

Abstract
A persistent dogma in neuroscience supported the idea that terminally differentiated neurons permanently withdraw from the cell cycle. However, since the late 1990s, several studies have shown that cell cycle proteins are expressed in post-mitotic neurons under physiological conditions, indicating that the cell cycle machinery is not restricted to proliferating cells. Moreover, many studies have highlighted a clear link between cell cycle-related proteins and neurological disorders, particularly relating to apoptosis-induced neuronal death. Indeed, cell cycle-related proteins can be upregulated or overactivated in post-mitotic neurons in case of acute or degenerative central nervous system disease. Given the considerable lack of effective treatments for age-related neurological disorders, new therapeutic approaches targeting the cell cycle machinery might thus be considered. This review aims at summarizing current knowledge about the role of the cell cycle machinery in post-mitotic neurons in healthy and pathological conditions.
Keywords: Cell cycle, Cell death, Neurodegenerative diseases, CDK, Cyclins, Stroke
Introduction
The cell cycle is an orderly set of events that results in the production of two daughter cells with identical genetic material. Cell cycle proteins tightly regulate precise execution and control of this process. Post-mitotic neuronal cells were long considered not to express these proteins. However, this view changed over the past decades with the identification of cell cycle machinery proteins in post-mitotic neurons of the healthy adult brain [1]. In addition, increasing evidence has highlighted a clear link between cell cycle re-entry and neurological diseases [2, 3]. Indeed, cell cycle proteins are often found to be overexpressed following acute neurological insults or in neurodegenerative disorders. Therefore, understanding the machinery that governs the cell cycle in neurons holds great potential for discovering new therapies for neurological disorders that remain at present mostly untreatable. Complementary to the existing literature on this topic (e.g. [3–11]), in this review, we aim to present an updated, concise but comprehensive overview of current knowledge about non-canonical functions of the core cell cycle machinery both in healthy and diseased post-mitotic neurons. Unless otherwise specified, results of the cited research papers were primarily obtained in mice.
Core cell cycle machinery: a quick overview
In eukaryotic organisms, the cell cycle comprises two major phases: interphase, consisting of Gap 1 (G1), synthesis (S), and Gap 2 (G2) phases during which the cell grows and duplicates its DNA, and the mitotic (M) phase of chromosome partition. The physical separation of all components of the cell to form two new daughter cells begins during mitosis and is called cytokinesis. When cells stop dividing, they enter a resting phase called G0 that corresponds to a (reversible) non-proliferative quiescent or (irreversible) differentiated status. Coordinated progression through all phases of the cell cycle is under the control of specific families of proteins (Fig. 1).
Fig. 1.

Core cell cycle machinery in proliferating cells. CDKs (orange) bind to their respective cyclins to form active complexes that allow the proliferation of the cell partly through pRb/E2F (brown) phosphorylation/release. Tight regulation of this process is possible thanks to CKIs (red) and other cell cycle regulators (blue)
Cyclin-dependent kinases and cyclins
Cyclin-dependent kinases (CDKs) are serine/threonine kinases that phosphorylate many cell cycle-related proteins. They are universal regulators of the cell cycle, present in all known dividing eukaryotic cells. As their name implies, they only become activated upon the binding of regulatory subunits called cyclins. Expression levels of the different cyclins oscillate throughout the cell cycle and favour the transition between certain cycle phases. In humans, around 30 cyclins and 21 Cdks have currently been described, but only a few of them are known to be directly involved in cell division. Core cell cycle CDKs, including CDK1, CDK2, CDK3, CDK4 and CDK6, are phosphorylated and activated by the CDK-activating kinase (CAK) complex, which is composed of CDK7, cyclin H and MNAT1 [12]. The other CDKs and associated cyclins have vital roles, among others, in DNA transcription [13].
Upon proliferative signaling, CDK3 associates with cyclin C and promotes G0 to G1 phase transition by phosphorylating retinoblastoma-associated protein (pRb; see below) [14]. Most strains of mice commonly used in the laboratory carry a mutation that abolishes CDK3 activity [15] that is efficiently replaced by CDK1 or CDK2 [16]. Next, members of the cyclin D family (cyclins D1, D2, and D3) interact with CDK4 and CDK6. Upon binding D-type cyclins, CDK4 and CDK6 are activated and in turn phosphorylate and inactivate pRb, releasing E2F transcription factors (see below) that allow progression through the G1 phase. CDK2 in association with cyclin E and A controls S phase entry, whereas G2 entry relies on CDK1/cyclin A complex formation. CDK1/cyclin B complex activation will induce M phase progression, and inactivation of this complex is necessary for mitotic exit and cytoplasmic division (cytokinesis) [13].
Another essential cyclin-dependent kinase is CDK5, which forms complexes with non-cyclin CDK5 regulatory subunits 1 and 2 (CDK5R1 and CDK5R2), more commonly called p35 and p39. Although complexes of CDK5 and cyclin I can be detected during G1, S and G2 phases [17], CDK5 is thought to mainly inhibit the cell cycle in healthy neurons. Indeed, CDK5/p35(CDK5R1) acts by binding the transcription factor E2F1 and preventing it from interacting with its cofactor DP1 (TFDP1), thereby disrupting the ability of E2F1-DP1 complexes to bind the promoters of cell cycle genes [18].
CDK inhibitors
CDKs are not only regulated by the presence of cyclins, but also by CDK inhibitors (CKIs). There are two main families of inhibitory proteins that negatively regulate the cell cycle machinery: CIP/KIP inhibitors [p21 (CDKN1A), p27 (CDKN1B) and p57 (CDKN1C)] which bind to all CDK/cyclin complexes involved in the cell cycle, and INK4 inhibitors [p15 (CDKN2B), p16 (CDKN2A), p18 (CDKN2C) and p19 (CDKN2D)] that exclusively control CDK4 and CDK6 [19]. Other CKIs include the tyrosine kinases WEE1 and Checkpoint kinase 1 (CHEK1), which are key G2/M phase regulators. They prevent cells from entering the M phase of the cell division cycle in case of unrepaired DNA damage [20]. WEE1 inhibits the CDK1/cyclin B complex [21, 22], while CHEK1 can phosphorylate various substrates that are involved in DNA damage checkpoints, cell cycle arrest and DNA repair [23].
pRb and E2F families
The retinoblastoma-associated protein (pRb) family encompasses pRb itself (also called p105), p107 (RBL1) and p130 (RBL2). In their hypophosphorylated state, these proteins directly bind to and inhibit the E2F family of transcription factors that are necessary for G1/S phase transition [24]. Upon mitogenic signaling, pRb proteins are hyperphosphorylated and inactivated, mostly through CDK4 or 6/cyclin D and CDK2/cyclin E activity. Inactivation of pRb releases E2Fs, which induce the expression of proteins involved in cell cycle progression such as cyclins A and E, facilitating further pRb phosphorylation through a positive feedback loop [25].
Other cell cycle modulators
Polo-like kinase (PLK) proteins are major cell cycle regulators, comprising PLK1–PLK5. These proteins are involved in centriole duplication (PLK2 and 4), DNA replication (PLK3), centrosome separation and maturation, mitotic entry, spindle formation, chromosome segregation and cytokinesis (PLK1) [26].
Cell division cycle 25 (CDC25) phosphatases, encompassing CDC25A, B and C, are involved in G1/S and G2/M phase transitions through activation of CDK2 and CDK1, respectively. They do so by dephosphorylating Thr14 and Tyr15. Phosphorylation of these residues maintains CDKs in an inactive state [27].
The anaphase-promoting complex/cyclosome (APC/C) is a multisubunit E3 ubiquitin ligase [28] that controls the cell cycle in close collaboration with two cofactors: cell division cycle 20 (CDC20) and CDC20 homolog 1 (Cdh1). APC/CCDC20 inhibits M phase mainly via ubiquitination of cyclin B, whereas APC/CCdh1 is involved in M and G1 phases. When coupled to Cdh1, APC/C can ubiquitinate several substrates such as PLK1, which is necessary for G0/G1 transition [29], or CDC25 [27]. APC/CCdh1 contributes to maintaining the cell in the G1 phase, and its inhibition by early mitotic inhibitor 1 irreversibly commits mammalian cells to the cell cycle [30].
Cell cycle machinery in post-mitotic neurons: non-canonical functions
In addition to their canonical roles in driving the cell from its entry into G1 phase to cytokinesis, increasing evidence suggests that cell cycle elements also have other functions. Indeed, following mitotic exit, there is no total degradation of cell cycle proteins. In post-mitotic neurons, numerous cell cycle proteins are present, including cyclins, CDKs and CKIs [31–33]. These proteins are often detected outside the nucleus [1]. Importantly, cytoplasmic CDK/cyclin complexes are functionally active in post-mitotic neurons [31], suggesting that these proteins fulfil non-canonical essential physiological functions (Table 1).
Role of cell cycle proteins in neuronal differentiation and migration
Upon their generation in the ventricular zone, neuronal precursors differentiate and exit the cell cycle before migrating to the cortical plate [34, 35]. Several studies have implicated cell cycle-related proteins in the regulation of both neuronal differentiation and migration (Fig. 2).
Fig. 2.
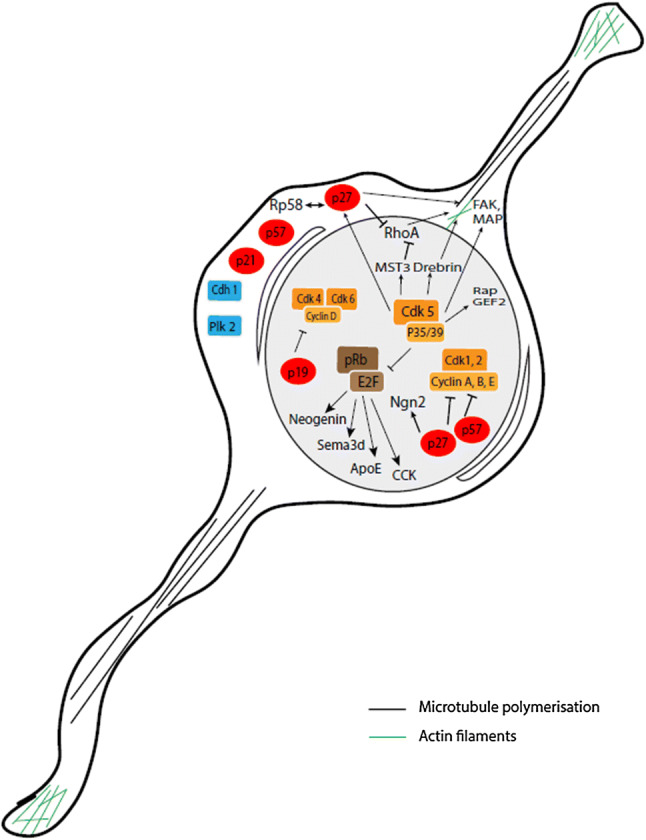
Overview of cell cycle-related proteins involved in differentiation/migration in post-mitotic neurons. Neuronal differentiation and migration are partly controlled by cell cycle-dependent and independent functions of CKIs (red), CDK/cyclin complexes (orange), pRb/E2F family members (brown) and by other cell cycle regulators (blue)
CDK5
CDK5 is an important actor of neuronal progenitor cell cycle arrest. As mentioned above, nuclear CDK5/p35(CDK5R1) complexes act as cell cycle suppressors by sequestering E2Fs, thereby inhibiting their ability to bind to the promoters of cell cycle genes [18]. Given the tight coupling of neuronal differentiation and cell cycle arrest, it is not surprising that CDK5 also has a pro-differentiation function. Indeed, CDK5-deficient embryonic neurons exhibit continued expression of nestin—an intermediate filament protein that is only present in progenitors—while they fail to express microtubule-associated proteins that mark mature neurons [36]. Increased activity of CDK5 during neonatal neuronal differentiation is crucial for neural network formation, and is caused by selective upregulation of its non-cyclin activator p39 (CDK5R2), which binds with CDK5 to become an active kinase [37].
Other studies have elucidated a role for CDK5 in neuronal migration through phosphorylation and regulation of numerous proteins that are critical for the organization of the microtubule network and actin cytoskeleton [38, 39]. During development, migration of cortical neurons is a tightly regulated process that relies mainly on three distinct migration modes: multipolar migration, glia-guided radial migration and somal translocation [35]. After multipolar migration to the upper intermediate zone, neurons switch to a bipolar morphology to initiate glia-guided radial migration. CDK5 plays an important role in radial migration [40, 41] by phosphorylating regulatory proteins such as Rap guanine nucleotide exchange factor 2 (RAPGEF2), mammalian Ste20-like kinase 3 (MST3 or STK24) and drebrin [42–44]. CDK5 has been shown to phosphorylate focal adhesion kinase (FAK or PTK2), which is vital for the organization of the microtubule network that promotes nuclear translocation along radial fibers [45, 46]. Neurons reach their final destination via somal translocation, a migration process in which the entire cell body follows a shortening leading process that remains attached to the pial surface, independently from radial glial cells [47]. CDK5 does not appear to be involved in somal translocation in the early phase of corticogenesis [48]. Finally, CDK5 regulates cytoplasmic microtubule motor protein dynein, which is essential for retrograde transport along neuronal axons and nuclear migration, through phosphorylation of nuclear distribution protein nudE neurodevelopment protein 1 like 1 (NDEL1) [49, 50].
CDK inhibitors
Neuronal differentiation is associated with a general reduction in CDK activity and an accumulation of CKIs such as p27 and p21 (CDKN1A&B) [31, 51]. However, CKIs also act as pro-differentiation factors through cell cycle-independent pathways. For example, p27 also triggers the stabilization of neurogenin-2, a transcription factor that promotes neuronal differentiation [52]. Experiments in mice and rats showed that p57 (CDKN1C) appears to be more efficient in promoting neuronal differentiation than p27, but requires interaction with cyclins and CDKs to exert its developmental functions [53]. Among the INK4 inhibitors, p19 (CDKN2D) plays a role in maintaining neurons in a post-mitotic state [54].
In addition to their roles in differentiation, CIP/KIP proteins regulate cell motility and migration. Both p27 and p57 are important for neuronal migration in the developing cortex [34, 52, 55]. This non-canonical role for CIP/KIP inhibitors relies on their expression outside the cell nucleus [4]. p27 promotes the radial migration of cortical projection neurons from the ventricular zone to the cortical plate, as well as the tangential migration of interneurons, by blocking the RHOA GTPase signaling pathway which is involved in cytoskeletal rearrangements. In addition, p27 promotes cortical interneuron migration through regulation of microtubule polymerization [56]. Interestingly, p27 is a specific substrate of CDK5. CDK5 phosphorylation of p27 at Ser10 is thought to regulate both the stability and cytoplasmic localization of p27 [55, 57], although other results indicate that phosphorylation at Ser10 is not essential for the latter [58]. Finally, the zinc finger transcriptional repressor RP58 (ZBTB18), a well-known coordinator of neuronal radial migration, has been shown to interact with p27 to mediate neuronal progenitor cell cycle exit as well as radial migration through suppression of RHOA signaling [59]. p57 participates in neuronal migration at a later stage than p27, controlling the last phase of proper positioning of neurons within the cortical plate [34].
pRb and E2F families
pRb and E2F transcription factors are two other core cell cycle regulators that are involved in neuronal differentiation. As previously mentioned, increased levels of hypophosphorylated (active) pRb result in E2F sequestration, thereby inhibiting transcription of genes necessary for cell cycle progression. Likewise, overexpression of p27 (CDKN1B) or pRb is sufficient to induce neuronal differentiation [60], although pRb may not be necessary to initiate differentiation of neurons [61].
In addition, pRb and E2F play an unexpected role in neuronal migration. Loss of pRb and/or p107 (RBL1) induces radial and tangential migration defects in cortical projection neurons and interneurons [62, 63]. This migration defect is rescued in the pRb-E2F3 but not in the pRb-E2F1 double knock-out mice. Among the molecules regulated by E2F3, several are involved in neuronal migration, such as neogenin and SEMA3D, two guidance molecules, and apolipoprotein E and cholecystokinin, two members of the reelin pathway [64].
Other
APC/CCdh1 is also involved in differentiation and plays essential roles in post-mitotic neurons [65]. APC/CCdh1 activity is necessary for in vitro differentiation of cortical neurons and for neurogenesis [66]. In addition, it has been found in Neuroscreen-1 cells that PLK2 silencing inhibits nerve growth factor-induced neuronal differentiation [67].
Role of cell cycle proteins in neuronal maturation and neuroplasticity
Upon reaching their final destination in the developing brain, post-mitotic neurons begin to mature. They extend their processes and become polarized with the extension of a single axon and the formation of dendrites. Cell cycle proteins have been shown to play a role in this maturation process, particularly in highly plastic cortical and hippocampal pyramidal neurons (Fig. 3).
Fig. 3.

Overview of cell cycle-related proteins involved in neuronal maturation and neuroplasticity in post-mitotic neurons. CDK/cyclin complexes (orange) are involved in neuronal maturation and neuroplasticity, as well as E2F (brown) and other cell cycle regulators such as APC/C members (blue)
Cyclins and CDKs
CDK1, cyclin B and E, and to a lesser extent, cyclin A and D, are expressed in dendrites [1]. CDK1, 2, and 4, as well as cyclin A, B, D, and E, are also expressed close to the axonal microtubule cytoskeleton [68]. Once again, these non-nuclear CDKs and cyclins form functional complexes and exhibit kinase activity. Moreover, these active complexes physically interact with the microtubule-associated protein tau [68]. Tau is known to localize to the axon to maintain proper microtubule stability, essential to axon integrity and efficient axonal transport [69]. Small interfering RNA (siRNA)-driven downregulation and pharmacological inhibition of CDK1/2 or cyclin B, D or E promote neurite outgrowth in mouse primary neurons [68], confirming that these proteins play a role in neuronal maturation.
A unique role is again reserved for CDK5. CDK5 is vital for axonal outgrowth and neuronal maturation through phosphorylation of numerous proteins, as shown in rat primary neurons [70]. Indeed, research in mice and rats showed that phosphorylation of synapsin III [71] or GRAB (guanine nucleotide exchange factor for Rab8) [72] regulates axonal outgrowth. Other CDK5 substrates that contribute to axonal growth include NGEF (ephexin1), Ras guanine nucleotide releasing factor 2 (RASGRF2), and numerous other upstream regulators and downstream effectors of the Rho GTPases (a family of G proteins that regulate actin dynamics) RHOA, Rac and Cdc42 [73]. Neuronal activity-dependent control of the phosphorylation of the scaffold protein liprinα1 by CDK5 is essential for maturation of excitatory synapses in mice and rats by regulating the localization of DLG4 (postsynaptic density protein 95 or PSD-95), a scaffold protein involved in postsynaptic densities [74]. Moreover, DLG4 (PSD-95) has been identified as a direct CDK5 substrate in mice and rats [75].
CDK5 also has a critical role in regulating synaptic plasticity [76]. It was shown in rat that CDK5 regulates synaptic vesicle exocytosis through phosphorylation of Munc-18 (thereby modulating Munc-18/syntaxin 1A interaction, resulting in increased neurotransmitter release) or voltage-dependent calcium channels (thereby modulating the interaction between SNARE proteins and voltage-dependent calcium channels, resulting in decreased neurotransmitter secretion) [77, 78]. While it has been reported in rats that CDK5 is vital for endocytosis through phosphorylation of dynamin I [79], other results rather suggest that CDK5 negatively regulates synaptic vesicle endocytosis [80, 81]. In addition, in rat hippocampal neurons CDK5 acts as a priming kinase for the phospho-dependent binding of PLK2 to its postsynaptic scaffolding substrate [82]. CDK5 has also been identified in mice and rats as a mediator of neuregulin signaling, required for the transcription of neurotransmitter receptors such as the acetylcholine receptor [83]. Efficiency and plasticity of synaptic transmission also rely on the regulation of dendritic spine morphology. Significant modulation of spine morphogenesis depends on actin dynamics and therefore also on Rho GTPases, as described above [73]. Finally, dopaminergic neurotransmission is also modulated by CDK5. CDK5 can either depress the dopamine system through phosphorylation of dopamine- and cAMP-regulated neuronal phosphoprotein PPP1R1B (DARPP-32) [84], or increase activity and stability of the tyrosine hydroxylase (TH) enzyme, which is responsible for dopamine synthesis [85]. However, the precise effects of CDK5 on synaptic plasticity and learning remain unclear, as both inhibitory [86] and stimulatory effects [87] have been reported.
Cyclin E plays a role in the formation of synapses in post-mitotic neurons through inhibition of CDK5 in a cell cycle-independent manner [33, 88]. Cyclin E regulates synaptic plasticity by forming cytoplasmic kinase-inactive complexes with CDK5 and sequestering it from its activators p35 and p39 (CDK5R1&2), thereby inhibiting the phosphorylation of CDK5 synaptic substrates. Ablation of cyclin E in post-mitotic neurons leads to a decreased number of synapses and dendritic spines in vitro [88].
Cyclin Y, originally identified as interacting with CDK14 and CDK16, has been shown in rats to inhibit synaptic plasticity of long-term potentiation, the most widely studied physiological substrate of memory and learning, by preventing plasticity-induced delivery of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor to synapses [89].
Recent evidence suggests that CDK7 is critical for the transcription of immediate-early genes that is triggered by neuronal activity in certain behavioural tasks, through phosphorylation of RNA polymerase II. In addition, CDK7 seems to be essential for long-term potentiation and for the gene transcription required for long-term memory formation in living mice [90].
CDK inhibitors
Not much is known about the role of CKIs in neuronal maturation, although p27 (CDKN1B) controls axonal transport of vesicles and organelles, which is an essential process in neuronal maturation and synapse formation, by promoting acetylation of microtubules [91].
pRb and E2F families
E2F1 has been detected both in neurites and close to synapses in the adult brain. E2F1 knockout mice show significant age-dependent olfactory and memory deficits [92], indicating a key role for this protein in synapse establishment and/or remodeling that remains to be elucidated.
Other
APC/CCdh1 and APC/CCDC20 complexes regulate neuronal maturation through degradation of specific proteins [4]. Nuclear APC/CCdh1 was found to control axonal growth and patterning in rats [93] through degradation of two axonal growth-promoting factors: ID2 and SKIL (SnoN) [94, 95]. Furthermore, APC/CCdh1 can regulate the size and activity of drosophila neuromuscular synapses [96].
It has been shown in rats that centrosomal APC/CCDC20 promotes presynaptic differentiation (an essential process for synapse formation) and dendrite morphogenesis by triggering the degradation of both NEUROD2, a neurogenic differentiation factor, and DNA-binding protein inhibitor ID1, a protein that inhibits dendrite growth [97, 98]. In line with this, loss of APC/CCDC20 impairs dendritic growth and branching in cerebellar granule neurons in vitro and in vivo [98], but has no effect on axonal growth.
Cell cycle proteins and neuronal DNA repair
Once they have undergone differentiation, migrated to their final position, and have fully matured and functionally integrated into the brain, neurons are particularly prone to DNA damage. Neurons are long-lived cells and consume a high level of oxygen. As a consequence, they are constantly exposed to reactive oxygen species (ROS), which are generated as a regular part of cellular metabolism and can damage many cell components, including DNA [99]. An efficient system for repair of DNA lesions is thus of particular importance for survival and normal function of neurons. However, repair in post-mitotic cells is not as effective as in dividing cells [100]. Furthermore, neuronal tissue displays low levels of antioxidant enzymes [101]. DNA lesions in neurons are, therefore, more likely to accumulate.
Among the most lethal DNA lesions are the double-strand breaks (DSBs). To fix DSBs, post-mitotic neurons rely on the error-prone non-homologous end joining (NHEJ) DNA repair system. Homologous recombination (HR), the other primary mechanism to repair DSBs, requires the presence of a sister chromatid, which is formed by DNA replication and is absent in neurons that have exited the cell cycle [102]. However, recent evidence showed that a replication-independent RNA-templated HR repair mechanism may exist in non-dividing post-mitotic rat neurons. This alternative repair system depends on RAD52, an HR protein involved in the repair of DSBs at active transcription sites during G0/G1 phase [103].
NHEJ repair in neurons requires cell cycle proteins (Fig. 4). Indeed, in rats, where CDK3 is present in contrast to many common mouse strains, it was found that exposure of cortical neurons to minor DNA damage is accompanied by activation of the CDK3/cyclin C complex and a subsequent increase in pRb phosphorylation and E2F1 expression. Concomitantly with this cell cycle activation, Ku70/80 (XRCC6/XRCC5), a heterodimeric protein complex that is a key mediator of NHEJ, is found in damaged neurons [104]. Increased pRb phosphorylation and a NHEJ response are also induced by the activation of ATM/p53 (TP53)-mediated CDK4 or 6/cyclin D complexes. ATM is a kinase that is activated by DSBs and phosphorylates the tumor suppressor protein p53, which in turn can induce DNA repair, as well as cell cycle arrest and apoptosis. It has been shown in rat cerebellar granule neurons that CDK5 directly phosphorylates and thereby activates ATM in response to DNA damage [105]. DNA-damaging ROS or X-ray irradiation induce expression of Cyclin D1, phosphorylation of pRb, but also expression of more global cell cycle regulators such as marker of proliferation Ki-67 and MCM2 (a vital component of the pre-replication complex that is formed during initiation of DNA replication) in mouse and rat neurons [106, 107]. Research with mice and rats indicates that CDK4/cyclin D complex activation is likely also mediated through DNA damage-induced stimulation of SERTAD1, a direct CDK4 activator [108]. Blocking CDK4/6 or cyclin C, thereby inhibiting cell cycle entry, increases DNA damage upon ROS exposure. On the contrary, forced entry in G1 phase through siRNA-mediated inhibition of p21 (CDKN1A) induces NHEJ response and DNA repair [104, 106]. Importantly, DNA damage that can be repaired never leads to S phase entry. Instead, neurons remain in the G1 phase.
Fig. 4.

Overview of cell cycle-related proteins involved in DNA repair and neuronal death in post-mitotic neurons. DNA repair in post-mitotic neurons is partly controlled by CDK/cyclin complexes (orange), pRb/E2F family members (brown) and by other proteins. Successful DNA repair does not result in cell cycle progression. An overload of irreparable DNA damage caused by pathological processes, however, can cause neurons to progress through the cell cycle, resulting in neuronal death
Core cell cycle machinery and neuronal death
The intracellular endpoint of many neurotoxic stimuli is the production of ROS and consequent oxidative stress. A massive ROS increase will induce an overload of DNA damage, a high amount of irrevocable DSBs, and a saturation of the repair system. This process will not only lead to cell cycle re-entry, but will induce progression of the neuron beyond G1 phase, through the different steps of the cell cycle, finally resulting in neuronal death (Fig. 4). Several studies have shown that inhibition of some cell cycle regulators could have potential neuroprotective effects [109, 110].
G1 cell cycle re-entry
Cell cycle re-entry mechanisms are identical for physiological and pathological ROS increase: ROS-induced DNA damage leads to ATM and SERTAD1 activation, resulting in activation of CDK4/cyclin D complexes [108, 111]. Silencing or pharmacological inhibition of ATM, cyclin D and/or CDK4 prevents DNA damage-induced apoptosis of mouse and rat neurons [111–114]. In addition, overexpression of a mutated form of pRb at CDK4/6 phosphorylation sites, which inhibits cell cycle entry, partially protects cortical mice and rat and sympathetic rat neurons from apoptosis following DNA damage [115]. Both ATM and CDK4/cyclin D activation induce E2F release and accumulation [116], which is likely responsible for cell cycle-induced neuronal death. Indeed, it was shown in different human immortal cell lines that E2F can bind the promoters of specific target genes, including numerous pro-apoptotic Bcl-2 family members such as BAK1, BID or BAD [117, 118]. Following UV-induced DNA damage, E2F binds to p53 (TP53). This interaction has been shown to stimulate the apoptotic function of p53 [119]. Experiments in rats and mice show that E2F inhibition may, therefore, protect neurons from apoptosis in several apoptotic paradigms [120–122].
In pathological conditions, neurons that re-enter the cell cycle can progress to the G1/S transition. E2F accumulation promotes a transition from G1 to S and G2 phases. S and G2 phase elements, such as cyclin E, CDK2 or cyclin B1, are found in suffering rat and mouse neurons [123–126]. E2F is also able to activate CDK1 in rat cerebellar granule neurons following activity-deprivation [121]. The activated CDK1/cyclin B complex induces death in rat cortical neurons by inhibitory phosphorylation of the BCL2L1 (Bcl-xL) anti-apoptotic protein [127]. In addition, the CDK1/cyclin B complex phosphorylates the transcription factor FOXO1, which disrupts cytoplasmic sequestration of FOXO1 and leads to FOXO1 accumulation in the nucleus. This leads to increased FOXO1-dependent transcription, which induces expression of the apoptotic activator BCL2L11 (BIM), thereby resulting in death of post-mitotic neurons [128].
If neurons did not die during G1 phase or after S phase, they do so when they pass the G2/M checkpoint [32]. Thus, although DNA damage-induced cell cycle re-entry is the first step towards DNA repair via recruitment of the NHEJ system, if the damage is too extensive and the system is overloaded, overactivation of E2F will induce progression through the cell cycle and activation of an irreversible apoptotic cascade.
CDK5
In mice and rats it was found that, following apoptotic stimuli, p35 (CDK5R1) is cleaved into p25 by calpains, preventing CDK5 from associating with p35 and resulting in the formation of CDK5/p25 hyperactive complexes [129]. CDK5 hyperactivation is linked to neuronal apoptosis through both cell cycle-independent and -dependent pathways. On the one hand, it was shown in different immortal cell lines and in mice that CDK5 can phosphorylate and directly activate several pro-apoptotic proteins, such as p53 (TP53) and chloride intracellular channel 4 [130, 131]. On the other hand, CDK5/p25 is also a key element in cell cycle re-entry, although normal CDK5/p35 complexes are thought to have an inhibitory effect on the cell cycle in healthy neurons. Indeed, CDK5-mediated phosphorylation of Cdh1 in rat cortical neurons, one of the main cofactors of APC/C, leads to sequestration of Cdh1 in the cytoplasm, and consequently to APC/C inactivation. The phosphorylation of Cdh1 also conducts to p27 (CDKN1B) depletion, S-phase entry and neuronal apoptosis [132]. Furthermore, results obtained in rat cerebellar granule and cortical neurons and SH-SY5Y cells show that CDK5/p25 can induce pRb phosphorylation, CDC25A, B and C expression and ATM phosphorylation [105, 133, 134], altogether leading to cell cycle re-entry.
Neuronal senescence
In addition to DNA repair and cell death, there exists an alternative fate for post-mitotic neurons that re-enter the cell cycle which is worth mentioning. In proliferating cells, the DNA damage response may result in permanent cell cycle arrest that is often accompanied by the acquisition of an immunogenic phenotype, a phenomenon called cellular senescence. Interestingly, it has been shown that non-dividing mature neurons may progress into a senescence-like state in response to a DNA damage response induced by DSBs or telomere dysfunction [135]. Just like senescent dividing cells, these senescence-like neurons produce and secrete ROS and pro-inflammatory cytokines, potentially contributing to age-related cognitive decline. The CIP/KIP cell cycle inhibitor p21 (CDKN1A) plays an essential role in driving post-mitotic neurons from a DNA damage response to a senescence-like phenotype, just like it does in proliferation-competent cells [135]. In addition to p21, the INK4 inhibitor p16 (CDKN2A), which inhibits CDK4 and CDK6, is another key marker of cellular senescence. Aberrant expression of p16 has been found in neurotoxicity-induced neuronal senescence in human SH-SY5Y cells and rat PC12 cells [136] and in several neurodegenerative diseases that are associated with cellular senescence [137].
Link between cell cycle machinery, neuronal death and central nervous system diseases
The endpoint of any neurological disease is the loss of a particular population of neurons. Protein aggregates or excitotoxicity are often found in neurodegenerative disorders and acute neurological insults, respectively, and induce accumulation of ROS and DSBs. Finally, cell death occurs within minutes (acute neurological insults) to many years (neurodegenerative disorders). Neuronal death is induced in part through cell cycle activation, as outlined above. We will discuss next some of the most common neurological disorders for which there is a clear link with cell cycle machinery.
Neurodegenerative diseases
Alzheimer’s disease
Alzheimer’s disease (AD) is characterized by a massive loss of neurons and atrophy in selective brain areas that are associated with memory and cognitive functions. Brains of AD patients show extracellular amyloid plaques and intracellular neurofibrillary tangles that mainly consist of amyloid β (Aβ) deposits and hyperphosphorylated tau, respectively. Aβ peptides are derived from the amyloid precursor protein (APP) following cleavage by gamma-secretase. Hyperphosphorylated tau protein is no longer connected to microtubules, leading to synaptic loss and neuronal death.
There is evidence for a role of aberrant neuronal cell cycle re-entry in AD. Neurons of human and mouse AD brains express cell cycle-specific antigens, such as marker of proliferation Ki-67, proliferating cell nuclear antigen (PCNA) [138–141] or Phospho-Histone H3, an M phase marker, aberrantly localized in the cytoplasm of human AD hippocampal neurons [142]. Neurons from human or murine AD brains, also show increased expression of cyclins and CDKs [123, 140, 143], in particular CDK1/Cyclin B complexes [144, 145], as well as increased expression of phospho-pRb and E2F [146]. CDK activation may be caused partly by downregulation of p21 (CDKN1A) by denticleless protein homolog (DTL, also known as CDT2), a protein which is upregulated in AD. Activated CDKs then phosphorylate tau and APP, resulting in tau hyperphosphorylation and Aβ toxicity, two hallmarks of AD [147]. Research using several mouse models of AD indicates that ectopic cell cycle events are present well before the first Aβ deposits appear [140].
Abnormal cell cycle re-entry of AD neurons leads to an altered temporal order of chromosome segregation, producing a premature division of centromeres. Therefore, duplicated chromosomes [148, 149], bi-nucleation [150] or premature centromere division [151] have consistently been observed in mouse and human AD brains. Experiments in mouse cortical neurons show that hyperploid neurons may survive for some time and contribute to synaptic dysfunction in AD [152].
Cellular senescence also plays a role in AD. Aberrant expression of p16 (CDKN2A), INK4 inhibitor and senescence marker, has been found in neurons in the brains of human patients with Alzheimer’s disease [153, 154].
AD and CDK5
There is a dysregulation of CDK5 activity in the brains of human AD patients [155]. Aβ aggregates likely induce the formation of CDK5/p25 complexes. The ensuing hyperactivation of CDK5 contributes to AD pathogenesis by affecting various intracellular pathways. For example, aberrant CDK5/p25 signaling participates in pathological cell cycle re-entry (see III.3). CDK5 is also involved in AD pathogenesis through cell cycle-independent roles. Indeed, CDK5 has been shown to aberrantly phosphorylate tau in rat and human neurons [156–158] and APP [159] in SH-SY5Y cells, contributing to protein aggregate formation. Moreover, a positive feedback loop has been demonstrated in rats and mice, where Aβ contributes to CDK5 abnormality by increasing intracellular calcium concentrations, which activates calpain-mediated cleavage of p35 (CDK5R1), leading to the formation of more hyperactive CDK5/p25 complexes [129, 160–162]. Recently it was also found in rats and mice that p27 (CDKN1B) promotes Aβ-induced neuronal apoptosis by promoting interaction between CDK5 and Cyclin D, thereby preventing CDK5 from associating with p35 and leading to cell cycle re-entry through aberrant activation of the MAPK/ERK pathway which is normally negatively regulated by CDK5/p35 [163]. Lastly, CDK5 may also contribute to neuronal apoptosis in AD by phosphorylation of p53 (TP53) and FOXO3 transcription factor, which are able to induce apoptosis [164, 165].
Pharmacological inhibition of CDK5 in rat hippocampal and cortical neurons or in transgenic AD mice thus protects against AD by blocking cell cycle re-entry but also by preventing tau hyperphosphorylation, Aβ production and neurofibrillary tangle accumulation [166–168]. Unfortunately, even though efforts towards the development of novel chemical products to specifically inhibit CDK5 function have been pursued, there is currently no specific CDK5 inhibitor. CDK5 inhibitors such as roscovitine or dinaciclib also inhibit other CDKs, while calpain inhibitors, such as MDL28170, exert only indirect CDK5 inhibition. A possibility might be to target CDK5/p25 specifically, without inhibiting CDK5/p35(CDK5R1), for example with CDK5 inhibitory peptide (CIP), p5 or p10, cleaved products of p35 that have been shown to efficiently inhibit CDK5/p25 activity in rat cortical neurons without influencing CDK5/p35 [169]. It has been shown that overexpression of CIP reduces tau hyperphosphorylation, amyloid pathology and neuroinflammation in HEK293 cells, rat cortical neurons and in transgenic mice, and that CIP may thus present a valuable tool to combat neurodegeneration [170–173]. However, it seems that CIP is too big to pass the blood–brain barrier, so that smaller peptides such as p5 may constitute more promising candidates for therapeutic interventions [169].
Parkinson’s disease
Parkinson’s disease (PD) is characterized by a progressive and selective loss of dopaminergic neurons in the substantia nigra and by the presence of Lewy bodies in surviving neurons, which are protein aggregates that are mostly composed of α-synuclein. Evidence suggests that numerous cell cycle proteins are linked to the specific loss of dopaminergic neurons. PLK2 may play a vital role in the phosphorylation of α-synuclein at Ser129, which is a hallmark of α-synuclein deposited in Lewy bodies [174, 175], although the precise implications of Ser129 phosphorylation in PD remains to be elucidated. In vivo studies have demonstrated the presence of cyclins as well as CDKs, phospho-pRb, and E2F in neurons of human PD patients or of rodents treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA), two neurotoxic drugs that selectively affect dopaminergic neurons, mimicking the pathophysiology of PD [176–180]. Moreover, knocking out E2F1 or pharmacologically inhibiting the CDK family using Flavopiridol protect neurons in mice treated with MPTP [176, 178]. On the contrary, p27 and p21 (CDKN1A&B) decrease the expression of α-synuclein, mediated by complexes of p130 (RBL2) and E2F4, indicating that this regulatory mechanism might be disrupted in PD [181]. Neurons from PD brains also display chromosomal abnormalities, as found in AD neurons. For instance, the presence of polyploid dopaminergic neurons in the substantia nigra has been demonstrated in PD patients, which is again indicative of cell cycle re-entry [178].
PD and CDK5
Hyperactive CDK5/p25 complexes are found in neurons of MPTP-treated rodents, but also in PD human brains tissue, where they are observed close to Lewy bodies [182–184]. Elevated levels of CDK5 in rodent PD models induce inhibitory phosphorylation and degradation of several substrates. For instance, experiments on rat neurons, human PD tissue and mice models showed that CDK5 can induce degradation of PEBP1 (Raf kinase inhibitory protein or RKIP), leading to overactivation of the MAPK/ERK signaling pathway that is involved in cell division, and S-phase entry [185]. CDK5 also degrades PRDX2, an anti-oxidant enzyme [186], and inactivates the transcription factor myocyte enhancer factor 2 (MEF2). The latter process may play a significant role in dopaminergic neuronal loss in vivo, since preventing CDK5-mediated phosphorylation of MEF2 is neuroprotective in the MPTP mouse model [187]. Consistent with the central role of CDK5 in PD, truncated peptides of p35 (CDK5R1) that specifically inhibit the CDK5/p25 complex rescue dopaminergic neurons and alleviate PD symptoms following MPTP treatment in mice [188–190].
Acute neurological insults
In addition to neurodegenerative diseases, cell cycle reactivation has also been linked with acute neurological insults. Excitotoxicity and ROS formation caused by ischemia or traumatic injury induce the expression of cell cycle proteins, once again linking neurological disease and neuronal death to the cell cycle.
Ischemia
Increasing evidence suggests a strong relationship between the cell cycle machinery and stroke-induced neuronal death [191, 192]. Following ischemia in humans or rodents, neurons undergo expression of cyclins and CDKs [109, 124, 191, 193, 194]. CDC25A has recently been shown in mice and rats to be a key mediator of ischemia-induced neuronal death through CDK4 activation [195]. In addition, cell cycle inhibitors such as p16 and p27 (CDKN2A and CDKN1B) have been found to be downregulated after ischemia [124, 194]. Interestingly, levels of p21 (CDKN1A) seem to be increased in surviving neurons that surround the ischemic area in rats [196]. Genetic or pharmacologic inhibition of cell cycle proteins has been shown to protect mouse and rat neurons from ischemic neuronal death [109, 110, 197].
Ischemia and CDK5
CDK5 has been directly linked to ischemic stroke in mice and rats [198, 199]. Ischemia-induced excitotoxicity activates calpain, leading to CDK5 hyperactivation and subsequent phosphorylation of NMDA receptors and amplification of intracellular calcium influx. These events result in mitochondrial dysfunction, membrane disruption, proteolysis and finally neuronal death [200, 201]. Moreover, CDK5 inhibition has been shown to be neuroprotective after ischemia in mice and rats [199, 202]. CDK5/p25 specific inhibitors should again be favored over CDK5 inhibitors such as roscovitine or calpain inhibitors since the CDK5/p35(CDK5R1) complex likely plays beneficial roles in neuronal protection and has essential physiological functions in post-mitotic neurons [203]. Moreover, following a stroke, the CDK5/p35 complex is overexpressed in endothelial cells where it may inhibit angiogenesis necessary for reperfusion [204, 205]. As mentioned above, p5, a specific inhibitor of CDK5/p25 is considered to be a promising neuroprotective agent for stroke [169].
Traumatic brain injury and spinal cord injury
Increasing evidence suggests that neuronal death following traumatic brain injury (TBI) and spinal cord injury (SCI) is also preceded by cell cycle re-entry [206, 207]. In rat and rabbit models of SCI and TBI, upregulation of cyclin D1, phospho-pRb, E2F, proliferating cell nuclear antigen, CDK4 and CDK1 has been observed [122, 208–212]. E2F may play a more significant role in neuronal cell death in SCI than in TBI [213]. Furthermore, inhibition of cell cycle machinery provides neuroprotection both in SCI and TBI [208, 210, 212].
Other neurological disorders
Consistent with the idea that aberrant cell cycle re-entry is likely to be a common feature associated with neuronal apoptosis in numerous diseases, several studies have highlighted an association between cell cycle machinery and other, less frequent but incapacitating, neurodegenerative diseases including amyotrophic lateral sclerosis, Pick’s disease, intractable temporal lobe epilepsy, progressive supranuclear palsy, corticobasal dementia and frontotemporal dementia [214–218]. In addition, increased levels of marker of proliferation Ki-67, but also CDKs are found in the brains of individuals with Down syndrome [138, 218]. Moreover, since obesity may be considered as a neurological disorder, it has been shown that inactivation or genetic ablation of pRb in pro-opiomelanocortin neurons leads to obesity [9].
Conclusions
Differentiated neurons appear to irreversibly exit the cell cycle, perhaps because cell division of neurons would result in cytoskeletal and synaptic disruptions. However, as shown in this review, cell cycle proteins still play critical roles in physiological processes in post-mitotic neurons. Evidence has pointed out that cell cycle factors are constitutively expressed in post-mitotic neuronal cells. An evolutionary process may have recycled cell cycle proteins to fulfill non-canonical functions such as neuronal differentiation, migration, maturation and DNA repair in post-mitotic neurons.
Importantly, in addition to their non-canonical physiological functions, research has strongly linked the cell cycle machinery to neurological disease and neuronal death (Table 1). Indeed, excitotoxicity and protein aggregation, resulting from the pathophysiology of acute neurological insults and neurodegenerative diseases, induce high production of ROS. As neuronal cells are major oxygen consumers, oxidative stress builds up, inducing high amounts of DNA damage. Because neurons do not own very efficient DNA repair machinery, DNA damage accumulates, and the DNA damage response will switch from DNA repair to apoptotic pathway induction. Several studies have highlighted the involvement of cell cycle machinery in DNA damage and apoptosis (Fig. 5). Interestingly, the role of the cell cycle machinery may be different depending on the cerebral region. DNA repair of cortical neurons may be less effective than DNA repair of cerebellar and hippocampal neurons [102] and may explain some specific cortical issues found for example in stroke.
Table 1.
Cell cycle proteins involved in physiological and pathological conditions in neurons

D differentiation, Mi migration, NP neuroplasticity, M maturation, Dr DNA repair, AD Alzheimer’s disease, PD Parkinson’s disease, Isch Ischemia, TB-SCI Traumatic brain or spinal cord injury
Fig. 5.

Link between neurological disorders, DNA damage and the cell cycle. Neurological disease may lead to excitotoxicity and protein aggregation, which both induce production of reactive oxygen species (ROS), in turn leading to double-strand DNA breaks (DSBs) that trigger cell cycle re-entry and activation of the DNA repair system. If the DNA damage cannot be repaired, however, overactivation of E2F will lead to cell cycle progression and apoptosis
However, a clear lack of knowledge regarding the precise molecular mechanisms that connect the cell cycle with neuronal death prevents the effective translation from research to clinical trials. Therefore, it is of relevance to try to understand precisely how the balance between DNA repair and apoptosis is regulated. Furthermore, it is important to avoid side effects of cell cycle inhibition. Since the cell cycle machinery is closely linked with DNA repair, depending on the amount of DNA damage, inhibition of cell cycle machinery may have deleterious effects. Moreover, although cell cycle arrest may protect neurons from apoptotic death, neurons that survive with continued activation of the DNA damage response may progress to a senescent-like state with equally detrimental consequences [219]. Further research is needed to elucidate potential therapeutic strategies targeting the cell cycle machinery to reach neuroprotection in a wealth of neurological disorders.
Acknowledgements
This work was supported by grants from the Belgian National Funds for Scientific Research (FRS-FNRS, Belgium), the Fondation Léon Fredericq, the Fondation Médicale Reine Elisabeth, the Fonds spéciaux (ULiège, Belgium), and L’Oréal-UNESCO For Women in Science.
Abbreviations
- G1 phase
Gap 1 phase
- S phase
Synthesis phase
- G2 phase
Gap 2 phase
- M phase
Mitotic phase
- CDK
Cyclin-dependent kinase
- CAK
CDK-activating kinase
- pRb
Retinoblastoma protein
- CDK5R1
Cyclin-dependent kinase 5 regulatory subunit 1
- CKI
CDK inhibitor
- CHEK1
Checkpoint kinase 1
- PLK
Polo-like kinase
- CDC25
Cell division cycle 25
- APC/C
Anaphase-promoting complex/cyclosome
- CDC20
Cell division cycle 20
- Cdh1
CDC20 homolog 1
- RAPGEF2
Rap guanine nucleotide exchange factor 2
- MST3
Mammalian Ste20-like kinase 3
- FAK
Focal adhesion kinase
- NDEL1
NudE neurodevelopment protein 1 like 1
- siRNA
Small interfering RNA
- RASGRF2
Ras guanine nucleotide releasing factor 2
- PSD-95
Postsynaptic density protein 95
- TH
Tyrosine hydroxylase
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ROS
Reactive oxygen species
- DSB
Double-strand break
- NHEJ
Non-homologous end joining
- HR
Homologous recombination
- AD
Alzheimer’s disease
- Aβ
Amyloid β
- APP
Amyloid precursor protein
- PCNA
Proliferating cell nuclear antigen
- DTL
Denticleless protein homolog
- CIP
CDK5 inhibitory peptide
- PD
Parkinson’s disease
- MPTP
1-Methyl-4-phenyl-1,2,3,6-Tetrahydropyridine
- 6-OHDA
6-Hydroxydopamine
- RKIP
Raf kinase inhibitory protein
- MEF2
Myocyte enhancer factor 2
- TBI
Traumatic brain injury
- SCI
Spinal cord injury
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Quentin Marlier and Tine D’aes contributed equally to this work.
References
- 1.Schmetsdorf S, Gärtner U, Arendt T. Constitutive expression of functionally active cyclin-dependent kinases and their binding partners suggests noncanonical functions of cell cycle regulators in differentiated neurons. Cereb Cortex. 2007;17:1821–1829. doi: 10.1093/cercor/bhl091. [DOI] [PubMed] [Google Scholar]
- 2.Herrup K, Yang Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci. 2007;8:368–378. doi: 10.1038/nrn2124. [DOI] [PubMed] [Google Scholar]
- 3.Yang Y, Herrup K. Cell division in the CNS: protective response or lethal event in post-mitotic neurons? Biochim Biophys Acta Mol Basis Dis. 2007;1772:457–466. doi: 10.1016/j.bbadis.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frank CL, Tsai LH. alternative functions of core cell cycle regulators in neuronal migration, neuronal maturation, and synaptic plasticity. Neuron. 2009;62:312–326. doi: 10.1016/j.neuron.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su SC, Tsai L-H. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- 6.Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawauchi T, Shikanai M, Kosodo Y. Extra-cell cycle regulatory functions of cyclin-dependent kinases (CDK) and CDK inhibitor proteins contribute to brain development and neurological disorders. Genes Cells Devoted Mol Cell Mech. 2013;18:176–194. doi: 10.1111/gtc.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawauchi T, Nabeshima Y. Growth arrest triggers extra-cell cycle regulatory function in neurons: possible involvement of p27kip1 in membrane trafficking as well as cytoskeletal regulation. Front Cell Dev Biol. 2019 doi: 10.3389/fcell.2019.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iqbal N, Li Z, Chua SC. Neuronal cell cycle events link caloric intake to obesity. Trends Endocrinol Metab. 2020;31:46–52. doi: 10.1016/j.tem.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godin JD, Nguyen L. Novel functions of core cell cycle regulators in neuronal migration. In: Nguyen L, Hippenmeyer S, editors. Cellular and molecular control of neuronal migration. Dordrecht: Springer; 2014. pp. 59–74. [DOI] [PubMed] [Google Scholar]
- 11.Herrup K. Post-mitotic role of the cell cycle machinery. Curr Opin Cell Biol. 2013;25:711–716. doi: 10.1016/j.ceb.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lolli G, Johnson LN. CAK-cyclin-dependent activating kinase: a key kinase in cell cycle control and a target for drugs? Cell Cycle Georget Tex. 2005;4:572–577. [PubMed] [Google Scholar]
- 13.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Ren S, Rollins BJ. Cyclin C/Cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117:239–251. doi: 10.1016/S0092-8674(04)00300-9. [DOI] [PubMed] [Google Scholar]
- 15.Ye X, Zhu C, Harper JW. A premature-termination mutation in the Mus musculus cyclin-dependent kinase 3 gene. Proc Natl Acad Sci. 2001;98:1682–1686. doi: 10.1073/pnas.041596198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li N, Fassl A, Chick J, et al. Cyclin C is a haploinsufficient tumour suppressor. Nat Cell Biol. 2014;16:1080–1091. doi: 10.1038/ncb3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagano T, Hashimoto T, Nakashima A, et al. Cyclin I is involved in the regulation of cell cycle progression. Cell Cycle Georget Tex. 2013;12:2617–2624. doi: 10.4161/cc.25623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Li H, Yabut O, et al. Cdk5 suppresses the neuronal cell cycle by disrupting the E2F1-DP1 complex. J Neurosci. 2010 doi: 10.1523/JNEUROSCI.5628-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 20.Elledge SJ, Zhou B-BS. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 21.Parker LL, Atherton-Fessler S, Piwnica-Worms H. p107weel is a dual-specificity kinase that phosphorylates p34cdc2 on tyrosine 15 (cell cycle/baculovirus expression) Cell Biol. 1992;89:2917–2921. doi: 10.1073/pnas.89.7.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:20. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci CMLS. 2013;70:4009–4021. doi: 10.1007/s00018-013-1307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 25.Stevens C, La Thangue NB. E2F and cell cycle control: a double-edged sword. Arch Biochem Biophys. 2003;412:157–169. doi: 10.1016/S0003-9861(03)00054-7. [DOI] [PubMed] [Google Scholar]
- 26.Lee S-Y, Jang C, Lee K-A. Polo-like kinases (plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev Reprod. 2014;18:65–71. doi: 10.12717/DR.2014.18.1.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lub S, Maes A, Maes K, et al. Inhibiting the anaphase promoting complex/cyclosome induces a metaphase arrest and cell death in multiple myeloma cells. Oncotarget. 2016;7:4062–4076. doi: 10.18632/oncotarget.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skaar JR, Pagano M. Cdh1: a master G0/G1 regulator. Nat Cell Biol. 2008;10:755–757. doi: 10.1038/ncb0708-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cappell SD, Mark KG, Garbett D, et al. EMI1 switches from being a substrate to an inhibitor of APC/CCDH1 to start the cell cycle. Nature. 2018;558:313–317. doi: 10.1038/s41586-018-0199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sumrejkanchanakij P, Tamamori-Adachi M, Matsunaga Y, et al. Role of cyclin D1 cytoplasmic sequestration in the survival of postmitotic neurons. Oncogene. 2003;22:8723–8730. doi: 10.1038/sj.onc.1206870. [DOI] [PubMed] [Google Scholar]
- 32.Frade JM, Ovejero-Benito MC. Neuronal cell cycle: the neuron itself and its circumstances. Cell Cycle. 2015;14:712–720. doi: 10.1080/15384101.2015.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odajima J, Wills ZP, Ndassa YM, et al. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell. 2011;21:655–668. doi: 10.1016/j.devcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itoh Y, Masuyama N, Nakayama K, et al. The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J Biol Chem. 2007;282:390–396. doi: 10.1074/jbc.M609944200. [DOI] [PubMed] [Google Scholar]
- 35.Marín O, Valiente M, Ge X, Tsai L-H. Guiding neuronal cell migrations. Cold Spring Harb Perspect Biol. 2010;2:a001834. doi: 10.1101/cshperspect.a001834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cicero S, Herrup K. Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci. 2005;25:9658–9668. doi: 10.1523/JNEUROSCI.1773-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li W, Allen ME, Rui Y, et al. p39 is responsible for increasing Cdk5 activity during postnatal neuron differentiation and governs neuronal network formation and epileptic responses. J Neurosci. 2016;36:11283–11294. doi: 10.1523/JNEUROSCI.1155-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohshima T, Ward JM, Huh CG, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohshima T, Gilmore EC, Longenecker G, et al. Migration defects of cdk5(-/-) neurons in the developing cerebellum is cell autonomous. J Neurosci. 1999;19:6017–6026. doi: 10.1523/JNEUROSCI.19-14-06017.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishimura YV, Sekine K, Chihama K, et al. Dissecting the factors involved in the locomotion mode of neuronal migration in the developing cerebral cortex. J Biol Chem. 2010;285:5878–5887. doi: 10.1074/jbc.M109.033761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishimura YV, Shikanai M, Hoshino M, et al. Cdk5 and its substrates, Dcx and p27kip1, regulate cytoplasmic dilation formation and nuclear elongation in migrating neurons. Dev Camb Engl. 2014;141:3540–3550. doi: 10.1242/dev.111294. [DOI] [PubMed] [Google Scholar]
- 42.Ye T, Ip JPK, Fu AKY, Ip NY. Cdk5-mediated phosphorylation of RapGEF2 controls neuronal migration in the developing cerebral cortex. Nat Commun. 2014;5:4826. doi: 10.1038/ncomms5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang J, Ip JPK, Ye T, et al. Cdk5-dependent Mst3 phosphorylation and activity regulate neuronal migration through RhoA inhibition. J Neurosci. 2014;34:7425–7436. doi: 10.1523/JNEUROSCI.5449-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanabe K, Yamazaki H, Inaguma Y, et al. Phosphorylation of drebrin by cyclin-dependent kinase 5 and its role in neuronal migration. PLoS ONE. 2014;9:e92291. doi: 10.1371/journal.pone.0092291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie Z, Tsai L-H. Cdk5 phosphorylation of FAK regulates centrosome-associated miocrotubules and neuronal migration. Cell Cycle Georget Tex. 2004;3:108–110. [PubMed] [Google Scholar]
- 46.Xie Z, Sanada K, Samuels BA, et al. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell. 2003;114:469–482. doi: 10.1016/S0092-8674(03)00605-6. [DOI] [PubMed] [Google Scholar]
- 47.Nadarajah B, Brunstrom JE, Grutzendler J, et al. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- 48.Kwon YT, Tsai LH. A novel disruption of cortical development in p35(-/-) mice distinct from reeler. J Comp Neurol. 1998;395:510–522. doi: 10.1002/(sici)1096-9861(19980615)395:4<510::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 49.Niethammer M, Smith DS, Ayala R, et al. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 50.Sasaki S, Shionoya A, Ishida M, et al. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous System. Neuron. 2000;28:681–696. doi: 10.1016/S0896-6273(00)00146-X. [DOI] [PubMed] [Google Scholar]
- 51.Cunningham JJ, Roussel MF. Cyclin-dependent kinase inhibitors in the development of the central nervous system. Cell Growth Differ. 2001;12:387–396. [PubMed] [Google Scholar]
- 52.Nguyen L, Besson A, Heng JI-T, et al. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tury A, Mairet-Coello G, DiCicco-Bloom E. The cyclin-dependent kinase inhibitor p57Kip2 regulates cell cycle exit, differentiation, and migration of embryonic cerebral cortical precursors. Cereb Cortex N Y N 1991. 2011;21:1840–1856. doi: 10.1093/cercor/bhq254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zindy F, Cunningham JJ, Sherr CJ, et al. Postnatal neuronal proliferation in mice lacking Ink4d and Kip1 inhibitors of cyclin-dependent kinases. Proc Natl Acad Sci USA. 1999;96:13462–13467. doi: 10.1073/pnas.96.23.13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat Cell Biol. 2006;8:17–26. doi: 10.1038/ncb1338. [DOI] [PubMed] [Google Scholar]
- 56.Godin JD, Thomas N, Laguesse S, et al. p27Kip1 is a microtubule-associated protein that promotes microtubule polymerization during neuron migration. Dev Cell. 2012;23:729–744. doi: 10.1016/j.devcel.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Rodier G, Montagnoli A, Di Marcotullio L, et al. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kotake Y, Nakayama K, Ishida N, Nakayama KI. Role of serine 10 phosphorylation in p27 stabilization revealed by analysis of p27 knock-in mice harboring a serine 10 mutation. J Biol Chem. 2005;280:1095–1102. doi: 10.1074/jbc.M406117200. [DOI] [PubMed] [Google Scholar]
- 59.Clément O, Hemming IA, Gladwyn-Ng IE, et al. Rp58 and p27(kip1) coordinate cell cycle exit and neuronal migration within the embryonic mouse cerebral cortex. Neural Dev. 2017;12:8. doi: 10.1186/s13064-017-0084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kranenburg O, Scharnhorst V, Van der Eb AJ, Zantema A. Inhibition of cyclin-dependent kinase activity triggers neuronal differentiation of mouse neuroblastoma cells. J Cell Biol. 1995;131:227–234. doi: 10.1083/jcb.131.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferguson KL, Vanderluit JL, Hébert JM, et al. Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J. 2002;21:3337–3346. doi: 10.1093/emboj/cdf338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferguson KL, McClellan KA, Vanderluit JL, et al. A cell-autonomous requirement for the cell cycle regulatory protein, Rb, in neuronal migration. EMBO J. 2005;24:4381–4391. doi: 10.1038/sj.emboj.7600887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Svoboda DS, Paquin A, Park DS, Slack RS. Pocket proteins pRb and p107 are required for cortical lamination independent of apoptosis. Dev Biol. 2013;384:101–113. doi: 10.1016/j.ydbio.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 64.McClellan KA, Ruzhynsky VA, Douda DN, et al. Unique requirement for Rb/E2F3 in neuronal migration: evidence for cell cycle-independent functions. Mol Cell Biol. 2007;27:4825–4843. doi: 10.1128/MCB.02100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eguren M, Manchado E, Malumbres M. Non-mitotic functions of the anaphase-promoting complex. Semin Cell Dev Biol. 2011;22:572–578. doi: 10.1016/j.semcdb.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 66.Delgado-Esteban M, García-Higuera I, Maestre C, et al. APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat Commun. 2013;4:2879. doi: 10.1038/ncomms3879. [DOI] [PubMed] [Google Scholar]
- 67.Draghetti C, Salvat C, Zanoguera F, et al. Functional whole-genome analysis identifies polo-like kinase 2 and poliovirus receptor as essential for neuronal differentiation upstream of the negative regulator? B-crystallin. J Biol Chem. 2009;284:32053–32065. doi: 10.1074/jbc.M109.009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmetsdorf S, Arnold E, Holzer M, et al. A putative role for cell cycle-related proteins in microtubule-based neuroplasticity. Eur J Neurosci. 2009;29:1096–1107. doi: 10.1111/j.1460-9568.2009.06661.x. [DOI] [PubMed] [Google Scholar]
- 69.Mandelkow E-M, Stamer K, Vogel R, et al. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 70.Nikolic M, Dudek H, Kwon YT, et al. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 71.Piccini A, Perlini LE, Cancedda L, et al. Phosphorylation by PKA and Cdk5 mediates the early effects of synapsin III in neuronal morphological maturation. J Neurosci. 2015;35:13148–13159. doi: 10.1523/JNEUROSCI.1379-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Furusawa K, Asada A, Urrutia P, et al. Cdk5 Regulation of the GRAB-mediated Rab8-Rab11 cascade in axon outgrowth. J Neurosci. 2017;37:790–806. doi: 10.1523/JNEUROSCI.2197-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shah K, Rossie S. Tale of the good and the bad Cdk5: remodeling of the actin cytoskeleton in the brain. Mol Neurobiol. 2018;55:3426–3438. doi: 10.1007/s12035-017-0525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang H, Lin X, Liang Z, et al. Cdk5-dependent phosphorylation of liprinα1 mediates neuronal activity-dependent synapse development. Proc Natl Acad Sci USA. 2017;114:E6992–E7001. doi: 10.1073/pnas.1708240114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morabito MA. Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci. 2004;24:865–876. doi: 10.1523/JNEUROSCI.4582-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cheung ZH, Fu AKY, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/J.NEURON.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 77.Cheng K, Ip NY. Cdk5: a new player at synapses. Neurosignals. 2003;12:180–190. doi: 10.1159/000074619. [DOI] [PubMed] [Google Scholar]
- 78.Fletcher AI, Shuang R, Giovannucci DR, et al. Regulation of exocytosis by cyclin-dependent kinase 5 via phosphorylation of Munc18. J Biol Chem. 1999;274:4027–4035. doi: 10.1074/jbc.274.7.4027. [DOI] [PubMed] [Google Scholar]
- 79.Tan TC, Valova VA, Malladi CS, et al. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol. 2003;5:701–710. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- 80.Tomizawa K, Sunada S, Lu Y-F, et al. Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J Cell Biol. 2003;163:813–824. doi: 10.1083/jcb.200308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nguyen C, Bibb JA. Cdk5 and the mystery of synaptic vesicle endocytosis. J Cell Biol. 2003;163:697–699. doi: 10.1083/jcb.200310038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seeburg DP, Feliu-Mojer M, Gaiottino J, et al. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fu AKY, Fu W-Y, Cheung J, et al. Cdk5 is involved in neuregulin-induced AChR expression at the neuromuscular junction. Nat Neurosci. 2001;4:374–381. doi: 10.1038/86019. [DOI] [PubMed] [Google Scholar]
- 84.Bibb JA. Role of Cdk5 in neuronal signaling, plasticity, and drug abuse. Neurosignals. 2003;12:191–199. doi: 10.1159/000074620. [DOI] [PubMed] [Google Scholar]
- 85.Moy LY, Tsai L-H. Cyclin-dependent kinase 5 phosphorylates serine 31 of tyrosine hydroxylase and regulates its stability. J Biol Chem. 2004;279:54487–54493. doi: 10.1074/jbc.M406636200. [DOI] [PubMed] [Google Scholar]
- 86.Posada-Duque RA, Ramirez O, Härtel S, et al. CDK5 downregulation enhances synaptic plasticity. Cell Mol Life Sci CMLS. 2017;74:153–172. doi: 10.1007/s00018-016-2333-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hernandez A, Tan C, Mettlach G, et al. Cdk5 modulates long-term synaptic plasticity and motor learning in dorsolateral striatum. Sci Rep. 2016;6:29812. doi: 10.1038/srep29812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghose A, Shashidhara LS. Cyclin beyond the cell cycle: new partners at the synapse. Dev Cell. 2011;21:601–602. doi: 10.1016/j.devcel.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 89.Cho E, Kim D-H, Hur Y-N, et al. Cyclin Y inhibits plasticity-induced AMPA receptor exocytosis and LTP. Sci Rep. 2015;5:12624. doi: 10.1038/srep12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He G, Yang X, Wang G, et al. Cdk7 is required for activity-dependent neuronal gene expression, long-lasting synaptic plasticity and long-term memory. Front Mol Neurosci. 2017;10:365. doi: 10.3389/fnmol.2017.00365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morelli G, Even A, Gladwyn-Ng I, et al. p27Kip1 modulates axonal transport by regulating α-tubulin acetyltransferase 1 stability. Cell Rep. 2018;23:2429–2442. doi: 10.1016/j.celrep.2018.04.083. [DOI] [PubMed] [Google Scholar]
- 92.Ting JH, Marks DR, Schleidt SS, et al. Targeted gene mutation of E2F1 evokes age-dependent synaptic disruption and behavioral deficits. J Neurochem. 2014;129:850–863. doi: 10.1111/jnc.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Konishi Y, Stegmüller J, Matsuda T, et al. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- 94.Lasorella A, Stegmüller J, Guardavaccaro D, et al. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature. 2006;442:471–474. doi: 10.1038/nature04895. [DOI] [PubMed] [Google Scholar]
- 95.Stegmüller J, Konishi Y, Huynh MA, et al. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50:389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 96.van Roessel P, Elliott DA, Robinson IM, et al. Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell. 2004;119:707–718. doi: 10.1016/j.cell.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 97.Kim AH, Puram SV, Bilimoria PM, et al. A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell. 2009;136:322–336. doi: 10.1016/j.cell.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang Y, Kim AH, Yamada T, et al. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science. 2009;326:575–578. doi: 10.1126/science.1177087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don’t divide what’s to repair? Mutat Res Mol Mech Mutagen. 2007;614:24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 100.Nouspikel T, Hanawalt PC. DNA repair in terminally differentiated cells. DNA Repair. 2002;1:59–75. doi: 10.1016/s1568-7864(01)00005-2. [DOI] [PubMed] [Google Scholar]
- 101.Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Narciso L, Parlanti E, Racaniello M, et al. The response to oxidative DNA damage in neurons: mechanisms and disease. Neural Plast. 2016;2016:1–14. doi: 10.1155/2016/3619274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Welty S, Teng Y, Liang Z, et al. RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J Biol Chem. 2018;293:1353–1362. doi: 10.1074/jbc.M117.808402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tomashevski A, Webster DR, Grammas P, et al. Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ. 2010;17:1189–1198. doi: 10.1038/cdd.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tian B, Yang Q, Mao Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat Cell Biol. 2009;11:211–218. doi: 10.1038/ncb1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schwartz EI, Smilenov LB, Price MA, et al. Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle Georget Tex. 2007;6:318–329. doi: 10.4161/cc.6.3.3752. [DOI] [PubMed] [Google Scholar]
- 107.Casafont I, Palanca A, Lafarga V, et al. Effect of ionizing radiation in sensory ganglion neurons: organization and dynamics of nuclear compartments of DNA damage/repair and their relationship with transcription and cell cycle. Acta Neuropathol (Berl) 2011;122:481–493. doi: 10.1007/s00401-011-0869-0. [DOI] [PubMed] [Google Scholar]
- 108.Biswas SC, Zhang Y, Iyirhiaro G, et al. Sertad1 plays an essential role in developmental and pathological neuron death. J Neurosci. 2010;30:3973–3982. doi: 10.1523/JNEUROSCI.6421-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wen Y, Yang S, Liu R, Simpkins JW. Cell-cycle regulators are involved in transient cerebral ischemia induced neuronal apoptosis in female rats. FEBS Lett. 2005;579:4591–4599. doi: 10.1016/j.febslet.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 110.Marlier Q, Jibassia F, Verteneuil S, et al. Genetic and pharmacological inhibition of Cdk1 provides neuroprotection towards ischemic neuronal death. Cell death Discover. 2018 doi: 10.1038/s41420-018-0044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kruman II, Wersto RP, Cardozo-Pelaez F, et al. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron. 2004;41:549–561. doi: 10.1016/s0896-6273(04)00017-0. [DOI] [PubMed] [Google Scholar]
- 112.Park DS, Levine B, Ferrari G, Greene LA. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Park DS, Morris EJ, Padmanabhan J, et al. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Padmanabhan J, Park DS, Greene LA, Shelanski ML. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19:8747–8756. doi: 10.1523/JNEUROSCI.19-20-08747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Park DS, Morris EJ, Bremner R, et al. Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci. 2000;20:3104–3114. doi: 10.1523/JNEUROSCI.20-09-03104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 117.Hershko T, Ginsberg D. Up-regulation of Bcl-2 Homology 3 (BH3)-only Proteins by E2F1 Mediates Apoptosis. J Biol Chem. 2004;279:8627–8634. doi: 10.1074/jbc.M312866200. [DOI] [PubMed] [Google Scholar]
- 118.Stanelle J, Stiewe T, Theseling CC, et al. Gene expression changes in response to E2F1 activation. Nucleic Acids Res. 2002;30:1859–1859. doi: 10.1093/nar/30.8.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hsieh J-K, Yap D, O’Connor DJ, et al. Novel function of the cyclin A binding site of E2F in regulating p53-induced apoptosis in response to DNA damage. Mol Cell Biol. 2002;22:78–93. doi: 10.1128/MCB.22.1.78-93.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hou ST, Cowan E, Walker T, et al. The transcription factor E2F1 promotes dopamine-evoked neuronal apoptosis by a mechanism independent of transcriptional activation. J Neurochem. 2001;78:287–297. doi: 10.1046/j.1471-4159.2001.00402.x. [DOI] [PubMed] [Google Scholar]
- 121.Konishi Y, Bonni A. The E2F-Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J Neurosci. 2003;23:1649–1658. doi: 10.1523/JNEUROSCI.23-05-01649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wu J, Sabirzhanov B, Stoica BA, et al. Ablation of the transcription factors E2F1-2 limits neuroinflammation and associated neurological deficits after contusive spinal cord injury. Cell Cycle Georget Tex. 2015;14:3698–3712. doi: 10.1080/15384101.2015.1104436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Copani A, Condorelli F, Caruso A, et al. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999;13:2225–2234. [PubMed] [Google Scholar]
- 124.Kuan C-Y. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci. 2004;24:10763–10772. doi: 10.1523/JNEUROSCI.3883-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Smith DS, Leone G, DeGregori J, et al. Induction of DNA replication in adult rat neurons by deregulation of the retinoblastoma/E2F G1 cell cycle pathway. Cell Growth Differ Mol Biol J Am Assoc Cancer Res. 2000;11:625–633. [PubMed] [Google Scholar]
- 126.Verdaguer E, García-Jordà E, Canudas AM, et al. Kainic acid-induced apoptosis in cerebellar granule neurons: an attempt at cell cycle re-entry. NeuroReport. 2002;13:413–416. doi: 10.1097/00001756-200203250-00010. [DOI] [PubMed] [Google Scholar]
- 127.Veas-Pérez de Tudela M, Delgado-Esteban M, Maestre C, et al. Regulation of Bcl-xL-ATP synthase interaction by mitochondrial cyclin B1-cyclin-dependent kinase-1 determines neuronal survival. J Neurosci. 2015;35:9287–9301. doi: 10.1523/JNEUROSCI.4712-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yuan Z, Becker EBE, Merlo P, et al. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science. 2008;319:1665–1668. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- 129.Lee MS, Kwon YT, Li M, et al. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 130.Zhang J, Krishnamurthy PK, Johnson GVW. Cdk5 phosphorylates p53 and regulates its activity. J Neurochem. 2002;81:307–313. doi: 10.1046/j.1471-4159.2002.00824.x. [DOI] [PubMed] [Google Scholar]
- 131.Guo D, Xie W, Xiong P, et al. Cyclin-dependent kinase 5-mediated phosphorylation of chloride intracellular channel 4 promotes oxidative stress-induced neuronal death. Cell Death Dis. 2018;9:951. doi: 10.1038/s41419-018-0983-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Veas-Pérez de Tudela M, Maestre C, Delgado-Esteban M, et al. Cdk5-mediated inhibition of APC/C-Cdh1 switches on the cyclin D1-Cdk4-pRb pathway causing aberrant S-phase entry of postmitotic neurons. Sci Rep. 2015;5:18180. doi: 10.1038/srep18180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hamdane M, Bretteville A, Sambo A-V, et al. p25/Cdk5-mediated retinoblastoma phosphorylation is an early event in neuronal cell death. J Cell Sci. 2005;118:1291–1298. doi: 10.1242/jcs.01724. [DOI] [PubMed] [Google Scholar]
- 134.Chang K-H, Vincent F, Shah K. Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci. 2012;125:5124–5137. doi: 10.1242/jcs.108183. [DOI] [PubMed] [Google Scholar]
- 135.Jurk D, Wang C, Miwa S, et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996–1004. doi: 10.1111/j.1474-9726.2012.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wan C, Liu J, Nie X, et al. 2, 3, 7, 8-Tetrachlorodibenzo-P-dioxin (TCDD) induces premature senescence in human and rodent neuronal cells via ROS-dependent mechanisms. PLoS ONE. 2014;9:e89811. doi: 10.1371/journal.pone.0089811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Martínez-Cué C, Rueda N. Cellular senescence in neurodegenerative diseases. Front Cell Neurosci. 2020 doi: 10.3389/fncel.2020.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nagy Z, Esiri MM, Smith AD. Expression of cell division markers in the hippocampus in Alzheimer’s disease and other neurodegenerative conditions. Acta Neuropathol (Berl) 1997;93:294–300. doi: 10.1007/s004010050617. [DOI] [PubMed] [Google Scholar]
- 139.Smith TW, Lippa CF. Ki-67 immunoreactivity in Alzheimer’s disease and other neurodegenerative disorders. J Neuropathol Exp Neurol. 1995;54:297–303. doi: 10.1097/00005072-199505000-00002. [DOI] [PubMed] [Google Scholar]
- 140.Yang Y, Varvel NH, Lamb BT, Herrup K. Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J Neurosci. 2006;26:775–784. doi: 10.1523/JNEUROSCI.3707-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Esteras N, Bartolomé F, Alquézar C, et al. Altered cell cycle-related gene expression in brain and lymphocytes from a transgenic mouse model of Alzheimer’s disease [amyloid precursor protein/presenilin 1 (PS1)] Eur J Neurosci. 2012;36:2609–2618. doi: 10.1111/j.1460-9568.2012.08178.x. [DOI] [PubMed] [Google Scholar]
- 142.Ogawa O, Zhu X, Lee H-G, et al. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta Neuropathol (Berl) 2003;105:524–528. doi: 10.1007/s00401-003-0684-3. [DOI] [PubMed] [Google Scholar]
- 143.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pei J-J, Braak H, Gong C-X, et al. Up-regulation of cell division cycle (cdc) 2 kinase in neurons with early stage Alzheimer’s disease neurofibrillary degeneration. Acta Neuropathol (Berl) 2002;104:369–376. doi: 10.1007/s00401-002-0565-1. [DOI] [PubMed] [Google Scholar]
- 145.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jordan-Sciutto KL, Malaiyandi LM, Bowser R. Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:20. doi: 10.1093/jnen/61.4.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huang F, Wang M, Liu R, et al. CDT2-controlled cell cycle reentry regulates the pathogenesis of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc. 2018 doi: 10.1016/j.jalz.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.López-Sánchez N, Fontán-Lozano Á, Pallé A, et al. Neuronal tetraploidization in the cerebral cortex correlates with reduced cognition in mice and precedes and recapitulates Alzheimer’s-associated neuropathology. Neurobiol Aging. 2017;56:50–66. doi: 10.1016/j.neurobiolaging.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 149.Yang Y, Geldmacher DS, Herrup K, et al. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhu X, Siedlak SL, Wang Y, et al. Neuronal binucleation in Alzheimer disease hippocampus. Neuropathol Appl Neurobiol. 2008;34:457–465. doi: 10.1111/j.1365-2990.2007.00908.x. [DOI] [PubMed] [Google Scholar]
- 151.Spremo-Potparević B, Živković L, Djelić N, et al. Premature centromere division of the X chromosome in neurons in Alzheimer’s disease. J Neurochem. 2008;106:2218–2223. doi: 10.1111/j.1471-4159.2008.05555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Barrio-Alonso E, Hernández-Vivanco A, Walton CC, et al. Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci Rep. 2018;8:14316. doi: 10.1038/s41598-018-32708-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Arendt T, Rödel L, Gärtner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer’s disease. NeuroReport. 1996;7:3047–3049. doi: 10.1097/00001756-199611250-00050. [DOI] [PubMed] [Google Scholar]
- 154.McShea A, Harris PL, Webster KR, et al. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 155.Lee KY, Clark AW, Rosales JL, et al. Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res. 1999;34:21–29. doi: 10.1016/s0168-0102(99)00026-7. [DOI] [PubMed] [Google Scholar]
- 156.Kimura T, Ishiguro K, Hisanaga S-I. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65. doi: 10.3389/fnmol.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Patrick GN, Zukerberg L, Nikolic M, et al. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 158.Wada Y, Ishiguro K, Itoh TJ, et al. Microtubule-stimulated phosphorylation of tau at Ser202 and Thr205 by cdk5 decreases its microtubule nucleation activity. J Biochem (Tokyo) 1998;124:738–746. doi: 10.1093/oxfordjournals.jbchem.a022174. [DOI] [PubMed] [Google Scholar]
- 159.Liu F, Su Y, Li B, et al. Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 2003;547:193–196. doi: 10.1016/s0014-5793(03)00714-2. [DOI] [PubMed] [Google Scholar]
- 160.Cruz JC, Tsai L-H. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 161.Shukla V, Skuntz S, Pant HC. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch Med Res. 2012;43:655–662. doi: 10.1016/j.arcmed.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wilkaniec A, Czapski GA, Adamczyk A. Cdk5 at crossroads of protein oligomerization in neurodegenerative diseases: facts and hypotheses. J Neurochem. 2016;136:222–233. doi: 10.1111/jnc.13365. [DOI] [PubMed] [Google Scholar]
- 163.Jaiswal S, Sharma P. Role and regulation of p27 in neuronal apoptosis. J Neurochem. 2017;140:576–588. doi: 10.1111/jnc.13918. [DOI] [PubMed] [Google Scholar]
- 164.Lapresa R, Agulla J, Sánchez-Morán I, et al. Amyloid-ß promotes neurotoxicity by Cdk5-induced p53 stabilization. Neuropharmacology. 2018;146:19–27. doi: 10.1016/j.neuropharm.2018.11.019. [DOI] [PubMed] [Google Scholar]
- 165.Shi C, Viccaro K, Lee H-G, Shah K. Cdk5-Foxo3 axis: initially neuroprotective, eventually neurodegenerative in Alzheimer’s disease models. J Cell Sci. 2016;129:1815–1830. doi: 10.1242/jcs.185009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Alvarez A, Toro R, Cáceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents β-amyloid-induced neuronal death. FEBS Lett. 1999;459:421–426. doi: 10.1016/S0014-5793(99)01279-X. [DOI] [PubMed] [Google Scholar]
- 167.Lopes JP, Oliveira CR, Agostinho P. Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-β and prion peptides. Cell Cycle. 2009;8:97–104. doi: 10.4161/cc.8.1.7506. [DOI] [PubMed] [Google Scholar]
- 168.Piedrahita D, Hernández I, López-Tobón A, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J Neurosci. 2010;30:13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zheng Y-L, Amin ND, Hu Y-F, et al. A 24-residue peptide (p5), derived from p35, the Cdk5 neuronal activator, specifically inhibits Cdk5-p25 hyperactivity and tau hyperphosphorylation. J Biol Chem. 2010;285:34202–34212. doi: 10.1074/jbc.M110.134643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zheng Y, Li B-S, Amin ND, et al. A peptide derived from cyclin-dependent kinase activator (p35) specifically inhibits Cdk5 activity and phosphorylation of tau protein in transfected cells. Eur J Biochem. 2002;269:4427–4434. doi: 10.1046/j.1432-1033.2002.03133.x. [DOI] [PubMed] [Google Scholar]
- 171.Zheng Y-L, Kesavapany S, Gravell M, et al. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sundaram JR, Poore CP, Sulaimee NHB, et al. Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J Neurosci. 2013;33:334–343. doi: 10.1523/JNEUROSCI.3593-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bk B, Skuntz S, Prochazkova M, et al. Overexpression of the Cdk5 inhibitory peptide in motor neurons rescue of amyotrophic lateral sclerosis phenotype in a mouse model. Hum Mol Genet. 2019;28:3175–3187. doi: 10.1093/hmg/ddz118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bergeron M, Motter R, Tanaka P, et al. In vivo modulation of polo-like kinases supports a key role for PLK2 in Ser129 α-synuclein phosphorylation in mouse brain. Neuroscience. 2014;256:72–82. doi: 10.1016/j.neuroscience.2013.09.061. [DOI] [PubMed] [Google Scholar]
- 175.Inglis KJ, Chereau D, Brigham EF, et al. Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J Biol Chem. 2009;284:2598–2602. doi: 10.1074/jbc.C800206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Alvira D, Tajes M, Verdaguer E, et al. Inhibition of cyclin-dependent kinases is neuroprotective in 1-methyl-4-phenylpyridinium-induced apoptosis in neurons. Neuroscience. 2007;146:350–365. doi: 10.1016/j.neuroscience.2007.01.042. [DOI] [PubMed] [Google Scholar]
- 177.El-Khodor BF, Oo TF, Kholodilov N, Burke RE. Ectopic expression of cell cycle markers in models of induced programmed cell death in dopamine neurons of the rat substantia nigra pars compacta. Exp Neurol. 2003;179:17–27. doi: 10.1006/exnr.2002.8047. [DOI] [PubMed] [Google Scholar]
- 178.Höglinger GU, Breunig JJ, Depboylu C, et al. The pRb/E2F cell-cycle pathway mediates cell death in Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104:3585–3590. doi: 10.1073/pnas.0611671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jordan-Sciutto KL, Dorsey R, Chalovich EM, et al. Expression patterns of retinoblastoma protein in Parkinson disease. J Neuropathol Exp Neurol. 2003;62:68–74. doi: 10.1093/jnen/62.1.68. [DOI] [PubMed] [Google Scholar]
- 180.Smith PD, O’Hare MJ, Park DS. CDKs: taking on a role as mediators of dopaminergic loss in Parkinson’s disease. Trends Mol Med. 2004;10:445–451. doi: 10.1016/j.molmed.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 181.Gallastegui E, Domuro C, Serratosa J, et al. p27Kip1 regulates alpha-synuclein expression. Oncotarget. 2018;9:16368–16379. doi: 10.18632/oncotarget.24687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Alvira D, Ferrer I, Gutierrez-Cuesta J, et al. Activation of the calpain/cdk5/p25 pathway in the girus cinguli in Parkinson’s disease. Parkinsonism Relat Disord. 2008;14:309–313. doi: 10.1016/j.parkreldis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 183.Smith PD, Crocker SJ, Jackson-Lewis V, et al. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA. 2003;100:13650–13655. doi: 10.1073/pnas.2232515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Crocker SJ, Smith PD, Jackson-Lewis V, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci. 2003;23:4081–4091. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wen Z, Shu Y, Gao C, et al. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson’s disease. Neurobiol Aging. 2014;35:2870–2880. doi: 10.1016/j.neurobiolaging.2014.05.034. [DOI] [PubMed] [Google Scholar]
- 186.Qu D, Rashidian J, Mount MP, et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron. 2007;55:37–52. doi: 10.1016/j.neuron.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 187.Smith PD, Mount MP, Shree R, et al. Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J Neurosci. 2006;26:440–447. doi: 10.1523/JNEUROSCI.2875-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Binukumar BK, Shukla V, Amin ND, et al. Peptide TFP5/TP5 derived from Cdk5 activator P35 provides neuroprotection in the MPTP model of Parkinson’s disease. Mol Biol Cell. 2015;26:4478–4491. doi: 10.1091/mbc.E15-06-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.He R, Huang W, Huang Y, et al. Cdk5 inhibitory peptide prevents loss of dopaminergic neurons and alleviates behavioral changes in an MPTP induced Parkinson’s disease mouse model. Front Aging Neurosci. 2018;10:162. doi: 10.3389/fnagi.2018.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang Q, Xie H, Ji Z, et al. Cdk5/p25 specific inhibitory peptide TFP5 rescues the loss of dopaminergic neurons in a sub-acute MPTP induced PD mouse model. Neurosci Lett. 2016;632:1–7. doi: 10.1016/j.neulet.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 191.Rashidian J, Iyirhiaro G, Aleyasin H, et al. Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc Natl Acad Sci USA. 2005;102:14080–14085. doi: 10.1073/pnas.0500099102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Rashidian J, Iyirhiaro GO, Park DS. Cell cycle machinery and stroke. Biochim Biophys Acta Mol Basis Dis. 2007;1772:484–493. doi: 10.1016/j.bbadis.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 193.Chen B, Wang W. The expression of Cyclins in neurons of rats after focal cerebral ischemia. J Huazhong Univ Sci Technol Med Sci. 2008;28:60–64. doi: 10.1007/s11596-008-0115-8. [DOI] [PubMed] [Google Scholar]
- 194.Katchanov J, Harms C, Gertz K, et al. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–5053. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Iyirhiaro GO, Im DS, Boonying W, et al. Cdc25A is a critical mediator of ischemic neuronal death in vitro and in vivo. J Neurosci. 2017 doi: 10.1523/JNEUROSCI.3017-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Schmidt-Kastner R, Truettner J, Zhao W, et al. Differential changes of bax, caspase-3 and p21 mRNA expression after transient focal brain ischemia in the rat. Brain Res Mol Brain Res. 2000;79:88–101. doi: 10.1016/s0169-328x(00)00104-2. [DOI] [PubMed] [Google Scholar]
- 197.Gendron TF, Mealing GA, Paris J, et al. Attenuation of neurotoxicity in cortical cultures and hippocampal slices from E2F1 knockout mice. J Neurochem. 2001;78:316–324. doi: 10.1046/j.1471-4159.2001.00423.x. [DOI] [PubMed] [Google Scholar]
- 198.Meyer DA, Torres-Altoro MI, Tan Z, et al. Ischemic stroke injury is mediated by aberrant Cdk5. J Neurosci. 2014;34:8259–8267. doi: 10.1523/JNEUROSCI.4368-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Menn B, Bach S, Blevins TL, et al. Delayed treatment with systemic (s)-roscovitine provides neuroprotection and inhibits in vivo CDK5 activity increase in animal stroke models. PLoS ONE. 2010;5:e12117. doi: 10.1371/journal.pone.0012117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wang J, Liu S, Fu Y, et al. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci Publ. 2003;6:1039. doi: 10.1038/NN1119. [DOI] [PubMed] [Google Scholar]
- 201.Liu S-L, Wang C, Jiang T, et al. The role of Cdk5 in Alzheimer’s disease. Mol Neurobiol. 2016;53:4328–4342. doi: 10.1007/s12035-015-9369-x. [DOI] [PubMed] [Google Scholar]
- 202.Gutiérrez-Vargas JA, Moreno H, Cardona-Gómez GP. Targeting CDK5 post-stroke provides long-term neuroprotection and rescues synaptic plasticity. J Cereb Blood Flow Metab. 2017;37:2208–2223. doi: 10.1177/0271678X16662476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shah K, Lahiri DK. Cdk5 activity in the brain—multiple paths of regulation. J Cell Sci. 2014;127:2391–2400. doi: 10.1242/jcs.147553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Slevin M, Krupinski J. Cyclin-dependent kinase-5 targeting for ischaemic stroke. Curr Opin Pharmacol. 2009;9:119–124. doi: 10.1016/j.coph.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 205.Mitsios N, Pennucci R, Krupinski J, et al. Expression of cyclin-dependent kinase 5 mRNA and protein in the human brain following acute ischemic stroke. Brain Pathol Zurich Switz. 2007;17:11–23. doi: 10.1111/j.1750-3639.2006.00031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dumont RJ, Okonkwo DO, Verma S, et al. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol. 2001;24:254–264. doi: 10.1097/00002826-200109000-00002. [DOI] [PubMed] [Google Scholar]
- 207.Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics. 2010;7:3–12. doi: 10.1016/j.nurt.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Byrnes KR, Stoica BA, Fricke S, et al. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain. 2007;130:2977–2992. doi: 10.1093/brain/awm179. [DOI] [PubMed] [Google Scholar]
- 209.Di Giovanni S, Knoblach SM, Brandoli C, et al. Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann Neurol. 2003;53:454–468. doi: 10.1002/ana.10472. [DOI] [PubMed] [Google Scholar]
- 210.Kabadi SV, Stoica BA, Loane DJ, et al. CR8, a novel inhibitor of CDK, limits microglial activation, astrocytosis, neuronal loss, and neurologic dysfunction after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2014;34:502–513. doi: 10.1038/jcbfm.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sakurai M, Hayashi T, Abe K, et al. Cyclin D1 and Cdk4 protein induction in motor neurons after transient spinal cord ischemia in rabbits. Stroke J Cereb Circ. 2000;31:200–207. doi: 10.1161/01.str.31.1.200. [DOI] [PubMed] [Google Scholar]
- 212.Skovira JW, Wu J, Matyas JJ, et al. Cell cycle inhibition reduces inflammatory responses, neuronal loss, and cognitive deficits induced by hypobaria exposure following traumatic brain injury. J Neuroinflammation. 2016;13:299. doi: 10.1186/s12974-016-0769-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Aubrecht TG, Faden AI, Sabirzhanov B, et al. Comparing effects of CDK inhibition and E2F1/2 ablation on neuronal cell death pathways in vitro and after traumatic brain injury. Cell Death Dis. 2018 doi: 10.1038/s41419-018-1156-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Stone JG, Siedlak SL, Tabaton M, et al. The cell cycle regulator phosphorylated retinoblastoma protein is associated with tau pathology in several tauopathies. J Neuropathol Exp Neurol. 2011;70:578–587. doi: 10.1097/NEN.0b013e3182204414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol (Berl) 1997;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- 216.Nagy Z, Esiri MM. Neuronal cyclin expression in the hippocampus in temporal lobe epilepsy. Exp Neurol. 1998;150:240–247. doi: 10.1006/exnr.1997.6753. [DOI] [PubMed] [Google Scholar]
- 217.Fiala M, Avagyan H, Merino JJ, et al. Chemotactic and mitogenic stimuli of neuronal apoptosis in patients with medically intractable temporal lobe epilepsy. Pathophysiology. 2013;20:59–69. doi: 10.1016/j.pathophys.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Husseman JW, Nochlin D, Vincent I. Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging. 2000;21:815–828. doi: 10.1016/S0197-4580(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 219.Fielder E, von Zglinicki T, Jurk D. The DNA damage response in neurons: die by apoptosis or survive in a senescence-like state? J Alzheimers Dis JAD. 2017;60:S107–S131. doi: 10.3233/JAD-161221. [DOI] [PubMed] [Google Scholar]