

Abstract
Transthyretin amyloidosis (ATTR) is a progressive life-threatening disease characterized by the deposition of transthyretin (TTR) amyloid fibrils. Several pathogenic variants have been shown to destabilize TTR tetramers, leading to aggregation of misfolded TTR fibrils. However, factors that underlie the differential age of disease onset amongst amyloidogenic TTR variants remain elusive. Here, we examined the biological properties of various TTR mutations and found that the cellular secretory pattern of the wild-type (WT) TTR was similar to those of the late-onset mutant (Ala97Ser, p. Ala117Ser), stable mutant (Thr119Met, p. Thr139Met), early-onset mutant (Val30Met, p. Val50Met), but not in the unstable mutant (Asp18Gly, p. Asp38Gly). Cytotoxicity assays revealed their toxicities in the order of Val30Met > Ala97Ser > WT > Thr119Met in neuroblastoma cells. Surprisingly, while early-onset amyloidogenic TTR monomers (M-TTRs) are retained by the endoplasmic reticulum quality control (ERQC), late-onset amyloidogenic M-TTRs can be secreted extracellularly. Treatment of thapsigargin (Tg) to activate the unfolded protein response (UPR) alleviates Ala97Ser M-TTR secretion. Interestingly, Ala97Ser TTR overexpression in Drosophila causes late-onset fast neurodegeneration and a relatively short lifespan, recapitulating human disease progression. Our study demonstrates that the escape of TTR monomers from the ERQC may underlie late-onset amyloidogenesis in patients and suggests that targeting ERQC could mitigate late-onset ATTR.
Electronic supplementary material
The online version of this article (10.1007/s00018-019-03357-1) contains supplementary material, which is available to authorized users.
Keywords: Amyloidosis, Drosophila melanogaster, Endoplasmic reticulum quality control, ERQC, Proteostasis, Transthyretin, TTR
Introduction
Transthyretin amyloidosis (ATTR) is a progressive degenerative disease resulting from deposition of misfolded transthyretin (TTR) as non-fibrillar aggregates or amyloid fibrils [1]. These deposits progressively lead to multi-organ failure and ultimately patient’s death 3–15 years after disease onset [2, 3]. Although senescent events associated with the wild-type (WT) TTR gene leads to ATTRwt (formerly known as senile systemic amyloidosis), hereditary ATTR due to mutations in the TTR gene lead to either familial amyloid polyneuropathy (ATTR-FAP), familial amyloid cardiomyopathy (ATTR-FAC), or a rare form of oculoleptomeningeal amyloidosis (OLMA) [4–6].
TTR is a soluble protein secreted mainly from the liver, retinal pigment epithelium and choroid plexus, and is circulated in the plasma and cerebral spinal fluid (CSF). It is responsible for transporting thyroxine (T4) and holo-retinol binding protein (RBP4) [7–9]. TTR is produced as a monomer and assembled into tetramers in the endoplasmic reticulum (ER) before its secretion [10], but most amyloidogenic mutations cause native TTR tetramers to destabilize and misfold into monomeric subunits. These subunits then self-assemble into insoluble amyloid fibrils, causing tissue damage [11]. Dissociated TTR monomers have also been shown to rapidly aggregate into smaller complexes that induce cytotoxicity in human neuroblastoma cell line [12].
Currently, more than 150 pathogenic variants in the TTR gene have been identified, with varying geographic distributions, disease onset, organ involvement and severity amongst variants [13, 14] (http://amyloidosismutations.com). These clinical variations result from differences in the secretion efficiency and amyloidogenecity of TTR variants, which can be predicted by a combination of their thermodynamic and kinetic stabilities in vitro [15, 16]. Val30Met (V30M), alternatively termed p. Val50Met according to the Human Genome Variation Society nomenclature which counts the first 20-amino acid signal peptide, is the most common pathogenic variant predominantly affecting patients originating from Portugal, Sweden and Japan. V30M TTR-FAP typically presents as an early-onset (at around 30 years old) with slow progressive polyneuropathy [17–19]. Interestingly, a protective transsuppressor mutant Thr119Met (T119M, p. Thr139Met) was also found to resist the dissociation of TTR into monomers [11].
Previously, our group identified Ala97Ser (A97S, p. Ala117Ser) as the major pathogenic variant of TTR-FAP in the Taiwanese population, accounting for more than 90% of the ATTR pedigrees [20, 21]. Compared to V30M carriers, patients with A97S TTR-FAP are characterized with a later onset (usually after 50 years old) and rapid disease progression in their clinical presentations, including paresthesia, muscle weakness and autonomic dysfunction [20]. It also accounts for frequent etiology of axonal degeneration type adult-onset pan-modality polyneuropathy [22]. However, it is not fully understood how the A97S-TTR variant leads to late-onset and relatively faster disease progression.
In this study, we delineate a cellular mechanism for late-onset ATTR phenotype using A97S-TTR as the focus. We examined the secretion pattern and cellular localization of wild type (WT), T119M, V30M, A97S, Asp18Gly (D18G, p. Asp38Gly) and their monomeric mutants (M-TTRs) using TTR secretion and immunofluorescence analysis. We also compare the cytotoxic effects of A97S-TTR on IMR-32 neuroblastoma cells alongside WT, T119M and V30M proteins. Additionally, we modelled A97S TTR-FAP in the Drosophila melanogaster alongside the common V30M variant and WT. We compared the impact of these mutants using crawling, lifespan, and climbing assays [23].
Materials and methods
Plasmid constructs and antibodies
TTR variants were generated from human TTR plasmid (Origene, RC204976, Origene Technologies) utilizing the quikChange Lightning site-directed mutagenesis kit procedure from Agilent (Santa Clara, CA, USA) using WT TTR DNA as template. Monomeric mutation (F87M/L110M) was introduced into TTR variants to make M-TTR variants. MCherry-ER-3 was a gift from Michael Davidson (Addgene plasmid # 55041).
Antibodies used are as follows: the polyclonal rabbit anti-human TTR (prealbumin; FL-147; Santa Cruz, CA, USA); polyclonal rabbit anti-human TTR (prealbumin; A0002; DAKO, Copenhagen, Denmark), mouse monoclonal anti-GAPDH (ProteinTech, Rosemont, IL, USA); mouse monoclonal anti-GM130 (BD Biosciences, San Jose, CA, USA); Alexa flour 488, 546 (Thermo Fisher Scientific, Waltham, MA, USA) and HRP-conjugated anti-rabbit, anti-mouse antibodies (ImmunoResearch laboratories, Westgrove, PA, USA).
Cell culture and transfection
Human embryonic kidney cells (HEK293) and human hepatoma cells (HepG2) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. IMR-32 cells were cultured in DMEM-F12 medium supplemented with 10% fetal bovine serum plus 100 U/ml penicillin and 100 µg/ml streptomycin. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. HEK293 cells were pretreated with DMSO or thapsigargin (2 µM; Sigma-Aldrich Cat#67526-95-8) 1 h (h) prior to M-TTRs transfection.
Transient transfection of TTR and M-TTR plasmids was performed with lipofectamine 3000 (Invitrogen) according to manufacturer’s recommendations. Cells were changed to serum-free phenol red free medium prior to transfection. Plasmids were transfected for 24 h for western blot studies and 48 h for immunofluorescence studies. For all transfection experiments, plasmid encoding GFP was co-transfected to monitor transfection efficiency. The transfection efficiency of HEK293 and HepG2 cells were about 85–90% and 40–50%, respectively. Cells were more than 95% viable 24 and 48 h post-transfection.
Immunoblotting
For the preparation of condition medium samples, media was collected and concentrated using Amicon Ultra-4 centrifugal filter units (3 kDa, Merck Millipore). Media was subsequently centrifuged at 15,000g for 20 min at 4 °C to remove debris and prepared as medium samples (extracellular forms of TTR). Cells were washed twice with ice-cold phosphate buffer saline (PBS) and lysed with RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM Nacl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) containing 10% protease and phosphatase inhibitors (Sigma-Aldrich) and centrifuged at 15,000g for 20 min at 4 °C. The supernatant was prepared as cell lysate samples (intracellular forms of TTR). Proteins were quantified with the Pierce BCA protein assay kit (Thermo Scientific). Protein samples were suspended in SDS sample buffer [250 mM Tris–HCl (pH 6.8), 50% glycerol, 0.1% Bromophenol blue, 5% SDS, 5% β-mercaptoethanol], boiled and resolved by SDS-PAGE on 15% gel.
For native or non-reduced and non-boiled SDS pages, cells were lysed in RIPA buffer (50 mM Tris–HCl, 150 mM Nacl, 1 mg/ml sodium deoxycholate and 1% Nonidet P-40) containing 10% protease and phosphatase inhibitors (Sigma-Aldrich) and centrifuged at 15,000g for 20 min at 4 °C. Protein samples were suspended in native sample buffer (250 mM Tris–HCl (pH 6.8), 50% glycerol, 0.1% Bromophenol blue) and left unboiled before loading onto gels (12%).
Proteins were then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore) and then probed with the indicated antibodies. Immunological bands were identified using HRP-conjugated secondary antibody.
Immunofluorescence microscopy
HEK293 cells were grown on glass coverslips coated with Poly-d-lysine and transfected with plasmid DNA encoding the WT- and mutant TTRs. For ER localization, cells were co-transfected with mCherry-ER-3 (Addgene plasmid #55041). Forty-eight hours after transfection, cells were fixed with 4% PFA, permeabilized in PBST solution (0.3% Triton-X100 in PBS) and processed for fluorescence imaging. Cells were incubated with mouse anti-GM130 (BD Biosciences) and rabbit anti-human TTR (Santa Cruz). Secondary antibodies conjugated with Alexa flour 488 (1:1000), 546 (1:1000) (Thermo Fisher Scientific) were used. Images were taken using confocal microscopy (ZEISS LSM 700) and analyzed with ZEN software (Zeiss).
TTR protein expression and purification
The production of recombinant wild-type TTR and variants followed the protocol described previously [24]. Briefly, the protein expression vector, GB1-His-His-His-His-His-His-His-TEV-TTR, contains GB1 tag, followed by 7x Histidine tag and the cleavage site of tobacco etch virus (TEV) protease. The GB1-fused TTR proteins were expressed in Escherichia coli BL21 (DE3) cells by induction with 0.5 mM IPTG (Gold Biotechnology) at 37 °C for 5–6 h. Cells were re-suspended in lysis buffer (50 mM NaPi, 300 mM NaCl, 10 mM imidazole, and 7 mM β-mercaptoethanol, pH 8.0), followed by sonication for lysis. Supernatants were centrifuged and purified using nickel affinity column with appropriate equilibration, wash and elution buffers. To obtain tetrameric TTR protein without his-tag, TEV protease was added to fused protein (GB1-TTR) at a ratio 5 to 1 (GB1-TTR − TEV protease = 5:1), at 30 °C and incubated for 48 h before further purification using size exclusion chromatography (Superdex 200, GE Healthcare).
Cell viability assay
IMR-32 cells (6 × 103 cells/well) were plated in 96-well microplates. The cells were treated with either vehicle control or recombinant TTR proteins (at final concentrations 0.22 and 0.44 mg/ml). Following a 72-h incubation period, cell viability was measured using MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; Sigma-Aldrich) assay as previously described [8].
Lactate dehydrogenase (LDH) cytotoxicity assay
Cellular damage following TTR treatment in IMR-32 cells was assessed by measurement of LDH released into the medium solution. During treatment, 6 × 103 cells/well were incubated at 37 °C with TTR proteins (0.22 and 0.44 mg/ml) for 72 h. At the end of treatment, 50 μl of medium solution was transferred to a 96-well plate and the LDH reaction was performed using Pierce LDH Cytotoxicity Assay Kit (Thermo Scientific) following the manufacturer’s instructions. To determine LDH activity, the absorbance at 680 nm (background signal) was subtracted from the absorbance at 490 nm. LDH activity results are shown as fold of controls.
Generation of transgenic hTTR flies
To generate UAS-TTR flies, the human TTR gene (hTTR) construct was synthesized by in vitro synthesis (GenScript Corporation). The synthetic gene was codon-optimized for Drosophila, including a kozak sequence (CAAA), human secretory signal peptide for TTR and a C-terminal HA-tag. The gene coding WT-hTTR was cloned into a pUAST-attb vector using both EcoR1 and Xho1. Lightning site-directed mutagenesis kit procedure from Agilent (Santa Clara, CA, USA) was used to create V30M and A97S variants using the pUAST-attb-hTTR-wt-HA vector as template. Transgenic Drosophila was generated by site-specific transformation via phiC3.1 integrase, followed with standard genetics to maintain the p element insertion [25]. UAS-Aβ42, D42-GAL4, nSyb-GAL4 fly lines were obtained from Bloomington stock center (BDSC).
Fly husbandry was performed following standard procedures. Flies stock were reared in 50-ml plastic vials containing standard Drosophila cornmeal food. The Gal4-UAS system was used for transgenic-expression in a tissue-specific manner. The flies were raised at 25 °C and transferred to 29 °C to increase Gal4 expression.
Western blot
Homogenates were prepared by homogenizing 20 fly heads (nSyb-gal4/+ , nSyb > hTTR-WT-HA, nSyb > hTTR-V30M-HA and nSyb > hTTR A97S-HA) in 50 µl RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM Nacl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) containing 10% protease inhibitor and centrifuged at 15000 g for 20 min at 4 °C. Proteins were quantified with the Pierce BCA protein assay kit (Thermo Scientific). Protein samples were resolved by gradient SDS-PAGE and then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore). Primary antibodies used are mouse anti-HA, 1:500 (Proteintech, cat # 66006-1-Ig), mouse anti-GAPDH, 1:5000 (Proteintech, cat # 60004-1-Ig). Anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibody (1:10,000, GeneTex) was used. Signals were generated using ECL-Plus reagent (Millipore) and detected using the Luminescence/Fluorescence imaging system LAS-4000 (Fujifilm).
Larva crawling assay
The crawling behavior assay was carried out at room temperature (25 °C). Wandering third-instar larvae of each genotype were picked from vials and rinsed in distilled water. Larvae were placed on a 1% black agarose gel for 30 s to recover from handling. Larva locomotion was recorded with a camera for 60 s. 10 larvae were tested for each group. Videos were transferred to the computer and analyzed using the Image-J plugin wrMTrck as previously described [26].
Immunnostaining and analysis of larvae neuromuscular junction (NMJs)
NMJ 6/7 phenotypes were analyzed at the A3 segment of wandering third instar larvae as previously described [27]. Larvae were dissected in cold calcium-free HL3 saline (70 mM NaCl, 5 mM KCl, 20 mM MgCl2, 10 mM NaHCO3, 5 mM trehalose, 115 mM sucrose, and 5 mM HEPES, pH 7.2). Larval body fillets were fixed in Bouin’s fluid (Sigma-Aldrich) for 5 min and washed in PBT (0.01% Triton-X-100) thrice, for 10 min each. Fixed fillets were incubated with primary antibodies overnight at 4 °C, rinsed thrice in PBT and incubated with secondary antibodies for 2 h at room temperature. Primary antibodies used are mouse anti-Dlg (4F3, 1:100; Developmental Studies Hybridoma Bank, DSHB, Iowa City, IA; RRID: AB_528203), goat anti-HRP conjugated FITC, rabbit anti-HRP conjugated TRITC or Cy5 (RRID: AB_2314647; RRID: AB_2340257; Jackson ImmunoResearch, West Grove, PA, USA). Larval fillets were mounted onto slides with PBS containing 87.5% glycerol and 0.22 M 1, 4-diaza-byciclo (2.2.2) octane (Dabco, Sigma-Aldrich). NMJ images were acquired by confocal Z-stack scanning (Zeiss LSM700, Germany) using 40 oil objectives and processed by LSM image examiner.
Climbing assay
The locomotor ability of adult flies with different genotypes was determined using the climbing assay (negative geotaxis) as described previously [28]. Ten (10) flies were placed in an empty vial marked 8 cm from the bottom. All flies were manually tapped to the bottom of the vial. The number of flies that crawl pass the 8 cm mark in 6 s in four repeats per genotype was recorded.
Survival assay
Transgenic UAS flies reared at 25 °C were crossed to the nSyb-Gal4 driver for pan-neuronal expression. After eclosion, a cohort of male flies from each genotype was sorted in setups of 20 flies in each vial. Flies were incubated at 29 °C and maintained on standard media. Fresh food media were changed every 2–3 days followed by mortality scoring. Statistical analysis was performed using GraphPad prism version 6.01 for Kaplan–Meier survival curves using the log-rank tests [29].
Statistics
Each experiment was repeated at least three times; combined data were expressed as mean ± SEM and compared using the Analysis of variance (ANOVA) followed by Fisher’s least significant difference test. A p < 0.05 was considered statistically significant and marked with an asterisk in the figures. * Indicates p < 0.05; ** indicates p < 0.01; *** indicates p < 0.001; **** indicates P < 0.0001.
Results
Secretion pattern of A97S-TTR
Since ATTR onset has been correlated with the secretion efficiency of TTR variants [15, 16], we first compared the secretory patterns of A97S-TTR with WT-TTR and other mutant TTRs, including the prevalent pathogenic V30M variant, the amyloidosis suppressor T119M variant, and the D18G variant, which has low secretion efficiency due to its structural instability [15, 16]. Cultured HEK293 and HepG2 cells were transfected with WT and mutant TTRs; then intracellular and secreted TTR proteins were blotted from both cell lysates and culture media, respectively. Samples were run under SDS-PAGE (Fig. 1) and native PAGE conditions (Fig S1). In both cells, intracellular TTR protein levels were comparable to WT and all mutant TTR (with slightly lower level for D18G) (Fig. 1a, e). Similar levels of secreted A97S-, WT-, T119M- and V30M-TTR were found in the media (Fig. 1b, f) while D18G-TTR was barely detected (Fig. 1b, f, S1b), consistent with previous reports [10, 15]. Secretion analysis revealed that A97S-TTR was secreted with a similar efficiency to WT-, V30M-, and T119M-TTR (Fig. 1c, g).
Fig. 1.

Secretion pattern of WT and mutant TTRs in HEK293 cells (a–d) and HepG2 cells (e–h). Cultured HEK293 and HepG2 cells were transiently transfected with plasmids encoding WT- and mutant- TTRs for 24 h. Intracellular and secreted TTRs were analysed from the cell lysates and culture media, respectively, using western blot. a, b, e, f Western blot of both cell lysate and medium samples were analyzed using anti-human TTR antibody. c, d, g, h T119M-, V30M- and A97S-TTR had secretion efficiency similar to that of WT-TTR in both HEK293 and HepG2 cells while the secretion efficiency of D18G was dramatically decreased. Data are presented as mean ± standard error of mean from at least three independent experiments. Statistical analysis was performed by one-way analysis of variance, followed by Fisher’s least significant difference test. **p < 0.01, ***p < 0.001
Subcellular localization of A97S-TTR
To characterize the intracellular localization of A97S-TTR within the secretory pathway, we assessed the distribution of A97S-,WT- and other TTR variants (V30M, D18G, and T119M) within the ER and Golgi apparatus of HEK293 cells using immunofluorescence (Fig. 2). We observed A97S-TTR colocalize with the ER marker (Calreticulin + KDEL), similar to WT-, V30M-, T119M-, and D18G-TTRs (Fig. 2a). In cells stained with the Golgi marker GM130, we found that A97S-, V30M- and T119M-TTR variants as well as WT-TTR colocalized well with the punctate of the Golgi apparatus, suggesting they are trafficked through the protein secretory pathway (Fig. 2b). In contrast, D18G-TTR exhibited minimal overlap with GM130 (Fig. 2b, bottom row), consistent with inefficient ER trafficking of this TTR variant to the Golgi apparatus [16]. Altogether, our data indicated that the A97S mutant could be trafficked in the secretory pathway similar to WT and V30M TTR, suggesting other mechanisms may underlie the late-onset and fast progression of A97S TTR-FAP.
Fig. 2.

Subcellular distribution WT and mutant TTRs in HEK293 cells. Cells were stained with the ER marker calreticulin + KDEL (a) or the Golgi marker GM130 (b). T119M-, V30M-, A97S- as well as WT-TTR were colocalized with both ER and Golgi markers. Notably, D18G-TTR was colocalized with ER but not Golgi maker. Scale bar = 5 µm
Cellular toxicity of A97S-TTR
Since we did not find major differences in the secretory pattern between A97S-, V30M- and WT-TTR, we next sought other potential mechanisms for A97S pathogenicity. We compared the cytotoxic effect of A97S-, WT-, T119M- and V30M-TTRs using cell viability assays by MTT and LDH release assays (Fig. 3). Purified TTR proteins were added to the medium of cultured IMR-32 cells at 0.22 mg/ml (4 μM) and 0.44 mg/ml (8 μM) for 72 h. In the MTT assay, WT-treated and T119M-treated cells both showed slight but insignificant decreases in cell viability compared to control cells treated with the vehicle, while A97S-treated cells showed an intermediate decrease at both doses (78.5% ± 3.60, p = 0.152 & 73.5% ± 0.81, p = 0.119; Fig. 3a, b). V30M-treated cells showed a severe decrease in viability (67.3% ± 1.20, p = 0.005 and 59.4% ± 0.87, p = 0.0009), consistent with a previous study [8] but less severe than another [12]. Similarly, treatment with both WT- and T119M-TTR led to slight but no significant increase in LDH release (a sign of significant cell membrane compromise) (Fig. 3c, d). IMR-32 cells exposed to V30M mutant protein caused the most significant increase in LDH release (4.2 ± 0.1614, p = 0.0004 & 5.2 ± 0.4239 p < 0.0001 folds of control), whereas A97S-TTR led to an intermediate increase (4.0 ± 0.2317, p = 0.0006 & 4.3 ± 0.0854, p = 0.0003 folds of control; Fig. 3c, d). Altogether, these results suggest that the A97S mutant exhibits less toxicity toward cultured cells than V30M-TTR, while WT- and T119M-TTR have a minimal toxicity.
Fig. 3.

Cytotoxicity of WT-TTR and mutant TTRs to neuroblastoma cells. IMR-32 cells were incubated with 0.22 and 0.44 mg/ml recombinant TTR proteins for 72 h. The cytotoxicity of the TTRs was measured by MTT (a, b) and LDH release assays (c, d). MTT assay showed no apparent change in cell survival for cell treated with WT- and T119M-TTRs. V30M-TTR showed the most severe toxicity while A97S-TTR showed intermediate toxicity at both concentrations (a, b). LDH release assay showed no obvious toxicity and mild toxicity for T119M- and WT-TTRs, respectively. Again, V30M-TTR showed the most toxicity while A97S-TTR had intermediate toxicity. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001
Monomeric late-onset TTR variants bypass ERQC and are secreted by cells
Previously, the secretory characteristics of monomeric TTRs (M-TTRs) in cells have been assessed by introducing a monomeric F87M/L110M mutation, which inhibits TTR tetramerization [16, 30]. This technique uncovered that amyloidogenic M-TTRs were retained by the ER and could not be secreted from cells [15]. To examine potential ER retention of A97S M-TTR (M-A97S), we introduced the F87M/L110M into A97S-, V30M-, D18G-, non-amyloidogenic T119M-, and WT-TTRs to inhibit tetramerization [30]. We confirmed previous findings that WT and non-amyloidogenic T119M M-TTRs could be secreted, while early-onset V30M and highly destabilized D18G M-TTRs were not present in the media [15] (Fig. S2). Surprisingly, A97S M-TTR, unlike other amyloidogenic M-TTRs, was secreted from the cells with a similar secretion efficiency such as WT M-TTR (M-WT; Fig. S2). To examine whether this phenomenon was the case with other late-onset mutants, we constructed monomeric TTRs of six other late-onset ATTR-associated mutants Pro24Ser (P24S, p. Pro44Ser), Ala45Gly (A45G, p. Ala65Gly), Phe64Ile (F64I, p. Phe84Ile), His88Arg (H88R, p. His108Arg), Ile107Val (I107V, p. Ile127Val), and Val122Ile (V122I, p. Val142Ile) (Fig. 4a). We ran the proteins under reducing SDS-PAGE and found that most of the monomeric TTR variants were secreted at similar or (even) slightly higher efficiencies than that of the WT M-TTR (Fig. 4a–c, S3). To confirm that these M-TTR variants were secreted as monomers, the proteins were separated under non-reduced SDS-PAGE (Fig. 4c, S3) and non-denaturing native PAGE (Fig. S4) gels. Both conditions revealed that late-onset M-TTRs were majorly secreted as monomers (Fig. 4c, S4b). Consistent with a previous report [15], T119M M-TTR (M-T119M) was detected in both monomeric and tetrameric forms (Fig. S4), suggesting its strong inhibitory effect against monomer formation. This finding reveals a novel ER quality control (ERQC) bypass mechanism for monomeric forms of amyloidogenic A97S and other late-onset TTR variants.
Fig. 4.

Secretion of amyloidogenic late-onset monomeric TTRs (M-TTRs). a Western blot of both cell lysate and medium samples were analyzed using anti-human TTR antibody. b Secretion efficiency of M-TTRs of WT, T119M and other mutants. The highly unstable D18G and early-onset V30M mutants were not secreted while all late-onset mutants were secreted to the medium. c Secretion pattern of amyloidogenic late-onset M-TTRs in non-reduced, non-boiled SDS PAGE. Only WT-TTR was secreted as tetrameric form. The highly unstable D18G and V30M mutants showed no secretion while all late-onset M-TTRs were secreted as monomers to the medium. Western blot for cell lysates is shown in Fig. S3. d Immunoblot of WT, V30M and A97S M-TTRs in the lysate and medium of HEK293 cells pretreated with 2 µM Tg. e Quantification of relative secretion of WT M-TTR and A97S M-TTR to WT M-TTR vehicle (DMSO) in HEK293 cells pretreated with Tg shows a significant decrease in A97S M-TTR. *p < 0.05; **p < 0.01
To test whether enhancing ERQC could inhibit the secretion of M-A9S, we used a sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor thapsigargin (Tg) to activate the unfolded protein response (UPR) and looked at its effect on M-A97S secretion. Remarkably, Tg dramatically reduces M-A97S secretion by > 45% (Fig. 4d, e), suggesting that enhancing ERQC could inhibit M-A97S secretion.
Impaired locomotion in the Drosophila model of ATTR
To gain more insight into the pathological properties of mutant A97S-TTR in vivo, we established a Drosophila model for ATTR-related amyloidosis by expressing human WT-, V30M-, or A97S-TTR using the Gal4-UAS system [31]. Wild-type and mutant human TTR genes under UAS enhancer control were each inserted into the genome of Drosophila and their expression was activated in the nervous system by nSyb-Gal4. All transgenic flies expressing human TTR (hTTR) were confirmed by immunoblotting (Fig. 5a). At the larval stage, expression of mutant V30M-hTTR resulted in a slight but significant reduction in crawling speed compared to wild type control (D42-Gal4/+) (Fig. 5b). To test whether expression of hTTR variants may affect synapse organization, neuromuscular junctions (NMJs) in third instar larvae [32] were monitored by double-labelling of the axonal membrane marker anti-HRP and the postsynaptic density marker anti-Dlg (the Drosophila PSD95 homolog) (Fig. 5c). Quantification of NMJ bouton number at A3 segment between muscle 6 and 7 did not reveal a significant difference between control, wild-type and hTTR mutants in all transgenic larvae driven by motor neuron-specific promoters (D42-Gal4) (Fig. 5d).
Fig. 5.

Expression of mutant V30M-hTTR in Drosophila melanogaster caused reduced crawling in larvae. a Western blot of homogenates from transgenic fly heads expressing either WT-, V30M- and A97S-hTTR in the nervous system. b Larvae expressing V30M-hTTR showed a minor decrease in crawling speed while others showed no significant changes. c Confocal images and quantifications of NMJ boutons wild type and mutant hTTR larvae showed no obvious changes in the bouton shape and numbers at this stage. d Quantification of bouton number in c. Scale bar = 50 µm. *p < 0.05
To assess the impact of mutant hTTR overexpression in adult flies, we employed the negative geotaxis (climbing) assay to assess the impairment of neuromuscular coordination in Drosophila [33]. The climbing ability was assayed on Days 7, 14 and 21 in flies overexpressing WT-, A97S-, and V30M-hTTR, or amyloid β (Aβ) 42 as positive control. At Day 7, flies overexpressing V30M-hTTR and Aβ 42 displayed early impairment in climbing activity compared to control (nSyb-gal4/+), WT- and A97S-hTTR flies (Fig. 6a). Interestingly, by Day 14, A97S-hTTR flies exhibited a rapidly progressive decline in climbing activity which was persistent until Day 21, while V30M-hTTR flies continuously exhibited gradual decline in their climbing activity (Fig. 6a). In contrast, WT-hTTR flies showed gradual progressive decline in climbing. Taken together, these results reveal a late-onset impaired climbing with fast progressivity in A97S-hTTR flies compared to an earlier age-dependent deterioration in V30M-hTTR flies.
Fig. 6.
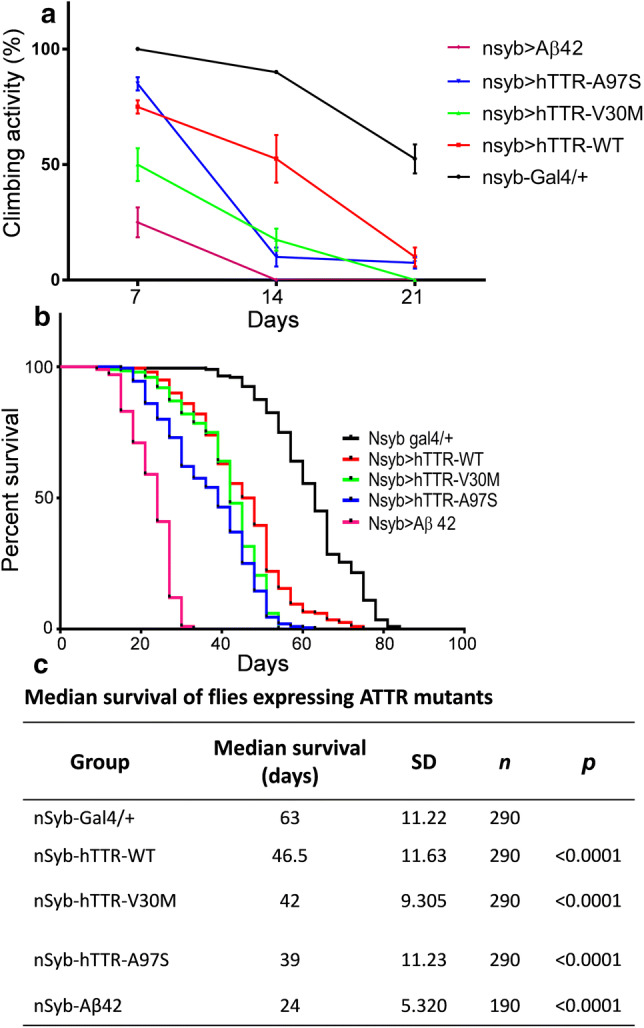
Climbing and survival assay of nSyb-gal4 driven transgenic Drosophila melanogaster. a Flies expressing V30M-hTTR and Aβ42 showed early progressive decline while those expressing A97S-hTTR had a late fast progressive decline in climbing activity. Cohorts of flies from each genotype were subjected to climbing assay for 3 weeks. b Survival curves of flies with pan-neuronal expression of either WT, V30M or A97S-hTTR. While flies expressing WT- and V30M-hTTR showed a decreased lifespan, A97S-hTTR decreased lifespan in a slightly more aggressive manner. c Results of Log-rank (Mantel-Cox) test of data in (b). All experiments were performed at 29 °C. ****p < 0.0001
Mutant hTTR expression results in shortened lifespan
To determine the effect of wild-type and mutant hTTR expression on the lifespan of transgenic flies, the Kaplan–Meier survival function was performed (Fig. 6b). Flies expressing WT and both mutant hTTR had significant shortened lifespan in comparison to control flies (T50 %: WT = 46.5 ± 0.82 days, V30M = 42.0 ± 0.66 days, A97S = 39.0 ± 0.79 days vs control = 63.0 ± 0.79 days; Fig. 6c). Interestingly, the A97S-hTTR flies had a slight but significant decrease in lifespan compared to the V30M and WT-hTTR flies (Fig. 6b, c).
Discussion
Using expression of WT-, A97S-, V30M-, D18G-, and T119M-TTR in cultured cells, we found that the A97S-TTR variant had protein secretion and subcellular localization patterns similar to the WT-TTR, as well as the common amyloidogenic V30M and the transsuppressor T119M mutants, but not the unstable D18G mutant (Fig. 1). Interestingly, A97S-TTR exhibited an intermediate toxicity to the cells compared to V30M-TTR (Fig. 3). Surprisingly, unlike monomeric V30M-TTR, which was trapped intracellularly by the ERQC, monomeric A97S-TTR and other late-onset TTR mutant monomers were able to escape ERQC and be secreted into the media similarly as monomeric WT and T119M-TTR (Fig. 4, S3, S4). This secretion could be blocked by the activation of UPR responses with Tg (Fig. 4). This result suggests a novel ERQC bypass mechanism and secretion of monomeric forms of late-onset TTR mutations for the formation of amyloids (Fig. 7). Furthermore, both A97S- and V30M-overexpressing Drosophila exhibited an age-dependent progressive decline in their motor activity (Fig. 6) and a drastic decrease in their lifespan (Fig. 6).
Fig. 7.

Schematic diagram depicting pathways for amyloidosis caused by WT and mutant TTR. For TTR mutants with intermediate changes in stability and early onset, such as V30M, the ERQC traps and prevent their monomeric secretion but allows their tetrameric secretion [15]. For highly unstable TTRs (e.g. D18G), they have minimal secretion of mutant protein, causing a late-onset disease. For A97S and other late-onset prone (LOP) TTRs, while their tetramers are stably secreted, their TTR monomers can also be secreted. This may contribute to their late-onset ATTR in the patients
Previously, the intrinsic thermodynamics and kinetics of secreted TTR tetramers have been proposed to predict the onset of ATTR in patients [34, 35]. According to this model, point mutations alter the native tetrameric structure of circulating TTR, promoting the release of amyloidogenic TTR monomers, which misfold, aggregate, then deposit as amyloid at target organs [36, 37]. In addition, secretion of TTR protein is also a primary determinant for ATTR since it determines the availability of amyloidogenic TTR for distal deposition [38]. Late-onset cases of V30M TTR-FAP have been reported in patients in non-endemic foci [18]; however, these patients present with symptoms such as a lower penetrance rate, mild autonomic symptoms, reduced number of myelinated fibers, tissue deposition of short, thin, mesh-like amyloid fibrils consisting predominantly of C-terminal TTR fragments and shorter disease duration [39–42]. Environmental and genetic factors may play important roles in this phenotypic variability for V30M FAP [43–45].
This study reveals the first evidence that the A97S- and other late-onset M-TTR variants can be secreted extracellularly, similar to the WT M-TTR (Fig. 4, S3, S4). Previously, amyloidogenic early-onset (V30M, E54K, L55P) and highly destabilized (D18G, A25T) M-TTRs have been reported to retain in the ER and subsequently degraded by ERAD [15]. Thus, while TTR tetramers may be thermodynamically stable in patients carrying the A97S and other late-onset-prone (LOP) TTR variants, their monomeric forms may be secreted at low levels, gradually initiating amyloidogenesis and proteotoxic aggregation leading to their late-onset in patients (Fig. 7). A similar mechanism has been linked to other late-onset neurodegenerative diseases such as Huntington’s disease and familial forms of Alzheimer’s and Parkinson’s disease [46]. This may also explain the source of WT-TTR fibrils detected in amyloid fibrils in young ATTR-FAP patients [47, 48].
Several strategies have been developed to treat ATTR, such as TTR tetramer stabilization, gene knockdown, anti-SAP therapy, and use of fibril disruptors against the disease [49]. Stabilization of TTR heterotetramers in compound heterozygotes for both V30M and T119 M TTR alleles inspired identification of TTR tetramer stabilizers [50], such as tafamidis, diflunisal and tolcapone, which bind to transthyretin’s T4 pockets and stabilize the tetramer [51–54]. Another approach aimed at reducing circulating TTR levels has led to the development and approval of patisiran, an RNAi therapeutic [55, 56]. Other small interfering RNA (siRNA) and anti-sense oligonucleotides are currently being evaluated [57, 58]. Amyloid disruptors or drugs acting as inhibitors of amyloid formation led to the identification of epigallocatechin-3-gallate (EGCG), which binds to soluble TTR at the interface of both dimers, increases TTR’s resistance to dissociation and inhibits TTR fibril formation [59–62].
Our results propose that therapies which enhance ERQC may be helpful for late-onset ATTR resulting from A97S mutation. Recent approaches aimed at improving ERQC via ER reprograming to drastically reduce secretion of aggregate prone proteins has led to the identification of some targets [63]. From our data, the ER stressor Tg could reduce A97S M-TTR secretion, suggesting that the ERQC pathway may be a potential target for ATTR induced by A97S and other TTR mutations that escape ERQC as monomers. Newer evidence also implicated the formation of pathogenic aggregates originating from secretory proteins to defective ER proteostasis [38, 64]. Remodelling ER proteostasis resulted in reduced secretion and extracellular aggregation of destabilized, aggregation-prone variants of amyloidogenic proteins [63, 65].
Finally, we used cell and Drosophila models to test the toxicity of A97S-TTR in comparison with WT- and other TTR variants. Our results showed that A97S-TTR is less toxic to the cells (Fig. 3), but causes a late-onset and relatively faster progression in neurodegeneration revealed by both motor activities and lifespan assay (Fig. 6). Expression of V30M-hTTR in the nervous system led to reduced crawling activity in the larvae (Fig. 5) and an earlier age-dependent climbing performance deficit in adult flies compared to A97S-hTTR flies (Fig. 6). This could be linked to its early onset phenotypic pattern as seen in humans [66]. Interestingly, expression of A97S-hTTR in the nervous system reduces fly’s lifespan compared to V30M-hTTR (Fig. 6). This could be associated with the fast progressive nature of A97S variant in patients [20]. We did not detect amyloid deposits in our Drosophila models using thioflavine S staining (data not shown), therefore, it is not clear whether the toxicity comes from amyloid plagues or non-fibrillar TTR amyloid precursors. However, these fly models still recapitulate some aspects of human ATTR pathology and may serve as good models for future studies and test for potential treatments.
In conclusion, our results suggest secretion of monomeric amyloidogenic TTRs via bypass of the ERQC as an important mechanism for late-onset ATTR pathogenesis. We hypothesize that within the ER, there are populations of stable wild-type and mutant TTR monomers which are folded into tetramers. In early-onset disease associated TTR mutants, although the ERQC prevents their monomeric secretion, [15] the influence of ER-assisted folding (ERAF) pathway allows their secretion as tetramers into the extracellular environment, where their inherent stabilities coupled with other factors determine their rate of dissociation into misfolded monomers for amyloid deposition [16, 67]. In contrast, both monomers and tetramers in late-onset disease-associated TTR mutants can bypass the ERQC and secrete into the extracellular environment. Our results suggested that late-onset ATTR pathogenesis is not necessarily due to the unfolding of their tetramers, but rather the escape of their monomeric amyloidogenic precursors that accumulate overtime, leading to high serum concentrations which can facilitate proteotoxic aggregation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Pei-Jun Chen and Yung-Yu Lu at NYMU Core Facility for confocal imaging assistance; Chien-Hsiang Wang, Pei-Yi Chen, Vanitha Nithianandam, and Dr. Cheng-Ting Chien (Institute of Molecular Biology, Academia Sinica) for assistance in fly larva assays; Yu-Han Yeh, Yu-Chien Hung (Graduate Institute of Physiology, National Taiwan University) for assistance in adult fly assays; Dr. Jenn-Yah Yu (Department of Life Sciences, NYMU) for fly facilities; Drs. Won-Jing Wang, Wei-Yi Chen (Institute of Biochemistry and Molecular Biology, NYMU), Chia-Hsiang Chang, Fang-Shin Nian, and Ms. Yi-Wen Ou (Institute of Brain Science, NYMU) for useful comments and suggestions. This work was supported by Ministry of Science and Technology (MOST 105-2314-B-075 -066 and 105-2113-M-001-021-MY2); Academia Sinica (AS-iMATE-107-31); National Health Research Institutes (NHRI-EX105-10507EC, NHRI-EX106-10507EC, NHRI-EX107-10507EC); Yen Tjing Ling Medical Foundation (CI-106-6); Taipei Veterans General Hospital (V104-B015, V105C-166, V106C-153, V107C-152, VGHUST106-G7-5-1, VGHUST107-G7-1-3); 2017 Global ASPIRE Transthyretin (TTR) Amyloidosis Competitive Research Grant Awards (WI229986) to YTL; the Ministry of Science and Technology, Taiwan (MOST 104-2745-B-075-001, MOST-105-2633-B-009-003, 106-2321-B-075-001, 106-2628-B-010-002-MY3, 107-2321-B-075-001, 107-2221-E-010-014, 108-2321-B-010-011-MY2, and 108-2638-B-010-001-MY2) to JWT; the Development and Construction Plan of NYMU School of Medicine from Yin Yen-Liang Foundation (107F-M01-0502); and the Brain Research Center, NYMU through the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE), Taiwan. All the above funding sources had no involvement in study design, collection, analysis/interpretation of data; the writing of the report; or the decision to submit the article for publication.
Author contributions
RBI, YTL and JWT conceptualized the project and designed the experiments. RBI, SYY and CCC conducted cell and fly experiments. RBI, KPL, TYY, CCC, RS, JWT and YTL analyzed and interpreted data. RBI, JWT, CCC and YTL wrote the manuscript. All authors read and revised the manuscript.
Compliance with ethical standards
Conflict of interest
Authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jin-Wu Tsai, Email: tsaijw@ym.edu.tw.
Yo-Tsen Liu, Email: ytliu2@vghtpe.gov.tw.
References
- 1.Finsterer J, Iglseder S, Wanschitz J, Topakian R, Loscher WN, Grisold W. Hereditary transthyretin-related amyloidosis. Acta Neurol Scand. 2018 doi: 10.1111/ane.13035. [DOI] [PubMed] [Google Scholar]
- 2.Castano A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163–178. doi: 10.1007/s10741-014-9462-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gertz MA. Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am J Manag Care. 2017;23(7 Suppl):S107–S112. [PubMed] [Google Scholar]
- 4.Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005;62(7):1057–1062. doi: 10.1001/archneur.62.7.1057. [DOI] [PubMed] [Google Scholar]
- 5.Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F, Branzi A. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7(7):398–408. doi: 10.1038/nrcardio.2010.67. [DOI] [PubMed] [Google Scholar]
- 6.Mathieu F, Morgan E, So J, Munoz DG, Mason W, Kongkham P. Oculoleptomeningeal amyloidosis secondary to the rare transthyretin c.381T > G (p.Ile127Met) mutation. World Neurosurg. 2018;111:190–193. doi: 10.1016/j.wneu.2017.12.096. [DOI] [PubMed] [Google Scholar]
- 7.Hamilton JA, Benson MD. Transthyretin: a review from a structural perspective. Cell Mol Life Sci CMLS. 2001;58(10):1491–1521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F, Hu C, Dong Y, Lin MS, Liu J, Jiang X, Ge Y, Guo Y. The impact of V30A mutation on transthyretin protein structural stability and cytotoxicity against neuroblastoma cells. Arch Biochem Biophys. 2013;535(2):120–127. doi: 10.1016/j.abb.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 9.Pfeffer BA, Becerra SP, Borst DE, Wong P. Expression of transthyretin and retinol binding protein mRNAs and secretion of transthyretin by cultured monkey retinal pigment epithelium. Mol Vis. 2004;10:23–30. [PubMed] [Google Scholar]
- 10.Batista AR, Sena-Esteves M, Saraiva MJ. Hepatic production of transthyretin L12P leads to intracellular lysosomal aggregates in a new somatic transgenic mouse model. Biochem Biophys Acta. 2013;1832(8):1183–1193. doi: 10.1016/j.bbadis.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Kamata M, Susanto MT, Chen IS. Enhanced transthyretin tetramer stability following expression of an amyloid disease transsuppressor variant in mammalian cells. J Gene Med. 2009;11(2):103–111. doi: 10.1002/jgm.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA. 2004;101(9):2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Plante-Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018;265(4):976–983. doi: 10.1007/s00415-017-8708-4. [DOI] [PubMed] [Google Scholar]
- 14.Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN, Obici L, Westermark P, Grateau G, Wechalekar AD. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35(9):E2403–E2412. doi: 10.1002/humu.22619. [DOI] [PubMed] [Google Scholar]
- 15.Sato T, Susuki S, Suico MA, Miyata M, Ando Y, Mizuguchi M, Takeuchi M, Dobashi M, Shuto T, Kai H. Endoplasmic reticulum quality control regulates the fate of transthyretin variants in the cell. EMBO J. 2007;26(10):2501–2512. doi: 10.1038/sj.emboj.7601685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121(1):73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 17.Conceicao I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late- and early-onset cases in Portugal. Muscle Nerve. 2007;35(1):116–118. doi: 10.1002/mus.20644. [DOI] [PubMed] [Google Scholar]
- 18.Koike H, Misu K, Ikeda S, Ando Y, Nakazato M, Ando E, Yamamoto M, Hattori N, Sobue G. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59(11):1771–1776. doi: 10.1001/archneur.59.11.1771. [DOI] [PubMed] [Google Scholar]
- 19.Koike H, Tanaka F, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, Fujitake J, Kawanami T, Kato T, Yamamoto M, Sobue G. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83(2):152–158. doi: 10.1136/jnnp-2011-301299. [DOI] [PubMed] [Google Scholar]
- 20.Liu YT, Lee YC, Yang CC, Chen ML, Lin KP. Transthyretin Ala97Ser in Chinese–Taiwanese patients with familial amyloid polyneuropathy: genetic studies and phenotype expression. J Neurol Sci. 2008;267(1–2):91–99. doi: 10.1016/j.jns.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Yang NC, Lee MJ, Chao CC, Chuang YT, Lin WM, Chang MF, Hsieh PC, Kan HW, Lin YH, Yang CC, Chiu MJ, Liou HH, Hsieh ST. Clinical presentations and skin denervation in amyloid neuropathy due to transthyretin Ala97Ser. Neurology. 2010;75(6):532–538. doi: 10.1212/WNL.0b013e3181ec7fda. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh ST. Amyloid neuropathy with transthyretin mutations: overview and unique Ala97Ser in Taiwan. Acta neurologica Taiwanica. 2011;20(2):155–160. [PubMed] [Google Scholar]
- 23.Ugur B, Chen K, Bellen HJ. Drosophila tools and assays for the study of human diseases. Dis Models Mech. 2016;9(3):235–244. doi: 10.1242/dmm.023762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y-T, Yen Y-J, Ricardo F, Chang Y, Wu P-H, Huang S-J, Lin K-P, Yu T-Y. Biophysical characterization and modulation of Transthyretin Ala97Ser. Ann Clin Transl Neurol (Accepted) 2019 doi: 10.1002/acn3.50887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA. 2007;104(9):3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks DS, Vishal K, Kawakami J, Bouyain S, Geisbrecht ER. Optimization of wrMTrck to monitor Drosophila larval locomotor activity. J Insect Physiol. 2016;93–94:11–17. doi: 10.1016/j.jinsphys.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang CH, Huang YC, Chen PY, Cheng YJ, Kao HH, Pi H, Chien CT. USP5/Leon deubiquitinase confines postsynaptic growth by maintaining ubiquitin homeostasis through Ubiquilin. eLife. 2017 doi: 10.7554/elife.26886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang CN, Wu MF, Liu CC, Jung WH, Chang YC, Lee WP, Shiao YJ, Wu CL, Liou HH, Lin SK, Chan CC. Differential protective effects of connective tissue growth factor against Abeta neurotoxicity on neurons and glia. Hum Mol Genet. 2017;26(20):3909–3921. doi: 10.1093/hmg/ddx278. [DOI] [PubMed] [Google Scholar]
- 29.Gronke S, Clarke DF, Broughton S, Andrews TD, Partridge L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010;6(2):e1000857. doi: 10.1371/journal.pgen.1000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40(38):11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 31.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118(2):401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 32.Keshishian H, Broadie K, Chiba A, Bate M. The drosophila neuromuscular junction: a model system for studying synaptic development and function. Annu Rev Neurosci. 1996;19:545–575. doi: 10.1146/annurev.ne.19.030196.002553. [DOI] [PubMed] [Google Scholar]
- 33.McGurk L, Berson A, Bonini NM. Drosophila as an in vivo model for human neurodegenerative disease. Genetics. 2015;201(2):377–402. doi: 10.1534/genetics.115.179457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421(2–3):185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunjes DL, Castano A, Clemons A, Rubin J, Maurer MS. Transthyretin cardiac amyloidosis in older americans. J Cardiac Fail. 2016;22(12):996–1003. doi: 10.1016/j.cardfail.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coelho T, Merlini G, Bulawa CE, Fleming JA, Judge DP, Kelly JW, Maurer MS, Plante-Bordeneuve V, Labaudiniere R, Mundayat R, Riley S, Lombardo I, Huertas P. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther. 2016;5(1):1–25. doi: 10.1007/s40120-016-0040-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen JJ, Genereux JC, Wiseman RL. Endoplasmic reticulum quality control and systemic amyloid disease: impacting protein stability from the inside out. IUBMB Life. 2015;67(6):404–413. doi: 10.1002/iub.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koike H, Misu K, Sugiura M, Iijima M, Mori K, Yamamoto M, Hattori N, Mukai E, Ando Y, Ikeda S, Sobue G. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63(1):129–138. doi: 10.1212/01.wnl.0000132966.36437.12. [DOI] [PubMed] [Google Scholar]
- 40.Koike H, Ikeda S, Takahashi M, Kawagashira Y, Iijima M, Misumi Y, Ando Y, Ikeda SI, Katsuno M, Sobue G. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology. 2016;87(21):2220–2229. doi: 10.1212/WNL.0000000000003362. [DOI] [PubMed] [Google Scholar]
- 41.Koike H, Nishi R, Ikeda S, Kawagashira Y, Iijima M, Sakurai T, Shimohata T, Katsuno M, Sobue G. The morphology of amyloid fibrils and their impact on tissue damage in hereditary transthyretin amyloidosis: an ultrastructural study. J Neurol Sci. 2018;394:99–106. doi: 10.1016/j.jns.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 42.Bergstrom J, Gustavsson A, Hellman U, Sletten K, Murphy CL, Weiss DT, Solomon A, Olofsson BO, Westermark P. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol. 2005;206(2):224–232. doi: 10.1002/path.1759. [DOI] [PubMed] [Google Scholar]
- 43.Dardiotis E, Koutsou P, Zamba-Papanicolaou E, Vonta I, Hadjivassiliou M, Hadjigeorgiou G, Cariolou M, Christodoulou K, Kyriakides T. Complement C1Q polymorphisms modulate onset in familial amyloidotic polyneuropathy TTR Val30Met. J Neurol Sci. 2009;284(1–2):158–162. doi: 10.1016/j.jns.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 44.Soares ML, Coelho T, Sousa A, Batalov S, Conceicao I, Sales-Luis ML, Ritchie MD, Williams SM, Nievergelt CM, Schork NJ, Saraiva MJ, Buxbaum JN. Susceptibility and modifier genes in Portuguese transthyretin V30M amyloid polyneuropathy: complexity in a single-gene disease. Hum Mol Genet. 2005;14(4):543–553. doi: 10.1093/hmg/ddi051. [DOI] [PubMed] [Google Scholar]
- 45.Soares ML, Coelho T, Sousa A, Holmgren G, Saraiva MJ, Kastner DL, Buxbaum JN. Haplotypes and DNA sequence variation within and surrounding the transthyretin gene: genotype-phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur J Hum Genet EJHG. 2004;12(3):225–237. doi: 10.1038/sj.ejhg.5201095. [DOI] [PubMed] [Google Scholar]
- 46.Shao S, Hegde RS. Target Selection during Protein Quality Control. Trends Biochem Sci. 2016;41(2):124–137. doi: 10.1016/j.tibs.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 47.Tsuchiya-Suzuki A, Yazaki M, Kametani F, Sekijima Y, Ikeda S. Wild-type transthyretin significantly contributes to the formation of amyloid fibrils in familial amyloid polyneuropathy patients with amyloidogenic transthyretin Val30Met. Hum Pathol. 2011;42(2):236–243. doi: 10.1016/j.humpath.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 48.Koike H, Ando Y, Ueda M, Kawagashira Y, Iijima M, Fujitake J, Hayashi M, Yamamoto M, Mukai E, Nakamura T, Katsuno M, Hattori N, Sobue G. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287(1–2):178–184. doi: 10.1016/j.jns.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 49.Nuvolone M, Merlini G. Emerging therapeutic targets currently under investigation for the treatment of systemic amyloidosis. Expert Opin Ther Targets. 2017;21(12):1095–1110. doi: 10.1080/14728222.2017.1398235. [DOI] [PubMed] [Google Scholar]
- 50.Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293(5539):2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 51.Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci USA. 1996;93(26):15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109(24):9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid Int J Exp Clin Investig. 2006;13(4):236–249. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 54.Sant’Anna R, Gallego P, Robinson LZ, Pereira-Henriques A, Ferreira N, Pinheiro F, Esperante S, Pallares I, Huertas O, Almeida MR, Reixach N, Insa R, Velazquez-Campoy A, Reverter D, Reig N, Ventura S. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat Commun. 2016;7:10787. doi: 10.1038/ncomms10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL, Lin KP, Vita G, Attarian S, Plante-Bordeneuve V, Mezei MM, Campistol JM, Buades J, Brannagan TH, 3rd, Kim BJ, Oh J, Parman Y, Sekijima Y, Hawkins PN, Solomon SD, Polydefkis M, Dyck PJ, Gandhi PJ, Goyal S, Chen J, Strahs AL, Nochur SV, Sweetser MT, Garg PP, Vaishnaw AK, Gollob JA, Suhr OB. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 56.Wood H. FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat Rev Neurol. 2018;14(10):570. doi: 10.1038/s41582-018-0065-0. [DOI] [PubMed] [Google Scholar]
- 57.Benson MD, Ackermann EJ, Monia BP. Treatment of transthyretin cardiomyopathy with a TTR-specific antisense oligonucleotide (IONIS-TTRRx) Amyloid Int J Exp Clin Investig. 2017;24(sup1):134–135. doi: 10.1080/13506129.2017.1280015. [DOI] [PubMed] [Google Scholar]
- 58.Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, Perez J, Chiesa J, Warrington S, Tranter E, Munisamy M, Falzone R, Harrop J, Cehelsky J, Bettencourt BR, Geissler M, Butler JS, Sehgal A, Meyers RE, Chen Q, Borland T, Hutabarat RM, Clausen VA, Alvarez R, Fitzgerald K, Gamba-Vitalo C, Nochur SV, Vaishnaw AK, Sah DW, Gollob JA, Suhr OB. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 59.Ferreira N, Cardoso I, Domingues MR, Vitorino R, Bastos M, Bai G, Saraiva MJ, Almeida MR. Binding of epigallocatechin-3-gallate to transthyretin modulates its amyloidogenicity. FEBS Lett. 2009;583(22):3569–3576. doi: 10.1016/j.febslet.2009.10.062. [DOI] [PubMed] [Google Scholar]
- 60.Ferreira N, Saraiva MJ, Almeida MR. Natural polyphenols as modulators of TTR amyloidogenesis: in vitro and in vivo evidences towards therapy. Amyloid Int J Exp Clin Investig. 2012;19(Suppl 1):39–42. doi: 10.3109/13506129.2012.668502. [DOI] [PubMed] [Google Scholar]
- 61.Ferreira N, Saraiva MJ, Almeida MR. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “in vivo” evidence from FAP mice models. PLoS One. 2012;7(1):e29933. doi: 10.1371/journal.pone.0029933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miyata M, Sato T, Kugimiya M, Sho M, Nakamura T, Ikemizu S, Chirifu M, Mizuguchi M, Nabeshima Y, Suwa Y, Morioka H, Arimori T, Suico MA, Shuto T, Sako Y, Momohara M, Koga T, Morino-Koga S, Yamagata Y, Kai H. The crystal structure of the green tea polyphenol (−)-epigallocatechin gallate-transthyretin complex reveals a novel binding site distinct from the thyroxine binding site. Biochemistry. 2010;49(29):6104–6114. doi: 10.1021/bi1004409. [DOI] [PubMed] [Google Scholar]
- 63.Plate L, Cooley CB, Chen JJ, Paxman RJ, Gallagher CM, Madoux F, Genereux JC, Dobbs W, Garza D, Spicer TP, Scampavia L, Brown SJ, Rosen H, Powers ET, Walter P, Hodder P, Wiseman RL, Kelly JW. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife. 2016 doi: 10.7554/elife.15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 65.Chen JJ, Genereux JC, Qu S, Hulleman JD, Shoulders MD, Wiseman RL. ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem Biol. 2014;21(11):1564–1574. doi: 10.1016/j.chembiol.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alves-Ferreira M, Coelho T, Santos D, Sequeiros J, Alonso I, Sousa A, Lemos C. A trans-acting factor may modify age at onset in familial amyloid polyneuropathy ATTRV30M in Portugal. Mol Neurobiol. 2018;55(5):3676–3683. doi: 10.1007/s12035-017-0593-4. [DOI] [PubMed] [Google Scholar]
- 67.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.