


Abstract
PpLSU3, a mobile group I intron found in the ribosomal RNA genes of Physarum polycephalum, encodes the I-PpoI homing endonuclease. This enzyme represents one of the rare cases in nature where a protein is expressed from an RNA polymerase I transcript. Our previous results showed that the full length intron, but not a further processed species, is the messenger for I-PpoI, implying a role of the untranslated region (UTR) in gene expression. To study the function of the 3′-UTR in expression of the endonuclease and in splicing of the intron, we replaced the I-PpoI gene in PpLSU3 with the gene for the α-fragment of Escherichia coli β-galactosidase, and then integrated this chimeric intron into all the chromosomal rDNA repeats of yeast. The resulting cells synthesized functional α-fragment, as evidenced by a complementation assay analogous to that used in E.coli. The β-galactosidase activity thus provides an unusual and potentially valuable readout for Pol I transcription from chromosomal rDNA. This is the first example in which a eucaryotic homing endonuclease gene has been successfully replaced by a heterologous gene. Using deletion mutagenesis and a novel randomization approach with the α-fragment as a reporter, we found that a small segment of the 3′-UTR dramatically influences both splicing and protein expression.
INTRODUCTION
Group I introns are self-splicing RNA elements that are found mostly in mitochondria and chloroplasts of lower eucaryotes, but that also occur in a range of other genomes including those in bacteria, viruses and nuclei. About 20% of all group I introns are nuclear, and all of these are located in rDNA, i.e. the repeated genes encoding ribosomal RNA. Many group I introns encode site-specific endonucleases that mediate intron mobility via the double-strand-break repair pathway, a process termed intron homing (reviewed in 1). Only six of the approximately 100 nuclear group I introns have been shown or inferred to be mobile: PpLSU3 from the myxomycete (acellular slime mold) Physarum polycephalum (2), DiSSU1 and DiSSU2 from the myxomycete Didymium iridis (3,4; A.Vader and S.Johansen, unpublished data), NaSSU1 and NaLSU1 from several species of the amoebae flagellate Naegleria (5,6) and PtSSU1 from the red algea Porphyra tenera (P.Haugen and S.Johansen, personal communication). The wide and erratic distribution of group I introns in nature has led to the belief that they have experienced horizontal transfer among different species during evolution (7,8). Homing endonucleases of group I introns are thought to have been acquired by some introns, independent of the intron as a mobile element (reviewed in 9). Once they became inserted into their new host gene, the endonuclease genes must have adapted to their new environment to allow proper expression.
Homing endonucleases from nuclear group I introns probably are derived from a common ancestor, since they form a distinct class by amino acid sequence (10). For their expression and function, these enzymes face two particular challenges. First, the endonucleases must avoid damaging the genome. The sequence complexity of nuclear genomes is vastly greater than that of organelles and bacteriophages. Although homing endonucleases have a recognition sequence that is long enough to be unique, for all the enzymes studied to date this sequence is variably degenerate (11–17). Thus, the specificity of cleavage in vivo is likely to be derived in part from a controlled and low level of expression, to prevent cleavage at chromosomal sites related in sequence to the bona fide rDNA target site. The second challenge is to be expressed from the rDNA locus, which is transcribed by RNA polymerase I. Normally, mRNAs for proteins are made by RNA polymerase II, which contain the 5′ methyl-guanosine cap and 3′ poly(A) tail that are important for the stability, export and efficient translation of mRNA (18–22). Pol I-derived RNAs, on the other hand, are not capped or polyadenylated, and they are usually not destined for translation.
Until recently, little was known about how the endonucleases of mobile nuclear group I introns are expressed. Using a yeast strain that carries PpLSU3 in all its approximately 150 rDNA repeats and that is also temperature sensitive for pol I, we showed previously that the I-PpoI endonuclease is indeed expressed from a pol I transcript (23). This is the first well-studied example of natural protein expression from an rDNA locus. Furthermore, we provided evidence that the full length, excised intron RNA serves as the mRNA for I-PpoI, while the equally stable processed RNA fragment that also contains the open reading frame (ORF) and the same 5′ end does not. This result implies that sequences downstream of the ORF somehow facilitate translation of the presumably capless and poly(A)-less RNA. In the present work, we have studied the role of the part of the 3′-untranslated region (3′-UTR) immediately downstream of the ORF in expression of the endonuclease and in splicing of the intron. To facilitate characterization of mutants, we replaced the I-PpoI ORF with the gene for the α-fragment of β-galactosidase as a reporter. Splicing of this chimeric intron from pre-rRNA was maintained, and functional α-fragment was expressed. The overall translation efficiency of the pol I-derived α-RNA was ∼3% compared with that of the α-RNA made from the Gal promoter. Using the gene for the α-fragment as a reporter, we found that the portion of the 3′-UTR immediately upstream from the intron internal processing site has a dramatic influence on splicing and protein expression.
MATERIALS AND METHODS
Yeast strains and plasmid construction
Yeast strains are listed in Table 1. Growth of yeast was performed according to standard procedures (24). Normally, yeast cells were grown in YEPD medium at 30°C. When selection of plasmids was required, cells were grown in synthetic minimal medium supplemented with amino acids.
Table 1. Yeast strains used in this study.
| Strain | Genotype | Description | Reference |
|---|---|---|---|
| 1. NOY505 | MATa ade2 ura3 leu2 trp1 his3 can1 | 51 | |
| 2. INVSc2/I3 | MATα his3-Δ200 ura3-167 25S::I3 | PpLSU3 integrated | 37 |
| 3. NOY505R | MATa ade2 ura3 leu2 trp1 his3 can1 | Insertion of T in I-PpoI target site | This study |
| 25S::1925T | |||
| 4. NOY505GalΩT | MATa ade2 ura3 leu2 trp1 his3 | Plasmid RS404GalΩT integrated into trp1 locus | This study |
| can trp1::RS404GalΩT | |||
| 5. NOY505GalΩT/Jlα | MATa ade2 ura3 leu2 trp1 his3 | JLI3α integrated into rDNA of NOY505GalΩT | This study |
| can trp1::RS404GalΩT 25S::I3α | |||
| 6. NOY505GalΩT/JlαΔ7 | MATa ade2 ura3 leu2 trp1 his3 | JLI3αΔ7 integrated, similar to #4 | This study |
| can trp1::RS404GalΩT 25S::I3αΔ7 |
NOY505GalΩT/JlαΔ8, NOY505GalΩT/JlαΔ9, NOY505GalΩT/JlαΔ10, NOY505GalΩT/JlαΔ1 and NOY505GalΩT/JlαΔ11 are identical to #6, except with the slightly longer deletion, as shown in Figure 5.
Plasmids were constructed by standard cloning methods (25). Mutations were introduced by PCR in two steps with the mutations included in the primers (26). Plasmid pCPIPpo was constructed by cloning the EcoRI–SphI fragment from pGal IPpo (27) into the EcoRI and SphI sites of the URA3-based plasmid yCP50 (28). Plasmid pYESMlacZ was constructed by cloning the lacZ gene into the BamHI and HindIII sites of pYESM (29), a slightly modified version of the expression plasmid pYES2 (Invitrogen). pYESMα was constructed by cloning the sequence for the first 85 amino acid residues of the lacZ gene into the HindIII and BamHI sites of pYESM. Plasmids pJLI3 contains PpLSU3 with 170 bp of 5′ yeast rDNA exon sequence and 130 bp of 3′ yeast rDNA exon sequence cloned into the EcoRI and SalI sites of the HIS3-based plasmid pRS423 (30). pJLI3delIPpo was constructed by deleting the I-PpoI ORF of pJLI3 and replacing it with a ClaI site; it retains the first seven and last eight amino acids of I-PpoI ORF. pJLI3α contains the sequence encoding the first 85 amino acid residues of the lacZ gene cloned into the ClaI site of pJLdelIPpo. Plasmids pJLαI3Δ10, pJLαI3Δ3, pJLαI3Δ7, pJLαI3Δ8, pJLαI3Δ9, pJLαI3Δ11 and pJLαI3Δ12 are the same as pJLαI3 except for the deletions as indicated in Figure 5. Plasmid pBSα was constructed by inserting the ClaI fragment of PJLI3α into the ClaI site of pBluescriptII KS+ (Promega). Plasmid pNOY103I3 was obtained by transforming plasmid pNOY103 (31) into the PpLSU3 integrated strain INVSc2 (Table 1) and selecting for colonies that grew on SD-ura plates. The plasmid in these colonies was recovered and the integration of the intron in the 25S rDNA on pNOY103 was checked by PCR analysis and restriction enzyme digestion. Plasmid pNOY103I3Δ10 was constructed as follows: the 4.5 kb SacI fragment of pNOY103I3 was cloned into pBluescript IIKS+ to create pBS/SacI which now has unique BamHI and NheI sites to facilitate cloning. PCR–mutagenesis was performed on pBS/SacI using standard technique to create PBS/SacIΔ10 which deletes the fifth 10 nt in the UTR1. The 4.5 kb SacI fragment of pBS/SacI Δ10 was cloned to pNOY103I3 to replace the wild-type fragment. pNOY103I3MP7 was created using the same strategy except in this construct, the G-binding site in PpLSU3 is changed from AG to TA.
Figure 5.

Constructs generated to study the role of UTR1 in splicing and protein expression. The numbers above the sequences are counted from the beginning of the UTR1. Dashes indicate that the nucleotides are deleted. Randomized nucleotides are indicated as N.
RNA analysis and β-galactosidase assays
RNA was prepared from yeast cells by standard techniques (23). For northern blotting, RNA was fractionated on a 1.5% agarose/formaldehyde gel and then transferred by capillary action to a Genescreenplus membrane (Du Pont NEN). The membrane was baked at 80°C for 2 h, prehybridized for 4 h, and then probed with antisense RNA for 16 h at 50°C in the presence of 50% formamide. The probe was generated by T3 or T7 RNA polymerase 40 µCi [α-32P]UTP. For detection of PpLSU3 RNA, plasmid pd55ΔSph-Xho (21) was used to make the riboprobe and, for detection of α-fragment RNA, plasmid pBSα was used.
β-Galactosidase activity was assayed by standard protocols (32), using o-nitrophenyl-β-d-galactoside (ONPG) as a substrate. Briefly, yeast cells were collected by centrifugation, resuspended in Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7.0), and then diluted at various concentrations in the same buffer to 1 ml. Thirty microliters of 0.1% SDS and 60 µl of chloroform were added to the cell suspension, and after incubation at 30°C for 15 min, ONPG was added to start the reaction.
Integration of intron into rDNA
The plasmid pCPIPpo and one of the pJL series plasmids were co-transformed into the yeast strain NOY505 by the lithium acetate method (32) and the cells were plated on SD-Ura-His plates. Transformants were streaked on SGal-Ura-His plates, on which the galactose induced the expression of I-PpoI. Cells that grew to form colonies on SGal-Ura-His plates either had point mutations at I-PpoI target site (23,27) or had the intron integrated, both of which confer resistance to I-PpoI. Single colonies on SGal-Ura-His plates were grown in SGal-Ura-His liquid medium. DNA was extracted from the above culture and PCR was performed to screen for intron-integrated colonies, using primers that flank the PpLSU3 insertion site. Cells were cured of plasmids by growing them in liquid YEPD for approximately 10 generations and plating on YEPD plates. The loss of the plasmids was checked by replica-plating colonies from YEPD plates onto SD-Ura and SD-His plates.
Construction of the randomized pools
Bsu36I and SphI are natural restriction sites in the chimeric PpLSU3-α intron, bracketing the region of interest. The Bsu36I–SphI fragment of pJLI3α was replaced by an unrelated 3 kb DNA fragment. The resulting plasmid is called pJLI3αstuffer and was used as the vector to create the randomized pools. The long primer used in the randomization in experiment A has the following structure: 5′-CCTGAGGCCG10 . . . G80CCACGGGCAA90AACG-CNNNNNNTACCAAGTA110 . . . AGTGGCATGC154-3′, where the underlined nucleotides are the restriction sites. A similar primer was generated for experiment B, in which the seven nucleotides from nucleotides 89 to 95 were randomized (Fig. 5). The PCR cassettes were created by amplifying with two short primers that include sequences from each end of the 154mer. The resulting PCR products were purified and cut with Bsu36I and SphI. The vector pJLI3αstuffer was digested with the same enzymes and repurified. To create the randomized pool of recombinant plasmids, 300 ng of the digested vector and 20 ng of PCR product were ligated in a 50 µl reaction, and the resulting DNA was transformed into DH5α E.coli cells by electroporation. About 5% of the cells was plated on 2YT/amp plates to determine the transformation efficiency. The remaining cells were added to 200 ml of 2YT liquid medium with ampicillin and grown overnight at 37°C. Plasmid was purified as a batch from the overnight culture and transformed into yeast strain NOY505GalΩT along with pCpIPpo. The plasmid pools from the first and second randomization experiments are called pJLI3αran1 and pJLI3αran2, respectively. Yeast transformants from approximately 50 plates were washed off the plates and combined. A small aliquot (0.5%) was grown in 100 ml SD-Ura-His medium to an OD600 of 1–1.5. Cells were then plated on SD-Ura-His and SG-Ura-His plates.
RESULTS
Deletion of 10 nucleotides in the 3′-UTR of the I-PpoI ORF affects splicing of the intron
PpLSU3 is found naturally in the slime mold P.polycephalum, in the approximately 200 extrachromosomal rDNA molecules of this organism (2). The 944 nucleotide self-splicing intron contains the ORF for the endonuclease I-PpoI inserted into the P1 loop of a group I ribozyme. Expression of PpLSU3 from a plasmid in Saccharomyces cerevisiae causes most cells to die due to double-strand breaks made by I-PpoI in the repeated rDNA genes on chromosome XII. Surviving cells that are resistant to the lethal effects of the endonuclease grow out at a frequency of ∼0.1–1%, and in the major class of these survivors PpLSU3 has homed into every rDNA copy (27). After ribozyme-mediated splicing has removed the intron from the pre-ribosomal RNA, the linear excised PpLSU3 RNA undergoes further self-processing (23) (Fig. 1A). It cleaves itself at either of two closely spaced internal processing sites (IPS1 and IPS2), yielding two 5′ fragments that contain the ORF for the I-PpoI endonuclease, and two 3′ fragments that comprise the ribozyme. The 5′ fragment generated by cleavage at IPS2 is unstable, but the other three fragments accumulate to high levels in cells.
Figure 1.

The trans-integration method. (A) Schematic drawing of a yeast 25S rDNA gene with PpLSU3 inserted in position 1925 (E.coli reference sequence). Exon sequences are indicated by filled boxes, and intron sequences are indicated by open boxes. IPS1, internal processing site observed in vitro and in P.polycephalum; IPS2, internal processing site observed in yeast only. The numbers indicate the nucleotides from the beginning of the intron. The I-PpoI ORF extends from nucleotides 14 to 503. The unstable 5′ intron RNA fragment is shown by dashed lines. (B) Schematic diagram of the trans-integration method. Mutations at I-PpoI target sites are indicated by an X over the rDNA repeats.
We recently described a method to cause integration of mutant forms of PpLSU3 into some or all of the chromosomal rDNA repeats of yeast (23). In this method, called transintegration, homing is initiated by expression of the endonuclease I-PpoI from the inducible Gal promoter on a separate plasmid (Fig. 1B). Intron variants that do not express I-PpoI, and thus would themselves be unable to home, can be efficiently integrated into rDNA repeats as long a they remain splicing competent. However, mutations that compromise splicing, thereby preventing or slowing proper maturation of ribosomal RNA, are selected against in this process. Thus, the inability of a particular mutant intron to become integrated is interpreted to mean that the mutation interferes with splicing. We have not sought to determine the minimal splicing rate in vivo that is required for proper processing of pre-rRNA and growth of cells.
Based on different mutational analyses, we concluded earlier that the full-length excised intron, but not the equally stable processed 5′ fragment, serves as the mRNA for I-PpoI (23). This result implies that sequences downstream of the ORF must play an important role in gene expression. In order to identify cis-acting elements that allow the full-length intron to be translated, we have focused on the part of the 3′-UTR that extends from the last codon of the ORF to the internal processing site (IPS1). This region is referred to here as UTR1 (Figs 1A and 2A). The 53 nt UTR1 can be folded into a stable secondary structure, and was proposed previously to play a role in I-PpoI expression (33). We used deletion mapping to analyze the functional importance of this region. Five serial 10 nt deletion mutants of PpLSU3 were constructed, and these plasmids were cotransformed into yeast along with plasmid pCPIPpo in order to cause integration of the intron into rDNA repeats (Fig. 2A). Four of the deletion mutants became integrated into every rDNA copy, indicating that they did not compromise splicing by this functional assay. In contrast, the mutant deleting the fifth and most distal 10 nt segment (boxed in Fig. 2A) failed to integrate into rDNA, indicating a defect in splicing. Northern blot analysis with the four integrated strains showed that deleting the first or the second 10 nt in the UTR1 had no effect on levels of any of the intron RNAs; deleting the third 10 nt and the fourth 10 nt lowered the steady state levels of the 5′-half RNA by 5- and 20-fold, respectively (data not shown), but did not alter the levels of the full-length intron. We interpret these phenotypes to mean that the latter deletions lowered the stability of the processed 5′ fragment of RNA. Consistent with our previous conclusion, both of these strains had the same expression level of I-PpoI as the wild-type, even though the levels of the 5′-half RNA were greatly decreased (data not shown).
Figure 2.
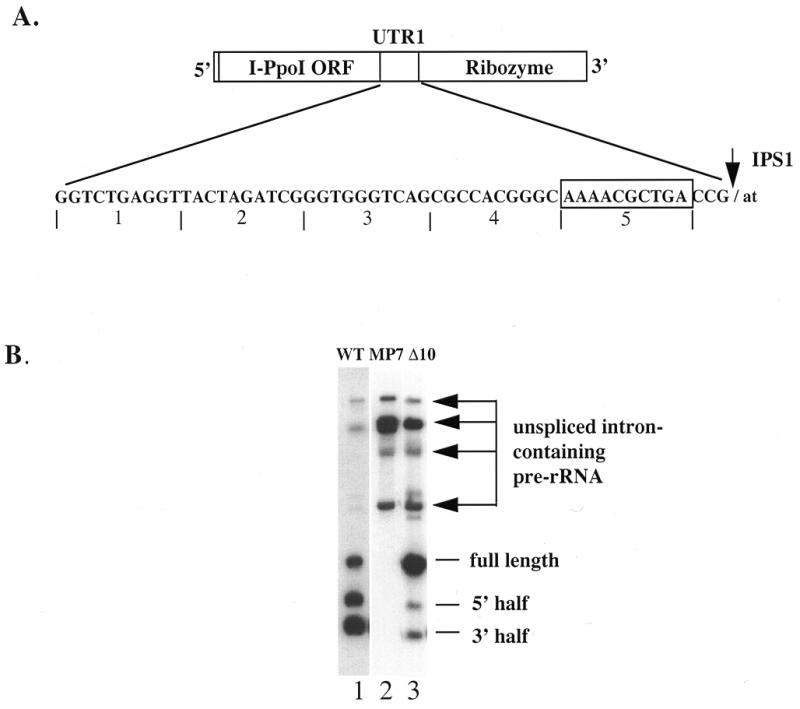
Deletion of a 10 nt fragment in the 3′-UTR of I-PpoI ORF affects splicing of the intron. (A) The sequence of the UTR1 is shown. The numbers underneath indicate regions that are deleted in the five serial deletion mutants. The boxed nucleotides are deleted in the plasmid pNOY103I3Δ10. (B) Northern blot of the 10 nt deletion mutant. PpLSU3 or its mutant forms were cloned into the intron insertion site in the 35S rDNA unit of the plasmid pNOY103 (34,35). RNA was prepared from yeast strain NOY505R transformed and expressing each of the three plasmids. Ten micrograms of total RNA were loaded for northern blot analysis. WT, wild-type PpLSU3; MP7, the G-binding site of PpLSU3 mutated; Δ10, the fifth 10 nt of PpLSU3 deleted. The arrows point to pre-rRNA species containing the unspliced intron.
To directly address whether deletion of the fifth and most distal 10 nt affects intron splicing, we cloned this mutant form of PpLSU3 into the intron insertion site in a complete 35S rDNA transcription unit driven by the Gal 7 promoter, in a yeast plasmid (34,35). The resulting plasmid is called pNOY103I3Δ10. As a parallel control, we introduced point mutations into the catalytic site of the ribozyme, i.e. the P7 stem, in the same context. The resulting plasmid, pNOY10I3MP7, should not allow splicing of the intron and therefore should lead to accumulation of precursor rRNA, but not free intron RNA species, after induction of cells with galactose. RNA was prepared from such yeast cells, and northern blot analysis was performed using a probe specific for the intron (Fig. 2B). Cells expressing the wild-type intron on the same backbone plasmid showed the expected distribution of bands, representing unspliced and partially spliced pre-ribosomal RNAs, full-length excised intron RNA, and processed intron RNA (Fig. 2B, lane 1). As a control, cells expressing the splicing defective intron showed an absence of spliced products, as expected (lane 2). Compared with wild-type, cells expressing the Δ10 mutant intron showed significantly higher amounts of unspliced and partially spliced RNAs (lane 3), confirming that this deletion compromises splicing. Also, in the Δ10 mutant the levels of both 3′ and 5′ processed RNA species were reduced, implying that cleavage of the excised full-length intron RNA was also affected by the deletion. The latter result is not surprising since one end of the deletion is only 3 nt from IPS1 (Fig. 2A).
The segment of 10 nt that is deleted in pNOY103I3Δ10 is in the P1 loop, an element in group I introns that is not directly involved in ribozyme core structure. Hence it is not obvious how these 10 nt could affect splicing. By inspection of the intron sequence, we noted the existence of a perfect 8 nt complementarity between a sequence near the beginning of the I-PpoI ORF (CGGUCAGU) and a sequence in the 3′-UTR partly overlapping the 10 nt segment found to be important for splicing (GCUGACCG). This base-pairing might help loop out the I-PpoI ORF, thus preventing it from interfering with the correct folding of the ribozyme core structure. To test this hypothesis, we changed all eight nucleotides in the I-PpoI ORF to their complementary sequence, and then tested the resulting mutant for its ability to integrate into rDNA. Contrary to expectations, the mutant showed no defect in its ability to integrate into all rDNA copies (data not shown). Therefore, the splicing defect caused by the 10 nt deletion cannot be attributed to this complementarity.
Expression of the α-fragment of β-galactosidase in place of I-PpoI
In the absence of an obvious explanation for the splicing defect in pNOY103I3Δ10, we decided to explore the importance of this 10 nt sequence in more detail. For this purpose, we decided to use the gene for the α-fragment of β-galactosidase as reporter in place of the I-PpoI ORF. Expression of E.coli β-galactosidase (the product of the lacZ gene) is widely used as a reporter in colorimetric assays in a variety of organisms. The tiny α-fragment itself can function as the reporter in the presence of the much larger Ω fragment of β-galactosidase that is usually expressed constitutively. This phenomenon is called alpha-complementation and is the basis of blue–white selection for cloning in E.coli and other organisms (36–39). To the best of our knowledge, alpha-complementation previously has not been described in yeast, except as a one sentence reference to unpublished results in a paper describing alpha-complementation in mammalian cells (40). We cloned the 255 bp gene encoding the α-fragment in place of the 489 bp I-PpoI ORF on a plasmid carrying PpLSU3. This chimeric intron, called PpLSU3-α, could be integrated into all rDNA repeats after expression of I-PpoI in trans, as evidenced by PCR analysis using flanking rDNA primers. Two size classes of colonies were observed on SGal-Ura-His plates. PCR and sequence analysis showed that the larger ones had rDNA point mutations at the I-PpoI target site, while the smaller ones carried the integrated chimeric intron. In liquid culture, the doubling time of the small colonies, after curing the plasmids in YEPD, was 4 h, compared with 3 h for cells carrying the wild-type PpLSU3 in rDNA, and with 1.5 h for the intronless strain (data not shown). We infer that the slower growth rates are due to inefficient splicing and, thus, that PpLSU3-α is inferior in splicing to the wild-type PpLSU3. The α gene sequences responsible for inefficient splicing are unknown. It seems likely that in the wild-type intron the ribozyme and ORF sequences have co-evolved to achieve maximally efficient splicing.
To test for alpha-complementation in yeast, we created the strain NOY505GalΩT, in which the gene for the Ω-fragment of β-galactosidase (which has amino acid residues 9–37 deleted) under the control of the Gal 1,10 promoter was inserted into the trp1 locus on chromosome IV (Fig. 3A). Crude extracts of this strain showed no β-galactosidase activity upon induction with galactose, as expected since Ω by itself is not functional (Fig. 3B). However, in presence of a 2 µ-based plasmid expressing the α-fragment from the same promoter, the crude extracts showed ∼500 U of activity. This value represents ∼25% of the activity of the intact β-galactosidase expressed from the plasmid pYESMlacZ under the same conditions (Fig. 3B). A similar experiment was carried out to test for expression of the α-fragment derived from the PpLSU3-α intron integrated strain NOY505GalΩT/JLα. In this case the crude extracts showed ∼15 U of β-galactosidase activity (Fig. 3B). We conclude that the functional α-fragment protein is expressed from pre-ribosomal RNA, though less efficiently than from the Gal promoter.
Figure 3.
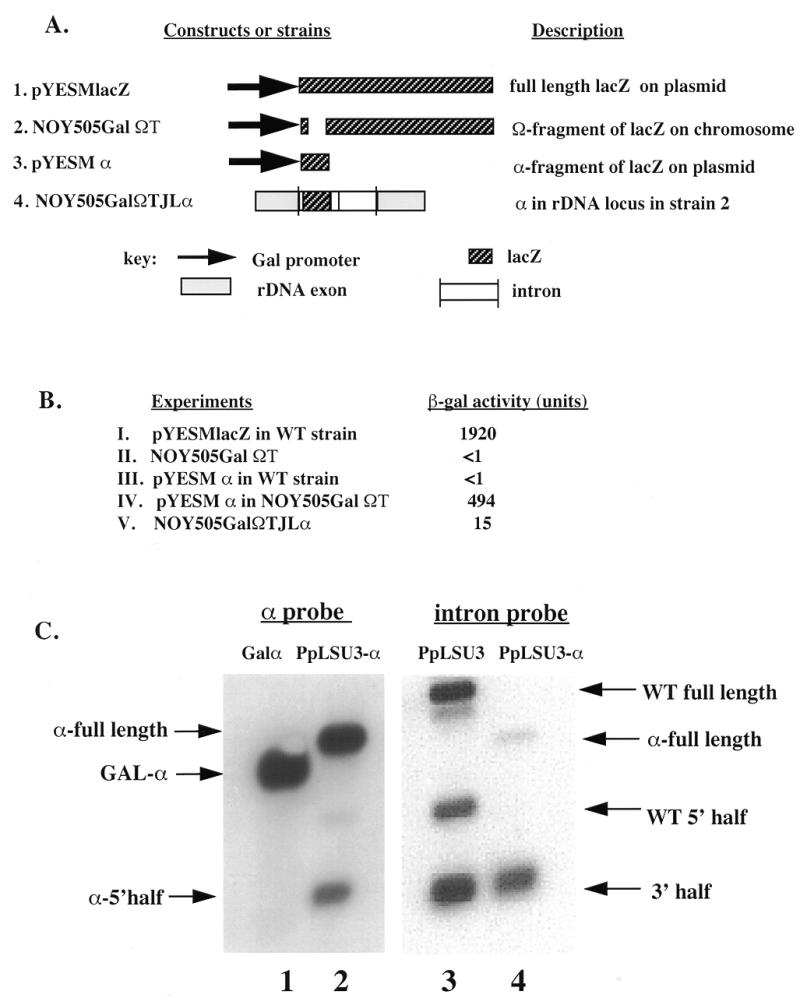
Expression of functional α-fragment from PpLSU3-α integrated into rDNA. (A) Constructs and strains used. (B) β-gal activity assay. I, pYESMlacZ transformed into strain NOY505; II, plasmid RS404GalΩT integrated into the trp1 locus of strain NOY505 to generate strain NOY505GalΩT; III, pYESMα transformed into NOY505; IV, pYESMα transformed into strain NOY505GalΩT; V, PpLSU3-α integrated into strain NOY505GalΩT to create strain NOY505GalΩT/JLα. (C) Northern blot analysis of the α-integrated strain. 1, 10 µg of total RNA from NOY505GalΩT transformed with pYESMα; 2, 10 µg of total RNA from strain NOY505GalΩT/JLα; 3, 1 µg of total RNA from strain INVSc2/I3; 4, 1 µg of total RNA from NOY505GalΩT/JLα.
To measure the steady state level of α-RNA derived from the intron, we performed northern blot analysis on RNA from strain NOY505Gal ΩT/JLα. Probing the membrane with an intron-specific riboprobe showed that the intron RNA containing the α sequences is fully spliced and further cleaved at IPS, similar to wild-type (Fig. 3C, right panel). However, due to low resolution, it was not possible to determine on this gel whether one or both of the two closely spaced IPS sequences were cleaved. Probing the membrane with an α-specific riboprobe revealed the presence of the full-length and the 5′-half intron RNA containing the α-fragment. Thus, it appears that of the two sites, at least IPS1 is utilized. The level of the α-RNA (full-length plus 5′ half) was similar to that from the Gal 1,10 promoter control after galactose induction (Fig. 3C, left panel). It is not known how much of the α-RNA is exported to the cytoplasm. However, in earlier studies using subcellular fractionation of a yeast lysate from a strain carrying the wild-type PpLSU3 in rDNA, we showed that all intron RNA species were similarly distributed in the nucleus and the cytoplasm (23). Thus, the same conclusion seems likely to hold for the α-RNA. Since β-galactosidase activity from the rDNA locus is ∼30-fold lower compared with that from the Gal promoter, we conclude that the overall translation efficiency (irrespective of subcellular localization) of the pol I-derived α-RNA is ∼3% of that of the pol II-derived α-mRNA. A similar estimate of translation efficiency was calculated for the I-PpoI endonuclease derived from wild-type PpLSU3 integrated into rDNA (data not shown).
Although many group I introns contain ORFs in their peripheral regions, there is only one previous report of insertion of a heterologous gene in place of endonuclease ORF where the chimeric intron was still spliced. Belfort and colleagues (41) made a construct in which the T4 phage td I-TevI gene was replaced by the chloramphenicol acetyltransferase (CAT) gene. This chimeric intron was able to splice itself from the primary transcript and the CAT gene was found to be expressed (41). However, in eucaryotic systems the PpLSU3-α integrated yeast strain is the first example where another gene replacing the endonuclease ORF is expressed from within a group I intron. The fact that expression is mediated by RNA polymerase I makes this especially notable.
We tested whether the splicing defect caused by the most distal 10 nt deletion in the context of I-PpoI ORF also is manifested in the context of PpLSU3-α. Expression of I-PpoI in cells carrying a plasmid with this deletion led to partial integration of the intron into rDNA. By Southern blotting, only 40% of all rDNA copies acquired the chimeric intron, while the other 60% of the copies presumably became resistant to cleavage by I-PpoI by virtue of point mutations, as found previously in similar experiments (23,27). These results suggest that splicing was comprised, although more mildly compared with the construct containing the I-PpoI ORF. Consistent with this conclusion, northern blot analysis of this integrated strain showed more accumulation of pre-rRNA containing unspliced intron, compared with a strain containing the wild-type PpLSU-α (see below).
A randomization approach to study splicing and gene expression of PpLSU3 at the rDNA locus
To gain more information on how the distal portion in UTR1 affects splicing of the intron, we decided to screen random sequences for ones that could restore splicing. The approach used, which is patterned after a study of a retrovirus RNA packaging signal (42), in principle allows tens of thousands of different sequences to be sampled in one experiment. A pool of plasmids carrying PpLSU3-α with many different sequences in the distal portion of UTR1 was created as follows (Fig. 4A). A long oligonucleotide was synthesized containing randomized nucleotides at the positions desired. The oligonucleotide served as the template in a PCR reaction with two external primers, each containing a unique restriction enzyme site, to create a randomized PCR cassette. After digestion with the two restriction enzymes, the cassette was ligated into the vector backbone, which had been prepared by enzymatic removal of an irrelevant stuffer fragment inserted in place of the wild-type sequence. The role of the stuffer is to ensure that no wild-type sequence is carried over into the pool. The ligation mixture was transformed into E.coli, and the resulting transformants were grown up in liquid medium as a batch culture. Plasmid DNA prepared from the batch culture thus consisted of a pool of molecules each having a different sequence at the randomized positions. The frequency of occurrence of a particular sequence in the plasmid pool can be estimated from the number of transformants and the number of randomized bases. For example, if there were 8 nt of randomized sequence, corresponding to 48 = approximately 64 000 different possible sequences, and 105 E.coli transformants, then each sequence would be represented about once in the pool. On the other hand, if 10 nucleotides were randomized, corresponding to about 1 million different possible sequences, then the probability that a particular sequence is present in the pool would be about 0.1.
Figure 4.

Schematic drawing of the randomization approach. (A) Construction of the randomized plasmid pool. The boxes and shadings representing exon, intron and lacZ sequences are the same as in Figure 3. The randomized region is indicated as NNN. The drawing is not to scale. Restriction sites: B, Bsu36I; S, SphI. (B) Identification of sequences that allow integration into rDNA repeats.
To identify sequences required for intron splicing, the plasmid pool was transformed into the yeast strain NOY505GalΩT, which expresses the Ω-fragment of lacZ, along with plasmid pCPIPpo encoding the I-PpoI homing endonuclease. Yeast transformants were combined and then plated on SGal-Ura-His plates to induce expression of I-PpoI. Colonies containing sequences that do not allow splicing of the intron will not allow the intron to integrate into chromosomal rDNA repeats, and most of these will not grow on SGal-Ura-His plates because they are killed by the action of the endonuclease. To identify the sequences that are compatible with splicing, we PCR-amplified DNA across the intron insertion site in chromosomal rDNA, followed by sequencing of PCR products. In parallel, the role of each sequence in expression of the α-fragment was assessed by patching the colonies onto X-gal plates or by a quantitative β-galactosidase assay (Fig. 4B).
Effects of randomized mutations in the 3′-UTR on protein synthesis and splicing
Because of the dramatic phenotype of the most distal 10 nt deletion near the end of the UTR1, we focused on this region with randomization mutagenesis, but decided to include in the mutagenesis window the last 3 nt of the UTR1, based on the assumption that these sequences might be important for intron processing. For simplicity these 13 nt were divided into two subregions. The last 6 nt were randomized in experiment A, and the 7 nt immediately upstream in experiment B (Fig. 5). In experiment A, 3.0 × 104 E.coli transformants were obtained, or seven times the calculated complexity of 46 = 4096, implying that the entire sequence space was sampled. DNA prepared as a batch was used to transform yeast strain NOY505GalΩT along with pCPIPpo. The resulting approximately 80 000 yeast transformants thus should include almost all of the possible E.coli clones. All yeast transformants were washed off the plates and combined. A small aliquot (0.5%) was grown in SD-Ura-His medium. To identify sequences that allow integration, these cells were subsequently plated on SD-Ura-His and SGal-Ura-His plates in parallel. If only the wild-type sequence or a few variant sequences allowed splicing of the intron, one would expect to see only a few colonies growing on SGal-Ura-His plates, since cells that do not splice the intron from the pre-rRNA will not grow. Contrary to our expectations, the number of colonies on SGal-Ura-His was ∼1% of the number that grew on SD-Ura-His plates, a ratio that is similar to that of the wild-type construct, indicating immediately that the wild-type sequence is not required for splicing. We PCR-amplified the integrated intron from 21 individual colonies and sequenced the PCR products. A single predominant sequence was found in each colony, with individual sequences bearing no significant resemblance to the wild-type and little resemblance to each other (Table 2B).
Table 2B. Sequence of the randomized region from experiment B.
| cgggc AAAACGC tgaccg/ta |
|---|
| ATATCAA |
| GGCTACG |
| TACTCGC |
| GTCTACG |
| ACGCGTG |
| TCATCAC |
| ATTACGC |
| ACTATCC |
| TTCAAGC |
| AGAATGT |
| CGGTGGC |
| GAATCGT |
| GTTTCGC |
| GGGCACC |
The first line is the wild-type sequence, with slash indicating IPS1. The sequence recovered after randomization is in uppercase.
To test the role of the randomized sequences in expression, 1000 colonies grown on SGal-Ura-His were patched onto SGal-Ura-His plates containing X-gal. Since the donor plasmid pJLI3α does not have a promoter upstream of the intron sequence and thus does not express any α-fragment (data not shown), alpha-complementation has to come from the integrated copies in rDNA. Of the 1000 colonies checked for expression, we found four classes: (i) the majority of colonies (760 out of 1000) were more blue than the wild-type strain NOY505GalΩT/JLα. A quantitative β-galactosidase activity assay of crude extracts from 12 colonies in this class revealed that they expressed 3–5-fold more functional β-galactosidase activity than wild-type. (ii) The next most abundant category, with about 150 colonies, appeared pale blue and expressed similar levels of the α-fragment as the wild-type. (iii) Less abundant still were 58 dark blue colonies. In quantitative β-galactosidase activity assays, crude extracts of 12 of these showed 20–30-fold higher enzymatic activity than wild-type. The highest activity was 516 U, or a 34-fold increase over the wild-type level. (iv) Least abundant were 32 very pale blue or white colonies. They expressed <10% of β-galactosidase activity compared with the wild-type. In summary, since ∼80% of colonies expressed more α-fragment than wild-type, we infer that the wild-type sequence functionally down-regulates the expression of the endonuclease.
The sequences of the randomized region were compared for 12 dark blue colonies and 12 white colonies. To rule out the possibility that the white phenotype was a consequence of mutations elsewhere on the chromosome, the 12 white colonies first were cured of plasmids, and then the cells were retransformed with pYESMα, in which the α-fragment gene is under control of the Gal promoter. Nine colonies expressed high levels of β-galactosidase activity from this plasmid, ruling out adventitious chromosomal mutations. The randomized sequences from these are listed in Table 2A. One common feature of the dark blue colonies is that the G at IPS1 (TGACCG/, slash indicates cleavage site) was changed from G to A or T in nine of 12 cases. Since mutating the IPS1 from G to A abolishes cleavage at IPS1 (23), presumably in these nine colonies cleavage at IPS1 also was abolished. However, in our earlier study, expression of I-PpoI increased only by 3-fold when the IPS1 was altered, while the dark blue colonies have at least a 20-fold increase in β-gal activity. Moreover, the three colonies that have AATAGG in the randomized positions might be expected to retain cleavage at the IPS1, and yet they also showed 20-fold more β-gal activity. Therefore, it seems that mechanisms other than the inability of cleavage at IPS1 contribute to high expression. In contrast to the dark blue colonies, seven of the nine white colonies retained the wild-type G residue at the IPS1. The reason why they do not express any α-fragment is unclear. Taken together, these data clearly show that the wild-type sequence of the 6 nt randomized in this experiment is not required for intron splicing, since a large number of sequences are compatible with splicing. Furthermore, the data suggest that this sequence plays an important role in expression of the endonuclease. The mechanism by which protein expression can be modulated by up to 300-fold (from 10-fold reduced to 30-fold enhanced over wild-type) is a topic of continuing investigation.
Table 2A. Sequences of the randomized region in experiment A.
| Dark blue | White |
|---|---|
| gcaaaacgcTGACCG/ta | gcaaaacgcTGACCG/ta |
| AATAGG (3) | TGTGTG |
| GTTTAA | TGTGTG |
| CCTCGA | TGTATG |
| GCTCAA | TGACGG |
| GGCATA | TGACGG |
| CCGCAT | CCTTCG |
| GTTTAT | GCCTCG |
| CAATTT | GGTACT |
| TAAATT (2) | TGGCTT |
Sequences of the randomized region from dark blue colonies and white colonies on X-gal plates. The second line in each column is the wild-type sequence with the slash indicating the processing site IPS1. The sequence recovered after randomization is in uppercase. The numbers next to the sequences show independent colonies with the same sequence.
In experiment B we randomized 7 nt further upstream (Fig. 5) using the same procedure. About 7 × 104 E.coli transformants were obtained, or about four times the theoretical complexity of 47 = 16 384. DNA was prepared as a batch and transformed into NOY505GalΩT along with pCPIPpo, yielding about 120 000 transformants. To our surprise, the ratio of colonies on SGal-Ura-His versus on SD-Ura-His was 1% again, similar to that when the wild-type construct was used. Fourteen individual colonies on SGal-Ura-His plates were picked and the integrated intron copies were PCR-amplified. Sequencing results revealed that multiple sequences were integrated, demonstrating that the wild-type sequence for the 7 nt is not essential for intron splicing (Table 2B). The colonies in this experiment were not further analyzed for protein expression on X-gal plates.
The distal segment of the UTR1 may serve as a spacer to facilitate intron splicing
Since the wild-type sequence or closely related sequences obviously are not essential to maintain functional splicing, we investigated if the length of this sequence plays a crucial role. Five further mutants were constructed, deleting 7, 8, 9, 11 or 12 nucleotides in the UTR1, as shown in Figure 5. The mutants were tested for their ability to integrate into rDNA repeats in the transintegration assay, in parallel with the original 10 nt deletion. While the 10 nt deletion yielded cells in which only ∼40% of the rDNA repeats had acquired a PpLSU3-α insertion, all five of the new mutants were able to integrate into every rDNA copy (data not shown). Northern blot analysis was carried out on RNAs of each of these strains. Consistent with the partial integration phenotype of the 10 nt deletion mutant, Δ10, significantly greater amounts of the pre-rRNA species containing unspliced intron were seen in that case (Fig. 6, lanes 3 and 4) compared with wild-type (lane 2). Of the other five deletion mutant strains, Δ7 and Δ8 showed a wild-type or nearly wild-type pattern on northern blots (Fig. 6, lanes 2, 5 and 6). In contrast, Δ9, Δ11 and Δ12 accumulated unspliced intron-containing pre-rRNA, like the original Δ10 mutant (Fig. 6, lanes 7–9; compare bands indicated by arrows). We interpret these results to mean that the 9–12 nt segment of RNA serves some function in promoting efficient splicing. But given the randomization results, this function apparently is largely sequence independent. The molecular basis of these effects are unknown.
Figure 6.
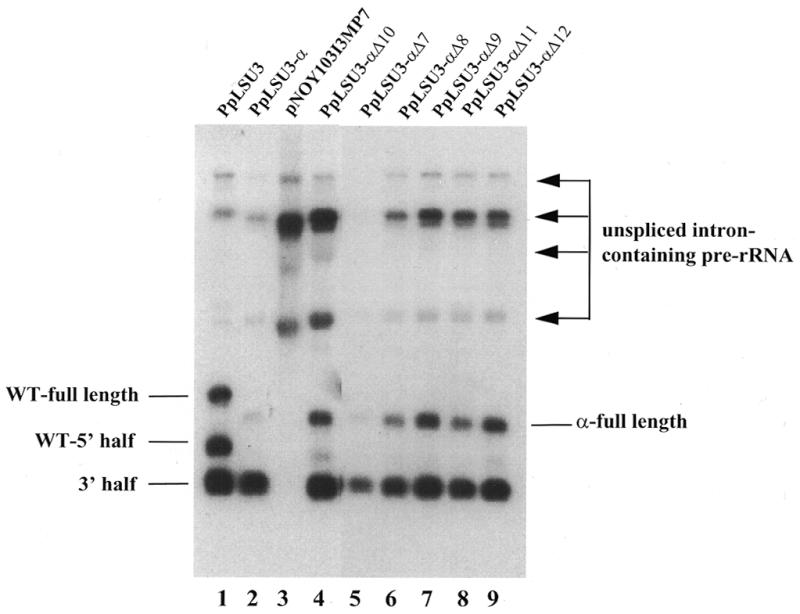
Northern blot analysis showing effect of deletions on splicing. Lane 1, wild-type PpLSU3 integrated in rDNA of strain INVSc2/I3; lane 2, PpLSU3-α integrated into rDNA of strain NOY505GalΩT/JLα; lane 3, pNOY103I3MP7 transformed into NOY505R; lanes 4–9, PpLSU3-α mutants deleting 10, 7, 8, 9, 11 or 12 nt in UTR1, respectively, were integrated into rDNA of strain NOY505GalΩT. One microgram of total RNA was loaded in each lane except for lanes 3 and 4 where 10 and 8 µg of total RNA were loaded, respectively. Arrows point to pre-rRNA species containing the unspliced intron.
DISCUSSION
Homing endonucleases of group I introns are thought to have been acquired independently of the intron as mobile genetic elements (reviewed in 9). Once they became inserted into their new host gene, the endonucleases must have adapted to their new environment to allow proper expression. Although there is as yet no detailed explanation of how a protein is translated from RNA spliced out of pre-ribosomal RNA, we have found several important clues in the yeast system. First, as shown previously for yeast cells carrying the wild-type PpLSU3 integrated into all rDNA repeats (23), the full-length excised intron RNA appears to serve as mRNA for I-PpoI, while the further processed 5′ half intron RNA does not. Thus, the self-cleavage of the intron functionally suppresses protein expression. Second, intron RNA species accumulate to high levels in these cells, making up ∼2% of total RNA, with the full-length species itself accounting for ∼0.5% of total RNA. Comparison of the translation efficiency of I-PpoI messenger from rDNA and from the Gal 1,10 promoter led to the conclusion that the pol I-derived I-PpoI mRNA is translated at ∼3% of the efficiency of the pol II-derived RNA. Third, in the present study we have demonstrated that the UTR1 plays a role both in splicing of the intron and in expression of the encoded protein. Deletions of 8–12 nt in the distal part of UTR1 compromise splicing. Sequence alterations in the last 6 nt of the UTR1 can alter protein expression by up to 300-fold. The mechanism by which this portion of the intron RNA acts remains to be elucidated.
Expression of other genes in place of the endonuclease ORF in a group I intron
Protein expression in eucaryotic cells is almost exclusively restricted to pol II-derived mRNAs. An outstanding exception appears to be the endonucleases encoded by mobile nuclear group I introns. We tested if genes normally transcribed by pol II could replace the I-PpoI ORF in the slime mold intron PpLSU3 when it is integrated into yeast rDNA. Chimeric PpLSU3 constructs with any of four of five genes tried (the neomycin resistance gene, TRP1, URA3 and HIS3) failed to integrate into chromosomal rDNA repeats (data not shown), indicating that splicing was compromised. It is likely that the RNA sequences of these genes, which are somewhat larger than the I-PpoI ORF, somehow interfere with the correct folding of the ribozyme. In contrast, a PpLSU3 chimeric intron carrying the gene for the α-fragment of β-galactosidase was found to integrate successfully into all rDNA copies. Furthermore, functional protein was expressed, presumably from the pol I-derived pre-ribosomal RNA, as inferred previously for the I-PpoI (23). The overall translation efficiency of the α-RNA from PpLSU3-α was estimated to be 3% of that of the pol II-made α-RNA, similar to that inferred earlier for the I-PpoI mRNA. These data are consistent with a recent analysis of a pol I-derived HIS4 RNA, which was shown to be translated 3% as well as the endogenous HIS4 RNA from a pol II promoter (43). The yeast strain with the PpLSU3-α integrated into every rDNA copy could be a useful tool for studying pol I expression in a chromosomal context.
Expression of homing endonucleases from other nuclear group I introns
Study of expression of nuclear group I intron-encoded endonucleases has provided new insights into the mechanisms of protein expression from the rDNA locus, i.e. the nucleolus. The strategies for expression of I-Dir and I-Nan, endonucleases encoded by DiSSU1 and NaSSU1, respectively, are quite different from that used by I-PpoI. DiSSU1 and NaSSU1 are twin-ribozyme introns that contain a small self-cleaving ribozyme (GIR1) embedded in the conventional group I ribozyme GIR2 (5,44). The ORFs for I-DirI and I-NanI are located downstream of GIR1. A hydrolysis reaction catalyzed by GIR1 is proposed to generate the 5′ end of the mRNA (5,44,45). The DiSSU1 intron has two other unusual features that no doubt figure in protein expression (45). The I-DirI ORF contains a 51 nt spliceosomal intron that is removed during maturation, implying that the spliceosome has access to the nucleolus. Furthermore, the 3′ end of the mature presumptive mRNA is polyadenylated. Removal of the spliceosomal intron appears to facilitate nucleo-cytoplasmic transport of the RNA, since only the intron-less species was found in the cytoplasm. Further experiments using an organism amenable to molecular genetic analysis are needed to address the details of the mechanism by which I-DirI and I-NanI endonucleases are synthesized.
Yeast strains carrying mobile group I introns integrated into rDNA should be powerful tools to study the expression of group I intron endonucleases from pre-rRNA. In the case of PpLSU3, expression of the α-fragment and blue–white screening allowed us to examine the phenotypes of large numbers of mutant sequences in the 3′-UTR. We are currently also attempting to express in place of I-PpoI the tiny yeast SOM1 gene, which encodes a peptide of only 74 amino acids that is essential for mitochondrial function (46,47). According to preliminary data, a chimeric PpLSU3-SOM1 intron can be efficiently integrated into rDNA without compromising splicing, and the Som1 protein also is expressed (J.Lin, H.-S.Nam and V.M.Vogt, unpublished data). Analogous chimeras of other nuclear group I introns may provide a method to identify more precisely cis-acting sequences as well as trans-acting cellular proteins involved in splicing of the intron and expression of the homing endonucleases.
Role of sequences between the I-PpoI ORF and the ribozyme
There are numerous examples of 3′-UTR sequences controlling mRNA stability and localization, and therefore controlling protein synthesis (reviewed in 20,48,49). The 53 nt UTR1 of I-PpoI ORF can be folded into a stable secondary structure (33), suggestive of some function. Cleavage at the IPS originally was hypothesized to play a role in I-PpoI expression, with the notion that the 5′-half PpLSU3 RNA might be more easily exported to the cytoplasm than the full-length intron RNA. Contrary to this hypothesis, while both the full-length and the 5′-half RNA are found in large quantities in the cytoplasm, only the full-length intron appears to act as a messenger (23). We have now examined the role of UTR1 in more detail, showing that the first 40 nt of this 53 nt sequence do not play a role either in splicing or protein expression, but that the last 13 nt do affect both of these processes. From a combinatorial sequence analysis with the α-fragment as a reporter, it appears that the wild-type sequence acts to reduce protein expression. We hypothesize that this down-regulation represents an evolutionary pressure to keep the expression of the endonuclease low. A further conclusion from these results is that mechanisms in addition to cleavage at IPS1 probably are involved, since some of the recovered sequences from the randomization experiment led to much higher levels of α-fragment than could be explained by reduction in intron processing.
In their natural environment, group I intron-encoded endonucleases are expressed at very low to undetectable levels, even under conditions where homing occurs (4,50–53; A.Vader and S.Johansen, unpublished data). In vitro homing endonucleases are able to cleave sequences that are related but not identical to the wild-type recognition site (11–17). Thus, overexpression of homing endonuclease probably is detrimental to host cells, leading to the expectation that down-regulation of expression was favored during evolution. In the natural host for PpLSU3, P.polycephalum, the levels of intron RNA species are much lower than those in yeast, with the full-length, the 5′-half and the 3′-half PpLSU3 RNAs being 250-, 30- and 200-fold lower than those in yeast, respectively. Consistent with the low RNA accumulation, I-PpoI protein is expressed 300-fold lower in Physarum than in yeast (23). Perhaps long-term growth of yeast cells carrying PpLSU3 would lead to the accumulation of mutations that reduce expression of the endonuclease.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Masayasu Nomura for providing yeast strain NOY505 and plasmid pNOY103. We thank Steinar Johansen for helpful discussions during the course of this work, and Robert Suran and Swati Joshi for critical reading of the manuscript. This work was supported by grant GM-51860 from the USPHS.
REFERENCES
- 1.Belfort M. and Perlman,P.S. (1995) J. Biol. Chem., 270, 30237–30234. [DOI] [PubMed] [Google Scholar]
- 2.Muscarella D.E. and Vogt,V.M. (1989) Cell, 56, 443–454. [DOI] [PubMed] [Google Scholar]
- 3.Johansen S. and Vogt,V.M. (1994) Cell, 76, 725–734. [DOI] [PubMed] [Google Scholar]
- 4.Johansen S., Elde,M., Vader,A., Haugen,P., Haugli,K. and Haugli,F. (1997) Mol. Microbiol., 24, 737–745. [DOI] [PubMed] [Google Scholar]
- 5.Einvik C., Decatur,W.A., Embley,T.M., Vogt,V.M. and Johansen,S. (1997) RNA, 3, 710–720. [PMC free article] [PubMed] [Google Scholar]
- 6.Elde M., Haugen,P., Willassen,N.P. and Johansen,S. (1999) Eur. J. Biochem., 259, 281–288. [DOI] [PubMed] [Google Scholar]
- 7.Cavalier-Smith T. (1991) Trends Genet., 7, 145–148. [PubMed] [Google Scholar]
- 8.Dujon B. (1989) Gene, 82, 91–114. [DOI] [PubMed] [Google Scholar]
- 9.Mueller J.E., Bryk,M., Loizos,N. and Belfort,M. (1993) In Linn,S.M., Lloyd,R.S. and Roberts,R.J. (eds), Nucleases. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 113–143.
- 10.Johansen S., Embley,T.M. and Willassen.N.P. (1993) Nucleic Acids Res., 21, 4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aaggaard C., Awayez,M.J. and Garrett,R.A. (1997) Nucleic Acids Res., 25, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Argast G.M., Stephens,K.M., Emond,M.J. and Monnat,R.J.,Jr (1998) J. Mol. Biol., 280, 345–353. [DOI] [PubMed] [Google Scholar]
- 13.Bryk M., Quirk,S.M., Mueller,J.E., Loizos,N., Lawrence,C. and Belfort,M. (1993) EMBO J., 12, 2141–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colleaux L., D’Auriol,L., Galibert,F. and Dujon,B. (1988) Proc. Natl Acad. Sci. USA, 85, 6022–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lykke-Anderson J., Thi-Ngoc,H. and Garrett,R. (1994) Nucleic Acids Res., 22, 4583–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wernette C., Saldanha,R., Smith,D., Ming,D., Perlman,P. and Butow,R. (1992) Mol. Cell. Biol., 12, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wittmayer P.K., McKenzie,J.L. and Raines,R.T. (1998) Gene, 206, 11–21. [DOI] [PubMed] [Google Scholar]
- 18.Decker C.J. and Parker,R. (1994) Trends Biochem. Sci., 29, 336–340. [DOI] [PubMed] [Google Scholar]
- 19.Gallie D.R. (1991) Genes Dev., 5, 2108–2116. [DOI] [PubMed] [Google Scholar]
- 20.Jacobson A. (1996) In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Translational Control. Cold Spring Habor Laboratory Press, Cold Spring Harbor, NY, pp. 451–480.
- 21.Lewis J.D., Gunderson,S.I. and Mattaj,I.W. (1995) J. Cell Sci., 19, 13–19. [Google Scholar]
- 22.Sachs A.B., Sarnow,P. and Hentze,M.W. (1997) Cell, 89, 831–838. [DOI] [PubMed] [Google Scholar]
- 23.Lin J. and Vogt,V.M. (1998) Mol. Cell. Biol., 18, 5809–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherman F., Fink,G.R. and Hicks,J.B. (1986) Laboratory Course Manual for Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Sambrook J.E., Fritsch,F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 26.Vallette F., Mege,E., Reiss,A. and Adesnik,M. (1988) Nucleic Acids Res., 17, 723–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muscarella D.E. and Vogt,V.M. (1993) Mol. Cell Biol., 13, 1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rose M.D., Novick,P., Thomas,J.H., Botstein,D. and Fink,G.D. (1987) Gene, 60, 237–243. [DOI] [PubMed] [Google Scholar]
- 29.Decatur W.A., Johansen,S. and Vogt,V.M. (2000) RNA, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sikorski R.S. and Hieter,P. (1989) Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nogi Y., Yano,R., Dodd,J., Carles,C. and Nomura,M. (1993) Mol. Cell. Biol., 13, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1990) Current Protocols in Molecular Biology. John Wiley and Sons, Inc. New York, NY.
- 33.Ruoff B., Johansen,S. and Vogt,V.M. (1992) Nucleic Acids Res., 20, 5899–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nogi Y., Yano,R. and Nomura,M. (1991) Proc. Natl Acad. Sci. USA, 88, 3962–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nogi Y., Vu,L. and Nomura,M. (1991) Proc. Natl Acad. Sci. USA, 88, 7026–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin S., Villarejo,M. and Zabin,I. (1970) Biochem. Biophys. Res. Commun., 40, 249–254. [DOI] [PubMed] [Google Scholar]
- 37.Karkhoff S.R.R. and Schweizer,H.P. (1994) Gene, 140, 7–15. [DOI] [PubMed] [Google Scholar]
- 38.Knipfer N., Nooruddin,L. and Shrader,T.E. (1998) Gene, 217, 69–75. [DOI] [PubMed] [Google Scholar]
- 39.West S.E.H., Romero,M.J.M., Regassa,L.B., Zielinski,N.A. and Welch,R.A. (1995) Gene, 160, 81–86. [DOI] [PubMed] [Google Scholar]
- 40.Moosmann P. and Rusconi,S. (1996) Nucleic Acids Res., 24, 1171–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan W.K., Belfort,G. and Belfort,M. (1988) Gene, 73, 295–304. [DOI] [PubMed] [Google Scholar]
- 42.Doria-Rose N.A. and Vogt,V.M. (1998) J. Virol., 72, 8073–8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lo H.-J., Huang,H.-K. and Donahue,T.F. (1998) Mol. Cell. Biol., 18, 665–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Decatur W.A., Einvik,C., Johansen,S. and Vogt,V.M. (1995) EMBO J., 14, 4558–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vader A., Nielsen,H. and Johansen,S. (1999) EMBO J., 18, 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bauerfeind M., Esser,K. and Michaelis,G. (1998) Mol. Gen. Genet., 257, 635–640. [DOI] [PubMed] [Google Scholar]
- 47.Esser K., Pratje,E. and Michaelis,G. (1996) Mol. Gen. Genet., 252, 437–445. [DOI] [PubMed] [Google Scholar]
- 48.Richter J.D. (1996) In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 481–504.
- 49.Wickens M., Kimble,J. and Strickland,S. (1996) In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 411–450.
- 50.Dalgaard J.Z., Garrett,R.A. and Belfort,M. (1993) Proc. Natl Acad. Sci. USA, 90, 5414–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dujon B., Cottarel,G., Colleaux,L., Bertermier,M., Jacquier,A., d’Auriol,L. and Galibert,G. (eds) (1985) Achievements and Perspectives in Mitochondrial Research. Elsevier, Amsterdam, The Netherlands.
- 52.Dürrenberger F. and Rochaix,J.D. (1991) EMBO J., 10, 3495–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wenzlau J.M., Saldanha,R.J., Butow,R.A. and Perlman,P.S. (1989) Cell, 56, 421–430. [DOI] [PubMed] [Google Scholar]