with the LAMP amplification carried out by standard laboratory procedures with benchtop equipment (5). Based on the threshold times of amplification curves (4) and (5) (Fig. 3) and the calibration curve (Fig. S6), we estimate that the recovery rate of our isolation membrane is about 70% of the recovery rate of the standard spin column. Generally, chitosan-based nucleic acid recovery rate surpasses that of comparable silica columns under similar conditions [28, 29]. In our hands, our chitosan membrane’s performance is slightly less than that of the standard spin column, perhaps because we do not have the benefits of centrifugation.
To further test our cassette’s performance, we pre-stored component B with primers for HBV, HCV, and HIV in the three reaction chambers and processed a sample comprised of a mixture of 10 μL HBV (3.0 × 105 IU) and 10 μL of HCV (6.9 × 105 IU) in plasma with our cassette. The HBV and HCV amplicons were detected both at the end of the process with the colorimetric dye HNB that changes color from violet (in the absence of dsDNA) to blue (in the presence of dsDNA) (Figure 3b) and in real time with the EvaGreen intercalating dye that emits fluorescence when excited with blue light from a LED (Figure 3c). The HIV chamber remained dark attesting to the absence of undesired primer diffusion among the reaction chambers.
3.5. Integrated molecular diagnosis from sample to answer.
We tested plasma samples containing HIV, HCV, and /or HBV with our cassette (Figure 4). When only one of the targets were present in the sample, only the reaction chamber specific to that target lighted up (Figure 4a), In the case of a co-infection with two or more pathogens, e.g., HBV and HCV, the reaction chambers specific to the pathogens present in the sample lighted up. The reaction chamber(s) specific to any absent pathogen(s) remained dark. When all three pathogens were present, all three chambers lighted up. Within 60 min incubation time, there was no cross-talk among the reaction chambers and there were no false positives.
Figure 4.
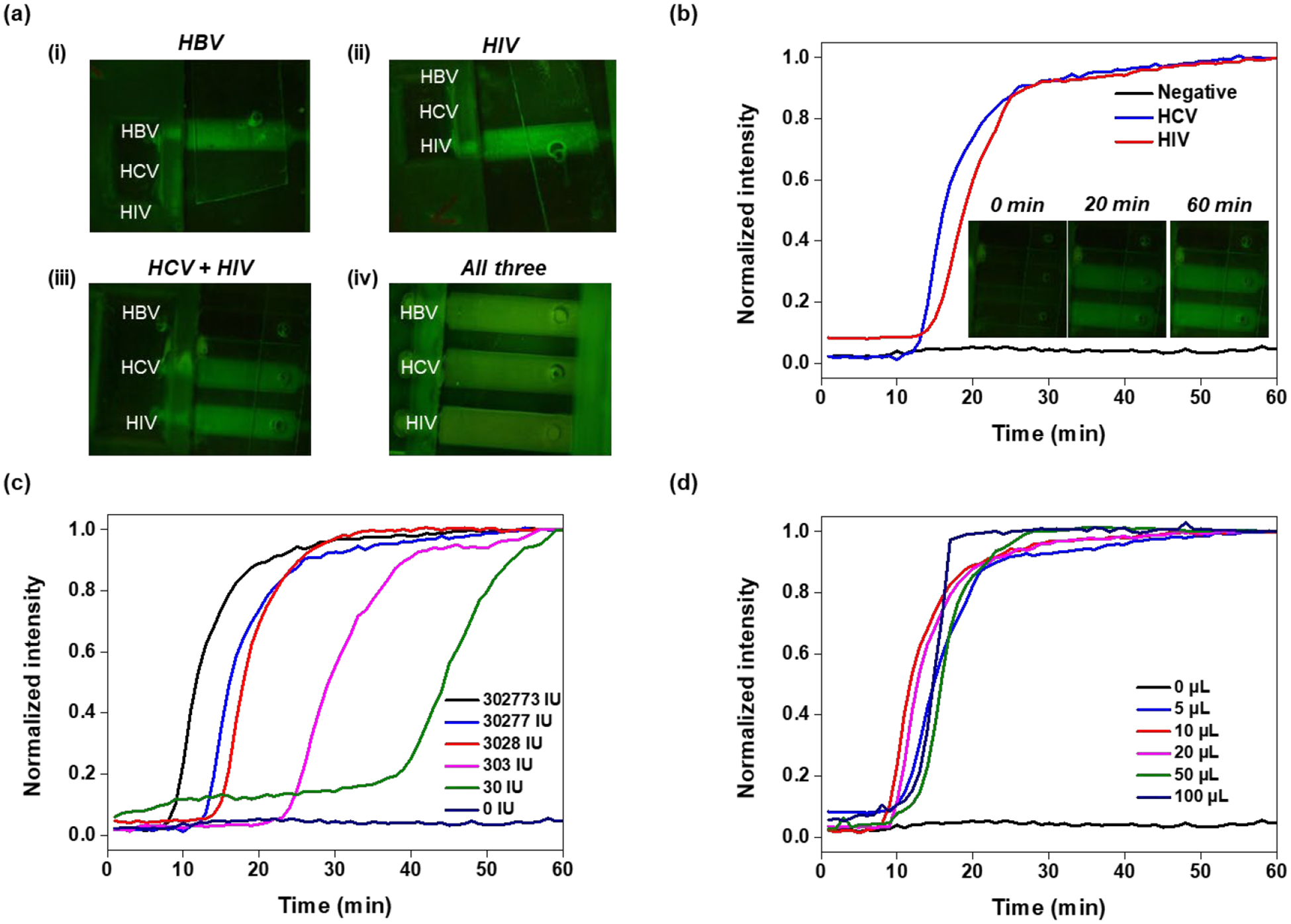
Co-testing for HBV, HCV, and HIV in a single human plasma sample with our handheld slider cassette. (a) Fluorescence emission from the reaction chambers when the sample contains (i) only HBV; (ii) only HCV; (iii) Both HCV and HIV; and (iv) All three targets: The sample volume is 10 μL for each. When present, the amounts of HBV, HCV, and HIV are, respectively, 3.0 × 105 IU, 6.9 × 105 IU, and 3.1 × 105 IU. (b) Real-time amplification curves detected with our custom processor from samples containing both HCV and HIV in human plasma and from non-template control. (c) Amplification curves from samples containing HBV at various concentrations as indicated. (d) Amplification curves obtained with HBV plasma volumes, ranging from 5 to 100 μL with concentration of 30,277,330 IU/mL.
Our custom-made processor detected real time amplification curves with the USB camera (Figure 4b) as exemplified with a 20 μL plasma sample laden with HCV (6.9 × 107 IU/mL) and HIV (3.1 × 107 IU/mL). The emission intensities were normalized with the maximum intensities in each case and depicted as functions of time. The inset includes images of the reaction chamber array at various times during the incubation.
Plasma samples laden with HBV at concentrations ranging from 30 IU (international unit) to 3 × 105 IU were tested successfully with our cassette (Figure 4c and Fig. S6) with good reproducibility (Table S5). Our experiments suggest that our system’s sensitivity is about 30 IU of HBV (168 copies of HBV DNA) per reaction volume. This corresponds to 17 copies per 1ml of sample, which betters previous reports [30]. Many rapid molecular tests do not include nucleic acid concentration and are limited to very small sample volumes. In contrast, our cassette can accommodate sample volumes ranging from 5 μL to 100 μL, enabling increased sensitivity by increasing sample volume.
4. Conclusions
We describe a handheld, manually-operated slider cassette for multiplex molecular co-detection of co-endemic pathogens such as HBV, HCV, and HIV in human plasma. Our slider cassette combines several novel features such as the integration of sample processing; nucleic acid concentration, amplification, and detection; refrigeration-free storage of reagents; ability to co-detect multiple pathogens, and minimal requirements from the user. Features that are not typically available in inexpensive, simple devices for point of care and home use (Table S3). Our cassette enables multiple unit operations, ranging from lysis to nucleic acid isolation, concentration, purification, elution, and amplification in a single device. Our device can be operated manually by pushing the slider from one station to another, reducing device cost. Alternatively, the cassette operation can be automated with the use of a stepper motor.
The reaction mixes are pre-stored in the cassette in dry form to reduce the number of necessary unit operations and to accommodate long, refrigeration-free shelf-life. The liquids such as lysis buffer, wash solution, and elution buffer are stored in blisters. The unit that contains the reactor array and stores the dry reagents is combined with the rest of the cassette body that contains the blisters via an elution well. The slider motion actuates the liquid-storing blisters, causing them to release and dispense the lysis buffer, wash solution, and elution buffer without a need for a separate actuation mechanism for each blister that would have required a few electrical motors and control circuity with added cost and complexity.
We use a chitosan-coated glass fiber membrane for solid-phase nucleic acid extraction and concentration– both DNA and RNA. The chitosan reverses its electrostatic charge upon changes in solution pH, enabling nucleic acid adsorption during lysis with low pH solution and nucleic acid desorption during elution with high pH solution. The use of a chitosan-coated glass fiber eliminates the need for alcohol-based wash solutions that inhibit polymerase and are difficult to remove in a microfluidic setting.
Our cassette relies on a manifold that utilizes capillary valves to aliquot the eluted, purified templates to multiple reaction chambers. Here, we employed cassettes with three reaction chambers. More are, however, possible. Multiplexing is achieved by spatial distribution of templates into individual reaction chambers, each specialized for a specific target by storing primers for that target. The dry reagents are hydrated and reconstituted when the elution buffer flows into the reaction chambers.
Our reaction array is self-sealing. We deposited solid wax (docosane) in the elution well upstream of the reaction array as well as downstream of the reactors. During sample preparation and reaction chambers filling, the wax allows free flow of liquids. Once the reaction chambers are heated to their LAMP incubation temperature, the wax melts, spreads over the aqueous solution and provides an evaporation barrier, preventing liquid loss during incubation. After the conclusion of the test, when the cassette cools to room temperature, the wax re-solidifies entombing the amplicon-rich reaction chambers and preventing potential contamination of the work specie.
We demonstrated the feasibility of both end point amplicon detection with colorimetric dye and real-time fluorescence detection of emission from intercalating dye. The colorimetric detection has the advantage that it can be done by eye without a need for a camera. The fluorescence detection that can be carried out with a USB camera (as done here) or with smartphone [31] enables us to monitor the amplification process in real time like in qPCR and use the threshold time to estimate template concentration. Furthermore, real time detection enables us to shorten the duration of the test when templates are abundant and the signal appears early. We have not experienced any false positives in our experiments. When needed, we can enhance our assay’s specificity by replacing the generic intercalating dye that we have used here with template-specific molecular beacons. Furthermore, the colorimetric dye can be used in tandem with the fluorescent dye, enabling both real-time and end point detection in one assay.
Our cassette is versatile. We demonstrated our system’s ability to process plasma samples ranging in volume from 5 to 100 μL and ranging in the number of templates from 30 to 300,000 IU HBV. Even larger sample volumes are possible. The ability to operate with relatively large sample volumes allows our device to achieve better sensitivity than possible with rapid molecular tests where unprocessed or minimally processed sample is added directly into the rection volume, which greatly limits sample volume.
Our cassette meets the need for the detection of co-endemic diseases that share similar symptoms but require diverse disease management strategies. Since our cassette’s specificity is dictated by the reagents pre-stored in the detachable reactor array, our cassette body can be matched with various reactor arrays to repurpose our system.
Our cassette is simple and inexpensive to manufacture and to operate. It can be either 3D-printed on-demand as we have done here or fabricated in large quantities by injection molding. Hence, it has the potential to meet the testing needs in resource-limited settings such as developing countries and rural areas. The cassette operation can be further simplified by replacing the electrical heating with chemical heating methods for electricity-free operation as we have previously demonstrated [32]. Chemical heating and colorimetric detection would enable instrument-free operation. Furthermore, a smartphone camera with either LED for fluorescence or filtered flash-based excitation [33] would allow real time detection and quasi-quantification.
In this work, we addressed human diseases, but the need for molecular testing outside laboratory settings is much broader and includes, among other things, food safety; plant, animal, and soil pathogens; water supply integrity; and environmental surveillance for non-native species (environmental nucleic acids).
Supplementary Material
Acknowledgement
This work has been supported, in part, by NIH grant R61 AI140484-01 to the University of Pennsylvania.
References
- [1].Rodriguez-Manzano J, Malpartida-Cardenas K, Moser N, Pennisi I, Cavuto M, Miglietta L, et al. , Handheld Point-of-Care System for Rapid Detection of SARS-CoV-2 Extracted RNA in under 20 min, ACS Cent Sci, 7(2021) 307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Seok Y, Batule BS, Kim M-G, Lab-on-paper for all-in-one molecular diagnostics (LAMDA) of zika, dengue, and chikungunya virus from human serum, Biosensors and Bioelectronics, 165(2020) 112400. [DOI] [PubMed] [Google Scholar]
- [3].Kellner MJ, Koob JG, Gootenberg JS, Abudayyeh OO, Zhang F, SHERLOCK: nucleic acid detection with CRISPR nucleases, Nat Protoc, 14(2019) 2986–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Razavi-Shearer D, Gamkrelidze I, Nguyen MH, Chen D-S, Van Damme P, Abbas Z, et al. , Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study, The lancet Gastroenterology & hepatology, 3(2018) 383–403. [DOI] [PubMed] [Google Scholar]
- [5].Blach S, Zeuzem S, Manns M, Altraif I, Duberg A-S, Muljono DH, et al. , Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study, The lancet Gastroenterology & hepatology, 2(2017) 161–76. [DOI] [PubMed] [Google Scholar]
- [6].Pai NP, Karellis A, Kim J, Peter T, Modern diagnostic technologies for HIV, The Lancet HIV, 7(2020) e574–e81. [DOI] [PubMed] [Google Scholar]
- [7].Revill PA, Tu T, Netter HJ, Yuen LKW, Locarnini SA, Littlejohn M, The evolution and clinical impact of hepatitis B virus genome diversity, Nat Rev Gastroenterol Hepatol, 17(2020) 618–34. [DOI] [PubMed] [Google Scholar]
- [8].Shen XX, Qiu FZ, Shen LP, Yan TF, Zhao MC, Qi JJ, et al. , A rapid and sensitive recombinase aided amplification assay to detect hepatitis B virus without DNA extraction, BMC Infect Dis, 19(2019) 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yamamoto C, Nagashima S, Isomura M, Ko K, Chuon C, Akita T, et al. , Evaluation of the efficiency of dried blood spot-based measurement of hepatitis B and hepatitis C virus seromarkers, Sci Rep, 10(2020) 3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Maasoumy B, Wedemeyer H, Natural history of acute and chronic hepatitis C, Best practice & research Clinical gastroenterology, 26(2012) 401–12. [DOI] [PubMed] [Google Scholar]
- [11].Soares RRG, Varela JC, Neogi U, Ciftci S, Ashokkumar M, Pinto IF, et al. , Sub-attomole detection of HIV-1 using padlock probes and rolling circle amplification combined with microfluidic affinity chromatography, Biosens Bioelectron, 166(2020) 112442. [DOI] [PubMed] [Google Scholar]
- [12].Saud LR, Chagas AL, Maccali C, Pinto PV, Horvat N, Alencar RS, et al. , Hepatocellular carcinoma in patients coinfected with hepatitis B or C and HIV: more aggressive tumor behavior?, European Journal of Gastroenterology & Hepatology, 33(2021) 583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Broughton JP, Deng X, Yu G, Fasching CL, Servellita V, Singh J, et al. , CRISPR-Cas12-based detection of SARS-CoV-2, Nat Biotechnol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sivakumar R, Dinh VP, Lee NY, Ultraviolet-induced in situ gold nanoparticles for point-of-care testing of infectious diseases in loop-mediated isothermal amplification, Lab Chip, 21(2021) 700–9. [DOI] [PubMed] [Google Scholar]
- [15].Zhao Y, Chen F, Li Q, Wang L, Fan C, Isothermal Amplification of Nucleic Acids, Chem Rev, 115(2015) 12491–545. [DOI] [PubMed] [Google Scholar]
- [16].Yi TT, Zhang HY, Liang H, Gong GZ, Cai Y, Betaine-assisted recombinase polymerase assay for rapid hepatitis B virus detection, Biotechnol Appl Biochem, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ahn H, Batule BS, Seok Y, Kim MG, Single-Step Recombinase Polymerase Amplification Assay Based on a Paper Chip for Simultaneous Detection of Multiple Foodborne Pathogens, Anal Chem, 90(2018) 10211–6. [DOI] [PubMed] [Google Scholar]
- [18].Tang R, Yang H, Gong Y, You M, Liu Z, Choi JR, et al. , A fully disposable and integrated paper-based device for nucleic acid extraction, amplification and detection, Lab Chip, 17(2017) 1270–9. [DOI] [PubMed] [Google Scholar]
- [19].Hongjaisee S, Doungjinda N, Khamduang W, Carraway TS, Wipasa J, Debes JD, et al. , Rapid visual detection of hepatitis C virus using a reverse transcription loop-mediated isothermal amplification assay, Int J Infect Dis, 102(2021) 440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Batule BS, Seok Y, Kim MG, Paper-based nucleic acid testing system for simple and early diagnosis of mosquito-borne RNA viruses from human serum, Biosens Bioelectron, 151(2020) 111998. [DOI] [PubMed] [Google Scholar]
- [21].Kong H, Zhang W, Yao J, Li C, Lu R, Guo Z, et al. , A RT-LAMP based hydrogen ion selective electrode sensing for effective detection HIV-1 RNA with high-sensitivity, Sensors and Actuators B: Chemical, 329(2021). [Google Scholar]
- [22].Sun W, Du Y, Li X, Du B, Rapid and Sensitive Detection of Hepatitis C Virus in Clinical Blood Samples Using Reverse Transcriptase Polymerase Spiral Reaction, J Microbiol Biotechnol, 30(2020) 459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xu Z, Yin K, Ding X, Li Z, Sun X, Li B, et al. , An integrated E-Tube cap for sample preparation, isothermal amplification and label-free electrochemical detection of DNA, Biosens Bioelectron, 186(2021) 113306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jiang X, Loeb JC, Manzanas C, Lednicky JA, Fan ZH, Valve-Enabled Sample Preparation and RNA Amplification in a Coffee Mug for Zika Virus Detection, Angew Chem Int Ed Engl, 57(2018) 17211–4. [DOI] [PubMed] [Google Scholar]
- [25].Park BH, Oh SJ, Jung JH, Choi G, Seo JH, Kim DH, et al. , An integrated rotary microfluidic system with DNA extraction, loop-mediated isothermal amplification, and lateral flow strip based detection for point-of-care pathogen diagnostics, Biosens Bioelectron, 91(2017) 334–40. [DOI] [PubMed] [Google Scholar]
- [26].Qiu X, Thompson JA, Chen Z, Liu C, Chen D, Ramprasad S, et al. , Finger-actuated, self-contained immunoassay cassettes, Biomed Microdevices, 11(2009) 1175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Byrnes SA, Bishop JD, Lafleur L, Buser JR, Lutz B, Yager P, One-step purification and concentration of DNA in porous membranes for point-of-care applications, Lab Chip, 15(2015) 2647–59. [DOI] [PubMed] [Google Scholar]
- [28].Cao W, Easley CJ, Ferrance JP, Landers JP, Chitosan as a Polymer for pH-Induced DNA Capture in a Totally Aqueous System, Analytical Chemistry, 78(2006) 7222–8. [DOI] [PubMed] [Google Scholar]
- [29].Hagan KA, Meier WL, Ferrance JP, Landers JP, Chitosan-Coated Silica as a Solid Phase for RNA Purification in a Microfluidic Device, Analytical Chemistry, 81(2009) 5249–56. [DOI] [PubMed] [Google Scholar]
- [30].Seok Y, Yin Q, Bai H, Bau HH, Sensitive, Single-Pot, Two-Stage Assay for Hepatitis Viruses, Analytical Chemistry, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nguyen HQ, Nguyen VD, Van Nguyen H, Seo TS, Quantification of colorimetric isothermal amplification on the smartphone and its open-source app for point-of-care pathogen detection, Sci Rep, 10(2020) 15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li RJ, Mauk MG, Seok Y, Bau HH, Electricity-free chemical heater for isothermal nucleic acid amplification with applications in COVID-19 home testing, Analyst, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Joung HA, Ballard ZS, Ma A, Tseng DK, Teshome H, Burakowski S, et al. , Paper-based multiplexed vertical flow assay for point-of-care testing, Lab Chip, 19(2019) 1027–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.