

Abstract
Platelets are anucleated cells that circulate in the blood as sentinels of tissue integrity. In fact, they are rich in a plethora of proteins and other factors stored in different granules which they selectively release upon stimulation. Moreover, platelets synthesize a vast number of lipids and release various types of vesicles, including exosomes which are rich in genetic material. Platelets possess a central function to interact with other cell types, including inflammatory cells and cancer cells. Recent findings have enlightened the capacity of platelets to induce changes in the phenotype of cancer cells which acquire invasiveness thus enhancing their metastatic potential. Thus, it has been hypothesized that targeting the platelet may represent a novel strategy to prevent the development and progression of cancer. This is supported by the efficacy of the antiplatelet agent low-dose aspirin. Studies are ongoing to verify whether other antiplatelet agents share the anticancer effectiveness of aspirin.
Keywords: Platelets, Cancer cells, Metastasis, Aspirin, Antiplatelet agents, Prostaglandin E2, Epithelial–mesenchymal transitions
Introduction
Cancer is a complex disease and represents a major health problem worldwide due to its growing incidence in the general population [1]. This phenomenon is dependent on several reasons, including the limited knowledge on the mechanisms involved in the initial events of cancer development. This is a significant deficiency which contributes to the development of inadequate preventive strategies.
World Cancer Report shows that action on smoking, diet, and infections can prevent one-third of cancers [2]. Interestingly, smoking and Western lifestyle resulting in an overall energy imbalance due to a highly caloric diet, rich in fat, combined with moderate physical activity, are also risk factors for the development of cardiovascular disease [3]. Another significant finding which has enlightened the similarities of cancer and heart disease is that the administration of the antiplatelet agent low-dose aspirin for some years is associated with a reduced incidence and mortality due to cancer, in particular, colorectal cancer (CRC), at long-term follow-up [4]. Collectively, this information has laid the basis for the hypothesis that platelet activation in response to tissue damage is a significant contributor to early events in tumorigenesis [5]. In fact, platelets, once activated, play a central role in cell–cell communication through the plethora of soluble factors and microvesicles, rich in genetic material, which they release [6]. Importantly, in cancer patients, platelets can present a different repertoire of proteins, mRNAs and microRNAs stored in their granules due to the capacity of platelets to uptake different molecules present in the environment (including plasma circulation) [7, 8].
The activation of platelets is now recognized as a central event linking tissue damage to the development of chronic inflammation [9]. Thus, activated platelets may contribute to the formation of the tumor microenvironment which participates in multistep tumorigenesis [10, 11]. In fact, it is noteworthy that tumors are not a collection of relatively homogeneous cancer cells, but instead, they are as complex organs constituted by individual specialized cell types scattered in the tumor microenvironment [12]. The new information opens the way to novel strategies to restrain cancer development by affecting chronic inflammation and possibly platelet activation. The chemopreventive data of low-dose aspirin against CRC in clinical studies were recently found appropriate by the US Preventive Services Task Force which recommends the use of the drug for primary prevention of cardiovascular disease and CRC [13].
The development of tumor metastases plays a significant contribution to the death of cancer patients. Thus, the prevention of an initial metastasis in high-risk patients and new metastases in patients with the limited disease are central therapeutic goals [14].
Clinical and experimental findings support the role of platelets in the multistep process of invasion and metastasis [15]. Tumor cells begin metastasis by the invasion of the healthy tissue surrounding the primary tumor; then, they enter the bloodstream, arrest at the first capillary bed encountered and extravasate to colonize distant tissues [15]. The initiation of metastasis can be partly attributed to the genetic heterogeneity among subpopulations of cells within primary tumors [16]. However, other events should occur to induce an invasive phenotype of cancer cells that involve the interplay of cancer cells with stromal cells and the environment. Importantly, cancer cells can change their genetic program under the pressure of the innumerable molecules produced in tumor microenvironment: (1) some cancer cells can acquire the features of a stem cell due to the epithelial mesenchymal transition (EMT) phenomenon [17, 18]; (2) cancer cells can acquire the expression of some megakaryocytic genes thus promoting the coagulation cascade or the activation of platelets (platelet mimicry) [19]; (3) some cancer cells have the ability to mimic the activities of endothelial cells and to participate in neovascularization (vasculogenic mimicry) [20]. These events are promoted by the capacity of cancer cells to acquire novel phenotypic features which are orchestrated by the direct crosstalk with cells of the stroma and platelets and/or by several mediators that they release during cellular activation.
In this Review, we provide an overview of platelet biology and novel functions, beyond hemostasis and thrombosis, which contribute to the development of cancer and its progression. We have summarized the knowledge available on the anticancer effect of conventional antiplatelet agents used in the setting of acute coronary syndrome. Also, we have described novel antiplatelet agents in clinical development which might be potentially efficacious for the prevention and treatment of cancer and metastases.
Platelet biology
Platelet structure
Platelets are small subcellular fragments (2–5 μm of diameter, 0.5 μm thick and 6–10 fl volume) with a mean half-life of 7–10 days [21]. They arise from the cytoplasm of megakaryocytes, unique hematological polyploid cells that undergo differentiation and maturation upon the action by interleukin IL-3, IL-6 and IL-11, and thrombopoietin [22]. Despite their small volume, platelets are complex cellular elements; their ultra-structure analysis allows to understand the functional aspects of these cells better. Platelets are made up of four structural regions. The peripheral zone, necessary for the release of α-granular content during platelet activation [23] which consists of a glycocalyx with adhesive properties, a plasmatic membrane rich in tissue factors [24] and an intricate cytoskeletal regulating the shape of platelets. The sol–gel zone, which is the corresponding cytoplasm part of the cellular fragment and has a pivotal role in providing a contracting system and supporting the open canalicular system and the dense tubular system [25]. It is responsible for changes in platelet morphology and in granule release [26]. The organelle zone which allows platelets to be unique and distinct compared to other blood cells due to the presence of secretory organelles, including lysosomes, dense granules, and α-granules [27] (Fig. 1). Lysosomes contain acid hydrolases, phospholipase, and kinases that act as proteolytic and hydrolytic enzymes [28]. Dense granules are the smallest granules in the cytoplasm, with a high electron density due to the presence of elevated levels of calcium, phosphate, magnesium, serotonin, adenosine triphosphate (ATP) and adenosine diphosphate (ADP) that play a central role in platelet activation, aggregation and thrombus formation [29]. The α-granules contain adhesive proteins, coagulation factors, fibrinolytic factors, growth factor mitogens, cytokines, and chemokines. Some proteins, including P-selectin and CD40 ligand (CD40L), translocate from these granules to the surface of platelet plasma membrane in response to stimulation and play a role in inflammation and atherosclerosis [30]. Importantly, α-granules contain a plethora of growth factors which contribute to the tissue repair process. Among them, there are platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and vascular endothelial growth factor (VEGF) [31]. Moreover, platelet α-granules contain transforming growth factor (TGF)-β, a molecule involved in cellular migration [31]. However, platelets release also antiangiogenic factors, including the platelet factor 4 (PF-4) [32]. Alpha-granules also contain β-thromboglobulin with chemotactic properties for fibroblasts and inflammatory cells [33]. Finally, there is the membrane zone, and plasma membrane makes extensive invaginations that form a network inside the platelet [23].
Fig. 1.
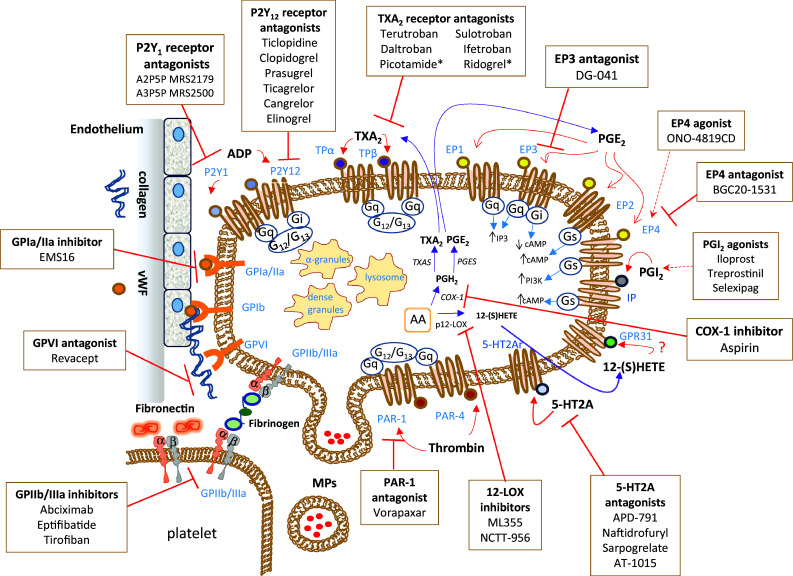
Platelet structure, primary receptors, eicosanoid machinery, and targeted therapeutics. Platelets are involved in the release of many mediators that are stored in different granules in the cytoplasm and microvesicles. A plethora of proteins are contained in α-granules; there are cell adhesion proteins, blood clotting factors, growth and angiogenic factors. The plasma membrane of platelets expresses several transmembrane receptors, involved in the crosstalk with other platelets and different cell types. Platelets adhere to the damaged vascular endothelium through the binding of integrin receptors (GP, glycoproteins) to the extracellular matrix proteins, such as collagen and VWF (von Willebrand factor). Possible strategies to inhibit the adhesion of platelets to a damaged endothelium involve the use of agents that interact with collagen binding sites, such as revacept, thus preventing the activation of the collagen receptors GPIa/IIa and GPVI expressed on the plasma membrane of platelets. Platelet aggregation, mediated by the binding of fibrinogen or fibronectin to GPIIb/IIIa on agonist-stimulated platelets, is inhibited by different antagonists of the GPIIb/IIIa receptor (abciximab, eptifibatide, and tirofiban). The activation of platelets by ADP (adenosine diphosphate) is mainly affected by antagonists of the P2Y12 receptor, among them, there is the pro-drug clopidogrel. An important antiplatelet agent is aspirin, which irreversibly inhibits the activity of cyclooxygenase (COX)-1 involved in the production of prostaglandin (PG) H2 that is then converted to the potent pro-aggregatory agent thromboxane (TX)A2 by the activity of TXA2 synthase (TXAS). A secondary product of COX-1-dependent pathway is PGE2 produced by the activity of different PGE synthases (PGES). TXA2 and PGE2 cause different platelet responses by the interaction with specific receptors. Various receptor antagonists in clinical development are reported. Picotamide and ridrogrel act by a dual mechanism involving the blockage of TP receptors and the inhibition of TXAS. There are four different receptors for PGE2 on the platelet surface: EP1, EP2, EP3 and EP4. The stimulation of EP3 leads to platelet activation, and specific antagonists are under clinical development, including DG-041. EP4 and EP2 signaling may increase intraplatelet cAMP (cyclic adenosine monophosphate) levels and thus possibly counteracting the platelet activation by EP3. Some agonists and antagonists of these receptors have been synthesized. An important receptor present on the platelet plasma membrane is the IP receptor for prostacyclin (PGI2), for which different commercially available agonists have been developed (Iloprost, Treprostinil, Selexipag). Another abundant eicosanoid produced by platelets is 12(S)-HETE [12(S)-hydroxyeicosatetraenoic acid] via the activity of the platelet-type lipoxygenase (p12-LOX). The mechanism of action is 12-(S)HETE has not been entirely understood. Recently, it has been proposed the activation of the orphan receptor GPR31 by 12-(S)HETE. The discovery of selective inhibitors of 12-LOX, such as ML355, will allow enhancing our knowledge of the role played by 12-LOX in health and disease. Protease-activated receptors (PARs) are involved in platelet activation by thrombin. There are two receptors, known as PAR-1 and PAR-4. However, PAR-1 possesses a higher affinity for thrombin. The PAR-1 antagonist vorapaxar was approved for clinical use in 2014. New antiplatelet agents include serotonin receptor antagonists (5-HT2A antagonists)
Activated platelets may also release two types of membrane vesicles (Fig. 1), such as microparticles (MPs) and exosomes. MPs are vesicles with a size of 0.1–1 μm and no nucleus while exosomes have a size of 40–100 nm and derive from multivesicular endosomes within the cell [34, 35]. The MPs recognize the target cell due to the expression of cell surface proteins [36].
Despite the fact that platelets lack the genomic DNA, they are not completely silent. Platelets, in fact, retain a small part of the RNA of megakaryocytes and the translation machinery. Thus, in response to platelet activation, the translation of proteins with relevant biologic activities has been reported [37]. In fact, thrombin-activated platelets synthesize B-cell lymphoma 3-encoded protein (Bcl-3), a member of the IkB-α family of regulatory proteins [38]. Evangelista et al. [39] have shown that the complete suppression of thromboxane (TX)A2 biosynthesis by aspirin in vitro was recovered in response to thrombin and fibrinogen due to the occurrence of de novo cyclooxygenase(COX)-1 synthesis in platelets. This phenomenon might interfere with the complete and persistent suppression of TXA2 biosynthesis by aspirin necessary for cardioprotection [40]. Moreover, fibrinogen via the interaction with β3 integrin provides a signal for de novo synthesis of P-selectin in platelets which is required for maintenance of the P-selectin content [41]. P-selectin is involved in the interaction of platelets with other cells, including carcinoma cells [42], and it also promotes the Th1-like immune response [43]. Tinoco et al. found that P-selectin glycoprotein ligand-1 (PSGL-1) expressed on the surface of T cells induces their dysfunction (a phenomenon called exhaustion) leading to the impediment to control and eliminate cancer cells [44].
Furthermore, platelets present a high number of mitochondria containing several copies of their circular genome. The development of appropriate techniques has allowed characterizing the platelet transcriptome and proteome [45, 46].
Platelet lipidomics of eicosanoids
In response to the stimulation of platelets by different platelet agonists, several phospholipases, including cytosolic phospholipase A2 (cPLA2) are activated. cPLA2 causes the hydrolysis of the sn2 acyl bond of glycerophospholipids to produce free fatty acids (FAs). The most significant FA regarding cell signaling is arachidonic acid (AA) (Fig. 2). AA is then transformed through enzymatic and nonenzymatic pathways to the formation of several biologically active compounds collectively known as eicosanoids [47, 48].
Fig. 2.

Major pathways for the biosynthesis of eicosanoids in platelets. Arachidonic acid (AA), esterified in membrane phospholipids, can be released upon platelet activation by different stimuli via the action of phospholipases (PLs), including the cytosolic phospholipase A2 (cPLA2). In platelets, AA is transformed to PGH2 by the activity of COX-1; then, PGH2 is the substrate of different synthases, thus leading to the formation of the prostanoids: TXA2, PGE2, PGD2, PGF2α. Thromboxane synthase (TXAS), cytosolic PGE synthase (cPGES), microsomal PGE synthase-2 (mPGES-2), lipocalin-type prostaglandin D synthase (L-PGDS), hematopoietic prostaglandin D synthase (H-PGDS) and PGF synthase (PGFS) are the downstream synthases involved in the production of prostanoids. AA is also transformed to 12(S)-HETE by the activity of p12-LOX. The enzyme produces 12(S)-hydroperoxy-eicosatetraenoic acid [12(S)-HPETE] which is, then, converted to 12(S)-HETE by glutathione reductase (GR). PGE2, PGD2 and 12(S)-HETE can be esterified into membrane phospholipids by the action of fatty acid CoA ligase (FACL) to produce new lipid mediators, i.e., the phospholipid esterified eicosanoids
Eicosanoids comprise a wide array of lipids generated by three major enzymatic pathways: the COX pathway; the lipoxygenase (LOX) pathway; and the cytochrome P-450 monooxygenase pathway. Additionally, free radical peroxidation can non-enzymatically convert AA to isoprostanes [47, 48].
In platelets, the activity of COX-1 catalyzes the conversion of AA to prostaglandin (PG) H2 via a two-step process (Fig. 2). In the first step, the COX activity introduces two molecules of oxygen to free AA, generating the bicyclic peroxide intermediate PGG2. In the second phase, the peroxidase activity of COX-1 reduces PGG2 to the unstable endoperoxide PGH2 [49]. Then, PGH2 is metabolized to the biologically active prostanoids by the activity of different and specific prostanoid synthases (Fig. 2). In the platelet, the primary product of the COX-1 pathway is
TXA2, produced from PGH2 through the action of TXA2 synthase (TXAS). TXA2 is a potent stimulus for platelet aggregation and vascular smooth muscle cell contraction via the activation of thromboxane receptors (TPs) [50, 51] (Fig. 1). In platelets, the biosynthesis of PGE2 from PGH2 is catalyzed by two different synthases: a cytosolic PGE synthase (cPGES) and microsomal PGE synthase type-2 (mPGES-2) [52] (Fig. 2). Finally, minor products of platelet COX-1 pathway are PGF2α [53] and PGD2 [54] (Fig. 2).
PGE2 binds with similar affinity to four receptors: EP1, EP2, EP3, and EP4 [55] (Fig. 1). Some isoforms of the EP3 receptor have been identified. EP3 receptors have been shown to couple to Gi and Gq proteins [55] (Fig. 1). At low concentrations (<10−6 M), PGE2 increases the sensitivity of platelets to aggregating agents markedly via the activation of the EP3 receptor Gi signaling, thus leading to the inhibition of cyclic adenosine monophosphate (cAMP) formation [56] (Fig. 1). Platelets also express EP2 and EP4 receptors which are coupled to Gs, thus inducing the increase of cAMP levels; this effect might translate into the inhibition of platelet activation. This is the mechanism by which the endothelial product of COX activity prostacyclin (PGI2) inhibits platelet activation via the binding to the PGI2 receptor IP expressed on platelets [57] (Fig. 1). However, it has been shown that PGE2 may inhibit platelet activation only at high concentrations through the activation of IP receptors [56].
LOXs constitute a family of non-heme iron dioxygenases that produce bioactive lipids such as leukotrienes, lipoxins, 12- hydroxy-eicosatetraenoic acids [HETE] and 15-HETE [58–60].
Three major types of LOX have been identified, 5-LOX, 12-LOX, and 15-LOX [59]. Human platelet-type 12-LOX (p12-LOX) was established in the early 1970s by Hamberg and Samuelsson [61] (Figs. 1, 2). The initial product formed from AA by the activity of p12-LOX is exclusively the biologically active metabolite 12(S)-hydroperoxy-eicosatetraenoic acid [12(S)-HPETE] [61], that upon reduction results in the production of 12(S)-HETE [62, 63] (Fig. 2). 12(S)-HETE potentiates platelet activation, thrombin generation, calcium mobilization and α-granule secretion [64]. Recent work using small molecule inhibitors supports a pro-thrombotic role for 12-LOX in human platelets [63]. The precise mechanism of action of 12-HETE on platelets is not completely understood. Guo et al. showed that GPR31, a plasma membrane orphan G protein-coupled receptor, displays high affinity for the human 12(S)-HETE [65] (Fig. 1). However, further studies should be performed to clarify its involvement in platelet function.
Recently, a new class of lipid mediators has been identified in human platelets, called oxidized phospholipids (oxPLs) [66]. OxPLs were initially characterized as non-enzymatically generated species, but recent studies have indicated that these mediators are also generated through enzymatic activity. In response to platelet activation, the enzymatic products of COX-1 and p12-LOX, i.e., PGE2, PGD2 and 12(S)-HETE can be reesterified possibly using fatty acid CoA ligase (FACL) [67] (Fig. 2) into membrane phospholipids thus changing the plasma membrane composition. In fact, eicosanoids are attached to phosphatidylethanolamine (PE) and phosphatidylcholine (PC). In activated platelets, both 12(S)-HETE and PG-containing phospholipids have been detected [66]. The administration of low-dose aspirin which is associated with the inhibition of COX-1 activity translates into the reduction of prostanoids incorporated into membrane PLs [66]. It has been shown that 12(S)-HETE-PE becomes externalized after its synthesis, suggesting that oxPLs may play a role in extracellular phospholipid-dependent signaling events, like coagulation, that is a process requiring the presence on the cell surface of negatively charged PE and phosphatidylserine [68]. Further studies are necessary to elucidate the biological functions of oxPLs in platelets completely. In particular, it remains to explore the possibility that lipid oxidation influences the dynamic behavior of the cell membrane.
LOX products are associated with carcinogenic processes such as tumor cell proliferation, differentiation, angiogenesis and apoptosis [60, 69]. The exposure to 12(S)-HETE or the overexpression of 12-LOX in cancer cells is associated with enhanced cell growth as well as migration [70].
Major platelet receptors
The platelet membrane contains many transmembrane receptors on their surface (Fig. 1). Most of these receptors have a direct role in hemostasis. In Fig. 1, the most important pharmacological tools to affect their activation are shown. Glycoprotein (GP) Ib-IX-V receptors are involved in platelet adhesion and aggregation, following interaction with von Willebrand factors(VWF), stored in α-granules; their activation leads to the release of both TXA2 and ADP by platelets [71].
Three immunoreceptor tyrosine-based activation motif (ITAM)-coupled receptors are expressed by platelets: (1) GPVI, a receptor for proteins of the extracellular matrix, such as collagen and laminin, that signals via the associated ITAM-containing Fc receptor γ chain (FcRγ); (2) FcγRIIA, an ITAM-containing receptor for immune complexes; (3) C-type lectin-like receptor (CLEC)-2, a receptor for podoplanin, a mucin-type transmembrane protein expressed in multiple tissues [72].
GPVI, the first platelet collagen receptor, is indispensable in the modulation of platelet adhesion and aggregation mediated by integrins [73]. When GPVI binds to collagen (Fig. 1) or other ligands such as the collagen-related peptide (CRP), rapidly induces the activation of the integrin GPIIb/IIIa (αIIbβ3) [74]. The receptor GPIIb/IIIa contributes to the formation of platelet aggregates via the binding of its primary ligand fibrinogen which enables the cross-linking of adjacent platelets [74] (Fig. 1). Other ligands, such as fibronectin, can mediate platelet aggregation in different physiological and pathological conditions [75, 76]. Plasma fibronectin is a dimer that contains two tripeptides Arg-Gly-Asp (RGD) sites that could potentially cross-link adjacent platelets similarly to fibrinogen. Indeed, early studies showed that plasma fibronectin might support platelet aggregation [77]. However, other findings showed the inhibitory effect of fibronectin on platelet aggregation [78, 79]. Recently, Reheman et al. [80] generated fibrinogen/VWF/conditional plasma fibronectin triple-deficient (TKO; Cre+, Fnflox/flox, Fg/VWF−/−) mice and their results show that fibronectin may play dual roles in thrombosis and hemostasis. Its soluble form is inhibitory possibly for competition with more potent integrin ligands such as fibrinogen [81]. In contrast, extracellular matrix-like fibrils of fibronectin contribute to thrombosis and hemostasis by either self-assembly or covalent interaction with fibrin or other matrix proteins [80, 82, 83].
FcγRIIA is best known for its role in immune-mediated thrombocytopenia and thrombosis. Moreover, FcγRIIA plays a major role in the outside-in signaling of integrin αIIbβ3 and, thus, contributes to thrombus stabilization at sites of vascular injury [84] (Fig. 3).
Fig. 3.
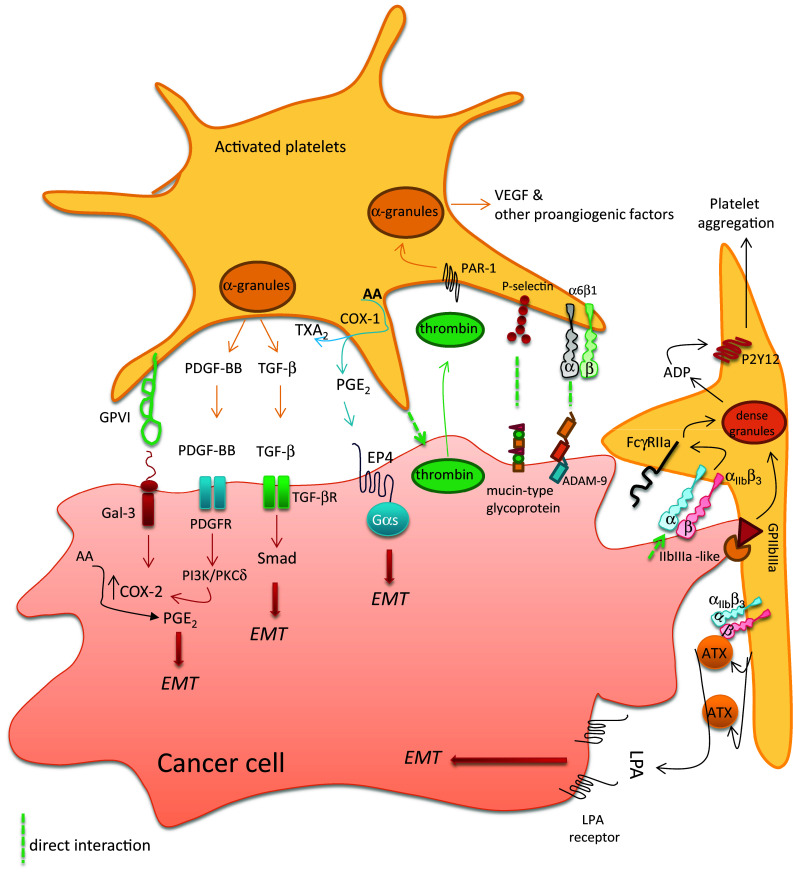
Molecular determinants involved in the crosstalk between platelets and cancer cells. Platelets interact with tumor cells through different receptors expressed on platelet surface (i.e., collagen receptor GPVI, P-selectin and the integrins α6β1 or αIIbβ3, and GPIIb/IIIa). The direct interaction between the two cell types triggers platelet activation, the secretion of platelet α-granule content, i.e., growth and angiogenic factors (such as PDGF, TGF-β, and VEGF). Moreover, platelets release ADP from dense granules and synthesize prostanoids, including TXA2 and PGE2. The interaction of platelets with cancer cells causes the induction of COX-2 in cancer cells, which contributes to a further increase in PGE2 production. Aberrant PGE2 generation is a hallmark of cancer. The overexpression of COX-2 in cancer cells involves both transcriptional and posttranscriptional mechanisms mediated by the release of PDGF. Among the numerous events triggered by platelet–cancer cell crosstalk, there is the induction of the epithelial–mesenchymal transition (EMT) phenomenon in cancer cells, which can be mediated by soluble mediators(proteins and lipids) (modified from Dovizio et al. [97] and Guillem-Llobat et al. [98])
In the family of G-protein-coupled receptors (GPCRs), we have to mention the thrombin protease-activated receptors (PAR)-1 and PAR-4, the ADP receptors P2Y1 and P2Y12 and TXA2 receptors TPs [85] (Fig. 1).
Thrombin is a serine protease generated during the coagulation cascade involved in fibrin formation and platelet aggregation and secretion. It acts through the activation of two GPCRs expressed on the platelet surface, i.e., PAR-1 and PAR-4 [86, 87] (Fig. 1). PARs are activated after thrombin-mediated proteolytic cleavage of their N-terminal exodomain. PAR-1 is the primary human platelet receptor. In fact, thrombin is 10–100 times more potent to activate PAR-1 than PAR-4. The stimulation of PAR receptors increases the levels of cytosolic Ca2+, induces a shape change in platelets, and stimulates TXA2 production and ADP secretion [88].
ADP, released from dense granules of platelets, induces platelet aggregation, but it is also involved in platelet granule secretion. These effects occur via the activation of the purinergic receptors [89]. Three different type of ADP receptors are expressed on platelets: a P2X-type ion channel-linked receptor and two P2Y-type GPCRs, P2Y1 and P2Y12. The receptors coupled with G proteins, Gq, and Gi, which activate PLC and inhibit adenylyl cyclase, respectively (Fig. 1). ADP-dependent activation of the P2Y1 receptor leads to an increase in intracellular calcium via Gq coupling; P2Y12 activation translates into Gi coupling thus resulting in the inhibition of adenylyl cyclase and the prevention of the phosphorylation of vasodilator-stimulated phosphoprotein (VASP) [89]. This protein has been recently identified as a microfilament- and a focal contact-associated protein whose phosphorylation correlates with the inhibition of platelet function [90]. Furthermore, the stimulation of P2Y12 leads to the activation of phosphoinositide 3-kinase (PI3K) and Akt kinase [89].
Human TP receptor exists in two isoforms TPα and TPβ, generated through an alternative splicing, which differ only at the C-terminal tails [55]. The Gq-coupled TP subtype, TPα, and Gi-coupled TPβ subtype have been shown in human platelets (Fig. 1). Both the TPα and TPβ subtypes mediate the stimulation of PLC associated with enhanced levels of intracellular inositol 1,4,5-triphosphate and diacylglycerol. The formation of inositol 1,4,5-triphosphate induces an increase in the cytosolic concentration of Ca2+, whereas the release of diacylglycerol activates PKC. In contrast, TPα and TPβ play an antagonist role towards the adenylate cyclase, stimulating and inhibiting it, respectively [91]. Moreover, TP activation is associated with the induction of the G12/13 pathway which involves the activation of RhoGTPase nucleotide exchange factors (RhoGEFs) [92] (Fig. 1). Using the TXA2 mimetic U46619, it has been shown that platelet TP activation causes platelet shape change and intracellular Ca2+ mobilization independently of secreted granule contents [93]. However, U46619 acts at the Gq-coupled TP receptor to cause secretion of granule contents. Finally, U46619-induced platelet aggregation depends on Gi stimulation by ADP and other released granule contents [93].
Platelet functions
In their resting state, platelets show a disc-shaped and circulate freely in the bloodstream. When there is a vascular endothelial damage/dysfunction, platelets are activated and adhere to the injured endothelium. Then, they release a plethora of growth factors and inflammatory mediators thus recruiting inflammatory cells [94]. In this scenario, platelets release ADP and synthesize prostanoids, mainly TXA2 which amplify the platelet response by activating further platelets and leading to the formation of platelet aggregates [94]. Although these events are a physiological response to repair of damaged endothelium, the induction of a chronic inflammatory response in the context of impaired vascular protective mechanisms translates into the development of atherothrombosis [9]. In addition to their canonical roles, platelets show a versatile phenotype, thus contributing to different processes as innate and adaptive immune responses, atherosclerosis and tumor metastasis [95].
Role of platelets in metastasis
Several findings have pointed out the central role of platelets in the metastatic dissemination of cancer cells. In fact, numerous clinical observations indicate a potential relationship between the blood coagulation system, platelet functions, and cancer spreading via the bloodstream [15].
Cancer cells that reach the bloodstream can interact with platelets. The crosstalk between platelets and cancer cells confers an advantage for metastatic progression through different mechanisms [15]. Platelet aggregates surrounding tumor cells promote their survival, protection from immune elimination and the adhesion of tumor cells to the endothelium which permits the arrest and extravasation of cancer cells [15].
However, recent findings have enlightened novel mechanisms by which platelets may promote the development of tumor metastasis. In fact, platelets can induce a more malignant phenotype in cancer cells characterized by enhanced migratory properties [96–98]. Platelets, through a direct contact with cancer cells and the release of mediators, including PDGF, TGF-β and PGE2, can promote EMT in cancer cells [96–98] (Fig. 3). This biological process consists of the capacity of cancer cells to acquire mesenchymal markers (such as vimentin, fibronectin and the transcription factors Twist, Snail, and Zeb) with the concomitant loss of epithelial markers (such as E-cadherin). These events translate into the acquisition of a disseminating phenotype that allows tumor cells to colonize distant organs [17]. In 2011, Labelle and collaborators [96] reported that platelets induce EMT in murine colon and breast cancer cells through the release of TGF-β1 and the activation of TGF-β/Smad signaling. This leads to the development of a more aggressive tumor phenotype (Fig. 3). Recently, we have shown that the crosstalk between human platelets and human adenocarcinoma cell line HT-29 leads to the induction of EMT in tumor cells [97] associated with an aberrant expression of COX-2, which is a recognized marker of tumor progression [99]. These events were triggered both by a direct interaction between the two cell types and by the release of platelet-derived PDGF (Fig. 3). In fact, using different pharmacological tools, we found that platelets interact with HT29 cells through the platelet collagen receptor GPVI and the galectin-3 expressed on the surface of cancer cells and which contains a collagen-like domain [97] (Fig. 3). Thus, the use of revacept, a new antiplatelet agent which blocks the activation of platelet GPVI [100] (Fig. 1), caused the reduction of platelet-induced COX-2 expression in tumor cells and also the changes in EMT-related genes (e.g., Zeb, Twist, vimentin, and E-cadherin) [97].
Recently, Guillem-Llobat et al. investigated whether platelets trigger colon cancer cells for metastasis and whether pharmacological inhibition of platelet function may prevent it. Platelets induced a mesenchymal-like phenotype in HT29 cells by the downregulation of E-cadherin and upregulation of Twist1, enhanced cell mobility and a pro-aggregatory action on platelets [98]. These changes were prevented by different antiplatelet agents, aspirin (an inhibitor of COX-1), DG-041 (an antagonist of the PGE2 EP3 receptor) and ticagrelor (a P2Y12 receptor antagonist) (Fig. 1). The authors also found that PGE2, released from platelets, triggered the molecular events in HT29 cells through the activation of EP4 receptor signaling [98] (Fig. 3). It was shown that the injection of HT29 cells with a mesenchymal-like phenotype, into the circulation of immunodeficient mice, activated platelets which released enhanced levels of TXA2 and PGE2 in vivo [98]. The prothrombotic phenotype of cancer cells undergoing EMT participated to the development of metastases. In fact, the administration of low-dose aspirin, which inhibited platelet activation and the biosynthesis of prostanoids, was associated with reduced formation of metastases [98].
The effect of different anticoagulants was investigated in the platelet/cancer cell cross-talk [101]: (1) heparin and low-molecular weight heparin (LMWH) which indirectly inhibit thrombin by strongly catalyzing the function of antithrombin; (2) the Xa inhibitor fondaparinux; and (3) warfarin which indirectly affects the coagulation cascade and thrombin generation by depleting the active form of the vitamin K [101].
Breast cancer cells (MCF-7) induced the secretion of growth factors, especially VEGF (Fig. 3), from platelets and this response was prevented by heparin and fondaparinux. Suppression of VEGF release was not seen from platelets of the patients who were exposed to the oral anticoagulant warfarin. In contrast, inhibition of VEGF release was shown in patients treated with LMWH and a lower extent in patients anticoagulated with fondaparinux [101]. Altogether these results suggest a thrombin-dependent mechanism in the tumor cell-mediated release of platelet angiogenic proteins and platelet angiogenic response. It has been shown that human adenocarcinomas of the colon (HCT-8 and LoVo) and one anaplastic murine tumor (Hut-20) cell lines can induce platelet activation via the generation of thrombin. This phenomenon may participate in tumor cell-mediated platelet responses [102]. Cancer cell-derived thrombin generation can depend on enhanced expression of tissue factor (TF) and cancer procoagulant (CP), i.e., a cysteine protease derived from a broad spectrum of malignant and embryonic (amnion-chorion) tissue [103].
Another study investigated the mechanisms involved in platelet secretion by the highly metastatic colorectal cancer cell line Caco-2 and the prostate carcinoma cell line PC3M-luc. These cells stimulated the release of platelet dense granules (Fig. 3), including the release of ADP; then, ADP caused the aggregation of platelets. Mitrugno et al. [104] showed a novel essential role for the platelet immune receptor FcγRIIa in cancer cell-induced platelet activation (Fig. 3). Using pharmacological tools, it was found that the inhibition of immune FcγRIIa abolished tumor cell-induced platelet secretion and aggregation [104]. The interaction between cancer cells and platelets activated FcγRIIa-spleen tyrosine kinase(Syk)-PLCγ signaling pathway and dense granule secretion. These effects caused a further platelet activation, and secretion of granule contents, thus contributing to platelet recruitment and protection of circulating tumor cells that ultimately facilitates tumor cell survival in the circulation and at metastatic sites [104] (Fig. 3). Thus, FcγRIIa could be a possible target for an antimetastatic therapy.
Mannori et al., examined the adhesion of six colon cancer cell lines to purify recombinant E-, P-, and L-selectin. Among them, LS 180, T84, and COLO 205 can bind to all three selectins. In contrast, the colon cancer cell line COLO 320 interacts with P- and L-selectin, but not E-selectin. HT29 cells can bind to E-selectin but not P- or L-selectin. They demonstrated that P-selectin mediates adhesive interactions of some colon cancer cells with thrombin-activated platelets. This interaction appeared to depend mainly on mucin-type glycoproteins expressed on the surface of cancer cells [105] (Fig. 3).
The implication of platelet receptor GPIIb/IIIa on the crosstalk between platelets and cancer cells has been investigated in some studies using different cancer cell types [106–108]. In particular, Boukerche and colleagues [106] showed that melanoma cells (M3Dau) interacted with platelets by the platelet receptor GPIIb/IIIa and the GPlIb/lIla-like complex expressed on tumor cells (Fig. 3). Platelets promote ADP release from melanomas by interacting with the tumor surface. Then, ADP induces platelet aggregation and degranulation leading to the formation of larger platelet-tumor aggregates [106].
Recently, using a genetic animal model of megakaryocyte-restricted knockout strategy, Mammadova-Bach and collaborators [109] showed that platelets interact with breast and colon tumor cells through the binding of platelet integrin α6β1 and tumor ADAM-9, a member of disintegrin and metalloproteinase family (Fig. 3). This interaction promoted platelet activation, granule secretion, and subsequent endothelial transmigration of tumor cells, which culminated in the promotion of tumor metastasis. In this study, in addition to genetic deletion of platelet α6β1, GoH3, an integrin α6-blocking antibody, was used that prevented platelet-tumor cell cross-talk and diminished lung metastasis in wild-type mice. This finding suggests the possibility of developing a therapeutic strategy to target this integrin and interfere with tumor metastasis in cancer patients [109].
The role of platelet-derived lysophosphatidic acid (LPA) in the metastatic process
Lysophosphatidic acid (LPA) is a bioactive phospholipid involved in numerous cellular responses through the activation of specific GPCRs. It has been reported that platelets represent the primary source of LPA identified so far. Activated platelets release a high amount of different molecular species of LPA [110] which promotes platelet shape change and aggregation through the stimulation of LPA receptor type 5 (LPA5) [111]. Several studies suggest that LPA plays a significant role in the development of cancer [112]. In breast and ovarian cancer, LPA, produced by blood platelets, is a major factor promoting bone metastases [113]. The mechanism involves the activation of the LPA receptor type 1 (LPA1) expressed in tumor cells [114]. The blockage of this receptor using a specific LPA1 antagonist prevented tumor cell–platelet interaction without affecting normal platelet functions [114].
It was recently demonstrated that platelets are involved in the biosynthesis of LPA through the release of Autotaxin (ATX), an LPA-producing enzyme. In fact, Leblanc and collaborators [115] reported that in tumor cell-induced platelet aggregation, platelets release ATX (both as free enzymes and/or bound to β3 integrins, αIIbβ3 and αVβ3) which hydrolyzed LPA precursors (phosphatidic acid, PC, phosphatidylserine, and PE) to form LPA, and thus promoting cancer cell invasion and metastasis [115]. One mechanism explaining the pro-metastatic action of LPA is the induction of EMT [116, 117] (Fig. 3). In ovarian cancer cells, LPA promoted the nuclear translocation of β-catenin with the transcriptional activation of Wnt/β-catenin target genes and hypoxia-induced factor-1α (HIF1α) thus inducing the expression of mesenchymal marker genes [116, 117].
Pharmacological targeting of the platelet to fight cancer metastasis
The evidence supporting the hypothesis that platelet activation is involved in the development of cancer and the promotion of metastasis opens the way to the possible chemopreventive use of antiplatelet agents. Here, we overview the results of the anti-cancer effects obtained with conventional antiplatelet agents used to prevent atherothrombosis and with novel agents in clinical development.
Low-dose aspirin
The analyses of the data from cardiovascular prevention randomized clinical trials (RCTs) with aspirin have shown that the use of the drug, even at the low-doses of 75–100 mg daily which target mainly the platelet, reduces incidence and mortality due to CRC and other types of cancer [4, 118]. Interestingly, the aspirin anticancer effect was associated with the prevention of the formation of distal metastases [119] (Table 1).
Table 1.
Experimental and clinical evidence of the anticancer effects of antithrombotic agents
| Drug | Target | Effect | References |
|---|---|---|---|
| Aspirin | COX-1 |
Reduction of cancer incidence and death, in RCTs Anti-metastatic effect in a mouse model of hematogenous metastasis |
[98, 118, 119] |
| Monoclonal antibody MoAb | GPIIb/IIIa receptors | Inhibition of platelet–melanoma interactions | [106] |
| Monoclonal antibody 10E5 and XV454 | GPIIb/IIIa receptors | Reduction of lung metastases in mice | [132, 133] |
| Revacept | Collagen-like binding sites | Prevention of the upregulation of COX-2 and EMT in platelet–tumor cell cocultures | [97] |
| Heparin/fondaparinux | Indirect inhibitors of thrombin and Factor Xa | Inhibition of the activation of platelets by breast cancer cells | [101] |
| Clopidrogel (active metabolite) | P2Y12 receptor | Its coadministration with aspirin prevents or delays the development of hepatocarcinoma and improves survival | [131] |
| DG-041 | Platelet EP3 receptor | Prevention of platelet-dependent induction of EMT and migration in colon cancer cells | [98] |
Aspirin, acetylsalicylic acid (ASA), is a member of nonsteroidal antiinflammatory drugs (NSAIDs) which inhibit prostanoid biosynthesis by an irreversible inactivation of COX-1 and COX-2. This occurs through the acetylation of a specific serine residue located in the cyclooxygenase active site, at position 529 and 516 of COX-1 and COX-2, respectively [120–122]. Despite the short pharmacological half-life (i.e., 20 min), the daily administration of aspirin at low-doses causes an antiplatelet effect because of irreversible COX-1 inactivation (occurring both in the pre-systemic and systemic circulation) in the anucleated platelets characterized by a low rate of protein synthesis. Chronic dosing with low-dose aspirin causes a virtually complete inhibition of platelet COX-1 activity (≥97%), maximal COX-1 acetylation (75%) and inhibition of platelet function throughout dosing interval (24 h) [123, 124].
Numerous studies using biomarkers of prostanoid biosynthesis in vivo have shown that low-dose aspirin affects profoundly platelet COX-1 activity while causing only a marginal effect on vascular prostanoid biosynthesis dependent on COX-2 activity [5, 47, 123, 125]. This selective effect is due to the capacity of nucleated cells to recover a functional COX-2 in the interval between aspirin doses. This knowledge of the pharmacodynamics of aspirin led to hypothesize that the anticancer effect of aspirin was due to its a selective inhibitory action on the platelet [5, 125, 126]. However, the use of a direct biomarker of aspirin effect which consists in the assessment of the extent of the acetylation of COX-1 in cells and tissues [124, 127] has allowed to detect the acetylation of COX-1 by aspirin on colorectal mucosa of individuals undergoing CRC screening. The effect on colorectal mucosa was lower than the acetylation of platelet COX-1. However, it translated into a significant inhibition of intestinal mucosal PGE2 associated with reduced phosphorylation of the S6 protein of the 40S ribosomal subunit (pS6) [127], a protein involved in the protein synthesis and cell growth [128]. Thus, low-dose aspirin, in addition to inhibiting platelet function and reducing the release of platelet-derived factors, may have a direct effect on the target tissue by preventing the activation of pro-tumorigenic pathways [5].
The antimetastatic effect of aspirin was recently demonstrated in vivo in a mouse model of hematogenous metastasis using human adenocarcinoma cell line HT29 [98] (Table 1). Guillem-Llobat et al. [98] showed that the administration of low-dose aspirin, which inhibited platelet activation and the biosynthesis of prostanoids, reduced the formation of lung metastases. The anti-metastatic effect of low-dose aspirin involved the inhibition of (1) the prothrombotic properties of cancer cells; (2) cancer cell EMT and migratory capacity.
Altogether these findings allow going a step further in the interpretation of the mechanisms of aspirin as an anticancer agent. This information is relevant for the selection of the appropriate dose of aspirin to use for the prevention of cancer in patients.
P2Y12 receptor antagonists
The ADP platelet receptor P2Y12 is the target of effective antithrombotic agents, including the thienopyridines (ticlopidine, clopidogrel, and prasugrel) that irreversibly inhibit the receptor and the novel direct and reversible antagonists (ticagrelor, cangrelor, and elinogrel) [129].
Despite several lines of evidence, obtained in experimental animal models, sustain a possible anticancer effect of these drugs, the proof that this strategy is effective in patients is still missing.
Here, we overview the results obtained by targeting P2Y12 receptor in experimental models of tumorigenesis/metastasis in vitro and in vivo. Wang et al. [130] demonstrated that tumor metastases are reduced in P2Y12-deficient mice. The coadministration of the antiplatelet drugs aspirin and clopidogrel prevented or delayed the development of hepatocellular carcinoma and improved survival in a mouse model of chronic immune-mediated hepatitis B (Table 1) [131].
Blockage of platelet GPIIb/IIIa receptors
As reported above, the involvement of platelet receptor GPIIb/IIIa in the crosstalk of platelets and a melanoma cell line was found [106]. In this study, Fab fragments of the monoclonal antibody MoAb (LYP18), directed against the platelet GPIIb/IIIa complex, inhibited platelet–melanoma cell interactions and platelet–platelet aggregation [106] (Table 1). In a murine model of metastasis, Nierodzik and colleagues [132] found that the blockage of the platelet GPIIb/IIIa receptor, using the monoclonal antibody 10E5, decreased lung colonization of cancer cells (Table 1). It has been reported that the oral inhibitor of GPIIb/IIIa, XV454, protected from metastasis formation in a murine model of lung cancer [133] (Table 1).
PAR antagonists
The importance of PAR receptors in platelet function was investigated by Italiano and collaborators [134] who showed that distinct populations of platelet α-granules, containing different angiogenesis influencing proteins, can be differentially released. The secretion of the different sets of α-granules from platelets is regulated by PAR-1 and PAR-4 activation [134]. Thus, these receptors can play a crucial role in regulating angiogenesis and, in turn, modulate the processes of wound healing and tumor growth [134, 135]. The involvement of PAR receptors in metastasis is shown by the results of in vivo studies conducted in a murine model of hematogenous metastasis. Melanoma cells were intravenously injected in Par4(−/−) mice, and the protection from lung metastasis was observed [136]. Also, a recent study focused on the crosstalk between breast cancer cells and platelets showed that heparin and fondaparinux reduced the activation of platelets by tumor cells through the prevention of PAR-1 activation by thrombin (Table 1) [101].
Blockage of platelet GPVI receptor
As reported in the section of platelet biology, GPVI is a key platelet receptor for collagen. It is a single span transmembrane receptor, with two immunoglobulin domains associated with the FcR-γ chain containing the ITAM subunit. The involvement of this platelet receptor in metastasis was shown by the results of studies conducted in vivo and in vitro. In a murine model of metastasis, using a Lewis lung carcinoma (D121) or melanoma (B16F10.1) cell line, an approximately 50% reduction in the number of visible tumor foci was found in GPVI-deficient mice versus control C57BL/6J mice [137].
Inhibition of GPVI-mediated platelet activation can be achieved both by anti-GPVI antibodies and by the soluble GPVI receptor revacept, a dimeric soluble GPVI-Fc fusion protein, which inhibits platelet aggregation without altering general hemostasis when administered to humans [100]. The results of studies performed in vitro showed that the exposure of HT29 colon cancer cells to revacept prevented the induction of COX-2 and the expression of EMT markers induced by the interaction with platelets [97]. Revacept interfered with the interaction of platelet collagen receptors with galectin-3 [97], a protein highly expressed in cancer cells which contains a collagen-like domain [138] (Table 1). The efficacy of revacept as anticancer agent should be verified in vivo in animal models of metastasis and if confirmed this agent should be tested in patients.
EP3 antagonists
PGE2 production, increased in inflamed atherosclerotic plaques, may participate in the activation of platelets via the EP3 signaling pathway [139]. A highly selective EP3 antagonist has been developed, i.e., DG-041 [140]. The administration of DG-041 to humans completely inhibited platelet aggregation with no concurrent increase in bleeding time (even at high doses) [141].
The effect of DG-041 in platelet–HT29 cell interaction was recently evaluated [98]. DG-041 prevented the induction of a mesenchymal-like phenotype in cancer cells with migratory properties, and these effects were associated with reduced biosynthesis of platelet TXA2 and PGE2 (Table 1). Since platelets, but not HT29 cells, express EP3 receptors, the effects of DG-041 were dependent on a specific inhibitory action on the platelet receptor [98]. This study has enlightened the role of platelet-derived PGE2 on the induction of a metastatic phenotype in cancer cells. PGE2 acted on HT29 cells via the activation of EP4 receptors (Fig. 3) [98]. PGE2-dependent activation of cancer cell EP4 was involved in the induction of EMT. However, the migratory properties of cancer cells required the direct interaction of platelets with cancer cells together with the activation of EP4 by PGE2 (Fig. 3) [98].
Conclusions
Platelets play an important role in the processes of hemostasis. However, new knowledge has enlightened that platelets orchestrate the activation of other cells involved in tissue repair, including inflammatory cells and stromal cells [135]. Moreover, platelets express and secrete many pro-inflammatory molecules that serve to initiate and modulate immune responses [142, 143]. In the context of cancer metastasis, the formation of circulating platelet–tumor cell aggregates protects malignant cells from immune elimination by natural killer (NK) cells [144]. It is noteworthy that platelets express Toll-like receptors (TLRs) [145] which participate in infectious processes through the induction of an antibacterial response [146]. Platelets might also promote autoimmune diseases, including multiple sclerosis [147].
If the platelet responses are unrestrained, they play a fundamental role in the development of pathological conditions such as atherothrombosis [9, 96]. Similarly, platelets promote tumorigenesis by the link existing between platelet activation and the development of chronic inflammation. Moreover, platelets contribute to cancer development through their crosstalk with cancer cells. Cancer cells acquire a prothrombotic phenotype, thus promoting the formation of platelet aggregates which surround the cancer cells; then, the exchange of factors turns on different signaling pathways which lead to the acquisition of an advantage for cancer cells in respect of survival and invasiveness. Thus, the final goal of platelet–cancer cell interaction is to facilitate tumor colonization to distant organs [15]. The occurrence of these pro-tumorigenic functions of platelets is sustained by the efficacy of the antiplatelet agent low-dose aspirin to reduce the incidence and mortality for cancer [5].
More basic and clinical research is necessary to develop a broad armamentarium of effective antiplatelet agents with the capacity to affect the multistep process of cancer metastasis. In fact, many pieces of evidence suggest that different cancer cell types have developed specific mechanisms to activate platelets. Thus, effective antimetastatic treatments should involve (1) the characterization of the molecular determinants of platelet–cancer cell interaction and (2) the selection of the appropriate drug which affects the specific platelet receptor. However, the efficacy of low-dose aspirin as an anticancer agent through the inhibition of the biosynthesis of prostanoids, suggests that the blockage of the amplification of the primary platelet response is sufficient to restrain cancer progression [5].
There is an urgent need to perform basic and clinical research focused on the study of the role played by platelet activation in the different steps of cancer development. The results of these studies will allow developing novel strategies to prevent cancer and to avoid that primary cancer cells metastasize through the blood vessels to various distant organs.
Acknowledgements
This work was supported by the Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) (Grant PRIN 2010–2011, Protocol Number 2010FHH32M), and Associazione Italiana per la Ricerca sul Cancro (Grant IG-12111) (to P. Patrignani).
Abbreviations
- AA
Arachidonic acid
- ADP
Adenosine diphosphate
- ATP
Adenosine triphosphate
- ATX
Autotaxin
- Bcl-3
B-cell lymphoma 3-encoded protein
- cAMP
Cyclic adenosine monophosphate
- CD40L
CD40 ligand
- CLEC
C-type lectin-like receptor
- COX
Cyclooxygenase
- CP
Cancer procoagulant
- cPGES
Cytosolic PGE synthase
- CRC
Colorectal cancer
- CRP
Collagen-related peptide
- EGF
Epidermal growth factor
- EMT
Epithelial–mesenchymal transition
- FA
Fatty acid
- FACL
Fatty acid-CoA ligase
- FcγRIIa
Fcγ receptor IIa
- GP
Glycoprotein
- GPCR
G-protein coupled receptor
- HETE
Hydroxy-eicosatetraenoic acid
- HIF
Hypoxia-induced factor
- HPETE
Hydroperoxy-eicosatetraenoic acid
- IL
Interleukin
- IP
Prostaglandin I2 receptor
- ITAM
Immunoreceptor tyrosine-based activation motif
- LMWH
Low-molecular weight heparin
- LOX
Lipoxygenase
- LPA
Lysophosphatidic acid
- mPGES
Microsomal PGE synthase
- MPs
Microparticles
- NK
Natural killer
- NSAID
Nonsteroidal anti-inflammatory drug
- OxPLs
Oxidized phospholipids
- PAR
Protease-activated receptor
- PC
Phosphatidylcholine
- PDGF
Platelet-derived growth factor
- PE
Phosphatidylethanolamine
- PF-4
Platelet factor 4
- PG
Prostaglandin
- PI3K
Phosphoinositide 3-kinase
- PL
Phospholipid
- PLA
Phospholipase A
- pS6
S6 protein
- PSGL-1
P-selectin glycoprotein ligand-1
- RCT
Randomized clinical trial
- RGD
Arginylglycylaspartic acid
- RhoGEF
RhoGTPase nucleotide exchange factor
- RNA
Ribonucleic acid
- Syk
Spleen tyrosine kinase
- TF
Tissue factor
- TGFβ
Transforming growth factor β
- TLR
Toll-like receptor
- TP
Thromboxane A2 receptor
- TX
Thromboxane
- TXAS
Thromboxane A2 synthase
- VASP
Vasodilator-stimulated phosphoprotein
- VEGF
Vascular endothelial growth factor
- VWF
Von Willebrand factor
Footnotes
Annalisa Contursi and Angela Sacco contributed equally.
References
- 1.Smittenaar CR, Petersen KA, Stewart K, Moitt N. Cancer incidence and mortality projections in the UK until 2035. Br J Cancer. 2016;115:1147–1155. doi: 10.1038/bjc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization (2009) WHO Report on the Global Tobacco Epidemic. Geneva, ISBN: 978 92 4 156391 8
- 3.Després JP. Body fat distribution and risk of cardiovascular disease: an update. Circulation. 2012;126(10):1301–1313. doi: 10.1161/CIRCULATIONAHA.111.067264. [DOI] [PubMed] [Google Scholar]
- 4.Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376(9754):1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- 5.Patrignani P, Patrono C. Aspirin and cancer. J Am Coll Cardiol. 2016;68(9):967–976. doi: 10.1016/j.jacc.2016.05.083. [DOI] [PubMed] [Google Scholar]
- 6.Dovizio M, Alberti S, Guillem-Llobat P, Patrignani P. Role of platelets in inflammation and cancer: novel therapeutic strategies. Basic Clin Pharmacol Toxicol. 2014;114(1):118–127. doi: 10.1111/bcpt.12156. [DOI] [PubMed] [Google Scholar]
- 7.Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, Schellen P, Verschueren H, Post E, Koster J, Ylstra B, Ameziane N, Dorsman J, Smit EF, Verheul HM, Noske DP, Reijneveld JC, Nilsson RJ, Tannous BA, Wesseling P, Wurdinger T. RNA-Seq of tumor-educated platelets enables blood-based pan-cancer, multiclass, and molecular pathway cancer diagnostics. Cancer Cell. 2015;28(5):666–676. doi: 10.1016/j.ccell.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nilsson RJ, Balaj L, Hulleman E, van Rijn S, Pegtel DM, Walraven M, Widmark A, Gerritsen WR, Verheul HM, Vandertop WP, Noske DP, Skog J, Würdinger T. Blood platelets contain tumor-derived RNA biomarkers. Blood. 2011;118(13):3680–3683. doi: 10.1182/blood-2011-03-344408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115(12):3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franco AT, Corken A, Ware J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood. 2015;126(5):582–588. doi: 10.1182/blood-2014-08-531582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan M, Jurasz P. The role of platelets in the tumor microenvironment: from solid tumors to leukemia. Biochim Biophys Acta. 2016;1863(3):392–400. doi: 10.1016/j.bbamcr.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18(6):884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bibbins-Domingo K, U.S. Preventive Services Task Force Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2016;164:836–845. doi: 10.7326/M16-0577. [DOI] [PubMed] [Google Scholar]
- 14.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16(4):201–218. doi: 10.1038/nrc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11(2):123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz-Cano SJ. Tumor heterogeneity: mechanisms and bases for a reliable application of molecular marker design. Int J Mol Sci. 2012;13(2):1951–2011. doi: 10.3390/ijms13021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalluri R, Weinberg RA. The basics of epithelial–mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talbot LJ, Bhattacharya SD, Kuo PC. Epithelial–mesenchymal transition, the tumor microenvironment, and metastatic behavior of epithelial malignancies. Int J Biochem Mol Biol. 2012;3(2):117–136. [PMC free article] [PubMed] [Google Scholar]
- 19.Tímár J, Tóvári J, Rásó E, Mészáros L, Bereczky B, Lapis K. Platelet-mimicry of cancer cells: epiphenomenon with clinical significance. Oncology. 2005;69(3):185–201. doi: 10.1159/000088069. [DOI] [PubMed] [Google Scholar]
- 20.Qiao L, Liang N, Zhang J, Xie J, Liu F, Xu D, Yu X, Tian Y. Advanced research on vasculogenic mimicry in cancer. J Cell Mol Med. 2015;19(2):315–326. doi: 10.1111/jcmm.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patel SR, Hartwig JH, Italiano JE., Jr The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. 2005;115(12):3348–3354. doi: 10.1172/JCI26891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norol F, Vitrat N, Cramer E, Guichard J, Burstein SA, Vainchenker W, Debili N. Effects of cytokines on platelet production from blood and marrow CD34+ cells. Blood. 1998;91(3):830–843. [PubMed] [Google Scholar]
- 23.Michelson AD. Platelets. London: Elsevier; 2013. [Google Scholar]
- 24.Lhermusier T, Chap H, Payrastre B. Platelet membrane phospholipid asymmetry: from the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J Thromb Haemost. 2011;9(10):1883–1891. doi: 10.1111/j.1538-7836.2011.04478.x. [DOI] [PubMed] [Google Scholar]
- 25.Cimmino G, Golino P. Platelet biology and receptor pathways. J Cardiovasc Transl Res. 2013;6(3):299–309. doi: 10.1007/s12265-012-9445-9. [DOI] [PubMed] [Google Scholar]
- 26.White JG. Tubular elements in platelet granules. Blood. 1968;32(1):148–156. [PubMed] [Google Scholar]
- 27.Harris JR. Blood cell biochemestry. New York: Springer Science + Business Media; 1991. [Google Scholar]
- 28.Fukuda M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem. 1991;266(32):21327–21330. [PubMed] [Google Scholar]
- 29.McNicol A, Israels SJ. Platelet dense granules: structure, function and implications for haemostasis. Thromb Res. 1999;95(1):1–18. doi: 10.1016/S0049-3848(99)00015-8. [DOI] [PubMed] [Google Scholar]
- 30.Fuentes QE, Fuentes QF, Andrés V, Pello OM, Font de Mora J, Palomo GI. Role of platelets as mediators that link inflammation and thrombosis in atherosclerosis. Platelets. 2013;24(4):255–262. doi: 10.3109/09537104.2012.690113. [DOI] [PubMed] [Google Scholar]
- 31.Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23(4):177–189. doi: 10.1016/j.blre.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatterjee M, Huang Z, Zhang W, Jiang L, Hultenby K, Zhu L, Hu H, Nilsson GP, Li N. Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood. 2011;117(14):3907–3911. doi: 10.1182/blood-2010-12-327007. [DOI] [PubMed] [Google Scholar]
- 33.Senior RM, Griffin GL, Huang JS, Walz DA, Deuel TF. Chemotactic activity of platelet alpha granule proteins for fibroblasts. J Cell Biol. 1983;96(2):382–385. doi: 10.1083/jcb.96.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hargett LA, Bauer NN. On the origin of microparticles: from “platelet dust” to mediators of intercellular communication. Pulm Circ. 2013;3(2):329–340. doi: 10.4103/2045-8932.114760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bobrie A, Colombo M, Raposo G, Théry C. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic. 2011;12(12):1659–1668. doi: 10.1111/j.1600-0854.2011.01225.x. [DOI] [PubMed] [Google Scholar]
- 36.Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res. 2010;107(9):1047–1057. doi: 10.1161/CIRCRESAHA.110.226456. [DOI] [PubMed] [Google Scholar]
- 37.Zimmerman GA, Weyrich AS. Signal-dependent protein synthesis by activated platelets: new pathways to altered phenotype and function. Arterioscler Thromb Vasc Biol. 2008;28(3):s17–s24. doi: 10.1161/ATVBAHA.107.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci USA. 1998;95(10):5556–5561. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evangelista V, Manarini S, Di Santo A, Capone ML, Ricciotti E, Di Francesco L, Tacconelli S, Sacchetti A, D’Angelo S, Scilimati A, Sciulli MG, Patrignani P. De novo synthesis of cyclooxygenase-1 counteracts the suppression of platelet thromboxane biosynthesis by aspirin. Circ Res. 2006;98(5):593–595. doi: 10.1161/01.RES.0000214553.37930.3e. [DOI] [PubMed] [Google Scholar]
- 40.Patrono C, Baigent C, Hirsh J, Roth G. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition) Chest. 2008;133(6 Suppl):199S–233S. doi: 10.1378/chest.08-0672. [DOI] [PubMed] [Google Scholar]
- 41.Yang H, Lang S, Zhai Z, Li L, Kahr WH, Chen P, Brkić J, Spring CM, Flick MJ, Degen JL, Freedman J, Ni H. Fibrinogen is required for maintenance of platelet intracellular and cell-surface P-selectin expression. Blood. 2009;114(2):425–436. doi: 10.1182/blood-2008-03-145821. [DOI] [PubMed] [Google Scholar]
- 42.Borsig L, Wong R, Feramisco J, Nadeau DR, Varki NM, Varki A. Heparin and cancer revisited: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc Natl Acad Sci USA. 2001;98(6):3352–3357. doi: 10.1073/pnas.061615598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Austrup F, Vestweber D, Borges E, Löhning M, Bräuer R, Herz U, Renz H, Hallmann R, Scheffold A, Radbruch A, Hamann A. P- and E-selectin mediate recruitment of T-helper-1 but not T-helper-2 cells into inflammed tissues. Nature. 1997;385(6611):81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- 44.Tinoco R, Carrette F, Barraza ML, Otero DC, Magaña J, Bosenberg MW, Swain SL, Bradley LM. PSGL-1 is an immune checkpoint regulator that promotes T cell exhaustion. Immunity. 2016;44(5):1190–1203. doi: 10.1016/j.immuni.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Macaulay IC, Carr P, Gusnanto A, Ouwehand WH, Fitzgerald D, Watkins NA. Platelet genomics and proteomics in human health and disease. J Clin Invest. 2005;115(12):3370–3377. doi: 10.1172/JCI26885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McRedmond JP, Park SD, Reilly DF, Coppinger JA, Maguire PB, Shields DC, Fitzgerald DJ. Integration of proteomics and genomics in platelets: a profile of platelet proteins and platelet-specific genes. Mol Cell Proteomics. 2004;3(2):133–144. doi: 10.1074/mcp.M300063-MCP200. [DOI] [PubMed] [Google Scholar]
- 47.Patrignani P, Patrono C. Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim Biophys Acta. 2015;1851(4):422–432. doi: 10.1016/j.bbalip.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 48.Patrignani P, Tacconelli S. Isoprostanes and other markers of peroxidation in atherosclerosis. Biomarkers. 2005;10(Suppl 1):S24–S29. doi: 10.1080/13547500500215084. [DOI] [PubMed] [Google Scholar]
- 49.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 50.Hourani SM, Cusack NJ. Pharmacological receptors on blood platelets. Pharmacol Rev. 1991;43(3):243–298. [PubMed] [Google Scholar]
- 51.Dorn GW, 2nd, Becker MW. Thromboxane A2 stimulated signal transduction in vascular smooth muscle. J Pharmacol Exp Ther. 1993;265(1):447–456. [PubMed] [Google Scholar]
- 52.Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol. 2006;119(3):229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 53.Smith JB, Willis AL. Formation and release of prostaglandins by platelets in response to thrombin. Br J Pharmacol. 1970;40(3):545P–546P. [PMC free article] [PubMed] [Google Scholar]
- 54.Song WL, Stubbe J, Ricciotti E, Alamuddin N, Ibrahim S, Crichton I, Prempeh M, Lawson JA, Wilensky RL, Rasmussen LM, Puré E, FitzGerald GA. Niacin and biosynthesis of PGD2 by platelet COX-1 in mice and humans. J Clin Invest. 2012;122(4):1459–1468. doi: 10.1172/JCI59262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79(4):1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 56.Fabre JE, Nguyen M, Athirakul K, Coggins K, McNeish JD, Austin S, Parise LK, FitzGerald GA, Coffman TM, Koller BH. Activation of the murine EP3 receptor for PGE2 inhibits cAMP production and promotes platelet aggregation. J Clin Invest. 2001;107(5):603–610. doi: 10.1172/JCI10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116(1):4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao L, Funk CD. Lipoxygenase pathways in atherogenesis. Trends Cardiovasc Med. 2004;14(5):191–195. doi: 10.1016/j.tcm.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 59.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 60.Honn KV, Tang DG, Gao X, Butovich IA, Liu B, Timar J, Hagmann W. 12-lipoxygenases and 12(S)-HETE: role in cancer metastasis. Cancer Metastasis Rev. 1994;13(3–4):365–396. doi: 10.1007/BF00666105. [DOI] [PubMed] [Google Scholar]
- 61.Hamberg M, Samuelsson B. Prostaglandin endoperoxides. Novel transformations of arachidonic acid in human platelets. Proc Natl Acad Sci USA. 1974;71(9):3400–3404. doi: 10.1073/pnas.71.9.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgan LT, Thomas CP, Kühn H, O’Donnell VB. Thrombin-activated human platelets acutely generate oxidized docosahexaenoic-acid-containing phospholipids via 12-lipoxygenase. Biochem J. 2010;431(1):141–148. doi: 10.1042/BJ20100415. [DOI] [PubMed] [Google Scholar]
- 63.Ikei KN, Yeung J, Apopa PL, Ceja J, Vesci J, Holman TR, Holinstat M. Investigations of human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation. J Lipid Res. 2012;53(12):2546–2559. doi: 10.1194/jlr.M026385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yeung J, Holinstat M. 12-lipoxygenase: a potential target for novel anti-platelet therapeutics. Cardiovasc Hematol Agents Med Chem. 2011;9(3):154–164. doi: 10.2174/187152511797037619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo Y, Zhang W, Giroux C, Cai Y, Ekambaram P, Dilly AK, Hsu A, Zhou S, Maddipati KR, Liu J, Joshi S, Tucker SC, Lee MJ, Honn KV. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem. 2011;286(39):33832–33840. doi: 10.1074/jbc.M110.216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aldrovandi M, O’Donnell VB. Oxidized PLs and vascular inflammation. Curr Atheroscler Rep. 2013;15(5):323. doi: 10.1007/s11883-013-0323-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Donnell VB. Mass spectrometry analysis of oxidized phosphatidylcholine and phosphatidylethanolamine. Biochim Biophys Acta. 2011;1811(11):818–826. doi: 10.1016/j.bbalip.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 68.O’Donnell VB, Murphy RC. New families of bioactive oxidized phospholipids generated by immune cells: identification and signaling actions. Blood. 2012;120(10):1985–1992. doi: 10.1182/blood-2012-04-402826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10(3):181–193. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klampfl T, Bogner E, Bednar W, Mager L, Massudom D, Kalny I, Heinzle C, Berger W, Stättner S, Karner J, Klimpfinger M, Fürstenberger G, Krieg P, Marian B. Up-regulation of 12(S)-lipoxygenase induces a migratory phenotype in colorectal cancer cells. Exp Cell Res. 2012;318(6):768–778. doi: 10.1016/j.yexcr.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ozaki Y, Asazuma N, Suzuki-Inoue K, Berndt MC. Platelet GPIb-IX-V-dependent signaling. J Thromb Haemost. 2005;3(8):1745–1751. doi: 10.1111/j.1538-7836.2005.01379.x. [DOI] [PubMed] [Google Scholar]
- 72.Boulaftali Y, Hess PR, Kahn ML, Bergmeier W. Platelet immunoreceptor tyrosine-based activation motif (ITAM) signaling and vascular integrity. Circ Res. 2014;114(7):1174–1184. doi: 10.1161/CIRCRESAHA.114.301611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nieswandt B, Watson SP. Platelet–collagen interaction: is GPVI the central receptor? Blood. 2003;102(2):449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- 74.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin alphaIIb beta3 signaling in platelets. J Thromb Haemost. 2005;3(8):1752–1762. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 75.Dunne E, Spring CM, Reheman A, Jin W, Berndt MC, Newman DK, Newman PJ, Ni H, Kenny D. Cadherin 6 has a functional role in platelet aggregation and thrombus formation. Arterioscler Thromb Vasc Biol. 2012;32(7):1724–1731. doi: 10.1161/ATVBAHA.112.250464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cho J, Mosher DF. Role of fibronectin assembly in platelet thrombus formation. J Thromb Haemost. 2006;4(7):1461–1469. doi: 10.1111/j.1538-7836.2006.01943.x. [DOI] [PubMed] [Google Scholar]
- 77.Arneson MA, Hammerschmidt DE, Furcht LT, King RA. A new form of Ehlers-Danlos syndrome. Fibronectin corrects defective platelet function. JAMA. 1980;244(2):144–147. [PubMed] [Google Scholar]
- 78.Santoro SA. Inhibition of platelet aggregation by fibronectin. Biochem Biophys Res Commun. 1983;116(1):135–140. doi: 10.1016/0006-291X(83)90391-1. [DOI] [PubMed] [Google Scholar]
- 79.Moon DG, Kaplan JE, Mazurkewicz JE. The inhibitory effect of plasma fibronectin on collagen-induced platelet aggregation. Blood. 1986;67(2):450–457. [PubMed] [Google Scholar]
- 80.Reheman A, Yang H, Zhu G, Jin W, He F, Spring CM, Bai X, Gross PL, Freedman J, Ni H. Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor. Blood. 2009;113(8):1809–1817. doi: 10.1182/blood-2008-04-148361. [DOI] [PubMed] [Google Scholar]
- 81.Gartner TK, Bennett JS. The tetrapeptide analogue of the cell attachment site of fibronectin inhibits platelet aggregation and fibrinogen binding to activated platelets. J Biol Chem. 1985;260(22):11891–11894. [PubMed] [Google Scholar]
- 82.Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, Wagner DD. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000;106(3):385–392. doi: 10.1172/JCI9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Reheman A, Spring CM, Kalantari J, Marshall AH, Wolberg AS, Gross PL, Weitz JI, Rand ML, Mosher DF, Freedman J, Ni H. Plasma fibronectin supports hemostasis and regulates thrombosis. J Clin Invest. 2014;124(10):4281–4293. doi: 10.1172/JCI74630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhi H, Rauova L, Hayes V, Gao C, Boylan B, Newman DK, McKenzie SE, Cooley BC, Poncz M, Newman PJ. Cooperative integrin/ITAM signaling in platelets enhances thrombus formation in vitro and in vivo. Blood. 2013;121(10):1858–1867. doi: 10.1182/blood-2012-07-443325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Woulfe DS. Platelet G protein-coupled receptors in hemostasis and thrombosis. J Thromb Haemost. 2005;10:2193–2200. doi: 10.1111/j.1538-7836.2005.01338.x. [DOI] [PubMed] [Google Scholar]
- 86.Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114(10):1070–1077. doi: 10.1161/CIRCULATIONAHA.105.574830. [DOI] [PubMed] [Google Scholar]
- 87.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 88.Smyth SS, Woulfe DS, Weitz JI, Gachet C, Conley PB, Goodman SG, Roe MT, Kuliopulos A, Moliterno DJ, French PA, Steinhubl SR, Becker RC. G-protein-coupled receptors as signaling targets for antiplatelet therapy. Arterioscler Thromb Vasc Biol. 2009;29(4):449–457. doi: 10.1161/ATVBAHA.108.176388. [DOI] [PubMed] [Google Scholar]
- 89.Fitzgerald DJ, Fitzgerald GA. Historical lessons in translational medicine: cyclooxygenase inhibition and P2Y12 antagonism. Circ Res. 2013;112(1):174–194. doi: 10.1161/CIRCRESAHA.111.300271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schwarz UR, Geiger J, Walter U, Eigenthaler M. Flow cytometry analysis of intracellular VASP phosphorylation for the assessment of activating and inhibitory signal transduction pathways in human platelets—definition and detection of ticlopidine/clopidogrel effects. Thromb Haemost. 1999;82(3):1145–1152. [PubMed] [Google Scholar]
- 91.Hirata T, Ushikubi F, Kakizuka A, Okuma M, Narumiya S. Two thromboxane A2 receptor isoforms in human platelets. Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J Clin Invest. 1996;97(4):949–956. doi: 10.1172/JCI118518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Siehler S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br J Pharmacol. 2009;158(1):41–49. doi: 10.1111/j.1476-5381.2009.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paul BZ, Jin J, Kunapuli SP. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J Biol Chem. 1999;274(41):29108–29114. doi: 10.1074/jbc.274.41.29108. [DOI] [PubMed] [Google Scholar]
- 94.Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482–2494. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- 95.Xu XR, Zhang D, Oswald BE, Carrim N, Wang X, Hou Y, Zhang Q, Lavalle C, McKeown T, Marshall AH, Ni H. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit Rev Clin Lab Sci. 2016;53(6):409–430. doi: 10.1080/10408363.2016.1200008. [DOI] [PubMed] [Google Scholar]
- 96.Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial–mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20(5):576–590. doi: 10.1016/j.ccr.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dovizio M, Maier TJ, Alberti S, Di Francesco L, Marcantoni E, Münch G, John CM, Suess B, Sgambato A, Steinhilber D, Patrignani P. Pharmacological inhibition of platelet–tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol Pharmacol. 2013;84(1):25–40. doi: 10.1124/mol.113.084988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guillem-Llobat P, Dovizio M, Bruno A, Ricciotti E, Cufino V, Sacco A, Grande R, Alberti S, Arena V, Cirillo M, Patrono C, FitzGerald GA, Steinhilber D, Sgambato A, Patrignani P. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget. 2016;7(22):32462–32477. doi: 10.18632/oncotarget.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang D, Dubois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2010;29(6):781–788. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ungerer M, Rosport K, Bültmann A, Piechatzek R, Uhland K, Schlieper P, Gawaz M, Münch G. Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation. 2011;123(17):1891–1899. doi: 10.1161/CIRCULATIONAHA.110.980623. [DOI] [PubMed] [Google Scholar]
- 101.Battinelli EM, Markens BA, Kulenthirarajan RA, Machlus KR, Flaumenhaft R, Italiano JE., Jr Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood. 2014;123:101–112. doi: 10.1182/blood-2013-02-485011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell. 2006;10(5):355–362. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 103.Gale AJ, Gordon SG. Update on tumor cell procoagulant factors. Acta Haematol. 2001;106(1–2):25–32. doi: 10.1159/000046586. [DOI] [PubMed] [Google Scholar]
- 104.Mitrugno A, Williams D, Kerrigan SW, Moran N. A novel and essential role for FcγRIIa in cancer cell-induced platelet activation. Blood. 2014;123(2):249–260. doi: 10.1182/blood-2013-03-492447. [DOI] [PubMed] [Google Scholar]
- 105.Mannori G, Crottet P, Cecconi O, Hanasaki K, Aruffo A, Nelson RM, Varki A, Bevilacqua MP. Differential colon cancer cell adhesion to E-, P-, and L-selectin: role of mucin-type glycoproteins. Cancer Res. 1995;55(19):4425–4431. [PubMed] [Google Scholar]
- 106.Boukerche H, Berthier-Vergnes O, Tabone E, Doré JF, Leung LL, McGregor JL. Platelet–melanoma cell interaction is mediated by the glycoprotein IIb–IIIa complex. Blood. 1989;74(2):658–663. [PubMed] [Google Scholar]
- 107.Nierodzik ML, Plotkin A, Kajumo F, Karpatkin S. Thrombin stimulates tumor-platelet adhesion in vitro and metastasis in vivo. J Clin Invest. 1991;87(1):229–236. doi: 10.1172/JCI114976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Karpatkin S, Pearlstein E, Ambrogio C, Coller BS. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J Clin Invest. 1988;81(4):1012–1019. doi: 10.1172/JCI113411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mammadova-Bach E, Zigrino P, Brucker C, Bourdon C, Freund M, De Arcangelis A, Abrams SI, Orend G, Gachet C, Mangin PH. Platelet integrin α6β1 controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight. 2016;1(14):e88245. doi: 10.1172/jci.insight.88245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gerrard JM. Robinson P (1989) Identification of the molecular species of lysophosphatidic acid produced when platelets are stimulated by thrombin. Biochim Biophys Acta. 1001;3:282–285. doi: 10.1016/0005-2760(89)90112-4. [DOI] [PubMed] [Google Scholar]
- 111.Williams JR, Khandoga AL, Goyal P, Fells JI, Perygin DH, Siess W, Parrill AL, Tigyi G, Fujiwara Y. Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J Biol Chem. 2009;284(25):17304–17319. doi: 10.1074/jbc.M109.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3(8):582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 113.Boucharaba A, Serre CM, Grès S, Saulnier-Blache JS, Bordet JC, Guglielmi J, Clézardin P, Peyruchaud O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J Clin Invest. 2004;114(12):1714–1725. doi: 10.1172/JCI200422123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boucharaba A, Serre CM, Guglielmi J, Bordet JC, Clézardin P, Peyruchaud O. The type 1 lysophosphatidic acid receptor is a target for therapy in bone metastases. Proc Natl Acad Sci USA. 2006;103(25):9643–9648. doi: 10.1073/pnas.0600979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Leblanc R, Lee SC, David M, Bordet JC, Norman DD, Patil R, Miller D, Sahay D, Ribeiro J, Clézardin P, Tigyi GJ, Peyruchaud O. Interaction of platelet-derived autotaxin with tumor integrin αVβ3 controls metastasis of breast cancer cells to bone. Blood. 2014;124(20):3141–3150. doi: 10.1182/blood-2014-04-568683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Burkhalter RJ, Westfall SD, Liu Y, Stack MS. Lysophosphatidic acid initiatesepithelial to mesenchymal transition and induces β-catenin-mediated transcription in epithelial ovarian carcinoma. J Biol Chem. 2015;290(36):22143–22154. doi: 10.1074/jbc.M115.641092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ha JH, Ward JD, Radhakrishnan R, Jayaraman M, Song YS, Dhanasekaran DN. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Gαi2, Src, and HIF1α signaling nexus. Oncotarget. 2016;7(25):37664–37679. doi: 10.18632/oncotarget.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377(9759):31–41. doi: 10.1016/S0140-6736(10)62110-1. [DOI] [PubMed] [Google Scholar]
- 119.Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M, Mehta Z, Meade TW. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. 2012;379(9826):1602–1612. doi: 10.1016/S0140-6736(11)61720-0. [DOI] [PubMed] [Google Scholar]
- 120.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56(3):387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 121.Loll PJ, Picot D, Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol. 1995;2(8):637–643. doi: 10.1038/nsb0895-637. [DOI] [PubMed] [Google Scholar]
- 122.Lecomte M, Laneuville O, Ji C, DeWitt DL, Smith WL. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J Biol Chem. 1994;269(18):13207–13215. [PubMed] [Google Scholar]
- 123.Patrignani P, Filabozzi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J Clin Invest. 1982;69(6):1366–1372. doi: 10.1172/JCI110576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Patrignani P, Tacconelli S, Piazuelo E, Di Francesco L, Dovizio M, Sostres C, Marcantoni E, Guillem-Llobat P, Del Boccio P, Zucchelli M, Patrono C, Lanas A. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J Thromb Haemost. 2014;12(8):1320–1330. doi: 10.1111/jth.12637. [DOI] [PubMed] [Google Scholar]
- 125.Patrono C, Patrignani P, García Rodríguez LA. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest. 2001;108(1):7–13. doi: 10.1172/JCI200113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Di Francesco L, López Contreras LA, Sacco A, Patrignani P. New insights into the mechanism of action of aspirin in the prevention of colorectal neoplasia. Curr Pharm Des. 2015;21(35):5116–5126. doi: 10.2174/1381612821666150915110706. [DOI] [PubMed] [Google Scholar]
- 127.Patrignani P, Sacco A, Sostres C, Bruno A, Dovizio M, Piazuelo E, Di Francesco L, Contursi A, Zucchelli M, Schiavone S, Tacconelli S, Patrono C, Lanas A (2017) Low-dose aspirin acetylates cyclooxygenase-1 in human colorectal mucosa: implications for the chemoprevention of colorectal cancer. Clin Pharmacol Ther (Epub ahead of print) [DOI] [PubMed]
- 128.Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19(18):2199–2211. doi: 10.1101/gad.351605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cattaneo M. New P2Y(12) inhibitors. Circulation. 2010;121(1):171–179. doi: 10.1161/CIRCULATIONAHA.109.853069. [DOI] [PubMed] [Google Scholar]
- 130.Wang Y, Sun Y, Li D, Zhang L, Wang K, Zuo Y, Gartner TK, Liu J. Platelet P2Y12 is involved in murine pulmonary metastasis. PLoS One. 2013;8(11):e80780. doi: 10.1371/journal.pone.0080780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sitia G, Aiolfi R, Di Lucia P, Mainetti M, Fiocchi A, Mingozzi F, Esposito A, Ruggeri ZM, Chisari FV, Iannacone M, Guidotti LG. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci USA. 2012;109(32):E2165–E2172. doi: 10.1073/pnas.1209182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nierodzik ML, Klepfish A, Karpatkin S. Role of platelets, thrombin, integrin IIb-IIIa, fibronectin and von Willebrand factor on tumor adhesion in vitro and metastasis in vivo. Thromb Haemost. 1995;74(1):282–290. [PubMed] [Google Scholar]
- 133.Amirkhosravi A, Mousa SA, Amaya M, Blaydes S, Desai H, Meyer T, Francis JL. Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb Haemost. 2003;90(3):549–554. doi: 10.1160/TH03-02-0102. [DOI] [PubMed] [Google Scholar]
- 134.Italiano JE, Jr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, Ryeom S, Folkman J, Klement GL. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111(3):1227–1233. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dovizio M, Sacco A, Patrignani P. Curbing tumorigenesis and malignant progression through the pharmacological control of the wound healing process. Vasc Pharmacol. 2017;89:1–11. doi: 10.1016/j.vph.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 136.Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104(2):397–401. doi: 10.1182/blood-2004-02-0434. [DOI] [PubMed] [Google Scholar]
- 137.Jain S, Russell S, Ware J. Platelet glycoprotein VI facilitates experimental lung metastasis in syngenic mouse models. J Thromb Haemost. 2009;7(10):1713–1717. doi: 10.1111/j.1538-7836.2009.03559.x. [DOI] [PubMed] [Google Scholar]
- 138.Nangia-Makker P, Balan V, Raz A. Regulation of tumor progression by extracellular galectin-3. Cancer Microenviron. 2008;1(1):43–51. doi: 10.1007/s12307-008-0003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schober LJ, Khandoga AL, Dwivedi S, Penz SM, Maruyama T, Brandl R, Siess W. The role of PGE(2) in human atherosclerotic plaque on platelet EP(3) and EP(4) receptor activation and platelet function in whole blood. J Thromb Thrombolysis. 2011;32(2):158–166. doi: 10.1007/s11239-011-0577-6. [DOI] [PubMed] [Google Scholar]
- 140.Singh J, Zeller W, Zhou N, Hategen G, Mishra R, Polozov A, Yu P, Onua E, Zhang J, Zembower D, Kiselyov A, Ramírez JL, Sigthorsson G. Antagonists of the EP3 receptor for prostaglandin E2 are novel antiplatelet agents that do not prolong bleeding. ACS Chem Biol. 2009;4:115–126. doi: 10.1021/cb8002094. [DOI] [PubMed] [Google Scholar]
- 141.Fox SC, May JA, Johnson A, Hermann D, Strieter D, Hartman D, Heptinstall S. Effects on platelet function of an EP3 receptor antagonist used alone and in combination with a P2Y12 antagonist both in vitro and ex vivo in human volunteers. Platelets. 2013;24(5):392–400. doi: 10.3109/09537104.2012.704648. [DOI] [PubMed] [Google Scholar]
- 142.Semple JW, Italiano JE, Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 143.Li C, Li J, Li Y, Lang S, Yougbare I, Zhu G, Chen P, Ni H. Crosstalk between platelets and the immune system: old systems with new discoveries. Adv Hematol. 2012;2012:384685. doi: 10.1155/2012/384685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999;59(6):1295–1300. [PubMed] [Google Scholar]
- 145.Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, Garraud O. The inflammatory role of platelets via their TLRs and Siglec receptors. Front Immunol. 2015;2(6):83. doi: 10.3389/fimmu.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yeaman MR. Platelets: at the nexus of antimicrobial defence. Nat Rev Microbiol. 2014;12(6):426–437. doi: 10.1038/nrmicro3269. [DOI] [PubMed] [Google Scholar]
- 147.Langer HF, Choi EY, Zhou H, Schleicher R, Chung KJ, Tang Z, Göbel K, Bdeir K, Chatzigeorgiou A, Wong C, Bhatia S, Kruhlak MJ, Rose JW, Burns JB, Hill KE, Qu H, Zhang Y, Lehrmann E, Becker KG, Wang Y, Simon DI, Nieswandt B, Lambris JD, Li X, Meuth SG, Kubes P, Chavakis T. Platelets contribute to the pathogenesis of experimental autoimmune encephalomyelitis. Circ Res. 2012;110(9):1202–1210. doi: 10.1161/CIRCRESAHA.111.256370. [DOI] [PMC free article] [PubMed] [Google Scholar]