


Abstract
Approximately 6000 specific DNA deletion events occur during development of the somatic macronucleus of the ciliate Tetrahymena. The eliminated Tlr1 element is 13 kb or more in length and has an 825 bp inverted repeat near the rearrangement junctions. A functional analysis of the cis-acting sequences required for Tlr1 rearrangement was performed. A construct consisting of the entire inverted repeat and several hundred base pairs of flanking DNA on each side was rearranged accurately in vivo and displayed junctional variability similar to the chromosomal Tlr1 rearrangement. Thus, 11 kb or more of internal element DNA is not required in cis for DNA rearrangement. A second construct with only 51 bp of Tetrahymena DNA flanking the right junction underwent aberrant rearrangement. Thus, a signal for determination of the Tlr1 junction is located in the flanking DNA, 51 bp or more from the right junction. Within the Tlr1 inverted repeat are 19 bp tandem repeats. A construct with the 19mer repeat region deleted from the right half of the inverted repeat utilized normal rearrangement junctions. Thus, despite its transposon-like structure, Tlr1 is similar to other DNA rearrangements in Tetrahymena in possessing cis-acting sequences outside the deleted DNA.
INTRODUCTION
During sexual reproduction in the ciliated protozoan, Tetrahymena, the somatic macronucleus develops from a mitotic product of the germ line micronucleus. Macronuclear development entails extensive DNA rearrangement resulting in a decrease in the DNA sequence complexity of ~10–20%. The remaining DNA is replicated to 45C, except rDNA which is amplified to ~10 000 copies per macronucleus (1). The vast majority of DNA rearrangements in Tetrahymena are breakage and rejoining events in which an internal sequence is eliminated and the flanking sequences are ligated (reviewed in 2). These events occur at an estimated 6000 different sites (3). At the majority of these sites, 0.5–3 kb of DNA is eliminated. However, longer stretches of micronuclear-limited DNA of 10 kb or more have been cloned (4,5). The deletion events are highly regular in the sense that a given site undergoes rearrangement in every developing macronucleus. Some of these rearrangements utilize alternative junctions, resulting in a limited repertoire of rearrangement products from that site (6–8).
Nine of the deleted elements in Tetrahymena, including the M, R, Tlr1 and mse2.9 elements, have been described at the sequence level. The M rearrangement deletes 600 or 900 bp of micronuclear-limited DNA. The two alternative rearrangements utilize different left boundaries, M1 and M2, which are 300 bp apart in the micronuclear genome, and the same right boundary, M3 (6). In vivo analysis of M region constructs showed that a cis-acting sequence consisting of a 10 bp polypurine tract, is located in the flanking DNA near the rearrangement junctions (9). This sequence motif is sufficient to specify a cut site ~45 bp away in an orientation-specific manner. However, the polypurine tract has not been found near the boundaries of any of the other deleted elements that have been analyzed to date.
Two other elements have been shown to have cis-acting sequences in the flanking DNA, although in these cases the critical sequences are not as well defined. One of these, mse2.9, is a 2.9 kb DNA element in the second intron of a gene of unknown function (10). A 10 bp inverted repeat is present in the critical region, 70–80 bp from the junction sites (11). The R element, a short deleted element close to M, also has cis-acting sequences on both sides of the element. These are evidently complex in nature, because clusters of base substitutions throughout the 70 bp region that is critical for rearrangement did not affect rearrangement efficiency or accuracy. Furthermore, flanking sequences on the right can substitute for those on the left, even though there is no sequence homology between them (12).
Tlr1 belongs to the less common class of rearrangements that delete large amounts of DNA (4,5). The Tlr1 element is 13 kb or more in length (Fig. 1). Tlr1 rearrangement occurs at 10–12 h of mating, the same time as the deletion of the shorter elements (Capowski and Karrer, unpublished data).
Figure 1.

Restriction map of Tlr1. The arrows represent the long inverted repeat. Within the inverted repeat are the 19mer tandem repeats: *, 19A (ATTATTTCTTTTTACATTT) and #, 19B (TTTCTCATTTTATGAAAAG). The bold lines represent macronucleus-destined DNA and the thin lines micronucleus-limited DNA. The lines above the map indicate the fragments used in cloning and for making probes. B, BglII; C, ClaI; E, EcoRI; H, HindIII; R, RsaI; S, Sau3A.
A discrete array of alternate junctions are observed for Tlr1 (7,13). One junction is favored, and the joining at that junction produces the ‘major’ rearrangement product. The left boundary of Tlr1 ranges over 296 bp in the minor variants and the right boundary over 196 bp. The Tlr1 major rearrangement also displays junctional microheterogeneity over 6–7 bp (13).
The most striking structural feature of Tlr1 is an 825 bp inverted repeat near the rearrangement junctions. The outermost half of the inverted repeat contains tandem repeats of two different 19mers, 19A and 19B. In the case of the major rearrangement product, the entire inverted repeat on the right side is within eliminated DNA, whereas the outermost 245 bp of the repeat, including the 19A tandem repeats, is retained at the left.
Southern analysis indicated that 19A and 19B are associated with each other at six or seven sites in the micronuclear genome and that the 19mers occur in pairs of similar sized restriction fragments (7). No macronuclear fragments apart from the one containing the left boundary of Tlr1 were detected, implying the 19mers are generally present within deleted DNA. The working model is that there is a small family of three or four rearrangements, including Tlr1, which contain the 19mer repeat sequences near the rearrangement junctions.
Southern hybridization with Tlr1.C-B (Fig. 1) showed that the innermost half of the inverted repeat belongs to a larger family of repeated, micronuclear-limited sequences with a copy number of about 30 (7). Thus the 19mer repeats of Tlr1 are associated with a larger family of repeated sequences that is also micronucleus-limited.
The objective of this study was to determine whether the cis-acting sequences for Tlr1 are contained within the element, or, as has been shown for two of the short elements that undergo developmentally regulated deletion in Tetrahymena, in the flanking DNA.
MATERIALS AND METHODS
Cell strains
Strains CU428, Mpr/Mpr (6-mp-s, VII) and CU427, Chx/Chx (cy-s, VI) of inbreeding line B were obtained from P. Bruns (Cornell University).
DNA constructs
All constructs were first built between the two NotI sites in the multiple cloning site of the plasmid pHSS6 (14). These clones are designated here as the pH series of constructs. The NotI fragments were subsequently excised from pHSS6 and ligated into the NotI site of the Tetrahymena processing vector pD5H8, producing the constructs designated here as the pD series.
pDWT.camRestriction fragments containing the junction sequences of Tlr1 were cloned previously (7). The right end fragment, Tlr.rB-H (Fig. 1), containing 750 of the 825 bp inverted repeat, 48 bp of DNA between the inverted repeat and the major junction site and 51 bp of flanking DNA, was cloned into pHSS6 to create pHMicR. The left end fragment, IIC7, containing 859 bp of mic-limited DNA including the entire inverted repeat and 1039 bp of flanking DNA, was released from pBR322 as an EcoRI–BamHI fragment including 346 bp of pBR322 DNA and ligated into pHSS6. To join the left and right end fragments, pHMicR and pHIIC7 were digested with EcoRI and NcoI. Fragments containing the Tlr1 repeats were gel purified from each digestion and ligated together. The resulting construct, pHIIC7+MicR, had the Tlr1 repeats in an inverted orientation in pHSS6. This construct was modified to contain the entire 825 bp inverted repeat from the right side and the adjacent HindII fragment, Tlr1.rH-H1. The right end EcoRI–NotI fragment of pHIIC7+MicR was replaced with a fragment from pBskMicRWT (a gift from D. Wexler) containing the complete inverted repeat, 125 bp of micronuclear DNA internal to the repeat and flanking sequences from the right side extending to the second HindIII site, to create pHWT (wild-type).
The presence of repeated sequences in the insert led to non-specific recombination events that deleted the entire insert or a part of it in Escherichia coli cells. To allow for selection of transformants with intact construct, a chloramphenicol resistance gene (cmR) was cloned within the inverted repeat sequences. A 3.8 kb SmaI fragment containing the cmR gene fragment was released from plasmid pMOB45 (15) and EcoRI linkers were added for ligation into the EcoRI site of pHWT to create pHWT.cam.
To allow for transformation into mating Tetrahymena cells, the NotI cassette was excised from pHSS6 and ligated into the NotI site of the Tetrahymena rDNA shuttle vector pD5H8 (16), producing pDWT.cam.
Construction of pDIR.campHWT.cam was digested with HindIII to release three HindIII fragments consisting of IIC7+MicR (6.74 kb), the right side flanking sequence (Tlr1.rH-H1, 730 bp) and the pHSS6 vector with pBR322 sequences (2.5 kb). The HindIII digestion mixture was purified and religated without separating the individual fragments. A clone which lacked the Tlr1.rH-H1 fragment (pHIR.cam) was identified and the orientation of the HindIII fragments was confirmed by restriction mapping. The 7.11 kb IR.cam NotI fragment was purified and ligated into the NotI site of pD5H8 to create pDIR.cam.
Construction of pDΔ.cam. A clone lacking the 19mer repeats from the right half of the Tlr1 inverted repeat was provided by D. Wexler. This MicRΔ construct contained an internal deletion of the 396 bp IIC7.1b fragment, from the ClaI to the RsaI site of the inverted repeat. The EcoRI–NotI MicR fragment from pHIIC7+MicR was replaced with MicRΔ to create pHΔ. The cmR gene fragment was added as described above. The 7.44 kb NotI Δ.cam insert fragment was ligated into the NotI site of pD5H8 to create pDΔ.cam.
Tetrahymena conjugation and transformation
Tetrahymena strains CU427 and CU428 were used in all the mating and transformation experiments. Matings were as described by Bruns and Brussard (17). Mating pairs were fixed by mixing 20 µl of the mating culture with 10 µl Schaudinn’s fixative (two parts saturated HgCl2, one part absolute ethanol) and examined under the microscope for the presence of developing macronuclei. Two hours past the point where 50% of the mating pairs showed anlagen, usually 8–10 h of mating, the mating cells were transformed by electroporation. The cells were electroporated according to the protocol developed by Gaertig and Gorovsky (18). Paromomycin was added to a final concentration of 100 µg/ml 20–24 h post-electroporation. Transformants were selected 2–4 days after adding paromomycin and were subsequently grown in 20 ml 2% PPYS with 100 mg/ml paromomycin for DNA isolation.
Whole cell DNA was isolated from Tetrahymena by a modification of the method of Austerberry and Yao (19). Briefly, the cells were pelleted, then lysed with SDS and proteinase K at 65°C. The DNA was precipitated with 12% polyethylene glycol in 1.2 M NaCl and spooled out with a pasteur pipette. It was rinsed in 70% ethanol, dried and redissolved in TE pH 8.0. The sample was treated with RNase, extracted with phenol:chloroform (24:1) and precipitated with 0.5 vol 7.5 M NH4OAC and 0.54 vol isopropanol. In some cases, the DNA was spun at 13 000 r.p.m. for 45 min in the microfuge to pellet contaminating carbohydrates before precipitation with isopropanol.
Southern analysis
Whole cell DNA was digested with the appropriate restriction enzymes, size fractionated by gel electrophoresis through 0.8–1% agarose and transferred to Genescreen nylon filters (NEN) by the downward capillary method (20). Probes were labeled using the random primer method (21). All blots were probed with a 726 bp HindIII–Sau3A fragment (IIC7.1a, Fig. 1) that is specific for DNA flanking Tlr1 on the left side.
Polymerase chain reaction (PCR)
A typical PCR reaction had 20 pmol of each primer, 100 ng of genomic template DNA, 2.5 mM MgCl2, 0.2 mM dNTPs, 10% glycerol, 1× PCR buffer and 0.5 ml Taq DNA polymerase (Promega). The primers and DNA template were denatured in the thermocycler at 95°C for 1 min and annealed at 55°C for 1 min before adding Taq polymerase on ice, then extended in the first cycle. A typical reaction had 35 cycles of 95°C for 1 min, 55–60°C for 1 min, 72°C for 1 min followed by the last cycle at 72°C for 7 min. The sequences of the primers are provided in Table 1.
Table 1. Primers.
| Primera | Sequence |
|---|---|
| Tet 1 | 5′-AAATGAGAAATTTTAAAAATTTTTAGAAACG-3′ |
| Tet 2 | 5′-ACGTGAGAAACTTTAGAAACTTGAGAAAAAT-3′ |
| Tet 3 | 5′-GTAGAATATTTTTTTTACCTGTACTGATCT-3′ |
| Tet 4 | 5′-AAATGCTCCGATTGTAAATTCTCTCTCTCG-3′ |
| Tet 5 | 5′-GCTTTACATATAATTATCTGCTTCTTATACGA-3′ |
| Tet 6 | 5′-ACTATGATTCCTCGTAAGCTTTCACTTACA-3′ |
| Tet 7 | 5′-TATATTTCTTATTTCTTTTTATTTTTCTCAAG-3′ |
| C1 | 5′-TACCTACCAGTTCTCCGCCT-3′ |
| C2 | 5′-GATGCAAAGCAGCTGGAAGG-3′ |
| C4 | 5′-TAGCAATTTAACTGTGATAAACTACCGCA-3′ |
| C4 | 5′-GATAAGCTGTCAAACATGAGAATTCCGG-3′ |
aTet, Tetrahymena DNA primers; C, construct-specific primers.
DNA sequencing
For sequencing the PCR products directly, the USB sequencing kit from Amersham Life Science was utilized following manufacturer’s protocols. Sequencing was typically done with end-labeled primer, in the thermocycler for 35 cycles consisting of 94°C for 1 min followed by 60°C for 1 min, beginning with a preheated thermocycler. Alternatively, direct sequencing was done in the thermocycler using [α-32P]dATP in the sequencing reaction. Sequenced products were run out on a 6% denaturing gel for 2–4 h. The gel was dried for 1 h and exposed to X-ray film.
RESULTS
The inverted repeat plus several hundred base pairs of flanking DNA is sufficient for Tlr1 rearrangement
Cis-acting sequences for DNA rearrangement in Tetrahymena can be tested in vivo by analysis of rearrangement of constructs in the rDNA vector pD5H8 (22). Micronuclear sequences cloned on this vector and introduced into the macronuclear anlagen of mating cells undergo DNA rearrangements similar to chromosomal sequences.
The salient features of the Tlr1 rearrangement are shown in Figure 1. In order to obtain a manageable construct for an in vivo assay of Tlr1 DNA rearrangement, a plasmid was built containing the inverted repeat along with 796 bp of DNA flanking the inverted repeat on the left and 830 bp on the right, omitting the 11 kb or more of DNA between the two halves of the inverted repeat. Early versions of the construct were unstable in a variety of recombination deficient E.coli strains, presumably due to the presence of the long inverted repeat. In order to overcome this problem, a gene conferring chloramphenicol resistance (cmR) was cloned within the insert. This allowed for selection of bacterial clones containing the intact construct. Since the resistance gene was placed within the deleted DNA, it was not expected to affect DNA rearrangement in Tetrahymena.
Mating cells were transformed with the construct by electroporation at 8 h after mixing the two mating types. The timing correlates with DNA rearrangement in the developing macronucleus, which is followed by rDNA amplification. About 30 Tetrahymena transformants were selected based on resistance to paromomycin, conferred by a mutation in the 17S rRNA gene of the vector. Whole cell DNA was isolated from six independent lines, digested with BamHI to release the construct from rDNA, and analyzed by Southern hybridization (Fig. 2). The blots were hybridized with a probe specific for the left junction of Tlr1. DNA from a line transformed with vector alone, included as a negative control (lane 8), showed no hybridization. At the exposures used, no hybridization was seen to the chromosomal macronuclear DNA. The construct is present on the rDNA, therefore it is at a 200-fold higher copy number than the chromosomal sequence.
Figure 2.
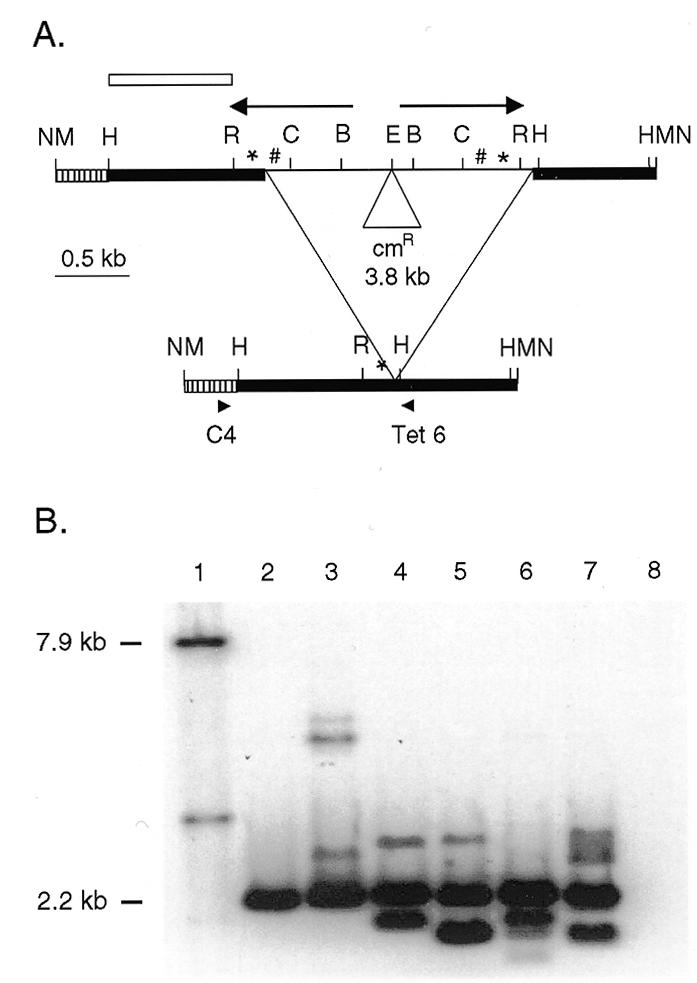
Rearrangement of a construct with the Tlr1 inverted repeat and flanking sequences. (A) The WT.cam construct and the major rearrangement product. Bold lines represent macronuclear DNA and the thin line is the micronucleus-limited DNA. The arrows show the inverted repeat. *, 19A tandem repeats; #, 19B tandem repeats; hatched bar, 346 bp of pBR322; cmR, chloramphenicol resistance gene; B, BglII; C, ClaI; E, EcoRI; H, HindIII; M, BamHI; N, NotI; R, RsaI. The NotI or BamHI fragment of the unrearranged construct is 7.9 kb and the rearranged product utilizing the major in vivo junction is expected to be 2.2 kb. (B) Southern analysis of pDWT.cam transformants. Whole cell DNA was digested with BamHI and probed with the probe indicated by the open bar in (A). Lane 1 contains pDWT.cam plasmid DNA digested with BamHI to release the 7.9 kb unrearranged construct fragment. Lanes 2–7 have DNA from pDWT.cam transformants C10, F5, A8, H3, F2 and G3 that show a 2.2 kb band corresponding to accurately rearranged construct. Lane 8 contains DNA from a cell line transformed with pD5H8 vector.
A 2.2 kb band was present in all the transformant lanes, indicating that the construct was rearranged faithfully in all six lines analyzed (Fig. 2). The majority (60–90%) of the DNA was in the 2.2 kb band. The rearranged product was PCR amplified from the Tetrahymena transformants using a Tetrahymena primer Tet 6 from the macronuclear-retained DNA at the right side and a primer specific to the bacterial plasmid pBR322 region of the construct (C4) (to prevent amplification of chromosomal macronuclear DNA). The expected 1.2 kb product was obtained from all the transformants. No PCR product was obtained from the DNA of untransformed cells. The 1.2 kb PCR product was gel purified and sequenced directly using radiolabeled Tet 6 primer. For two of the transformants, a unique sequence was determined for the junction (Table 2). For four of the transformants, the film showed clean sequence up to a point at which multiple bands appeared. Since the sequence of the Tlr1 rearrangement is known and much of the microheterogeneity is within a track of A residues, it was possible to follow the sequences of two or even three rearranged products within a single transformant. The sequence analysis revealed that the rearranged products showed junctional diversity or microheterogeneity over a range of 8–10 bp, similar to that for the chromosomal Tlr1 rearrangement (13).
Table 2. Sequence of the Tlr1 WT.cam construct rearrangement junctions.
| Transformant | Sequencea |
|---|---|
| F5 | CTAAAaaaaaagatt |
| A8 | CTAAAGTTaaaagatt |
| CTAAAGTaaaagatt | |
| B3 | CTAAAGTTaaaagatt |
| CTAAAGTTaaaaagatt | |
| B6 | CTAAAGTaaaagatt |
| G3 | CTAAAGTaaagatt |
| CTAAAGTaaaagatt | |
| CTAAAGTaaaaagatt | |
| C10 | CTAAAGTTTCTCaagatt |
aUpper case represents sequence from the left side and lower case from the right side. Underlined sequence may be from either side. PCR products were purified and sequenced directly.
In addition to the band representing the major rearrangement, there were also variant bands. The sizes of some of the fragments corresponded to the naturally occurring variants previously seen in the deletion of the chromosomal Tlr1 element (7). Other minor bands detected in the transformants that did not correspond to the chromosomal Tlr1 rearrangement are likely to be artefactual, since some aberrant rearrangement is not uncommon in this system (9).
No unrearranged DNA was detected even after long exposures. Thus, rearrangement of the construct mimicked rearrangement of the chromosomal element in efficiency, junction variability and junction microheterogeneity. The experiment demonstrated that the entire inverted repeat and several hundred base pairs of flanking DNA are sufficient for an accurate DNA rearrangement of Tlr1 in vivo. The internal 11 kb or more of micronuclear-limited DNA is not required.
Flanking DNA is required for accurate Tlr1 rearrangement
To determine whether the flanking region of Tlr1 contained cis-acting signals for DNA rearrangement, a construct was built that lacked some of the flanking sequences. The left end of Tlr1 was not a suitable target for this experiment because deletion of the left flanking sequences would involve deletion of a substantial portion of the inverted repeat and thus not permit distinction of the roles of the flanking sequences and the inverted repeat. The right flanking sequences were chosen because the right half of the inverted repeat is entirely deleted in the major rearrangement and all the naturally occurring variants. The construct pDIR.cam was similar to the first construct, with the difference that the Tlr1.rH-H1 fragment was removed, leaving only 51 bp of mac destined sequences to the right of the major rearrangement junction site.
Mating Tetrahymena cells undergoing DNA rearrangement were transformed with pDIR.cam by electroporation. Twelve paromomycin resistant Tetrahymena transformants were obtained. Whole cell DNA was isolated from nine of these, digested with NotI to release the insert and analyzed on Southern blots probed with sequences specific to the left of the Tlr1 element (Fig. 3A).
Figure 3.
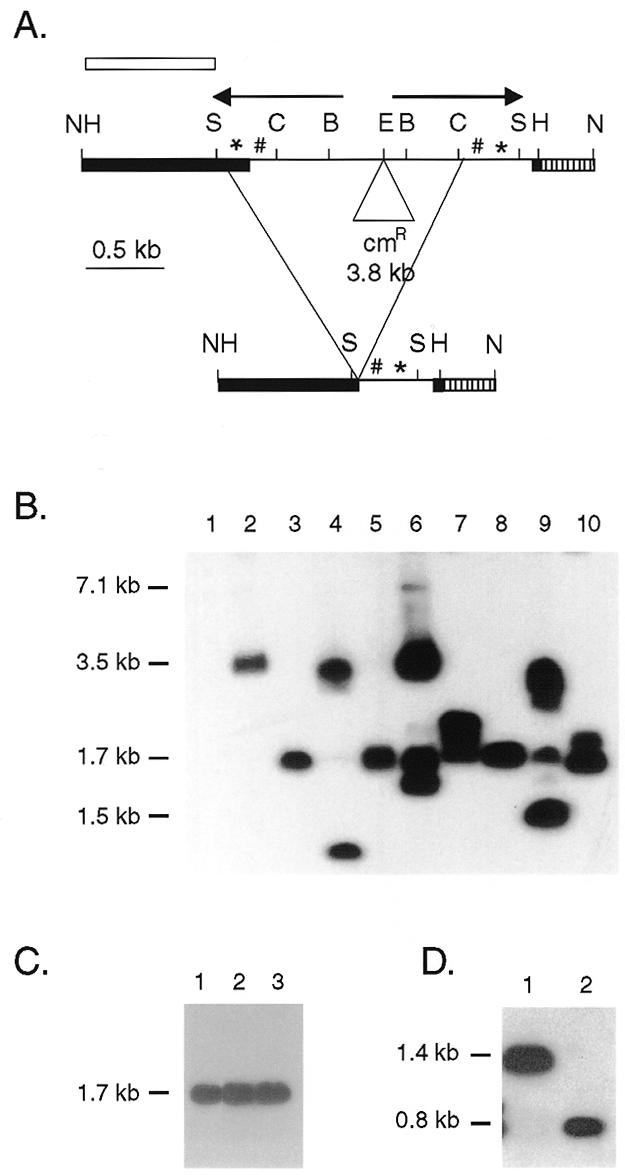
Rearrangement of a construct without the HindIII fragment containing flanking sequences to the right of the element. (A) Restriction map of the IR.cam construct and the predominant rearrangement product, ap1.7. The bold lines represent macronuclear DNA and the thin line micronuclear-limited DNA for the major chromosomal rearrangement. Arrows, inverted repeat; *, 19A tandem repeats; #, 19B tandem repeats; hatched bar, 346 bp of pBR322; cmR, chloramphenicol resistance gene; B, BglII; C, ClaI; E, EcoRI; H, HindIII; N, NotI; S, Sau3A. The open bar indicates the probe for the blots in (B), (C) and (D). The NotI fragment of the unrearranged construct is 7.1 kb and the rearranged product utilizing the junctions used in vivo is expected to be 1.5 kb. (B) Southern analysis of pDIR.cam transformants. Whole cell DNA was digested with NotI to release the construct from the rDNA. Lane 1, negative control lane containing DNA from a pD5H8 transformant. Lanes 2–10, DNA from independent pDIR.cam transformants. Seven of the nine transformants show an aberrant rearrangement that produced a 1.7 kb NotI fragment. (C) Whole cell DNA from pDIR.cam transformant A8 digested with NotI in lane1, NotI and ClaI in lane 2, and NotI and BglII in lane 3. (D) Whole cell IR.cam transformant A8 DNA digested with HindIII in lane 1, and HindIII and Sau3A in lane 2.
If flanking sequences are not essential for Tlr1 rearrangement, micronucleus-limited sequences would be deleted from the 7.11 kb Tlr1 NotI fragment of pDIR.cam to generate a 1.5 kb NotI fragment. If sequences in Tlr1r.H-H1 are required for Tlr1 rearrangement, the Tlr1 sequences in the construct were expected to remain unrearranged or undergo aberrant rearrangement. The pDIR.cam construct underwent aberrant DNA rearrangement in all the transformant cell lines (Fig. 3B). Multiple bands ranging from 0.75 to 3.1 kb were detected. A 1.5 kb NotI fragment was seen in only one of nine transformant lines examined.
A 1.7 kb band was detected in seven of nine pDIR.cam transformants. The presence of a common rearrangement product in multiple transformants suggests that, in the absence of the wild-type cis-acting signal, a cryptic signal may have been utilized to determine the junctions. PCR amplification of this product was attempted several times under varying conditions without success. Aberrant amplification was observed which may have been due to the presence of hairpin structures during annealing. Therefore, the junctions of the 1.7 kb aberrant product (ap1.7) were mapped using Southern and PCR analyses.
Southern analysis was performed on a transformant that contained only ap1.7 and no other rearrangement variants (Fig. 3B, lane 8). To map the left boundary, whole cell DNA was digested with NotI alone or NotI together with ClaI or BglII. A 1.7 kb fragment was detected in all three lanes (Fig. 3C). This showed that both of the ClaI sites, together with the intervening DNA, were deleted in the rearrangement to ap1.7.
In another Southern experiment, whole cell DNA was digested with HindIII or HindIII and Sau3A (Fig. 3D). An ~1.4 kb fragment was observed with the HindIII digestion and a 0.8 kb fragment was seen in the HindIII and Sau3A double digest, suggesting that ap1.7 retained the HindIII and the Sau3A sites from the left side. Thus, the left junction of ap1.7 lies between the Sau3A and the ClaI sites (Fig. 3A).
A PCR experiment was performed to map the junctions of ap1.7 more closely (Fig. 4). A series of oligonucleotides from within the inverted repeat were used against construct-specific oligonucleotides from both sides to determine which micronuclear sequences were retained in ap1.7. As a positive control, the inverted repeat primers were used against a Tetrahymena DNA primer Tet 5 (Fig. 4B, lanes 1, 4 and 7). All the products expected for amplification of the chromosomal DNA were obtained from the control reactions (159 bp with Tet 4, 191 bp with Tet 3 and 388 bp with Tet 2), indicating that the primers and the template DNA were compatible under these PCR conditions.
Figure 4.
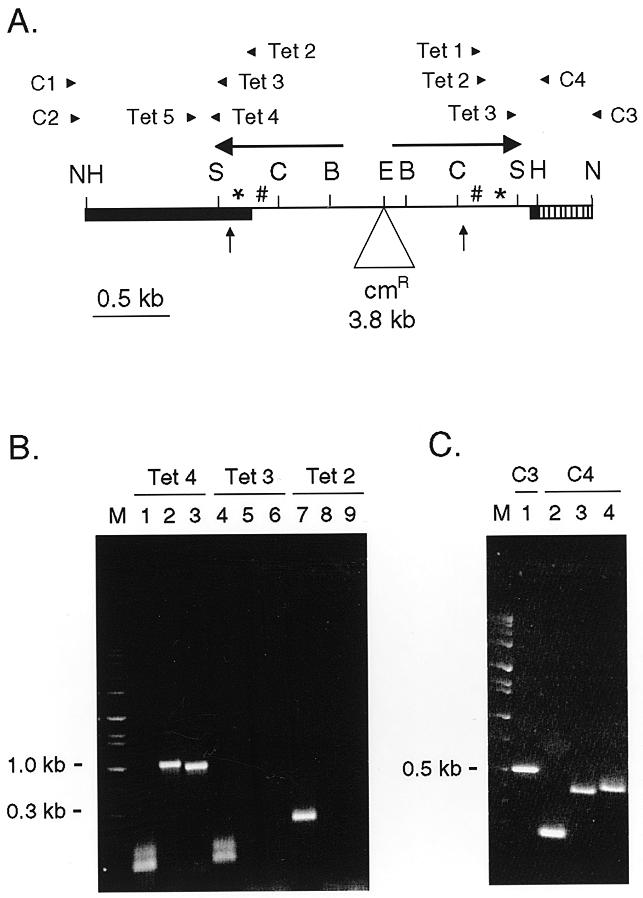
PCR mapping of ap1.7. (A) Diagrammatic representation of the pDIR.cam construct showing the location of the oligonucleotide primers. Vertical arrows indicate the boundaries of the deleted region for ap1.7 (B and C) PCR amplification of whole cell DNA from the IR.cam A8 transformant. M, marker lanes. (B) Left end: lanes 1–3 were primed with Tet 4, lanes 4–6 with Tet 3 and lanes 7–9 with Tet 2. The first of each set was against Tetrahymena DNA specific oligonucleotide Tet 5; the second and third sets with construct-specific oligonucleotides C1 and C2, respectively. (C) Right end: lane 1 was primed with Tet 3 and construct-specific oligonucleotide C3. Lanes 2, 3 and 4 were primed with Tet 3, Tet 2 and Tet 1, respectively, against construct-specific oligonucleotide C4.
To map the left boundary of ap1.7, two construct-specific primers (C1 and C2) were used with the same set of Tlr1 primers (Tet 2, Tet 3 and Tet 4). PCR products of 921 and 861 bp were obtained with the oligonucleotide Tet 4 and construct-specific primers C1 and C2, respectively. No products were obtained with oligonucleotides Tet 3 or Tet 2 (Fig. 4B). Since Tet 3 did not produce a PCR product with the construct-specific primer, some part of the sequence corresponding to Tet 3 primer is deleted from the rearranged aberrant product. This suggested that the left boundary of ap1.7 is within 27 bp of the Sau3A site.
Southern analysis indicated the junction of the ap1.7 rearrangement is within a 1.4 kb HindIII fragment. The PCR analysis of the left boundary suggests that only ~854 bp of that can be derived from the left side (826 bp of IIC7.1a + 27 bp or less beyond the Sau3A site). Thus, ~550 bp of DNA inside the right HindIII site of the construct must be retained in ap1.7. This was supported by PCR analysis of the right boundary of the rearrangement. A set of Tlr1 primers (Tet 1, Tet 2 and Tet 3) were used against two construct-specific primers C3 and C4. Negative control experiments verified that these primer pairs did not amplify the macronuclear chromosomal DNA of a non-transformed cell line. PCR products were obtained with all three primer sets from within the inverted repeat, suggesting that the sequence corresponding to those primers was retained in ap1.7 (Fig. 4C). Their sizes (539 bp with Tet 3 + C3, 188 bp with Tet 3 + C4, 385 bp with Tet 2 + C4 and 414 bp with Tet 1 + C4) agreed with the Southern data. Thus, the right junction of ap1.7 can be narrowed to 175 bp between the ClaI site and the Tet 1 primer sequence (Fig. 4A), and is likely to be within 20–30 bp of the ClaI site.
To verify that the right boundary of the rearrangement in the other transformants was within the inverted repeat, a PCR was performed on DNA from two additional transformants which showed only the 1.7 kb NotI band on the Southern blot (Fig. 3, lanes 3 and 5). Using oligonucleotides C4 and Tet 3 as primers, the expected PCR product of 188 bp was obtained from both, suggesting that the right junction was within the inverted repeat in these two transformants as well (data not shown).
In conclusion, Southern hybridization and PCR data show that the left boundary of the predominant aberrant rearrangement, ap1.7, is ~32–60 bp within the inverted repeat, close to the boundary of the naturally occurring variant rv0.9 (13). The aberrant characteristic of ap1.7 is the right boundary, which is within the inverted repeat, unlike any Tlr1 chromosomal rearrangement seen to date. The inaccurate rearrangement of pDIR.cam suggests that the 0.7 kb HindIII right side flanking DNA fragment contains cis-acting sequences that are required for determination of the junction site.
19mer sequences are not required on both sides of the construct
The most striking structural feature of Tlr1 is the inverted repeat sequence. The inverted repeat contains tandem repeats of two different 19mer sequences, 19A and 19B. Southern analysis indicates that the 19mer repeats are conserved amongst other putative Tlr1 family members (7). The next objective was to determine whether the 19mer tandem repeat sequences were required for Tlr1 rearrangement. The 19mer sequences cannot be removed from the left side of the construct because that region contains the rearrangement junction. The 19mer region (fragment IIC7.1b) was removed from the right side of the inverted repeat. The resulting construct, pDΔ.cam, was identical to the control construct with the exception that the 393 bp ClaI–RsaI fragment, IIC7.1b (Fig. 1), was deleted from the right half of the inverted repeat. This left 432 bp of the 825 bp inverted repeat in the construct, including the terminal 33 bp and 399 bp internal to IIC7.1b.
The pDΔ.cam construct was introduced into mating cells by electroporation and 42 paromomycin resistant Tetrahymena transformants were obtained. Whole cell DNA was isolated from seven transformants, digested with NotI to release the construct from the rDNA and analyzed by Southern hybridizations (Fig. 5). In all seven transformants there was a 2.2 kb NotI fragment, the size expected for accurate deletion at the junctions of the major chromosomal rearrangement.
Figure 5.
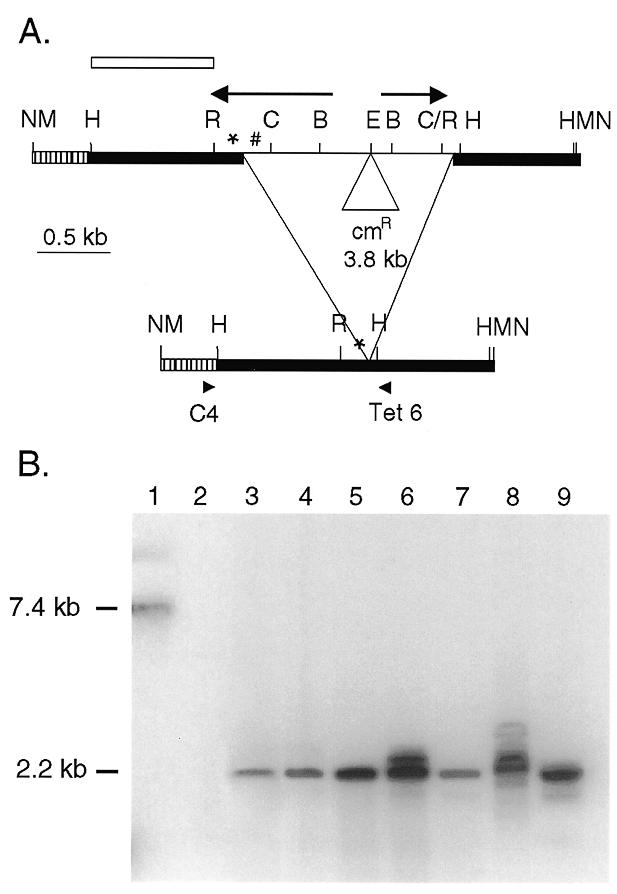
Rearrangement of a construct with deletion of the 19mer repeats from the right inverted repeat. (A) Restriction map of the pDΔ.cam construct and the rearrangement. The bold lines represent macronuclear DNA and the thin line micronuclear-limited DNA. Arrows, inverted repeat; *, 19A tandem repeats; #, 19B tandem repeats; hatched bar, 346 bp of pBR322; cmR, chloramphenicol resistance gene; B, BglII; C, ClaI; E, EcoRI; H, HindIII; M, BamHI; N, NotI; R, RsaI. The open bar indicates the probe for the blot in (B). The unrearranged construct is within a 7.4 kb BamHI fragment and for the accurately rearranged product the fragment is expected to be 2.2 kb. (B) Southern blot of whole cell DNA digested with BamHI to release the construct from the rDNA and probed with sequences from the left side of the element. Lane 1 contains the pDΔ.cam plasmid DNA restricted with BamHI to release the 7.44 kb unrearranged construct. Lane 2 contains DNA from a pD5H8 transformant as a negative control. Lanes 3–9 contain DNA from pDΔ.cam transformants, and show a 2.2 kb band corresponding to an accurately rearranged construct.
In order to verify the location of the rearrangement junction, the rearranged product was PCR amplified from the transformants. Whole cell genomic DNA was amplified using one primer specific to the construct, C4, and the other primer specific to Tetrahymena DNA, Tet 6 (Fig. 5). This prevented amplification of the chromosomal macronuclear DNA. The expected 1.2 kb product was obtained from all the transformants (data not shown). No PCR product was seen from a non-transformed line, confirming the construct specificity of the primers.
Thus, the Southern and PCR results concur and show that the pDΔ.cam construct was rearranged accurately, similar to the Tlr1 chromosomal rearrangement. This shows that the 19mers are not required on both sides of the Tlr1 construct for correct rearrangement.
DISCUSSION
A series of plasmid constructs containing the Tlr1 element were built and assayed for DNA rearrangement in vivo, in order to identify sequences required for DNA rearrangement. The first construct showed that the entire inverted repeat, together with 796 bp of flanking DNA on the left and 831 bp on the right is sufficient for accurate rearrangement. The internal 11 kb or more of the element is not required. This is comparable to the correct rearrangement of the M element construct that lacked 395 of the 600 bp M element (9). The rearranged WT.cam construct products showed junction variability and microheterogeneity similar to those in the macronuclear Tlr1 products of the chromosomal rearrangement.
The significance of the flanking region in Tlr1 rearrangement was revealed by aberrant rearrangement of the second construct, pDIR.cam, which had only 51 bp of sequence flanking the major Tlr1 junction. Instead of the 1.5 kb NotI fragment expected for joining as in the major chromosomal rearrangement of Tlr1, the NotI fragments containing the rearrangement junction ranged from 0.75 to 3 kb. Analysis of the most common rearrangement product (ap1.7) obtained from pDIR.cam revealed that the left junction was close to that found in the minor Tlr1 variant rv0.9 (13). The right junction, however, was ~400 bp inside of the right inverted repeat. The right junction is outside of the inverted repeat in all the Tlr1 chromosomal rearrangements analyzed to date. The fact that the right joining site of ap1.7 was so far inside the inverted repeat suggests the right flanking sequence contains important cis-acting sequences that designate the right boundary of the rearrangement. This is reminiscent of the M rearrangement in which short repeats in the flanking DNA determine the rearrangement junction. No well-conserved motif for Tlr1 or its cryptic partner was identified despite extensive scrutiny of the flanking Tlr1 sequence.
Another construct was built to test the role of the 19mer tandem repeats located within the Tlr1 inverted repeats, which are conserved at other sites in micronuclear-limited DNA. In the pDΔ.cam construct, the 19mer sequences were deleted from one of the inverted repeats. Surprisingly, pDΔ.cam underwent rearrangement and utilized the same junctions as the major Tlr1 rearrangement product. Thus, removal of the 19mer sequences from one side did not affect DNA rearrangement. There are several possible explanations for this result. First, the 19 bp repeats may be required at only one end of the element. That would be consistent with the postulated redundancy of rearrangement promoting sequences within the M element (23). In principle, it would be interesting to delete the 19mer region from both ends of the construct. However, since the major chromosomal junction for the Tlr1 rearrangement is within this region (Fig. 1), the results of that experiment would be difficult to interpret.
Second, the 19mer sequences may be required for rearrangement on the chromosome but not on the construct. For example, the inverted repeats may be involved in bringing the macronuclear junctions of this large element in close proximity to allow the rearrangement reaction to occur. In the construct there is less internal DNA and the junctions are closer together than they are on the chromosome, therefore the repeat sequences may not be required.
Third, the inverted repeats may be involved in some process other than developmentally regulated DNA rearrangment, such as transposition within the micronuclear genome. We do not favor this hypothesis because the 19mer region binds developmentally regulated proteins that are detected only at the time of DNA rearrangement (Ellingson and Karrer, unpublished data).
The Tlr1 element with its long inverted repeat bears a structural resemblance to transposable elements in Drosophila (24), sea urchins (25) and the hypotrichous ciliates (26,27). Critical sequences for the transposition of the elements in sea urchins and the ciliates have not yet been identified. The subterminal region of the P element of Drosophila binds the transposase protein and is required for P element transposition (28,29). Although the 19mer sequences of Tlr1 bind developmental stage specific proteins in vitro, this region is not required at both ends of the element for Tlr1 rearrangement.
This study revealed that cis-acting sequences defining the junction site of Tlr1 are present outside of the deleted DNA. Transposable elements, on the other hand, are self-contained in their sequence requirements and have no dependence on flanking DNA. Thus, though Tlr1 structurally resembles transposable elements, it is similar to other micronuclear-limited elements in Tetrahymena with respect to its cis-acting sequence requirements.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Michael Cox and Sergei Saveliev, in whose laboratory some of the sequencing work was done, for providing reagents, guidance and support. We also thank Daniel Wexler for help in building some of the DNA constructs. This work was supported by grant MCB-9631404 from the National Science Foundation. N.S.P. was supported in part by an Arthur J. Schmitt fellowship and by a Marquette University fellowship.
REFERENCES
- 1.Yao M.-C. and Gorovsky,M. (1974) Chromosoma, 48, 1–18. [DOI] [PubMed] [Google Scholar]
- 2.Karrer K.M., (1999) In Asai,D.J. and Forney,J.D. (eds), Tetrahymena thermophila. Academic Press, San Diego, pp. 127–186.
- 3.Austerberry C.F., Allis,C.D. and Yao,M.-C. (1984) Proc. Natl Acad. Sci. USA, 81, 7383–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard E.A. and Blackburn,E.H. (1985) Mol. Cell. Biol., 5, 2039–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yokoyama R.W. and Yao,M.-C. (1982) Chromosoma, 85, 11–22. [DOI] [PubMed] [Google Scholar]
- 6.Austerberry C.F. and Yao,M.-C. (1988) Mol. Cell. Biol., 8, 3947–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells J.M., Ellingson,J.L.E., Catt,D.M., Berger,P.J. and Karrer,K.M. (1994) Mol. Cell. Biol., 14, 5939–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chau M.F. and Orias,E. (1996) Biol. Cell, 86, 111–120. [DOI] [PubMed] [Google Scholar]
- 9.Godiska R. and Yao,M.-C. (1990) Cell, 61, 1237–1246. [DOI] [PubMed] [Google Scholar]
- 10.Heinonen T.Y.K. and Pearlman,R.E. (1994) J. Biol. Chem., 269, 17428–17433. [PubMed] [Google Scholar]
- 11.Li J. and Pearlman,R.E. (1996) Nucleic Acids Res., 24, 1943–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chalker D.L., La Terza,A., Wilson,A., Kroenke,C.D. and Yao,M.-C. (1999) Mol. Cell. Biol., 19, 5631–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patil N.S., Hempen,P.M., Udani,R.A. and Karrer,K.M. (1997) J. Eukaryot. Microbiol., 44, 518–522. [DOI] [PubMed] [Google Scholar]
- 14.Siefert H.S., Chen,E.Y., So,M. and Heffron,F. (1986) Proc. Natl Acad. Sci. USA, 83, 735–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bittner M. and Vapnek,D. (1981) Gene, 15, 319–329. [DOI] [PubMed] [Google Scholar]
- 16.Yao M.-C. and Yao,C.-H. (1989) Mol. Cell. Biol., 9, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruns P.J. and Brussard,T.B. (1974) J. Exp. Zool., 188, 337–344. [DOI] [PubMed] [Google Scholar]
- 18.Gaertig J. and Gorovsky,M. (1992) Proc. Natl Acad. Sci. USA, 89, 9196–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Austerberry C.F. and Yao,M.-C. (1987) Mol. Cell. Biol., 7, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) Current Protocols in Molecular Biology. Greene Publishing Associates, Inc. and John Wiley & Sons, Inc., New York, NY.
- 21.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- 22.Godiska R., James,C. and Yao,M.-C. (1993) Genes Dev., 7, 2357–2365. [DOI] [PubMed] [Google Scholar]
- 23.Coyne R.S., Chalker,D.L. and Yao,M.-C. (1996) Annu. Rev. Genet., 30, 557–578. [DOI] [PubMed] [Google Scholar]
- 24.Engels W.R. (1996) Curr. Top. Microbiol. Immunol., 204, 103–123. [DOI] [PubMed] [Google Scholar]
- 25.Liebermann D., Hoffman-Liebermann,B., Troutt,A.B., Kedes,L. and Cohen,S.N. (1986) Mol. Cell. Biol., 6, 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaraczewski J.W. and Jahn,C.L. (1993) Genes Dev., 7, 95–105. [DOI] [PubMed] [Google Scholar]
- 27.Hunter D.J., Williams,K., Cartinhour,S. and Herrick,G. (1989) Genes Dev., 3, 2101–2112. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman P.D., Doll,R.F. and Rio,D.C. (1989) Cell, 59, 359–371. [DOI] [PubMed] [Google Scholar]
- 29.Mullins M.C., Rio,D.C. and Rubin,G.M. (1989) Genes Dev., 3, 729–738. [DOI] [PubMed] [Google Scholar]