

Abstract
Cell communication is needed for organ function and stress responses, especially in the heart. Cardiac fibroblasts, cardiomyocytes, immune cells, and endothelial cells comprise the major cell types in ventricular myocardium that together coordinate all functional processes. Critical to this cellular network is the non-cellular extracellular matrix (ECM) that provides structure and harbors growth factors and other signaling proteins that affect cell behavior. The ECM is not only produced and modified by cells within the myocardium, largely cardiac fibroblasts, it also acts as an avenue for communication among all myocardial cells. In this Review, we discuss how the development of therapeutics to combat cardiac diseases, specifically fibrosis, relies on a deeper understanding of how the cardiac ECM is intertwined with signaling processes that underlie cellular activation and behavior.
Keywords: cardiac fibrosis, heart development, cardiac fibroblast, cardiomyocyte, macrophage, extracellular matrix, collagen
Introduction
The cardiac extracellular matrix (ECM) is a dynamic protein network that integrates foundational cellular processes in the heart. It not only provides structural support to cells within the myocardium, but also functions as a reservoir for growth factors and other proteins that underlie cell-cell communication. Beginning in early development, dynamic changes in the composition of the ECM network facilitate proper cell behavior and ventricular development as the heart matures to achieve full contractile rigor1–3. ECM composition is also critical for maintaining heart function after injury or disease in adults, as the fully mature myocardium lacks regenerative healing capacity. However, adult ECM reorganization is frequently associated with chronic injury or pathology that impedes cardiac function4,5.
Cardiac fibroblasts (CFs) can activate to a contractile cell type known as myofibroblasts, which are responsible for the bulk of ECM production, maintenance and remodeling in the heart6. A healthy adult heart is largely devoid of myofibroblasts, but once activated they facilitate cardiac wound healing and ventricular remodeling7–9. Myofibroblast formation and function can be beneficial in terms of providing mechanical support to an acutely injured tissue, but their sustained activity and proliferation contributes to pathological fibrosis and tissue stiffening9–11.
Developing therapeutics to combat cardiac diseases, specifically fibrosis, relies on a deeper understanding of how the cardiac ECM is intertwined with signaling processes for cellular activation and behavior. In this Review, we highlight the concept that the composition and dynamic state of the cardiac ECM is tied to fibroblast activation, which is also regulated by signals from cardiomyocytes and immune cells, illustrating a dynamic feedback system among myocardial cells and their surrounding ECM (Figure 1).
Figure 1. The ECM facilitates communication among fibroblasts, cardiomyocytes and macrophages in the heart.
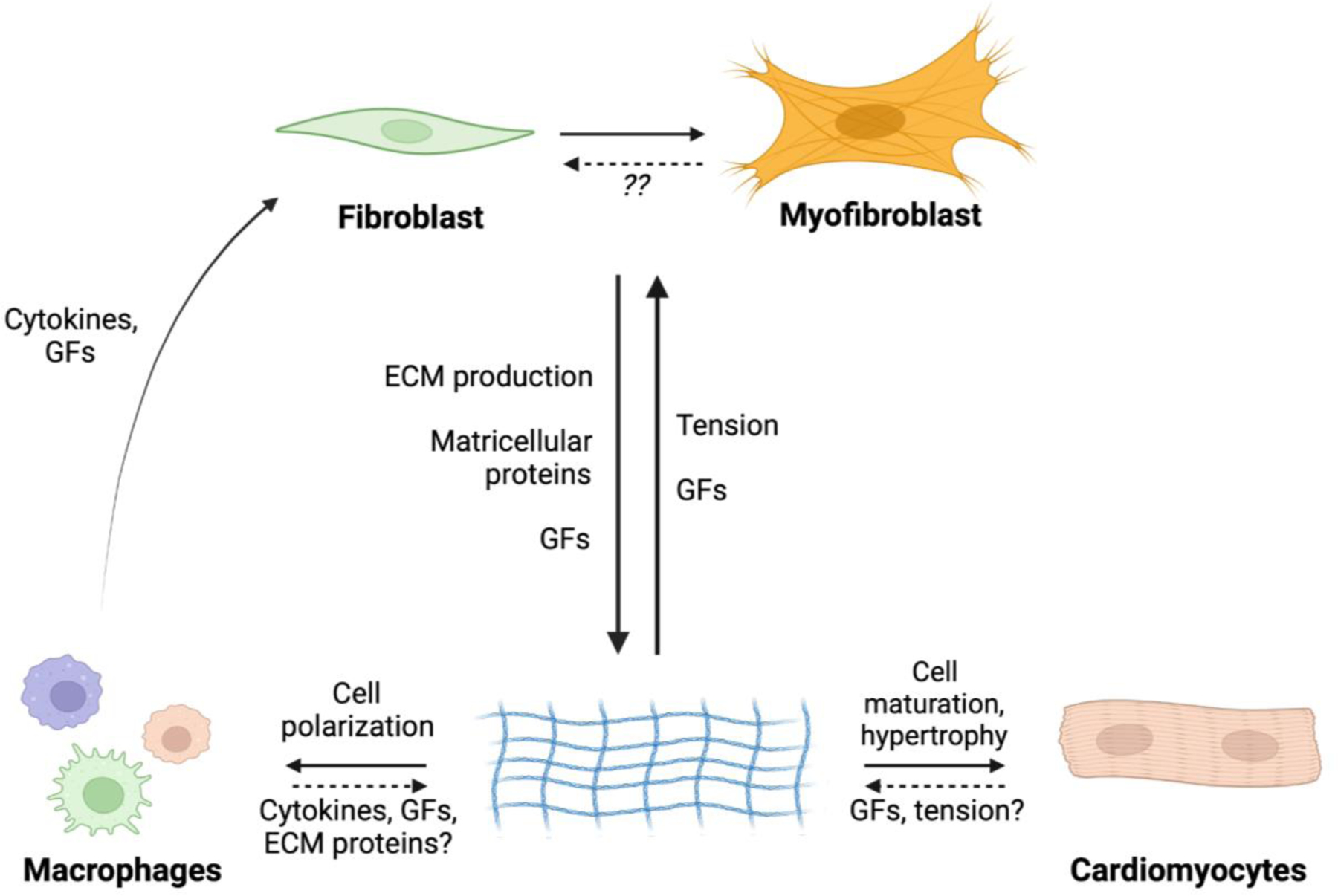
Fibroblasts are activated by cues such as mechanical tension and inflammatory cytokines and chemokines that are typical of injury. Activated fibroblasts (myofibroblasts) modify the ECM, which in turn regulates behaviors of myocardial cells such as myocytes and macrophages. The ECM affects these cell populations through changes in mechanical tension, as well as availability of growth factors and other matricellular proteins. It remains unclear exactly how macrophages and cardiomyocytes might exert influences on the ECM, although growth factor signaling is likely key. It is also unknown the degree to which myofibroblasts convert back to a quiescent/homeostatic phenotype. Abbreviations: GF, growth factors; ECM, extracellular matrix.
The cardiac ECM
The cardiac ECM facilitates force transmission and impacts cardiac ventricular wall stiffness, and also plays a critical role in transducing molecular signals and mediating cell-cell crosstalk4. The ECM network harbors latent growth factors and proteins that are rapidly released during injury or ECM remodeling4. Active proteins can then engage cell surface receptors on multiple cardiac cell types that initiate intracellular signal transduction cascades to alter cell behavior12. Prime examples of proteins that localize to the ECM matrix are transforming growth factor-β (TGFβ), platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF). Alterations in the conformation and organization of the ECM can affect the amount and accessibility of these growth factors to cells within the myocardium.
The protein composition of the ECM, activity of crosslinking enzymes, and connections between ECM and cells all contribute to the overall stiffness of a tissue. Assessing stiffness in the heart is not trivial, as has been recently well-reviewed13. While some ECM proteins have been characterized, such as fibrillar collagens, a catalogue for the spatiotemporal expression of specific ECM proteins in homeostasis and stages of disease progression has not been established. Ongoing research and refinement of specialized techniques13 are starting to untangle how ECM composition and stiffness affect heart function.
Fibronectin is a foundational ECM protein that is laid down during early heart development, which is replaced with “priming” collagens in the neonatal period (P0 to P7), and by early adulthood >80% of the cardiac ECM is collagen type I and 11% is type III1–3. With aging, the adult mammalian heart becomes stiffer over time in coordination with increased hypertrophy1,14. Experimental evidence suggests that increased collagen I levels in the adult heart generates a stiffer environment for supporting greater contractility but with aging this might also impair relaxation, leading to the observed hypertrophy14. In an analysis of CFs and ECM from human heart failure patients, Perestrelo et al., described differential ECM organization that accompanied altered tissue stiffness and CF activation15. Variations in collagen organization and alignment have been shown to regulate CF differentiation in vitro16.
With respect to the adult heart and predisposition to disease, studies have investigated the contributions of a variety of ECM components to cardiac stiffness and cardiomyocyte biology. Proper fibronectin polymerization is required for cardiac fibrosis post ischemia/reperfusion injury33, although mechanisms for this improvement are unclear. Loss of α2 fibrils of type I collagen led to decreased overall collagen I content, a more fragile collagen fiber and thus a more compliant ventricle in response to passive inflation17. While proper secretion of type I collagen fibrils is critical, crosslinking these fibrils to form a mature collagen network is also required for augmenting cardiac stiffness in response to hypertension18,19. The essential role of fibrillar collagen in cardiac stiffness has been supported by observations in human patients with severe aortic stenosis20. Furthermore, overexpression of the collagen reinforcing protein periostin in the mouse heart promoted greater pressure overload-induced hypertrophy with increased ECM content21.
Since fibrillar collagen crosslinking is essential for ECM maturation and cardiac stiffness, it is reasonable to hypothesize that proteins regulating ECM crosslinking could also affect myocardial organization and the cellular response to injury. For example, the lysyl oxidase family of essential collagen and elastin crosslinking proteins are upregulated in cardiac hypertrophy in both human disease and mouse models22–24. Overexpression of lysyl oxidase-like 1 (Loxl1) from murine cardiomyocytes results in myocyte hypertrophy, increased ventricular wall thickness, and interstitial fibrosis25; interestingly, cardiac function was preserved. On the other hand, pharmacologic inhibition or genetic deletion of lysyl oxidase-like protein 2 (Loxl2) significantly reduced levels of cardiac fibrosis in response to overload hypertrophy in mice, which correlated with better cardiac compliance and improved cardiac function26.
Various matricellular proteins are also involved with the regulation of ECM stiffness, but their involvement in cardiac fibrosis and myocyte regulation is less straightforward. For example, both thrombospondin (Thbs)-1-27 and Thbs-4-28 null mice showed augmented cardiac hypertrophy after aortic transaortic constriction (TAC), which was associated with increased cardiac fibrosis, although it remains unclear how the Thbs1 or 4 proteins might regulate interstitial fibrosis in the heart. Deletion of Thbs-3, however, protected mice from TAC-induced cardiac hypertrophy through maintaining cardiomyocyte integrin expression and sarcolemmal stability.29 Deletion of tenascin-C, which regulates ECM quality and cardiomyocyte attachment, reduced cardiac fibrosis and decreased cardiomyocyte hypertrophy after pressure overload30. SPARC-null mice developed less fibrosis but without changes in cardiomyocyte size after pressure overload stimulation31. Furthermore, deletion of transglutaminase-2 led to reduced collagen content after injury to the heart but augmented cardiac hypertrophy32. These studies suggest that select matricellular proteins can regulate ECM properties and remodeling directly, which secondarily impacts cardiomyocyte hypertrophic potential.
Fibroblast-derived ECM during cardiac development
One of the most impactful scientific advancements over the past several years has been single-cell RNA (scRNA) sequencing, which allows for precise characterization of cell populations within a heterogeneous tissue34. A number of scRNA-seq methodologies have been optimized for analysis of cells within the heart, and their applications and limitations have recently been discussed35,36. Using such sequencing approaches have suggested ways in which fibroblast activation and/or differentiation might impact cardiomyocyte biology, although additional mechanistic research is needed.
Fibroblast heterogeneity has long been appreciated37, but recent RNA sequencing studies have provided a higher resolution of the gene expression differences between similar cell populations, including cardiac fibroblasts. Wang et al., uncovered five distinct fibroblast gene expression states during postnatal cardiac development, some of which were identified as responsible for ECM modifications that supported cardiomyocyte growth and maturation38. Farbehi et al., examined mouse hearts three and seven days after myocardial infarction and identified 11 subpopulations within platelet-derived growth factor receptor (PDGFR)-α expressing fibroblasts, distinguishable by variable expression of smooth muscle α-actin (αSMA), Wnt pathway genes, and ECM modifiers39, which are characteristic indicators of fibroblast activation. In addition to fibroblasts with unique gene expression signatures, subsets of cardiomyocytes with transcription signatures specific to regions of the heart have also been identified40, which are speculated to underlie neonatal regenerative responses41. It is not yet clear whether these cellular "subtypes" represent bonafide stable cell types or are a snapshot of a cells transitioning from one differentiated state to another42,43.
Studies have elucidated cardiomyocyte differentiation during neonatal development44, but the field is only now starting to uncover how the ECM and cellular microenvironment affects cardiac myocyte maturation (Figure 2). Wang et al., performed scRNA-seq on mouse hearts from several developmental stages to analyze regulatory signaling networks of different cell populations and how they might interact with one another38. Their work demonstrated that CFs drive cardiomyocyte maturation through changes in their transcriptional program that includes ECM maturation. Importantly, adult CFs uniquely contributed to induced pluripotent stem cell-derived cardiomyocyte maturation that was not achieved with neonatal CFs38. This has profound implications not only for congenital diseases, but also for research aimed at inducing cardiomyocyte regeneration after injury. Clearly the conditioning or maturation of the ECM by CFs can directly impact cardiomyocyte differentiation and disease responsiveness.
Figure 2. Cardiomyocytes and fibroblasts sense each other through the dynamic ECM environment.
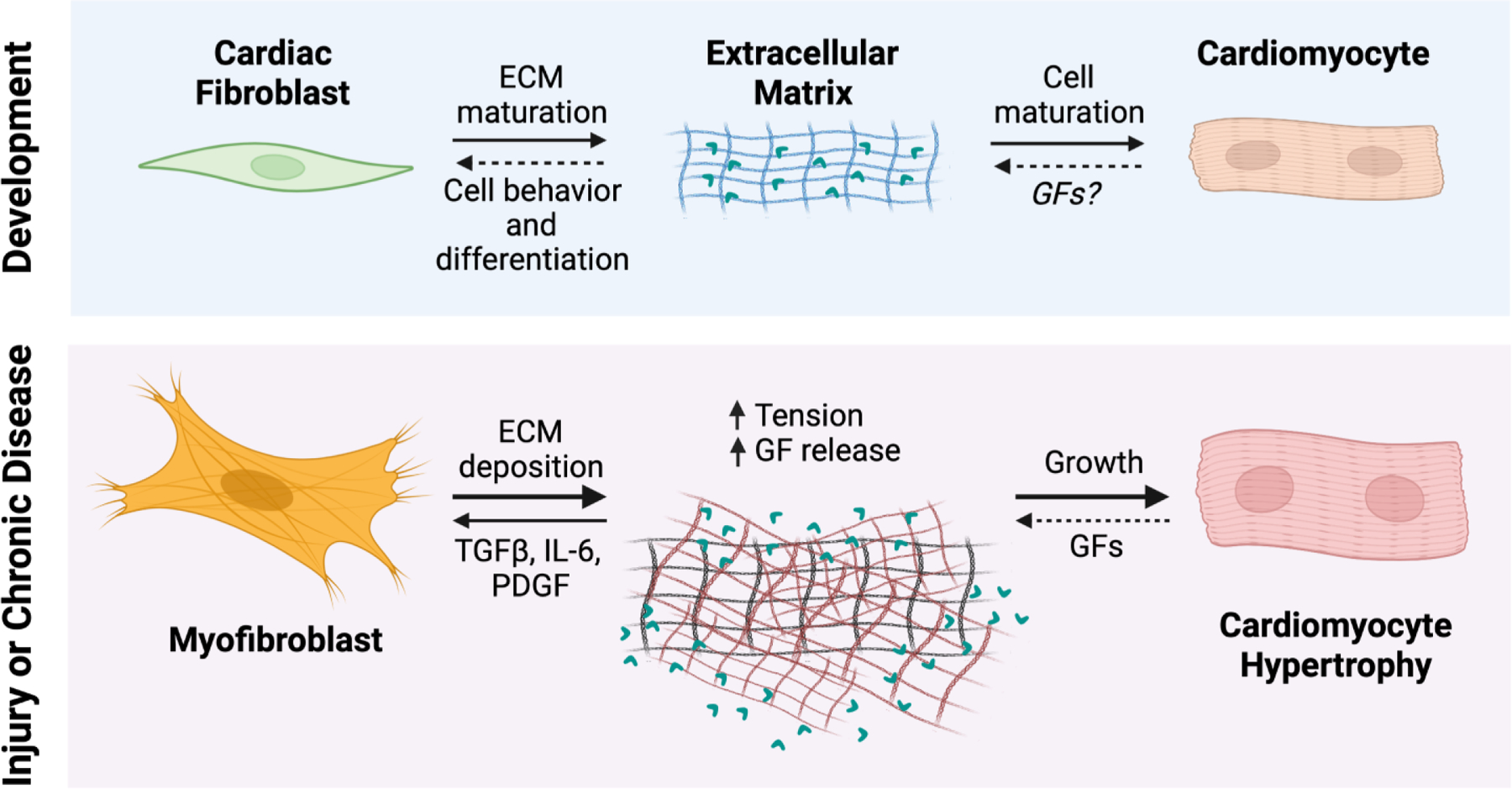
CFs are the main producers of core ECM proteins during cardiac development, and thus affect cardiomyocyte differentiation and maturation. During cardiac injury or chronic disease conditions, the ECM is altered, and fibroblasts interpret changes in mechanical tension and cytokine/chemokine signaling. They then differentiate to myofibroblasts that secrete the bulk of ECM proteins during adult cardiac wound repair and remodeling. This increased matrix deposition is sensed by cardiomyocytes, causing changes in cell phenotype such as hypertrophy. Abbreviations: GF, growth factors; ECM, extracellular matrix; TGFβ, transforming growth factor-β; IL-6, interleukin-6; PDGF, platelet derived growth factor.
While scRNA sequencing highlights cellular gene expression heterogeneity within a sample, one drawback of these studies is the loss of information about regional tissue heterogeneity. To this end, Lacraz et al., used cryosections to assess transcriptional differences (RNA tomography) in cells across different regions of the heart following ischemia/reperfusion injury45. By specifically analyzing transcripts isolated from infarcted tissue versus regions remote to injury, they uncovered Sox9 as a regulator of fibrotic gene expression, including collagen and other ECM proteins45. Likewise, Mantri et al., used chick embryo hearts to combine scRNA-seq with spatial RNA-seq to show that developmental gene expression programs are region-specific within the heart across select cell types, demonstrating that cellular differentiation corresponds with morphologic changes within a tissue46. For example, migration of epicardial-derived progenitor cells into the myocardium coincided with expression of ECM factors known to induce cell migration46. In another study, Liu et al., utilized fate-mapping studies of cell cycle activity to show regional differences in cardiomyocyte regulation of cell division and response to injury47, corroborating reports of heterogeneous cardiomyocyte populations. Although not tested directly, the authors proposed that changes in ECM composition could be a driver of heterogenic cell behavior in this setting.
Periostin is a matricellular protein involved with collagen fiber maturation and is a known marker for CF activation after injury21. However, a periostin-expressing fibroblast lineage was recently found to exist transiently during postnatal heart growth, which was distinct from the Tcf21+ fibroblast population that arises from the epicardium48. Ablating periostin-expressing fibroblasts had detrimental effects on cardiomyocyte maturation and sympathetic innervation in the first seven days after birth, which coincided with the appearance of a more mature ECM network. Further investigation into the temporal expression of integrins, adhesion proteins, and other cellular machinery related to mechanosensitivity in fibroblasts is needed for full appreciation of the signaling networks that balance normal cardiac growth in coordination with the ECM.
Activated fibroblasts and cardiac fibrosis
As we have discussed, the process of ECM maturation and remodeling is primarily regulated by CFs, which directly secrete ECM components during development and in the adult heart49,50. When stimulated by mechanical stress or secreted factors, CFs activate into a cell type known as a myofibroblast, which functions as a hybrid fibroblast/smooth muscle cell that expresses αSMA and other contractile proteins 10,51,52. Myofibroblasts synthesize and secrete high levels of ECM and matricellular proteins, including collagens type I and III, fibronectin splice variants (e.g., Fn-EDa), periostin, tenascin C and matrix metalloproteases52,53. Mechanical and secreted signals activate fibroblasts to remodel their surrounding matrix in three main ways: secreting ECM components, degrading existing ECM structures, and reorganizing and refining newly formed ECM networks4. First, fibroblasts actively synthesize and secrete fibrillar ECM proteins, which are the basic components of cardiac ECM9,51,54,55. Secondly, fibroblasts also secrete metalloproteinases, which include proteins in the matrix metalloprotease (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) families, all of which are critical for ECM degradation and remodeling56. Lastly, fibroblasts secrete proteins that refine and crosslink collagen, such as SPARC, osteopontin, tenascin C, periostin, and lysyl oxidase12. Collectively, CFs can modify the ECM in a variety of ways to impact cells throughout the myocardium, including cardiomyocytes and resident/activated immune cells.
For an in-depth discussion of the process of cardiac fibrosis and mechanisms of cell activation, we refer the reader to other reviews 10,11,57–59. Regarding the present Review, cardiac fibrosis occurs when myofibroblasts increase the total net deposition of ECM proteins in the myocardium. Fibrotic ECM is stiffer and less compliant than normal matrix, and results in altered biophysical dynamics within the myocardial wall. As discussed, fibroblast activation can occur after an overt injury such as MI, or in response to mechanosensitive protein stimulation, inflammatory cytokines, or a neuroendocrine/chemical stimulus11,49,58. In response to these signals, CFs activate and proliferate, depositing collagens and other matrix proteins to alter ECM support function and ventricular wall dynamics. Importantly, fibroblast-generated new ECM deposition prevents cardiac wall rupture and maintenance of chamber geometries after MI60–62. We recently explored the temporal dynamics of fibroblast activation and persistence in the heart after MI injury in mice63. Fibroblasts within the injured area of a mouse heart are initially depleted after MI, but by three days activated fibroblasts proliferate and migrate into the injured myocardium to restore total fibroblast cellular content63. This cellular reorganization is accompanied by the compaction of remaining basal lamina structures along with new ECM formation, leading to scar formation63. Interestingly, the myofibroblast cell state is transient in the acutely injured heart as these cells are either lost after scar formation or they change into a more differentiated state, known as a matrifibrocyte, which expresses markers of bone and cartilage63. Thus, the initially adaptative function of CF activation leads to deposition of permanent scar tissue and its initial remodeling, which is then maintained by a particular subpopulation of more highly differentiated fibroblasts (matrifibrocytes).
Using cell-specific cre-LoxP genetic systems in the mouse, a variety of genes have been examined for their contribution to cardiac fibrosis and adaptive remodeling. Cardiomyocyte-specific deletion of the TGFβ receptors 1 and 264,65, endothelial cell-specific deletion of endothelin-166, and macrophage-specific deletion of collagen IV67 have all resulted in alterations in the ECM and changes in cardiac function at baseline or with injury. These data suggest that non-fibroblast cell types can influence the extracellular environment, although evidence has shown the primacy of the CF or myofibroblast in directly regulating ECM content in the heart. For example, fibroblast- or myofibroblast-specific deletion of Smad2/368, TGFβ receptors 1 and 268–70, mitogen-activated protein kinase (MAPK) p3860 and β-catenin71 each showed compromised ECM production in response to cardiac injuries, which secondarily resulted in less cardiomyocyte hypertrophy. Indeed, we showed that fibroblast-specific deletion of the collagen chaperone protein heat shock protein-47 (HSP47) resulted in significantly less cardiac fibrosis and collagen type I, III and V deposition in the cardiac ECM with pressure overload stimulation, which compromised cardiomyocyte hypertrophy6. Importantly, deletion of Hsp47 from cardiomyocytes or endothelial cells did not reduce the induction of collagen expression and interstitial fibrosis after pressure overload6. Collectively, these results indicate that fibroblasts or myofibroblasts are the primary cell type in the heart responsible for fibrillar collagen production and augmentation of ECM after injury, which is required for effective cardiomyocyte hypertrophy with pressure overload and other injury-based stimuli (Figure 2).
An important point of emphasis is that cardiac fibrosis does not arise from a common etiologic origin. It is a heterogeneous process that manifests from a variety of pathologic stimuli, making it unlikely that a single therapeutic intervention will alleviate all fibrotic conditions of the heart. For example, “replacement fibrosis” is defined by the acute formation of collagenous fibrotic material, or a scar, that replaces dead cardiomyocytes, and this process is physiologically important in maintaining ventricular wall integrity and geometry59,72. The pathways driving replacement fibrosis in myofibroblasts seem to be more robust and less therapeutically amenable to inhibition. By comparison, "interstitial fibrosis” is a thickening or expansion of interstitial ECM that is not due to myocardial cell death, but instead due to inflammation, pathological neuroendocrine signaling or diverse types of cardiomyopathies59,72. Many types of interstitial fibrosis progress slowly and are likely driven by select signaling pathways, such as the local renin-angiotensin system in the heart73, sustained β-adrenergic signaling and catecholamine overload74,75, and inflammation76,77. These diverse yet more singular etiologies underlying progressive interstitial fibrosis are arguably more therapeutically tangible than replacement fibrosis. “Perivascular fibrosis” occurs around the vascular adventitia throughout the heart, and is frequently observed in hypertensive disease where it can impair coronary blood flow78. Studies are attempting to focus on context-dependent treatments for cardiovascular diseases with unique fibrotic signatures, which accompanies progress in imaging cardiac fibrosis79, drug delivery strategies80 and use of circulating biomarkers81 to enable personalized therapeutic strategies.
TGFβ signaling in cardiac fibroblast activation
A variety of growth factors and cytokines induce fibroblast activation, including angiotensin II, endothelin-1, TGFβ, FGF, PDGF, Interleukin (IL)-6, IL-4, IL-182–85. However, each of these factors converge on some aspect of the TGFβ signaling pathway to affect CF activity (reviewed in86). In fact, TGFβ-induced fibroblast activation has been shown to override activation induced by changes in matrix stiffness87. These data, among other reports, have implicated TGFβ as a central factor in the activation and regulation of fibroblasts that leads to the development of cardiac fibrosis.
The precursors of TGFβ ligands (1, 2 and 3) are sequestered in the cardiac ECM network as a latent complex. Cardiac stress, inflammation, or changes in mechanical stretch leads to the release of the ligands where they can signal to CFs and other myocardial cells. While there is speculation that cardiomyocytes might be the major source of TGFβ ligands during cardiac remodeling65, it is still unclear which cell type(s) within the heart produce TGFβ that resides within the ECM. A neutralizing antibody treatment that primarily targeted interstitial TGFβ was shown to reduce cardiac fibrosis in an HCM model65 and following pressure overload injury88. However, the antibody treatment failed to rescue adverse cardiac remodeling in response to pressure overload88, suggesting that interstitial cells are the main effector cell type of TGFβ signaling underlying cardiac fibrosis. This was further supported by studies that specifically ablated TGFβ receptors 1 and 2 in CFs, which also attenuated cardiac fibrosis and improved heart function after injury68–70.
Unfortunately, therapies that target TGFβ signaling to combat cardiovascular disease have not been promising. In several preclinical animal models, systemic administration of both small molecule inhibitors and neutralizing antibodies to TGFβ isoforms or their receptors have resulted in toxicities that include hemorrhage, heart valve degeneration, and histopathologic changes in various organs89–91. In addition, pirfenidone is an FDA-approved treatment for patients with idiopathic pulmonary fibrosis that is thought to largely act through inhibition of TGFβ signaling and has been shown to decrease fibrosis in several pre-clinical models of cardiac injury92,93. However, in a recent clinical trial to assess whether it could improve outcomes in a cohort of HFpEF patients94, pirfenidone treatment significantly decreased cardiac fibrosis as measured by MRI but did not improve diastolic dysfunction. While TGFβ signaling is undoubtedly critical to the development of cardiac fibrosis and myofibroblast formation in the heart, current clinical studies collectively illustrate a need for alternate targets, or at least more selective therapeutic targets within the TGFβ pathway.
One molecular target downstream of TGFβ activation is MAPK p38, which is involved with CF differentiation16,60. Indeed, use of select p38 inhibitors have shown reduced cardiac fibrosis and even reduced skeletal muscle fibrosis with disease, suggesting a viable treatment approach in chronic and progressive cardiac fibrotic diseases95,96. Recently, a study showed that treating mice with the small molecule salinomycin, which is a mesenchymal cancer cell inhibitor, diminished injury following MI and reverted angiotensin II-induced fibrosis97. The same study showed that salinomycin decreased activation of human CFs in culture, which are also mesenchymal cells. Other studies have shown that the expression of IL-11 also occurs downstream of TGFβ activation and appears to be specifically related to fibroblast activation, which makes for an attractive therapeutic target98,99. In addition, the ST2/IL-33 signaling axis has been implicated in cardiac fibrosis through direct signaling at the level of the myofibroblast100,101. Additional studies are needed to ascertain whether systemic IL-11 inhibition, ST2 inhibition or salinomycin treatment will have anti-fibrotic effects, either with or without TGFβ inhibition. As IL-11 and IL-33 are also implicated in the pathobiology of renal failure, their regulation presents an attractive possibility for treating patients with multi-organ disease such as cardiorenal and chronic kidney diseases99.
Mechanical signals in cardiac fibroblast activation
CFs not only process signals from growth factors and cytokines, but also sense and respond to mechanical signals (Figure 1)52,102. It is well established in vitro that culture substrate stiffness affects the CF transition to myofibroblasts55,103. While CFs maintain a quiescent phenotype when cultured on a hyaluronic acid (HA) gels that have a physiologically-relevant stiffness, they are activated and express a full spectrum of ECM proteins when cultured on stiffer HA gels that mimic a pathological environment, such as the scar region following MI103. Similarly, fibroblasts can be activated through sensed changes in mechanical stretch from their surrounding environment103. These in vitro findings match changes of in vivo gene expression profiles post-MI discussed above63.
Several molecular signaling pathways are involved in the ability of CFs to sense and interpret mechanical changes in their environment. For an in-depth discussion, we refer the reader to a recent review104. Briefly, these pathways include: 1) cell surface integrin complexes that bind specific ECM proteins; 2) regulation of actin and other cytoskeletal proteins, including those within focal adhesions that directly respond to changes in cell shape; 3) ion channels such as TRPV4 and TRPC6 that are critical for the activation of downstream signaling pathways and intracellular kinases; and 4) the nuclear mechano-sensing complexes such as the LINC complex, which directly affects gene expression in response to mechanical stimuli104.
While changes in ECM stiffness and tension are known to activate fibroblasts through signaling cascades initiated by integrin receptor activation105, only recently have researchers determined that the Hippo signaling pathway is also essential for the mechanosensing ability of CFs during development and disease conditions102. Using an epicardial-specific Wt-1-CreERT2 mouse line, deletion of YAP/TAZ106 or LATS1/2107 led to defects in the coronary vasculature during development, illustrating that the Hippo pathway is critical for epicardial cell differentiation during development, from which cardiac interstitial fibroblasts are derived. Indeed, Hippo signaling is required for CFs to maintain a quiescent stage in the postnatal heart. Lats1/2 deletion in Tcf21-expressing fibroblasts led to their spontaneous transition to a myofibroblast state, leading to cardiac fibrosis without injury108. Furthermore, deletion of YAP using the same Tcf21-MerCreMer system attenuated injury-induced cardiac fibrosis either post-MI or in response to chronic AngII/PE treatment109, while YAP overexpression with adeno-associated virus infection in mice augmented stress-induced fibrosis in vivo110. Collectively, studies suggest that identifying critical components of the Hippo signaling pathway may offer more specific therapeutic targets to quell cardiac fibrosis.
Cardiac ECM directly affects cardiomyocytes in disease
While some evidence suggests that cardiomyocytes and fibroblasts can directly contact each other, which would have to occur through the restrictive basal lamina that surrounds each cardiomyocyte111, the majority of studies have focused on the interactions between these two cell types through paracrine signaling or ECM modifications. One primary means of such signaling could be related to the composition of the ECM itself. As discussed earlier, defective collagen production by CFs due to deletion of Hsp47 resulted in less cardiomyocyte hypertrophy with pressure overload6. We also previously showed that Postn-null mice have altered ECM characteristics and reduced ability to expand the ECM with pressure overload, which similarly reduced cardiomyocyte hypertrophy with disease-inducing stimulation21. Cardiomyocytes directly attach to ECM generated by the fibroblasts through both integrins and the dystroglycan-sarcoglycan complex located within the cell membrane, which provides cytoskeletal support and allows for stress and mechanical sensing112. Indeed, deletion of β1 integrin from the heart results in fibrosis and dilated cardiomyopathy, and these mice were intolerant to pressure overload stimulation113, similar to mice in which the integrin-linking proteins Talin-1/2 were deleted114. In contrast, overexpression of α7/β1d integrin in the hearts of transgenic mice was protective and reduced the degree of cardiomyopathy following MI injury115. Interestingly, a recent study demonstrated that mechanical cues perceived by cardiomyocytes lead to altered nuclear localization and chromatin reorganization, affecting cell fate116. Thus, cardiomyocytes sense the underlying ECM and its integrity in formulating a disease response, and it is reasonable to speculate that cardiomyocytes and fibroblasts communicate to each other through this dynamic ECM environment and its mechanical properties (Figure 2).
Macrophages in fibroblast activation and ECM composition
While CFs sense cell-ECM interactions as part of their activation cues, they are also activated by inflammatory cytokines and chemokines that are typical of the diseased heart. Several excellent reviews have covered the ways in which immune responses affect fibroblast activation and disease progression77,117,118,119. Immune cells are among the first responders to an injury where they actively mediate clearance of dead cells and initiate inflammatory processes that underlie tissue healing and ECM remodeling (reviewed in119). Among other cell types, macrophages, neutrophils, T cells, B cells, mast cells, and dendritic cells all contribute to these processes120,121. However, sustained inflammatory states and prolonged activity of select immune cell types continually activate fibroblasts, exacerbating pathological fibrosis and cardiomyopathy. Thus, regulating the inflammatory response and activity of immune cell subtypes has been another therapeutic strategy in attempting to better control cardiac wound healing and pathological fibrosis77,117,118. In this section, we focus on a discussion of macrophage communication with fibroblasts and cardiomyocytes, and macrophage-derived ECM modifications (Figure 3).
Figure 3. Promising therapeutic targets to mediate CF activation and fibrosis.
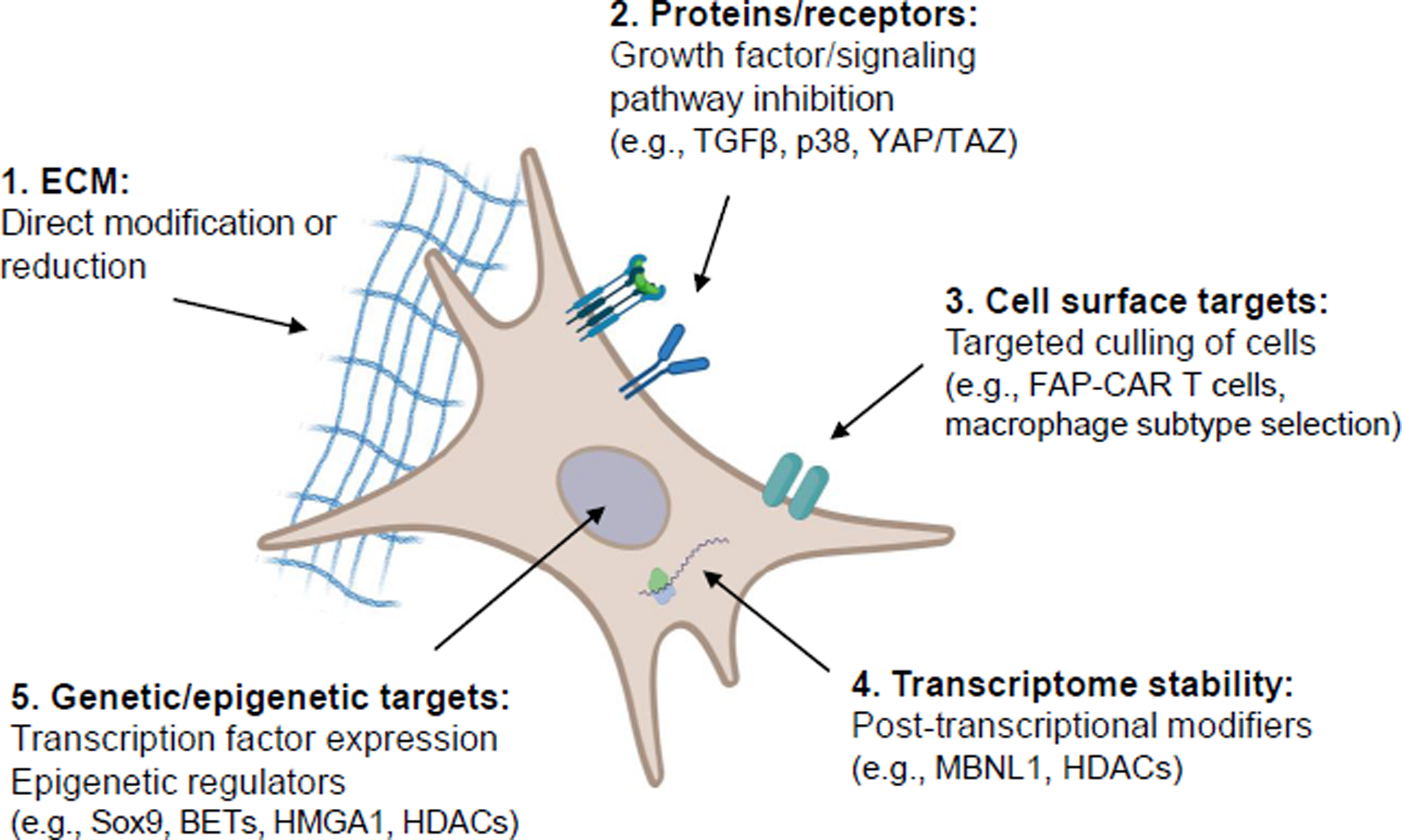
Based on recent studies, there are several avenues that can be further explored for their potential to alleviate cardiac fibrosis. It is likely that a combination of these approaches will efficiently combat the progression of fibrosis after injury or disease. 1) The extracellular matrix (ECM) can be targeted with inhibitors of crosslinking and fibrillogenesis, or indirectly through changing cell-derived expression of ECM factors. 2) Cell surface receptors and downstream signaling cascades can be used for targeted pathway inhibition, including signaling cascades like transforming growth factor-beta, p38/MAPK (mitogen-activated protein kinase) and Yap/Taz. 3) Cell surface markers can be used to identify and target unwanted cells, such as FAP recognition of CAR (chimeric antigen receptor) T cells. This approach may also be utilized to target macrophage subpopulations. 4) Post-transcriptional modifiers, such as muscleblind-like 1 (MBNL1) and histone deacetylases (HDACs), could be exploited to alter RNA transcript stability and subsequent cell activation. 5) Cell activation may be modified by regulating epigenetic modifications and transcription factor expression (HMGA1, high-mobility group A1).
Like fibroblasts, cardiac macrophage populations are heterogeneous during development, homeostasis and disease122–124. The traditional definitions of “M1” and “M2” macrophages as pro- or anti-inflammatory are evolving to include specific cell surface markers that more closely define a particular subtype of macrophage and its functions125. For example, cardiac tissue resident macrophages that are of embryonic origin and are CCR2-, MHCIIlow , TIMD4+, and CX3CR1+ are generally protective to the heart, while recruited circulating monocyte-derived macrophages that are CCR2+ (MHCIIhigh, Ly6Chigh) are generally inflammatory and detrimental to the long term healing response126–129. Different types of macrophages and their cell surface receptors provide unique targets for modulating tissue responses and the activity of fibroblasts.
Depletion of CD206+ macrophages attenuated injury after MI in mice, which contributed to altered fibroblast activation (through macrophage-derived IL-1α and osteopontin) and decreased collagen deposition130. Yan et al., classified monocytes and macrophage populations based on their level of expression of Ly6C, CD11b and F4/80 each, observing differential upregulation of genes involved with ECM remodeling, including collagen organization131. Recently, our lab showed that CCR2+ monocytes and macrophages are recruited and expand in number within the central core of the MI area of injury in the heart, while the tissue resident CCR2- CX3CR1+ macrophages expand most notably around the periphery of the injury area132. Mechanistically, CFs co-cultured with MI-derived CCR2+ cardiac macrophages showed induction of markers of myofibroblast formation and activation, whereas co-culturing with cardiac-derived CX3CR1+ macrophages did not elicit such an activation response132. These results corroborate studies showing divergent functions for different macrophage sub-populations in the heart. For example, tissue resident (CCR2-) macrophages, previously denoted as “M2”, are associated with enhanced cardiac healing after injury and less fibrosis, while CCR2+ macrophages, previously termed “M1” macrophages, are typically pro-inflammatory and promote fibrosis and maladaptation126,127. Studies suggest that CCR2+ macrophages secrete a large amount of pro-inflammatory cytokines, including TGFβ, IL-1β, IFN and IL-6133,134, which act upon local/recruited CFs. In addition to cytokine and growth factor secretion, macrophage phagocytosis of debris and ECM is critical to the healing process, paving the way for remodeling of the myocardium with angiogenesis125. An in-depth discussion of the function of different populations of macrophages in the heart was recently published, and provides additional considerations for ways in which they may affect fibroblast function after injury135.
There are also data linking immune cell regulation of the fibroblast and its control of ECM composition to the regenerative response of cardiomyocytes136–138. This again suggests that myocytes and fibroblasts communicate in the heart through the ECM, but that macrophages are important regulators of fibroblast activity during this process. Indeed, macrophages are known to be required for neonatal heart regeneration136. Godwin et al., used clodronate-loaded liposomes to deplete macrophages after cryo injury of axolotl hearts in a model of adult heart regeneration137. In this model, depletion of macrophages accelerated CF activation and decreased tissue regeneration, with altered expression of matrix metalloproteases and lysyl oxidase enzymes that was associated with poor ECM architecture137. Furthermore, Simoes et al., recently demonstrated that cardiac macrophages themselves may be able to deposit several collagen isoforms, as well as proteins that bind and modify collagen67. Collectively, these studies suggest that macrophages are critical to the initial wound healing response through modifying fibroblast activation, impacting ECM composition directly (Figure 1). However, it is still unclear in any of the discussed models whether macrophage-derived ECM deposition and modifications provide long-lasting contributions to the matrix directly, or if they set up the tissue microenvironment to properly attract and activate CFs and other cells.
Emerging therapies to treat cardiac fibrosis
Anti-fibrotic therapies for cardiovascular disease have remained largely unsuccessful. Given ongoing discoveries about the temporal regulation and heterogeneity of cell populations in the heart, successful modulation of fibrosis will likely require a combination of approaches. Figure 3 provides a summary of therapeutic targets of fibroblast activation that are discussed in this Review.
It is reasonable to speculate that modifying the ECM directly may attenuate disease progression. To this point, studies have targeted myofibroblasts or ECM proteins to decrease cardiac fibrosis. Genetically blocking myofibroblast-specific TGFβ signaling during fibrotic progression led to partial injury resolution in a chronic HCM mouse model70, suggesting that cardiac fibrosis is reversable under certain conditions. Lineage tracing studies revealed that periostin-expressing myofibroblasts could convert back to a quiescent fibroblast state after cessation of AngII/PE treatment61. Furthermore, directly targeting fibronectin fibrillogenesis with a small molecule inhibitor, pUR4, attenuated the development of fibrosis and improved heart function following ischemia/reperfusion injury33. Even considering results from these studies, it is still unclear the extent to which ECM modifications can truly be reversed, which depends on other aspects of the injury or disease. For example, if fibrosis develops to replace dead cardiomyocytes, removing fibrotic tissue without replenishing myocardial cell populations would be detrimental to heart function and could lead to ventricular wall rupture. Likewise, if interstitial fibrosis is providing structural support to an ailing heart, alleviating the fibrosis without providing another source of structure would impact whole organ function. In this regard, future studies should investigate a multifaceted therapeutic approach that addresses cell and organ function in coordination with beneficial ECM composition.
In the past several years, molecular targets have been identified that affect fibroblast activity in the heart. A non-histone chromatin-binding protein, high-mobility group A1 (HMGA1), was shown to regulate CF activation through modulation of FOXO1139, which was previously connected to the development of cardiac fibrosis140,141. Overexpression of HMGA1 in periostin-expressing fibroblasts increased, and knockdown diminished, isoproterenol- and angiotensin II- induced cardiac fibrosis and dysfunction in vivo, and directly modulated CF activation in vitro139. Concomitant inhibition of FOXO1 reversed the effects of HMGA1 overexpression, without obvious drawbacks in vitro or in vivo. However, it is not entirely clear how HMGA1 might regulate other essential biologic and cellular processes, including inflammatory signaling in sepsis-induced cardiac injury142. In humans, HMGA1 was identified as a candidate gene for susceptibility to MI143 and as a genetic variant in some patients with type 2 diabetes144, providing rationale for further investigation into its therapeutic potential.
Another emerging target of CF activation is the family of histone deacetylases (HDACs), which are classically known to modulate gene expression and extranuclear signaling transduction cascades145. HDAC inhibition with Givinostat improved heart function in several models of diastolic dysfunction146,147. Recently, Travers et al., showed that Givinostat seems to work by directly modulating CF activation148. Interestingly, although overt fibrosis did not manifest in their model of HFpEF, fibroblast activation and expression of ECM modifying genes were repressed by HDAC inhibition, leading to improvement in cardiac function. These studies not only point to possible benefits of targeting HDACs to regulate CF phenotypes, but also illustrate advantages to monitoring the status of the cardiac ECM, even outside of overt fibrotic disease.
The BET family of transcriptional coactivators have also been identified as mediators of cardiac pathology. Using small molecule inhibitors to interfere with their function have lessened heart failure in mouse injury models149,150. Alexanian et al., recently used the BET inhibitor JQ1 to reversibly mediate CF activation in response to pressure overload hypertrophy151. Their experiments showed that JQ1 treatment mitigated cardiac fibrosis and improved survival by decreasing Meox1-mediated CF activation151. Interestingly, the researchers observed phenotypic reversion upon removal of BET inhibition151, again demonstrating the persistence of fibrotic signals independent of traditional CF activation states.
Another promising avenue for controlling CF activation is through transcriptome modulation. Muscleblind-like 1 (MBNL1) is an RNA binding protein that can affect RNA transcript stability and localization, and was recently shown to modulate the CF differentiation in vivo152. Fibroblast-specific knockout of MBNL1 significantly reduced the fibroblast transition to myofibroblasts after injury, as well as further progression to the matrifibrocyte phenotype that resides within a mature scar63,152. Interestingly, the authors found that this effect was, at least in part, due to Sox9 transcript stability by MBNL1152. Furthermore, they again observed a disconnect between absolute gene expression and the progression of fibrosis, as knocking out MBNL1 in periostin-expressing fibroblasts did not alter αSMA expression, but still protected against injury-induced cardiac dysfunction152. While more research is needed, post-transcriptional modulation of RNA translation provides another targeted approach to curbing fibroblast activation.
A major recent advance in cancer therapeutics is engineering chimeric antigen receptor (CAR)-cytotoxic T cells that cause them to recognize proteins on the surface of cancer cells to effectively target and eliminate them153. Aghajanian et al., treated mice with CAR T cells targeted to a subpopulation of CFs that express fibroblast activation protein (FAP), thereby decreasing fibrosis and maladaptive tissue remodeling in response to injury154. More recently, the same group used CD5-targeted lipid nanoparticles to generate FAP-specific CAR T cells in vivo155, successfully reducing angiotensin II-induced hypertrophy in mice. While clinical data are needed, a major advantage to treating human patients would be the temporal control of targeting a specific cell population after injury. Additional studies are needed to determine: a) if FAP is indeed the best cellular marker for targeting human CFs with chronic disease; b) the temporal regulation of any CAR T cell target of interest, keeping in mind the heterogeneity of each cell population; and c) full assessment of toxicities associated with depleting activated fibroblasts in other organs154,155. The development of these and other techniques for reprogramming and drug delivery can easily be applied to different macrophage populations in the heart as well. These exciting advancements in targeting a specific cell population at a specific time provide favorable avenues to alter the course of pathologic tissue scarring and heart repair in humans.
Concluding remarks
The ECM is critical for proper cell communication in the heart. Different cell populations, largely CFs, condition the ECM to provide structural support, growth factors and other signaling proteins that regulate the behavior of all cells within the heart. Because of the inherent heterogeneity in tissue architecture, it is becoming clear that a much larger systems biology approach is necessary to understand both homeostasis and disease progression in the heart. Untangling the multitude of interactions among the ECM and myocardial cell populations will prove beneficial in our efforts to advance therapeutic options to address pathological remodeling of the heart that occurs with disease and aging.
Acknowledgements
We would like to acknowledge the researchers and studies that have contributed to our knowledge of cardiac development and fibrosis, but were not discussed here due to space and reference limitations. This work was supported by grants from the NIH (R01HL105924, R01HL142217) to J.D.M and a Career Development Award from the American Heart Association (20CDA35310504) to Q.M.
Footnotes
Competing Interests
All authors declare no conflicts of interest.
References
- 1.Burgess ML, McCrea JC & Hedrick HL Age-associated changes in cardiac matrix and integrins. Mech. Ageing Dev. 122, 1739–1756, doi: 10.1016/s0047-6374(01)00296-2 (2001). [DOI] [PubMed] [Google Scholar]
- 2.de Souza RR Aging of myocardial collagen. Biogerontology 3, 325–335, doi: 10.1023/a:1021312027486 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Weber KT, Pick R, Jalil JE, Janicki JS & Carroll EP Patterns of myocardial fibrosis. J. Mol. Cell. Cardiol. 21 Suppl 5, 121–131, doi: 10.1016/0022-2828(89)90778-5 (1989). [DOI] [PubMed] [Google Scholar]
- 4.Lu P, Takai K, Weaver VM & Werb Z Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 3, doi: 10.1101/cshperspect.a005058 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frangogiannis NG Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 65, 70–99, doi: 10.1016/j.mam.2018.07.001 (2019). [DOI] [PubMed] [Google Scholar]
- 6. Khalil H et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight 4, e128722, doi: 10.1172/jci.insight.128722 (2019). By ablating a critical collagen chaperone (Hsp47), this study shows that collagen production specifically from cardiac fibroblasts is essential to the development of cardiac fibrosis following injury.
- 7.Sun Y & Weber KT Infarct scar: a dynamic tissue. Cardiovasc. Res. 46, 250–256, doi: 10.1016/s0008-6363(00)00032-8 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Weber KT Fibrosis in hypertensive heart disease: focus on cardiac fibroblasts. J. Hypertens. 22, 47–50, doi: 10.1097/00004872-200401000-00011 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Willems IE, Havenith MG, De Mey JG & Daemen MJ The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am. J. Pathol. 145, 868–875 (1994). [PMC free article] [PubMed] [Google Scholar]
- 10.Hinz B Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537, doi: 10.1038/sj.jid.5700613 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Wynn TA Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210, doi: 10.1002/path.2277 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hynes RO & Naba A Overview of the matrisome--an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 4, a004903, doi: 10.1101/cshperspect.a004903 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villalobos Lizardi JC et al. A guide for assessment of myocardial stiffness in health and disease. Nat. Cardiovasc. Res. 1, 8–22 (2022). [Google Scholar]
- 14.Varagic J, Susic D & Frohlich E Heart, aging, and hypertension. Curr. Opin. Cardiol. 16, 336–341, doi: 10.1097/00001573-200111000-00004 (2001). [DOI] [PubMed] [Google Scholar]
- 15. Perestrelo AR et al. Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart. Circ. Res. 128, 24–38, doi: 10.1161/CIRCRESAHA.120.317685 (2021). In this paper, the authors combined a number of imaging and analysis techniques to observe differential ECM organization and fibroblast activation in mouse hearts after myocardial infarction, which was reflected in experiments with isolated fibroblasts from human heart failure patients.
- 16.Bugg D et al. Infarct Collagen Topography Regulates Fibroblast Fate via p38-Yes-Associated Protein Transcriptional Enhanced Associate Domain Signals. Circ. Res. 127, 1306–1322, doi: 10.1161/CIRCRESAHA.119.316162 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weis SM et al. Myocardial mechanics and collagen structure in the osteogenesis imperfecta murine (oim). Circ. Res. 87, 663–669, doi: 10.1161/01.res.87.8.663 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Mukherjee D & Sen S Alteration of cardiac collagen phenotypes in hypertensive hypertrophy: role of blood pressure. J. Mol. Cell. Cardiol. 25, 185–196, doi: 10.1006/jmcc.1993.1021 (1993). [DOI] [PubMed] [Google Scholar]
- 19.Norton GR et al. Myocardial stiffness is attributed to alterations in cross-linked collagen rather than total collagen or phenotypes in spontaneously hypertensive rats. Circulation 96, 1991–1998, doi: 10.1161/01.cir.96.6.1991 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Echegaray K et al. Role of Myocardial Collagen in Severe Aortic Stenosis With Preserved Ejection Fraction and Symptoms of Heart Failure. Rev. Esp. Cardiol. (Engl. Ed.) 70, 832–840, doi: 10.1016/j.rec.2016.12.038 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Oka T et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 101, 313–321, doi: 10.1161/CIRCRESAHA.107.149047 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyazaki H et al. Comparison of gene expression profiling in pressure and volume overload-induced myocardial hypertrophies in rats. Hypertens. Res. 29, 1029–1045, doi: 10.1291/hypres.29.1029 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Sivakumar P, Gupta S, Sarkar S & Sen S Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol. Cell. Biochem. 307, 159–167, doi: 10.1007/s11010-007-9595-2 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Zibadi S, Vazquez R, Larson DF & Watson RR T lymphocyte regulation of lysyl oxidase in diet-induced cardiac fibrosis. Cardiovasc. Toxicol. 10, 190–198, doi: 10.1007/s12012-010-9078-7 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Ohmura H et al. Cardiomyocyte-specific transgenic expression of lysyl oxidase-like protein-1 induces cardiac hypertrophy in mice. Hypertens. Res. 35, 1063–1068, doi: 10.1038/hr.2012.92 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Yang J et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat. Commun. 7, 13710, doi: 10.1038/ncomms13710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia Y et al. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension 58, 902–911, doi: 10.1161/HYPERTENSIONAHA.111.175323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frolova EG et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. FASEB J. 26, 2363–2373, doi: 10.1096/fj.11-190728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schips TG et al. Thrombospondin-3 augments injury-induced cardiomyopathy by intracellular integrin inhibition and sarcolemmal instability. Nat. Commun. 10, 76, doi: 10.1038/s41467-018-08026-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Podesser BK et al. Tenascin-C promotes chronic pressure overload-induced cardiac dysfunction, hypertrophy and myocardial fibrosis. J. Hypertens. 36, 847–856, doi: 10.1097/HJH.0000000000001628 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Bradshaw AD et al. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 119, 269–280, doi: 10.1161/CIRCULATIONAHA.108.773424 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shinde AV et al. Tissue transglutaminase induction in the pressure-overloaded myocardium regulates matrix remodelling. Cardiovasc. Res. 113, 892–905, doi: 10.1093/cvr/cvx053 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valiente-Alandi I et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 138, 1236–1252, doi: 10.1161/CIRCULATIONAHA.118.034609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta RK & Kuznicki J Biological and Medical Importance of Cellular Heterogeneity Deciphered by Single-Cell RNA Sequencing. Cells 9, doi: 10.3390/cells9081751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M, Gu M, Liu L, Liu Y & Tian L Single-Cell RNA Sequencing (scRNA-seq) in Cardiac Tissue: Applications and Limitations. Vasc. Health Risk Manag. 17, 641–657, doi: 10.2147/VHRM.S288090 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamada S & Nomura S Review of Single-Cell RNA Sequencing in the Heart. Int. J. Mol. Sci. 21, doi: 10.3390/ijms21218345 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tallquist MD Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 82, 63–78, doi: 10.1146/annurev-physiol-021119-034527 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat. Commun. 11, 2585, doi: 10.1038/s41467-020-16204-w (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farbehi N et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 8, doi: 10.7554/eLife.43882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLaughter DM et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 39, 480–490, doi: 10.1016/j.devcel.2016.10.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui M et al. Dynamic Transcriptional Responses to Injury of Regenerative and Non-regenerative Cardiomyocytes Revealed by Single-Nucleus RNA Sequencing. Dev. Cell 55, 665–667, doi: 10.1016/j.devcel.2020.11.006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skelly DA et al. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 22, 600–610, doi: 10.1016/j.celrep.2017.12.072 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Kovacic JC, Mercader N, Torres M, Boehm M & Fuster V Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation 125, 1795–1808, doi: 10.1161/CIRCULATIONAHA.111.040352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Padula SL, Velayutham N & Yutzey KE Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration. Int. J. Mol. Sci. 22, doi: 10.3390/ijms22063288 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lacraz GPA et al. Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis During Ischemic Injury. Circulation 136, 1396–1409, doi: 10.1161/CIRCULATIONAHA.117.027832 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Mantri M et al. Spatiotemporal single-cell RNA sequencing of developing chicken hearts identifies interplay between cellular differentiation and morphogenesis. Nat. Commun. 12, 1771, doi: 10.1038/s41467-021-21892-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat. Commun. 12, 5784, doi: 10.1038/s41467-021-25933-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hortells L, Meyer EC, Thomas ZM & Yutzey KE Periostin-expressing Schwann cells and endoneurial cardiac fibroblasts contribute to sympathetic nerve fasciculation after birth. J. Mol. Cell. Cardiol. 154, 124–136, doi: 10.1016/j.yjmcc.2021.02.001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis J & Molkentin JD Myofibroblasts: trust your heart and let fate decide. J. Mol. Cell. Cardiol. 70, 9–18, doi: 10.1016/j.yjmcc.2013.10.019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tallquist MD & Molkentin JD Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 14, 484–491, doi: 10.1038/nrcardio.2017.57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frangogiannis NG, Michael LH & Entman ML Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc. Res. 48, 89–100, doi: 10.1016/s0008-6363(00)00158-9 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C & Brown RA Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell. Biol. 3, 349–363, doi: 10.1038/nrm809 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Serini G et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 142, 873–881, doi: 10.1083/jcb.142.3.873 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cleutjens JP, Verluyten MJ, Smiths JF & Daemen MJ Collagen remodeling after myocardial infarction in the rat heart. Am. J. Pathol. 147, 325–338 (1995). [PMC free article] [PubMed] [Google Scholar]
- 55.van Putten S, Shafieyan Y & Hinz B Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 93, 133–142, doi: 10.1016/j.yjmcc.2015.11.025 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Cawston TE & Young DA Proteinases involved in matrix turnover during cartilage and bone breakdown. Cell Tissue Res. 339, 221–235, doi: 10.1007/s00441-009-0887-6 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Prabhu SD & Frangogiannis NG The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 119, 91–112, doi: 10.1161/CIRCRESAHA.116.303577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Umbarkar P, Ejantkar S, Tousif S & Lal H Mechanisms of Fibroblast Activation and Myocardial Fibrosis: Lessons Learned from FB-Specific Conditional Mouse Models. Cells 10, doi: 10.3390/cells10092412 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frangogiannis NG Cardiac fibrosis. Cardiovasc. Res. 117, 1450–1488, doi: 10.1093/cvr/cvaa324 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Molkentin JD et al. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 136, 549–561, doi: 10.1161/CIRCULATIONAHA.116.026238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanisicak O et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260, doi: 10.1038/ncomms12260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maruyama S et al. Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol. Med. 8, 949–966, doi: 10.15252/emmm.201506151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu X et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Invest. 128, 2127–2143, doi: 10.1172/JCI98215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rainer PP et al. Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ. Res. 114, 1246–1257, doi: 10.1161/CIRCRESAHA.114.302653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teekakirikul P et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-beta. J. Clin. Invest. 120, 3520–3529, doi: 10.1172/JCI42028 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adiarto S et al. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci. 91, 651–657, doi: 10.1016/j.lfs.2012.02.006 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Simoes FC et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 11, 600, doi: 10.1038/s41467-019-14263-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khalil H et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 127, 3770–3783, doi: 10.1172/JCI94753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhandary B et al. Cardiac Fibrosis in Proteotoxic Cardiac Disease is Dependent Upon Myofibroblast TGF -beta Signaling. J. Am. Heart Assoc. 7, e010013, doi: 10.1161/JAHA.118.010013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meng Q et al. Myofibroblast-Specific TGFbeta Receptor II Signaling in the Fibrotic Response to Cardiac Myosin Binding Protein C-Induced Cardiomyopathy. Circ. Res. 123, 1285–1297, doi: 10.1161/CIRCRESAHA.118.313089 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiang FL, Fang M & Yutzey KE Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 8, 712, doi: 10.1038/s41467-017-00840-w (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giordano C, Francone M, Cundari G, Pisano A & d’Amati G Myocardial fibrosis: morphologic patterns and role of imaging in diagnosis and prognostication. Cardiovasc. Pathol. 56, 107391, doi: 10.1016/j.carpath.2021.107391 (2022). [DOI] [PubMed] [Google Scholar]
- 73.Zou Y et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 6, 499–506, doi: 10.1038/ncb1137 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Kim J, Eckhart AD, Eguchi S & Koch WJ Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J. Biol. Chem. 277, 32116–32123, doi: 10.1074/jbc.M204895200 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Travers JG et al. Pharmacological and Activated Fibroblast Targeting of Gbetagamma-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression. J. Am. Coll. Cardiol. 70, 958–971, doi: 10.1016/j.jacc.2017.06.049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rurik JG, Aghajanian H & Epstein JA Immune Cells and Immunotherapy for Cardiac Injury and Repair. Circ. Res. 128, 1766–1779, doi: 10.1161/CIRCRESAHA.121.318005 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smolgovsky S, Ibeh U, Tamayo TP & Alcaide P Adding insult to injury - Inflammation at the heart of cardiac fibrosis. Cell. Signal. 77, 109828, doi: 10.1016/j.cellsig.2020.109828 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dai Z, Aoki T, Fukumoto Y & Shimokawa H Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J. Cardiol. 60, 416–421, doi: 10.1016/j.jjcc.2012.06.009 (2012). [DOI] [PubMed] [Google Scholar]
- 79.Giordano C, Francone M, Cundari G, Pisano A & d'Amati G Myocardial fibrosis: morphologic patterns and role of imaging in diagnosis and prognostication. Cardiovasc. Pathol. 56, 107391, doi: 10.1016/j.carpath.2021.107391 (2021). [DOI] [PubMed] [Google Scholar]
- 80.Liu M, Lopez de Juan Abad B & Cheng K Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies. Adv. Drug Deliv. Rev. 173, 504–519, doi: 10.1016/j.addr.2021.03.021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tarbit E, Singh I, Peart JN & Rose'Meyer RB Biomarkers for the identification of cardiac fibroblast and myofibroblast cells. Heart Fail. Rev. 24, 1–15, doi: 10.1007/s10741-018-9720-1 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Bujak M et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 173, 57–67, doi: 10.2353/ajpath.2008.070974 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kanellakis P, Ditiatkovski M, Kostolias G & Bobik A A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc. Res. 95, 77–85, doi: 10.1093/cvr/cvs142 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Ma F et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One 7, e35144, doi: 10.1371/journal.pone.0035144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peng H et al. Profibrotic Role for Interleukin-4 in Cardiac Remodeling and Dysfunction. Hypertension 66, 582–589, doi: 10.1161/HYPERTENSIONAHA.115.05627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leask A Getting to the heart of the matter: new insights into cardiac fibrosis. Circ. Res. 116, 1269–1276, doi: 10.1161/CIRCRESAHA.116.305381 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Cho N, Razipour SE & McCain ML Featured Article: TGF-beta1 dominates extracellular matrix rigidity for inducing differentiation of human cardiac fibroblasts to myofibroblasts. Exp. Biol. Med. (Maywood) 243, 601–612, doi: 10.1177/1535370218761628 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koitabashi N et al. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J. Clin. Invest. 121, 2301–2312, doi: 10.1172/JCI44824 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anderton MJ et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 39, 916–924, doi: 10.1177/0192623311416259 (2011). [DOI] [PubMed] [Google Scholar]
- 90.Mitra MS et al. A Potent Pan-TGFbeta Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 175, 24–34, doi: 10.1093/toxsci/kfaa024 (2020). [DOI] [PubMed] [Google Scholar]
- 91.Herbertz S et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 9, 4479–4499, doi: 10.2147/DDDT.S86621 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee KW et al. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation 114, 1703–1712, doi: 10.1161/CIRCULATIONAHA.106.624320 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nguyen DT, Ding C, Wilson E, Marcus GM & Olgin JE Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm 7, 1438–1445, doi: 10.1016/j.hrthm.2010.04.030 (2010). [DOI] [PubMed] [Google Scholar]
- 94.Lewis GA et al. Pirfenidone in heart failure with preserved ejection fraction: a randomized phase 2 trial. Nat. Med. 27, 1477–1482, doi: 10.1038/s41591-021-01452-0 (2021). [DOI] [PubMed] [Google Scholar]
- 95.Kojonazarov B et al. p38 MAPK Inhibition Improves Heart Function in Pressure-Loaded Right Ventricular Hypertrophy. Am. J. Respir. Cell Mol. Biol. 57, 603–614, doi: 10.1165/rcmb.2016-0374OC (2017). [DOI] [PubMed] [Google Scholar]
- 96.Wissing ER et al. P38alpha MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum. Mol. Genet. 23, 5452–5463, doi: 10.1093/hmg/ddu270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burke RM et al. Prevention of Fibrosis and Pathological Cardiac Remodeling by Salinomycin. Circ. Res. 128, 1663–1678, doi: 10.1161/CIRCRESAHA.120.317791 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fernandez-Ruiz I Cardioprotection: IL-11 is a potential therapeutic target in cardiovascular fibrosis. Nat. Rev. Cardiol. 15, 1, doi: 10.1038/nrcardio.2017.197 (2018). [DOI] [PubMed] [Google Scholar]
- 99.Corden B, Adami E, Sweeney M, Schafer S & Cook SA IL-11 in cardiac and renal fibrosis: Late to the party but a central player. Br. J. Pharmacol. 177, 1695–1708, doi: 10.1111/bph.15013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ghali R et al. IL-33 (Interleukin 33)/sST2 Axis in Hypertension and Heart Failure. Hypertension 72, 818–828, doi: 10.1161/HYPERTENSIONAHA.118.11157 (2018). [DOI] [PubMed] [Google Scholar]
- 101.Vianello E, Dozio E, Tacchini L, Frati L & Corsi Romanelli MM ST2/IL-33 signaling in cardiac fibrosis. Int. J. Biochem. Cell Biol. 116, 105619, doi: 10.1016/j.biocel.2019.105619 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Meng F, Xie B & Martin JF Targeting the Hippo pathway in heart repair. Cardiovasc. Res., doi: 10.1093/cvr/cvab291 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herum KM, Choppe J, Kumar A, Engler AJ & McCulloch AD Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 28, 1871–1882, doi: 10.1091/mbc.E17-01-0014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD & Omens JH Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 16, 361–378, doi: 10.1038/s41569-019-0155-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.MacKenna DA, Dolfi F, Vuori K & Ruoslahti E Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Invest. 101, 301–310, doi: 10.1172/JCI1026 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Singh A et al. Hippo Signaling Mediators Yap and Taz Are Required in the Epicardium for Coronary Vasculature Development. Cell Rep. 15, 1384–1393, doi: 10.1016/j.celrep.2016.04.027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xiao Y et al. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell. 45, 153–169 e156, doi: 10.1016/j.devcel.2018.03.019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiao Y et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 33, 1491–1505, doi: 10.1101/gad.329763.119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Francisco J et al. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 5, 931–945, doi: 10.1016/j.jacbts.2020.07.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Francisco J et al. AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci. Rep. 11, 10553, doi: 10.1038/s41598-021-89989-5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schultz F et al. Cardiomyocyte-myofibroblast contact dynamism is modulated by connexin-43. FASEB J. 33, 10453–10468, doi: 10.1096/fj.201802740RR (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lapidos KA, Kakkar R & McNally EM The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 94, 1023–1031, doi: 10.1161/01.RES.0000126574.61061.25 (2004). [DOI] [PubMed] [Google Scholar]
- 113.Shai SY et al. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ. Res. 90, 458–464, doi: 10.1161/hh0402.105790 (2002). [DOI] [PubMed] [Google Scholar]
- 114.Manso AM et al. Loss of mouse cardiomyocyte talin-1 and talin-2 leads to beta-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 114, E6250–E6259, doi: 10.1073/pnas.1701416114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Okada H et al. Integrins protect cardiomyocytes from ischemia/reperfusion injury. J. Clin. Invest. 123, 4294–4308, doi: 10.1172/JCI64216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seelbinder B et al. Nuclear deformation guides chromatin reorganization in cardiac development and disease. Nat. Biomed. Eng., doi: 10.1038/s41551-021-00823-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ferrari I & Vagnozzi RJ Mechanisms and strategies for a therapeutic cardiac immune response. J. Mol. Cell. Cardiol. 158, 82–88, doi: 10.1016/j.yjmcc.2021.05.013 (2021). [DOI] [PubMed] [Google Scholar]
- 118.Zaidi Y, Aguilar EG, Troncoso M, Ilatovskaya DV & DeLeon-Pennell KY Immune regulation of cardiac fibrosis post myocardial infarction. Cell. Signal. 77, 109837, doi: 10.1016/j.cellsig.2020.109837 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lai SL, Marin-Juez R & Stainier DYR Immune responses in cardiac repair and regeneration: a comparative point of view. Cell. Mol. Life Sci. 76, 1365–1380, doi: 10.1007/s00018-018-2995-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pinto AR et al. Revisiting Cardiac Cellular Composition. Circ. Res. 118, 400–409, doi: 10.1161/CIRCRESAHA.115.307778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Litvinukova M et al. Cells of the adult human heart. Nature 588, 466–472, doi: 10.1038/s41586-020-2797-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Bajpai G et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 124, 263–278, doi: 10.1161/CIRCRESAHA.118.314028 (2019). This study uncovered foundational differences betweenthe injury-induced responses of tissue-resident macrophages that were CCR2- and recruited monocyte-derived CCR2+ macrophages.
- 123.Bajpai G et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245, doi: 10.1038/s41591-018-0059-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wong NR et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 54, 2072–2088 e2077, doi: 10.1016/j.immuni.2021.07.003 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nahrendorf M & Swirski FK Abandoning M1/M2 for a Network Model of Macrophage Function. Circ. Res. 119, 414–417, doi: 10.1161/CIRCRESAHA.116.309194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lindsey ML, Saucerman JJ & DeLeon-Pennell KY Knowledge gaps to understanding cardiac macrophage polarization following myocardial infarction. Biochim. Biophys. Acta 1862, 2288–2292, doi: 10.1016/j.bbadis.2016.05.013 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Dick SA et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39, doi: 10.1038/s41590-018-0272-2 (2019). Single cell RNA sequencing was used to identify a number of transcriptionally-distinct macrophage populations in the heart in response to injury, including genetic characterization of resident macrophages that are beneficial to heart function.
- 128.Epelman S, Lavine KJ & Randolph GJ Origin and functions of tissue macrophages. Immunity 41, 21–35, doi: 10.1016/j.immuni.2014.06.013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Revelo XS et al. Cardiac Resident Macrophages Prevent Fibrosis and Stimulate Angiogenesis. Circ. Res. 129, 1086–1101, doi: 10.1161/CIRCRESAHA.121.319737 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shiraishi M et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Invest. 126, 2151–2166, doi: 10.1172/JCI85782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yan X et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 62, 24–35, doi: 10.1016/j.yjmcc.2013.04.023 (2013). [DOI] [PubMed] [Google Scholar]
- 132.Vagnozzi RJ et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577, 405–409, doi: 10.1038/s41586-019-1802-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Martinez FO, Sica A, Mantovani A & Locati M Macrophage activation and polarization. Front. Biosci. 13, 453–461, doi: 10.2741/2692 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Mouton AJ et al. Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 113, 26, doi: 10.1007/s00395-018-0686-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gao Y, Qian N, Xu J & Wang Y The Roles of Macrophages in Heart Regeneration and Repair After Injury. Front. Cardiovasc. Med. 8, 744615, doi: 10.3389/fcvm.2021.744615 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Aurora AB et al. Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 124, 1382–1392, doi: 10.1172/JCI72181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Godwin JW, Debuque R, Salimova E & Rosenthal NA Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen. Med. 2, doi: 10.1038/s41536-017-0027-y (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maron BA et al. Individualized interactomes for network-based precision medicine in hypertrophic cardiomyopathy with implications for other clinical pathophenotypes. Nat. Commun. 12, 873, doi: 10.1038/s41467-021-21146-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xie Q et al. High-Mobility Group A1 Promotes Cardiac Fibrosis by Upregulating FOXO1 in Fibroblasts. Front. Cell Dev. Biol. 9, 666422, doi: 10.3389/fcell.2021.666422 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Arcidiacono B et al. HMGA1 is a novel transcriptional regulator of the FoxO1 gene. Endocrine 60, 56–64, doi: 10.1007/s12020-017-1445-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xin Z et al. FOXO1/3: Potential suppressors of fibrosis. Ageing Res. Rev. 41, 42–52, doi: 10.1016/j.arr.2017.11.002 (2018). [DOI] [PubMed] [Google Scholar]
- 142.Cai ZL et al. The effect of HMGA1 in LPS-induced Myocardial Inflammation. Int. J. Biol. Sci. 16, 1798–1810, doi: 10.7150/ijbs.39947 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.De Rosa S et al. HMGA1 is a novel candidate gene for myocardial infarction susceptibility. Int. J. Cardiol. 227, 331–334, doi: 10.1016/j.ijcard.2016.11.088 (2017). [DOI] [PubMed] [Google Scholar]
- 144.Chiefari E et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA 305, 903–912, doi: 10.1001/jama.2011.207 (2011). [DOI] [PubMed] [Google Scholar]
- 145.Gillette TG & Hill JA Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ. Res. 116, 1245–1253, doi: 10.1161/CIRCRESAHA.116.303630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jeong MY et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci. Transl. Med. 10, doi: 10.1126/scitranslmed.aao0144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wallner M et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci. Transl. Med. 12, doi: 10.1126/scitranslmed.aay7205 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Travers JG et al. HDAC Inhibition Reverses Preexisting Diastolic Dysfunction and Blocks Covert Extracellular Matrix Remodeling. Circulation 143, 1874–1890, doi: 10.1161/CIRCULATIONAHA.120.046462 (2021). This paper demonstrated that the HDAC inhibitor, Givinostat, modulated fibroblast activation and improved cardiac function in a mouse model of HFpEF; their findings also illustrate advantages to monitoring cellular expression of ECM-modifying genes and proteins, even outside of overt fibrotic disease.
- 149.Anand P et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 154, 569–582, doi: 10.1016/j.cell.2013.07.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Spiltoir JI et al. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 63, 175–179, doi: 10.1016/j.yjmcc.2013.07.017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Alexanian M et al. A transcriptional switch governs fibroblast activation in heart disease. Nature 595, 438–443, doi: 10.1038/s41586-021-03674-1 (2021). This study used the BET inhibitor JQ1 to reversibly mediate Meox1-induced cardiac fibroblast activation in response to pressure overload hypertrophy, highlighting the dynamics of fibroblast differentiation and activation.
- 152.Bugg D et al. MBNL1 drives dynamic transitions between fibroblasts and myofibroblasts in cardiac wound healing. Cell Stem Cell, doi: 10.1016/j.stem.2022.01.012 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.June CH, O'Connor RS, Kawalekar OU, Ghassemi S & Milone MC CAR T cell immunotherapy for human cancer. Science 359, 1361–1365, doi: 10.1126/science.aar6711 (2018). [DOI] [PubMed] [Google Scholar]
- 154.Aghajanian H et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433, doi: 10.1038/s41586-019-1546-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Rurik JG et al. CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96, doi: 10.1126/science.abm0594 (2022). In this paper, the authors utilized recent advances in lipid nanoparticle technology to engineer chimeric antigen receptor T cells in vivo that effectively targeted FAP-expressing fibroblasts to diminish angiotensin II-induced cardiac injury in mice.