

SUMMARY
The severity of T cell-mediated gastrointestinal (GI) diseases such as graft-versus-host disease (GVHD) and inflammatory bowel diseases correlate with a decrease in the diversity of the host gut microbiome composition characterized by loss of obligate anaerobic commensals. The mechanisms underpinning these changes in the microbial structure remain unknown. Here we show in multiple specific pathogen free (SPF), gnotobiotic and germ-free murine models of GI GVHD that the initiation of the intestinal damage by the pathogenic T cells altered ambient oxygen levels in the GI tract and caused dysbiosis. The change in oxygen levels contributed to the severity of intestinal pathology in a host intestinal HIF-1α– and a microbiome-dependent manner. Regulation of intestinal ambient oxygen levels with oral iron chelation mitigated dysbiosis and reduced the severity of the GI GVHD. Thus targeting ambient intestinal oxygen levels may represent a novel, non-immunosuppressive strategy to mitigate T-cell driven intestinal diseases.
One Sentence Summary:
Oxygen levels control microbiota changes and GVHD severity after BMT.
eTOC Blurb
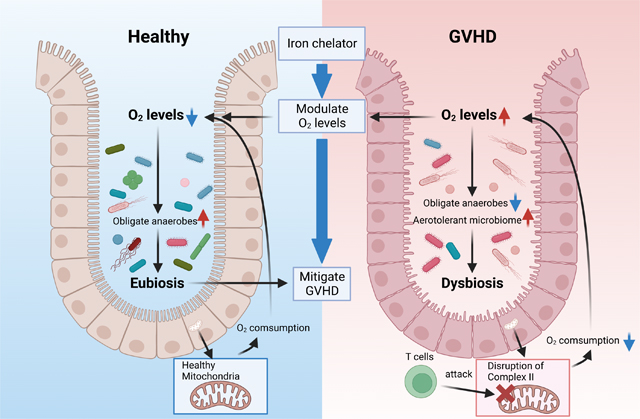
Whether dysbiosis is a cause or consequence of pathology in intestinal diseases is not understood. Here, Seike et al. show that defective oxygen utilization by intestinal epithelial cells lead to a loss of intestinal hypoxia and concomitant dysbiosis. Oxygen modulation rescues intestinal hypoxia and alleviates gastro-intestinal graft-versus-host disease.
Graphical abstract
INTRODUCTION
The composition of host intestinal microbiome directly contributes to human health and diseases. Correspondingly, a healthy gut is inhabited by a diverse community of mostly obligate anaerobic bacteria (eubiosis) that is influenced by the host genetics, diet and immunity 1 2. The breakdown of this balance, called dysbiosis and often characterized by a shift in the microbial community structure from obligate to facultative anaerobes and aerotolerant microbes, is associated with several diseases, including immune mediated intestinal diseases such as graft-versus-host disease (GVHD)3 4 5 6. Whilst a strong correlation exists between disease severity and dysbiosis, the mechanisms that lead to dysbiosis are unknown. Furthermore, whether dysbiosis is a cause, an amplifier, a regulator or a mere consequence of the disease process remains poorly understood.
Gastrointestinal (GI) GVHD is a potentially fatal complication of allogeneic hematopoietic stem cell transplantation (allo-HSCT)7. Reduction in intestinal microbial diversity with loss of obligate anaerobes relative to other bacteria is associated with increased GVHD mortality 3 8 9 . However, seminal experimental studies performed before the advent of modern germ-free facilities and sequencing technologies demonstrated that absence of microbiome mitigated GVHD severity 10. Thus, while microbial dysbiosis prognosticates outcomes after allo-HSCT, the role of microbiome in GVHD remains unclear. Specifically, (a) whether dysbiosis is caused by or is a consequence of severe GVHD, (b) the mechanisms that underpin the development of dysbiosis, and (c) whether dysbiosis before or after HSCT directly amplifies or negatively regulates GVHD severity remain unknown.
The mammalian GI tract is hypoxic with an oxygen (O2) gradient that traverses the surface of the colonic mucosa to the center of gut lumen. This hypoxic gradient shapes the intestinal microbial community structure, promoting colonization with predominantly obligate anaerobes at homeostasis 11 12. The host intestinal epithelial cells (IECs) are thus uniquely adapted to the hypoxic environment, i.e., ‘‘physiologic hypoxia’’. The IECs depend on microbial metabolites, specifically short-chain fatty acids (SCFAs), as their primary source of energy for oxidative phosphorylation (OXPHOS) despite low oxygen 13 14. Thus, microbial-derived SCFAs regulate the barrier function at homoeostasis 15 and in GI GVHD 16 17. Moreover, epithelial metabolism and the cellular O2 sensor, hypoxia-inducible factor1α (HIF), are key determinants of intestinal function under conditions of ‘‘physiologic hypoxia’’1 18. In the context of enteric infections, intestinal ambient oxygen level is a critical ecological driver of dysbiosis 1 2 19 20. In the context of non-infectious immune mediated GI damage, pathogenic T cells target OXPHOS in the IEC leading to deficient O2 utilization 21. However, whether the change in the luminal oxygen due to poor consumption of O2 from the metabolic defect impacts dysbiosis remains unknown.
Herein we showed that dysbiosis was a consequence of the loss of ‘physiologic hypoxia’ caused by poor oxygen utilization by the IECs 21. We also showed that the microbiome by itself is a critical negative regulator of GI GVHD. In contrast to the current paradigm, germ-free animals demonstrated greater GVHD while promotion of eubiosis after transplant mitigated the severity of GVHD. However, dysbiosis before HSCT did not impact GVHD severity. Rescue of physiological hypoxia by iron chelation mitigated the severity of GI GVHD. Thus, our data provide a mechanism for induction of dysbiosis and demonstrate that promotion of eubiosis by regulating ambient oxygen reduces the severity of intestinal damage after allo-HCT.
RESULTS
Allogeneic dysbiosis is not pathogenic to naïve animals
We first determined whether dysbiosis associated with GVHD following allogeneic HSCT is toxic in the absence of HSCT. Naïve C57BL/6 (B6) animals were irradiated with 10 Gy and transplanted with splenic T cells and T cell-depleted bone marrow (TCD BM) from allogeneic BALB/c donors (Allo B6) to induce GVHD and dysbiosis (Fig. 1A). Dysbiosis in stool from Allo B6 animals was confirmed at 2 weeks (Fig. 1B to D, Fig.S1A and B). Allo B6 stools contained more Proteobacteria, particularly facultative anaerobes in the Enterobacteriaceae family such as Escherichia (Fig. 1A and B). Allo B6 animals were cohoused with a cohort of naïve, un-transplanted B6 hosts for 6 weeks (B6 co-housed with Allo B6), while a cohort of naïve B6 animals from the same colony were not co-housed (Not co-housed B6) and served as control animals (see Fig. 1A). Microbiomes of B6 co-housed with Allo B6 changed towards Allo B6 at 6 weeks (Fig.S1C to G). Despite the shift in community structure and reduction in diversity, both the cohoused and non-cohoused cohorts of un-transplanted naïve B6 mice showed similar weight gain with no signs of GVHD (Fig. 1E and F).
Figure 1. Allogeneic dysbiosis is not pathogenic to naïve animals.

(A to F), naïve C57BL/6 mice (B6) 7–8 weeks old were co-housed with allogeneic recipient mice (Allo B6) two weeks after post-transplant. Mice were co-housed in a ratio of 1:1 naïve mice to BMT mice respectively (A). Data are from 3 independent experiments (N=9). Stool from from Allo B6 before co-house or B6 were analyzed by 16S rRNA gene sequencing. (B to D) Microbiome composition (left) with the percent abundance of obligate anaerobes and other bacteria (right) (B), PCoA (C), and inverse Simpson alpha diversity index of microbiome (D) in stool were shown. (E and F) The body weigh change (H) and the clinical GVHD score (F) of B6 co-housed with Allo B6, not co-housed B6, and not co-housed Allo B6 are shown (co-housed with Allo B6 and not co-housed B6: N=9, not co-housed Allo B6; N=6). (G to L) B6 mice 6–8 weeks old were treated with 2 weeks of 4 antibiotics cocktail (ampicillin 1gr/L, kanamycin 1gr/L, metronidazole 1gr/L and vancomycin 0.5gr/L plus 3% stevia) in filtered double distillated drinking water. Antibiotics treated mice (B6Ab) were co-housed in ratio of 1:1 antibiotic treated mice to BMT mice respectively (G). Data are from 3 independent experiments. (H) Stool from B6Ab, B6 and Germ free (GF) mice were analyzed by qPCR. Relative quantification of bacteria in stool from B6, antibiotics treated B6 at day7, and GF mice were analyzed (B6: N=10, Antibiotics d7: N=10, GF: N=8). (I) Inverse Simpson alpha diversity index of microbiome of B6 and B6Ab are shown (B6N: N=4, B6Ab, N=3). (J) Inverse Simpson alpha diversity index of microbiome of B6Ab, B6Ab co-housed with Syn or Allo B6 at day14 after co-house were shown (B6Ab N=4, B6Ab co-housed with Syn or Allo B6 N=2). (K) The body weight changes of co-housed B6Ab mice are shown (Co-housed with Syn: N=14, Co-housed with Allo: N=15). (L) Ileal and colonic histopathological GVHD score of co-housed B6Ab mice are shown (N=3). (M and N) B6 mice were treated for 2 weeks with 4 antibiotics cocktail (ampicillin 1 mg/ml, neomycin 1mg/ml, metronidazole 1mg/ml and vancomycin 0.5mg/ml) in filtered double distillated drinking water, followed by 10 doses of intestinal content gavage from BMT recipient mice 2weeks after BMT. Each gavage day one BMT mouse whole gut content was collected. Stool solution was gavaged to each recipient mouse (M). Data are from 3 independent experiments (B6Ab with Allo stool: N=15, B6Ab with PBS: N=8). (N) The body weight change of gavaged B6Ab mice are shown. The horizontal line in box (B, D, I, J) represents the median with the box bounding the interquartile range. The ends of the whisker lines represent the minimum and maximum values. Two-tailed paired Wilcoxon test (B) and Two-tailed unpaired t-test (D, I, L), and one-way ANOVA analysis with Tukey post hoc test (H, J) were used to determine significance (mean ± s.e.m.). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. See also Figure S1 and S2.
It is possible that some organisms in the endogenous normal microbiome of the cohoused mice may have prevented the pathogenic effects of dysbiosis. Therefore, we examined the impact of dysbiosis in naïve B6 animals treated with a cocktail of four antibiotics to effectively clear endogenous GI microbiome in B6 hosts3. The naïve B6 animals were treated with the four-antibiotic cocktail for 2 weeks (B6Ab, Fig. 1G), and near complete loss of microbiome was confirmed (Fig. 1H and I, Fig.S2A and B) before cohousing with Allo B6 as above. To further control for the ability of microbiota from transplanted mice to colonize naïve antibiotic treated B6 hosts, we also cohoused another cohort of these animals with B6 syngeneic BMT (Syn B6, Fig. 1G). We confirmed that B6 and Syn B6 showed similar microbiome and eubiosis (Fig.S2C and D). The naïve, un-transplanted B6 animals were cohoused and monitored for 8 weeks for signs of weight loss and GI damage. Co-housing with Allo B6 caused dysbiosis (Fig. 1J). As shown in Fig. 1K and L, cohousing of the un-transplanted antibiotic treated naïve B6 hosts with either dysbiotic Allo B6 or the control Syn B6 animals did not impact weight gain or cause GVHD.
It is formally possible that cohousing alone might not be sufficient to cause complete microbiota alterations and cause dysbiosis in un-transplanted naïve B6 animals, despite pre-treatment with antibiotic cocktails. Therefore, to mitigate any potential inefficiencies of cohousing on changes to the microbiome, we treated the naïve B6 un-transplanted hosts with the antibiotics or diluent control as above and then directly gavaged them with intestinal content from Allo B6 animals (Fig. 1M) and monitored as above. Gavage of antibiotic treated naïve B6 animals with Allo B6 stools caused dysbiosis (Fig. S2E and F). Un-transplanted antibiotic treated naïve B6 animals that received dysbiotic stool gavage from Allo B6 animals showed similar body weight gain as control un-transplanted antibiotic treated naïve B6 controls that received PBS gavage (Fig. 1N).
While clinical features of GI damage were not apparent, it is possible that gavage of the dysbiotic stool into the antibiotic treated, naïve un-transplanted B6 mice might cause changes in host immunity. We therefore gavaged Allo B6 stool, the control Syn B6 stool, PBS, or sham into antibiotic treated animals as above and performed systematic phenotyping of T cells (phenotype, cytokine, regulatory T cells) and dendritic cells in distant secondary lymph nodes (LN), spleen and regional (mesenteric) LNs on days 14 and 42 after the stool gavage (Fig. S3A and B). No significant differences were noted the in the CD4+ and CD8+ subsets, PD1, CD69, CD62L, CD44, CD4+Foxp3+ Tregs, T cell IFNγ, IL-4 and IL-17A, or the CD80, CD86, 40, PDL1, IL-6 and TNFα expression in the CD11c+ cells) between the cohorts. Furthermore, the local tissue T cell phenotype (in colon and ileum) and numbers were also similar between all cohorts (Fig. S3C).
When we analyzed histopathological changes in the small and large intestines that may have been caused by transfer of dysbiotic stool, we found no evidence of damage or significant differences between the groups at 2 weeks (Fig. S3D) or 6 weeks (Fig. S3E) after intestinal content gavage. These data collectively demonstrate that a shift towards dysbiosis did not affect the intestinal health of the un-transplanted naïve B6 animals and that allo-dysbiosis by itself is not deleterious in the absence of preceding damage. Thus, dysbiosis after allo-HCT is not a cause, but is a consequence of GVHD.
Post-transplant but not pre-transplant dysbiosis regulates GVHD
We next addressed whether changes to the microbiome after allo-HCT influence GVHD severity. To this end, we explored whether host dysbiosis before transplant impacts GVHD outcomes. B6 animals were treated with either antibiotics or diluent controls and reconstituted with stool microbiome from Allo B6 animals (Fig. 2A). They were then lethally irradiated (10Gy) and transplanted with 5x106 BM cells and 2.5x106 splenic T cells from either B6 syngeneic or allogeneic BALB/donors. Allogeneic recipients with or without antibiotics and stool gavage showed dysbiosis and similar microbiome between allogeneic groups, but changes from B6 mice before HCT (Fig. 2B and C). However, all of the allogeneic recipients with or without dysbiosis prior to the transplant showed similar mortality and clinical GVHD severity (Fig. 2D and E). Histopathology of the intestine confirmed similar pathological GVHD severity (Fig. 2F). These data suggest that the status of the pre-transplant microbiome does not substantially impact GVHD outcomes.
Figure 2. Pre-transplant dysbiosis or eubiosis does not have significant impact on GVHD.

(A) B6 mice were treated for 2 weeks with 4 antibiotics cocktail (Fig.1G) or PBS, followed by gavaged Allo B6 intestinal content or PBS. After antibiotics treatment and intestinal content gavage, mice received BMT. (B and C) Stool from BMT recipients and B6 mice were analyzed by 16S rRNA gene sequencing. PCoA (B) and inverse Simpson alpha diversity index of microbiome (C) are shown. (B6 N=5, Allo N=3, Allo with Ab N=4, Allo with Ab + stool N=4). Survival rate (D), clinical GVHD score (E), and pathological GVHD score of colon (F) are shown. (D) Allo: N=3, Allo with Ab: N=4, Allo with Ab+stool: N=4, B6: N=5. (E) Syn with Ab+stool: N=6, Allo: N=5, Allo with Ab: N=5, Allo with stool: N=5, Allo with Ab+stool: N=6. (F) Syn with Ab+stool: N=3, Allo: N=3, Allo with Ab: N=3, Allo with stool: N=3, Allo with Ab+stool: N=2. Two independent experiments were performed. One-way ANOVA analysis with Tukey post hoc test (C) and one-way ANOVA analysis with Dunn`s post hoc test (F) was used to determine significance (mean ± s.e.m.). **P < 0.01, ***P < 0.001. See also Figure S3.
Pretransplant dysbiosis does not address the issue of the impact of the change from pre-BMT eubiosis to post-BMT dysbiosis. Because GVHD is associated with post-transplant dysbiosis, we examined whether prevention of post-transplant dysbiosis would mitigate GI GVHD (Fig. 3A). B6 animals received allogeneic BMT (described in Methods). Each individual recipient mouse was cohoused with an individual naïve B6 WT mouse in a single cage from day 14 after HSCT. Simpson diversity and PCoA analyses of stools from these recipient mice revealed that cohoused allogeneic mice (Allo B6 co-housed with B6 for 2weeks) had higher diversity and different microbiota compositions from single housed allogeneic mice (Allo B6 not co-housed) (Fig. 3B to F) demonstrating that the cohousing shifted the stool microbiome of Allo B6 recipients towards the healthy microbiome. Co-housed Allo B6 recipients demonstrated greater survival, reduced weight loss and milder clinical GVHD than not co-housed Allo B6 (Fig. 3G to I). Taken together, the data demonstrate that dysbiosis pre-transplant per se are not disease enhancing, but that promoting post-BMT eubiosis ameliorates the severity of GVHD.
Figure 3. Post-transplant eubiosis ameliorates the severity of GVHD.

Allo B6 were co-housed with B6 at ratio of Allo B6: B6 = 1:1 from day 14 after BMT (A). Data are from three independent experiments. (B-F) Stool from Allo B6 co-housed with B6 for 2weeks, B6 co-housed with Allo B6 for 2weeks, Allo B6 not co-housed, and B6 were analyzed by 16S rRNA gene sequencing. (B) Inverse Simpson alpha diversity index were shown. (Allo B6 not co-housed: N=6, Allo B6 co-housed with B6: N=4, B6 co-housed with Allo B6: N=5, B6: N=4). (C) PCoA were shown (Allo B6 not co-housed: N=9, Allo B6 co-housed with B6: N=6, B6 co-housed with Allo B6: N=5, B6: N=7). Microbiome composition (D) and taxa differentially abundant by LEfSe analysis (E & F) are shown. Survival rate (G), body weight change (H), and clinical GVHD score (I) of BMT recipients were shown. (Allo B6 were co-housed with B6: N=18, Allo B6 not co-housed: N=6). The horizontal line in box (B) represents the median with the box bounding the interquartile range. The ends of the whisker lines represent the minimum and maximum values. One-way ANOVA analysis with Tukey post hoc test (B), log-rank test (G), and two-tailed Mann-Whitney test (I) was used to determine (mean ± s.e.m.). *P < 0.05.
The above effects could be secondary to pre-existing microbiota or from the experimental effects of gavage and antibiotic use. Therefore, to determine whether dysbiotic stool has any intrinsic pathogenic effects and eubiosis has beneficial effects in the absence of above variables, we next compared weight gain of GF mice with littermate GF mice that were conventionalized with either dysbiotic allo-stool or eubiotic syngeneic stool (Fig. 4A). GF animals that received eubiotic stool from syngeneic mice and those that were conventionalized with dysbiotic allo-stools demonstrated weight gain when compared with GF controls confirming that dysbiotic microbiome by itself is not pathogenic in the absence of prior damage (Fig. 4A, 4B, C and Fig. S4A).
Figure 4. GF mice transplanted with healthy microbiome showed reduced GVHD.

(A to C) Germ free B6 (GF) mice were gavaged intestinal content from Syn and Allo BMT recipient mice. (A) Body weight change is shown (GF: N=2, GF with Syn B6 stool: N=4, GF with Allo B6 stool: N=5). Stool from GF with Syn or Allo B6 stool 4weeks after stool gavage analyzed by 16S rRNA gene sequencing. PCoA (B) and microbiome composition (C) are shown (GF with Syn B6 stool: N=7, GF with Allo B6 stool: N=10). (D to F) SPF B6 and GF B6 mice received BMT from B6 or BALB/c donor. Survival rate (D), body weight change (E), and clinical GVHD score (F) are shown (GF Syngeneic: N=2, GF Allogeneic: N=18, SPF Syngeneic: N=2, SPF Allogeneic: N=17). (G to J) GF B6 mice received BMT from B6 or BALB/c donor. Then GF mice were gavaged intestinal content from SPF Syn and SPF Allo BMT recipient mice (G). Survival rate (H) and clinical GVHD score 6 weeks after BMT (I) are shown (Allo GF with Syn B6 stool N=6, Allo GF with Allo B6 stool N=4). (J) Ileum and colonic histopathological score at day7 after BMT are shown (n=4). Two-tailed unpaired t-test (A, I, J), log-rank test (D, H), and two-tailed Mann-Whitney test (F) was used to determine (mean ± s.e.m.). *P < 0.05, ***P<0.001, ****P<0.0001. See also Figure S4.
Absence of host microbiome aggravates GVHD
Our data demonstrate that changes in endogenous GI microbiome structure after allo-HCT are not a cause but a consequence of GVHD and suggest that they might play a salutary role after BMT 3 4 16 22 23. However, before the advent of stringent germ-free (GF) facilities, and before technologies to confirm presence of microbes such as 16S sequencing, GF mice exhibited reduced GVHD 10, suggesting an overall pathogenic role for host microbiome. Therefore, to elucidate the importance of microbiota definitively, and directly, in the gut before BMT, we next hypothesized that in contrast to the current paradigm, GF recipients will demonstrate greater GVHD than normal SPF animals. To rule out any potential confounding littermate effects at birth between GF and SPF conditions, age-matched GF B6 littermate mice were first separated into two cohorts at 4–6 weeks of age. One group was continued under stringent GF conditions and the second group was aged under SPF conditions up to age 12–14 weeks. These GF and SPF cohorts were then irradiated and transplanted with either syngeneic B6 or allogeneic BALB/c donors and were followed post-BMT under stringent GF or SPF conditions respectively. All the syngeneic recipients from both the GF and SPF cohorts survived and showed no signs of GVHD, demonstrating that absence of microbiome did not cause mortality in the absence of allo-reactive T cell mediated damage. All allogeneic recipients under SPF conditions, as expected, demonstrated signs of GVHD with a median survival of 7 weeks (Fig. 4D to F). However, the GF mice that received allogeneic HSCT demonstrated greater mortality, weight loss and clinical GVHD (Fig. 4D to F).
Because allo-stool (dysbiotic microbiome) neither caused disease nor improved weight gain in GF mice, while eubiotic microbiome improved weight gain in GF animals, we next surmised that GF mice transplanted with healthy microbiome will show reduced GVHD. To this end, GF animals received allo-stool or syngeneic stool (Fig. 4G). The engraftment and fidelity of the transplanted microbiome was confirmed (Fig. S4B to D). These animals were then irradiated and utilized as recipients of either syngeneic or allogeneic BMT as in Methods (Fig. 4G). Syngeneic recipients that received dysbiotic microbiome survived, demonstrating, once again, no inherent toxicity from the dysbiotic stool. The allogeneic recipients of dysbiotic stool demonstrated expected GVHD mortality and clinical/pathological GVHD (Fig. 4H to J). By contrast, allo-recipients that were transplanted with eubiotic healthy microbiome demonstrated greater survival and reduced weight loss and clinical/pathological GVHD (Fig. 4H to J). Thus, the structure of the host microbiome is dispensable for induction of GVHD, but is critical for regulation of GVHD after it has been initiated.
Loss of intestinal physiologic hypoxia causes dysbiosis
We next explored the mechanisms for development of dysbiosis following allo-BMT. The physiologic hypoxia (low oxygen) present at homeostasis in the intestinal milieu regulates the composition and structure of the intestinal microbiome 1. In the context allo-BMT, host IECs demonstrate a metabolic defect in OXPHOS due to a T cell-induced disruption of mitochondrial complex II 21. We therefore hypothesized that lack of utilization of cellular O2 caused by disruption of OXPHOS in the IECs after allo-HCT will lead to loss of physiologic hypoxia (increase in O2) and thus cause dysbiosis by making the GI tract more permissive for growth of aerotolerant and facultative anaerobes at the expense of obligate anaerobes. To this end, we systematically measured oxygen consumption of IECs, the changes in the intestinal luminal and mucosal levels of O2 by colonoscopy, and intestinal cellular hypoxia by pimonidazole after HSCT. B6 received either syngeneic B6 or allogeneic BALB/c transplants as in Methods. Consistent with previous reports, CD326+ IECs harvested from the allogeneic mice on day 21 after HSCT demonstrated a reduction in oxygen consumption rate (OCR) when compared with IECs from syngeneic controls (Fig. 5A). Furthermore, IECs from allogeneic animals did not respond to treatment with carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), a mitochondrial uncoupler, when compared with IECs from syngeneic animals demonstrating the reduction in allo-IEC mitochondrial ETC functions and utilization of O2 (Fig. 5A). However, both syngeneic and allogeneic IECs showed similar extracellular acidification rates (ECAR, an indicator of glycolysis), with reduced OCR/ECAR ratio (Fig. S5A). This suggests that the defect in O2 utilization should increase O2 levels, leading to a loss of intestinal luminal and cellular physiological hypoxia.
Figure 5. The defect in O2 utilization in IECs leading to a loss of intestinal luminal and cellular physiological hypoxia.

(A) Representative bio-energetic profiles of isolated colonic IECs from syngeneic and allogeneic mice (BALB/c→B6) under basal conditions and following treatment with mitochondrial inhibitors (oligomycin, FCCP, rotenone/antimycin A) by Seahorse analyzer. Oxygen consumption rate (OCR) of day21 after BMT is shown (N=4). Four independent experiments were performed. (B) Quantification of O2 levels in intestine from recipients 7days and 21days after BMT (BALB/c→B6). Naive: N=5, Syngeneic: N=4 (Day7 outer and Day21 outer), N=5(Day7 inner), Allogeneic: N=4 (Day7 outer and Day21 outer), N=5 (Day7 inner). (C) B6 received BMT from B6 or BALB/c donor. Representative images of Hypoxyprobe staining and relative fluorescent intensity in colon from recipients 21days after BMT (scale bar= 50μm). (D) Unirradiated B6D2F1 mice received 10 × 107 splenocytes from syngeneic B6D2F1 or allogeneic B6 donors. Representative images of Hypoxyprobe staining and relative fluorescent intensity in colon from recipients at 21days after BMT (scale bar= 50μm) (N=4). (E) B6 mice received chemotherapy and received 1x107 T cells along with 1x107 TCD-BM cells from either syngeneic B6 or allogeneic BALB/c donors. Representative images of Hypoxyprobe staining and relative fluorescent intensity in colon from recipients 21days after BMT (scale bar= 50μm) (N=4). (F) B6 mice received 10 Gy total body irradiation without T-cell and BM cells. Representative images of Hypoxyprobe staining relative fluorescent intensity in colon from recipients 7days after BMT (scale bar= 50μm) (N=4). (G and H) CD4++CD25-CD4+4-CD4+5RBhi (CD4+5RB high) T cells or CD4++CD25-CD4+4-CD4+5RBlow (CD4+5RB low) T cells from B6 mice were transferred to Rag1-/- SPF (G) or Rag1-/- GF mice (H). Hypoxyprobe staining and relative fluorescent intensity in colon from recipients 8weeks after T cell transferring (scale bar= 50μm) (SPF: N=4, GF CD4+5RB low: N=4, GF CD4+5RB high N=5). (C to H) At least, four independent experiments were performed. One-way ANOVA analysis with Tukey post hoc test (B) and two-tailed unpaired t-test (C to H) was used to determine significance (mean ± s.e.m.). *P < 0.05, **P < 0.01. See also Figure S5.
To confirm the increased O2, we performed colonoscopy with probes that directly measure oxygen levels in the colonic mucosa (adjacent to IECs) and in the center the intestinal lumen of the HSCT recipients. The O2 concentrations in deeper locations, in the inner mucus of the colon, immediately adjacent and closer to the IECs was greater in the allo-recipients than the controls (Fig. 5B). The oxygen levels in the lumen, further away from the IECs initially demonstrated similar levels of oxygen in allo-recipients as in the controls naïve or syngeneic B6 controls early after BMT (Fig. 5B). However, 3 weeks after BMT, the allo-recipients demonstrated greater levels of O2 in the intestinal lumen when compared with the controls demonstrating a loss of hypoxia in the lumen farther away from the IECs (Fig. 5B). Finally, loss of intestinal cellular hypoxia with pimonidazole was also documented in the allo-BMT recipients at days 7 and 21 after HSCT (Fig. 5C). Thus, underutilization of the O2 by the IECs because of T cell-induced metabolic defect leads to loss of intestinal physiological hypoxia in GVHD.
Next, to validate whether the loss of physiological intestinal hypoxia is exclusively from T cell mediated GVHD and not a consequence of strain-dependent artifacts or damage from conditioning, we determined the status of intestinal hypoxia in the non-irradiated parent into F1 model (B6→B6D2F1), where alloreactive donor T cells cause GVHD despite absence of conditioning (Fig.S5B and C). Allogeneic IECs demonstrated increased levels of oxygen (Fig. 5D). Similar loss of hypoxia was also observed when allogeneic B6 animals were conditioned with chemotherapy (busulfan and cyclophosphamide) prior to HSCT (Fig. 5E). By contrast, when animals were conditioned with irradiation alone hypoxia was maintained (Fig. 5F). To demonstrate that the loss of physiological hypoxia is independent of microbiome composition, we utilized mouse strains from a different mouse supplier (Taconic, JAX). Allogeneic Taconic B6, Taconic BALB/c and JAX BDF1 recipients showed similar loss of hypoxia at day 7 after BMT (Fig.S5D to F). These results extend previous observations and demonstrate that the increase in O2 concentration in the intestines is observed only in the context of allo-T cell-mediated damage of GI tract 21. To further determine whether this can be extended to other T cell-mediated GI damage, we determined intestinal oxygen concentration in the CD45RBhi adoptive T cell-mediated autoimmune model of IBD. Induction of T cell-mediated autoimmune colitis also led to loss of hypoxia in both SPF (Fig. 5G, Fig.S5G) and GF animals (Fig. 5H, Fig.S5H). Thus, T cell-mediated disruption of intestinal cell OXPHOS in the IECs leads to poor utilization of cellular O2 resulting in loss of physiologic hypoxia that promotes an environment permissive for dysbiosis.
Next, to enhance the generalizability and demonstrate that loss of physiological hypoxia is not an artifact of the specific vivarium conditions, we performed the similar experiments with different strain combinations at a second institution (MSKCC, NY Center). Similar to the data from University of Michigan, HCT recipients with GVHD showed loss of hypoxia in colon (Fig. 6A). Changes in microbiome composition (Fig. 6B) and decreased ratio of obligate/facultative anaerobes in recipients with GVHD (Fig. 6C) were observed. These data demonstrated GVHD induced loss of hypoxia and dysbiosis in colon is independent of the institution, vendors and strains.
Figure 6. Loss of hypoxia in colon and dysbiosis are induced by GVHD with independent of the different institution and microbiome.

Lethally irradiated (1000cGy) 129 mice received bone marrow (5x106 cells) and purified CD5+T cells (2x106 cells) from C57BL/6J donors in different institution (Memorial Sloan Kettering Cancer Center). (A) Tissue hypoxia was determined with Pimonidazole (left, scale bar= 50μm). Quantification of intestinal O2 levels in intestine from recipients (right). Samples were collected 4, 8, 14, and 21days after BMT (Naïve: N=3, BM only: N=4, BM+T: day4, 8, 14 N=4, day21 N=3). (B) Stool microbial composition was determined by 16S rRNA gene sequencing. (C) The classification of obligate/facultative (O/F) anaerobe ratio was determined. Two-tailed unpaired t-test (A) was used to determine significance (mean ± s.e.m.). **P < 0.01.
Intestinal O2 levels regulate GVHD in a microbiome-dependent and -independent manner
We determined whether the loss of physiologic hypoxia has a direct effect on IECs that could impact GI GVHD. To analyze the impact of increase in O2 we determined the expression of cellular O2 sensor, hypoxia inducible factor (HIF-1α). B6 animals received syngeneic or allogeneic BMT (see Methods). Hif-1α gene expression was similar in the IECs harvested from allogeneic and syngeneic animals (Fig. 6A). However, the HIF-1α protein was reduced in the IECs from allogeneic animals when compared to syngeneic animals on day 21 after BMT (Fig. 7B). By contrast, the expression of proryl hydroxylase 3 (PHD3), a protease that regulate the protein level of HIF-1α, was increased in the allo-IECs (Fig. 7C and D)
Figure 7. O2 modulation after allo-HSCT improves intestinal physiologic hypoxia and GVHD.

(A to D) B6 received BMT from syngeneic B6 or allogeneic BALB/c donor. (A) Hif1α mRNA expression of isolated colonic IECs from BMT recipients 21days after BMT (N=4). (B) Representative image and the relative intensity of HIF1α immunohistochemical staining with intestinal tissue from BMT recipients 21days after BMT are shown (Scale bar=50μm, N=4). (C) Egln3 (Phd3) mRNA expression of isolated colonic IECs from BMT recipients on day21 after BMT (N=3). (D) Representative image of immunoblot and the relative protein density for PHD3 and β-Actin with colonic IECs from BMT recipients 21days after BMT are shown (N=4). (E and F) Hif1αfl/fl mice and Hif1αfl/fl Vil1cre mice received BMT from BALB/c donor. Survival rate (E) and clinical GVHD (F) score are shown (Hif1αfl/fl allogeneic: N=6, Hif1αfl/fl Vil1cre: N=4). (G to L) B6 received BMT from BALB/c donor mice. BMT recipients were orally treated with deferasirox (20mg/kg) and vehicle every day. (G) Hypoxyprobe staining and relative fluorescent intensity in colon from recipients 21days after BMT (scale bar= 50μm). Four independent experiments were performed. Stool from BMT recipients were analyzed by 16S rRNA gene sequencing. PCoA (H), inverse Simpson alpha diversity index of microbiome (I), and microbiome composition (J) in stool from recipients 21days after BMT were shown (Allo B6 vehicle: N=7, Allo B6 deferasirox: N=9). Survival rate (K) and clinical GVHD score (L) of BMT recipients (Allo B6 vehicle: N=24, Allo B6 deferasirox: N=10). Representative plots and a graph summarizing the results of at least two independent experiments are shown. Two-tailed unpaired t-test (A, C, G, I), two-tailed Mann-Whitney test (B, D, F, L), or log-rank test (E, K) (mean ± s.e.m.) were used to determine significance. *P < 0.05, **P<0.01. See also Figure S6.
Next, to determine whether the IEC cell autonomous loss of physiologic hypoxia after allo-HSCT has a microbiome independent effect on GI GVHD, we utilized littermate congenic IEC specific Hif-1a gene knock out mice (Hif1afl/fl Villin1cre B6) or the WT Hif1afl/fl B6 animals (to harmonize pre-HCT microbiome) as allogeneic HSCT recipients. The animals were cohoused post-BMT to harmonize microbiome changes. The IEC-specific Hif1afl/fl Villin1cre B6 allogeneic recipients showed greater GVHD severity and mortality, compared to the littermate Hif1afl/fl B6 mice despite being cohoused (Fig. 7E and F). Taken together these data demonstrate that changes in O2 after allo-HSCT directly affect host IECs, enhancing GVHD severity independent of changes in the microbiome.
We next determined the direct functional link between the increase in O2, the loss of physiologic hypoxia, observed after allo-HSCT to the resultant dysbiosis and GVHD severity. To mitigate the impact of excess luminal O2 by reducing the Fenton reaction in the IECs, we administered the iron chelator, deferasirox, by oral gavage after BMT (see Methods) 24 25. We determined the experimental dose and timing by measuring the level of iron in the stool from both small and large intestines following gavage with Fe chelator (Fig. S6A). We first determined whether gavaging with deferasirox altered the level of O2 concentration in the intestines after HSCT and found that it mitigated loss of hypoxia in the allogeneic recipients (Fig. 7G). The PCoA analysis showed different microbial composition between vehicle and deferasirox treated allogeneic animals (Fig. 7H), with an increase in microbial diversity (Fig. 7I and J) thus demonstrating that changes in O2 directly contributed to dysbiosis after allo-HSCT. We next hypothesized that squelching of O2 to ameliorate dysbiosis after allo-HSCT will lead to attenuation of GVHD mortality. Consistent with the hypothesis, deferasirox treated allogeneic animals demonstrated reduced severity of clinical GVHD and survival (Fig. 7K and L). The iron chelator did not show any effect on inflammatory cytokine secretion and Treg ratio in spleen (Fig.S6B and C).
Because alteration of O2 affects both host IECs and the microbiome, we next determined whether restoring intestinal hypoxia by Fe chelation after allo-HSCT mitigated GVHD independent of its impact on dysbiosis. To determine microbiome independent effects, we utilized GF B6 animals as allo-HSCT recipients and treated them with deferasirox or diluent control (see Methods). All transplanted mice were maintained in stringent GF environment post BMT. Treatment with Fe chelation ameliorated loss of hypoxia after BMT in the GF recipients (Fig. S6D). All syngeneic GF mice treated with either diluent control or deferasirox survived without GVHD. The allogeneic GF mice treated with diluent control demonstrated severe GVHD and died within 2 weeks after BMT (Fig. S6E and F). By contrast, deferasirox treated allogeneic GF mice (Fig. S6E and F) demonstrated improved survival when compared with diluent treated allo-recipients, but eventually succumbed to GVHD. These data collectively demonstrate that O2 modulation, to improve intestinal physiologic hypoxia has direct salutary effects both on host IECs and the host microbiome to improve GI GVHD.
DISCUSSION
Colonic dysbiosis is associated with human intestinal diseases, including GI GVHD and IBD1 9. However, whether dysbiosis is a cause or consequence, an amplifier or a mollifier, and the mechanisms remain elusive. Herein we address these gaps and demonstrate that pre-transplant dysbiosis by itself is not pathogenic, but rather post-HCT dysbiosis is a consequence of the changes in intestinal luminal oxygen level from the tissue injury caused by allogeneic donor T cells after allo-HCT. We additionally demonstrate that correction of post-transplant dysbiosis has beneficial impact. We show that in contrast to the existing paradigm, absence of microbiome after HCT aggravates GVHD, and thus provide clarity to the emerging data that the structure of the microbiome after HCT, regulates the severity of GI GVHD 3 10 16 17 21 22. These data extend previous observations that T cell mediated disruption of intestinal OXPHOS leads to the loss of luminal physiological hypoxia (increase in O2) 21, which functions as a control switch to shift the intestinal microbiome from predominantly obligate anaerobes to facultative anaerobes and aerotolerant microbes, thus providing mechanistic insights into the cause of dysbiosis. Finally, targeting excess O2 with iron chelation promoted physiologic hypoxia, mitigated dysbiosis and attenuated GVHD severity, thus suggesting that Fe chelation with available oral drugs may be a novel strategy to clinically mitigate GVHD severity.
During homeostasis, the host intestinal epithelial cell metabolism is dependent on OXPHOS resulting in high epithelial oxygen consumption and as a consequence epithelial and luminal hypoxia 1. Epithelial hypoxia helps to maintain a microbial community dominated by obligate anaerobic bacteria at homeostasis 1 20. We build on prior observation that the disruption of host IEC metabolism pathogenic T effector cells results in an increase in intestinal oxygen, thereby driving an expansion of facultative anaerobic and aerotolerant bacteria, a hallmark of dysbiosis GVHD 4 22 23. Thus, one critical mechanism for dysbiosis, the loss of physiologic hypoxia in a non-infectious intestinal diseases like GVHD and IBD are similar to that observed following infection by enteric infectious pathogens 1 22. This shared mechanism linked to tissue oxygenation in causing dysbiosis allows for a better understanding of other diseases associated with dysbiosis. It is however possible that the dominant mechanisms of dysbiosis may be distinct in different systemic diseases.
Seminal studies suggested that germ-free hosts had less severe GVHD9 10. These studies formed a paradigm for the field of HCT, even as they appeared to stand in contrast to emerging data implicating strong correlation between microbiome and GVHD. The modern-day studies, however, showing shifts in microbiome or use of antibiotics as potential regulators of GVHD, do not address the notion of stringent germ-free status on GVHD. We evaluated the role of stringent germ-free status on GVHD and found that complete absence of microbiome in transplanted mice that are housed in germ-free conditions show greater severity and mortality from GVHD than normal SPF recipients. These data directly demonstrate an overall salutary role for microbiome in GVHD. The reason for the different results from the studies by van Bekkum et al, is likely because those earlier studies were performed in an era when sequencing for microbiome was not yet available and as such the ability to confirm true germ-free status or the reliability of the colony conditions to be maintained in a germ-free state was likely not optimal 10 26. Furthermore, our data demonstrate that adding back enteric microbiome mitigated GVHD severity in GF hosts, thus definitively demonstrating a critical role for microbiome in regulation of GVHD.
Our data utilizing cohousing SPF, antibiotic treated, and GF animals provide several insights into the role of dysbiosis before and after allo-HCT. First, dysbiosis by itself, in the absence of initial damage by alloreactive T cells, is not pathogenic, at least in the period that acute GVHD mortality typically occurs in these systems. Second, the presence of dysbiosis prior to HCT did not aggravate GVHD. However, promotion of eubiosis after HCT mitigated GVHD. Similarly, GF mice conventionalized with healthy stools mitigated GVHD. These data, when taken together with observations that outcomes are worse in humans with use of antibiotics that primarily target anaerobes after HCT demonstrate that microbiome primarily has a salutary role in GVHD. Thus, promotion of eubiosis after HCT (i.e prebiotic or probiotic approaches with appropriate antibiotic stewardship for growth of healthy microbiome, i.e., obligate anaerobe microbes) as opposed to treating dysbiosis before (eliminating facultative anaerobes or aerobes, i.e. only antibiotic approaches) may be a rational strategy to therapeutically modulate microbiome to ameliorate GVHD. The mechanisms by which eubiosis after HCT may mitigate GVHD is likely (but not limited) to generation of metabolites by obligate aerobeic commensals that nourish the IECs, or promote immune and tissue tolerance, or provide nutrients to healthy commensals and regulate the microbial ecology by promoting a milieu that is not permissive for pathobionts 16 17 21 27 28 29 30. However, the biology of GVHD is complex with several studies demonstrating a role for DAMPs and PAMP production by tissue damage as amplifiers of a GVH response 31 32. Therefore it is possible that dysbiosis when characterized by expansion of pathobionts that gain access to hosts circulation and tissues amplifies GVHD 4 22 33.
The increase in, and the type of pre-transplant dysbiosis noted in the clinical studies that correlated with outcomes suggest that factors that lead to dysbiosis could predict worse GVHD, but do not directly demonstrate that dysbiosis by itself directly caused GVHD. Nonetheless, it is possible, even if unlikely based on our GF reconstitution experiments, that there might be a threshold for the specific type, or degree of dysbiosis, beyond which pre-transplant dysbiosis may cause or promote GVHD. The change in specific bacteria (Enterococcus and reduction in Blautia), has been hard to compare between different studies, but nonetheless all of the studies are broadly consistent with reduction in obligate anaerobes and an increase in proportion of aerotolerant microbiome following GVHD. Thus the nature of specific microbes and their impact in the context of the wider ecology will need to be assessed in future studies with GF mice monocolonized or polycolonized with depletion of specific microbe of interest to directly assess their pathogenic or beneficial effects, alone, and in the context of other microbes. However, our observations, regarding hypoxia loss in GVHD and development of dysbiosis are consistent across vendor source, different strains, multiple model systems and under different experimental conditions from different institutions.
It remains unknown whether the degree of GVHD severity affects microbiome contents in a specific manner. However, our data, consistent with previous data, have shown that there is a general loss of diversity. It remains to be determined whether the measurement like the specific and absolute loss of diversity can be consistently quantified and compared across different patients and contexts, and to formally assess and rule out that the degree of dysbiosis may directly cause GVHD. Additionally, the improvement of survival in co-housing experiments could be confounded by the social, behavioral or neurological effects of cohabitation, besides, or in addition to, changes in microbiome. These potential variables will need to be investigated in carefully designed future studies.
During homeostasis, despite being in a relative hypoxic environment, intestinal metabolism is directed toward oxidative phosphorylation (OXPHOS), resulting in high epithelial oxygen consumption. Disruption of OXPHOS in the IECs by alloreactive T cells, reduces oxygen consumption 21. Our data now extends these observations and provides a potential novel therapeutic approach. It demonstrates that increase in luminal oxygen makes the intestinal environment less permissive for obligate anaerobes, thus promoting dysbiosis. The excess tissue oxygen levels is also corroborated by lower expression of the O2 sensor HIF-1α in the IECs 34 35 36. It is likely that the lack of O2 consumption might also contribute by direct free radical injury to the IECs and increase GVHD severity. Administration of oral iron chelator reduced excess luminal O2, promoted physiological hypoxia, improved microbial diversity, reduced dysbiosis and ameliorated GI GVHD, but did not alter donor T cell immune responses in the host. Moreover, oral Fe chelation also modestly improved GVHD survival in GF animals demonstrating microbiome independent effects. Thus, squelching of excess O2 by Fe chelation improved GI GVHD largely in a microbiome dependent, but also, to a modest degree, in a microbiome independent manner. The specific mechanisms for microbiome independent effects of Fe chelation on host tissues, such as ferroptosis, will need further investigation 37 38. Furthermore, Fe chelation likely impacts Fe availability to microbes directly affecting the structure of microbiome39. Nonetheless, our data thus provide a novel role for tissue O2 as a mechanism for dysbiosis and Fe chelation as a therapeutic target for amelioration of non-infectious immune mediated intestinal diseases.
LIMITATIONS OF THE STUDY
The study did not address the impact of alterations of oxygen levels on the changes in specific bacterial genus and species. This will need to be carefully dissected in future studies. The lack of deleterious effects of dysbiosis on the GI tract of the hosts were limited to a relatively short duration (6 weeks). The impact on long-term GI or general health of the host will need to be addressed. The effect of the absence or variations in key host immune cell functions such as cytokines, effector responses that affect the host response to dysbiosis and on GVHD severity was not addressed in this study. Iron chelation likely directly impacts the host microbial ecology, such as alterations in siderophore producers. Our study demonstrates a cause for dysbiosis in murine models, but the direct relevance of intestinal ambient oxygen levels to human GI GVHD will need to be analyzed in prospective human studies.
STAR METHODS
RESOURCE AVALABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Reddy Pavan (Pavan.Reddy@bcm.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Unprocessed 16s rRNA sequencing reads were deposited at the NCBI Short Read Archive (SRA) and are accessible via BioProject PRJNA910578.
The code used for analysis for 16S rRNA sequencing is available at https://doi.org/10.5281/zenodo.7401507.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice:
C57BL/6 (027, B6, H-2Kb, CD4+5.2), BALB/c (028, H-2Kd), and BDF1 (099, H-2Kb/d) were purchased from Charles River Laboratories. B6.129S7-Rag1tm1Mom/J(002216, Rag1-/-) mice, BDF1 (100006, B6D2F1/J), B6.Cg-Tg(Vil1cre)1000Gum/J mice(021504), and 129 (002448, 129S1/SvImJ) were purchased from the Jackson Laboratory. Taconic B6 (B6-F, C57BL/6NTac) and Taconic BALB/c (BALB-F, BALB/cAnNTac) were purchased from Taconic. Hif1a-floxed mice (Hif1afl/fl, C57BL/6 background) 40 containing the loxP site were crossed with Vil1cre mice (C57BL/6 background) to generate intestinal epithelial cells specific HIF1a-null mice (Hif1afl/fl Vil1-cre mice). Germ free (GF) C57BL/6 mice and Rag1-/- GF mice were raised and housed in ISOcage Positive isolators (Techniplast) at the germ-free mouse facility at the University of Michigan. Germ-free status was verified by aerobic, anaerobic cultures, and gram stain. 6–12 weeks old female mice used for experiments. All mice were kept under specific pathogen-free (SPF) conditions or GF conditions and cared for according to regulations reviewed and approved by the University of Michigan Committee on the Use and Care of Animals (PRO00009494), which are based on the University of Michigan Laboratory Animal Medicine guidelines. Mouse studies from MSKCC followed the respective Institutional Animal Care and Use Committee guidelines (99-07-025) and were kept under specific pathogen-free (SPF) conditions. B6 mice 6–8 weeks old were treated with 2 weeks of antibiotics cocktail (ampicillin 1mg/ml (A9393, Sigma Aldrich) + kanamycin 1mg/l (60615, Sigma Aldrich) + metronidazole 1mg/ml (M1547, Sigma Aldrich) + vancomycin 0.5mg/l (SBR00001, Sigma Aldrich) plus 3% stevia or ampicillin 1 mg/ml + neomycin 1mg/ml + metronidazole 1mg/ml + vancomycin 0.5mg/ml in filtered double distillated drinking water. BMT recipients were orally treated with deferasirox (20mg/kg, SML2673–50, Sigma Aldrich) and vehicle every day until day21 after BMT. For the co-housing experiments, mice were co-housed in a ratio of 1:1 naïve mice, B6 Ab, and BMT mice respectively. For Fig. 1A, Allo B6 and B6 were co-housed. For Fig. 1G, SynB6 or Allo B6 were co-housed with B6Ab. For Fig. 3A, the design of the experiment is same as Fig. 1A. For the fecal microbiota transplant, mice were gavaged by 10 doses of intestinal content from recipient mice for 2weeks. Each gavage day one BMT mouse whole intestinal content was collected and homogenized in sterile PBS. 200ul of the solution was gavaged to each recipient mouse.
Hematopoietic cell transplantation model:
Transplantations were performed as previously described 31. Briefly, Splenic T cells from donors were enriched, and T-cell-depleted BM (TCD-BM) was depleted of T cells by autoMACS (Miltenyi Biotec) utilizing CD90.2 microbeads (130-121-278, Miltenyi Biotec) or CD5 microbeads (130-049-301, Miltenyi Biotec). The details of HCT model were described in Table S1. The mice were randomly assigned to syngeneic, allogeneic or treatment groups in each experiment.
Colitis models:
For the T-cell transfer induced colitis model, isolated splenic T cells from B6 mice were stained with DAPI (#422801, 1μM, Biolenged), APC-Cy7 anti-CD4+ (560246, GK1.5, 1:100, BD Biosciences, San Jose, CA), APC anti-CD25 (101910, 3C7, 1:100, Biolegend), FITC anti-CD4+4 (103006, IM7, 1:100, Biolegend) and PE anti-CD4+5RB (103308, C363–16A, 1:100, Biolegend). CD4++CD25-CD4+4-CD4+5RBhi cells were sorted with the MoFlo Astrios cell sorter (Beckman Coulter) and intraperitoneally injected into Rag-1-/- SPF or GF recipients.
METHOD DETAILS
Systemic and histopathological analysis of GVHD:
Survival after HCT was monitored daily and assessed the degree of clinical GVHD weekly, as described in Table S241. Histopathological analysis of the liver, gastrointestinal (GI) tract, and lung, which are the primary GVHD target organs, was performed as described utilizing a semi-quantitative scoring system implemented in a blinded manner by a single pathologist (C.L.) 42. A pathology scoring of GVHD was used to assess the following abnormalities known to be associated with GVHD. Small intestine: villous blunting, crypt regeneration, loss of enterocyte brush border, luminal sloughing of cellular debri, crypt cell apoptosis, outright crypt destruction, and lamina propria lymphocytic infiltrate; colon: crypt regeneration, surface coloncytes, colonocyte vacuolization, surface colonocyte attenuation, crypt cell apoptosis, outright crypt destruction, and lamina propria lymphocytic infiltrate. The scoring system denoted 0 as normal, 0.5 as focal and rare, 1.0 as focal and mild, 2.0 as diffuse and mild, 3.0 as diffuse and moderate, and 4.0 as diffuse and severe. Scores were added to provide a total score for each specimen. Only after scoring was performed were codes broken and data compiled. After scoring, the codes were broken, and the data compiled.
DNA extraction, 16S rRNA gene sequencing and data analysis:
The University of Michigan Microbiome Core extracted DNA and prepared and sequenced the amplicon libraries. DNA was extracted using an Eppendorf EpMotion liquid handling system and the Qiagen MagAttract PowerMicrobiome kit (previously MoBio PowerMag Microbiome, 27500-4-EP, Qiagen) kit and protocol. DNA (1 μl) was quantified with the Quant-iT PicoGreen dsDNA Assay kit (p7589, Invitrogen).
The University of Michigan Microbiome Core prepared and sequenced the amplicon libraries. Extracted DNA was amplified with dual-index primers targeting the V4 region of the 16S rRNA gene, as previously described 43 with the following PCR conditions: 2 min at 95°C, 30 cycles x [95°C for 20 s, 55°C for 15 s, and 72°C for 5 min], followed by 72°C for 10 min. Libraries were prepared as previously described44, with minor modifications: the final library concentration was 5.5 pM and 15% PhiX spike-in was added to increase diversity. Sequencing was performed on an Illumina MiSeq using the 500 cycles MiSeq Reagent Kit V2 (catalog no. MS-102-2003) with modifications described in the Schloss MiSeq SOP 43.
Paired-end 16S V4 sequences were processed using the software Mothur 45 (version 1.40.2 for first sequencing run, version 1.42.3 for subsequent run). The Schloss MiSeq SOP43as of August 2019 was followed to reduce PCR and sequencing errors. The sequences were aligned to a reference alignment based on SILVA release 132. After pre-clustering and chimera removal with vsearch, the remaining sequences (as well as the OTUs later) were classified to RDP taxonomy46 based on RDP training set no 16 47. The bacterial 16S V4 sequences were phylotyped into genus bins to make community composition bar plots and clustered into 97% identity OTUs, with OTU abundance compiled for subsequent statistical analysis. These steps also followed the Schloss MiSeq SOP. Based on OTU abundance, PCoA was plotted and alpha diversity estimated with inverse Simpson. Differentially abundant taxa were determined with LEfSe version 1.1.2 48 ; cladograms and LDA scores of LEfSe results were plotted using the same software.
The relative abundance of bacterial OTUs grouped by oxygen sensitivity were compared by summing the relative abundance of known obligate anaerobes (Actinomyces, Bacteroides, Clostridium, Faecalibacterium, Blautia, Ruminococcus, Parabacteroides, and Bifidobacterium), facultative anaerobes (Escherichia/Shigella, Klebsiella, Salmonella, Enterococcus, Lactobacillus, and Staphylococcus), and unclassified. Differences in relative abundance between experimental groups were evaluated with Wilcoxon signed-rank tests using the ggpubr (version 0.4.0) and ggplot2 49 (version 3.3.5) R packages in R version 4.1.3. Unprocessed 16s rRNA sequencing reads were deposited at the NCBI Short Read Archive (SRA) and are accessible via BioProject PRJNA910578. The code used for analysis for 16S rRNA sequencing is available at https://doi.org/10.5281/zenodo.7401507.
Intestinal epithelial cells and intraepithelial cells isolation:
Luminal contents from dissected colon and ileum were flushed with CMF buffer; Ca2+/Mg2+ free HBSS (14185052, Thermo Fisher Scientific) supplemented with 25mM sodium bicarbonate (S6014, Sigma-Aldrich) and 2% FBS (100–106, Gemini Bio Products, USA). Intestines were then minced into 5mm pieces, washed with CMF buffer four times, transferred to CMF with 5mM EDTA (51201, Lonza), and incubated at 37°C for 40 minutes (shaking tubes every 10 minutes). Supernatants containing IECs were then transferred through 100 μM cell filter followed by incubation on ice for 10 minutes to allow sedimentation. Supernatants were again transferred through a 75 μM cell filter.
Preparation of lymphocytes single cell suspension and flow cytometry:
Systemic lymph nodes (axillary and inguinal), mesenteric lymph nodes and spleens were mechanically disrupted. Red blood cells were lysed (R7757, Sigma-Aldrich). For intra epithelial lymphocytes (IEL) isolation, intestines were minced in HBSS buffer (141850, Gibco) supplemented with 2.5% heat-inactivated FBS (100–106, Gibco) (HBSS+) and washed with magnetic stirring at 37°C. Intestine pieces were then incubated in HBSS+/1 mM DTT (DTT, Gold Biotechnology) at 37°C followed by additional washes and incubation in HBSS+/1mM EDTA (51201, Lonza). The supernatant was then layered on a 75%/40% Percoll Plus (17–5445, GE Healthcare) gradient to collect enriched IELs.
Single cell suspensions were resuspended in FACS wash buffer (2% bovine serum albumin in PBS). Cells were stained with conjugated monoclonal antibodies (mAbs): fluorescein isothiocyanate (FITC)–conjugated mAbs to CD8+ (100705, clone 53–6.7, BioLegend), IL-17A (506907, clone TC11–18H10.1, BioLegend), CD25 (101907, clone 3c7, BioLegend), CD11c (117305, clone N418, BioLegend) and CD8+0 (104705, clone 16–10A1, Biolegend); phycoerythrin (PE)-conjugated mAbs to CD62L (104407, clone MEL-14, BioLegend), CD69 (104507, clone H1.2F3, BioLegend), IFNγ (505807, clone XMG1.2,BioLegend), FoxP3 (126403, clone MF-14, BioLegend), CD4+0 (553791, clone 3/23, BD Pharmingen), CD274 (124307, PD-L1, clone 10F.9G2, BioLegend), IL-6 (504503, clone MP5–20F3, BioLegend); PerCP-Cy5.5 to CD4+5.2 (109827, clone 104, BioLegend); APC-conjugated mAbs to mouse CD4+4 (103011, clone IM7, BioLegend), CD279 (PD-1, 135209, clone 29F.1.A12, BioLegend), IL4 (504105, clone 11B11, BioLegend), RORγt (17-6988-82, clone AFKJS-9, Invitrogen), CD11c (117309, clone N418, BioLegend), TNFα (506307, clone MP6-XT22, BioLegend); and APC-Cy7 to CD4+ (100413, clone GK1.5, BioLegend). For cytokine analysis, cells were treated with RPMI containing 10% FBS, phorbol 12-myr-istate 13-acetate (PMA) (10 ng/mL, P1585, Sigma-Aldrich)/Ionomycin (1mM, I3909, Sigma-Aldrich) cocktail or lipopolysaccharide (LPS, 500ng/mL, L2654, Sigma-Aldrich) in the presence of protein transport inhibitor cocktail (X500, 00-4980-03, eBioscience) at 37°C for 6 hours. For intracellular staining, cells were fixed with FoxP3 staining buffer set (FoxP3 and RORγt) or IC fixation buffer (for cytokines, 00-8222-49, eBioscience) and permeabilized with permeabilization buffer (00-8333-56, eBioscience) according to the manufacturer protocol. Cells were analyzed using the Attune NxT flow cytometer.
Immunoblot analysis:
Isolated mitochondria or IECs were lysed in RIPA buffer (89901, Thermo Scientific). Equal amounts of proteins were loaded on 4–12% SDS-PAGE gel (NP0321, Invitrogen), electrophoresed and subsequently transferred to a PVDF membrane (ISEQ85R, Millipore) using a Bio-Rad semi-dry transfer cell (20 V, 1 h). Blots were incubated with anti-HIF1a (GTX127309, polyclonal, 1:1000, GeneTex), anti-PHD3 (NB100–139, 1:1000, Novus Biologicals), and anti-β actin (8226, mAbcam8226, 1:3000, Abcam) primary antibodies overnight at 4oC. Incubation with secondary anti-rabbit-HRP (7074S, Cell Signaling Technology) was performed at room temperature for 1 hour. Bound antibody was detected using Super Signal ECL substrate (32106, Thermo Scientific) and quantitated using ChemiDoc MP Imaging system (BioRad). Densitometric analysis was performed using Image J 50 (v 1.53c).
Immunohistochemistry staining:
For Hif1α staining, colonic tissues were processed, embedded in paraffin, and cut into 5 μm sections. Slides were de-paraffinized, and heat-induced antigen retrieval was performed with 10 mM sodium citrate buffer. Endogenous peroxidases were quenched with 3.0% hydrogen peroxide for 15 min. Primary anti-Hif1 α (NB100–479, Novus Biologicals) was diluted 1:200 in PBST containing 10% goat serum (16210064, Thermo Fisher Scientific) and incubated for 60 min at room temperature. Bound anti-body was detected using an anti-rabbit HRP labeled polymer (ab214880, abcam) incubated for 30 min and ImmPACT DAB (SK-4105, VECTOR labolatories). Slides were then counterstained with hematoxylin, dehydrated, and covered. For hypoxia staining, recipient mice were administered pimonidazole (PMDZ, HP7) from Hypoxyprobe, Inc. by intraperitoneal injection 30 min prior to sacrifice. Colon and ileum samples were paraffin-embedded and stained according to the manufacturer’s instructions and counterstained with DAPI (P36931, Thermo Scientific) 51 .
Seahorse analysis:
IECs were resuspended with complete seahorse XF assay medium (103335–100, Aglient) with 17.5 mM glucose (G7021, Sigma-Aldrich), 1 mM sodium pyruvate (S8636, Sigma-Aldrich), 2 mM glutamine (GLL02, Caisson Labs), 2 %BSA (BP1600–100, Fisher Scientific), 10uM Y-27632 and 1% penicillin-streptomycin (516106, Sigma-Aldrich) adjusted to pH 7.4. Cells were plated at 8 × 104 cells per well in a Seahorse assay plate, pretreated with matrigel (354262, Corning). Cells were equilibrated to 37 °C for 30 min before assay. Respiration profile was assessed in 96XF instrument with Mitostress assay as indicated upon cell treatment with 5 μM FCCP (ab120081, abcam), 7.5μM oligomycin A (75351, Sigma-Aldrich), 4μM Rotenone (3616, Tocris), and 4μM antimycin A (A8674, Sigma-Aldrich). Seahorse Wave Desktop Software (version 2.6.1.53) was used for data analysis.
Measurements of oxygen concentration in colonic mucosa:
C57BL/6 mice post HCT day7 and day 21 were subject to be determined oxygen concentration levels in the colonic mucosa. The oxygen Pst1 optode microsensors (Presens) at the tip of a fiber optic cable were used. The optode was inserted into the mouse’s colon at a depth of one to three cm via an endoscope (Karl Storz). Before oxygen measurements, mice were anesthetized with 4% isoflurane and then received 2% isoflurane during the oxygen measurement. The colon was insufflated with nitrogen gas to expunge both extraneous oxygen and oxygen that had leached into the rectum when insertion of the endoscope. Visual assessments of when the microsensor contacted with the colonic mucosa using a camera attached to the endoscope. Oxygen concentrations were recorded with a PreSens Microx TX3 Trace Micro fiber optic O2 transmitter and Presens’ Oxyview TX3 v. 5.31 software using default parameters. Prior to measurements, a two-point calibration of the microsensor was conducted with air-saturated water and 100 ml of anoxic water that was attained by the addition of one gram of sodium sulfite (Acros Organics) and 50 μL of 500 mM cobalt nitrate solution (Acros Organics). Mouse body was kept on a 37˚C heating pad with a Gaymar T-pump system. To prepare the colon for imaging, the colon was flushed with water. Oxygen concentration readings from the mucosa that remained stable for at least 30 seconds were recorded. Readings from three to five locations in the colon were then obtained for each mouse; readings from each location were subsequently averaged to obtain a mean oxygen concentration for each mouse. 52.
RNA isolation and RT-PCR:
RNA isolation and RT-PCR: Total RNA from single-cell suspensions was isolated using the RNeasy Kit (74104, QIAGEN) and reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (4374966, Applied Biosystems). The following primers and PowerUP SYBR green polymerase were used to detect the following transcripts: ; 5′- TGACCTCAACTACATGGTCTACA-3′ and 5′-CTTCCCATTCTCGGCCTTG-3′ (Gapdh), 5′- CAGTCACCTGGTTGCTGCAA -3′ and 5′-CAGTCACCTGGTTGCTGCAA -3′ (Hif1α), 5′- TGCTGAAGAAAGGGCAGAAG -3′ and 5′-GCACACCACAGTCAGTCTTTA-3′ (Egln: Phd3). All reactions were performed according to manufacturer’s instructions. All primers were verified for the production of a single specific PCR product via melting curve analysis
Quantification of iron in intestinal content:
The intestinal content in colon and ileum from allogeneic recipient mice were collected after euthanized. Then, the intestinal contents were processed according to the manufacturer`s instruction of Iron Assay Kit (MAK025, Sigma-Aldrich).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was performed using Graph Pad Prism (v8.0.0, Graph Pad Software Inc) and Excel2016 (version2105) to do the graph figures and statistics. P values <0.05 were considered as significant: P values >0.05 were considered as non-significant (* p<0.05, ** p<0.01, *** p<0.001 and **** p<0.0001). All sample sizes and statistical tests used are detailed in each figure legend. All replicates are biological replicates. No data were excluded. All experiments in vitro and in vivo were performed twice or more than twice. Data shown as ±SEM according to figure legend.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD90.2 MicroBeads, mouse | Miltenyi Biotec | Cat# 130-121-278 |
| CD5 (Ly-1) MicroBeads, mouse | Miltenyi Biotec | Cat# 130-049-301 |
| DAPI | BioLegend | Cat# 422801 |
| Rat Anti-Mouse CD4 Antibody, APC-H7 Conjugated | BD Biosciences | Cat# 560246, RRID:AB_1645236 |
| APC anti-mouse CD25 | BioLegend | Cat# 101910, RRID: AB_2280288 |
| FITC anti-mouse/human CD44 antibody | BioLegend | Cat# 103006, RRID:AB_312957 |
| PE anti-mouse CD45RB antibody | BioLegend | Cat# 103308, RRID:AB_313015 |
| FITC anti-mouse CD8a antibody | BioLegend | Cat# 100705, RRID:AB_312744 |
| FITC anti-mouse IL-17A antibody | BioLegend | Cat# 506907, RRID:AB_536009 |
| FITC anti-mouse CD25 antibody | BioLegend | Cat# 101907, RRID:AB_961210 |
| FITC anti-mouse CD11c antibody | BioLegend | Cat# 117305, RRID:AB_313774 |
| FITC anti-mouse CD80 antibody | BioLegend | Cat# 104705, RRID:AB_313126 |
| PE anti-mouse CD62L antibody | BioLegend | Cat# 104407, RRID:AB_313094 |
| PE anti-mouse CD69 antibody | BioLegend | Cat# 104507, RRID:AB_313110 |
| PE anti-mouse IFN-gamma antibody | BioLegend | Cat# 505807, RRID:AB_315401 |
| PE anti-mouse FOXP3 antibody | BioLegend | Cat# 126403, RRID:AB_1089118 |
| Rat Anti-CD40 Monoclonal Antibody, Phycoerythrin Conjugated, Clone 3/23 | BD Biosciences | Cat# 553791, RRID:AB_395055 |
| PE anti-mouse CD274 (B7-H1, PD-L1) antibody | BioLegend | Cat# 124307, RRID:AB_2073557 |
| PE anti-mouse IL-6 antibody | BioLegend | Cat# 504503, RRID:AB_315337 |
| PerCP/Cyanine5.5 anti-mouse CD45.2 antibody | BioLegend | Cat# 109827, RRID:AB_893352 |
| APC anti-mouse/human CD44 antibody | BioLegend | Cat# 103011, RRID:AB_312962 |
| APC anti-mouse CD279 (PD-1) antibody | BioLegend | Cat# 135209, RRID:AB_2251944 |
| APC anti-mouse IL-4 antibody | BioLegend | Cat# 504105, RRID:AB_315319 |
| ROR gamma (t) Monoclonal Antibody (AFKJS-9), APC, | Thermo Fisher Scientific | Cat# 17-6988-82, RRID:AB_10609207 |
| APC anti-mouse CD11c antibody | BioLegend | Cat# 117309, RRID:AB_313778 |
| APC anti-mouse TNF-alpha antibody | BioLegend | Cat# 506307, RRID:AB_315428 |
| APC/Cyanine7 anti-mouse CD4 antibody | BioLegend | Cat# 100413, RRID:AB_312698 |
| HIF1A-human antibody | GeneTex | Cat# GTX127309, RRID:AB_2616089) |
| HIF Prolyl Hydroxylase 3 Antibody | Novus Biologicals | Cat# NB100-139, RRID:AB_2096716 |
| Mouse Anti-Actin, beta Monoclonal Antibody, Unconjugated, Clone mAbcam 8226 | Abcam | Cat# ab8226, RRID:AB_306371 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7074, RRID:AB_2099233 |
| HIF-1 alpha Antibody | Novus Biologicals | Cat# NB100-479, RRID:AB_10000633 |
| Goat Anti-Rabbit IgG H&L (HRP polymer) | abcam | Cat# ab214880 |
| Chemicals, peptides, and recombinant proteins | ||
| Ampicillin | Sigma Aldrich | Cat# A9393 |
| Kanamycin sulfate | Sigma Aldrich | Cat# 60615 |
| Metronidazole | Sigma Aldrich | Cat# M1547 |
| Vancomycin | Sigma Aldrich | Cat# SBR00001 |
| Neomycin | Sigma Aldrich | Cat# N1876 |
| Deferasirox | Sigma Aldrich | Cat# SML2673-50 |
| HBSS (10X), no calcium, no magnesium, no phenol red | Gibco |
Cat# 14185052 |
| Sodium bicarbonate | Sigma Aldrich | Cat# S6014 |
| FBS | GeminiBio | Cat# 100-106 |
| 0.5 M EDTA Solution | Lonza | Cat# 51201 |
| Red blood cell lysing buffer | Sigma Aldrich | Cat# R7757 |
| DTT | Gold Biotechnology | Cat# DTT |
| Percoll PLUS | GE Healthcare | Cat# 17-5445 |
| PMA | Sigma Aldrich | Cat# P1585 |
| Ionomycin | Sigma Aldrich | Cat# I3909 |
| LPS | Sigma Aldrich | Cat# L2654 |
| Protein Transport Inhibitor Cocktail | eBioscience | Cat# 00-4980-03 |
| IC Fixation Buffer | eBioscience | Cat# 00-8222-49 |
| Permeabilization Buffer (10X) | eBioscience | Cat# 00-8333-56 |
| RIPA Lysis and Extraction Buffer | Thermo Scientific | Cat# 89901 |
| ECL Western Blotting Substrate | Thermo Scientific | Cat# 32106 |
| Goat Serum, New Zealand origin | Gibco | Cat# 16210064 |
| ImmPACT® DAB Substrate Kit, Peroxidase (HRP) | VECTOR labolatories | Cat# SK-4105 |
| ProLong™ Gold Antifade Mountant with DAPI | Invitrogen | Cat# P36931 |
| Seahorse XF Media | Agilent | Cat# 102353-100 |
| D-(+)-Glucose | Sigma Aldrich | Cat# G7021 |
| FCCP | abcam | Cat# ab120081 |
| Oligomycin A | Sigma Aldrich | Cat# 75351 |
| Rotenone | Tocris | Cat# 3616 |
| Antimycin A from Streptomyces sp. | Sigma Aldrich | Cat# A8674 |
| Critical commercial assays | ||
| MagAttract PowerMicrobiome DNA/RNA EP | QIAGEN | Cat# 27500-4-EP |
| Quant-iT™ PicoGreen™ dsDNA Assay Kits and dsDNA Reagents | Invitrogen | Cat# P7589 |
| MiSeq Reagent Kits v2 | illumina | Cat# MS-102-2003 |
| NuPAGE™ 4 to 12%, Bis-Tris, 1.0-1.5 mm, Mini Protein Gels | Invitrogen | Cat# NP0321BOX |
| Immobilon®-PSQ Membrane, PVDF, 0.2 m, 8.5 cm x 10 m roll | Millipore | Cat# ISEQ85R |
| Hypoxyprobe-Red549 Kit | Hypoxyprobe | Cat# HP7 |
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor | Applied Biosystems | Cat# 4374966 |
| Iron Assay Kit | Sigma Aldrich | Cat# MAK025 |
| Deposited data | ||
| Code used for analysis for 16S rRNA sequencing: https://doi.org/10.5281/zenodo.7401507. | zenodo | https://doi.org/10.5281/zenodo.7401507. |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Charles River Laboratories | Cat# 027, RRID:IMSR_CRL:02 7 |
| Mouse: BALB/c | Charles River Laboratories | Cat# 028, RRID:IMSR_CRL:02 8 |
| Mouse: BDF1 | Charles River Laboratories | Cat# 099, RRID:IMSR_CRL:09 9 |
| Mouse: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratories | Cat# 002216, RRID:IMSR_JAX:00 2216 |
| Mouse: BDF1 | The Jackson Laboratories | Cat# 100006, RRID:IMSR_JAX:10 0006 |
| Mouse: B6.Cg-Tg(Vil1-cre)1000Gum/J | The Jackson Laboratories | Cat# 021504, RRID:IMSR_JAX:02 1504 |
| Mouse: 129S1/SvImJ | The Jackson Laboratories | Cat# 002448, RRID:IMSR_JAX:00 2448 |
| Mouse: C57BL/6NTac | Taconic Biosciences | Cat# B6 F, RRID:IMSR_TAC:b6 |
| Mouse: BALB/cAnNTac | Taconic Biosciences | Cat# BALB-F, RRID:IMSR_TAC:bal b |
| Mouse: Hif1afl/fl | Gift from Yatrik M. Shah | N/A |
| Oligonucleotides | ||
| RNA sequence, mouse Gapdh:5′- TGACCTCAACTACATGGTCTACA-3′ and 5′- CTTCCCATTCTCGGCCTTG-3′ |
This paper | N/A |
| RNA sequence, mouse Hif1α:5′- CAGTCACCTGGTTGCTGCAA -3′ and 5′- CAGTCACCTGGTTGCTGCAA -3′ |
This paper | N/A |
| RNA sequence, mouse Egln(Phd3):5′- TGCTGAAGAAAGGGCAGAAG -3′ and 5′- GCACACCACAGTCAGTCTTTA-3′ |
This paper | N/A |
| Software and algorithms | ||
| FlowJo | BD |
https://www.flowjo.com, RRID:SCR_008520, version 10.2 |
| mothur | Schloss et al., 200945 |
https://mothur.org/, RRID:SCR_011947, version 1.40.2 and 1.42.3 |
| LEfSe | Segata et al., 201148 |
http://huttenhower.sph.harvard.edu/galaxy , RRID:SCR_014609, version 1.1.2 |
| ggpubr | https://cloud.r-project.org/web/packages/ggpubr/index.html |
https://CRAN.R-project.org/package=ggpubr, RRID:SCR_021139, version 0.4.0 |
| ggplot2 | Wickham, 2016 49 |
https://cran.r-project.org/web/packages/ggplot2/index.html, RRID:SCR_014601v ersion 3.3.5 |
| R | R Foundation |
http://www.r-project.org/, RRID:SCR_001905, version 4.1.3 |
| ImageJ | Schindelin et al., 201250 |
https://imagej.net/software/fiji/, RRID:SCR_003070 ,version 1.53c |
| Seahorse Wave Desktop Software | Agilent |
https://www.agilent.com/en/products/cell-analysis/software-download-for-wave-desktop, RRID:SCR_014526, version 2.6.1.53 |
| Graph Pad Prism | Graph Pad Software Inc |
http://www.graphpad.com/, RRID:SCR_002798, version 8.0.0, |
| Excel2016 | Microsoft |
https://www.microsoft.com/en-us/microsoft-365/excel, RRID:SCR_016137, version 2105 |
Highlights.
Allogeneic intestinal dysbiosis is not pathogenic to naïve animals
Loss of intestinal physiologic hypoxia causes dysbiosis
The defect in oxygen utilization in IECs leads to a loss of intestinal hypoxia
Oxygen modulation after allo-HSCT improves intestinal hypoxia and GVHD
Acknowledgments:
This work was supported by the US National Institutes of Health grants P01HL149633 (P.R. and G.D.), HL152605 (P.R), CA217156 (P.R.), R01CA148828 (Y.M.), R01CA245546 (Y.M.), and R01DK095201 (Y.M.). This work in Memorial Sloan Kettering Cancer Center was supported by National Cancer Institute award numbers, R01-CA228358, R01-CA228308, P30 CA008748 MSK Cancer Center Support Grant/Core Grant and P01-CA023766 ; National Heart, Lung, and Blood Institute (NHLBI) award number R01-HL123340 and R01-HL147584; and TriInstitutional Stem Cell Initiative (M.R.M. vdB.).
We acknowledge use of the Microbiome Core, the Microscopy & Image-analysis Laboratory (MIL) and Germ-free facility of the University of Michigan’s Biomedical Research Core Facilities for the preparation of samples and images. Support for the MIL core is provided by the University of Michigan Rogel Cancer Center (NIH grant CA46592).
Footnotes
Declaration of Interests: Authors declare that they have no competing interests.
Inclusion and Diversity Statement:
In all the key murine experiments we worked to ensure sex balance in the selection of non-human subjects. Our authors list is multi-racial, but none of the authors of this paper self-identify as a member of a minority community. While citing references scientifically relevant for this work, we actively worked to promote gender balance in our reference list.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Litvak Y, Byndloss MX, and Baumler AJ. (2018). Colonocyte metabolism shapes the gut microbiota. Science 362. 10.1126/science.aat9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byndloss MX, Pernitzsch SR, and Baumler AJ. (2018). Healthy hosts rule within: ecological forces shaping the gut microbiota. Mucosal Immunol 11, 1299–1305. 10.1038/s41385-018-0010-y. [DOI] [PubMed] [Google Scholar]
- 3.Shono Y, Docampo MD, Peled JU, Perobelli SM, Velardi E, Tsai JJ, Slingerland AE, Smith OM, Young LF, Gupta J, et al. (2016). Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med 8, 339ra371. 10.1126/scitranslmed.aaf2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peled JU, Gomes ALC, Devlin SM, Littmann ER, Taur Y, Sung AD, Weber D, Hashimoto D, Slingerland AE, Slingerland JB, et al. (2020). Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N Engl J Med 382, 822–834. 10.1056/NEJMoa1900623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caruso R, Lo BC, and Nunez G. (2020). Host-microbiota interactions in inflammatory bowel disease. Nat Rev Immunol 20, 411–426. 10.1038/s41577-019-0268-7. [DOI] [PubMed] [Google Scholar]
- 6.Halfvarson J, Brislawn CJ, Lamendella R, Vazquez-Baeza Y, Walters WA, Bramer LM, D'Amato M, Bonfiglio F, McDonald D, Gonzalez A, et al. (2017). Dynamics of the human gut microbiome in inflammatory bowel disease. Nat Microbiol 2, 17004. 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeiser R, and Blazar BR. (2017). Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med 377, 2167–2179. 10.1056/NEJMra1609337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bowerman KL, Varelias A, Lachner N, Kuns RD, Hill GR, and Hugenholtz P. (2020). Continuous pre- and post-transplant exposure to a disease-associated gut microbiome promotes hyper-acute graft-versus-host disease in wild-type mice. Gut Microbes 11, 754–770. 10.1080/19490976.2019.1705729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shono Y, and van den Brink MRM. (2018). Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat Rev Cancer 18, 283–295. 10.1038/nrc.2018.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Bekkum DW, Roodenburg J, Heidt PJ, and van der Waaij D. (1974). Mitigation of secondary disease of allogeneic mouse radiation chimeras by modification of the intestinal microflora. J Natl Cancer Inst 52, 401–404. 10.1093/jnci/52.2.401. [DOI] [PubMed] [Google Scholar]
- 11.Espey MG. (2013). Role of oxygen gradients in shaping redox relationships between the human intestine and its microbiota. Free Radic Biol Med 55, 130–140. 10.1016/j.freeradbiomed.2012.10.554. [DOI] [PubMed] [Google Scholar]
- 12.Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, et al. (2014). Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology 147, 1055–1063 e1058. 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, and Bultman SJ. (2011). The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13, 517–526. 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blouin JM, Penot G, Collinet M, Nacfer M, Forest C, Laurent-Puig P, Coumoul X, Barouki R, Benelli C, and Bortoli S. (2011). Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int J Cancer 128, 2591–2601. 10.1002/ijc.25599. [DOI] [PubMed] [Google Scholar]
- 15.Colgan SP, and Taylor CT. (2010). Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol 7, 281–287. 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, Oravecz-Wilson K, Wu SR, Sun Y, Rossi C, et al. (2016). Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol 17, 505–513. 10.1038/ni.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujiwara H, Docampo MD, Riwes M, Peltier D, Toubai T, Henig I, Wu SJ, Kim S, Taylor A, Brabbs S, et al. (2018). Microbial metabolite sensor GPR43 controls severity of experimental GVHD. Nat Commun 9, 3674. 10.1038/s41467-018-06048-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al. (2015). Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 17, 662–671. 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byndloss MX, and Baumler AJ. (2018). The germ-organ theory of non-communicable diseases. Nat Rev Microbiol 16, 103–110. 10.1038/nrmicro.2017.158. [DOI] [PubMed] [Google Scholar]
- 20.Litvak Y, Byndloss MX, Tsolis RM, and Baumler AJ. (2017). Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol 39, 1–6. 10.1016/j.mib.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Fujiwara H, Seike K, Brooks MD, Mathew AV, Kovalenko I, Pal A, Lee HJ, Peltier D, Kim S, Liu C, et al. (2021). Mitochondrial complex II in intestinal epithelial cells regulates T cell-mediated immunopathology. Nat Immunol 22, 1440–1451. 10.1038/s41590-021-01048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stein-Thoeringer CK, Nichols KB, Lazrak A, Docampo MD, Slingerland AE, Slingerland JB, Clurman AG, Armijo G, Gomes ALC, Shono Y, et al. (2019). Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 366, 1143–1149. 10.1126/science.aax3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, Liu C, West ML, Singer NV, Equinda MJ, et al. (2012). Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med 209, 903–911. 10.1084/jem.20112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glover LE, Lee JS, and Colgan SP. (2016). Oxygen metabolism and barrier regulation in the intestinal mucosa. J Clin Invest 126, 3680–3688. 10.1172/JCI84429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ibrahim AS, Gebermariam T, Fu Y, Lin L, Husseiny MI, French SW, Schwartz J, Skory CD, Edwards JE Jr., and Spellberg BJ. (2007). The iron chelator deferasirox protects mice from mucormycosis through iron starvation. J Clin Invest 117, 2649–2657. 10.1172/JCI32338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Packey CD, Shanahan MT, Manick S, Bower MA, Ellermann M, Tonkonogy SL, Carroll IM, and Sartor RB. (2013). Molecular detection of bacterial contamination in gnotobiotic rodent units. Gut Microbes 4, 361–370. 10.4161/gmic.25824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toubai T, Fujiwara H, Rossi C, Riwes M, Tamaki H, Zajac C, Liu C, Mathew AV, Byun J, Oravecz-Wilson K, et al. (2019). Host NLRP6 exacerbates graft-versus-host disease independent of gut microbial composition. Nat Microbiol 4, 800–812. 10.1038/s41564-019-0373-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, and Kasper DL. (2020). Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 577, 410–415. 10.1038/s41586-019-1865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato Y, Atarashi K, Plichta DR, Arai Y, Sasajima S, Kearney SM, Suda W, Takeshita K, Sasaki T, Okamoto S, et al. (2021). Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 599, 458–464. 10.1038/s41586-021-03832-5. [DOI] [PubMed] [Google Scholar]
- 30.Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin WB, Guo CJ, Violante S, et al. (2020). Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479. 10.1038/s41586-020-2193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, Maeda Y, Tawara I, Krijanovski O, Gatza E, et al. (2008). Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest 118, 2562–2573. 10.1172/JCI34712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toubai T, Hou G, Mathewson N, Liu C, Wang Y, Oravecz-Wilson K, Cummings E, Rossi C, Evers R, Sun Y, et al. (2014). Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood 123, 3512–3523. 10.1182/blood-2013-12-545335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamburini FB, Andermann TM, Tkachenko E, Senchyna F, Banaei N, and Bhatt AS. (2018). Precision identification of diverse bloodstream pathogens in the gut microbiome. Nat Med 24, 1809–1814. 10.1038/s41591-018-0202-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semenza GL. (2012). Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408. 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, and Haase VH. (2004). Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 114, 1098–1106. 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, and Gonzalez FJ. (2008). Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology 134, 2036–2048, 2048 e2031–2033. 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu M, Tao J, Yang Y, Tan S, Liu H, Jiang J, Zheng F, and Wu B. (2020). Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis 11, 86. 10.1038/s41419-020-2299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu S, He Y, Lin L, Chen P, Chen M, and Zhang S. (2021). The emerging role of ferroptosis in intestinal disease. Cell Death Dis 12, 289. 10.1038/s41419-021-03559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellermann M, and Arthur JC. (2017). Siderophore-mediated iron acquisition and modulation of host-bacterial interactions. Free Radic Biol Med 105, 68–78. 10.1016/j.freeradbiomed.2016.10.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah YM, Matsubara T, Ito S, Yim SH, and Gonzalez FJ. (2009). Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab 9, 152–164. 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J Jr., and Ferrara JL. (1998). Tumor necrosis factor-alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest 102, 1882–1891. 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill GR, Cooke KR, Teshima T, Crawford JM, Keith JC Jr., Brinson YS, Bungard D, and Ferrara JL. (1998). Interleukin-11 promotes T cell polarization and prevents acute graft-versus-host disease after allogeneic bone marrow transplantation. J Clin Invest 102, 115–123. 10.1172/JCI3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, and Schloss PD. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79, 5112–5120. 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koenigsknecht MJ, Theriot CM, Bergin IL, Schumacher CA, Schloss PD, and Young VB. (2015). Dynamics and establishment of Clostridium difficile infection in the murine gastrointestinal tract. Infect Immun 83, 934–941. 10.1128/IAI.02768-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541. 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Garrity GM, Tiedje JM, and Cole JR. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267. 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, and Tiedje JM. (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42, D633–642. 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C. (2011). Metagenomic biomarker discovery and explanation. Genome Biol 12, R60. 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wickham H. (2016). ggplot2 : Elegant Graphics for Data Analysis. Use R!,. 2nd ed. Springer International Publishing; : Imprint: Springer,. [Google Scholar]
- 50.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, et al. (2014). Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40, 66–77. 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim YG, Sakamoto K, Seo SU, Pickard JM, Gillilland MG 3rd, Pudlo NA, Hoostal M, Li X, Wang TD, Feehley T, et al. (2017). Neonatal acquisition of Clostridia species protects against colonization by bacterial pathogens. Science 356, 315–319. 10.1126/science.aag2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed 16s rRNA sequencing reads were deposited at the NCBI Short Read Archive (SRA) and are accessible via BioProject PRJNA910578.
The code used for analysis for 16S rRNA sequencing is available at https://doi.org/10.5281/zenodo.7401507.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.