

Summary
The ch12q13 locus is among the most significant childhood obesity loci identified in genome-wide association studies. This locus resides in a non-coding region within FAIM2; thus, the underlying causal variant(s) presumably influence disease susceptibility via cis-regulation. We implicated rs7132908 as a putative causal variant by leveraging our in-house 3D genomic data and public domain datasets. Using a luciferase reporter assay, we observed allele-specific cis-regulatory activity of the immediate region harboring rs7132908. We generated isogenic human embryonic stem cell lines homozygous for either rs7132908 allele to assess changes in gene expression and chromatin accessibility throughout a differentiation to hypothalamic neurons, a key cell type known to regulate feeding behavior. The rs7132908 obesity risk allele influenced expression of FAIM2 and other genes and decreased the proportion of neurons produced by differentiation. We have functionally validated rs7132908 as a causal obesity variant that temporally regulates nearby effector genes and influences neurodevelopment and survival.
Keywords: childhood obesity, obesity, genome-wide association study, risk variant, hypothalamus, stem cells, neuron differentiation
Graphical abstract
Highlights
-
•
rs7132908 is a causal variant at the chr12q13 obesity locus
-
•
rs7132908 regulates nearby effector genes with allele and cell-type specificity
-
•
Obesity risk allele decreases generation of neurons that regulate appetite
A locus on chr12q13 is strongly associated with childhood obesity by genome-wide associate studies. Littleton et al. identified a causal variant at this locus, which regulates gene expression in neural cell types. The obesity risk allele also decreased the proportion of appetite-regulating hypothalamic neurons generated by stem cell differentiation.
Introduction
Childhood obesity affects approximately 14.7 million individuals aged 2–19 years in the United States, corresponding to approximately one in five children and adolescents.1 The global prevalence of childhood obesity has increased substantially, rising from less than 1% to more than 7% in recent decades.2 Obesity increases the risk of leading causes of poor health and early death via hypertension, metabolic disorders, cardiovascular disease, and common cancers.3 Common cases of obesity result from both environmental and genetic factors.4 The genetic component explains a large portion of obesity risk, with heritability estimates ranging from 40% to 85%,5 but remains incompletely understood. However, it is known that neuronal pathways in the hypothalamus control food intake and are key regulators for obesity.4 Several human stem cell-derived hypothalamic neuron models have been developed6,7,8,9,10 to investigate the molecular basis of body weight regulation.6,10,11,12,13,14,15,16,17,18
Genome-wide association studies (GWAS) have identified genomic regions that harbor susceptibility variants conferring adult19,20 and childhood obesity21,22,23,24 risk. An ongoing challenge is to translate GWAS loci into meaningful discoveries that can expand our knowledge of complex traits. Most variants identified by GWASs are non-coding, so their underlying mechanism is not obvious.4 These non-coding variants likely influence disease risk by functioning within cis-regulatory elements and altering expression of effector genes within their topologically associating domain (TAD). These effector genes are not necessarily the most proximal gene, as cis-regulatory elements can influence gene expression up to megabases away. Therefore, functional characterization must be conducted to determine which variants are causal and which effector genes, near or far, confer susceptibility to disease. Most attention has been focused on only the very strongest GWAS loci, such as FTO,25,26,27 while many other loci that rank lower in the signal list remain understudied.
Our latest childhood obesity trans-ancestral GWAS meta-analysis identified a locus on chr12q13 named after its nearest gene, FAIM2.21,22 This signal has also been independently reported for obesity risk in children28,29 and adults30,31,32,33 across several ancestral populations. Crucially, this locus is more pronounced in children and ranks among more well-studied loci such as FTO, MC4R, TMEM18, and BDNF in the pediatric setting22; as such, it has been less studied given its less obvious role in adult obesity pathogenesis.
To implicate candidate causal non-coding variants at the chr12q13 obesity locus, our trans-ancestral fine-mapping refined this signal to a 99% credible set of six single nucleotide polymorphisms (SNPs).22 More recently, Bayesian fine-mapping further refined this locus to one signal with 95% credible sets of 1–4 SNPs depending on which body weight trait definition was used (e.g., maximum weight, maximum BMI, mean weight).34 These credible sets consistently implicate rs7132908 as the variant with the highest computed probability of being causal.22,34 The obesity risk A allele is common, with frequencies ranging from 10.25% to 60.53% across ethnicities35 and 28.86% globally.36 In addition to childhood obesity, this locus is also associated with related traits: increased BMI in adults, increased weight in adults, elevated type 2 diabetes susceptibility, increased body fat percentage in children and adults,37 increased risk of problematic alcohol use,38 BMI variance,39 increased waist circumference,40 and earlier age at menarche.41
We used our established variant-to-gene mapping approach that implicates potential cis-regulatory elements at GWAS loci using assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) to identify regions of accessible chromatin and high-resolution promoter-focused Capture-C/Hi-C to identify distal promoter interactions with those open regions.11,42,43,44,45,46,47 rs7132908 resides within a putative cis-regulatory element in several human neural cell types,11,46,47 consistent with data from the Encyclopedia of DNA Elements (ENCODE) consortium’s “Registry of candidate cis-Regulatory Elements” (version 3), which has annotated a cell-type-agnostic candidate distal enhancer encompassing rs7132908 (candidate cis-regulatory element EH38E3015886).48
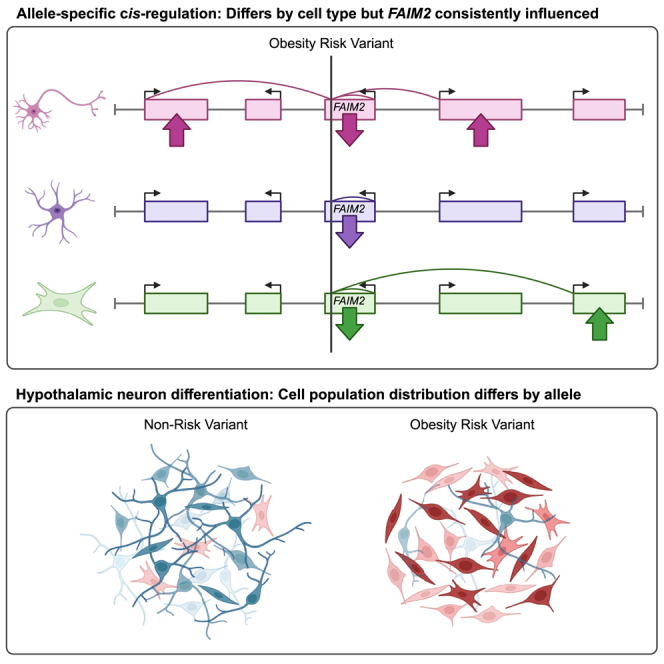
The rs7132908 region contacts promoters of FAIM2 and several other genes within its TAD.11,46,47 We nominated these genes as candidate effector genes. FAIM2 has additional support via colocalization with expression quantitative trait loci (eQTL) data49 but has not been directly implicated in obesity pathogenesis. We used reporter assays in astrocytes to characterize the cis-regulatory activity of rs7132908 and found that this variant regulated FAIM2 expression with allele specificity. Next, we generated hypothalamic neural progenitors and a heterogeneous population of hypothalamic neurons that were homozygous for either rs7132908 allele. We used bulk ATAC-seq pre-differentiation and single-nucleus ATAC-seq post-differentiation, when the cells were heterogeneous, to assess chromatin accessibility. The rs7132908 region transitioned from closed to open chromatin during differentiation from ESCs to hypothalamic neurons. We also used bulk or single-nucleus RNA-seq to characterize changes in gene expression at three time points throughout differentiation, finding that rs7132908 genotype regulated expression of FAIM2 and other genes in multiple cell types. Finally, we report the striking observation that the rs7132908 obesity risk A allele decreased the proportion of neurons from 61% to 11%. Our data strongly implicate rs7132908 as a causal variant at the chr12q13 obesity locus and nominates FAIM2 as a candidate effector gene for further study.
Results
The chr12q13 locus more strongly influences childhood BMI
The effect size of the chr12q13 locus has been shown to decrease as age increases using longitudinal data from children 3–17 years old.29 We sought to determine if the association between this locus and childhood BMI is stronger than adult BMI using results from the most recent childhood50 and adult20 BMI GWAS. We found that the effect of the chr12q13 locus on childhood BMI (β = 0.0704, standard error = 0.008) was significantly stronger than its effect on adult BMI (β = 0.0303, standard error = 0.0018) (p value = 1.01 × 10−6).
FAIM2 is the lead candidate effector gene at the chr12q13 childhood obesity locus
Chromosome conformation capture methods identify physical interactions between genomic regions and can nominate possible functional relationships, such as enhancer-promoter interactions. The putative cis-regulatory element harboring rs7132908 interacted variably with promoters of 11 candidate effector genes (AC025154.2, AQP2, AQP5, AQP6, ASIC1, BCDIN3D, FAIM2, LIMA1, LINC02395, LINC02396, and RACGAP1) across neural cell types and their progenitors (Figure 1A; Table S1),11,46,47 suggesting potential temporal and cell-type-specific control of multiple genes in the region, similar to the FTO locus.25,26,27 We performed colocalization analysis to intersect eQTL signals from the Genotype-Tissue Expression (GTEx) project with our variant-to-gene mapping results.49 With the conservative overlap of the two approaches, we found that only FAIM2 was implicated by both analyses (Table S1). We therefore nominated FAIM2 as the primary candidate effector gene at this locus.
Figure 1.

rs7132908 regulates FAIM2 expression with allele and cell-type specificity
(A) Chromatin accessibility represented by ATAC-seq tracks depicting normalized reads and chromatin loops at the TAD containing rs7132908 in neural cell types. Chromatin loops represent significant contacts between regions of open chromatin that harbored rs7132908 and a gene promoter. Gray dashed vertical line indicates rs7132908 position.
(B) Graphic representation of firefly luciferase reporter vectors used in luciferase reporter assays.
(C‒F) Fold change of firefly luciferase fluorescence normalized to the promoter only control vector driven by the FAIM2 promoter in primary astrocytes (n = 7 biological replicates) (C), FAIM2 promoter in HEK293Ts (n = 7 biological replicates) (D), LIMA1 promoter in primary astrocytes (n = 8 biological replicates) (E), and RACGAP1 promoter in primary astrocytes (n = 9 biological replicates) (F). Data are represented as mean ± SD. ∗p value <0.05, ∗∗p value <0.01, ∗∗∗p value <0.001 by one-way ANOVA with Tukey’s correction for multiple comparisons.
We sought to identify if loss-of-function mutations in any of the coding, candidate effector genes (AQP2, AQP5, AQP6, ASIC1, BCDIN3D, FAIM2, LIMA1, and RACGAP1) are associated with cases of obesity in the Penn Medicine BioBank. We found that none were significantly associated with obesity in either European or African populations. We note that there were relatively few rare coding variants in FAIM2 and no individuals were homozygous for FAIM2 mutations.
Hypothalamic neurons and astrocytes are relevant in vitro models to study the effects of rs7132908 genotype
rs7132908 is in the 3′ untranslated region (UTR) of FAIM2 and 34,612 base pairs (bp) from the FAIM2 transcription start site. The interaction between rs7132908 and the FAIM2 promoter was observed in three neural cell types: primary astrocytes, iPSC-derived cortical neural progenitors, and ESC-derived hypothalamic neurons (Figure 1A; Table S1).11,46,47 We measured gene expression to aid in prioritizing in vitro models for our study. FAIM2 expression was 2.26 transcripts per million (TPM) in iPSC-derived cortical neural progenitors, 42.85 TPM in primary astrocytes, and 136.75 TPM in ESC-derived hypothalamic neurons (Table S1).11,46,47 We previously identified that BMI-associated variants are significantly enriched in cis-regulatory elements in a hypothalamic neuron model.11 While this significant enrichment has not been detected in primary astrocytes,47 seven of nine candidate effector genes (AC025154.2, AQP5, AQP6, FAIM2, LINC02395, LINC02396, and RACGAP1) nominated at the chr12q13 locus in ESC-derived hypothalamic neurons were also nominated in primary astrocytes (Figure 1A; Table S1), suggesting similar genomic architecture in this region in these two cellular settings. Therefore, ESC-derived hypothalamic neurons and primary astrocytes were selected as in vitro models for studying the putative cis-regulatory relationship between rs7132908 and genes within its TAD.
rs7132908 regulates FAIM2 expression with allele and cell-type specificity
Many commonly used reporter assays to assess cis-regulatory function require a cell model that can be efficiently transfected. Neuron-like cells produced by stem cell differentiation are post-mitotic and transfection of these cells is very inefficient. For this reason, and given the comparable observations described above, we used primary astrocytes to characterize the cis-regulatory function of the region harboring rs7132908 with luciferase reporter assays. We used vectors containing either rs7132908 allele and promoters of interest and control vectors (Figure 1B).
The putative enhancer sequence with the non-risk allele significantly increased luciferase expression 1.75-fold (adjusted p value <0.001) (Figure 1C). In contrast, the same vector with a single base change to the obesity risk A allele significantly decreased luciferase expression 0.53-fold (adjusted p value = 0.003) (Figure 1C). We then used HEK293Ts to determine if this cis-regulatory activity occurs in a non-neural cell type. In HEK293Ts, the putative enhancer sequence harboring the non-risk G allele did not significantly increase luciferase expression, while the obesity risk allele decreased luciferase expression by 0.60-fold (adjusted p value = 0.037) (Figure 1D). We conclude that rs7132908 regulates expression from the FAIM2 promoter in astrocytes but displays weaker effects in non-neuronal HEK293Ts.
In addition to FAIM2, our variant-to-gene mapping efforts in primary astrocytes also nominated LIMA1 and RACGAP1 as possible effector genes (Figure 1A; Table S1). However, when we assessed the cis-regulatory activity of this region with the LIMA1 and RACGAP1 promoter sequences, we observed no significant changes in luciferase expression with either rs7132908 allele, although we note the results for the risk A allele with the RACGAP1 promoter were highly variable (Figures 1E and 1F).
Transcription factors bind at regulatory sequences, such as enhancers, and mediate the regulation of gene expression. We predicted the impact of the obesity risk A allele on transcription factor binding, identifying 12 transcription factors potentially regulating gene expression at this chr12q13 locus: HNF4A, HNF4G, PRD14, PRDM14, SRBP2, SREBF1, SREBF2, ZN143, ZN423, ZN554, ZN768, and ZNF416.
rs7132908 genotype influences gene expression in ESC-derived hypothalamic neural progenitors
After characterizing the cis-regulatory activity of rs7132908 in astrocytes, we characterized the effect of the rs7132908 childhood obesity risk allele at multiple time points throughout differentiation to hypothalamic neurons. We used the H9 ESC line, which is homozygous for the rs7132908 non-risk G allele and leveraged CRISPR-Cas9 homology-directed repair to generate three isogenic, clonal lines that were homozygous for the rs7132908 risk A allele.
To characterize chromatin accessibility in homogeneous ESCs, we performed bulk ATAC-seq. The first principal component was due to genotype at rs7132908 (Figure 2A); 286 peaks were differentially accessible (Figure 2B; Table S2). However, rs7132908 itself was not found in a peak of accessible chromatin in these undifferentiated ESCs (Figure 2C).
Figure 2.

The putative cis-regulatory region harboring rs7132908 is inactive in ESCs
(A) PCA plot of ESC ATAC-seq libraries (GG n = 3, AA n = 3 lines).
(B) Volcano plot of adjusted p values (−log10) and fold change (log2) of ATAC-seq peaks tested for differential accessibility due to the rs7132908 obesity risk allele in ESCs. Red dots indicate differentially accessible peaks and black dots indicate peaks with no significant differences in accessibility.
(C) Chromatin accessibility represented by ATAC-seq tracks depicting normalized reads across FAIM2 in ESCs homozygous for either rs7132908 allele. Red vertical line indicates rs7132908 position.
(D) PCA plot of ESC RNA-seq libraries (GG n = 2, AA n = 3 lines).
(E) Volcano plot of adjusted p values (−log10) and fold change (log2) of genes tested for differential expression due to the rs7132908 obesity risk allele in ESCs. Blue dots indicate down-regulated genes and red dots indicate up-regulated genes. Gray dots indicate genes with no significant differences in expression.
(F) Heatmap depicting differentially expressed due to the rs7132908 obesity risk allele in ESCs.
To identify any transcriptional differences due to rs7132908 genotype in homogeneous ESCs, we performed bulk RNA-seq. The first principal component, explaining 44.5% of the variation between samples, was due to genotype at rs7132908 (Figure 2D). Forty-four genes were differentially expressed (Figures 2E and 2F; Table S3). Forty-two genes were significantly down-regulated in the rs7132908 risk A allele homozygote ESCs, while just two genes were up-regulated. As most enhancer-promoter interactions occur within the same TAD, we wanted to determine if rs7132908 regulated expression of genes within its TAD. However, none of the genes in the TAD harboring rs713290851 were differentially expressed in ESCs. We observed relatively small changes in expression and accessibility due to the introduction of the obesity risk allele in ESCs, consistent with the notion that rs7132908 primarily functions in neural cells.
To generate hypothalamic neural progenitors and characterize the effects of rs7132908 at this stage, we differentiated ESCs for 14 days using an established protocol (Figure 3A).6 Day 14 was selected given it was the last day after direction toward ventral diencephalon hypothalamic identity and cell cycle exit but before neuron maturation.6 We compared the transcriptomic profile of the hypothalamic neural progenitors homozygous for the rs7132908 non-risk allele to profiles of primary human tissues in the GTEx RNA-seq database49 (donor ages 20–71 years old, with 68.1% 50 years or older) and primary human pediatric hypothalamus tissue from three donors homozygous for the rs7132908 non-risk allele (donor ages 4–14 years old, average age = 8.67). The non-risk hypothalamic neural progenitors most highly correlated with the primary human pediatric hypothalamus tissue (correlation coefficient = 0.80, p value <0.001) (Figure S1A).
Figure 3.

rs7132908 genotype influences gene expression in ESC-derived hypothalamic neural progenitors
(A) Schematic of differentiation of ESCs to hypothalamic neurons, including duration, phases, and key small molecules to direct cell fates.
(B) Volcano plot of adjusted p values (−log10) and fold change (log2) of a total of genes tested for differential expression in hypothalamic neural progenitors. Blue dots indicate down-regulated genes and red dots indicate up-regulated in hypothalamic neural progenitors homozygous for the obesity risk allele. Gray dots indicate genes with no significant differences in expression.
(C) Boxplots of gene expression (normalized log2 cpm) for genes in the rs7132908 TAD that were differentially expressed.
(D) Heatmap depicting module 4 genes up-regulated due to the rs7132908 obesity risk allele in hypothalamic neural progenitors.
(E) Heatmap depicting module 5 genes down-regulated due to the rs7132908 obesity risk allele in hypothalamic neural progenitors. See also Figure S1.
To identify transcriptional differences due to rs7132908 genotype in homogeneous hypothalamic neural progenitors, we performed bulk RNA-seq. The first principal component, explaining 86.2% of the variation between samples, was due to batch as we differentiated pairs of non-risk and risk allele cells at two separate times (Figures S1B and S1C). Additionally, principal variance component analysis determined that the expected proportion of variance attributed to batch was 85.2% (Figure S1D). Therefore, we incorporated batch information as a covariate in our linear model to adjust for this effect for our differential expression analysis and used corrected expression data for visualizing the effects of batch correction, following best practices. As a result, the first principal component corresponded to rs7132908 genotype (Figures S1B and S1C) and the expected proportion of variance attributed to batch was decreased to 0% (Figure S1D); 6,494 genes were differentially expressed (Figure 3B; Table S4). Five genes in the TAD harboring rs713290851 were differentially expressed. FAIM2 and three other genes (TMBIM6, LARP4, and COX14) were down-regulated in the neural progenitors homozygous for the rs7132908 risk A allele and AQP2 was up-regulated (Figure 3C).
To explore global changes in gene expression, we clustered the differentially expressed genes into five modules with hierarchical clustering (Figure S1E). Even after batch correction, approximately 40% of the remaining variance between samples was attributed to genes that were variable across genotype and batch (Figure S1D), which comprised three modules (modules 1, 2, and 3) (Figure S1E). Therefore, we selected the two modules (modules 4 and 5) representing the genes differentially expressed due to genotype at rs7132908 and unaffected by batch for downstream analysis (Figure S1E). Module 4 consisted of 216 genes consistently up-regulated in neural progenitors homozygous for the rs7132908 risk A allele (Figure 3D). Functional enrichment analysis of the module 4 up-regulated genes identified significantly enriched Gene Ontology terms,52,53 with top-ranking biological processes involving blood vessel development, while other significant biological processes included programmed cell death, apoptotic process, and intrinsic apoptotic signaling pathway in response to endoplasmic reticulum stress (Table S5). Module 5 consisted of 152 genes consistently down-regulated in neural progenitors homozygous for the rs7132908 risk allele (Figure 3E). The module 5 down-regulated genes were also used to identify any enriched Gene Ontology terms,52,53 however, no significantly enriched biological processes were identified (Table S5).
ESC-derived hypothalamic neurons molecularly resemble the human hypothalamus
Next, we generated hypothalamic-like neurons by differentiating for 40 days using an established protocol6 and then collected nuclei (Figure 3A). Day 40 was selected given a previous characterization of this protocol found that this duration was sufficient to produce heterogeneous populations of functional neurons that closely resemble those found in the human hypothalamus.6 These nuclei were used to simultaneously profile gene expression and open chromatin in each cell using a multi-omic single-nucleus RNA-seq and ATAC-seq approach.
A previously published human hypothalamus single-cell RNA-seq reference dataset54 was used to identify cell types in our dataset (Figure S2A). To ensure that the cell type identifications were likely to be accurate, we prioritized cells with high-confidence annotations using a classification score threshold (≥0.8) that was previously demonstrated to increase accuracy.55 This method identified cells as neurons, oligodendrocyte precursors (OPCs), or fibroblasts based on their transcriptional profile with classification scores above our threshold (Figure 4A). These annotations are further supported by expression patterns of known marker genes for each cell type, including MAP2 and TUBB3 for neurons and COL1A1, COL1A2, and COL6A2 for fibroblasts (Figure 4B). We note that the OPC population did not highly or uniformly express conventional marker genes, such as PDGFRA, CSPG4, OLIG1, OLIG2, and SOX10 (Figure S2B), although this population did express cell cycle genes, such as CENPF and TOP2A, which have been observed in OPCs56 and neural intermediate progenitors57 (Figure 4B).
Figure 4.

ESC-derived hypothalamic neurons molecularly resemble the human hypothalamus
(A) UMAP depicting all cells clustered by single-nucleus RNA-seq profile and annotated by cell type.
(B) Dot plot depicting average expression (scaled and log2 normalized counts) and percent of cells that expressed neuron (MAP2 and TUBB3), fibroblast (COL1A1, COL1A2, and COL6A2), and OPC (CENPF and TOP2A) marker genes, split by cell type.
(C) UMAP depicting all neurons clustered by single-nucleus RNA-seq profile and annotated by cluster identity.
(D) Dot plot depicting average expression (scaled and log2 normalized counts) and percent of cells that expressed inhibitory (GAD1), excitatory (SLC17A6), GABAergic (SLC32A1), and hypothalamic (POMC, NPY, OTP, and SST) neuron marker genes, split by cluster identity.
(E) Heatmap showing average module scores across all neuron clusters for each human prenatal hypothalamic nucleus gene set published in the Allen Brain Atlas database, plotted as the column Z score per neuron cluster. See also Figure S2.
Additionally, we compared the transcriptomic signatures of each cell type to expression data in the GTEx RNA-seq database49 (donor ages 20–71 years old, with 68.1% 50 years or older) and primary human pediatric hypothalamus tissue from three donors homozygous for the rs7132908 non-risk allele (donor ages 4–14 years old, average age = 8.67). We found that the neurons were most strongly correlated with pediatric hypothalamus and adult hypothalamus (correlation coefficients = 0.56 and 0.54, respectively, p values <0.001), the OPCs correlated most strongly with fibroblasts and pediatric hypothalamus (correlation coefficients = 0.57 and 0.52, respectively, p values <0.001), and the fibroblasts most strongly correlated with fibroblasts and tibial artery (correlation coefficients = 0.66 and 0.63, respectively, p values <0.001) (Figures S2C‒S2E).
Within the neuron population (Figure 4C), there were distinct expression patterns of markers for several neuron types, including inhibitory (GAD1), excitatory (SLC17A6), and GABAergic (SLC32A1) neurons (Figure 4D). We also identified neuronal clusters expressing known hypothalamus genes, such as POMC, NPY, OTP, and SST (Figure 4D). Next, we compared the transcriptomic signatures of each neuronal cluster (Figure 4C) to human prenatal hypothalamic subregion gene sets published in the Allen Brain Atlas database,58,59,60,61 given that the neuron population displayed expression patterns most similar to pediatric hypothalamus tissue. We found that each cluster closely resembled the hypothalamic arcuate nucleus, which regulates feeding behavior and energy expenditure,62 the dorsomedial hypothalamic nucleus, which regulates food intake and body weight,63 and the anterior hypothalamic nucleus, which regulates defensive behaviors64 (Figure 4E).
The putative cis-regulatory region harboring rs7132908 is active in ESC-derived hypothalamic cell types
We used single-nucleus ATAC-seq to characterize chromatin accessibility in the heterogeneous ESC-derived hypothalamic cells. Unlike in the ESCs, the cis-regulatory element containing rs7132908 was open in all cell types (Figure 5A). When comparing chromatin accessibility globally between rs7132908 genotypes across all annotated cells, 12,586 ATAC-seq peaks were differentially accessible (Figure 5B; Table S6). We found that 565 transcription factor motifs were significantly enriched in peaks more accessible with the rs7132908 non-risk G allele and 446 were enriched in peaks more accessible with the risk A allele (Table S7). The peak harboring rs7132908 at chr12:49,868,731–49,869,775 (GRCh38) displayed decreased accessibility with the risk A allele by 27.62% (adjusted p value = 1.08 × 10−88) when considering all annotated cells. We also repeated these analyses in each annotated cell type and detected 3,406, 12,386, and 7,543 significantly differentially accessible regions in neurons, OPCs, and fibroblasts, respectively (Figures 5C–5E; Table S6). The peak surrounding rs7132908 was less accessible with the risk A allele by 40.74% in fibroblasts (adjusted p value = 1.35 × 10−14), but more accessible in neurons with the risk A allele by 78.92% (adjusted p value = 2.31 × 10−21) and not significantly different in OPCs. We then identified significantly differentially accessible regions that were consistent between analyses when considering each individual cell type and all annotated cells combined (Figure 5F) and their top enriched transcription factor motifs (Figure 5G). We conclude that rs7132908 is in an active chromatin region post-differentiation and that the rs7132908 risk A allele influences accessibility locally and globally.
Figure 5.

The putative cis-regulatory region harboring rs7132908 is active in ESC-derived hypothalamic cell types
(A) Chromatin accessibility represented by ATAC-seq tracks depicting normalized reads across FAIM2 in ESC-derived neurons, OPCs, and fibroblasts. Red vertical line indicates rs7132908 position.
(B‒E) Volcano plots of adjusted p values (−log10) and fold change (log2) of ATAC-seq peaks tested for differential accessibility due to the rs7132908 obesity risk allele in total cells (B), neurons (C), OPCs (D), and fibroblasts (E). Black or colored dots indicate differentially accessible peaks and gray dots indicate peaks with no significant differences in accessibility.
(F) Bar plot of numbers of differentially accessible regions from (B)–(E) that overlapped between analyses.
(G) ATAC-seq read enrichment heatmaps for groups of regions categorized in (F) and their corresponding top-most enriched transcription factor binding motifs. Windows indicate which cell type(s) yielded such groups of differentially accessible regions.
The rs7132908 obesity risk allele dramatically decreases the proportion of neurons produced by hypothalamic neuron differentiation
As expected, during each hypothalamic neuron differentiation, we began to observe neuron morphology with brightfield microscopy once the cells were exposed to BDNF in the neuron maturation phase (days 14–40) (Figure 3A). Strikingly, there were fewer cells exhibiting neuron morphology for those homozygous for the rs7132908 risk A allele (Figure 6A). To confirm this observation, we stained day 40 cells from each genotype to detect MAP2, a marker of mature neuron dendrites. Indeed, although each well was seeded at the same density and cultured simultaneously, fewer MAP2+ cells were observed in the risk A allele condition (Figure S3).
Figure 6.

The rs7132908 obesity risk allele dramatically decreases the proportion of neurons produced by hypothalamic neuron differentiation
(A) Representative brightfield images of hypothalamic neurons mid-differentiation on day 29 (scale bar, 100 μm). Cells were homozygous for either the rs7132908 non-risk allele (left) or obesity risk allele (right).
(B and C) Proportion of total cells homozygous for the rs7132908 non-risk allele annotated as each cell type (n = 4 differentiation replicates) (B) and homozygous for the rs7132908 obesity risk allele annotated as each cell type (n = 4 differentiation replicates) (C). Data are represented as mean ± SD. See also Figure S3.
We further confirmed this result using our annotated single-nucleus RNA-seq dataset. We partitioned the annotated cells by genotype at rs7132908 and differentiation replicate sample, then quantified the proportions of cells from each replicate identified as neurons, OPCs, or fibroblasts in each condition, which controlled for the number of nuclei sequenced per sample. On average, the cells homozygous for the rs7132908 non-risk G allele were composed of 60.90% neurons, 18.33% OPCs, and 20.77% fibroblasts (Figure 6B). In contrast, the cells homozygous for the rs7132908 risk A allele were composed of 10.69% neurons, 12.78% OPCs, and 76.53% fibroblasts (Figure 6C). A single base change from the rs7132908 non-risk G allele to the obesity risk A allele in the same genetic background was sufficient to substantially decrease the proportion of neurons produced by hypothalamic neuron differentiation.
rs7132908 genotype influences gene expression in ESC-derived hypothalamic cell types
We identified changes in gene expression due to genotype at rs7132908 in the ESC-derived hypothalamic cells. First, we included all cells that passed our quality control and determined that 85% of the variation between replicate samples was explained by the rs7132908 genotype (Figure S4A). We then identified that 6,409 genes were differentially expressed (Figures 7A and 7B; Table S8). Four genes in the TAD harboring rs7132908,51 were differentially expressed; two were down-regulated in cells homozygous for the rs7132908 risk A allele (FAIM2 and ASIC1) and two were up-regulated (FMNL3 and LIMA1) (Figure 7C).
Figure 7.

rs7132908 genotype influences gene expression in ESC-derived hypothalamic cell types
(A, D, F, H) Heatmaps depicting differentially expressed genes due to the rs7132908 risk allele in all cells (A), neurons (D), OPCs (F), and fibroblasts (G).
(B, E, G, I) Volcano plots of adjusted p values (−log10) and fold change (log2) of genes tested for differential expression due to the rs7132908 obesity risk allele in all cells (B), neurons (E), OPCs (G), and fibroblasts (I). Colored dots indicate differentially expressed genes and gray dots indicate genes with no significant differences in expression.
(C, J, K) Boxplots of gene expression (log10 normalized counts) for genes in the rs7132908 TAD that were differentially expressed in all cells (C), OPCs (J), and fibroblasts (K). See also Figures S4 and S5.
We identified genes differentially expressed within each annotated cell type. rs7132908 genotype explained 21%, 84%, and 78% of the variation between replicate samples in the neurons, OPCs, and fibroblasts, respectively (Figures S4B‒S4D). Fifty-two, 2,678, and 1,911 genes were differentially expressed in neurons (Figures 7D and 7E; Table S8), OPCs (Figures 7F and 7G; Table S8), and fibroblasts (Figures 7H and 7I; Table S8), respectively. When considering genes located in the same TAD as rs7132908,51 no genes were differentially expressed in neurons, while one gene was differentially expressed in OPCs (LIMA1 up-regulated) (Figure 7J), and two genes were differentially expressed in fibroblasts (FAIM2 down-regulated; FMNL3 up-regulated) (Figure 7K). Functional enrichment analyses of up-regulated genes in both OPCs and fibroblasts identified similar Gene Ontology terms,52,53 including the biological processes of cell death and apoptosis (Table S9), while processes such as nervous system development, neuron differentiation, and neuron projection development were enriched among down-regulated genes (Table S9). However, the comparably shorter lists of significantly up- and down-regulated genes in neurons did not identify any significantly enriched biological processes.
As our sequencing efforts only captured transcriptional differences at three time points, we were therefore motivated to quantify FAIM2 expression in all cells throughout the 40-day hypothalamic neuron differentiation using real-time qPCR. FAIM2 expression peaked around day 14 in cells homozygous for the rs7132908 non-risk allele and around day 12 in cells homozygous for the risk allele (Figures S5A and S5B), which represents the hypothalamic neural progenitor phase of the differentiation (Figure 3A). Average FAIM2 expression was also higher with the non-risk allele on all measured days from day 14–40 (Figures S5A and S5B). We also characterized FAIM2 expression in vivo using our primary human pediatric (donor ages 4–14 years old, average age = 7.5) hypothalamus tissue RNA-seq data and determined that FAIM2 was highly expressed (median TPM = 415.66, n = 4) (Figure S5C; Table S10).
Discussion
The chr12q13 locus was first associated with variation in adult BMI and weight in 2009,65 BMI as a longitudinal trait during childhood (ages 3–17) in 2012,29 and childhood obesity as a dichotomous trait in 2012.21 The genotypic risk effect at the chr12q13 locus during childhood decreased as age increased,29 which suggests this locus may regulate age-dependent pathways in early childhood and could explain why this locus is more pronounced in childhood. More than 1,000 independent loci are now associated with measurements of obesity24 and only a few have been studied extensively enough to pinpoint a causal variant and implicate effector genes, such as the FTO25,26,27 and 2q24.3 loci.66
Fine-mapping by our group22 and others34 has refined the chr12q13 locus to credible sets of 1–6 SNPs, depending on methods. While rs7132908 is the strongest variant detected with colocalization analysis using multiple ancestral populations,34 we cannot rule out additional causal signals at this locus. A global functional investigation of BMI-associated SNPs in 3′ UTRs found that the rs7132908 obesity risk allele disrupted miRNA binding activity of miR-330-5p in hamster ovary cells and human subcutaneous preadipocytes, leading to an increase in FAIM2 expression.67 These results may, however, not accurately reflect regulation of FAIM2 expression in vivo as this gene is primarily expressed in the brain; furthermore, this microRNA (miRNA) product is a passenger strand that is typically found in lower abundance due to degradation during miRNA processing.68 More recently, others carrying out global analyses have implicated an enhancer in the region harboring rs7132908 with a luciferase reporter assay and found that, in mouse neuronal hypothalamus cells, the obesity risk allele significantly decreased enhancer activity with a minimal promoter,69 consistent with our results.
The rs7132908 obesity risk A allele led to differential expression of zero TAD genes in ESCs, five TAD genes in hypothalamic neural progenitors (AQP2, COX14, FAIM2, LARP4, and TMBIM6), one TAD gene in OPCs (LIMA1), and two TAD genes in fibroblasts (FAIM2 and FMNL3). These results, in combination with our observation that rs7132908 is not accessible in ESCs, suggest that rs7132908 does not regulate gene expression in stem cells. These results also implicate different effector genes depending on cell type, in agreement with the luciferase assay results where enhancer activity was observed in primary astrocytes but not HEK293Ts. Only FAIM2 was implicated in more than one cell type and its expression was consistently down-regulated with the obesity risk allele. Taken together, we demonstrated that rs7132908 resides within a cis-regulatory element that confers allele-specific and cell-type-specific effects on the expression of FAIM2 and other genes within its TAD. This result mirrors the well-studied FTO locus, where the rs1421085 obesity risk allele decreases the expression of IRX3 and IRX5 during early differentiation of mesenchymal progenitors to adipocytes26 and increases the expression of Fto in brown adipocytes.27
We did not observe large differences in accessibility at rs7132908 due to genotype in any cell type. Therefore, significant changes in effector gene expression are likely due to differences in transcription factor binding affinity. We predicted that the rs7132908 risk allele disrupts binding of 12 transcription factors, many of which are known to be both activators and repressors and are ubiquitously expressed. Further investigation is warranted to determine which specific transcription factors regulate gene expression at the chr12q13 locus.
We made the striking observation that the rs7132908 obesity risk A allele decreased the proportion of hypothalamic neurons produced by stem cell differentiation. We also observed that the obesity risk allele led to up-regulation of cell death and apoptosis gene sets and down-regulation of neuron development gene sets. However, orexigenic neurons were underrepresented in our model and we could not detect if any orexigenic or anorexigenic neuronal cell cluster or subpopulation was more severely decreased, highlighting the need for more experiments to determine how the rs7132908 obesity risk allele could increase appetite and childhood obesity risk. We observed that the rs7132908 obesity risk allele most significantly led to up-regulation of blood vessel development gene sets. One possible explanation is that these neural progenitors gave rise to mostly fibroblasts and fibroblasts play a significant role in the formation of new blood vessels by secreting angiogenic growth factors.70
Our working hypothesis is that rs7132908 regulates FAIM2 and possibly other genes that are required for normal anorexigenic neuron development or survival at a crucial time point in development and prior to adulthood. Genes downstream of rs7132908 may be less important in adulthood or a compensatory mechanism could arise later in life to decrease the effect of the rs7132908 risk allele. This is supported by our finding that FAIM2 expression was highest in neural progenitors homozygous for the rs7132908 non-risk allele after only 14 days of differentiation and that the obesity risk allele caused an approximate 50% decrease in FAIM2 expression at this time. Mice with reduced Faim2 expression or Faim2 null mice have reduced cerebellar size, internal granular layer thickness, and Purkinje neuron development, which are more severe in early developmental stages with substantial recovery over time.71 These observations could help explain the difference in the magnitude of the effect of the chr12q13 risk genotype on BMI with increasing age that is reported in this study and by others.29
FAIM2 protects neurons from Fas-induced apoptosis72,73 and regulates neurite outgrowth,74 neuroplasticity,75 and synapse formation.76 While Faim2 null mice have only been previously used to study neurological71,77,78,79 and immune80 phenotypes, one study reported that Faim2 null mice at 10–12 weeks of age and fed a standard diet ad libitum displayed subtle increases in fat content.77 Rodent studies have also demonstrated that Faim2 expression increased in the hypothalamic arcuate nucleus in response to restricted food intake81 and food deprivation.82 As for a mechanism that could explain the relationship between the rs7132908 risk allele and obesity, we propose that the resulting decrease in FAIM2 expression could cause altered proportions of orexigenic and anorexigenic neurons in the hypothalamus. If an individual had fewer anorexigenic POMC neurons, they would experience an increased appetite and higher risk of becoming overweight. FAIM2 is expressed in neurons outside of the hypothalamus, and while no associations between the chr12q13 locus or FAIM2 and neurological traits in children have been reported, further exploration into possible neurological comorbidities should be explored.
The rs7132908 risk allele has remained common in most human ancestral populations. This may be due to it previously providing an evolutionary advantage when food was scarcer and the risk of starvation was higher. Conversely, the impact of the rs7132908 risk allele, which like all GWAS variants should have a modest effect, may be exacerbated by our current obesogenic environment with higher caloric foods and more food availability than ever before.
We acknowledge that our methods nominate FAIM2 as a strong candidate effector gene at this locus, but do not rule out other potentially causal genes. Future work must also be dedicated to directly test our hypothesis that FAIM2 is a causal gene for childhood obesity. Using human exonic sequencing data from the Penn Medicine BioBank, we observed relatively few rare FAIM2 variants and no individuals homozygous for any given FAIM2 mutations, suggesting that FAIM2 mutations may be strongly deleterious. FAIM2 knockout and over expression stem cell lines could be differentiated to hypothalamic neurons to test if changes in FAIM2 expression is responsible for our observation of decreased neurons in vitro. The use of Faim2 knockout mice would also aid in determining if decreased Faim2 expression leads to changes in appetite, body fat, hypothalamic neuron composition, or neurodevelopment, which would make progress toward identifying the precise mechanism by which the rs7132908 genotype increases childhood obesity risk.
Overall, we functionally validated rs7132908 as a causal SNP at one of the strongest but commonly overlooked childhood obesity GWAS loci, implicated FAIM2 and other cell-type-specific effector genes, and nominated pathways acting downstream of the SNP involving nervous system development and cell death. We have also generated datasets from primary astrocytes and multiple time points throughout hypothalamic neuron differentiation that will serve as a resource to aid investigation of other loci and traits. This progress toward characterizing the precise mechanism underlying the association between the chr12q13 genomic region and obesity should enable future work with this key locus and guide comparable efforts to ultimately identify therapeutic targets.
Limitations of the study
There are several other limitations to our study to consider. First, although our ESC-derived in vitro model of hypothalamic neurogenesis expresses some appropriate marker genes, it likely does not fully recapitulate the hypothalamus during childhood. All the neuron clusters most closely resembled human hypothalamic tissue from the arcuate nucleus, anterior nucleus, and dorsomedial nucleus. While we intentionally used a differentiation protocol established to generate arcuate neurons, other hypothalamic nuclei, such as the paraventricular nucleus, also play key roles in appetite regulation and we were unable to represent all relevant neuron sub-types in our model. We also generated non-neuronal cell types (OPCs and fibroblasts) that correlated most highly with cultured fibroblasts in the GTEx RNA-seq database49 but still expressed neuronal markers (MAP2 and TUBB3) at some level, likely due to exposure to neuron maturation cell culture medium for 26 days. While we reported changes in gene expression and chromatin accessibility in these additional cell types, they may not be as biologically relevant. Second, we performed independent stem cell differentiations that led to batch effects, especially in the hypothalamic neural progenitor RNA-seq dataset. To reduce these effects, we included batch as a covariate in the linear model when detecting differentially expressed genes in this cell type. We also quantified the expected proportion of variance attributable to genotype and batch using principal variance component analysis and determined that post-batch correction, 40% of the remaining variance was attributable to both batch and genotype. Therefore, some genes that we detected to be significantly differentially expressed were still influenced by batch. As a result, we only included gene sets from modules that were consistent across the two differentiation batches in our downstream enrichment analysis. Third, we used the female H9 ESC line which prevented us from detecting sex-specific differences. Fourth, we did not investigate the effects of the rs7132908 obesity risk A allele in vivo. We were able to obtain four pediatric hypothalamus tissue samples, but with just three homozygous for the rs7132908 non-risk allele and only one heterozygote, this sample size was insufficient for allele-specific expression or eQTL analyses. In the future, increased accessibility to human pediatric hypothalamus tissue would aid investigation at the chr12q13 obesity locus.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MAP2 (Chicken, Polyclonal) | Abcam | Cat# ab5392; Lot# GR3450786-1; RRID: AB_2138153 |
| Anti-TTF-1/NKX2.1 (Mouse, Monoclonal) | Cell Marque | Cat# 343M-95; Lot# 0000051910; RRID: AB_1158934 |
| Anti-NeuN (Mouse, Monoclonal) | Millipore Sigma | Cat# MAB377; RRID: AB_2298772 |
| Anti-Chicken Alexa Fluor 488 (Goat, Polyclonal) | Abcam | Cat# ab150169; RRID: AB_2636803 |
| Anti-Mouse Alexa Fluor 488 (Goat, Polyclonal) | Invitrogen | Cat# A-11001; RRID: AB_2534069 |
| Biological samples | ||
| Frozen hypothalamus region, left hemisphere. 1674, 8 years old, male | NIH NeuroBioBank | N/A |
| Frozen hypothalamus region, left hemisphere. 5309, 14 years old, female | NIH NeuroBioBank | N/A |
| Frozen hypothalamus region, left hemisphere. 5976, 4 years old, female | NIH NeuroBioBank | N/A |
| Frozen hypothalamus region, left hemisphere. 6032, 4 years old, male | NIH NeuroBioBank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 1M HEPES | Gibco | Cat# 15630-080 |
| 0.25% Trypsin-EDTA | Gibco | Cat# 25200-056 |
| Dulbecco’s Phosphate-Buffered Saline (DPBS) without calcium and magnesium | Corning | Cat# 21-031-CV |
| Heat-Inactivated FBS | Gibco | Cat# 10082-147 |
| Dulbecco’s Modified Eagle’s Medium (DMEM) | ATCC | Cat# 30-2002 |
| 100X Antibiotic-Antimycotic | Gibco | Cat# 15240062 |
| 200 mM L-Glutamine | Corning | Cat# 25-005-CI |
| PBS, pH 7.4 | Gibco | Cat# 10010023 |
| ROCK Inhibitor Y-27632 | Stemcell Technologies | Cat# 72304 |
| Matrigel hESC-Qualified Matrix, LDEV-free | Corning | Cat# 354277 |
| Versene Solution | Gibco | Cat# 15040-066 |
| DMSO | Sigma | Cat# D2650-100ML |
| mFreSR | Stemcell Technologies | Cat# 05854 |
| Accutase | Stemcell Technologies | Cat# 07920 |
| Opti-MEM Reduced Serum Media | Gibco | Cat# 31985-062 |
| 4% Paraformaldehyde | Biotium | Cat# 22023 |
| XhoI | NEB | Cat# R0146S |
| Miller’s LB Broth | Corning | Cat# 46-050-CM |
| LB + Ampicillin (100 μg/mL) Agar Plates | University of Pennsylvania Cell Center Service Facility | Cat# 6005 |
| Ampicillin | Corning | Cat#61-238-RH |
| AflII | NEB | Cat# R0520S |
| Lipofectamine Stem Transfection Reagent | Invitrogen | Cat# STEM00003 |
| BfaI | NEB | Cat# R0568S |
| TRIzol Reagent | Invitrogen | Cat# 15596018 |
| DNase I | Zymo | Cat# E1009-A |
| 100% Ethanol | Electron Microscopy Sciences | Cat#15055 |
| TrypLE Express Enzyme | ThermoFisher | Cat# 12605036 |
| DMEM/F12 | Gibco | Cat# 11320-033 |
| Matrigel Growth Factor Reduced Basement Membrane Matrix | Corning | Cat# 354230 |
| Iscove’s Dulbecco’s Medium (IMDM) | Gibco | Cat# 12440053 |
| KnockOut Serum Replacement | Gibco | Cat# 10828-010 |
| MEM Non-Essential Amino Acids Solution | Gibco | Cat# 11140050 |
| 100 mM Sodium Pyruvate | Gibco | Cat# 11360070 |
| Penicillin-Streptomycin (10,000 U/mL) | Gibco | Cat# 15140122 |
| β-Mercaptoethanol | Sigma Life Science | Cat# M3148-25ML |
| Recombinant Human FGF basic/FGF2/bFGF (146 aa) Protein | R&D Systems | Cat# 233-FB |
| 1M Tris-HCl Buffer, pH 7.5 | Invitrogen | Cat# 15567027 |
| Tween 20, 10% (w/v) | Roche | Cat# 11332465001 |
| JumpStart Taq DNA Polymerase | Sigma Aldrich | Cat# D6558-50UN |
| AMPure XP Beads | Beckman Coulter | Cat# A63881 |
| IGEPAL CA-630 | Sigma | Cat# I8896-50ML |
| Triton X-100 | Sigma | Cat# X100-5ML |
| 10X PBST Buffer Strength Solution | Bio Basic | Cat# PW004 |
| DAPI (4′,6-Diamidino-2-Phenylindole, Dilactate) | Invitrogen | Cat# D3571 |
| ProLong Gold Antifade Mountant | Invitrogen | Cat# P36930 |
| Corticosterone | Sigma | Cat# 27840 |
| Linoleic Acid | Sigma | Cat# L1376 |
| Linolenic Acid | Sigma | Cat# L2376 |
| (±)-α-Lipoic Acid | Sigma | Cat# T5625 |
| Progesterone | Sigma | Cat# P0130 |
| Retinyl Acetate | Sigma | Cat# 46958 |
| (±)-α-Tocopherol | Sigma | Cat# T3251 |
| DL-α-Tocopherol Acetate | Sigma | Cat# T3376 |
| Bovine Serum Albumin | Sigma | Cat# A4919 |
| Sodium Bicarbonate | Sigma | Cat# S5761 |
| L-Ascorbic Acid | Sigma | Cat# A8960 |
| Putrescine Dihydrochloride | Sigma | Cat# P5780 |
| D(+)-Galactose | Sigma | Cat# G5388 |
| Holo-Transferrin | Sigma | Cat# T0665 |
| Catalase | Sigma | Cat# C1345 |
| L-Carnitine Hydrochloride | Sigma | Cat# C0283 |
| Glutathione | Sigma | Cat# G4251 |
| Sodium Selenite | Sigma | Cat# S5261 |
| Ethanolamine | Sigma | Cat# E9508 |
| Triiodo-L-Thyronine Sodium Salt | Sigma | Cat# T6397 |
| Insulin Solution, Human | Sigma | Cat# 19278 |
| Superoxide Dismutase | Sigma | Cat# S5395 |
| LDN-193189 (hydrochloride) | Cayman Chemical | Cat# 19396 |
| SB-431542 (hydrate) | Cayman Chemical | Cat# 13031 |
| SAG | Cayman Chemical | Cat# 11914 |
| Purmorphamine | Tocris | Cat# 4551 |
| IWR-1-Endo | Cayman Chemical | Cat# 13659 |
| DAPT | Cayman Chemical | Cat# 13197 |
| All-Trans Retinoic Acid | Cayman Chemical | Cat# 11017 |
| Human BDNF | Miltenyi Biotec | Cat# 130-093-811 |
| Laminin | Sigma | Cat# L2020-1MG |
| Hanks' Balanced Salt Solution | Sigma | Cat# H8264-100ML |
| 7.5% Bovine Serum Albumin Solution | Sigma | Cat# A8412-100ML |
| 1M Trizma Hydrochloride, pH 7.4 | Sigma | Cat# T2194-100ML |
| 5M Sodium Chloride Solution | Sigma | Cat# 59222C-500ML |
| 1M Magneisum Chloride Solution | Sigma | Cat# M1028-100ML |
| Protector RNase Inhibitor | Roche | Cat# 03335402001 |
| 100 mM DTT | Agilent | Cat# 600089-53 |
| GlutaMAX | Gibco | Cat# 35050061 |
| Critical commercial assays | ||
| AGM Astrocyte Growth Medium BulletKit | Lonza | Cat# CC-3186 |
| mTeSR1 Complete Kit | Stemcell Technologies | Cat# 85850 |
| LookOut Mycoplasma PCR Detection Kit | Sigma Aldrich | Cat# MP0035-1KT |
| Lipofectamine LTX Reagent with PLUS Reagent | Invitrogen | Cat# 15338030 |
| Monarch DNA Gel Extraction Kit | NEB | Cat# T1020S |
| Gibson Assembly HiFi HC 1-Step Kit | Codex | Cat# GA1100-4X10 |
| NEB Stable Competent E. coli (High Efficiency) | NEB | Cat# C3040H |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27106 |
| EndoFree Plasmid Maxi Kit | Qiagen | Cat# 12362 |
| Q5 Site-Directed Mutagenesis Kit | NEB | Cat# E0552S |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat# M0541S |
| Lipofectamine 3000 Transfection Reagent | Invitrogen | Cat# L3000001 |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1960 |
| Direct-zol RNA Miniprep Kit | Zymo | Cat# R2050 |
| Qubit RNA High Sensitivity Assay Kit | Invitrogen | Cat# Q32855 |
| SuperScript IV VILO Master Mix with ezDNase Enzyme | Invitrogen | Cat# 11766050 |
| Phusion High-Fidelity DNA Polymerase | NEB | Cat# M0530S |
| NucleoSpin Gel and PCR Clean-Up Kit | Takara | Cat# 740609 |
| Gibson Assembly Kit | NEB | Cat# E2611 |
| FAIM2 TaqMan Gene Expression Assay | ThermoFisher Scientific | Cat# 4331182, Assay ID Hs00392345_m1 |
| 18S TaqMan Gene Expression Assay | ThermoFisher Scientific | Cat# 4331182, Assay ID Hs99999901_s1 |
| TaqMan Fast Advanced Master Mix | Applied Biosystems | Cat# 4444557 |
| Quick-DNA Miniprep Plus Kit | Zymo | Cat# D4068 |
| Infinium Global Screening Array-24 v3.0 Kit | Illumina | Cat# 20030770 |
| Infinium OmniExpressExome-8 v1.6 Kit | Illumina | Cat# 20024676 |
| Quick-DNA/RNA Miniprep Plus Kit | Zymo | Cat# D7003 |
| RNA 6000 Nano Kit | Agilent | Cat# 5067-1511 |
| QIAseq FastSelect RNA Removal Kit | Qiagen | Cat# 333180 |
| NEBNext Ultra II Directional RNA Library Prep for Illumina Kit | NEB | Cat# E7760S |
| NEBNext Oligos for Illumina (Dual Index Primers Set 1) | NEB | Cat# E7600S |
| Qubit dsDNA High Sensitivity Assay Kit | Invitrogen | Cat# Q32851 |
| DNA 1000 Kit | Agilent | Cat# 5067-1504 |
| Tagment DNA TDE1 Enzyme and Buffer Kit | Illumina | Cat# 20034197 |
| Nextera DNA CD Indexes Kit | Illumina | Cat# 20018708 |
| MinElute PCR Purification Kit | Qiagen | Cat# 28004 |
| High Sensitivity D1000 ScreenTape Assay | Agilent | Cat# 5067–5587, 5067–5585, 5067–5603, 5067-5584 |
| High Sensitivity D5000 ScreenTape Assay | Agilent | Cat# 5067–5594, 5067–5593, 5067-5592 |
| Chromium Next GEM Chip J Single Cell Kit | 10X Genomics | Cat# 1000230 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle | 10X Genomics | Cat# 1000285 |
| Dual Index Kit TT Set A | 10X Genomics | Cat# 1000215 |
| Arima-HiC Kit | Arima Genomics | Cat# A510008 |
| Swift Accel-NGS 2S Plus DNA Library Kit | Swift Biosciences | Cat# 21024/21096 |
| Swift 2S Indexing Kit | Swift Biosciences | Cat# 28096 |
| KAPA Library Quantification Kit | Roche | Cat# 07960140001 |
| Deposited data | ||
| Raw and processed Hi-C data | This paper | GEO: GSE241592; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241592 |
| Raw and processed RNA-seq data | This paper | GEO: GSE241050; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241050 |
| Raw and processed ATAC-seq data | This paper | GEO: GSE241591; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241591 |
| Raw and processed single-nucleus RNA-seq data | This paper | GEO: GSE241594; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241594 |
| Raw and processed single-nucleus ATAC-seq data | This paper | GEO: GSE241593; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241593 |
| Experimental models: Cell lines | ||
| NHA - Human Astrocytes | Lonza | Cat# CC-2565 |
| HEK293T Cells | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| WA09 (H9) Human Embryonic Stem Cells (NIH Approval Number: NIHhESC-10-0062) | WiCell Research Institute | Lot# DL-05; RRID: CVCL_9773 |
| WA09 (H9) Human Embryonic Stem Cells (NIH Approval Number: NIHhESC-10-0062), rs7132908 AA Clone 2.1 | This paper | N/A |
| WA09 (H9) Human Embryonic Stem Cells (NIH Approval Number: NIHhESC-10-0062), rs7132908 AA Clone 9.1 | This paper | N/A |
| WA09 (H9) Human Embryonic Stem Cells (NIH Approval Number: NIHhESC-10-0062), rs7132908 AA Clone 10.1 | This paper | N/A |
| Oligonucleotides | ||
| Oligonucleotides | See Table S13 | N/A |
| Recombinant DNA | ||
| LentiCRISPRv2-mCherry plasmid | Agata Smogorzewska | Addgene Cat# 99154; RRID: Addgene_99154 |
| FAIM2 miRNA 3′ UTR target clone in pEZX-MT05 reporter vector | GeneCopoeia | Cat# HmiT096491-MT05 |
| FAIM2 promoter clone in pEZX-PG02 reporter vector | GeneCopoeia | Cat# HPRM47354-PG02 |
| LIMA1 promoter clone in pEZX-PG02 reporter vector | GeneCopoeia | Cat# HPRM34453-PG02 |
| RACGAP1 promoter clone in pEZX-PG02 reporter vector | GeneCopoeia | Cat# HPRM34625-PG02 |
| pGL4.10[luc2] reporter vector | Promega | Cat# E6651 |
| pRL-TK reporter vector | Promega | Cat# E2241 |
| pGL4.10[luc2]-rs7132908G-FAIM2 | This paper | N/A |
| pGL4.10[luc2]-rs7132908A-FAIM2 | This paper | N/A |
| pGL4.10[luc2]-FAIM2 | This paper | N/A |
| pGL4.10[luc2]-rs7132908G-LIMA1 | This paper | N/A |
| pGL4.10[luc2]-rs7132908A-LIMA1 | This paper | N/A |
| pGL4.10[luc2]-LIMA1 | This paper | N/A |
| pGL4.10[luc2]-rs7132908G-RACGAP1 | This paper | N/A |
| pGL4.10[luc2]-rs7132908A-RACGAP1 | This paper | N/A |
| pGL4.10[luc2]-RACGAP1 | This paper | N/A |
| gRNA_Cloning vector | Mali et al.83 | Addgene Cat# 41824; RRID: Addgene_41824 |
| gRNA_Cloning-rs7132908gRNA vector | This paper | N/A |
| pCas9_GFP | Ding et al.84 | Addgene Cat# 44719; RRID: Addgene_44719 |
| Software and algorithms | ||
| SnapGene v6.0.2 | SnapGene | https://www.snapgene.com/; RRID: SCR_015052 |
| Prism v10.0.0 | GraphPad | https://www.graphpad.com/features; RRID: SCR_002798 |
| CRISPOR v5.01 | Concordet85 | http://crispor.tefor.net/; RRID: SCR_015935 |
| Excel v2202 | Microsoft | RRID: SCR_016137 |
| AriaMx v1.5 | Agilent | https://www.agilent.com/en/product/real-time-pcr-%28qpcr%29/real-time-pcr-%28qpcr%29-instruments/ariamx-software-download |
| 2100 Bioanalyzer Expert vB.02.11.SI824 | Agilent | https://explore.agilent.com/Software-Download-2100-Expert?productURL=https%3A%2F%2Fwww.agilent.com%2Fen%2Fproduct%2Fautomated-electrophoresis%2Fbioanalyzer-systems%2Fbioanalyzer-software%2F2100-expert-software-228259; RRID: SCR_019715 |
| FastQC v0.11.9 | Andrews86; FASTQC87 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; RRID: SCR_014583 |
| Kallisto v0.48.0 | Bray et al.88; Bray et al.89 | https://pachterlab.github.io/kallisto/; RRID: SCR_016582 |
| bcl2fastq2 Conversion v2.20 | Illumina | https://sapac.support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html; RRID: SCR_015058 |
| R v4.2.2 and v4.2.3 | Comprehensive R Archive Network | https://cran.r-project.org/; RRID: SCR_001905 |
| RStudio v2022.07 and v2023.06.0 + 421 | Posit | https://posit.co/download/rstudio-desktop/ |
| cellSens Standard v2.3 | Olympus | https://www.olympus-lifescience.com/en/software/cellsens/ |
| ImageJ v1.54days | Schneider et al.90 | https://imagej.nih.gov/ij/download.html; RRID: SCR_003070 |
| PennCNV v1.0.5 | GitHub | https://penncnv.openbioinformatics.org/en/latest/user-guide/download/; RRID: SCR_002518 |
| PLINK v1.90b6.18 | Harvard University | https://zzz.bwh.harvard.edu/plink/download.shtml; RRID: SCR_001757 |
| Cell Ranger ARC v2.0.2 | 10X Genomics | https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/pipelines/latest/installation; RRID: SCR_023897 |
| Scrublet v0.2.3 | Wolock et al.91 | https://github.com/swolock/scrublet; RRID: SCR_018098 |
| SoupX v1.6.2 | Young et al.92 | https://github.com/constantAmateur/SoupX; RRID: SCR_019193 |
| Harmony v0.1.1 | Korsunsky et al.93 | https://cran.r-project.org/web/packages/harmony/index.html; RRID: SCR_022206 |
| TapeStation Analysis Software v4.1.1 | Agilent | https://www.agilent.com/en/product/automated-electrophoresis/tapestation-systems/tapestation-software/tapestation-software-379381 |
| Seurat v4.3.0 | Hao et al.94 | https://satijalab.org/seurat/articles/install.html; RRID: SCR_016341 |
| SCTransform v0.3.5 | Choudhary et al.95; Hafemeister et al.96 | https://cran.r-project.org/web/packages/sctransform/index.html; RRID: SCR_022146 |
| gprofiler2 v0.2.1 | Raudvere et al.97; Kolberg et al.98 | https://cran.r-project.org/web/packages/gprofiler2/vignettes/gprofiler2.html; RRID: SCR_018190 |
| tximport v1.24.0 | Soneson et al.99 | https://bioconductor.org/packages/release/bioc/html/tximport.html; RRID: SCR_016752 |
| edgeR v3.38.4 and v3.40.2 | Robinson et al.100 | https://bioconductor.org/packages/release/bioc/html/edgeR.html; RRID: SCR_012802 |
| ensembldb v2.20.2 | Rainer et al.101 | https://bioconductor.org/packages/release/bioc/html/ensembldb.html; RRID: SCR_019103 |
| EnsDb.Hsapiens.v86 v2.99.0 | Bioconductor | https://bioconductor.org/packages/release/data/annotation/html/EnsDb.Hsapiens.v86.html |
| limma v3.52.4 | Ritchie et al.102 | https://bioconductor.org/packages/release/bioc/html/limma.html; RRID: SCR_010943 |
| bowtie2 v2.2.6 | Langmead et al.103 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml; RRID: SCR_016368 |
| Picard v2.7.1 | GitHub | https://github.com/broadinstitute/picard; RRID: SCR_006525 |
| SAMtools v1.7 | Danecek et al.104 | http://www.htslib.org/; RRID: SCR_002105 |
| MACS2 v2.1.1 | Zhang et al.105 | https://pypi.org/project/MACS2/ |
| csaw v1.32.0 | Lun et al.106 | https://bioconductor.org/packages/release/bioc/html/csaw.html |
| ggplot2 v3.4.2 | Wickham107 | https://cran.r-project.org/web/packages/ggplot2/index.html; RRID: SCR_014601 |
| pheatmap v1.0.12 | Comprehensive R Archive Network | https://cran.r-project.org/web/packages/pheatmap/index.html; RRID: SCR_016418 |
| plotly v4.10.1 | Comprehensive R Archive Network | https://cran.r-project.org/web/packages/plotly/index.html |
| MACS3 v3.0.0b2 | GitHub | https://github.com/macs3-project/MACS |
| Signac v1.10.0 | Stuart et al.108 | https://stuartlab.org/signac/articles/install.html; RRID: SCR_021158 |
| DESeq2 v1.38.3 | Love et al.109 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID: SCR_015687 |
| SingleCellExperiment v1.20.1 | Amezquita et al.110; Amezquita et al.111 | https://bioconductor.org/packages/release/bioc/html/SingleCellExperiment.html |
| Matrix.utils v0.9.8 | Comprehensive R Archive Network | https://rdrr.io/cran/Matrix.utils/ |
| apeglm v1.20.0 | Zhu et al.112 | https://bioconductor.org/packages/release/bioc/html/apeglm.html |
| gplots v3.1.3 | Comprehensive R Archive Network | https://cran.r-project.org/web/packages/gplots/index.html |
| UCSC Genome Browser (GRCh37/hg19) | University of California Santa Cruz | https://genome.ucsc.edu/cgi-bin/hgGateway; RRID: SCR_005780 |
| HICUP pipeline v0.7.4 | Wingett et al.113 | https://www.bioinformatics.babraham.ac.uk/projects/hicup/; RRID: SCR_005569 |
| Pairtools v0.3.0 | Open2C et al.114 | https://pairtools.readthedocs.io/en/latest/installation.html; RRID: SCR_023038 |
| Pairix v0.3.7 | Lee et al.115 | https://github.com/4dn-dcic/pairix |
| Cooler v0.8.11 | Abdennur et al.116 | https://pypi.org/project/cooler/ |
| Mustache v1.0.1 | Roayaei Ardakany et al.117 | https://github.com/ay-lab/mustache |
| Fit-Hi-C2 v2.0.7 | Kaul et al.118 | https://github.com/ay-lab/fithic |
| BSgenome v1.68.0 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/BSgenome.html |
| SNPlocs.Hsapiens.dbSNP155.GRCh38 0.99.24 | Bioconductor | https://bioconductor.org/packages/release/data/annotation/html/SNPlocs.Hsapiens.dbSNP155.GRCh38.html |
| MotifDb v1.42.0 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/MotifDb.html |
| motifbreakR v2.14.2 | Coetzee et al.119 | https://bioconductor.org/packages/release/bioc/html/motifbreakR.html |
| ColoQuiaL | Chen et al.120 | https://github.com/bvoightlab/ColocQuiaL |
| STAR v2.7.9a | Dobin et al.121 | https://github.com/alexdobin/STAR; RRID: SCR_004463 |
| HTSeq-count v0.11.3 | Anders et al.122 | https://shicheng-guo.github.io/research/1941/01/08/HTseq; RRID: SCR_011867 |
| SKAT v2.2.5 | Comprehensive R Archive Network | http://cran.nexr.com/web/packages/SKAT/index.html; RRID: SCR_009396 |
| PVCA v3.18 | Bioconductor | https://www.bioconductor.org/packages/release/bioc/html/pvca.html; RRID: SCR_001356 |
| Other | ||
| Falcon Round-Bottom Polystyrene Test Tubes with Cell Strainer Snap Cap, 35 μm, 5 mL | Fisher Scientific | Cat# 08-771-23 |
| ZR BashingBead Lysis Tubes, 2 mm | Zymo | Cat# 56003-50 |
| #1.5 Acid-Treated Coverslips, 12 mm diameter | Fisher Scientific | Cat# NC0706236 |
| Nunc Cell-Culture Treated 6-well Plates | ThermoFisher Scientific | Cat# 140675 |
| Costar 24-well Clear TC-Treated Multiple Well Plates | Corning | Cat# 3524 |
| 75 cm2 U-Shaped Canted Neck Cell Culture Flask with Plug Seal Cap | Corning | Cat# 430720U |
| Falcon 25 cm2 Rectangular Canted Neck Cell Culture Flask with Blue Plug Seal Screw Cap | Corning | Cat# 353014 |
| Falcon 100 mm TC-Treated Cell Culture Dish | Corning | Cat# 353003 |
| White 96-well Immuno Microlite 1+ Plates | Thermo Scientific | Cat# 7571 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Struan F. A. Grant (grants@chop.edu).
Materials availability
Vectors (pGL4.10[luc2]-rs7132908G-FAIM2, pGL4.10[luc2]-rs7132908A-FAIM2, pGL4.10[luc2]-FAIM2, pGL4.10[luc2]-rs7132908G-LIMA1, pGL4.10[luc2]-rs7132908A-LIMA1, pGL4.10[luc2]-LIMA1, pGL4.10[luc2]-rs7132908G-RACGAP1, pGL4.10[luc2]-rs7132908A-RACGAP1, pGL4.10[luc2]-RACGAP1, and gRNA_Cloning-rs7132908gRNA) and cell lines (WA09 (H9) rs7132908 AA human embryonic stem cell clones 2.1, 9.1, and 10.1) generated in this study will be available from the lead contact with a completed Materials Transfer Agreement. This study did not generate any other new unique reagents.
Data and code availability
Hi-C, RNA-seq, ATAC-seq, single-nucleus RNA-seq, and single-nucleus ATAC-seq data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table. Human embryonic stem cell and tissue genotyping data reported in this study cannot be deposited in a public repository to protect donor confidentiality. To request access, contact the lead contact. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Primary astrocyte model
Primary Normal Human Astrocytes (NHA) of unknown sex were obtained from Lonza as cryopreserved cells. The cells were obtained at passage 1 and used before passage 10, as recommended. They were cultured following Lonza technical instructions in Lonza Astrocyte Growth Medium and in a humidified incubator at 37°C with 5% CO2. For thawing, cells were thawed quickly at 37°C, resuspended, and added slowly to an excess of warmed medium to seed at approximately 6,500 cells/cm2 in a T75 flask. For passaging, 70–80% confluent cells were washed with 30 mM HEPES buffered saline solution in water, incubated at 37°C with 0.025% trypsin-EDTA in DPBS for 3–4 min or until 90% of the cells rounded up, treated with 2 volumes of 5% FBS in DPBS to neutralize the trypsin, rinsed off the culture vessel with gentle pipetting, pelleted by centrifugation at 160 rcf for 5 min at 4°C, and then resuspended and seeded at the desired density. The cells were cultured in T75 flasks, 6-well plates, and 24-well plates. For freezing, cells were lifted as for passaging, resuspended to 1,000,000 cells/mL in FBS with 10% DMSO, frozen in 1 mL aliquots at −1 °C/min, and stored long-term in liquid nitrogen. The cells tested negative for mycoplasma contamination (Figure S6A).
HEK293T model
293T human female cells were obtained from ATCC as cryopreserved cells (ATCC Cat# CRL-3216; RRID: CVCL_0063). They were cultured following ATCC product information in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FBS, 1X Antibiotic-Antimycotic, and 2 mM L-glutamine and in a humidified incubator at 37°C with 5% CO2. For thawing, cells were thawed quickly at 37°C, resuspended, added slowly to an excess of warmed medium, pelleted by centrifugation at 125 rcf for 7 min at 25°C, resuspended in warmed medium, and seeded at approximately 17,500 cells/cm2 in a 10 cm dish. For passaging, 90% confluent cells were washed with PBS, incubated at 37°C with 0.25% trypsin-EDTA for 4–5 min, treated with 2 volumes of medium to neutralize the trypsin, pelleted by centrifugation at 1,200 rcf for 2 min at 25°C, and then resuspended and seeded at the desired density. The cells were cultured in 10 cm dishes, 6-well plates, and 24-well plates. For freezing, cells were lifted as for passaging, resuspended to 1,000,000 cells/mL in medium with 5% DMSO, frozen in 1 mL aliquots at −1 °C/min, and stored long-term in liquid nitrogen. The cells tested negative for mycoplasma contamination (Figure S6A).
ESC model
WA09 (H9) human female embryonic stem cells were obtained from the WiCell Research Institute as cryopreserved cells (WiCell Lot# DL-05; RRID: CVCL_9773). Before use, the cells were authenticated with short tandem repeat analysis to confirm cell line identity. They were cultured following WiCell protocols in mTeSR1 medium, on Matrigel hESC-qualified matrix, and in a humidified incubator at 37°C with 5% CO2. During CRISPR editing, the cells were briefly cultured on Matrigel Growth Factor Reduced Basement Membrane Matrix diluted in IMDM and mouse embryonic fibroblasts (MEFs) and in DMEM/F12 medium supplemented with 15% volume KnockOut Serum Replacement, 100 μM non-essential amino acids, 1 mM sodium pyruvate, 2 mM L-glutamine, 50 U/mL penicillin-streptomycin, 0.1 mM β-mercaptoethanol, and 10 ng/mL human bFGF. For thawing, cells were thawed quickly at 37°C, resuspended, added slowly to an excess of warmed medium, pelleted by centrifugation at 200 rcf for 5 min at 25°C, resuspended in warmed medium, and seeded into 1 well of a 6-well plate. For passaging as colonies, cells in large colonies were washed with Versene, incubated at room temperature with Versene for 6–9 min, rinsed off the culture vessel with medium and gentle pipetting, and then split across new culture vessels, generally using a 1:12 ratio. For passaging as single cells, cells in large colonies were washed with DPBS, incubated at 37°C with Accutase for 2–5 min, treated with 2 volumes of medium to neutralize the Accutase, pelleted by centrifugation at 200 rcf for 4 min at 25°C, and then resuspended and seeded at the desired density. For passaging when cultured on MEFs, MEFs were removed by incubating with TrypLE Express Enzyme for 3 min at room temperature. 10 μM ROCK Inhibitor Y-27632 was added to the medium for 24 h after thawing or passaging as single cells. The cells were cultured in 10 cm dishes, T25 flasks, 6-well plates, and 24-well plates. For freezing, cells were lifted as colonies as for passaging, pelleted by centrifugation at 200 rcf for 4 min at 25°C, resuspended in 2 mL mFreSR medium/lifted well of a 6-well plate, frozen in 1 mL aliquots at −1 °C/min, and stored long-term in liquid nitrogen. The cells were validated with karyotyping (Figure S6B) and tested negative for mycoplasma contamination (Figure S6A).
Pediatric postmortem brain tissue
Frozen human pediatric hypothalamus tissue from 4 postmortem individuals were obtained. The tissue donors included a 4-year-old male, 8-year-old male, 4-year-old female, and 14-year-old female, all classified as white and with no clinical diagnoses. The number of samples was limited by tissue availability.
Method details
Mycoplasma contamination testing
Cells were cultured in the absence of antibiotics for several days and until 90–100% confluent. Medium was then collected and used to detect mycoplasma by PCR using the LookOut Mycoplasma PCR Detection kit with JumpStart Taq DNA polymerase, following manufacturer’s instructions. PCR products, including positive and negative controls, were visualized with gel electrophoresis. Band sizes from experimental samples were compared to the negative control to determine that all cell cultures were negative for mycoplasma contamination (Figure S6A).
Bulk ATAC-seq library preparation
ATAC-seq libraries were prepared from primary astrocytes with 3 technical replicates, the rs7132908 non-risk G allele ESCs with 3 technical replicates and the rs7132908 risk A allele ESCs with 3 biological replicates. 50,000–100,000 cells from each replicate were centrifuged at 550 rcf for 5 min at 4°C to pellet. Each cell pellet was washed with cold PBS and resuspended in 50 μL cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, and 0.1% IGEPAL CA-630) then immediately centrifuged at 550 rcf for 10 min at 4°C. Nuclei were resuspended in transposition reaction mix (25 μL 2X Tagment DNA Buffer, 2.5 μL TDE1 Tagment DNA Enzyme, and 22.5 μL nuclease-free water) on ice, then incubated for 45 min at 37°C. The tagmented DNA was then purified using the Qiagen MinElute PCR Purification kit and eluted in 10.5 μL elution buffer. 10 μL of each purified tagmented DNA sample was amplified with PCR using the Nextera DNA CD Indexes kit and NEBNext High-Fidelity PCR Master Mix for 12 cycles to generate each library. The libraries were purified using AMPure XP beads at a 1.8X concentration. Library concentrations were measured with Qubit dsDNA High Sensitivity Assays. The completed libraries were assessed with the Agilent Bioanalyzer DNA 1000 kit and 2100 Bioanalyzer Expert software (RRID: SCR_019715). Completed libraries were pooled and sequenced on the Illumina NovaSeq 6000 platform using paired-end 51 bp reads.
Hi-C library preparation
Hi-C libraries were prepared from primary astrocytes with two technical replicates using the Arima-HiC kit, following manufacturer’s instructions and as previously described.43 In brief, cells were crosslinked with formaldehyde and then chromatin was digested with multiple restriction enzymes. The purified proximally-ligated DNA was then sheared and 200–600 bp DNA fragments were selected with AMPure XP beads. The size-selected fragments were then enriched using Enrichment Beads and then converted to Illumina-compatible sequencing libraries using the Swift Accel-NGS 2S Plus DNA Library kit and Swift 2S Indexing kit. The libraries were assessed using the Agilent Bioanalyzer DNA 1000 kit and 2100 Bioanalyzer Expert software (RRID: SCR_019715) and the KAPA Library Quantification kit. Completed libraries were pooled and sequenced on the Illumina NovaSeq 6000 platform using paired-end 101 bp reads.
RNA extraction from cells
To extract RNA from cultured cells for RNA-seq or real-time qPCR, cells were lifted and resuspended in TRIzol. RNA was extracted from each TRIzol sample with the Zymo Direct-zol RNA Miniprep kit, following manufacturer’s instructions, with recommended DNase I treatment.
DNA and RNA extraction from tissue
DNA and RNA were extracted from frozen human pediatric hypothalamus tissue samples in parallel. Each tissue sample was homogenized in DNA/RNA Shield in 2 mm ZR BashingBead Lysis Tubes with a FastPrep-24 5G high-speed benchtop homogenizer at 10 m/s at room temperature for 45 s. DNA and RNA were then extracted using the Zymo Quick-DNA/RNA Miniprep Plus kit, following manufacturer’s instructions.
Bulk RNA-seq library preparation
RNA extracted from each cell line and tissue sample was quantified and assessed with the Agilent Bioanalyzer RNA 6000 Nano kit and 2100 Bioanalyzer Expert software (RRID: SCR_019715). Cell line samples with an RNA integrity number (RIN) greater than 7 and tissue samples with a RIN greater than 5 were used for RNA-seq library preparation. RNA-seq libraries were prepared from each tissue sample with 3 technical replicates, primary astrocytes with 3 technical replicates, the rs7132908 non-risk G allele ESCs with 2 technical replicates, the rs7132908 risk A allele ESCs with 3 biological replicates, and hypothalamic neural progenitors with either allele from two independent differentiations (biological replicates) with 3 technical replicates. 40 ng to 1 μg of each RNA sample was used as input, depending on RNA extraction yield. Ribosomal RNA was depleted using the QIAseq FastSelect RNA Removal kit, following manufacturer’s instructions. Libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep for Illumina kit, NEBNext Oligos for Illumina (Dual Index Primers Set 1), and AMPure XP beads, following manufacturer’s instructions. Library concentrations were quantified with Qubit dsDNA High Sensitivity Assays. 5 ng of each library was used for assessment with the Agilent Bioanalyzer DNA 1000 kit and 2100 Bioanalyzer Expert software (RRID: SCR_019715). If the electropherogram did not display a narrow sample distribution around 300 bp, an additional bead cleanup or column purification was used to remove any contaminating primers, adapter-dimers, or large fragments generated by over-amplification. Completed libraries were pooled and sequenced on the Illumina NovaSeq 6000 platform using paired-end 51 bp reads.
Primary astrocyte transfection optimization
To optimize transfection of the primary astrocytes, we transfected with varying amounts of LentiCRISPRv2-mCherry vector DNA, which was a gift from Agata Smogorzewska (Addgene Cat# 99154; http://n2t.net/addgene:99154; RRID: Addgene_99154), Lipofectamine LTX, and PLUS Reagent and then quantified transfection efficiency and cell viability with flow cytometry in two separate experiments. Primary astrocytes were seeded at 50,000 cells/well in a 24-well plate and maintained until they reached 70–80% confluence. Lipofectamine LTX-DNA complexes with PLUS Reagent were prepared following manufacturer’s instructions in Opti-MEM so that each well would receive either 0 ng, 250 ng, 500 ng, or 750 ng vector DNA, 1 μL PLUS Reagent/1 μg of vector DNA, and either a 1:1, 1:2, 1:2.5, 1:3, 1:4, or 1:5 vector DNA (μg):Lipofectamine LTX (μL) ratio.
Approximately 22 h post-transfection, the cells were lifted, resuspended in PBS, fixed in 2% paraformaldehyde for 10 min at room temperature, resuspended in PBS, strained using a 35 μm strainer, and then counted using a CytoFLEX S N2-V3-B5-R3 Flow Cytometer. 10,000 events were collected for each condition and gating was set using the non-transfected control condition. Percent single cell events was calculated by dividing the number of single cell events by all events (10,000). Percent cell viability was then calculated by dividing the percent single cell events for each condition by the average percent single cell events for 2 replicates of non-transfected controls. Transfection efficiency was calculated by dividing the number of mCherry+ single cell events by the number of single cell events in each condition. This optimization experiment determined that ideal conditions for transfecting primary astrocytes at 70–80% confluence in a 24-well plate for 22 h are 750 ng vector DNA, 0.75 μL PLUS Reagent, and 1.875 μL Lipofectamine LTX (1:2.5 ratio) diluted in Opti-MEM for a total volume of 50 μL/well, which was used for all future primary astrocyte transfection experiments. These transfection conditions yielded high transfection efficiency (11.26%) when considering that the expected efficiency is 5–12%123 and high cell viability (85.69%) (Figures S7A and S7B).
Generation of luciferase assay vectors
The ENCODE consortium’s ‘Registry of candidate cis-Regulatory Elements’ (version 1) (RRID: SCR_006793) annotated a cell type-agnostic regulatory element with a distal enhancer-like signature surrounding rs7132908 at chr12:50,262,620–50,263,581 (GRCh37).48 To generate a DNA fragment containing this sequence with an additional 50 bp flanking each side for cloning, we designed PCR primers (Table S13) to amplify this region of interest and used a FAIM2 3′ UTR miRNA target clone (purchased from GeneCopoeia) as the PCR template and NEBNext High-Fidelity PCR Master Mix. Candidate effector genes were selected using the criteria that the promoters of these genes interacted with rs7132908, the promoters of these genes and rs7132908 were both in open chromatin, and that these genes were expressed (TPM >1) in primary astrocytes. To generate DNA fragments containing the FAIM2, LIMA1, and RACGAP1 promoter sequences, we also designed PCR primers (Table S13) to amplify these regions and used promoter clones (purchased from GeneCopoeia) as PCR templates. The promoterless pGL4.10[luc2] firefly luciferase reporter vector (purchased from Promega) was linearized at the multiple cloning site upstream of the luc2 reporter gene using the XhoI restriction enzyme. Each PCR product and the linearized plasmid were extracted after visualization with gel electrophoresis with the NEB Monarch DNA Gel Extraction kit to ensure that a fragment of correct length was purified. The putative enhancer region containing rs7132908 and each promoter were inserted at the multiple cloning site of pGL4.10[luc2] using the Codex Gibson Assembly HiFi HC 1-Step kit to generate pGL4.10[luc2]-rs7132908G-FAIM2, pGL4.10[luc2]-rs7132908G-LIMA1, and pGL4.10[luc2]-rs7132908G-RACGAP1 vectors. Each promoter alone was also inserted at the multiple cloning site to generate pGL4.10[luc2]-FAIM2, pGL4.10[luc2]-LIMA1, and pGL4.10[luc2]-RACGAP1 control vectors. Each Gibson Assembly product was used to transform NEB Stable Competent E. coli which were then plated on LB agarose plates with 100 μg/mL ampicillin to select for successfully transformed colonies. Bacterial plates were incubated overnight at 37°C and then individual colonies were selected for overnight growth in LB broth with 100 μg/mL ampicillin at 30°C with shaking at 250 rpm. Vector DNA was extracted from each overnight culture using the Qiagen QIAprep Spin Miniprep kit and then Sanger sequenced (Table S13) on both strands throughout the modified region to confirm successful insertion and sequence. Electropherograms and sequence files produced from Sanger sequencing were analyzed using SnapGene software (RRID: SCR_015052). Once vectors with perfect sequences were identified, we used the NEB Q5 Site-Directed Mutagenesis kit and primers (Table S13) to introduce the childhood obesity risk A allele at rs7132908 and generate pGL4.10[luc2]-rs7132908A-FAIM2, pGL4.10[luc2]-rs7132908A-LIMA1, and pGL4.10[luc2]-rs7132908A-RACGAP1 vectors. We used Sanger sequencing (Table S13) on both strands throughout the modified region to confirm successful mutagenesis and lack of polymerase errors. Bacteria glycerol stocks were prepared to store each transformed strain with verified sequences long-term. Each experimental vector, the unmodified pGL4.10[luc2] control vector, and pRL-TK (purchased from Promega) co-transfection control vector were then purified for transfection using the Qiagen EndoFree Plasmid Maxi kit. Each purified vector was used for three transfections and purification from glycerol stock was repeated, as needed.
Transfection of primary astrocytes
Primary astrocytes were seeded in three 24-well plates at varying densities so that they would reach 70–80% confluence on three different days for independent transfections. Once each plate reached 70–80% confluence, the cells were transfected in triplicate using optimized conditions to deliver 750 ng pGL4.10[luc2] firefly luciferase reporter vector DNA (unmodified, modified with promoter only, or modified with putative enhancer region and promoter) and 75 ng pRL-TK renilla luciferase reporter vector DNA. Three wells were also treated with only Opti-MEM and transfection reagents to serve as a mock transfected control. The cells were then cultured for approximately 22 h in a humidified incubator at 37°C with 5% CO2. This transfection process was repeated two more times with freshly thawed primary astrocytes with matched passage numbers and freshly purified vectors so that 9 independent transfections were completed.
Transfection of HEK293Ts
HEK293Ts were seeded in three 24-well plates at varying densities so that they would reach 70–90% confluence on three different days for independent transfections. Once each plate reached 70–90% confluence, the cells were transfected in triplicate with 500 ng pGL4.10[luc2] firefly luciferase reporter vector DNA (unmodified, modified with promoter only, or modified with putative enhancer region and promoter) and 50 ng pRL-TK renilla luciferase reporter vector DNA with 1 μL P3000 Reagent and 0.75 μL Lipofectamine 3000 diluted in Opti-MEM for a total volume of 50 μL/well. Three wells were also treated with only Opti-MEM and transfection reagents to serve as a mock transfected control. The cells were then cultured for approximately 24 h in a humidified incubator at 37°C with 5% CO2. This transfection process was repeated two more times with freshly thawed HEK293Ts with matched passage numbers and freshly purified vectors so that 9 independent transfections were completed.
Luciferase assay
Luciferase assay reagents were prepared using the Promega Dual-Luciferase Reporter Assay System, according to manufacturer’s instructions. After transfection with luciferase reporter vectors, primary astrocytes were washed with PBS, incubated in 500 μL Passive Lysis Buffer/well with rocking at room temperature for 15 min, and then gently pipetted to aid lysis with mechanical force. After transfection with luciferase reporter vectors, HEK293Ts were washed with PBS and lysed in 500 μL Passive Lysis Buffer/well with rocking at room temperature for 10 min. Each lysate was then collected and vortexed for 10 s. 20 μL of each lysate was added to a white, flat-bottom 96-well plate in triplicate for a total of 9 wells/condition. 20 μL Passive Lysis Buffer was also added to 9 wells to serve as a negative control. Each well was assayed using a SpectraMax iD5 Multi-Mode Microplate Reader by injecting 100 μL Luciferase Assay Reagent II, waiting 2 s, measuring firefly luciferase fluorescence for 10 s, injecting 100 μL Stop & Glo Reagent, waiting 2 s, and measuring renilla luciferase fluorescence for 10 s.
Generation of rs7132908 risk allele ESCs
A guide RNA and homology-directed repair template (Table S13) were designed to change the rs7132908 non-risk G allele to the obesity risk A allele with CRISPR-Cas9 in the ESC model. These methods were adapted from a previously published protocol for highly efficient CRISPR-Cas9 editing in human stem cells.124 The guide RNA was designed with the help of the CRISPOR program (RRID: SCR_015935).85 The guide RNA was prepared by incorporating the 20 bp target sequence into two 60-mer oligos (Table S13) purchased as 25 nmol DNA oligos from IDT which were then annealed, amplified with PCR using Phusion High-Fidelity DNA polymerase, and purified with extraction with the Takara NucleoSpin Gel and PCR Clean-Up kit after visualization with gel electrophoresis. The guide RNA was then cloned into the gRNA_Cloning vector,83 which was a gift from George Church (Addgene Cat# 41824; http://n2t.net/addgene:41824; RRID: Addgene_41824), at the AflII restriction site with the NEB Gibson Assembly kit to generate the gRNA_Cloning-rs7132908gRNA vector. The homology-directed repair template (Table S13) was prepared by designing a 100 bp single-stranded oligonucleotide centered around the gRNA sequence and with the desired base change, which was then purchased as a 4 nmol Ultramer DNA oligo from IDT. 0.5 μg gRNA_Cloning-rs7132908gRNA vector, 0.5 μg pCas9_GFP vector,84 which was a gift from Kiran Musunuru (Addgene Cat# 44719; http://n2t.net/addgene:44719; RRID: Addgene_44719), and 1 μg homology-directed repair template/well were transfected into 70–80% confluent ESCs on irradiated MEFs in a 6-well plate with 3 μL/well Lipofectamine Stem in 50 μL DMEM/F12. The cells were cultured in a humidified incubator at 37°C with 5% CO2 for 48 h. After transfection, single cells were lifted and 5,000–15,000 GFP+ cells were sorted into a 10 cm dish coated with Matrigel Growth Factor Reduced Basement Membrane Matrix diluted in IMDM and MEFs with fluorescence-activated cell sorting. After 10–15 days of maintenance, individual clones were manually picked and used for both screening and expansion. Some cells from each clone were used for Proteinase K DNA extraction. This DNA was used as a template for PCR across the edited region using the Phusion High-Fidelity DNA polymerase and the PCR products were then used for both restriction digestion screening and Sanger sequencing to confirm the base change (Figure S6C and S6D). Restriction digestion was a possible screening method because the change from the rs7132908 non-risk G allele to obesity risk A allele generated a unique BfaI restriction site. Electropherograms and sequence files produced from Sanger sequencing were analyzed using SnapGene software (RRID: SCR_015052). Clones confirmed to be homozygous for the rs7132908 obesity risk A allele underwent further validation with karyotyping (Figure S6B), de novo CNV analysis (Table S11), mycoplasma contamination testing (Figure S6A), and Sanger sequencing at the top 10 most likely off-target sites (Table S12).
Karyotyping
ESCs were passaged into a T25 flask and cultured under normal conditions until the cells reached 60–70% confluence. The flask was then packaged and shipped to Cell Line Genetics for G-band karyotyping of live cultures. Karyotyping reports indicated that all ESC lines had a normal human female karyotype (Figure S6B).
DNA extraction from cells
To extract DNA from cultured cells for genotyping, PCR, or Sanger sequencing, cells were lifted and then DNA was extracted with the Zymo Quick-DNA Miniprep Plus kit, following manufacturer’s instructions.
SNP genotyping
Genome-wide genotyping of DNA from ESC lines for de novo CNV analysis was performed using the Illumina Infinium Global Screening Array v3.0 BeadChip genotyping array. Genome-wide genotyping of DNA from human pediatric hypothalamus tissue was performed using the Illumina OmniExpressExome v1.6 BeadChip genotyping array. Genotyping arrays consist of many thousands of short invariant 50mer oligonucleotide probes conjugated to silica beads. Sample DNA is hybridized to the probes and a single-base, hybridization-dependent extension reaction is performed at each target SNP. Arrays are loaded onto an iScan System and scanned to extract data. DMAP files enable identification of bead locations on the BeadChip and quantification of the signal associated with each bead. Alternate alleles (herein denoted A and B) are labeled with different fluorophores. Raw fluorescence intensity from the two-color channels is processed into a discrete genotype call (normalized to continuous value 0-1 B-Allele Frequency (BAF)) and the total intensity from both channels (normalized to continuous value with median = 0 Log R Ratio (LRR)) at each SNP which are informative for copy number.
Screening for CRISPR off-target effects
The CRISPOR program (RRID: SCR_015935)85 was used to identify potential off-target sites for the guide RNA designed to change the rs7132908 non-risk G allele to the obesity risk A allele. Each potential off-target site was ranked by Cutting Frequency Determination score which is used to measure guide RNA specificity. Primers were designed to PCR amplify and Sanger sequence the top 10 potential off-target sites. Six potential off-target sites were excluded from screening because primers could not be designed in these regions with a melting temperature between 56°C and 70°C, likely because these regions were too repetitive. Each potential off-target site was amplified using the Phusion High-Fidelity DNA polymerase. Each PCR product was extracted after visualization with gel electrophoresis with the NEB Monarch DNA Gel Extraction kit to ensure that a fragment of correct length was purified. Each purified PCR product was then Sanger sequenced on both strands. Electropherograms and sequence files produced from Sanger sequencing were analyzed using SnapGene software (RRID: SCR_015052). Sequences from each CRISPR clone were compared to sequences from the parent H9 ESC line to determine that there were no off-target effects in all clones at the top 10 most likely off-target sites (Table S12).
Preparation of differentiation medium
Differentiation medium was prepared as previously published6 with some modifications. This medium is an optimized, serum-free reformulation of B27 which supports high quality neuronal cultures and overcomes quality variability of B27 due to different sources of bovine serum albumin. A 50X differentiation supplement was prepared containing DMEM/F12 with 1 μg/mL corticosterone, 50 μg/mL linoleic acid, 50 μg/mL linolenic acid, 2.35 μg/mL (±)-α-lipoic acid, 0.32 μg/mL progesterone, 5 μg/mL retinyl acetate, 50 μg/mL (±)-α-tocopherol, 50 μg/mL DL-α-tocopherol acetate, 125 mg/mL bovine serum albumin, 27.15 mg/mL sodium bicarbonate, 3.2 mg/mL L-ascorbic acid, 805 μg/mL putrescine dihydrochloride, 750 μg/mL D(+)-galactose, 250 μg/mL holo-transferrin, 125 μg/mL catalase, 100 μg/mL L-carnitine hydrochloride, 50 μg/mL glutathione, 0.7 μg/mL sodium selenite, 50 μg/mL ethanolamine, 0.1 μg/mL triiodo-L-thyronine sodium salt, and 200 μg/mL insulin. Differentiation medium was then prepared containing DMEM/F12 with 1X differentiation supplement, 1X Antibiotic-Antimycotic, 1X GlutaMAX, and 2.5 μg/mL superoxide dismutase.
Differentiation to neural progenitors
ESCs were plated as single cells at 1 million cells/well in a matrigel-coated 6-well plate or 200,000 cells/well in a matrigel-coated 24-well plate and cultured in mTeSR1 medium with 10 μM ROCK Inhibitor Y-27632 for 24 h in a humidified incubator at 37°C with 5% CO2. After 24 h, on day 0, the medium was changed to differentiation medium with 1 μM LDN-193189 and 10 μM SB-431542 for dual SMAD inhibition. On days 2, 4, 6, and 8, the medium was changed to differentiation medium with 1 μM LDN-193189, 10 μM SB-431542, 1 μM SAG, 1 μM Purmorphamine, and 10 μM IWR-1-endo for dual SMAD and Wnt signaling inhibition and Shh activation. This method directed the ESCs toward ventral diencephalon forebrain cell identity. On days 9, 11, and 13, the medium was changed to differentiation medium with 10 μM DAPT and 0.01 μM retinoic acid to direct the cells to exit cell cycle. Hypothalamic neural progenitors were collected for downstream experiments on day 14 (Figure 3A). These methods were previously optimized and validated.6 To confirm hypothalamic neural progenitor identity, we performed immunohistochemistry and observed expected expression of NKX2-1, which is a marker for the developing hypothalamus125 (Figure S1F), and NeuN, which is a marker for post-mitotic neurons (Figure S1G).
Differentiation to neurons
On day 14, hypothalamic neural progenitors were washed with DPBS, incubated at 37°C with Accutase for up to 7 min, treated with 2 volumes of medium to neutralize the Accutase, pelleted by centrifugation at 200 rcf for 3 min at 25°C, resuspended in differentiation medium with 10 ng/mL BDNF, and seeded at 1 million cells/well in a laminin-coated 6-well plate or 200,000 cells/well in a laminin-coated 24-well plate. Laminin-coated plates were prepared by diluting laminin to 0.05 mg/mL in cold Hanks’ Balanced Salt Solution, distributing 10 mL laminin solution across each plate, incubating overnight at 4°C, incubating at 37°C for 2 h before use, and washing with PBS 3 times before use. The medium was replaced with fresh differentiation medium with 10 ng/mL BDNF every 2–3 days until day 40 to promote hypothalamic neuron maturation. These methods were previously optimized and validated.6
Fluorescent immunohistochemistry
Cells for immunohistochemistry were cultured on acid-treated #1.5 glass coverslips. The cells were washed with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, and then incubated with PBS for 5 min at room temperature three times to wash. The cells were incubated in blocking solution (PBS with 5% (w/v) bovine serum albumin and 0.3% Triton X-100) for 1 h at room temperature. After blocking, primary antibodies (Anti-MAP2 (Abcam Cat# ab5392; RRID: AB_2138153), Anti-NKX2-1 (Cell Marque Cat# 343M-95; RRID: AB_1158934), and Anti-NeuN (Millipore Sigma Cat# MAB377; RRID: AB_2298772)) diluted in blocking solution (1:500) were added to the cells, then incubated overnight at 4°C with gentle rocking. After the primary antibody incubation, the cells were incubated with PBST for 10 min at room temperature three times to wash. Appropriate secondary antibodies (Anti-Chicken (Abcam Cat# ab150169; RRID: AB_2636803) and Anti-Mouse (Invitrogen Cat# A-11001; RRID: AB_2534069)) diluted in blocking solution (1:500) were added to the cells, then incubated for 1 h at room temperature, protected from light. After the secondary antibody incubation, the cells were incubated with PBST for 5 min at room temperature three times to wash. The cells were then washed with PBS for 3 min at room temperature and incubated with 300 nM DAPI for 5 min at room temperature to stain nuclei. After DAPI incubation, the cells were washed with PBS three times. The glass coverslips were mounted on glass slides with ProLong Gold Antifade Mountant. The cells were visualized with an Olympus DP74 camera using appropriate fluorescent filters and Olympus cellSens Standard software. Images for each fluorescent channel were merged using ImageJ (RRID: SCR_003070).90
Nuclei isolation
After hypothalamic neuron differentiation, the cells were washed with PBS, incubated at 37°C with Accutase for up to 7 min, treated with 2 volumes of medium to neutralize the Accutase, and pelleted by centrifugation at 300 rcf for 5 min at 4°C. The cell pellet was resuspended in PBS with 0.04% bovine serum albumin. 1 million cells or less were pelleted by centrifugation at 300 rcf for 5 min at 4°C and then resuspended in 100 μL chilled lysis buffer (water with 10 mM Trizma hydrochloride, 10 mM sodium chloride, 3 mM magnesium chloride, 1% bovine serum albumin, 0.1% Tween 20, 1 mM DTT, 1 U/μL RNase inhibitor, and 0.1% IGEPAL CA-630). The cells were incubated in lysis buffer on ice for 1 min and then 500 μL chilled wash buffer (water with 10 mM Trizma hydrochloride, 10 mM sodium chloride, 3 mM magnesium chloride, 1% bovine serum albumin, 0.1% Tween 20, 1 mM DTT, and 1 U/μL RNase inhibitor) was added. The nuclei were pelleted by centrifugation at 500 rcf for 5 min at 4°C. Addition of chilled wash buffer and pelleting were repeated two more times. The nuclei were then resuspended in chilled nuclei buffer (water with 1X Nuclei Buffer, 1 mM DTT, and 1 U/μL RNase inhibitor) to a concentration of 8,000 nuclei/μL in at least 25 μL and strained using a 35 μm strainer.
Single-nucleus library preparation
Single-nucleus RNA-seq and ATAC-seq libraries were prepared using the 10X Genomics Chromium Single Cell Multiome ATAC + Gene Expression workflow. Libraries were prepared from the rs7132908 non-risk G allele cells from two independent differentiations (biological replicates) for a total of 4 technical replicates and from the rs7132908 risk A allele cells from two CRISPR clones (biological replicates) and three independent differentiations (biological replicates) for a total of 4 technical replicates. In brief, isolated nuclei in chilled nuclei buffer were transposed in bulk which simultaneously fragmented DNA in regions of open chromatin and added adapter sequences to the ends of the DNA fragments. The transposed nuclei were then loaded onto a microfluidic chip which was run in the Chromium Controller instrument. In the instrument, nuclei were individually partitioned with Gel Beads-in-emulsion (GEMs). Each Gel Bead contains oligonucleotides with a unique 16 bp 10X Barcode sequence, a poly(dT) sequenced to capture mRNA, and a Spacer sequence that enables barcode attachment to transposed DNA fragments. The GEMs were then incubated to attach unique 10X Barcodes to mRNA and transposed DNA fragments which served to associate mRNA and transposed DNA fragments back to the same nucleus. Unique molecular identifiers (UMIs) were also used to distinguish individual, captured mRNA molecules for quantification. A reverse transcription reaction converted the mRNA into full-length cDNA. The GEMs were then broken and pooled fractions were recovered and purified. The products were taken through a pre-amplification PCR step to fill gaps and ensure maximum recovery of barcoded ATAC and cDNA fragments. The pre-amplified products were then used as input for both ATAC-seq library preparation and cDNA amplification for RNA-seq library preparation. Completed RNA-seq libraries were quantified and assessed with Agilent High Sensitivity D1000 ScreenTape assays and ATAC-seq libraries were quantified and assessed with Agilent High Sensitivity D5000 ScreenTape assays. RNA-seq libraries were then pooled and sequenced on the Illumina NovaSeq 6000 platform to reach a minimum of 20,000 paired-end reads/nucleus. ATAC-seq libraries were then pooled and sequenced on the Illumina NovaSeq 6000 platform to reach a minimum of 25,000 paired-end reads/nucleus.
cDNA generation
RNA samples were quantified with Qubit RNA High Sensitivity Assays. 30 ng of each RNA sample was used for cDNA generation using SuperScript IV VILO Master Mix after treatment with ezDNase to remove any DNA contamination. No reverse transcriptase controls were also generated using SuperScript IV VILO ‘No RT’ Control Master Mix.
Real-time qPCR
TaqMan Gene Expression Assays for FAIM2 and human 18S ribosomal RNA were validated with standard curves generated by pooling all cDNA samples quantified in an experiment to represent average conditions of all samples. The FAIM2 standard curve consisted of 5 points generated by a 1:5 serial dilution ranging from 0.0024 to 1.5 ng in triplicate. The 18S standard curve consisted of 8 points generated by a 1:5 serial dilution ranging from 0.0000192 to 1.5 ng in triplicate. Each sample was quantified with TaqMan Fast Advanced Master Mix and the Agilent AriaMX Real-Time PCR System. After assay validation, 0.5 ng of each experimental cDNA sample and no reverse transcriptase control were assayed in duplicate. Additionally, no template controls were assayed in triplicate.
Quantification and statistical analysis
Effect size comparison
Effect size (β) values and standard errors for the chr12q13 locus were obtained from the most recent childhood50 and adult20 BMI GWAS. A two-tailed two-sample z-test was used to determine if the β values were significantly different. A p value <0.05 was considered significant.
GWAS-eQTL colocalization
Childhood obesity GWAS summary statistics from the European ancestry population in the EGG consortium were used. Common variants (minor allele frequency ≥0.01) from the 1000 Genomes Project (v3)126 were used as a reference panel. SNP-gene sets from our variant-to-gene mapping efforts were used as leads. We used ColoQuiaL120 to test genome-wide colocalization of each lead against GTEx eQTLs (v8) (RRID: SCR_013042)49 from all 49 available tissues. Evidence of colocalization between a given childhood obesity GWAS signal and eQTL signal was identified by a conditional posterior probability of colocalization ≥0.8.
Gene burden testing
The Penn Medicine BioBank includes 18,573 European and 7,950 African ancestry adult individuals with recorded measurements of BMI that could be used for gene burden testing. Cases of obesity were defined as BMI ≥30 and controls were defined as BMI ≤25. There were 9,748 cases of obesity in the European population and 6,045 in the African population. We used the R package SKAT (RRID: SCR_009396)127 to detect associations between rare variants in each candidate effector gene and obesity as a dichotomous trait while adjusting for covariates such as age, sex, and the first 5 genome-wide principal components.
Luciferase assay data analysis
All fluorescence values were reduced by the average signal in the 9 negative control wells to correct for background fluorescence in the Passive Lysis Buffer and 96-well plate. The firefly luciferase fluorescence signal was then divided by the renilla luciferase fluorescence signal in each well to adjust for sample-to-sample variability due to differences in cell numbers, transfection efficiency, and pipetting. Normalized firefly luciferase fluorescence values were averaged for each condition (n = 9). Normalized fold change was calculated by dividing the average normalized firefly luciferase fluorescence values for each condition by this value produced by the promoter only vector (pGL4.10[luc2]-FAIM2, pGL4.10[luc2]-LIMA1, or pGL4.10[luc2]-RACGAP1).
Assays were excluded from statistical analysis if there was fluorescence detected (normalized fold change >0.1) in the negative control condition or if at least one normalized fold change value was greater than 2 standard deviations away from the mean of all other assays performed. Multiple independent transfections and assays were performed and are stated in the figure legend. All data are represented as mean ± standard deviation. Statistical analyses and visualization were performed using GraphPad Prism (RRID: SCR_002798) and ordinary one-way ANOVA tests with Tukey’s correction for multiple comparisons. p-values <0.05 were considered significant. ∗p-value <0.05, ∗∗p-value <0.01, ∗∗∗p-value <0.001.
Transcription factor binding prediction
The genomic position and alternative allele of rs7132908 (determined using SNPlocs.Hsapiens.dbSNP155.GRCh38 and BSgenome R packages) were used to scan through all position frequency matrix databases using the R package MotifDb to identify potential transcription factor binding disruption effects. The motifbreakR function119 was used with parameters filter = TRUE, threshold = 0.0005, method = ’ic’, bkg = c(A = 0.25, C = 0.25, G = 0.25, T = 0.25), and BPPARAM = BiocParallelSerialParam().
CNV detection
Samples must meet minimum quality control standards of call rate >98% and LRR standard deviation <0.3 to be used for CNV detection. We used PennCNV (RRID: SCR_002518) as our main CNV detection algorithm of the Illumina Infinium Global Screening Array v3.0 data due to its widespread usage. We filtered PennCNV calls to include CNVs with number of SNPs supporting ≥20, length ≥100,000, and Segmental Duplication track coverage <0.5. Related cell line clone CNV calls were compared to ensure consistency in CNV calling. All genomic coordinates are in human genome build version GRCh37.
De novo CNV detection
The related cell line clones annotated for each sample were verified by pairwise comparison of genome-wide SNP genotyping content using PLINK (RRID: SCR_001757). The "child" cell line CNVs were compared to their corresponding "parent" cell line CNVs using bedtools and if at least 50% reciprocal overlap is not observed, annotated as de novo. Such putative de novo calls were BAF LRR plotted for each pair of "child" and "parent" to allow for side-by-side comparison to ensure the de novo was not an erroneous call.
Bulk RNA-seq analysis
Sequencing data was demultiplexed to generate FASTQ files using Illumina bcl2fastq2 Conversion Software (RRID: SCR_015058). FASTQ files were assessed with FastQC (RRID: SCR_014583)86,87 to verify that there was high sequence quality, expected sequence length, and no adapter contamination. Paired-end FASTQ files for each replicate of primary astrocytes were mapped to the human reference genome (GRCh38) using STAR (RRID: SCR_004463).121 Genes were annotated using GENCODE human release 40 (RRID: SCR_014966).128 Raw read counts were calculated using HTSeq-count (RRID: SCR_011867).122 Paired-end FASTQ files for each replicate of all other cell types and tissue were mapped to the Ensembl human reference transcriptome (GRCh38)129 using Kallisto (RRID: SCR_016582).88,89 Abundance data generated with Kallisto was read into R (RRID: SCR_001905) using the package tximport (RRID: SCR_016752),99 annotated with Ensembl human gene annotation data (version 86)129 using ensembldb (RRID: SCR_019103)101 and EnsDb.Hsapiens.v86, and summarized as counts per million (cpm) at the gene level using edgeR (RRID: SCR_012802).100 Genes with less than 1 cpm in 2 or 3 samples, depending on the smallest set of replicates in the analysis, were removed to increase statistical power to detect differentially expressed genes. Samples within each analysis were normalized with the trimmed mean of M values (TMM) method.130 If batch effects were detected by principal component analysis, removeBatchEffects from the R package limma (RRID: SCR_010943)102 was used to correct count values to adjust for batch effects. Pre- and post-adjustment matrices were used for principal component analysis and principal variance component analysis using the R package PVCA (RRID: SCR_001356) to visualize the effects of batch correction. The R package limma (RRID: SCR_010943)102 was used to identify differentially expressed genes by first applying precision weights to each gene based on its mean-variance relationship using the voom function and then linear modeling and Bayesian statistics were employed to detect genes that were up- or down-regulated in each condition. If batch effects were detected by principal component analysis, batch information was included as a covariate in the linear model to adjust for batch effects. Genes with an adjusted p-value <0.05 and |log2 fold change| > 0.58 were considered significantly differentially expressed. Throughout the study, this fold change threshold was used for all differential expression analyses to detect genes that increased by approximately 50% or decreased by approximately 33%. Coordinates for the rs7132908 TAD were determined using the TADKB database51 and considering the most conservative region documented in all reported human cell lines (GRCh37). A list of genes in the rs7132908 TAD region were exporting using the UCSC Genome Browser (GRCh37) (RRID: SCR_005780).131,132 Significantly differentially expressed genes were clustered using Pearson correlation and the R function hclust. The clustered genes were cut into 2 modules in ESCs and 5 modules in hypothalamic neural progenitors. Significantly enriched Gene Ontology terms52,53 in each module were identified using gprofiler2 (RRID: SCR_018190).97,98 Results were visualized using ggplot2 (RRID: SCR_014601),107 gplots, and plotly.
Bulk ATAC-seq analysis
Sequencing data was demultiplexed to generate FASTQ files using Illumina bcl2fastq2 Conversion Software (RRID: SCR_015058). ATAC-seq peaks were called following the ENCODE ATAC-seq pipeline (https://www.encodeproject.org/atac-seq/). Briefly, paired-end reads from three replicates for each cell type were aligned to the human reference genome (GRCh38) using bowtie2 (RRID: SCR_016368),103 and duplicate reads were removed from the alignment using Picard (RRID: SCR_006525) MarkDuplicates and SAMtools (RRID: SCR_002105).104 Narrow peaks were called independently for each replicate using MACS2105 with parameters -p 0.01 --nomodel --shift −75 --extsize 150 -B --SPMR --keep-dup all --call-summits. Reproducible peaks, peaks called in at least 2 replicates (with at least 1 bp overlap), were used to generate a consensus set of peaks. Signal peaks were normalized using csaw106 in 10 kilobase (kb) bin background regions. A threshold of cpm >1 was used to exclude peaks with low abundance from the analysis. Tests for differential accessibility between rs7132908 genotypes were conducted with the glmQLFit approach implemented in edgeR (RRID: SCR_012802)100 using the normalization factors calculated by csaw. Open chromatin regions with adjusted p-value <0.05 and |log2 fold change| > 1 were considered differentially accessible. Results were visualized using ggplot2 (RRID: SCR_014601).107
Hi-C analysis
Hi-C analysis was performed as previously described.43 In brief, sequencing data was demultiplexed to generate FASTQ files using Illumina bcl2fastq2 Conversion Software (RRID: SCR_015058). Paired-end reads from each replicate were pre-processed using the HiCUP pipeline (RRID: SCR_005569)113 and aligned to the human reference genome (GRCh38) with bowtie2 (RRID: SCR_016368).103 The alignments files were parsed to pairtools (RRID: SCR_023038)114 to process and pairix115 to index and compress, then converted to Hi-C matrix binary format (.cool) by cooler116 at multiple resolutions (500 bp, 1, 2, 4, 10, 40, 500 kb and 1 megabase (Mb)) and normalized with the ICE method.133 The matrices from different replicates were merged at each resolution using cooler.116 Mustache117 and Fit-Hi-C2118 were used to call significant intra-chromosomal interaction loops from merged replicates matrices at three resolutions (1 kb, 2 kb, and 4 kb), with significance thresholds of q-value <0.1 and p-value < 1 × 10−6. The identified interaction loops were merged between both tools at each resolution. Lastly, interaction loops from all three resolutions were merged with preference for smaller resolution if there was overlap.
Single-nucleus pre-processing
Cell Ranger ARC analysis pipelines were used to process sequencing data generated with the 10X Genomics Chromium Single Cell Multiome ATAC + Gene Expression workflow. Sequencing data was demultiplexed to generate FASTQ files using mkfastq. The FASTQ files were aligned to the GRCh38 human reference genome with the Cell Ranger ARC package (RRID: SCR_023897) and cells were called using parameters -count --min-atac-count = 2000 --min-gex-count = 1000.
66,120 cells homozygous for the rs7132908 non-risk G allele representing two separate differentiations were sequenced. 45,916 cells homozygous for the rs7132908 obesity risk A allele representing two different clonal lines and three different differentiations were also sequenced. All 112,036 cells then underwent quality control to remove ambient RNA using SoupX (RRID: SCR_019193)92 with the contamination fraction automatically estimated for each sample and the count matrices were re-adjusted after removal. Doublets were detected and removed using the Python package Scrublet (RRID: SCR_018098),91 and cells with >10% mitochondrial reads were filtered out using Seurat (RRID: SCR_016341).94 After quality control, we retained 71,818 cells for downstream analyses.
RNA-seq data from all samples was SCTransformed (RRID: SCR_022146),95,96 integrated using the IntegrateData function, and then batch corrected using Harmony (RRID: SCR_022206)93 for differentiation, biological, and technical replicates. PCA and UMAP reduction were performed using the first 30 empirically selected principal components with standard pipelines (Figures S2F‒S2H).
We ran peak calling using MACS3 (https://macs3-project.github.io/MACS/) for each sample with their corresponding ATAC-seq fragments files. Peaks from all samples were pooled and reduced to a final set of 383,029 peaks accessible in at least one sample. This peak set was used to create a ChromatinAssay using Signac (RRID: SCR_021158).108 The peaks were filtered through ENCODE hg38 blacklist regions (https://github.com/Boyle-Lab/Blacklist/blob/master/lists/hg38-blacklist.v2.bed.gz) and annotated with EnsDb.Hsapiens.v86. We performed quality control following metrics recommended by Signac,108 including nucleosome banding pattern, TSS enrichment score, total number of fragments in peaks, fraction of fragments in peaks, and ratio of reads in genomic blacklist regions; we removed cells that were outliers by these metrics. We performed term frequency-inverse document frequency normalization with the RunTFIDF function and feature selection and dimension reduction using singular value decomposition (SVD) on the TD-IDF matrix with the RunSVD function, which produced latent semantic indexing components (LSI).134 Uniform manifold approximation and projection embedding was computed based on the first 29 LSI components (second to the 30th) for visualization in two-dimensional space with the RunUMAP function. The first component, being in strong correlation with total counts, was not used. Results were visualized using Seurat94 and ggplot2 (RRID: SCR_014601).107
Cell type identification
A previously published human hypothalamic arcuate nucleus single-cell RNA-seq dataset54 was used as a reference dataset to identify cell types in our single-nucleus RNA-seq dataset. Pairwise correspondences or ‘anchors’ between individual cells in each dataset were defined using the Seurat (RRID: SCR_016341) function FindTransferAnchors.55 Then each cell in our dataset was classified as one of the cell types in the reference dataset (neuron, astrocyte, OPC, mature oligodendrocyte, microglia, ependymal, pericyte, immature oligodendrocyte, fibroblast, choroid, and tanycyte) using the Seurat function TransferData,55 where the reference cell type with the highest observed classification score was assigned. As a result, neuron, astrocyte, OPC, ependymal, fibroblast, and tanycyte annotations were added to our dataset (Figure S2A). We then prioritized cells with a classification score ≥0.8 for downstream analyses as this threshold has been previously demonstrated to increase accuracy.55 In summary, we identified 38,044 cells as neurons, OPCs, or fibroblasts with a classification score above our threshold. PCA and UMAP reduction were performed using the first 20 empirically selected principal components with standard pipelines (Figure 4A). All cells annotated as neurons were then subset and reclustered with PCA and UMAP reduction using the first 15 empirically selected principal components (Figure 4C). Results were visualized using Seurat94 and ggplot2 (RRID: SCR_014601).107
Transcriptome correlation
Pseudobulk TPMs were calculated for each annotated cell type and replicate sample in the single-nucleus RNA-seq dataset by normalizing SoupX-corrected counts by gene size using gene annotation data from GENCODE human release 38 (GRCh37) (RRID: SCR_014966)128 and previously published code.135 TPMs from all rs7132908 non-risk allele replicate samples for each annotated cell type were then averaged. Similarly, average TPMs were also calculated for the rs7132908 non-risk allele replicate samples in the bulk RNA-seq datasets generated from the hypothalamic neural progenitors and human pediatric hypothalamus tissue sequenced inhouse. Median gene-level TPM data by tissue was downloaded from the GTEx Analysis RNA-seq database (v8) (RRID: SCR_013042).49 Ensembl gene IDs with version suffixes were converted to gene names using gene annotation data from GENCODE human release 26 (GRCh37) (RRID: SCR_014966).128 Average TPMs for each cell type of interest were merged with average TPMs from the human pediatric hypothalamus tissue and GTEx data. Then, the spearman rank correlation of genes expressed at greater than 5 TPMs in at least 2 samples were calculated using the R (RRID: SCR_001905) cor function. p-values for each correlation were calculated using the R cor.test function. Results were visualized in dot plots using ggplot2 (RRID: SCR_014601).107
To compare the transcriptome of the cells annotated as neurons in the single-nucleus RNA-seq dataset to human prenatal hypothalamic nuclei, data from the Allen Brain Atlas58,59,60,61 was downloaded as upregulated gene sets from the Harmonizome database.136 Left and right hemisphere gene sets for each hypothalamic nucleus were combined and used for downstream analysis. To infer the average expression of each gene set per single cell in the neuron dataset compared to random control genes, module scores for each gene set were calculated using the Seurat (RRID: SCR_016341) function AddModuleScore.137 Average module scores per neuron cluster were plotted as the column Z score for visualization. Results were visualized using ggplot2 (RRID: SCR_014601).107
Single-nucleus differential expression
Differential expression analysis of single-nucleus RNA-seq data was performed with DESeq2 (RRID: SCR_015687),109 following the standard workflow. In brief, raw counts and appropriate metadata for cell aggregation and comparison were extracted and used to create a SingleCellExperiment object using the R package SingleCellExperiment.110,111 Counts were aggregated to the sample level for each cell type using the Matrix.utils function aggregate.Matrix. DESeq2 objects were created from the raw counts, appropriate metadata, and design formula to compare the rs7132908 obesity risk allele to the non-risk allele in each cell type using the DESeq2 function DESeqDataSetFromMatrix.109 Differential expression analysis in each cell type was run using the DESeq2 function results109 and an adjusted p-value threshold of 0.05. The resulting log2 fold changes were shrunk using the apeglm method.112 Genes with an adjusted p-value <0.05 and |log2 fold change| > 0.58 were considered significantly differentially expressed. Throughout the study, this fold change threshold was used for all differential expression analyses to detect genes that increased by approximately 50% or decreased by approximately 33%. Results were visualized in volcano plots using ggplot2 (RRID: SCR_014601).107 Significantly differentially expressed genes were clustered using the R (RRID: SCR_001905) function hclust and plotted in heatmaps using the R package pheatmap (RRID: SCR_016418). Significantly enriched Gene Ontology terms52,53 in each set of genes significantly up- or down-regulated in each cell type were identified using gprofiler2 (RRID: SCR_018190).97,98
Single-nucleus differential accessibility
To find differentially accessible regions due to rs7132908 genotype, we performed differential accessibility tests between cells homozygous for either rs7132908 allele. We implemented logistic regression using the FindMarkers function from Signac (RRID: SCR_021158),108 with the total number of fragments in peaks as a latent variable to mitigate the effect of differential sequencing depth and using a min.pct threshold of 0.01 due to sparse single-nucleus ATAC-seq data. To ensure data correspondence, we used only the 38,044 annotated cells that had a classification score ≥0.8 by the RNA-seq analysis for this differential accessibility analysis. p-value adjustment was performed internally using Bonferroni correction based on the total number of peaks in the dataset. We repeated this analysis for each annotated cell type: neurons, OPCs, and fibroblasts.
We performed DNA motif analysis to identify potentially important genotype-specific regulatory sequences in different groups of differentially accessible peaks. We used motif position frequency matrices from the JASPAR 2022 CORE collection database.138 We detected transcription factor motifs enriched in differentially accessible peaks with an adjusted p-value <0.005 and |log2 fold change| ≥ 1. The FindMotifs function from Signac108 performed hypergeometric test on these differentially accessible peaks to test the probability of observing the motif at the given frequency by chance, compared to a background set of peaks matched for GC content. Motifs with an adjusted p-value <0.05 were considered significantly enriched.
Real-time qPCR analysis
Cq values for each sample were determined with the Agilent Aria software. To validate each TaqMan Gene Expression Assay using a standard curve, Cq values from each triplicate of samples were averaged and then plotted against the log of their corresponding mass of cDNA input (ng) using Microsoft Excel (RRID: SCR_016137). A linear trendline was then added to each graph and the R2 values and linear equations were displayed. Primer efficiency was calculated with . Percent primer efficiency was calculated by dividing the primer efficiency by 2. TaqMan Gene Expression Assays passed standard curve validation if the R2 value was greater than 0.99 and the percent primer efficiency was between 90 and 110%. Assays were used to calculate normalized relative expression if the no reverse transcriptase and no template control samples did not generate a Cq value. Normalized relative expression was calculated using , where E is primer efficiency. Results were visualized using GraphPad Prism (RRID: SCR_002798). Independent differentiations were performed and are represented by individual points on each graph. All data are represented as mean ± standard deviation.
Acknowledgments
We thank Dr. Bill Manley, Madeleine Salvatore, and Danny Frederick for training in stem cell culture; Dr. Guo-li Ming and Dr. Sarshan Pather for sharing data and code; Dr. Dhruv Sareen and Andrew Gross for sharing protocols and technical support; Gina Pacella for experiment design guidance; Dr. Jonathan Schug and Mitchell Conery for providing bioinformatic guidance; Dr. Alexis Crockett for providing mouse brain tissue for a pilot study; Dr. Shaon Sengupta for sharing equipment; Children’s Hospital of Philadelphia Human Pluripotent Stem Cell Core; Children’s Hospital of Philadelphia Center for Applied Genomics; Children’s Hospital of Philadelphia Flow Cytometry Core; University of Pennsylvania Genomic and Sequencing Core DNA Sequencing Laboratory; and University of Pennsylvania Cell Center Services. We acknowledge the Penn Medicine BioBank for providing data and thank the patient-participants of Penn Medicine who consented to participate in this research program. We would also like to thank the Penn Medicine BioBank team and Regeneron Genetics Center for providing genetic variant data for analysis. The Penn Medicine BioBank is approved under IRB protocol #813913 and is supported by the Perelman School of Medicine at the University of Pennsylvania, a gift from the Smilow family, and the National Center for Advancing Translational Sciences of the National Institutes of Health under CTSA award number UL1TR001878. Human tissue was obtained from the NIH Neurobiobank at the University of Maryland, Baltimore, MD. Some figures were created with BioRender.com. S.H.L is supported by the NICHD (F31 HD105404). S.F.A.G. is supported by the NICHD (R01 HD056465), the NIDDK (UM1 DK126194), and the Daniel B. Burke Endowed Chair for Diabetes Research. Biobank-specific acknowledgments are included in the supplemental information.
Author contributions
S.H.L. and S.F.A.G. conceived the project. S.H.L. designed the experiments. S.H.L., C.M.V., N.D., and K.C. performed cell culture. S.H.L. and C.M.V. collected cell materials. S.H.L. processed human tissue. S.H.L. and C.M.V. conducted cell line validation. S.H.L. optimized transfection. S.H.L., C.M.V., and N.D. performed luciferase assays. S.H.L. performed luciferase assay analysis. J.A.M. performed CRISPR. S.H.L. and K.C. performed immunocytochemistry and imaging. S.H.L., K.B., and S.L. prepared bulk RNA-seq libraries. J.A.P. and K.M.H. prepared bulk ATAC-seq libraries. J.A.P. and S.H.L. prepared Hi-C libraries. S.H.L. prepared nuclei for sequencing. J.A.P. sequenced bulk RNA-seq, ATAC-seq, and Hi-C libraries. J.P.B. performed effect size comparison. M.C.P. performed gene burden testing. S.H.L., K.B.T, A.C., and M.C.P. performed bulk RNA-seq analyses. M.C.P., K.B.T., and A.C. performed bulk ATAC-seq analyses. K.B.T. performed transcription factor, colocalization, and Hi-C analyses. S.H.L., K.B.T., and M.A.H. performed single-nucleus RNA-seq analyses. K.B.T. and S.H.L. performed single-nucleus ATAC-seq analyses. S.H.L. performed real-time qPCR. S.F.A.G., M.C.P., A.D.W., S.A.A., and J.A.P. provided critical feedback and supervision. S.H.L. and S.F.A.G. wrote the original manuscript draft. All authors reviewed and edited the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100556.
Supplemental information
References
- 1.Bryan S., Afful J., Carroll M., Te-Ching C., Orlando D., Fink S., Fryar C. National Health and Nutrition Examination Survey 2017–March 2020 Prepandemic Data Files Development of Files and Prevalence Estimates for Selected Health Outcomes. National Health Statistics Reports. 2021 doi: 10.15620/cdc:106273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.NCD Risk Factor Collaboration NCD-RisC Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet. 2017;390:2627–2642. doi: 10.1016/S0140-6736(17)32129-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lobstein T., Baur L., Uauy R., IASO International Obesity TaskForce Obesity in children and young people: a crisis in public health. Obes. Rev. 2004;5(Suppl 1):4–104. doi: 10.1111/j.1467-789X.2004.00133.x. [DOI] [PubMed] [Google Scholar]
- 4.Loos R.J.F., Yeo G.S.H. The genetics of obesity: from discovery to biology. Nat. Rev. Genet. 2022;23:120–133. doi: 10.1038/s41576-021-00414-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silventoinen K., Jelenkovic A., Sund R., Hur Y.M., Yokoyama Y., Honda C., Hjelmborg J.v., Möller S., Ooki S., Aaltonen S., et al. Genetic and environmental effects on body mass index from infancy to the onset of adulthood: an individual-based pooled analysis of 45 twin cohorts participating in the Collaborative project of Development of Anthropometrical measures in Twins (CODATwins) study. Am. J. Clin. Nutr. 2016;104:371–379. doi: 10.3945/ajcn.116.130252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajamani U., Gross A.R., Hjelm B.E., Sequeira A., Vawter M.P., Tang J., Gangalapudi V., Wang Y., Andres A.M., Gottlieb R.A., Sareen D. Super-Obese Patient-Derived iPSC Hypothalamic Neurons Exhibit Obesogenic Signatures and Hormone Responses. Cell Stem Cell. 2018;22:698–712.e9. doi: 10.1016/j.stem.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L., Egli D., Leibel R.L. Efficient Generation of Hypothalamic Neurons from Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2016;90:21.5.1–21.5.14. doi: 10.1002/cphg.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L., Meece K., Williams D.J., Lo K.A., Zimmer M., Heinrich G., Martin Carli J., Leduc C.A., Sun L., Zeltser L.M., et al. Differentiation of hypothalamic-like neurons from human pluripotent stem cells. J. Clin. Invest. 2015;125:796–808. doi: 10.1172/JCI79220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merkle F.T., Maroof A., Wataya T., Sasai Y., Studer L., Eggan K., Schier A.F. Generation of neuropeptidergic hypothalamic neurons from human pluripotent stem cells. Development. 2015;142:633–643. doi: 10.1242/dev.117978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirwan P., Jura M., Merkle F.T. Generation and Characterization of Functional Human Hypothalamic Neurons. Curr. Protoc. Neurosci. 2017;81:33.31–33.33.24. doi: 10.1002/cpns.40. [DOI] [PubMed] [Google Scholar]
- 11.Pahl M.C., Doege C.A., Hodge K.M., Littleton S.H., Leonard M.E., Lu S., Rausch R., Pippin J.A., De Rosa M.C., Basak A., et al. Cis-regulatory architecture of human ESC-derived hypothalamic neuron differentiation aids in variant-to-gene mapping of relevant complex traits. Nat. Commun. 2021;12:6749. doi: 10.1038/s41467-021-27001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L., Liu Y., Stratigopoulos G., Panigrahi S., Sui L., Zhang Y., Leduc C.A., Glover H.J., De Rosa M.C., Burnett L.C., et al. Bardet-Biedl syndrome proteins regulate intracellular signaling and neuronal function in patient-specific iPSC-derived neurons. J. Clin. Invest. 2021;131 doi: 10.1172/JCI146287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torz L., Niss K., Lundh S., Rekling J.C., Quintana C.D., Frazier S.E.D., Mercer A.J., Cornea A., Bertelsen C.V., Gerstenberg M.K., et al. NPFF Decreases Activity of Human Arcuate NPY Neurons: A Study in Embryonic-Stem-Cell-Derived Model. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms23063260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joslin A.C., Sobreira D.R., Hansen G.T., Sakabe N.J., Aneas I., Montefiori L.E., Farris K.M., Gu J., Lehman D.M., Ober C., et al. A functional genomics pipeline identifies pleiotropy and cross-tissue effects within obesity-associated GWAS loci. Nat. Commun. 2021;12:5253. doi: 10.1038/s41467-021-25614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sobreira D.R., Joslin A.C., Zhang Q., Williamson I., Hansen G.T., Farris K.M., Sakabe N.J., Sinnott-Armstrong N., Bozek G., Jensen-Cody S.O., et al. Extensive pleiotropism and allelic heterogeneity mediate metabolic effects of IRX3 and IRX5. Science. 2021;372:1085–1091. doi: 10.1126/science.abf1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirwan P., Kay R.G., Brouwers B., Herranz-Pérez V., Jura M., Larraufie P., Jerber J., Pembroke J., Bartels T., White A., et al. Quantitative mass spectrometry for human melanocortin peptides in vitro and in vivo suggests prominent roles for beta-MSH and desacetyl alpha-MSH in energy homeostasis. Mol. Metab. 2018;17:82–97. doi: 10.1016/j.molmet.2018.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L., Sui L., Panigrahi S.K., Meece K., Xin Y., Kim J., Gromada J., Doege C.A., Wardlaw S.L., Egli D., Leibel R.L. PC1/3 Deficiency Impacts Pro-opiomelanocortin Processing in Human Embryonic Stem Cell-Derived Hypothalamic Neurons. Stem Cell Rep. 2017;8:264–277. doi: 10.1016/j.stemcr.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao L., Liu Y., Qiu Z., Kumar S., Curran J.E., Blangero J., Chen Y., Lehman D.M. Molecular Profiling of Human Induced Pluripotent Stem Cell-Derived Hypothalamic Neurones Provides Developmental Insights into Genetic Loci for Body Weight Regulation. J. Neuroendocrinol. 2017;29 doi: 10.1111/jne.12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Locke A.E., Kahali B., Berndt S.I., Justice A.E., Pers T.H., Day F.R., Powell C., Vedantam S., Buchkovich M.L., Yang J., et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yengo L., Sidorenko J., Kemper K.E., Zheng Z., Wood A.R., Weedon M.N., Frayling T.M., Hirschhorn J., Yang J., Visscher P.M., GIANT Consortium Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum. Mol. Genet. 2018;27:3641–3649. doi: 10.1093/hmg/ddy271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradfield J.P., Taal H.R., Timpson N.J., Scherag A., Lecoeur C., Warrington N.M., Hypponen E., Holst C., Valcarcel B., Thiering E., et al. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat. Genet. 2012;44:526–531. doi: 10.1038/ng.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradfield J.P., Vogelezang S., Felix J.F., Chesi A., Helgeland Ø., Horikoshi M., Karhunen V., Lowry E., Cousminer D.L., Ahluwalia T.S., et al. A trans-ancestral meta-analysis of genome-wide association studies reveals loci associated with childhood obesity. Hum. Mol. Genet. 2019;28:3327–3338. doi: 10.1093/hmg/ddz161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frayling T.M., Timpson N.J., Weedon M.N., Zeggini E., Freathy R.M., Lindgren C.M., Perry J.R.B., Elliott K.S., Lango H., Rayner N.W., et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smemo S., Tena J.J., Kim K.H., Gamazon E.R., Sakabe N.J., Gómez-Marín C., Aneas I., Credidio F.L., Sobreira D.R., Wasserman N.F., et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Claussnitzer M., Dankel S.N., Kim K.H., Quon G., Meuleman W., Haugen C., Glunk V., Sousa I.S., Beaudry J.L., Puviindran V., et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015;373:895–907. doi: 10.1056/NEJMoa1502214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z., Chen N., Yin N., Liu R., He Y., Li D., Tong M., Gao A., Lu P., Zhao Y., et al. The rs1421085 variant within FTO promotes brown fat thermogenesis. Nat. Metab. 2023;5:1337–1351. doi: 10.1038/s42255-023-00847-2. [DOI] [PubMed] [Google Scholar]
- 28.Ntalla I., Panoutsopoulou K., Vlachou P., Southam L., William Rayner N., Zeggini E., Dedoussis G.V. Replication of established common genetic variants for adult BMI and childhood obesity in Greek adolescents: the TEENAGE study. Ann. Hum. Genet. 2013;77:268–274. doi: 10.1111/ahg.12012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mei H., Chen W., Jiang F., He J., Srinivasan S., Smith E.N., Schork N., Murray S., Berenson G.S. Longitudinal replication studies of GWAS risk SNPs influencing body mass index over the course of childhood and adulthood. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hotta K., Nakamura M., Nakamura T., Matsuo T., Nakata Y., Kamohara S., Miyatake N., Kotani K., Komatsu R., Itoh N., et al. Association between obesity and polymorphisms in SEC16B, TMEM18, GNPDA2, BDNF, FAIM2 and MC4R in a Japanese population. J. Hum. Genet. 2009;54:727–731. doi: 10.1038/jhg.2009.106. [DOI] [PubMed] [Google Scholar]
- 31.Hong K.W., Oh B. Recapitulation of genome-wide association studies on body mass index in the Korean population. Int. J. Obes. 2012;36:1127–1130. doi: 10.1038/ijo.2011.202. [DOI] [PubMed] [Google Scholar]
- 32.Jääskeläinen T., Paananen J., Lindström J., Eriksson J.G., Tuomilehto J., Uusitupa M., Finnish Diabetes Prevention Study Group Genetic predisposition to obesity and lifestyle factors--the combined analyses of twenty-six known BMI- and fourteen known waist:hip ratio (WHR)-associated variants in the Finnish Diabetes Prevention Study. Br. J. Nutr. 2013;110:1856–1865. doi: 10.1017/S0007114513001116. [DOI] [PubMed] [Google Scholar]
- 33.Poveda A., Ibáñez M.E., Rebato E. Common variants in BDNF, FAIM2, FTO, MC4R, NEGR1, and SH2B1 show association with obesity-related variables in Spanish Roma population. Am. J. Hum. Biol. 2014;26:660–669. doi: 10.1002/ajhb.22576. [DOI] [PubMed] [Google Scholar]
- 34.Verma A., Huffman J.E., Rodriguez A., Conery M., Liu M., Ho Y.-L., Kim Y., Heise D.A., Guare L., Panickan V.A., et al. Diversity and Scale: Genetic Architecture of 2,068 Traits in the VA Million Veteran Program. medRxiv. 2023 doi: 10.1101/2023.06.28.23291975. [DOI] [PubMed] [Google Scholar]
- 35.National Library of Medicine (US) National Center for Biotechnology Information. dbSNP. https://www.ncbi.nlm.nih.gov/snp/
- 36.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin S., Cule M., Basty N., Tyrrell J., Beaumont R.N., Wood A.R., Frayling T.M., Sorokin E., Whitcher B., Liu Y., et al. Genetic Evidence for Different Adiposity Phenotypes and Their Opposing Influences on Ectopic Fat and Risk of Cardiometabolic Disease. Diabetes. 2021;70:1843–1856. doi: 10.2337/db21-0129. [DOI] [PubMed] [Google Scholar]
- 38.Zhou H., Sealock J.M., Sanchez-Roige S., Clarke T.K., Levey D.F., Cheng Z., Li B., Polimanti R., Kember R.L., Smith R.V., et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci. 2020;23:809–818. doi: 10.1038/s41593-020-0643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H., Zhang F., Zeng J., Wu Y., Kemper K.E., Xue A., Zhang M., Powell J.E., Goddard M.E., Wray N.R., et al. Genotype-by-environment interactions inferred from genetic effects on phenotypic variability in the UK Biobank. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aaw3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tachmazidou I., Süveges D., Min J.L., Ritchie G.R.S., Steinberg J., Walter K., Iotchkova V., Schwartzentruber J., Huang J., Memari Y., et al. Whole-Genome Sequencing Coupled to Imputation Discovers Genetic Signals for Anthropometric Traits. Am. J. Hum. Genet. 2017;100:865–884. doi: 10.1016/j.ajhg.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Common Metabolic Diseases Knowledge Portal (cmdkp.org). rs7132908 Variant page. https://hugeamp.org/variant.html?variant=rs7132908 (RRID:SCR_020937).
- 42.Pahl M.C., Le Coz C., Su C., Sharma P., Thomas R.M., Pippin J.A., Cruz Cabrera E., Johnson M.E., Leonard M.E., Lu S., et al. Implicating effector genes at COVID-19 GWAS loci using promoter-focused Capture-C in disease-relevant immune cell types. Genome Biol. 2022;23:125. doi: 10.1186/s13059-022-02691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su C., Gao L., May C.L., Pippin J.A., Boehm K., Lee M., Liu C., Pahl M.C., Golson M.L., Naji A., et al. 3D chromatin maps of the human pancreas reveal lineage-specific regulatory architecture of T2D risk. Cell Metab. 2022;34:1394–1409.e4. doi: 10.1016/j.cmet.2022.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lasconi C., Pahl M.C., Pippin J.A., Su C., Johnson M.E., Chesi A., Boehm K., Manduchi E., Ou K., Golson M.L., et al. Variant-to-gene-mapping analyses reveal a role for pancreatic islet cells in conferring genetic susceptibility to sleep-related traits. Sleep. 2022;45 doi: 10.1093/sleep/zsac109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chesi A., Wagley Y., Johnson M.E., Manduchi E., Su C., Lu S., Leonard M.E., Hodge K.M., Pippin J.A., Hankenson K.D., et al. Genome-scale Capture C promoter interactions implicate effector genes at GWAS loci for bone mineral density. Nat. Commun. 2019;10:1260. doi: 10.1038/s41467-019-09302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su C., Argenziano M., Lu S., Pippin J.A., Pahl M.C., Leonard M.E., Cousminer D.L., Johnson M.E., Lasconi C., Wells A.D., et al. 3D promoter architecture re-organization during iPSC-derived neuronal cell differentiation implicates target genes for neurodevelopmental disorders. Prog. Neurobiol. 2021;201 doi: 10.1016/j.pneurobio.2021.102000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trang K.B., Pahl M.C., Pippin J.A., Su C., Littleton S.H., Sharma P., Kulkarni N.N., Ghanem L.R., Terry N.A., O’Brien J.M., et al. 3D genomic features across >50 diverse cell types reveal insights into the genomic architecture of childhood obesity. medRxiv. 2024 doi: 10.1101/2023.08.30.23294092. [DOI] [Google Scholar]
- 48.ENCODE Project Consortium. Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogelezang S., Bradfield J.P., Ahluwalia T.S., Curtin J.A., Lakka T.A., Grarup N., Scholz M., van der Most P.J., Monnereau C., Stergiakouli E., et al. Novel loci for childhood body mass index and shared heritability with adult cardiometabolic traits. PLoS Genet. 2020;16 doi: 10.1371/journal.pgen.1008718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu T., Porter J., Zhao C., Zhu H., Wang N., Sun Z., Mo Y.Y., Wang Z. TADKB: Family classification and a knowledge base of topologically associating domains. BMC Genom. 2019;20:217. doi: 10.1186/s12864-019-5551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49:D325–D334. doi: 10.1093/nar/gkaa1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang W.K., Wong S.Z.H., Pather S.R., Nguyen P.T.T., Zhang F., Zhang D.Y., Zhang Z., Lu L., Fang W., Chen L., et al. Generation of hypothalamic arcuate organoids from human induced pluripotent stem cells. Cell Stem Cell. 2021;28:1657–1670.e10. doi: 10.1016/j.stem.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marques S., Zeisel A., Codeluppi S., van Bruggen D., Mendanha Falcão A., Xiao L., Li H., Häring M., Hochgerner H., Romanov R.A., et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 2016;352:1326–1329. doi: 10.1126/science.aaf6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Franjic D., Skarica M., Ma S., Arellano J.I., Tebbenkamp A.T.N., Choi J., Xu C., Li Q., Morozov Y.M., Andrijevic D., et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron. 2022;110:452–469.e14. doi: 10.1016/j.neuron.2021.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones A.R., Overly C.C., Sunkin S.M. The Allen Brain Atlas: 5 years and beyond. Nat. Rev. Neurosci. 2009;10:821–828. doi: 10.1038/nrn2722. [DOI] [PubMed] [Google Scholar]
- 59.Sunkin S.M., Ng L., Lau C., Dolbeare T., Gilbert T.L., Thompson C.L., Hawrylycz M., Dang C. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucleic Acids Res. 2013;41:D996–D1008. doi: 10.1093/nar/gks1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller J.A., Ding S.L., Sunkin S.M., Smith K.A., Ng L., Szafer A., Ebbert A., Riley Z.L., Royall J.J., Aiona K., et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen E.H., Overly C.C., Jones A.R. The Allen Human Brain Atlas: comprehensive gene expression mapping of the human brain. Trends Neurosci. 2012;35:711–714. doi: 10.1016/j.tins.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 62.Cowley M.A., Pronchuk N., Fan W., Dinulescu D.M., Colmers W.F., Cone R.D. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- 63.Bellinger L.L., Bernardis L.L. The dorsomedial hypothalamic nucleus and its role in ingestive behavior and body weight regulation: lessons learned from lesioning studies. Physiol. Behav. 2002;76:431–442. doi: 10.1016/s0031-9384(02)00756-4. [DOI] [PubMed] [Google Scholar]
- 64.Xie Z., Gu H., Huang M., Cheng X., Shang C., Tao T., Li D., Xie Y., Zhao J., Lu W., et al. Mechanically evoked defensive attack is controlled by GABAergic neurons in the anterior hypothalamic nucleus. Nat. Neurosci. 2022;25:72–85. doi: 10.1038/s41593-021-00985-4. [DOI] [PubMed] [Google Scholar]
- 65.Thorleifsson G., Walters G.B., Gudbjartsson D.F., Steinthorsdottir V., Sulem P., Helgadottir A., Styrkarsdottir U., Gretarsdottir S., Thorlacius S., Jonsdottir I., et al. Genome-wide association yields new sequence variants at seven loci that associate with measures of obesity. Nat. Genet. 2009;41:18–24. doi: 10.1038/ng.274. [DOI] [PubMed] [Google Scholar]
- 66.Glunk V., Laber S., Sinnott-Armstrong N., Sobreira D.R., Strobel S.M., Batista T.M., Kubitz P., Moud B.N., Ebert H., Huang Y., et al. A non-coding variant linked to metabolic obesity with normal weight affects actin remodelling in subcutaneous adipocytes. Nat. Metab. 2023;5:861–879. doi: 10.1038/s42255-023-00807-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar P., Traurig M., Baier L.J. Identification and functional validation of genetic variants in potential miRNA target sites of established BMI genes. Int. J. Obes. 2020;44:1191–1195. doi: 10.1038/s41366-019-0488-8. [DOI] [PubMed] [Google Scholar]
- 68.Medley J.C., Panzade G., Zinovyeva A.Y. microRNA strand selection: Unwinding the rules. Wiley Interdiscip. Rev. RNA. 2021;12 doi: 10.1002/wrna.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nguyen H.P., Chan C.S., Cintron D.L., Sheng R., Harshman L., Nobuhara M., Ushiki A., Biellak C., An K., Gordon G.M., et al. Integrative single-cell characterization of hypothalamus sex-differential and obesity-associated genes and regulatory elements. bioRxiv. 2022 doi: 10.1101/2022.11.06.515311. [DOI] [Google Scholar]
- 70.Newman A.C., Nakatsu M.N., Chou W., Gershon P.D., Hughes C.C.W. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol. Biol. Cell. 2011;22:3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hurtado de Mendoza T., Perez-Garcia C.G., Kroll T.T., Hoong N.H., O'Leary D.D.M., Verma I.M. Antiapoptotic protein Lifeguard is required for survival and maintenance of Purkinje and granular cells. Proc. Natl. Acad. Sci. USA. 2011;108:17189–17194. doi: 10.1073/pnas.1114226108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Urresti J., Ruiz-Meana M., Coccia E., Arévalo J.C., Castellano J., Fernández-Sanz C., Galenkamp K.M.O., Planells-Ferrer L., Moubarak R.S., Llecha-Cano N., et al. Lifeguard Inhibits Fas Ligand-mediated Endoplasmic Reticulum-Calcium Release Mandatory for Apoptosis in Type II Apoptotic Cells. J. Biol. Chem. 2016;291:1221–1234. doi: 10.1074/jbc.M115.677682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Somia N.V., Schmitt M.J., Vetter D.E., Van Antwerp D., Heinemann S.F., Verma I.M. LFG: an anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc. Natl. Acad. Sci. USA. 1999;96:12667–12672. doi: 10.1073/pnas.96.22.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Merianda T.T., Vuppalanchi D., Yoo S., Blesch A., Twiss J.L. Axonal transport of neural membrane protein 35 mRNA increases axon growth. J. Cell Sci. 2013;126:90–102. doi: 10.1242/jcs.107268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tauber S.C., Harms K., Falkenburger B., Weis J., Sellhaus B., Nau R., Schulz J.B., Reich A. Modulation of hippocampal neuroplasticity by Fas/CD95 regulatory protein 2 (Faim2) in the course of bacterial meningitis. J. Neuropathol. Exp. Neurol. 2014;73:2–13. doi: 10.1097/NEN.0000000000000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schweitzer B., Suter U., Taylor V. Neural membrane protein 35/Lifeguard is localized at postsynaptic sites and in dendrites. Brain Res. Mol. Brain Res. 2002;107:47–56. doi: 10.1016/s0169-328x(02)00445-x. [DOI] [PubMed] [Google Scholar]
- 77.Komnig D., Gertz K., Habib P., Nolte K.W., Meyer T., Brockmann M.A., Endres M., Rathkolb B., Hrabě de Angelis M., German Mouse Clinic Consortium Faim2 contributes to neuroprotection by erythropoietin in transient brain ischemia. J. Neurochem. 2018;145:258–270. doi: 10.1111/jnc.14296. [DOI] [PubMed] [Google Scholar]
- 78.Reich A., Spering C., Gertz K., Harms C., Gerhardt E., Kronenberg G., Nave K.A., Schwab M., Tauber S.C., Drinkut A., et al. Fas/CD95 regulatory protein Faim2 is neuroprotective after transient brain ischemia. J. Neurosci. 2011;31:225–233. doi: 10.1523/JNEUROSCI.2188-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pawar M., Busov B., Chandrasekhar A., Yao J., Zacks D.N., Besirli C.G. FAS apoptotic inhibitory molecule 2 is a stress-induced intrinsic neuroprotective factor in the retina. Cell Death Differ. 2017;24:1799–1810. doi: 10.1038/cdd.2017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hurtado de Mendoza T., Liu F., Verma I.M. Antiapoptotic Role for Lifeguard in T Cell Mediated Immune Response. PLoS One. 2015;10 doi: 10.1371/journal.pone.0142161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Boender A.J., van Rozen A.J., Adan R.A.H. Nutritional state affects the expression of the obesity-associated genes Etv5, Faim2, Fto, and Negr1. Obesity. 2012;20:2420–2425. doi: 10.1038/oby.2012.128. [DOI] [PubMed] [Google Scholar]
- 82.Chen R., Wu X., Jiang L., Zhang Y. Single-Cell RNA-Seq Reveals Hypothalamic Cell Diversity. Cell Rep. 2017;18:3227–3241. doi: 10.1016/j.celrep.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ding Q., Regan S.N., Xia Y., Oostrom L.A., Cowan C.A., Musunuru K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Concordet J.P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Andrews S. 2010. FastQC: A quality control tool for high throughput sequence data.http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- 87.2015. FastQC.https://qubeshub.org/resources/fastqc [Google Scholar]
- 88.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 89.Bray N.L., Pimentel H., Melsted P., Pachter L. Erratum: Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:888. doi: 10.1038/nbt0816-888d. [DOI] [PubMed] [Google Scholar]
- 90.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wolock S.L., Lopez R., Klein A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Young M.D., Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 2020;9 doi: 10.1093/gigascience/giaa151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Choudhary S., Satija R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 2022;23:27. doi: 10.1186/s13059-021-02584-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kolberg L., Raudvere U., Kuzmin I., Vilo J., Peterson H. gprofiler2 -- an R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Res. 2020;9 doi: 10.12688/f1000research.24956.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rainer J., Gatto L., Weichenberger C.X. ensembldb: an R package to create and use Ensembl-based annotation resources. Bioinformatics. 2019;35:3151–3153. doi: 10.1093/bioinformatics/btz031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lun A.T.L., Smyth G.K. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016;44 doi: 10.1093/nar/gkv1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 108.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Amezquita R.A., Lun A.T.L., Becht E., Carey V.J., Carpp L.N., Geistlinger L., Marini F., Rue-Albrecht K., Risso D., Soneson C., et al. Orchestrating single-cell analysis with Bioconductor. Nat. Methods. 2020;17:137–145. doi: 10.1038/s41592-019-0654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Amezquita R.A., Lun A.T.L., Becht E., Carey V.J., Carpp L.N., Geistlinger L., Marini F., Rue-Albrecht K., Risso D., Soneson C., et al. Publisher Correction: Orchestrating single-cell analysis with Bioconductor. Nat. Methods. 2020;17:242. doi: 10.1038/s41592-019-0700-8. [DOI] [PubMed] [Google Scholar]
- 112.Zhu A., Ibrahim J.G., Love M.I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019;35:2084–2092. doi: 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wingett S., Ewels P., Furlan-Magaril M., Nagano T., Schoenfelder S., Fraser P., Andrews S. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 2015;4:1310. doi: 10.12688/f1000research.7334.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Open2C. Abdennur N., Fudenberg G., Flyamer I.M., Galitsyna A.A., Goloborodko A., Imakaev M., Venev S.V. Pairtools: from sequencing data to chromosome contacts. bioRxiv. 2023 doi: 10.1101/2023.02.13.528389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lee S., Bakker C.R., Vitzthum C., Alver B.H., Park P.J. Pairs and Pairix: a file format and a tool for efficient storage and retrieval for Hi-C read pairs. Bioinformatics. 2022;38:1729–1731. doi: 10.1093/bioinformatics/btab870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abdennur N., Mirny L.A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020;36:311–316. doi: 10.1093/bioinformatics/btz540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roayaei Ardakany A., Gezer H.T., Lonardi S., Ay F. Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome Biol. 2020;21:256. doi: 10.1186/s13059-020-02167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaul A., Bhattacharyya S., Ay F. Identifying statistically significant chromatin contacts from Hi-C data with FitHiC2. Nat. Protoc. 2020;15:991–1012. doi: 10.1038/s41596-019-0273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Coetzee S.G., Coetzee G.A., Hazelett D.J. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015;31:3847–3849. doi: 10.1093/bioinformatics/btv470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen B.Y., Bone W.P., Lorenz K., Levin M., Ritchie M.D., Voight B.F. ColocQuiaL: a QTL-GWAS colocalization pipeline. Bioinformatics. 2022;38:4409–4411. doi: 10.1093/bioinformatics/btac512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Alabdullah A.A., Al-Abdulaziz B., Alsalem H., Magrashi A., Pulicat S.M., Almzroua A.A., Almohanna F., Assiri A.M., Al Tassan N.A., Al-Mubarak B.R. Estimating transfection efficiency in differentiated and undifferentiated neural cells. BMC Res. Notes. 2019;12:225. doi: 10.1186/s13104-019-4249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maguire J.A., Gadue P., French D.L. Highly Efficient CRISPR/Cas9-Mediated Genome Editing in Human Pluripotent Stem Cells. Curr. Protoc. 2022;2:e590. doi: 10.1002/cpz1.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xie Y., Dorsky R.I. Development of the hypothalamus: conservation, modification and innovation. Development. 2017;144:1588–1599. doi: 10.1242/dev.139055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.1000 Genomes Project Consortium. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cunningham F., Allen J.E., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Austine-Orimoloye O., Azov A.G., Barnes I., Bennett R., et al. Ensembl 2022. Nucleic Acids Res. 2022;50:D988–D995. doi: 10.1093/nar/gkab1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nassar L.R., Barber G.P., Benet-Pagès A., Casper J., Clawson H., Diekhans M., Fischer C., Gonzalez J.N., Hinrichs A.S., Lee B.T., et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2023;51:D1188–D1195. doi: 10.1093/nar/gkac1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Srivatsan S.R., McFaline-Figueroa J.L., Ramani V., Saunders L., Cao J., Packer J., Pliner H.A., Jackson D.L., Daza R.M., Christiansen L., et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science. 2020;367:45–51. doi: 10.1126/science.aax6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Elgamal R.M., Kudtarkar P., Melton R.L., Mummey H.M., Benaglio P., Okino M.-L., Gaulton K.J. An integrated map of cell type-specific gene expression in pancreatic islets. bioRxiv. 2023 doi: 10.1101/2023.02.03.526994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rouillard A.D., Gundersen G.W., Fernandez N.F., Wang Z., Monteiro C.D., McDermott M.G., Ma'ayan A. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database. 2016;2016 doi: 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Castro-Mondragon J.A., Riudavets-Puig R., Rauluseviciute I., Lemma R.B., Turchi L., Blanc-Mathieu R., Lucas J., Boddie P., Khan A., Manosalva Pérez N., et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–D173. doi: 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Hi-C, RNA-seq, ATAC-seq, single-nucleus RNA-seq, and single-nucleus ATAC-seq data have been deposited at Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the Key resources table. Human embryonic stem cell and tissue genotyping data reported in this study cannot be deposited in a public repository to protect donor confidentiality. To request access, contact the lead contact. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.