

Abstract
Female fertility and offspring health are critically dependent on the maintenance of an adequate supply of high-quality oocytes. Like somatic cells, oocytes are subject to a variety of different types of DNA damage arising from endogenous cellular processes and exposure to exogenous genotoxic stressors. While the repair of intentionally induced DNA double strand breaks in gametes during meiotic recombination is well characterised, less is known about the ability of oocytes to repair pathological DNA damage and the relative contribution of DNA repair to oocyte quality is not well defined. This review will discuss emerging data suggesting that oocytes are in fact capable of efficient DNA repair and that DNA repair may be an important mechanism for ensuring female fertility, as well as the transmission of high-quality genetic material to subsequent generations.
Keywords: Ovary, Primordial follicles, Folliculogenesis, Base excision repair, Mismatch repair, Nucleotide excision repair, Homologous recombination, Non-homologous end joining, Detection and response
Introduction
DNA is under constant assault by endogenous and exogenous agents that damage and alter the chemical structure and genetic sequence. It is predicted that every cell in the human body receives tens of thousands of DNA lesions per day, including thousands generated by endogenous alkylating agents, hydrolytic deamination, non-enzymatic methylations and oxidation [1–3]. Similarly, many chemicals and agents present in the environment (such as in food, air or water) have carcinogenic or mutagenic properties. These exogenous and endogenous reactions can lead to various types of lesions on the nucleotide heterocyclic base or phosphate backbone and can turn a legitimate base into either a mutagenic, miscoding or noncoding lesion and can even change one base into another [1, 2, 4, 5]. These adducts can impair base pairing, block DNA replication and result in nucleotide loss and DNA single-strand breaks (SSBs). Furthermore, when two SSBs arise in close proximity, or when the DNA-replication apparatus encounters a SSB, double-strand breaks (DSBs) can form. Therefore, to ensure viability, cells have evolved multiple systems to detect and repair these different types of DNA damage.
Females are born with a finite supply of oocytes and it is imperative that the health of these oocytes is maintained throughout reproductive life to ensure that only the “best” possible oocytes are available for ovulation, fertilisation and subsequent embryonic development [6]. One mechanism by which oocyte number and quality may be safeguarded is the implementation of strategies to rapidly detect and repair DNA damage sustained as a consequence of normal metabolic activities, or through exposure to exogenous factors. Indeed, the ability of a cell to repair DNA damage is vital to the integrity of its genome and thus, to its normal functionality. It is particularly important for gametes to correct damage to their DNA, to avoid apoptosis and prevent the transmission of genetic mutations to offspring [6]. Surprisingly, however, detailed information on the ability of oocytes to undertake efficient DNA repair, and the contribution of DNA repair to oocyte quality, is only recently emerging as an essential area of research. This review will give an overview of the DNA repair pathways available to oocytes and discuss evidence from various animal models indicating that the capacity for DNA repair is vital for female fertility.
The female germline is stored in structures called primordial follicles
The female germline is stored in the ovary in the form of primordial follicles, which are structures comprising an immature oocyte, surrounded by a single layer of supporting somatic cells, known as granulosa cells (Fig. 1) [7]. The enclosed oocytes are non-growing, non-proliferative and arrested part way through the first meiotic prophase at the diplotene stage. They may remain in this growth and meiotic arrest for several months (mouse) or up to 50 years (human) before being activated to begin the process of folliculogenesis, which is characterised by dramatic growth of the oocyte and resumption of meiosis, as well as the extensive proliferation of the granulosa cells (Fig. 1) [8]. The coordinated development of the oocytes and granulosa cells during folliculogenesis ultimately results in the ovulation of a meiotically mature and developmentally competent oocyte (often referred to as MII stage), which can be fertilized and is able to support embryonic development (Fig. 1) [9, 10]. From birth onwards, the number of primordial follicles found in the ovary steadily declines until the pool becomes exhausted, leading to the end of fertility, followed by the beginning of menopause. Importantly, the entire complement of gametes available to the female is formed during fetal (human) or perinatal (mouse) development and new oocytes cannot be made after this point, even if the supply is prematurely depleted [11]. Thus, repair of those that are damaged is likely an important mechanism for ensuring that adequate numbers of primordial follicles are maintained in the ovary to support fertility and perpetuation of the species.
Fig. 1.
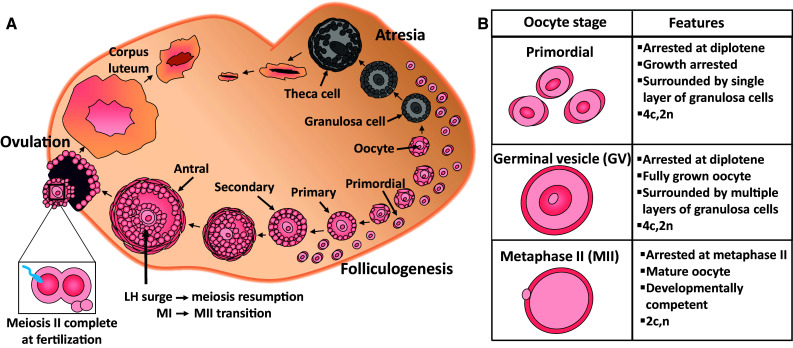
Schematic representation of folliculogenesis in the mammalian ovary. a Follicles form the functional unit of the ovary and are comprised of the central germ cell (oocyte), surrounded by specialized somatic granulosa cells, as well as a theca cell layer, surrounding mature growing follicles only. A finite supply of non-growing, meiotically arrested primordial follicle oocytes form the ovarian reserve and once lost from the follicle pool, these cells cannot be replenished. From birth onwards, a limited number of primordial follicles are periodically activated to undergo folliculogenesis; a process in which primordial follicles mature through primary, secondary, and antral stages under the influence of follicle-stimulating hormone (FSH) which is produced at puberty. From the growing follicle pool in women, one dominant follicle is ovulated under the influence of luteinizing hormone (LH), leaving behind remnants to form the corpus luteum, required to hormonally support a potential conceptus. The remaining growing follicles undergo atresia, a hormonally regulated apoptotic process, and this is the fate of the majority of follicles. This cyclical pattern continues until the pool of primordial follicles drops below a critical threshold and menopause ensues. b Schematic representation of different oocyte developmental stages. Primordial follicle oocytes are the most abundant form of oocyte in the ovary and are meiotically arrested at diplotene. They are non-dividing, so they can remain arrested in the ovary for months in mice, or decades in humans. They are distinguished morphologically by a surrounding single layer of squamous granulosa cells. Primordial follicle oocytes contain four chromatids (c) and are diploid (2n). Germinal vesicle (GV) oocytes remain meiotically arrested at diplotene, however, represent a fully grown oocyte. GV oocytes are 4c, 2n. Germinal vesicle breakdown indicates that meiosis has resumed and the extrusion of the first polar body represents completion of meiosis I in humans. The oocyte is then arrested at metaphase II (MII), at which stage it is developmentally competent and haploid (2c, n), but will only complete meiosis if fertilization occurs
In comparison to somatic cells, oocytes in primordial follicles exhibit a number of unique properties that should be considered when evaluating their ability to undertake DNA repair: (1) they are meiotically arrested, with a diploid nucleus containing four copies of each chromosome (2n, 4c), (2) their condensed chromosomes have relaxed slightly to allow some transcription, but homologs remain in close association with each other, (3) they are non-growing but metabolically active and (4) they are amongst the most long-lived cells in the body and remain in this unusual stasis for many months or decades, depending on the species. During this time, they may be subjected to ongoing exogenous and endogenous stressors. Notably, oocytes in primordial follicles appear to be more susceptible to DNA-damaging agents compared to surrounding somatic cells and growing oocytes, and have a greater tendency to undergo apoptosis [12, 13]. Recent work described below provides compelling evidence that this is likely to be due to the evolution of a highly sensitive apoptotic response, rather than lack of DNA repair capacity.
Expression profiling indicates that oocytes should be well-equipped to repair DNA damage
Early studies demonstrated that damaged DNA is efficiently repaired when transferred to mammalian MII oocytes and zygotes, or Xenopus oocyte extracts, showing that factors accumulated in the mature oocyte are vital for maintaining genome integrity [14–18]. Since that time, advances in methods for RNA amplification, and the development of sequencing and microarray platforms have enabled the transcriptomic analysis of DNA repair factors in oocytes and preimplantation embryos. Several studies have used microarrays to profile RNA expression in germinal vesical (GV) oocytes, MII stage oocytes and embryos in mice [19–21] and humans [22–24]. Interestingly, components from all DNA repair pathways including direct lesion reversal, base excision repair (BER), mismatch repair (MMR), nucleotide excision repair (NER), homologous recombination (HR) and non-homologous end-joining repair (NHEJ) are represented in mouse, monkey and human MII oocytes and embryos [23–26]. At least two groups have successfully performed global gene expression studies using mouse oocytes from earlier stages of folliculogenesis, including primordial, primary, secondary, small antral and large antral follicles [27–29]. The result of these works indicate that there is an over-representation of transcripts involved in DNA repair observed throughout oocyte development, with at least 80% of the genes commonly detected between all developmental stages [27]. Proteomic analysis using liquid chromatography–mass spectrometry (LC–MS/MS) of 7000 GV, MII, and zygotes identified a total of 53 proteins involved in the DNA repair in both MII oocytes and zygotes [30]. Of these proteins, 35 were upregulated in the MII oocyte and included single-strand break repair and nucleotide-excision repair proteins [30]. Combined, these studies provide strong evidence that oocytes from primordial follicles through to MII have the capacity to repair damaged DNA and maintain genome integrity (Table 1). Interestingly, a recent RNA-seq analysis suggested that there may be species differences in the ability of GV and MII oocytes to undertake DNA repair [31]. In this study, the overall expression patterns of genes involved in the repair of DNA double strand breaks were different between primates and mouse. Additionally, following in vitro exposure to etoposide, RAD51 (a key player in the homologous recombination repair pathway) was expressed and recruited to double strand break sites at much lower levels in primate oocytes than in mouse oocytes. Based on these data, it was proposed that rodent oocytes have a superior DNA repair competence to that of primates [31]. Of note, however, the rhesus monkeys used in these analyses were aged between 7 and 14 years, and females begin the process of maternal ageing as early as 10 years in this species [32]. Maternal ageing is associated with a decline in the expression of repair factors, including RAD51, and reduced DNA repair capacity in oocytes [29, 33–35], and it is not clear if maternal age was adequately controlled between species in this study. Thus, whether true inter-species differences in DNA repair capacity exist in oocyte requires further exploration.
Table 1.
DNA repair pathways identified in oocytes
| Repair mechanism | Type of damage | Common causes | Key components | Pathway overview | Oocyte function |
|---|---|---|---|---|---|
| Direct lesion reversal | Small base adducts | Alkylating agents | MGMT, ALKBH | [4] | Unknown |
| Mismatch repair (MMR) | DNA mismatches and insertion/deletion loops | DNA replication and recombination | MSH2-6, PMS1 and 2, PMS2L3 and 4 and MLH1 and 3 | [179] | [18] |
| Base excision repair (BER) | Small DNA lesions, such as oxidized or reduced bases, non-bulky adducts and DNA single-strand breaks | Endogenous oxidative and hydrolytic decay | DNA glycosylases initiate this process by releasing the modified base. The DNA repair enzymes X-ray repair cross-complementing group 1 (XRCC1), 8-Oxoguanine DNA glycosylase (OGG1) and apurinic/apyrimidinic endonuclease (APE1) play a central role in the BER pathway | [180] | [17, 63–65] |
| Nucleotide excision repair (NER) | Repairs bulky DNA adducts, cross-links and oxidative damages | Environmental agents including UV radiation | XPA, XPC and XPD | [97] | [15, 16] |
| Trans-lesion bypass mechanisms | Base damage and gaps blocking replication forks | Unrepaired DNA damage | Mutagenic DNA polymerase such as pol η (XPV) four specialized Y‐family DNA polymerases and six additional low‐fidelity polymerases | [181, 182] | [117] |
| Non- homologous end-joining (NHEJ) | Double strand breaks (DSBs) | Ionizing radiation (IR) or chemically induced | Ku70/80, DNA-PKcs, end-processing enzymes, polymerases and DNA ligase IV | [183] | [139, 184] |
| Homologous recombination (HR) | DSB, stalled replication forks and interstrand DNA crosslinks | Meiotic recombination, ionizing radiation (IR) | MRN complex, BRAC2, RAD51 | [185] | [31, 131, 141] |
| Fanconi anaemia (FANC) pathway | DNA interstrand crosslinks (ICLs) |
Mitomycin C (MMC) or diepoxybutane (DEB) UV radiation, IR, hydroxyurea, replication |
FA core complex (FANCA-G, FANCL, FANCM FAAP24 and FAAP100), FANCD1/BRCA2, FANCD2, FANCJ/BRIP1/BACH1, FANCN and FANCI | [108, 109, 186] | Unknown |
DNA damage and repair mechanisms
Specific repair pathways have evolved to correct the many different types of DNA damage that can occur. These pathways employ a suite of different enzymes, including topoisomerases, nucleases, ligases, phosphatases, polymerases and helicases that work in a lesion-dependent manner, although some proteins may have important roles in more than one pathway (Fig. 2). Major lesions and repair mechanisms are described below, with relevance to oocytes and pathological consequence for fertility discussed where the data are available.
Fig. 2.
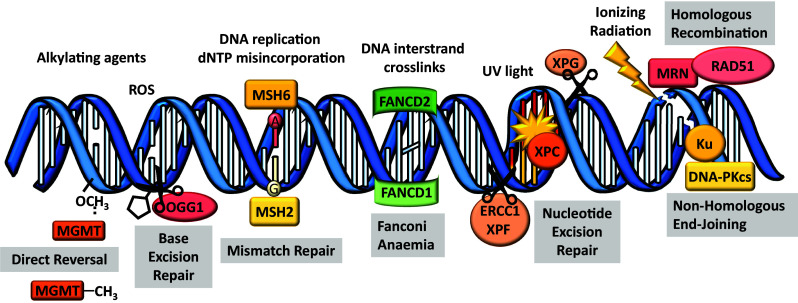
Types of DNA damage and repair mechanisms. Alkylating agents can transfer alkyl carbon groups (CH3) onto DNA and can lead to base-mismatched pairing. Some alkylated bases can be repaired by direct lesion reversal using dioxygenase enzymes e.g., MGMT. Other agents can cause more complex abducts such as reactive oxygen species (ROS) which causes 8-oxoguanine DNA lesions. The base excision repair pathway, via DNA glycosylases (e.g., OGG1), are responsible for replacing a single damaged nucleotide that cannot be repaired by direct reversal. Spontaneous DNA alterations can arise during DNA replication due to dNTP misincorporation, which are primarily repaired by the mismatch repair (MMR) pathway (e.g., MSH2–MSH6 complex initiates the repair of small nucleotide mismatches). DNA interstrand crosslinks are very toxic covalent links within the double helix that prevent DNA unwinding and affect both strands of DNA. The Fanconi anaemia pathway works to coordinate several distinct repair activities belonging to different classic repair pathways. Complex (CPLX) 1 is formed in response to DNA damage recruits FANCD2 which, via interactions with FANCD1, promotes loading of complex 2 that includes components from other repair pathways. When DNA is severely damaged (e.g., UV radiation) and large sections of DNA need to be replaced the nucleotide excision repair pathway is activated, XPC recognizes and binds the helix-distorting lesion and then recruits endonucleases XPG and ERCC1–XPF to excise the damaged DNA fragment and allow polymerase to resynthesis the strand. DNA double strand breaks are caused following exposure to exogenous stressors (e.g., ionizing radiation). Homologous recombination (HR) and non-homologous end joining (NHEJ) are the two main pathways responsible for DNA DSB repair. KU70/KU80 and MRN complex and are involved in the detection of double strand breaks and recruitment of various repair factors (e.g., RAD51 is essential for HR repair and DNA-PKcs is essential for NHEJ repair
Alkylation of DNA, direct lesion reversal and base excision repair (BER)
Alkylating agents are reactive chemicals that transfer alkyl carbon groups (adducts) onto a broad range of biological molecules, including DNA, consequently altering their structure and function. Eukaryotic cells harbor endogenous alkylating agents and metabolites, including MGT1 O6-methylguanine DNA methyltransferase, MAG 3-methyladenine DNA glycosylase, and APN1 apurinic/apyrimidinic (AP) endonuclease that alkylate nuclear DNA at both oxygens and nitrogens [36]. Reactive species such as free radicals and oxidants are formed inside living cells during normal metabolic activities and can also react with different components of DNA and modify bases [37]. Reactive oxygen species (ROS) are produced within growing follicles and are required for oocyte maturation, the resumption of meiosis and ovulation in rats [38, 39]. Although they play important roles within the ovary, the cyclic production of these DNA damaging agents may lead to an increased cumulative risk of ovarian pathology if ROS-induced DNA damage is not repaired. Indeed, increased ROS levels in follicular fluid have been associated with poor oocyte and embryo quality, low conception and pregnancy rate in women [40–42].
In the clinical setting, alkylating agents are commonly used as anti-cancer compounds and exposure to these compounds is associated with damage to oocyte DNA and depletion of the ovarian oocyte supply, leading to infertility and menopause in female cancer survivors [43–48]. Notably, in the case where female cancer patients are fertile after treatment with alkylators, studies have convincingly shown that there is no increase in birth defects or chromosomal abnormalities in their children [49, 50]. Such studies provide indirect evidence that the oocytes that survive exposure to these compounds are able to repair any damage they sustained.
Some adducts can be removed through direct lesion reversal by the AlkB homologue (ALKBH) family of dioxygenase enzymes that use an oxidative dealkylation reaction or O6-alkylguanine-DNA alkyltransferase (MGMT), which serves as an alkyl transfer protein [4, 51–53] (Table 1; Fig. 2). In mouse and human oocytes and early embryos, MGMT and other similar enzymes are highly expressed at the transcript level [23, 24, 27], suggesting that oocytes may be able to repair these adducts via direct lesion reversal, but direct functional studies to confirm this have not been undertaken.
Other bulky adducts must be repaired via the base excision repair (BER) pathway (Table 1), which removes and replaces a single damaged nucleotide by targeting the damaged base (such as an alkylated base). The initiation of BER occurs by the recognition and excision of a damaged DNA base by a DNA glycosylase (Fig. 2). Different DNA glycosylases recognize and remove different kinds of damaged bases (e.g., uracil glycosylase, 3-methyladenine glycosylase and UV-endonucleases), and the specificity of the repair pathway is determined by the type of glycosylase involved [54, 55]. In humans, there are currently 11 known DNA glycosylases [56]. 8-Oxoguanine is one of the most common DNA lesions resulting from reactive oxygen species (ROS) and is primarily repaired by the DNA glycosylase, OGG1 (Fig. 2). DNA glycosylases function by cleaving the N-glycosidic bond between the base and the backbone of the nucleotide residue, leaving an AP site, also known as an abasic site. Once the base is removed, the AP site is removed by an AP endonuclease or an AP lyase, which nicks the DNA strand 5′ or 3′ to the AP site, respectively. The remaining nucleotide backbone is excised by a phosphodiesterase and the gap is filled by a DNA polymerase and sealed by DNA ligase [55, 57].
During development from primordial follicle to mature MII stage, it is proposed that the oocyte accumulates an abundance of DNA repair factor transcripts and proteins to repair any DNA damage encountered at fertilisation and during subsequent early embryogenesis. This is likely necessary for two reasons, (1) transcription is restricted in a conceptus until the two-cell stage in mice [58], or the four-cell stage in humans [59] and (2) sperm DNA is prone to oxidative damage and 8-Oxoguanine lesions [60]. In support of this hypothesis, all genes involved in the BER repair pathway are expressed in human and monkey MII oocytes and embryos [23, 24, 26]. In contrast, although OGG1 protein is present in human sperm, the subsequent enzymes of the BER pathway, APE1 and XRCC1, cannot be detected at the protein level [61]. Interestingly, OGG1 protein is expressed at very low levels in mouse oocytes, while APE1 and XRCC1 protein are readily detectable [62]. This inverse relationship suggests that some degree of functional co-operation between the oocyte and sperm may be required to repair 8-Oxoguanine lesions after fertilisation in mice [62]. While the efficiency of BER repair remains to be fully explored, the fact that oxidative damage to sperm DNA is associated with both miscarriage and developmental abnormalities in offspring [60] suggests that there may be inherent limits to the DNA repair capacity of the oocyte in this instance. In addition to the repair of oxidative damage to sperm DNA, new evidence also points to a functional role for BER, and oocyte-derived XRCC1 in particular, in the repair of lesions in the paternal DNA that are generated during zygotic reprogramming [63]. Importantly, the repair of these lesions is linked with timely mitotic entry as the zygote transitions into a two-cell embryo. Thus, oocyte-derived BER appears to be important for at least two different types of DNA damage post fertilisation.
In the case of other types of damage, functional studies in mature Xenopus oocytes and nuclear extracts have demonstrated that bulky DNA lesions and AP sites are efficiently repaired by alternative BER mechanisms, including the proliferating cell nuclear antigen-(PCNA) dependent pathway and the DNA polymerase β-(pol β)-dependent pathway [17, 64, 65]. These same pathways are conserved in mammalian cells [66, 67], although they remain to be investigated in mammalian oocytes. Moreover, whether the BER pathway functions in oocytes at the primordial follicle stage has not been determined.
DNA replication-induced errors and mismatch repair (MMR)
Spontaneous DNA alterations can arise during DNA replication due to dNTP misincorporation, which can distort the helical structure of DNA. These replication-dependant errors are predicted to occur at the rate of 10−6–10−10 mutations/base pair/cell generation [68–71]. Notably, base–base mismatches and insertion/deletion mispairs can also be generated during recombination, which has important implications for oocytes during meiotic recombination. The mismatch repair (MMR) pathway is primarily utilized to repair these dNTP misincorporations, as well as insertion and deletion loops (IDLs) that occur during DNA replication and recombination (Table 1; Fig. 2). Moreover, some of the MMR components have been reported to play a significant role in DNA damage signaling via direct interaction with ATM in response to non-replication-related DNA damage [72, 73]. Heterodimeric complexes of MutS-related proteins (MutSα and MutSβ) target and repair base:base and IDL mispairs. MutSα (MSH2–MSH6 complex) initiates the repair of small 1–12 nucleotide mismatches and IDLs, whereas the repair of larger IDLs (2–16 nucleotides) is initiated by MutSβ (MSH2–MSH3 complex) [74, 75]. The partial redundancy between MutSα and MutSβ helps to explain the different phenotypes of the various MutS-related protein-null mice. MSH2, MSH3 and MSH6 mRNA are highly expressed in human GV oocytes [23]. Although MutS-related protein levels have not been measured in human oocytes, the presence of transcripts indicates that MMR may be required to maintain genome integrity. Indeed, Xenopus oocyte extracts have been shown to effectively repair different mismatches in vitro [18]. The other two eukaryotic MutS-related proteins, MSH4 and MSH5, are not involved in MMR, but have essential roles regulating chromosome pairing during meiotic recombination [76, 77]. In mice, loss of MSH4 and MSH5 leads to failed chromosome pairing during prophase I of meiosis, resulting in apoptosis and sterility in both males and females [78]. Indeed, in humans, mutations in MSH5 are associated with ovarian insufficiency [79, 80]. Of the four MutL homologs, MLH1, MLH3, PMS1 and PMS2, three are involved in mismatch repair and MLH3, PMS2 and MLH1 are required for mammalian meiosis [81–86]. Mice null for Mlh3, Pms2 and Mlh1 are infertile, spermatocytes fail to progress normally beyond the pachytene stage of meiosis, while oocytes fail to complete meiosis after fertilization [81–83]. Combined, these findings suggest that MMR and its related proteins are vital for coordinating oocyte DNA damage signaling initially not only during recombination but also after meiotic arrest.
Ultraviolet radiation and nucleotide excision repair (NER)
The most pervasive environmental DNA-damaging agent is ultraviolet light, which can induce thousands of lesions per exposed cell, per hour. There are two major classes of mutagenic DNA lesions induced by UV radiation, including cyclobutane–pyrimidine dimers (CPDs), which are the most abundant and probably most cytotoxic lesions and 6–4 photoproducts (6–4 PPs, pyrimidine adducts), which exert potentially lethal, mutagenic effects [2]. Many organisms use the photolyase enzyme that specifically binds to CPDs (CPD photolyase) or 6–4 PPs (6–4 photolyase) and reverses the damage using the energy of light, a process known as photoreactivation (Table 1). These photolyase enzymes are either absent or non-functional in humans and are considered to be ancient repair proteins [2, 87]. Thus, the nucleotide excision repair (NER) and to some extent, BER and MMR are involved in repairing DNA lesions induced by UV radiation in humans (Table 1).
While oocytes are not normally exposed to UV light, the development of assisted reproductive technologies, such as in vitro maturation and in vitro fertilisation, mean that there are new situations in which oocytes are exposed to potentially damaging visible light [88]. Similarly, some environmental factors such as chemotherapeutic drugs can also cause bulky abducts and have been shown to activate NER [89, 90]. Furthermore, studies have shown overlapping functions of some components of the BER and NER pathways, especially the repair of oxidative DNA damage [91–93], suggesting that NER may also be involved in ROS-associated ovarian pathology. Therefore, NER is also likely to be essential for oocyte genome integrity. The NER repair pathway acts on a wide variety of lesions, but is especially important for bulky, helix-distorting lesions [94]. In eukaryotes, NER functions by excising an oligonucleotide fragment of ~ 30 nucleotides that contains the DNA lesion. Briefly, XPC recognizes and binds to the DNA lesion, then recruits endonucleases XPG and XPF, as well as TFIIH, which is a subcomplex of polymerase II transcription machinery that operates to uncoil the DNA fragment. XPA and XPD are recruited at the same time by TFIIH to coordinate single strand DNA-binding protein replication protein A and helicase activity, respectively [95, 96]. XPG endonuclease and ERCC1–XPF heterodimeric protein then make cuts 3′ and 5′ of the damaged strand, respectively, releasing the damaged DNA fragment (Fig. 2). The gap that is generated is then repaired by DNA synthesis using the opposite, normal DNA strand as a template.
Two different NER pathways exist, with one that operates on transcriptionally active DNA and the other on transcriptionally silent DNA [97]. Significantly, in humans and mice, transcription-coupled NER (TCNER) can proceed in the absence of XPC, thus XPC may not be required for TCNER [98, 99]. In GV and MII, oocytes which are transcriptionally quiescent, Xpc is highly expressed along with most of the other genes involved in NER [23]. Studies utilizing the thymidine incorporation assay led to an increase in the number of radioactive [3H]thymidine nuclear incorporation in oocytes after UV irradiation [15, 16], demonstrating excision and replacement of damaged bases by repair enzymes in resting primordial and growing oocytes [15, 16]. Importantly, incorporation of [3H]thymidine was six times greater in growing oocytes versus primordial oocytes [15]. This suggests that repair mechanisms are not as efficient in primordial oocytes and may partially explain why they are more susceptible to DNA damage compared to growing oocytes. Although Xpa- and Xpc-deficient mice are fertile [100, 101], mice deficient in Ercc1 or those carrying a point mutations in XPD, show decreases in fertility over time, indicative of premature ovarian failure [102–104]. Together, these findings suggest that some components of NER pathway are vital for protecting the ovarian reserve, and warrant further investigation.
Interstrand crosslinks (ICLs) and Fanconi anaemia (FA) pathway
DNA interstrand crosslinks (ICLs) are highly toxic covalent links within the double helix that prevent DNA unwinding, thereby blocking both DNA replication and transcription [105]. ICLs are caused by a variety of endogenous metabolites (i.e., nitric oxide, malondialdehyde), environmental exposures (i.e., acrolein, crotonaldehyde, formaldehyde and acetaldehyde) and cancer chemotherapeutic agents (i.e., chlorambucil, cisplatin, mitomycin C, psoralen) that have two reactive groups [106]. Indeed, female patients exposed to ICL-inducing drugs (i.e., chlorambucil, cisplatin, carboplatin and ifosfamide) have a significantly reduced rate of successful pregnancy, possibly due to direct damage to the primordial follicles [107]. As ICLs affect both strands of DNA, the Fanconi anaemia (FA) pathway works to coordinate several distinct repair activities belonging to different classic repair pathways, including NER, translesion synthesis (TLS), and HR, to remove crosslinks [108]. The FA pathway was identified by cloning genes commonly mutated in Fanconi anaemia patients, which is an autosomal-recessive chromosomal instability disorder. Cells derived from FA patients are hypersensitive to DNA cross-linking agents, ionizing irradiation and oxygen radicals, which lead to increased apoptosis or growth arrest. Clinical manifestations of FA include various hematological deficiencies, developmental abnormalities, infertility and increased cancer susceptibility.
There are 11 identified FA proteins that form into two core complexes in response to stalled replication forks [109]. Complex 1 is formed in response to DNA damage and monoubiquitinates and recruits FANCD2 to chromatin containing the cross-link. Monoubiquitinated FANCD2 then promotes chromatin loading of the second complex via interactions with FANCD1/BRCA2, which includes components from other repair pathways including HR (e.g., MRN complex and Rad51). Recruitment of both complexes is required for the repair of the cross-link, and is presumably repaired through HR and translesion synthesis (TLS) [109]. Interestingly, FANCD2 has been shown to bind to synaptonemal complexes of meiotic chromosomes during spermatogenesis [110]. In particular, this pathway appears to have important roles in the repair of DNA during the proliferation of the embryonic precursors of oocytes (known as primordial germ cells and oogonia) and during meiotic recombination [111–113]. Moreover, male and female mice genetically null for FA factors Fac and Fanca have significantly reduced fertility, with premature reproductive senescence in females attributed to premature ovarian failure [111–113], thus demonstrating a role for FA in maintaining the ovarian follicular reserve.
Translesion synthesis and template switching
If the cell is to survive in a situation where repair cannot occur in time for replication, the only option is to bypass the lesion using polymerases with less stringent base-pairing requirements than replicative polymerases. At the site of a lesion, the high-fidelity DNA replication polymerase is transiently switched for low-fidelity DNA polymerase, but is returned once the replication fork passes the site of the lesion to continue as normal. Alternatively, the DNA lesion is completely bypassed at the replication fork, leaving a single-strand gap in the synthesized sequence, which is subsequently repaired using the sister chromatid as a template, as in HR repair [114]. TLS is unlikely to play a significant role in maintaining the genome integrity of diplotene-arrested oocytes because of their lack of mitotic activity. However, sperm production is associated with many rounds of DNA replication and cell division, and in agreement with this, all known TLS polymerases show higher expression in testis compared with tissues that contain only somatic cells, with high levels of polymerase expression particularly evident in round spermatids [115, 116]. Interestingly, growing oocytes accumulate polymerases involved in TLS [23], which may point to an important role for this pathway in the development embryo after fertilization [117].
DSB repair: homologous recombination (HR) and non-homologous end-joining (NHEJ) repair
Similar to somatic cells, DNA DSBs are generated in oocytes following exposure to exogenous stressors, such as ionizing radiation, chemotherapeutic drugs, and environmental toxicants [13, 118–120]. Recent studies have also reported the spontaneous induction and accumulation of DSBs in oocytes as a consequence of endogenous oxidative stress and maternal aging [34]. Furthermore, unlike somatic cells, DNA DSBs are intentionally induced and repaired in oocytes during meiotic recombination.
Unrepaired or incorrectly repaired DSBs can result in chromosome breaks, translocations, deletions and point mutations [121], ultimately leading to oocyte apoptosis, impaired oocyte growth and maturation, infertility, miscarriage or genetic defects in offspring [6, 119, 122, 123]. Indeed, chromosomal aberrations such as deletions, translocations and inversions are detected in ~ 2–6% of human oocytes [124, 125] and miscarriages [6, 126, 127]. While large deletions are lethal, small chromosomal losses underlie numerous rare human genetic disorders characterised by intellectual disability, growth retardation and/or congenital malformations. One example is 1p36 deletion syndrome, which affects 1/5000–10,000 births, making it one of the most common mental retardation syndromes in humans [128]. The majority of the deletions contributing to this syndrome arise de novo in the oocyte, possibly resulting from unequal recombination or erroneous repair of spontaneously occurring DNA DSBs [128]. Clearly, it is essential for oocytes to undertake efficient repair of DNA DSBs to avoid infertility and transmission of germline defects to the next generation. Indeed, studies in apoptosis-deficient mice showing that oocytes that have sustained DSBs as a consequence of chemotherapy or radiation can give rise to healthy offspring and provide strong evidence that oocytes can repair significant levels of DNA damage despite being predisposed to activate cell death [119, 129].
HR and NHEJ are the two main pathways responsible for DNA DSB repair [130]. In somatic cells, DNA DSBs lead to the phosphorylation of histone H2AX (γH2AX) by ATM kinase [130]. This results in the formation of discrete foci at the sites of DNA damage that facilitate the recruitment and targeting of repair factors. HR is considered to be the most error-free pathway for DSB as it uses the undamaged sister-chromatid as a template. As such, HR is likely to be the pathway of choice for oocytes because sister chromatids are present (at least before the resumption of meiosis), which can serve as a template for accurate, error-free repair. RAD51 and BRCA2 are essential for HR repair and are among the few specific repair factors that have been identified to be transcribed in primordial follicle oocytes [34, 131]. Interestingly, Rad51 is detected at higher levels in primordial follicle oocytes from young mice compared to old mice [29, 33, 34, 131], suggesting that primordial follicle oocytes lose the capacity to undertake efficient DSB repair as they age. In mammals, there is some evidence that GV and MII oocytes may utilize HR-repair enzymes (i.e., RAD51) in response to exogenous DNA-damaging agents in culture, possibly to modulate cell death [31, 131]. Indeed, microinjection of mature oocytes with a neutralizing RAD51 antibody increases their susceptibility to doxorubicin-induced death, presumably by reducing the ability of the oocyte to undertake timely DNA repair [31, 131]. Another key player in the repair response is the MRN complex, comprising MRE11, RAD50 and NBS1 [132]. It is one of the earliest responders to DNA DSB, acting as a sensor of DNA damage and mediating the activation of ATM. MRE11 also possesses nuclease activity and directly participates in the repair process. In mice, MRE11 is essential for DNA DSB repair during early meiosis I, but its expression and function has not been previously characterised within oocytes of primordial follicles [133–135]. However, evidence suggests that MRE11 is required to detect DNA damage in mature oocytes, as blocking MRE11 in the presence of DNA damage increases ɣH2AX foci and results in compromised DNA integrity in MII oocytes [135].
NHEJ repair tends to be error prone because the break ends are directly ligated without the use of a homologous template. Repair involves the binding of KU70/KU80 to each break end. DNA-PKcs then forms a connecting bridge, while XRCC4 acts as a scaffolding protein that allows Ligase IV to ligate the ends. While it is not yet clear what role NHEJ plays in oocytes. Ku70 and Ku80 mutants have been described as fertile, but rarely produce offspring [136, 137]. As these mice exhibit similar early aging without substantially increased cancer levels [136], it would be interesting to determine if these animals also suffer premature ovarian failure.
Interestingly, studies in somatic cells suggest that the choice between DSB repair pathways differs with increasing age [138]. The transcription of gene-encoding proteins involved in both HR and NHEJ have been detected in mammalian oocytes, although it is not clear under which circumstances they might be differentially activated [23, 24, 26, 131, 139]. One hypothesis, based on evidence from Xenopus oocytes, is that pathway choice is influenced by stage of oocyte development, with diplotene-arrested oocytes employing HR, while fully grown, meiotically mature oocytes may preferentially store NHEJ proteins for use in the developing embryo after fertilisation [140, 141].
Detection and response to DNA damage
In somatic cells, although different lesions are detected and repaired by different repair pathways, they all trigger common upstream signaling pathways. These pathways include slowing or arresting of cell-cycle progression [142, 143], changes in chromatin structure at the damage site and the transcriptional induction and posttranslational modification of various proteins involved in DNA repair [144, 145]. The protein kinases ATM, ATR and DNA-PKcs, and members of the poly(ADP-ribose) polymerase (PARP) family are considered to be the master regulators of these signaling pathways, acting together or independently to coordinate the responses to specific types of DNA damage [3, 146].
DNA-PKcs is essential for DSB repair, as deletion in mice results in severe immunodeficiency and radiation hypersensitivity [147]. Specifically, DNA-PKcs mediates the formation of the synaptic complex that brings together the two DNA molecules at the site of DNA DSBs and its kinase activity is only activated once this complex is formed [148]. Interestingly, both male and female DNA-PKcs-deficient mice are fertile [149], suggesting that DNA-PKcs has a non-essential role during meiosis.
Both Atm and Atr are highly expressed in mammalian oocytes [23, 24, 26] and mutation of ATM in humans gives rise to ataxia telangiectasia, a debilitating neurodegenerative disease with symptoms including hypersensitivity to agents that cause DSBs and infertility [150, 151]. Studies using mice and arabidopsis suggest conserved roles in meiosis, particularly during homologous chromosome recombination, and disruption results in abnormal chromosomal synapsis and subsequent chromosome fragmentation [152–156]. Indeed, the failure to repair the developmentally induced DSBs generated during meiotic recombination in ATM-deficient mice results in the complete absence of ovarian follicles and infertility [153, 154, 157]. Phosphorylated-ATM (activated) has also been identified in primordial follicle oocytes within 3 h of exposure to radiation, highlighting its role in the response of oocytes to exogenous sources of DNA damage [119]. Interestingly, low to moderate levels of DNA damage in fully grown prophase-arrested mouse oocytes do not prevent the resumption of meiosis because ATM is not effectively activated [158]. This means that oocytes can re-enter meiosis, even in the presence of DNA damage. However, an ATM-independent pathway involving the spindle assembly checkpoint (SAC) is activated prior to anaphase, which blocks the segregation of chromosomes and the development of mature oocytes [159–161]. In contrast, high levels of DSBs do activate an ATM/Chk1-dependent checkpoint, which maintains oocytes in G2/prophase arrest until repair of the damage occurs [158]. Thus, while ATM is essential for detecting DNA damage in immature oocytes, its role in fully grown GV oocytes appears to be dependent on the extent of damage.
Less is known about the function of PARPs in immature and mature oocytes. PARP-1 is a nuclear enzyme that is activated in response to DNA damage and has a key role in the organization of DNA repair by regulating chromatin structure and DNA metabolism by the poly(ADP-ribosyl)ation of numerous repair proteins and histones [162, 163]. Parp-1 knockout cells and mice are hypersensitive to DNA-damaging agents, including alkylating agents and γ-irradiation [164], but are resistant to oxidative stress-induced cell death [165]. Parp-1 null mice also show enhanced genomic instability and high frequencies of chromosome aberrations, sister chromatid exchanges and telomere shortening, but are still fertile [166–169]. PARP-2 is also activated in response to DNA damage and is required for the repair of single strand DNA breaks [162, 170, 171]. Interestingly, PARP-proteins are expressed in oocytes and have a critical role in the maintenance of chromosome stability during meiosis [172, 173]. Parp-1-deficient oocytes exhibit incomplete homologous chromosome synapsis and persistent histone H2AX phosphorylation [172] and Parp-2-deficient mice exhibiting severely impaired spermatogenesis [173]. Thus, PARP-proteins may also respond to and initiate repair of DNA damage in both resting and growing oocytes.
Repair or apoptosis?
After the initial detection of DNA damage, the cell must ‘decide’ whether to undergo apoptosis, or to repair the damaged DNA. The p53 protein is a transcription factor that is a key regulator of this ‘decision’ and in somatic cells it can initiate four main outcomes after activation; DNA repair, cell-cycle arrest, senescence and apoptosis [174]. In response to DNA damage ATM, CHK1 and CHK2 phosphorylate and activate p53, which then can initiate transcription of, amongst other targets, the CDK inhibitor, p21, to regulate cell-cycle and/or the pro-apoptotic BAX/BAK and PUMA proteins [174]. The choice between these outcomes depends on a number of other variables, indicating that the p53 pathway is responsive to activities of other signaling pathways within the stressed cell [174]. Notably, immature oocytes are already arrested, so presumably can only initiate apoptosis or DNA repair. Interestingly, a number of studies have failed to detect p53 in oocytes [119, 175, 176]. However, the expression of TAp63α, an isoform of p53’s homolog, p63, can be detected at high levels in the immature oocyte nucleus [175, 176]. TAp63α is vital for monitoring the integrity of the female germline, and DNA damage induces its phosphorylation [177] and results in its binding to p53-related DNA sites [175]. Thus, TAp63α represents a specific responder to DNA damage in immature prophase-arrested oocytes [178].
Conclusions
In summary, transcriptional profiling, the study of DNA repair-deficient knockout mouse models and in vitro assays, suggest that oocytes are equipped to execute the repair of a variety of different DNA lesions. However, further understanding of the functional roles of specific proteins at each stage of oocyte development, and the overall contribution of the DNA repair to the maintenance of oocyte quality throughout reproductive life, require further investigation. Such studies will provide insight into the role that DNA repair plays in determining the number and quality of oocytes available to females, which has implications for the length of the fertile lifespan, the age the menopause begins or reproductive senescence and the mechanisms utilized by the female germline to ensure transmission of high fidelity genetic information to future generations.
Funding
This work was supported by funding from the National Health and Medical Research Council (NHMRC) of Australia Project Grant to KH (1100219); AW was supported by a NHMRC Early Career Fellowship (1120300).
Footnotes
Jessica M. Stringer and Amy Winship are contributed equally.
References
- 1.Lindahl T, Barnes D. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–134. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 2.Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 3.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mishina Y, Duguid EM, He C. Direct reversal of DNA alkylation damage. Chem Rev. 2006;106(2):215–232. doi: 10.1021/cr0404702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461(7267):1071–1078. http://www.nature.com/nature/journal/v461/n7267/suppinfo/nature08467_S1.html [DOI] [PMC free article] [PubMed]
- 6.van den Berg MM, van Maarle MC, van Wely M, Goddijn M. Genetics of early miscarriage. Biochem Biophys Acta. 2012;1822(12):1951–1959. doi: 10.1016/j.bbadis.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Wear HM, McPike MJ, Watanabe KH. From primordial germ cells to primordial follicles: a review and visual representation of early ovarian development in mice. J Ovarian Res. 2016;9:36. doi: 10.1186/s13048-016-0246-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rimon-Dahari N, Yerushalmi-Heinemann L, Alyagor L, Dekel N. Ovarian folliculogenesis. Results Probl Cell Differ. 2016;58:167–190. doi: 10.1007/978-3-319-31973-5_7. [DOI] [PubMed] [Google Scholar]
- 9.Zhang H, Liu K. Cellular and molecular regulation of the activation of mammalian primordial follicles: somatic cells initiate follicle activation in adulthood. Hum Reprod Update. 2015;21(6):779–786. doi: 10.1093/humupd/dmv037. [DOI] [PubMed] [Google Scholar]
- 10.McGee EA, Raj RS. Regulators of ovarian preantral follicle development. Semin Reprod Med. 2015;33(3):179–184. doi: 10.1055/s-0035-1552584. [DOI] [PubMed] [Google Scholar]
- 11.Findlay JK, Hutt KJ, Hickey M, Anderson RA. How is the number of primordial follicles in the ovarian reserve established? Biol Reprod. 2015;93(5):111. doi: 10.1095/biolreprod.115.133652. [DOI] [PubMed] [Google Scholar]
- 12.Hanoux V, Pairault C, Bakalska M, Habert R, Livera G. Caspase-2 involvement during ionizing radiation-induced oocyte death in the mouse ovary. Cell Death Differ. 2007;14(4):671–681. doi: 10.1038/sj.cdd.4402052. [DOI] [PubMed] [Google Scholar]
- 13.Kerr JB, Brogan L, Myers M, Hutt KJ, Mladenovska T, Ricardo S, Hamza K, Scott CL, Strasser A, Findlay JK. The primordial follicle reserve is not renewed after chemical or gamma-irradiation mediated depletion. Reproduction. 2012;143(4):469–476. doi: 10.1530/REP-11-0430. [DOI] [PubMed] [Google Scholar]
- 14.Matsuda Y, Tobari I. Chromosomal analysis in mouse eggs fertilized in vitro with sperma exposed to ultraviolet light (UV) and methyl and ethyl methanesulfonate (MMS and EMS) Mutat Res Fundam Mol Mech Mutagen. 1988;198(1):131–144. doi: 10.1016/0027-5107(88)90048-6. [DOI] [PubMed] [Google Scholar]
- 15.Pedersen RA, Brandriff B (1980) Radiation- and drug-induced DNA repair in mammalian oocytes and embryos. In: Generoso WM, Shelby MD, de Serres FJ (eds) DNA repair and mutagenesis in eukaryotes. Basic Life Sciences, vol 15. Springer, Boston, MA, pp 389–410 [DOI] [PubMed]
- 16.Masui Y, Pedersen R. Ultraviolet light-induced unscheduled DNA synthesis in mouse oocytes during meiotic maturation. Nature. 1975;257(5528):705–706. doi: 10.1038/257705a0. [DOI] [PubMed] [Google Scholar]
- 17.Matsumoto Y (1999) Base excision repair assay using Xenopus laevis oocyte extracts. In: Henderson DS (ed) DNA repair protocols. Methods in Molecular Biology™, vol 113. Humana Press, pp 289–300 [DOI] [PubMed]
- 18.Varlet I, Radman M, Brooks P. DNA mismatch repair in Xenopus egg extracts: repair efficiency and DNA repair synthesis for all single base-pair mismatches. Proc Natl Acad Sci. 1990;87(20):7883–7887. doi: 10.1073/pnas.87.20.7883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang QT, Piotrowska K, Ciemerych MA, Milenkovic L, Scott MP, Davis RW, Zernicka-Goetz M. A genome-wide study of gene activity reveals developmental signaling pathways in the preimplantation mouse embryo. Dev Cell. 2004;6(1):133–144. doi: 10.1016/S1534-5807(03)00404-0. [DOI] [PubMed] [Google Scholar]
- 20.Cui XS, Li XY, Yin XJ, Kong IK, Kang JJ, Kim NH. Maternal gene transcription in mouse oocytes: genes implicated in oocyte maturation and fertilization. J Reprod Dev. 2007;53(2):405–418. doi: 10.1262/jrd.18113. [DOI] [PubMed] [Google Scholar]
- 21.Su Y-Q, Sugiura K, Woo Y, Wigglesworth K, Kamdar S, Affourtit J, Eppig JJ. Selective degradation of transcripts during meiotic maturation of mouse oocytes. Dev Biol. 2007;302(1):104–117. doi: 10.1016/j.ydbio.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gasca S, Pellestor F, Assou S, Loup V, Anahory T, Dechaud H, De Vos J, Hamamah S. Identifying new human oocyte marker genes: a microarray approach. Reprod Biomed Online. 2007;14(2):175–183. doi: 10.1016/S1472-6483(10)60785-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Menezo Y, Jr, Russo G, Tosti E, El Mouatassim S, Benkhalifa M. Expression profile of genes coding for DNA repair in human oocytes using pangenomic microarrays, with a special focus on ROS linked decays. J Assist Reprod Genet. 2007;24(11):513–520. doi: 10.1007/s10815-007-9167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaroudi S, Kakourou G, Cawood S, Doshi A, Ranieri DM, Serhal P, Harper JC, SenGupta SB. Expression profiling of DNA repair genes in human oocytes and blastocysts using microarrays. Hum Reprod. 2009;24(10):2649–2655. doi: 10.1093/humrep/dep224. [DOI] [PubMed] [Google Scholar]
- 25.Zeng F, Baldwin DA, Schultz RM. Transcript profiling during preimplantation mouse development. Dev Biol. 2004;272(2):483–496. doi: 10.1016/j.ydbio.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Zheng P, Schramm RD, Latham KE. Developmental regulation and in vitro culture effects on expression of DNA repair and cell cycle checkpoint control genes in rhesus monkey oocytes and embryos. Biol Reprod. 2005;72(6):1359–1369. doi: 10.1095/biolreprod.104.039073. [DOI] [PubMed] [Google Scholar]
- 27.Pan H, O’Brien MJ, Wigglesworth K, Eppig JJ, Schultz RM. Transcript profiling during mouse oocyte development and the effect of gonadotropin priming and development in vitro. Dev Biol. 2005;286(2):493–506. doi: 10.1016/j.ydbio.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 28.Yoon S-J, Kim K-H, Chung H-M, Choi D-H, Lee W-S, Cha K-Y, Lee K-A. Gene expression profiling of early follicular development in primordial, primary, and secondary follicles. Fertil Steril. 2006;85(1):193–203. doi: 10.1016/j.fertnstert.2005.07.1296. [DOI] [PubMed] [Google Scholar]
- 29.Govindaraj V, Krishnagiri H, Chakraborty P, Vasudevan M, Rao AJ. Age-related changes in gene expression patterns of immature and aged rat primordial follicles. Syst Biol Reprod Med. 2017;63(1):37–48. doi: 10.1080/19396368.2016.1267820. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Kou Z, Jing Z, Zhang Y, Guo X, Dong M, Wilmut I, Gao S. Proteome of mouse oocytes at different developmental stages. Proc Natl Acad Sci. 2010;107(41):17639. doi: 10.1073/pnas.1013185107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Liu D, He D, Suo S, Xia X, He X, Han J-DJ, Zheng P. Transcriptome analyses of rhesus monkey preimplantation embryos reveal a reduced capacity for DNA double-strand break repair in primate oocytes and early embryos. Genome Res. 2017;27(4):567–579. doi: 10.1101/gr.198044.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gagliardi C, Liukkonen JR, Phillippi-Falkenstein KM, Harrison RM, Kubisch HM. Age as a determinant of reproductive success among captive female rhesus macaques (Macaca mulatta) Reproduction. 2007;133(4):819–826. doi: 10.1530/REP-06-0323. [DOI] [PubMed] [Google Scholar]
- 33.Govindaraj V, Keralapura Basavaraju R, Rao AJ. Changes in the expression of DNA double strand break repair genes in primordial follicles from immature and aged rats. Reprod Biomed Online. 2015;30(3):303–310. doi: 10.1016/j.rbmo.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Titus S, Li F, Stobezki R, Akula K, Unsal E, Jeong K, Dickler M, Robson M, Moy F, Goswami S, Oktay K. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Science translational medicine. 2013;5(172):172ra121. doi: 10.1126/scitranslmed.3004925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grøndahl ML, Yding Andersen C, Bogstad J, Nielsen FC, Meinertz H, Borup R. Gene expression profiles of single human mature oocytes in relation to age. Hum Reprod. 2010;25(4):957–968. doi: 10.1093/humrep/deq014. [DOI] [PubMed] [Google Scholar]
- 36.Xiao W, Samson L. In vivo evidence for endogenous DNA alkylation damage as a source of spontaneous mutation in eukaryotic cells. Proc Natl Acad Sci USA. 1993;90(6):2117–2121. doi: 10.1073/pnas.90.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jena N. DNA damage by reactive species: mechanisms, mutation and repair. J Biosci. 2012;37(3):503–517. doi: 10.1007/s12038-012-9218-2. [DOI] [PubMed] [Google Scholar]
- 38.Takami M, Preston S, Toyloy V, Behrman HR. Antioxidants reversibly inhibit the spontaneous resumption of meiosis. Am J Physiol Endocrinol Metab. 1999;276(4):E684–E688. doi: 10.1152/ajpendo.1999.276.4.E684. [DOI] [PubMed] [Google Scholar]
- 39.Takami M, Preston S, Behrman H. Eicosatetraynoic and eicosatriynoic acids, lipoxygenase inhibitors, block meiosis via antioxidant action. Am J Physiol Cell Physiol. 2000;278(4):C646–C650. doi: 10.1152/ajpcell.2000.278.4.C646. [DOI] [PubMed] [Google Scholar]
- 40.Karuputhula NB, Chattopadhyay R, Chakravarty B, Chaudhury K. Oxidative status in granulosa cells of infertile women undergoing IVF. Syst Biol Reprod Med. 2013;59(2):91–98. doi: 10.3109/19396368.2012.743197. [DOI] [PubMed] [Google Scholar]
- 41.Elizur SE, Lebovitz O, Orvieto R, Dor J, Zan-Bar T. Reactive oxygen species in follicular fluid may serve as biochemical markers to determine ovarian aging and follicular metabolic age. Gynecol Endocrinol. 2014;30(10):705–707. doi: 10.3109/09513590.2014.924100. [DOI] [PubMed] [Google Scholar]
- 42.Oyawoye O, Abdel Gadir A, Garner A, Constantinovici N, Perrett C, Hardiman P. Antioxidants and reactive oxygen species in follicular fluid of women undergoing IVF: relationship to outcome. Hum Reprod. 2003;18(11):2270–2274. doi: 10.1093/humrep/deg450. [DOI] [PubMed] [Google Scholar]
- 43.Desmeules P, Devine PJ. Characterizing the ovotoxicity of cyclophosphamide metabolites on cultured mouse ovaries. Toxicol Sci. 2006;90(2):500–509. doi: 10.1093/toxsci/kfj086. [DOI] [PubMed] [Google Scholar]
- 44.Nozaki Y, Furubo E, Matsuno T, Fukui R, Kizawa K, Kozaki T, Sanzen T. Collaborative work on evaluation of ovarian toxicity. 6) Two- or four-week repeated-dose studies and fertility study of cisplatin in female rats. J Toxicol Sci. 2009;34(Suppl 1):SP73–SP81. doi: 10.2131/jts.34.s73. [DOI] [PubMed] [Google Scholar]
- 45.Oktem O, Oktay K. A novel ovarian xenografting model to characterize the impact of chemotherapy agents on human primordial follicle reserve. Cancer Res. 2007;67(21):10159–10162. doi: 10.1158/0008-5472.CAN-07-2042. [DOI] [PubMed] [Google Scholar]
- 46.Petrillo SK, Desmeules P, Truong TQ, Devine PJ. Detection of DNA damage in oocytes of small ovarian follicles following phosphoramide mustard exposures of cultured rodent ovaries in vitro. Toxicol Appl Pharmacol. 2011;253(2):94–102. doi: 10.1016/j.taap.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 47.Yucebilgin MS, Terek MC, Ozsaran A, Akercan F, Zekioglu O, Isik E, Erhan Y. Effect of chemotherapy on primordial follicular reserve of rat: an animal model of premature ovarian failure and infertility. Aust N Z J Obstet Gynaecol. 2004;44(1):6–9. doi: 10.1111/j.1479-828X.2004.00143.x. [DOI] [PubMed] [Google Scholar]
- 48.Yuksel A, Bildik G, Senbabaoglu F, Akin N, Arvas M, Unal F, Kilic Y, Karanfil I, Eryilmaz B, Yilmaz P, Ozkanbas C, Taskiran C, Aksoy S, Guzel Y, Balaban B, Ince U, Iwase A, Urman B, Oktem O. The magnitude of gonadotoxicity of chemotherapy drugs on ovarian follicles and granulosa cells varies depending upon the category of the drugs and the type of granulosa cells. Hum Reprod. 2015;30(12):2926–2935. doi: 10.1093/humrep/dev256. [DOI] [PubMed] [Google Scholar]
- 49.Nielsen BF, Schmidt AA, Mulvihill JJ, Frederiksen K, Tawn EJ, Stovall M, Johansen C, Boice JD, Jr, Winther JF. Chromosomal abnormalities in offspring of young cancer survivors: a population-based cohort study in Denmark. J Natl Cancer Inst. 2017 doi: 10.1093/jnci/djx248. [DOI] [PubMed] [Google Scholar]
- 50.Seppanen VI, Artama MS, Malila NK, Pitkaniemi JM, Rantanen ME, Ritvanen AK, Madanat-Harjuoja LM. Risk for congenital anomalies in offspring of childhood, adolescent and young adult cancer survivors. Int J Cancer. 2016;139(8):1721–1730. doi: 10.1002/ijc.30226. [DOI] [PubMed] [Google Scholar]
- 51.Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73(1):39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 52.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair. 2007;6(8):1079–1099. doi: 10.1016/j.dnarep.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 53.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12(2):104–120. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakumi K, Sekiguchi M. Structures and functions of DNA glycosylases. Mutat Res DNA Repair. 1990;236(2–3):161–172. doi: 10.1016/0921-8777(90)90003-N. [DOI] [PubMed] [Google Scholar]
- 55.Seeberg E, Eide L, Bjørås M. The base excision repair pathway. Trends Biochem Sci. 1995;20(10):391–397. doi: 10.1016/S0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- 56.Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal. 2011;14(12):2491–2507. doi: 10.1089/ars.2010.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinha RP, Hader DP. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci. 2002;1(4):225–236. doi: 10.1039/b201230h. [DOI] [PubMed] [Google Scholar]
- 58.Flach G, Johnson MH, Braude PR, Taylor RA, Bolton VN. The transition from maternal to embryonic control in the 2-cell mouse embryo. EMBO J. 1982;1(6):681–686. doi: 10.1002/j.1460-2075.1982.tb01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Braude P, Bolton V, Moore S. Human gene expression first occurs between the four-and eight-cell stages of preimplantation development. Nature. 1988;332(6163):459–461. doi: 10.1038/332459a0. [DOI] [PubMed] [Google Scholar]
- 60.Aitken RJ, Gibb Z, Baker MA, Drevet J, Gharagozloo P. Causes and consequences of oxidative stress in spermatozoa. Reprod Fertil Dev. 2016;28(1–2):1–10. doi: 10.1071/RD15325. [DOI] [PubMed] [Google Scholar]
- 61.Smith TB, Dun MD, Smith ND, Curry BJ, Connaughton HS, Aitken RJ. The presence of a truncated base excision repair pathway in human spermatozoa that is mediated by OGG1. J Cell Sci. 2013;126(6):1488–1497. doi: 10.1242/jcs.121657. [DOI] [PubMed] [Google Scholar]
- 62.Lord T, Aitken RJ. Fertilization stimulates 8-hydroxy-2′-deoxyguanosine repair and antioxidant activity to prevent mutagenesis in the embryo. Dev Biol. 2015;406(1):1–13. doi: 10.1016/j.ydbio.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 63.Ladstätter S, Tachibana-Konwalski K. A surveillance mechanism ensures repair of DNA lesions during zygotic reprogramming. Cell. 2016;167(7):1774.e1713–1787.e1713. doi: 10.1016/j.cell.2016.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsumoto Y, Kim K, Bogenhagen DF. Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: an alternative pathway of base excision DNA repair. Mol Cell Biol. 1994;14(9):6187–6197. doi: 10.1128/mcb.14.9.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oda N, Saxena JK, Jenkins TM, Prasad R, Wilson SH, Ackerman EJ. DNA polymerases α and β are required for DNA repair in an efficient nuclear extract from Xenopus oocytes. J Biol Chem. 1996;271(23):13816–13820. doi: 10.1074/jbc.271.23.13816. [DOI] [PubMed] [Google Scholar]
- 66.Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J Biol Chem. 1996;271(16):9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- 67.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16(11):3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Can Res. 1991;51(12):3075–3079. [PubMed] [Google Scholar]
- 69.Drake JW. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci. 1991;88(16):7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schaaper RM. Base selection, proofreading, and mismatch repair during DNA replication in Escherichia coli. J Biol Chem. 1993;268(32):23762–23765. [PubMed] [Google Scholar]
- 71.Kunkel TA. DNA replication fidelity. J Biol Chem. 2004;279(17):16895–16898. doi: 10.1074/jbc.R400006200. [DOI] [PubMed] [Google Scholar]
- 72.Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R. The mismatch repair system is required for S-phase checkpoint activation. Nat Genet. 2003;33(1):80. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- 73.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair. 2004;3(8–9):1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 74.Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9(1):89–96. doi: 10.1016/S0959-437X(99)80013-6. [DOI] [PubMed] [Google Scholar]
- 75.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 76.Edelmann W, Cohen PE, Kneitz B, Winand N, Lia M, Heyer J, Kolodner R, Pollard JW, Kucherlapati R. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat Genet. 1999;21(1):123–127. doi: 10.1038/5075. [DOI] [PubMed] [Google Scholar]
- 77.Kneitz B, Cohen PE, Avdievich E, Zhu L, Kane MF, Hou H, Kolodner RD, Kucherlapati R, Pollard JW, Edelmann W. MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 2000;14(9):1085–1097. doi: 10.1101/gad.14.9.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei K, Kucherlapati R, Edelmann W. Mouse models for human DNA mismatch-repair gene defects. Trends Mol Med. 2002;8(7):346–353. doi: 10.1016/S1471-4914(02)02359-6. [DOI] [PubMed] [Google Scholar]
- 79.Guo T, Zhao S, Zhao S, Chen M, Li G, Jiao X, Wang Z, Zhao Y, Qin Y, Gao F, Chen ZJ. Mutations in MSH5 in primary ovarian insufficiency. Hum Mol Genet. 2017;26(8):1452–1457. doi: 10.1093/hmg/ddx044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mandon-Pepin B, Touraine P, Kuttenn F, Derbois C, Rouxel A, Matsuda F, Nicolas A, Cotinot C, Fellous M. Genetic investigation of four meiotic genes in women with premature ovarian failure. Eur J Endocrinol. 2008;158(1):107–115. doi: 10.1530/EJE-07-0400. [DOI] [PubMed] [Google Scholar]
- 81.Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85(7):1125–1134. doi: 10.1016/S0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- 82.Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82(2):309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 83.Lipkin SM, Moens PB, Wang V, Lenzi M, Shanmugarajah D, Gilgeous A, Thomas J, Cheng J, Touchman JW, Green ED. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. 2002;31(4):385. doi: 10.1038/ng931. [DOI] [PubMed] [Google Scholar]
- 84.Wang T-F, Kleckner N, Hunter N. Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci. 1999;96(24):13914–13919. doi: 10.1073/pnas.96.24.13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hunter N, Borts RH. Mlh1 is unique among mismatch repair proteins in its ability to promote crossing-over during meiosis. Genes Dev. 1997;11(12):1573–1582. doi: 10.1101/gad.11.12.1573. [DOI] [PubMed] [Google Scholar]
- 86.Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie D-M, Monell C, Arnheim N, Bradley A. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13(3):336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- 87.Thoma F. Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J. 1999;18(23):6585. doi: 10.1093/emboj/18.23.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hirao Y, Yanagimachi R. Detrimental effect of visible light on meiosis of mammalian eggs in vitro. J Exp Zool Part A Ecol Genet Physiol. 1978;206(3):365–369. doi: 10.1002/jez.1402060308. [DOI] [PubMed] [Google Scholar]
- 89.Colton SL, Xu XS, Wang YA, Wang G. The involvement of ataxia-telangiectasia mutated protein activation in nucleotide excision repair-facilitated cell survival with cisplatin treatment. J Biol Chem. 2006;281(37):27117–27125. doi: 10.1074/jbc.M602826200. [DOI] [PubMed] [Google Scholar]
- 90.Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Can Res. 2002;62(17):4899–4902. [PubMed] [Google Scholar]
- 91.Vermeulen W, Jaeken J, Jaspers NG, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum complementation group G associated with Cockayne syndrome. Am J Hum Genet. 1993;53(1):185–192. [PMC free article] [PubMed] [Google Scholar]
- 92.Cooper PK, Nouspikel T, Clarkson SG, Leadon SA. Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science. 1997;275(5302):990–993. doi: 10.1126/science.275.5302.990. [DOI] [PubMed] [Google Scholar]
- 93.Nouspikel T, Lalle P, Leadon SA, Cooper PK, Clarkson SG. A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Proc Natl Acad Sci USA. 1997;94(7):3116–3121. doi: 10.1073/pnas.94.7.3116. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 94.Lans H, Marteijn JA, Schumacher B, Hoeijmakers JH, Jansen G, Vermeulen W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet. 2010;6(5):e1000941. doi: 10.1371/journal.pgen.1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sugitani N, Sivley RM, Perry KE, Capra JA, Chazin WJ. XPA: a key scaffold for human nucleotide excision repair. DNA Repair (Amst) 2016;44:123–135. doi: 10.1016/j.dnarep.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu Rev Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- 97.Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer. 2001;1(1):22–33. doi: 10.1038/35094000. [DOI] [PubMed] [Google Scholar]
- 98.Tsutakawa SE, Cooper PK. Transcription-coupled repair of oxidative DNA damage in human cells: mechanisms and consequences. Cold Spring Harb Symp Quant Biol. 2000;65:201–215. doi: 10.1101/sqb.2000.65.201. [DOI] [PubMed] [Google Scholar]
- 99.Sugasawa K, Ng JM, Masutani C, Iwai S, van der Spek PJ, Eker AP, Hanaoka F, Bootsma D, Hoeijmakers JH. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2(2):223–232. doi: 10.1016/S1097-2765(00)80132-X. [DOI] [PubMed] [Google Scholar]
- 100.Cheo DL, Ruven HJ, Meira LB, Hammer RE, Burns DK, Tappe NJ, van Zeeland AA, Mullenders LH, Friedberg EC. Characterization of defective nucleotide excision repair in XPC mutant mice. Mutat Res. 1997;374(1):1–9. doi: 10.1016/S0027-5107(97)00046-8. [DOI] [PubMed] [Google Scholar]
- 101.de Vries A, van Oostrom CT, Hofhuis FM, Dortant PM, Berg RJ, de Gruijl FR, Wester PW, van Kreijl CF, Capel PJ, van Steeg H, Verbeek SJ. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature. 1995;377(6545):169–173. doi: 10.1038/377169a0. [DOI] [PubMed] [Google Scholar]
- 102.Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7(6):427–439. doi: 10.1016/S0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 103.de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, Meradji M, Hoeijmakers JH. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296(5571):1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- 104.de Boer J, de Wit J, van Steeg H, Berg RJ, Morreau H, Visser P, Lehmann AR, Duran M, Hoeijmakers JH, Weeda G. A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol Cell. 1998;1(7):981–990. doi: 10.1016/S1097-2765(00)80098-2. [DOI] [PubMed] [Google Scholar]
- 105.Clauson C, Schärer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol. 2013;5(10):a012732. doi: 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schärer OD. DNA interstrand crosslinks: natural and drug-induced DNA adducts that induce unique cellular responses. ChemBioChem. 2005;6(1):27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 107.Chow EJ, Stratton KL, Leisenring WM, Oeffinger KC, Sklar CA, Donaldson SS, Ginsberg JP, Kenney LB, Levine JM, Robison LL, Shnorhavorian M, Stovall M, Armstrong GT, Green DM. Pregnancy after chemotherapy in male and female survivors of childhood cancer treated between 1970 and 1999: a report from the Childhood Cancer Survivor Study cohort. Lancet Oncol. 2016;17(5):567–576. doi: 10.1016/S1470-2045(16)00086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moldovan G-L, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kennedy RD, D’Andrea AD. The Fanconi anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19(24):2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 110.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7(2):249–262. doi: 10.1016/S1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 111.Chen M, Tomkins DJ, Auerbach W, McKerlie C, Youssoufian H, Liu L, Gan O, Carreau M, Auerbach A, Groves T. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat Genet. 1996;12(4):448–451. doi: 10.1038/ng0496-448. [DOI] [PubMed] [Google Scholar]
- 112.Cheng NC, van de Vrugt HJ, van der Valk MA, Oostra AB, Krimpenfort P, de Vries Y, Joenje H, Berns A, Arwert F. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum Mol Genet. 2000;9(12):1805–1811. doi: 10.1093/hmg/9.12.1805. [DOI] [PubMed] [Google Scholar]
- 113.Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003;12(16):2063–2076. doi: 10.1093/hmg/ddg219. [DOI] [PubMed] [Google Scholar]
- 114.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481(7381):287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 115.McDonald JP, Rapić-Otrin V, Epstein JA, Broughton BC, Wang X, Lehmann AR, Wolgemuth DJ, Woodgate R. Novel human and mouse homologs of Saccharomyces cerevisiae DNA polymerase η. Genomics. 1999;60(1):20–30. doi: 10.1006/geno.1999.5906. [DOI] [PubMed] [Google Scholar]
- 116.Yamada A, Masutani C, Iwai S, Hanaoka F. Complementation of defective translesion synthesis and UV light sensitivity in xeroderma pigmentosum variant cells by human and mouse DNA polymerase η. Nucleic Acids Res. 2000;28(13):2473–2480. doi: 10.1093/nar/28.13.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yagi Y, Ogawara D, Iwai S, Hanaoka F, Akiyama M, Maki H. DNA polymerases η and κ are responsible for error-free translesion DNA synthesis activity over a cis–syn thymine dimer in Xenopus laevis oocyte extracts. DNA Repair. 2005;4(11):1252–1269. doi: 10.1016/j.dnarep.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 118.Tatone C, Amicarelli F, Carbone MC, Monteleone P, Caserta D, Marci R, Artini PG, Piomboni P, Focarelli R. Cellular and molecular aspects of ovarian follicle ageing. Hum Reprod Update. 2008;14(2):131–142. doi: 10.1093/humupd/dmm048. [DOI] [PubMed] [Google Scholar]
- 119.Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH, Bouillet P, Mills A, Scott CL, Findlay JK, Strasser A. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell. 2012;48(3):343–352. doi: 10.1016/j.molcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kerr JB, Hutt KJ, Cook M, Speed TP, Strasser A, Findlay JK, Scott CL. Cisplatin-induced primordial follicle oocyte killing and loss of fertility are not prevented by imatinib. Nat Med. 2012;18(8):1170–1172. doi: 10.1038/nm.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25(5):409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Green DM, Kawashima T, Stovall M, Leisenring W, Sklar CA, Mertens AC, Donaldson SS, Byrne J, Robison LL. Fertility of female survivors of childhood cancer: a report from the childhood cancer survivor study. J Clin Oncol. 2009;27(16):2677–2685. doi: 10.1200/JCO.2008.20.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shaffer LG, Lupski JR. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet. 2000;34:297–329. doi: 10.1146/annurev.genet.34.1.297. [DOI] [PubMed] [Google Scholar]
- 124.Ravel C, Letur H, Le Lannou D, Barthelemy C, Bresson JL, Siffroi JP, Genetics Commission of the French Federation of C High incidence of chromosomal abnormalities in oocyte donors. Fertil Steril. 2007;87(2):439–441. doi: 10.1016/j.fertnstert.2006.06.053. [DOI] [PubMed] [Google Scholar]
- 125.Lim AS, Tsakok MF. Age-related decline in fertility: a link to degenerative oocytes? Fertil Steril. 1997;68(2):265–271. doi: 10.1016/S0015-0282(97)81513-0. [DOI] [PubMed] [Google Scholar]
- 126.McFadden DE, Friedman JM. Chromosome abnormalities in human beings. Mutat Res. 1997;396(1–2):129–140. doi: 10.1016/S0027-5107(97)00179-6. [DOI] [PubMed] [Google Scholar]
- 127.Zhang YX, Zhang YP, Gu Y, Guan FJ, Li SL, Xie JS, Shen Y, Wu BL, Ju W, Jenkins EC, Brown WT, Zhong N. Genetic analysis of first-trimester miscarriages with a combination of cytogenetic karyotyping, microsatellite genotyping and arrayCGH. Clin Genet. 2009;75(2):133–140. doi: 10.1111/j.1399-0004.2008.01131.x. [DOI] [PubMed] [Google Scholar]
- 128.Heilstedt HA, Ballif BC, Howard LA, Lewis RA, Stal S, Kashork CD, Bacino CA, Shapira SK, Shaffer LG. Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am J Hum Genet. 2003;72(5):1200–1212. doi: 10.1086/375179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC. Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science. 2014;343(6170):533–536. doi: 10.1126/science.1247671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27(3):247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 131.Kujjo LL, Laine T, Pereira RJ, Kagawa W, Kurumizaka H, Yokoyama S, Perez GI. Enhancing survival of mouse oocytes following chemotherapy or aging by targeting Bax and Rad51. PLoS One. 2010;5(2):e9204. doi: 10.1371/journal.pone.0009204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Williams RS, Williams JS, Tainer JA. Mre11-Rad50–Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol. 2007;85(4):509–520. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 133.Cherry SM, Adelman CA, Theunissen JW, Hassold TJ, Hunt PA, Petrini JHJ. The Mre11 complex influences DNA repair, synapsis, and crossing over in murine meiosis. Curr Biol. 2007;17(4):373–378. doi: 10.1016/j.cub.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Inagaki A, Roset R, Petrini JH. Functions of the MRE11 complex in the development and maintenance of oocytes. Chromosoma. 2016;125(1):151–162. doi: 10.1007/s00412-015-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mayer A, Baran V, Sakakibara Y, Brzakova A, Ferencova I, Motlik J, Kitajima TS, Schultz RM, Solc P. DNA damage response during mouse oocyte maturation. Cell Cycle. 2016;15(4):546–558. doi: 10.1080/15384101.2015.1128592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li H, Vogel H, Holcomb VB, Gu YS, Hasty P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol Cell Biol. 2007;27(23):8205–8214. doi: 10.1128/Mcb.00785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li GC, Ouyang HH, Li XL, Nagasawa H, Little JB, Chen DJ, Ling CC, Fuks Z, Cordon-Cardo C. Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol Cell. 1998;2(1):1–8. doi: 10.1016/S1097-2765(00)80108-2. [DOI] [PubMed] [Google Scholar]
- 138.Gorbunova V, Seluanov A, Mao Z, Hine C. Changes in DNA repair during aging. Nucleic Acids Res. 2007;35(22):7466–7474. doi: 10.1093/nar/gkm756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yukawa M, Oda S, Mitani H, Nagata M, Aoki F. Deficiency in the response to DNA double-strand breaks in mouse early preimplantation embryos. Biochem Biophys Res Commun. 2007;358(2):578–584. doi: 10.1016/j.bbrc.2007.04.162. [DOI] [PubMed] [Google Scholar]
- 140.Goedecke W, Vielmetter W, Pfeiffer P. Activation of a system for the joining of nonhomologous DNA ends during Xenopus egg maturation. Mol Cell Biol. 1992;12(2):811–816. doi: 10.1128/mcb.12.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hagmann M, Adlkofer K, Pfeiffer P, Bruggmann R, Georgiev O, Rungger D, Schaffner W. Dramatic changes in the ratio of homologous recombination to nonhomologous DNA-end joining in oocytes and early embryos of Xenopus laevis. Bio Chem Hoppe-Seyler. 1996;377(4):239–250. doi: 10.1515/bchm3.1996.377.4.239. [DOI] [PubMed] [Google Scholar]
- 142.Lowndes NF, Murguia JR. Sensing and responding to DNA damage. Curr Opin Genet Dev. 2000;10(1):17–25. doi: 10.1016/S0959-437X(99)00050-7. [DOI] [PubMed] [Google Scholar]
- 143.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15(17):2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 144.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 145.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR (2008) HP1-[bgr] mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453(7195):682–686. http://www.nature.com/nature/journal/v453/n7195/suppinfo/nature06875_S1.html [DOI] [PubMed]
- 146.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 147.Kurimasa A, Kumano S, Boubnov NV, Story MD, Tung C-S, Peterson SR, Chen DJ. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol Cell Biol. 1999;19(5):3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.DeFazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002;21(12):3192–3200. doi: 10.1093/emboj/cdf299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kurimasa A, Ouyang H, Dong LJ, Wang S, Li XL, Cordon-Cardo C, Chen DJ, Li GC. Catalytic subunit of DNA-dependent protein kinase: impact on lymphocyte development and tumorigenesis. Proc Natl Acad Sci USA. 1999;96(4):1403–1408. doi: 10.1073/pnas.96.4.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, WynshawBoris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86(1):159–171. doi: 10.1016/S0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 151.Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 152.Culligan KM, Britt AB. Both ATM and ATR promote the efficient and accurate processing of programmed meiotic double-strand breaks. Plant J. 2008;55(4):629–638. doi: 10.1111/j.1365-313X.2008.03530.x. [DOI] [PubMed] [Google Scholar]
- 153.Barlow C, Liyanage M, Moens PB, Tarsounas M, Nagashima K, Brown K, Rottinghaus S, Jackson SP, Tagle D, Ried T, Wynshaw-Boris A. Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development. 1998;125(20):4007–4017. doi: 10.1242/dev.125.20.4007. [DOI] [PubMed] [Google Scholar]
- 154.Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10(19):2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- 155.Keegan KS, Holtzman DA, Plug AW, Christenson ER, Brainerd EE, Flaggs G, Bentley NJ, Taylor EM, Meyn MS, Moss SB. The Atr and Atm protein kinases associate with different sites along meiotically pairing chromosomes. Genes Dev. 1996;10(19):2423–2437. doi: 10.1101/gad.10.19.2423. [DOI] [PubMed] [Google Scholar]
- 156.Moens PB, Tarsounas M, Morita T, Habu T, Rottinghaus ST, Freire R, Jackson SP, Barlow C, Wynshaw-Boris A. The association of ATR protein with mouse meiotic chromosome cores. Chromosoma. 1999;108(2):95–102. doi: 10.1007/s004120050356. [DOI] [PubMed] [Google Scholar]
- 157.Di Giacomo M, Barchi M, Baudat F, Edelmann W, Keeney S, Jasin M. Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proc Natl Acad Sci USA. 2005;102(3):737–742. doi: 10.1073/pnas.0406212102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Marangos P, Carroll J. Oocytes progress beyond prophase in the presence of DNA damage. Curr Biol. 2012;22(11):989–994. doi: 10.1016/j.cub.2012.03.063. [DOI] [PubMed] [Google Scholar]
- 159.Collins JK, Lane SI, Merriman JA, Jones KT. DNA damage induces a meiotic arrest in mouse oocytes mediated by the spindle assembly checkpoint. Nat Commun. 2015;6:8553. doi: 10.1038/ncomms9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lin F, Ma X-S, Wang Z-B, Wang Z-W, Luo Y-B, Huang L, Jiang Z-Z, Hu M-W, Schatten H, Sun Q-Y. Different fates of oocytes with DNA double-strand breaks in vitro and in vivo. Cell Cycle. 2014;13(17):2674–2680. doi: 10.4161/15384101.2015.945375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yuen WS, Merriman JA, O’Bryan MK, Jones KT. DNA double strand breaks but not interstrand crosslinks prevent progress through meiosis in fully grown mouse oocytes. PLoS One. 2012;7(8):e43875. doi: 10.1371/journal.pone.0043875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schreiber V, Dantzer F, Ame J-C, de Murcia G (2006) Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7(7):517–528. http://www.nature.com/nrm/journal/v7/n7/suppinfo/nrm1963_S1.html [DOI] [PubMed]
- 163.Masutani M, Nozaki T, Nakamoto K, Nakagama H, Suzuki H, Kusuoka O, Tsutsumi M, Sugimura T. The response of Parp knockout mice against DNA damaging agents. Mutat Res Rev Mutat Res. 2000;462(2):159–166. doi: 10.1016/S1383-5742(00)00033-8. [DOI] [PubMed] [Google Scholar]
- 164.De Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M. Requirement of poly (ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci. 1997;94(14):7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Heller B, Wang Z-Q, Wagner EF, Radons J, Burkle A, Fehsel K, Burkart V, Kolb H. Inactivation of the poly (ADP-ribose) polymerase gene affects oxygen radical and nitric oxide toxicity in islet cells. J Biol Chem. 1995;270(19):11176–11180. doi: 10.1074/jbc.270.19.11176. [DOI] [PubMed] [Google Scholar]
- 166.Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9(5):509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 167.di Fagagna FdA, Hande MP, Tong W-M, Lansdorp PM, Wang Z-Q, Jackson SP. Functions of poly (ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat Genet. 1999;23(1):76–80. doi: 10.1038/12680. [DOI] [PubMed] [Google Scholar]
- 168.Simbulan-Rosenthal CM, Haddad BR, Rosenthal DS, Weaver Z, Coleman A, Luo R, Young HM, Wang Z-Q, Ried T, Smulson ME. Chromosomal aberrations in PARP −/− mice: genome stabilization in immortalized cells by reintroduction of poly (ADP-ribose) polymerase cDNA. Proc Natl Acad Sci. 1999;96(23):13191–13196. doi: 10.1073/pnas.96.23.13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang Z-Q, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11(18):2347–2358. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Amé J-C, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Höger T, Ménissier-de Murcia J, de Murcia G. PARP-2, a novel mammalian DNA damage-dependent poly (ADP-ribose) polymerase. J Biol Chem. 1999;274(25):17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 171.de Murcia JM, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Amé JC, Dierich A, LeMeur M. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22(9):2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Yang F, Baumann C, De La Fuente R. Persistence of histone H2AX phosphorylation after meiotic chromosome synapsis and abnormal centromere cohesion in poly (ADP-ribose) polymerase (Parp-1) null oocytes. Dev Biol. 2009;331(2):326–338. doi: 10.1016/j.ydbio.2009.05.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dantzer F, Mark M, Quenet D, Scherthan H, Huber A, Liebe B, Monaco L, Chicheportiche A, Sassone-Corsi P, De Murcia G. Poly (ADP-ribose) polymerase-2 contributes to the fidelity of male meiosis I and spermiogenesis. Proc Natl Acad Sci. 2006;103(40):14854–14859. doi: 10.1073/pnas.0604252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9(5):402–412. http://www.nature.com/nrm/journal/v9/n5/suppinfo/nrm2395_S1.html [DOI] [PubMed]
- 175.Suh EK, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z, Elvin JA, Bronson RT, Crum CP, McKeon F. p63 protects the female germ line during meiotic arrest. Nature. 2006;444(7119):624–628. doi: 10.1038/nature05337. [DOI] [PubMed] [Google Scholar]
- 176.Livera G, Petre-Lazar B, Guerquin M-J, Trautmann E, Coffigny H, Habert R. p63 null mutation protects mouse oocytes from radio-induced apoptosis. Reproduction. 2008;135(1):3–12. doi: 10.1530/rep-07-0054. [DOI] [PubMed] [Google Scholar]
- 177.Tuppi M, Kehrloesser S, Coutandin DW, Rossi V, Luh LM, Strubel A, Hötte K, Hoffmeister M, Schäfer B, De Oliveira T, Greten F. Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat Struct Mol Biol. 2018;25(3):261–269. doi: 10.1038/s41594-018-0035-7. [DOI] [PubMed] [Google Scholar]
- 178.Gebel J, Tuppi M, Krauskopf K, Coutandin D, Pitzius S, Kehrloesser S, Osterburg C, Dötsch V. Control mechanisms in germ cells mediated by p53 family proteins. J Cell Sci. 2017;130(16):2663–2671. doi: 10.1242/jcs.204859. [DOI] [PubMed] [Google Scholar]
- 179.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 180.David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447(7147):941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Friedberg EC, Wagner R, Radman M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science. 2002;296(5573):1627–1630. doi: 10.1126/science.1070236. [DOI] [PubMed] [Google Scholar]
- 182.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 183.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Labhart P. Ku-dependent nonhomologous DNA end joining in Xenopus egg extracts. Mol Cell Biol. 1999;19(4):2585–2593. doi: 10.1128/mcb.19.4.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 186.Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]