

Abstract
We have analyzed the binding of the liver-specific microRNA-122 (miR-122) to three conserved target sites of hepatitis C virus (HCV) RNA, two in the non-structural protein 5B (NS5B) coding region and one in the 3′ untranslated region (3′UTR). miR-122 binding efficiency strongly depends on target site accessibility under conditions when the range of flanking sequences available for the formation of local RNA secondary structures changes. Our results indicate that the particular sequence feature that contributes most to the correlation between target site accessibility and binding strength varies between different target sites. This suggests that the dynamics of miRNA/Ago2 binding not only depends on the target site itself but also on flanking sequence context to a considerable extent, in particular in a small viral genome in which strong selection constraints act on coding sequence and overlapping cis-signals and model the accessibility of cis-signals. In full-length genomes, single and combination mutations in the miR-122 target sites reveal that site 5B.2 is positively involved in regulating overall genome replication efficiency, whereas mutation of site 5B.3 showed a weaker effect. Mutation of the 3′UTR site and double or triple mutants showed no significant overall effect on genome replication, whereas in a translation reporter RNA, the 3′UTR target site inhibits translation directed by the HCV 5′UTR. Thus, the miR-122 target sites in the 3′-region of the HCV genome are involved in a complex interplay in regulating different steps of the HCV replication cycle.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-016-2377-9) contains supplementary material, which is available to authorized users.
Keywords: microRNA, Accessibility, Ago2, Translation, Regulation
Introduction
microRNAs (miRNAs) are small RNAs that regulate mRNA expression mostly at a post-transcriptional level. To this end, the miRNAs are bound by Argonaute (Ago) proteins that constitute key components of miRNA-protein (miRNP) complexes. The miRNA is then used as a sequence-specific guide that directs the miRNP complex to target sites in mRNAs [1] where the complex mediates the regulation of mRNA half-life or activity.
Initially, miRNAs were reported to bind to the 3′ untranslated region (3′UTR) of mRNAs, while later it became clear that miRNAs also frequently bind to coding sequences [2–5] and rarely in the 5′UTRs of mRNAs [5, 6]. In the 3′UTR, miRNP complexes can exert their regulatory functions without blocking the passage of scanning or translating ribosomes. The activity of the mRNA is regulated by inhibiting translation initiation, sequestering the mRNAs to P-bodies for temporal storage, initiating degradation of the mRNAs via decapping and deadenylation, or by other mechanisms [3, 5, 7–9]. It was proposed that miRNA binding to 3′UTRs preferentially regulates mRNA stability, whereas binding to coding regions may preferentially regulate mRNA translation [10].
miRNA precursors are expressed in the nucleus, partially processed and transported to the cytosol where they are further processed into imperfect miRNA duplexes of about 22 bp with 3′-overhangs. This miRNA duplex is then unwound. One strand remains bound in the Ago protein in the complex and is used as the guide strand, while the opposite (passenger) strand is discarded. In humans, four Ago proteins are known (numbered 1–4), of which Ago2 is the most abundant [11]. In the Ago2 protein, the miRNA guide strand is anchored with one or two nucleotides at its 3′-end in the PAZ domain and with the 5′-phosphate in the MID domain [12–14]. Nucleotides 2–8 (the seed region) of the miRNA are accessible to hybridization with the target mRNA, of which miRNA nucleotides 2–5 appear to be particularly important for the initiation of seed binding [14]. In many cases, also nucleotides beyond miRNA position 14 (the supplementary binding region) contribute to target RNA binding [1].
Hepatitis C virus (HCV) employs the liver-specific miRNA-122 (miR-122) to promote its own replication [15]. Since miR-122 makes up 60–70 % of all miRNA in hepatocytes but is expressed hardly in other tissues [16, 17], it is considered to significantly contribute to the liver tropism of HCV. HCV is a member of the genus Hepacivirus in the family of Flaviviridae and has a single-strand RNA genome of positive orientation that replicates in the cytoplasm of hepatocytes [18, 19]. The single open reading frame (ORF) of the viral genome is translated under the control of an internal ribosome entry site (IRES) element in the 5′UTR [20]. The 3′UTR of the HCV RNA genome is not only involved in replication but also stimulates HCV translation [21, 22]. The HCV genome contains several miR-122 target site sequences (see Fig. 1a). Two of them (S1 and S2) are located in the very 5′-region of the 5′UTR. Ago protein mediated binding of miR-122 to these sites [23] is involved in HCV RNA stabilization against exonuclease degradation [23–26] and stimulation of HCV translation [23, 27–33]. Another potential miR-122 target site (S3) is located in the variable region (VR) of the HCV 3′UTR. Moreover, three potential miR-122 target sites are found in the non-structural 5B (NS5B) protein coding region of the HCV ORF, of which the first site (5B.1) is not conserved among all isolates, whereas the second (5B.2) and third site (5B.3) are conserved (Fig. 1b, Supplementary Figures S1–S4). However, the functions of these miR-122 target sites in the 3′-region of the HCV genome are largely unknown.
Fig. 1.

Conserved miR-122 target sites in the hepatitis C virus (HCV) RNA genome. a The HCV genome with the 5′- and 3′-untranslated regions (UTRs), the polyprotein open reading frame (ORF) and the gene segments encoding structural and non-structural (NS) proteins. In the 5′UTR, there are two conserved miR-122 target sites (S1 and S2, boxed) and the internal ribosome entry site (IRES). In the NS5B region (encoding the viral replicase) there are three miR-122 binding sites (numbered 5B.1, 2 and 3), of which sites 5B.2 and 5B.3 are highly conserved among isolates. One additional site (S3) is located in the variable region (VR) of the 3′UTR. The cis-acting replication element (CRE) in the NS5B region, the poly(U/C) tract and the highly conserved 3′X region in the 3′UTR are indicated. b miR-122 and conserved miR-122 target sites in the NS5B coding sequences and in the 3′UTR of two HCV isolates, Con1 (genotype 1b) and JFH-1 (genotype 2a). Upper part The miR-122 sequence with the seed sequence (nucleotides 2–8) and the potential supplementary binding region (involving nucleotides 14–20). Below Sequences of the conserved miR-122 target sites 5B.2, 5B.3 and S3 in the NS5B region and the 3′UTR in the Con1 (genotype 1b) and JFH-1 (genotype 2a) isolates. HCV genotype (GT) and NCBI nucleotide database accession numbers are given. Nucleotide numbers indicate numbering in the genotype 1b Con1 isolate. miR-122 target sequences are boxed. Alignment of 106 selected isolates and miR-122 seed and potential supplementary binding sites in the NS5B region are shown in Supplementary Figures S1, S2 and S5
Binding of a microRNA to its target site depends on target site accessibility. The HCV genome RNA has some regions of highly conserved structures, while others appear to be quite flexible. This fact is evident from SHAPE reactivities (which are a measure for the structural flexibility of a RNA region) that are conserved among isolates only for some selected regions of the HCV genome [34]. For example, the very 5′-region of the 5′UTR and the 3′X region of the 3′UTR have very low SHAPE reactivities in solution, indicating the presence of highly structured RNA regions. Accordingly, entropy values are very low in these regions. In contrast, SHAPE reactivities are very diverse among isolates over most regions of the HCV genome coding sequence [34], and entropy values largely vary among isolates, indicating that most RNA secondary structures in the open reading frame may be quite dynamic. These findings suggest that, except for some conserved cis-elements, there is a high degree of flexibility in RNA structure in the coding region of the HCV genome. This raises the question of what are the structural preconditions for miR-122 binding to the conserved target sites in the NS5B coding region of the HCV genome.
We investigated the binding of miR-122 to the two conserved target sites in the NS5B coding region of HCV and, in comparison, to the target site in the 3′UTR. We analyzed the impact of local RNA secondary structure on miRNA target site accessibility and on the intensity of miRNA-Ago2 complex binding. Moreover, we analyzed the function of these miR-122 target sites in the HCV genome on viral genome translation and replication.
Results
Conserved miR-122 target sites in the HCV NS5B coding region and 3′UTR
Two highly conserved miR-122 target sites in the NS5B coding region were identified in 106 selected HCV sequences, including two isolates of each subtype from each genotype from full-length or near-full-length HCV sequences if available (sequences are detailed in the MAFFT alignment file in the supplement to [35] on the website http://www.rna.uni-jena.de/supplements/hcv/SELECTED/selected_hcv.stk.html). The first site (5B.2) is located about 570 nucleotides upstream of the NS5B stop codon (Fig. 1b, Supplementary Figure S1). In site 5B.2, the seed target sequence (A)CACUCC is conserved in 99/106 isolates. A potential supplementary miR-122 binding sequence located 4–8 nucleotides upstream of the seed target sequence is conserved in 106/106 isolates (Supplementary Figure S1). A second highly conserved miR-122 target site (5B.3) in the NS5B coding sequence is located about 165 nucleotides upstream of the stop codon (Fig. 1b, Supplementary Figure S2). Its seed target sequence CACUCC is conserved in 106/106 isolates, and a potential supplementary binding site is conserved 10–12 nucleotides upstream of the seed target site. In both sites the seed sequences are conserved beyond coding sequence requirements (Supplementary Figures S3 and S4). A third seed target site (CACUCC) (5B.1) located about 30 nucleotides upstream of site 5B.2 is found in only 25/106 selected isolates [35] and, therefore, was not further analyzed. In the Variable Region of the 3′UTR, a miR-122 seed target sequence ([A]CACUCC) is conserved among 83/94 isolates, and a potential supplementary site can be suspected to overlap with the NS5B stop codon (Supplementary Figure S5). An adenosine (A) residue directly downstream of a miRNA seed target sequence was reported to be overrepresented in many miRNA target sites and shown to bind directly to Ago2 protein [1, 2, 14]. An A residue directly downstream of the miR-122 seed target sequence is present in 70/106 isolates for site 5B.2 (Supplementary Figure S1), in 101/106 isolates downstream of site 5B.3 (Supplementary Figure S2) and in 56/88 isolates for the 3′UTR site (Supplementary Figure S5). Taken together, the degree of conservation suggests a functional role of these miR-122 target sites in the HCV NS5B coding region and 3′UTR.
miR-122/Ago2 complexes bind to all conserved target sites
To analyze the binding of miR-122 to the conserved target sites in the HCV RNA, we transfected HeLa cells with 32P-labeled HCV RNAs containing each one of the miR-122 target sites, along with unlabeled miR-122 (or miR-124 as a control). The lengths of the in vitro-transcribed RNAs were chosen in a range of about 150–270 nucleotides, in case of the NS5B sites thereby mimicking local segments of RNA between migrating ribosomes but long enough to allow formation of local secondary structures. Cells were lysed 6 h post-transfection, and Ago2/miR-122/HCV RNA complexes were recovered from the cell lysates by co-immunoprecipitation (co-IP) using anti-Ago2 antibodies [23]. Following RNA isolation and gel electrophoresis, the amount of recovered 32P-labeled HCV RNA was detected by a phosphorimager, normalized to input RNA, compared to negative control Flag-co-IP readouts, and then served as readout for the intensity of miR-122 binding to the HCV RNA. For miR-122 target sites 5B.2, 5B.3 and the 3′UTR site (S3), we could detect binding of miR-122, while miR-124 yielded only background binding comparable to the negative Flag antibody control (Fig. 2).
Fig. 2.
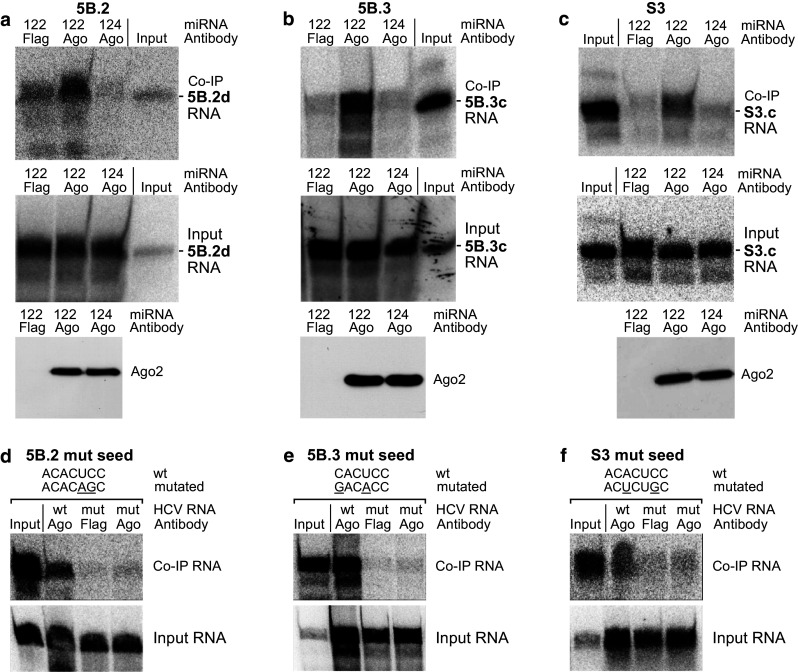
miR-122 binding to example NS5B and 3′UTR target sites. a Co-immunoprecipitation (co-IP) results with RNA 5B.2d (see Supplementary Figure S6) containing the first highly conserved miR-122 binding site in NS5B. HeLa cells were transfected with 32P-labeled HCV RNA 5B.2d along with either miR-122 duplex RNA or with miR-124 as a control. Cells were lysed 6 h post-transfection, and Ago2/miRNA/target RNA complexes were precipitated with an anti-Ago2 antibody. The “Input” lane shows a small aliquot of the in vitro-transcript used for transfections as a size marker for the RNAs recovered from co-IPs. RNA was extracted and analyzed by denaturing gel electrophoresis and phosphorimaging (upper panel). RNA was isolated from an aliquot of the cell extract prior to co-IP to check RNA integrity after cell lysis (middle panel). Ago2 western blots show the pull-down of Ago2 protein by the immunoprecipitation (lower panel). b Co-IP results and controls as in a, but with RNA 5B.3c. c Co-IP results and controls as in a, but with RNA S3c containing the 3′UTR miR-122 binding site. d–f Mutants of the binding sites knock out miR-122 binding. The seed sequences of the miR-122 binding sites were mutated as indicated, and the RNAs were subjected to co-IP as in a–c
The intensity of miR-122/Ago2 binding depends on target site accessibility
We then varied the range of flanking sequences upstream and downstream of the miR-122 target sites in the RNAs used for in vitro-transcription. The structures of the RNAs were predicted using the Vienna RNAfold WebServer programs. The predicted structures of the RNAs containing one particular target site revealed to change largely when varying the range of upstream and downstream flanking sequences (Figs. 3, 4, 5, left panels, Supplementary Figures S7–S15). From the predicted RNA structure outputs, the single strand probability (ssp) of the seed target sequence was calculated for each base from the Vienna RNA fold minimum free energy (MFE) output EPS files for “MFE structure drawing encoding base-pair probabilities” as a measure for its accessibility and averaged for all bases in the respective miR-122 seed target (for details please see “Materials and methods” section). Along with these changes in predicted RNA structures, also the intensity of binding of miR-122 to its target sites strongly varied (Figs. 3, 4, 5, middle panels). In RNAs 5B.2a and b, the miR-122 target site is largely hidden in double-stranded regions, and the intensity of miR-122/Ago2 binding ranges from low to moderate (Fig. 3a, b). In contrast, in RNA 5B.2c, the miR-122 target site is predicted to be more accessible, and concordantly the intensity of miR-122/Ago2 binding is high (Fig. 3c). Likewise with site 5B.3, miR-122 binding intensity is low when the predicted target site accessibility is low (RNA 5B.3a, Fig. 4a), whereas miR-122 binding intensity is increased when a part of the target sequence is predicted to be well accessible (Fig. 4b, c). With the 3′UTR RNAs, the same effect was observed (Fig. 5). Low target site accessibility (a and b) resulted in inefficient miR-122 binding, whereas a high target site accessibility returned highly efficient binding (c).
Fig. 3.
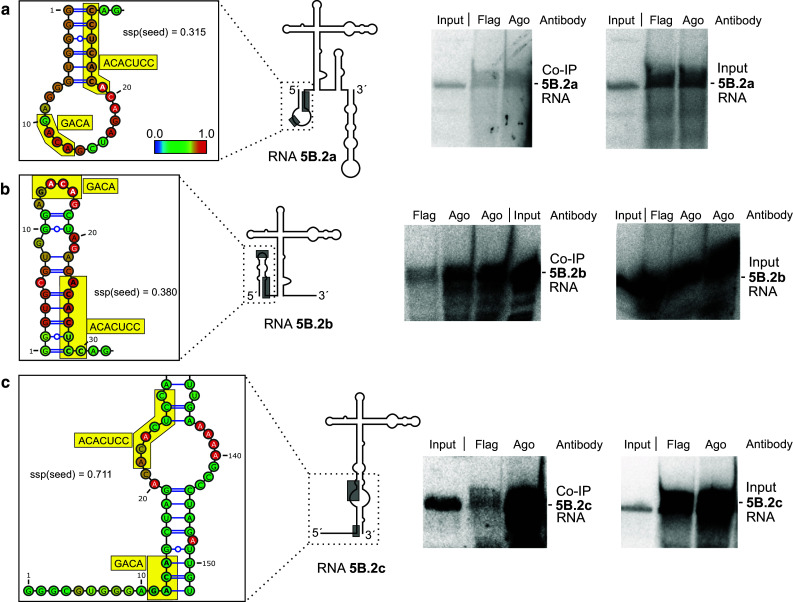
miR-122 dependent interaction of Ago2 protein with target RNAs containing miR-122 target site 5B.2. Left panels The structures of the RNAs are shown schematically with the miR-122 target sites boxed (the complete sequences and predicted structures of RNAs are shown in Supplementary Figures S6 to S15). In the enlargement, miR-122 target sites are shown in detail (boxes). The average single strand probability (ssp) for the miR-122 seed target site is indicated. Middle panels miR-122 dependent Ago2 protein binding to target RNAs. HeLa cells were transfected with miR-122 duplex RNA and with 32P-labeled target RNAs including the miR-122 target site sequence (named RNA 5B.2a, 5B.2b or 5B.2c, respectively) but with different lengths of upstream and downstream flanking regions. 6 h post transfection, Ago2/miRNA/target RNA complexes were subjected to co-IP with either an anti-Ago2 or a Flag antibody as negative control. The “Input” lane shows a small aliquot of the in vitro-transcript used for transfections as a size marker for the RNAs recovered from co-IPs. Right panels RNAs recovered after cells lysis but prior to co-IP to check RNA integrity after cell lysis
Fig. 5.
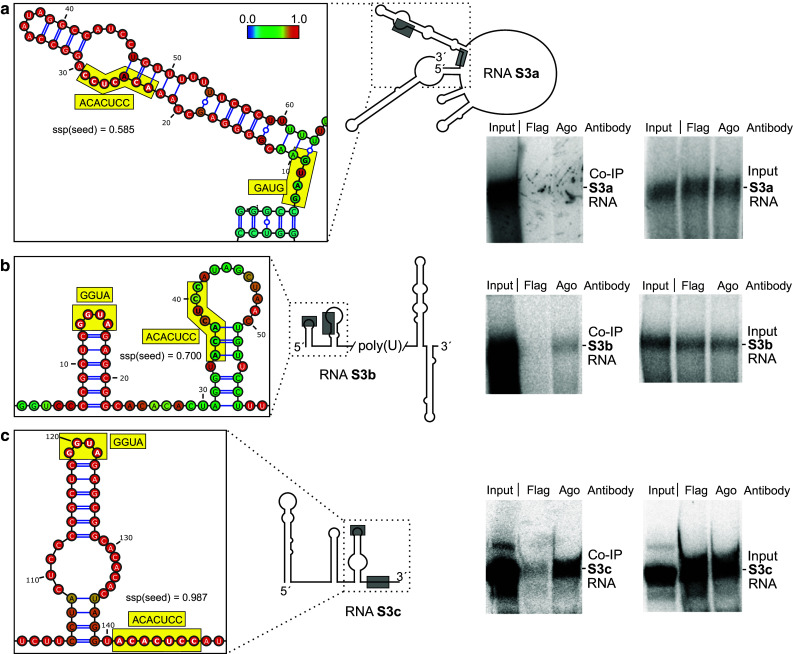
miR-122 dependent interaction of Ago2 with the 3′UTR miR-122 target site (“S3”). RNA structures, co-IP and control experiments are shown similar as in Fig. 3
Fig. 4.
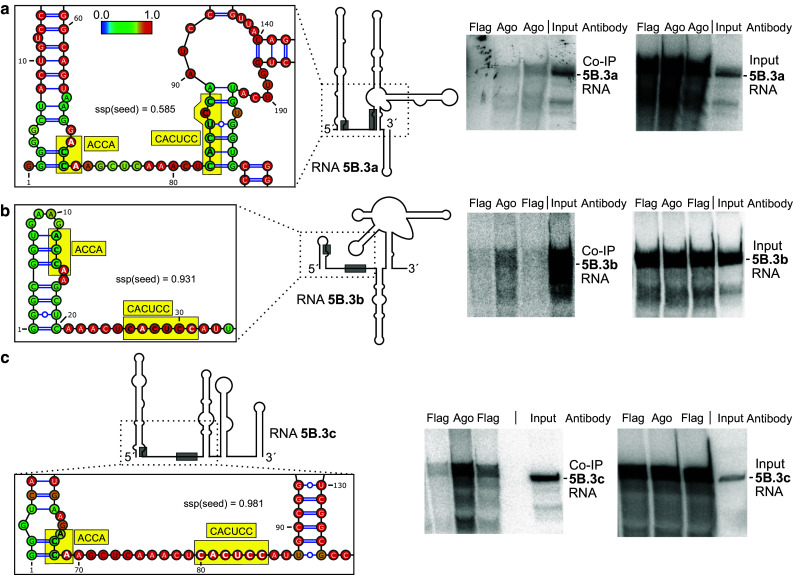
miR-122 dependent interaction of Ago2 with target site 5B.3. RNA structures, co-IP and control experiments are shown similar as in Fig. 3
To illustrate that target site accessibility undergoes dynamic changes which depend on the availability of the flanking RNA sequences, we used a bioinformatic in silico approach exemplary for site 5B.3. A window of 200 nucleotides was moved over the RNA, and the calculated target site accessibility (in terms of the predicted probability to be single-stranded) was plotted over the left position of the window (Fig. 6). The result shows that target site accessibility largely varies, even when the window of fixed length was moved only slightly. This approach even did not take into account that (for the target sites in the NS5B coding region) the distance of two migrating ribosomes—each of which forces the sequence occupied to be single-stranded—may migrate with different distances and even with different speed according to RNA secondary structures that must be overcome.
Fig. 6.

Accessibility of the miR-122 target sites in the NS5B coding region depends on availability of surrounding sequences forming local RNA structure. A window of 200 nts was moved in silico over the sequence containing target site 5B.3, and the predicted average single strand probability of the seed target sequence was plotted over the position of the upstream edge of the window
To analyze the binding strength found with the NS5B and 3′UTR target sites, we used the RNAs shown in Figs. 3, 4 and 5 as well as several additional different RNAs for each site that come with various lengths of flanking sequences (see Supplementary Figure 6). co-IPs were performed with all these RNAs (Figs. 3, 4, 5 show examples thereof). The single-strand probabilities of the miR-122 target sites were calculated as detailed in the “Materials and methods” section, and average binding strength was plotted over predicted target site accessibility for all these RNAs.
The results (Supplementary Figures 16 and 17) essentially show that the three target sites appear to be different, and that for each target site a different feature contribute most to the correlation between predicted accessibility and observed binding strength. Target site 5B.2 shows a surprisingly low correlation between predicted accessibility and experimental miR-122/Ago2 binding strength. This correlation is higher with site 5B.3 and is very high with the target site S3 in the 3′UTR. For site 5B.2, which has largely varying predicted accessibilities for most RNAs used, inclusion of the Adenosine residue directly downstream of the target site [2, 14] considerably improved the correlation between predicted accessibility and binding strength, whereas this effect was negligible for the other target sites. In site 5B.2, all features contribute to the binding in a way that the correlation between predicted accessibility and binding is best for the complete binding site. In contrast, in target sites 5B.3 and S3 the accessibility of the seed target sequence appears most important, among which the “nucleation sequence” CUCC (nucleotides opposite to miRNA nucleotides 2–5) appears not to contribute to this correlation in binding site S3. Taken together, each binding site appears to have its own characteristics possibly due to unique features of the flanking sequences, which may also depend on the binding of other proteins which in turn may alter the secondary structure folding of these RNAs.
Functional analysis of the miR-122 target sites
We used full-length HCV genomes to elucidate the function of the miR-122 binding sites in the NS5B and 3′UTR regions (Fig. 7). Since the potential target site 5B.1 is not conserved in most HCV isolates [35], this site was disabled in all used constructs by mutation. The target sites 5B.2, 5B.3 and S3 were mutated as shown in Fig. 2d–f, either alone, the sites 5B.2 and 5B.3 in combination (5B.2/3) or all three sites in combination (“triple”). The RNA genomes were transfected into HuH-7 cells, and cells or cell extracts were analyzed 3 days after transfection for expression of HCV NS3 and Core proteins by Western blot (Fig. 7b) and immune fluorescence (Fig. 7d) as well as HCV plus strand genome abundancy (Fig. 7c). Counting of cells expressing HCV proteins in large areas of the coverslips (not shown) revealed that single mutations in all sites slightly reduced the number of cells that showed detectable expression of HCV proteins (% values at the bottom of Fig. 7d). Mutations in all sites have a negative effect on protein production in the overall HCV replication cycle (which includes genome translation, minus strand synthesis, plus strand synthesis and further translation of progeny plus strand genomes), and the protein production detected with full-length systems largely originates from progeny plus strands [18]. Expression of NS3 and Core protein was mostly reduced with mutant 5B.2 and also 5B.3, whereas the S3 mutant, the NS5B double mutant 5B.2/3 and the triple mutant showed only slightly reduced viral protein expression (Fig. 7b). Plus strand abundancy was significantly reduced with the 5B.2 mutant (Fig. 7c). Thus, we conclude that the reduced protein production with mutant 5B.2 largely results from reduced progeny plus strand availability. In contrast, plus strand abundancy was only slightly reduced with the 5B.3 mutant but not significantly affected by the S3, NS5B double and the triple mutants. Thus, site 5B.2 clearly has a positive effect on overall genome replication and related viral protein expression, whereas the exact role of the other sites and of combination of sites is less clear. At least with site 5B.3 we assume also a positive effect on HCV RNA replication, whereas the role of site S3 in the 3′UTR appears less clear since its mutation appears to slightly reduce overall translation of HCV proteins but appears to have no effect on overall genome RNA replication and by that on RNA stability. As a consequence, we speculate that sites 5B.3 and S3 may have a negative effect on translation efficiency.
Fig. 7.
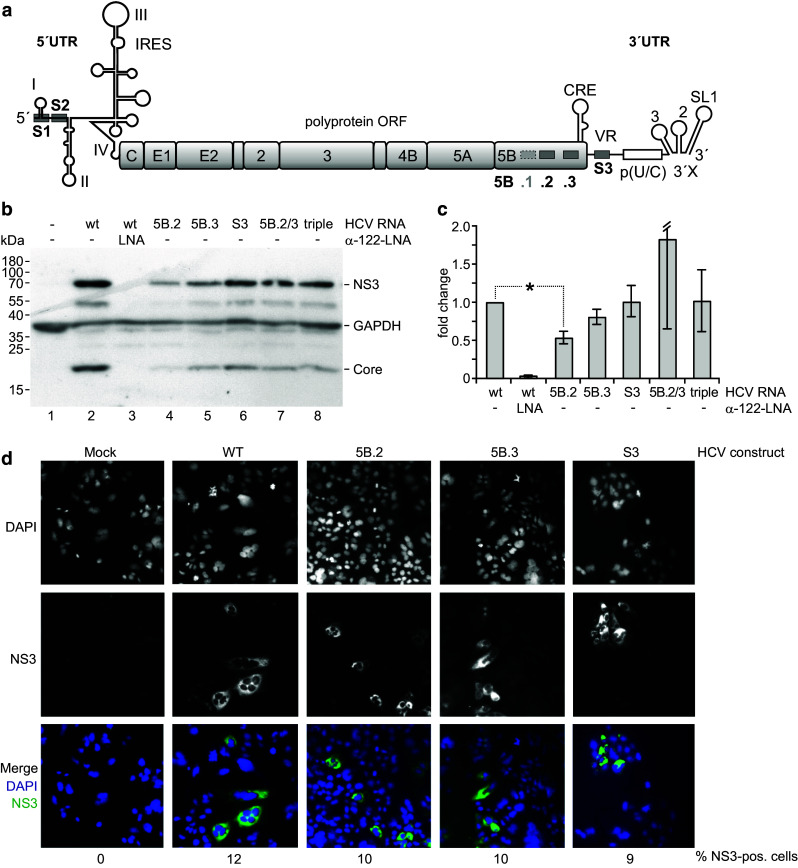
Effects of mutations in the miR-122 binding sites in the NS5B coding region and 3′UTR on HCV overall replication. a The HCV full-length genome with the miR-122 binding sites S1 and S2 in the 5′UTR, 5B.1, .2 and .3 in the NS5B coding region and S3 in the 3′UTR. Site 5b.1 was disabled in all constructs used here since this site is conserved only in a minor fraction of HCV isolates. b–d Cells were transfected with the wild-type HCV RNA or mutated variants as indicated. Cells were harvested 3 days after transfection, and aliquots of the cells extracts were used for analysis of protein expression (NS3 and Core) (b) and HCV plus strand genome abundancy by qPCR (c). d Shows expression of NS3 after 2 days by immune fluorescence. Nuclear staining by DAPI and NS3 staining each are shown in b/w, while the merge is shown in color (blue DAPI, green NS3). “5B.2/3” labels the double mutant with sites 5B.2 and 5B.3 mutated, “triple” the triple mutant with sites 5B.2, 5B.3 and S3 mutated in combination. The counted % of cells expressing NS3 is indicated below
For site S3 in the 3′UTR we separately analyzed its effect on translation using a specialized translation reporter system in which the HCV 3′UTR with the wt or mutated target site S3 was located downstream of the reporter gene (Fig. 8a). With this system, we found that mutation of the miR-122 target site S3 in the 3′UTR and addressing the site with a compensatorily mutated miR-122 variant significantly reduced translation efficiency (Fig. 8b). Thus, we conclude that the 3′UTR target site S3 has a negative effect on HCV genome translation and may have contrary effects on RNA replication and translation.
Fig. 8.

Effect of the 3′UTR miR-122 binding site on translation. a The HCV translation reporter RNA contains the complete HCV 5′UTR plus the AUG and 30 nucleotides of the core coding region to allow complete folding of the IRES, the humanised Renilla luciferase gene (hRLuc), the 3′-terminal 21 nucleotides of the HCV NS5B coding region including the stop codon and the HCV 3′UTR. b Translation assays. The wild-type HCV reporter RNA shown in a with either the wild-type (wt) S3 binding site or the S3 site mutated (mut) as shown in Fig. 2f was transfected into HuH-7.5 cells along with miRNA duplexes as indicated (either miR-122 wild-type or a mutated miR-122 variant with compensating mutations). Cells were lysed 6 h after transfection and luciferase activity measured
Discussion
In this study, we have analyzed the conservation features of the presumed miR-122 binding sites in the NS5B coding region and the 3′UTR of the HCV RNA genome, the binding of miR-122/Ago2 complexes to the target sites and the function of the target sites in HCV translation and replication.
Analysis of the correlation of miRNA binding efficiency with the accessibility of the target sites in the 3′UTRs of cellular mRNAs revealed that effective miRNA targets are preferentially located in regions of high accessibility [36–39]. Also in coding sequences, structural accessibility is the main feature common to all analyzed target sites [40]. Here we show in living cells that the efficiency of miR-122 binding to three highly conserved binding sites in the NS5B coding region and the 3′UTR of the HCV RNA genome largely depends on target site accessibility under conditions when the range of flanking sequences available for the formation of local RNA secondary structures changes. Most importantly, our results indicate that the sequence feature that contributes most to the correlation between target site accessibility and binding strength varies from site to site. In site 5B.2 it is the overall sequence of the site including an additional Adenosine residue directly downstream of the target site, in site 5B.3 it is mainly the seed target sequence, and in the 3′UTR site it is the complete seed target sequence and the supplementary sequence, while the so-called “nucleation sequence” (target nucleotides opposite to miRNA nucleotides 2–5) plays a negligible role. This suggests that the dynamics of miRNA/Ago2 binding not only depends on the target site itself but also may depend on flanking sequence context to a considerable extent, since different flanking sequences interacting with each miRNA target site may modulate the main target site feature that is most important for accessibility of each site to various extents. Moreover, the availability of the flanking sequences may be modulated by RNA binding proteins in an unpredictable manner.
The idea that binding of miR-122 to the target sites in the NS5B coding region and the 3′UTR may depend on dynamic changes in accessibility is supported by previous findings that miR-122/Ago2 complexes bind very strongly to the two 5′UTR target sites but much weaker to the downstream sites [23]. Analysis of miR-122 binding to the target sites in the HCV NS5B coding region by CLIP assays [41] showed that miR-122 binds to both sites (5B.2 and 5B.3) as well as to the non-conserved site 5B.1 located 30 nucleotides upstream of site 5B.2 to a much lower extent compared with the two miR-122 target sites S1 and S2 in the HCV 5′UTR. Another study showed that miR-122 binds with moderate efficiency to site 5B.2 in in vitro binding competition assays, linking miR-122 binding with a slightly negative effect on HCV replication [42].
Our results also can also be analyzed with respect to two special aspects of the target RNA recognition by microRNA/Ago2 complexes which both take into account the contribution of features of the Ago protein. The first aspect is the significance of an A residue at the t1 position, the target position opposite to miRNA nucleotide 1, and the second aspect is the role of the so-called nucleation step in the initial phase of miRNA binding to its target which depends on Ago protein structure and dynamics.
A computational study found an overrepresentation of A residues directly downstream of the target site [2], and also experimental support for the importance of this A residue in improving miRNP complex binding to a target mRNA was reported [43]. Another study evaluated stronger down-regulation of target mRNAs when an A residue was present [44]. A mechanistic explanation for these results was recently reported by a structural study: an A residue (but no other base) downstream of the target site fits into a pocket in the Ago2 protein and thereby increases the affinity of Ago2 protein to such targets [14]. However, our results obtained with the three conserved miR-122 target sites in the HCV RNA confirm that an A residue downstream of the target site substantially improves the correlation between calculated target site accessibility and the intensity of miR-122/Ago2 complex binding only for target site 5B.2 which shows the highest variability in target site accessibility among the sites analyzed. Interestingly, an A residue is also present downstream of the first miR-122 binding site S1 in the HCV 5′UTR in 70/83 isolates (see alignment in supplement to [35]), making this first binding site stronger than 5′UTR site S2 in vivo (ANR and MN, unpublished observations), in contrast to the situation in in vitro binding assays lacking Ago protein [45].
In contrast, we could not detect a leading role of the accessibility of the target nucleotides opposite of the nucleation sequence [46–48]. Ago2 protein first exposes nucleotides 2–5 of the miRNA (the so-called nucleation sequence), and upon target binding the protein rearranges its conformation to also expose the rest of the miRNA seed sequence to the target [14]. In contrast, in our experiments the accessibility of the entire seed target sequence appears to be more important. This discrepancy may be based on our test system, since the co-IP procedure detects miRNA/Ago2 complexes binding to their target sites rather stably over extended time, while transient steps of the target recognition procedure cannot be detected.
A previous study [42] proposed a slight inhibitory effect of site 5B.2 (called MRE 4 in that study) on HCV replication. In contrast, our functional analysis of the miR-122 binding sites by mutation revealed a significant positive effect of site 5B.2 on overall genome replication. Also the amounts of translated HCV proteins are reduced by the mutation in site 5B.2, implying that HCV translation may also be stimulated by site 5B.2. However, since by far most HCV proteins are produced not in the pilot round of translation of the infecting genome but on progeny plus strand genomes [18], we cannot make definite conclusions if the step of genome translation is stimulated by site 5B.2; it may also be possible that the positive effect on translation may be an indirect consequence of the positive effect on genome replication.
For the target site S3 in the HCV 3′UTR, we clearly show a significant reduction in translation efficiency when the site is placed in a specialized translation reporter construct (Fig. 8). In contrast, this mutation has no detectable effect on the overall amplification of plus strand HCV genomes (Fig. 7c). Thus, we can only speculate if site S3 in the 3′UTR may have a positive effect on RNA replication which may compensate for the negative effect on translation. Also for the double mutation 5B.2/3 and the triple site mutation, we can only speculate that a complicated interplay of different functions is involved in the HCV replication cycle, while each mutation finally results in reduced overall replication which is here shown by the reduced production of NS3 and Core proteins (Fig. 7b).
Materials and methods
Transcription templates and RNA synthesis
The templates for target RNAs were generated by PCR or plasmid Maxiprep. Plasmid pFK\I389/NS3-39/Hygubi/5.1 [49] was used to generate PCR templates from HCV genotype 1b (Con1) sequences (NCBI nucleotide database accession No. AJ238799) [50]. Plasmid pFL-J6-JFH1 [51] was used for PCR templates for HCV genotype 2a (JFH-1) (accession No. AB047639) [52] sequences. PCR forward and reverse primer sequences are specified in Supplementary Figure S6, along with the templates used. The forward primer sequences additionally contain the sequence 5′-ACAGTCGCATGGCAGGCTTAATACGACTCACTATAggg-3′ (which includes the T7 RNA polymerase promoter sequence) at their 5′-ends fused to the 5′-end of each specific sequence that hybridizes to the respective template DNA. Thus, the G in TATAG is the first nucleotide of the in vitro-transcribed RNAs. The resulting RNA sequences are specified in Supplementary Figure S6. As full-length genome the Jc1 chimera was used (genotype 2a) [53]. In this HCV genome, the miR-122 target sites in the NS5B region and the 3′UTR were mutated as indicated. In all mutated constructs, the non-conserved miR-122 target site 5B.1 upstream of site 5B.2 which is conserved only in some isolates was mutated to routinely disable this site. The templates for in vitro-transcribed RNAs used for the translation assays were generated by PCR from plasmid pUC18-J6/JFH1 IRES-hRluc-3′-UTR and its derivatives. This plasmid contains a T7 promoter, followed by the 340 nucleotide HCV genotype 2a 5′UTR and 33 nucleotides of core coding sequence from plasmid pFL-J6/JFH1 [51], a 9 nucleotide linker sequence, the coding sequence for humanised Renilla luciferase (hRLuc), 12 linker nucleotides, the 3′-terminal 21 nucleotides of the HCV NS5B coding region including the stop codon, and the HCV 3′UTR.
The PCR fragments or plasmids were used for in vitro-transcription using T7 RNA polymerase. To generate radioactively labeled RNA, transcription was performed using T7 RNA polymerase in the presence of 500 µM ATP, GTP and CTP, 100–300 µM UTP and 0.625 or 0.5 µM [α-32P] UTP (800 Ci/mmol; PerkinElmer). Aliquots of the radiolabeled RNAs were run on 7 M urea/6 % polyacrylamide gels and checked by autoradiography. RNA concentrations were determined by gel images and photometric analyses. The concentrations of the labeled RNAs were calculated from the amount of the limiting radiolabeled nucleotide used in the in vitro-transcription and considering the estimated efficiency of label incorporation (% nucleotides incorporated into the RNA versus % non-incorporated nucleotides). For full-length transcriptions 3750 µM cold rNTPs were used.
RNA and LNA/DNA mixmer oligonucleotides
RNA oligonucleotides were supplied by biomers.net, LNA/DNA mixmer oligos from Exiqon. Duplexes were formed between the guide (mat) and its complementary passenger strand (*). The sequences were:
miR-122 mat, 5′-(phos)UGGAGUGUGACAAUGGUGUUUG-3′;
miR-122*, 5′-(phos)AACGCCAUUAUCACACUAAAUA-3′;
miR-124mat, 5′-(phos)UAAGGCACGCGGUGAAUGCC-3′;
miR-124*, 5′-(phos)CGUGUUCACAGCGGACCUUGAU-3′,
miR-122 S3mutmat, 5′-(phos)UGCAGAGUGACAAUGGUGUUUG-3′;
miR-122 S3mut*, 5′-(phos)AACGCCAUUAUCACUCUAAAUA-3′.
LNA/DNA mixmer oligonucleotides
Anti-miR-122 LNA for sequestering mir122, +C*C*A* + T*T*G* + T*C*A* + C*A*C* + T*C* + C; where a (+) indicates a following LNA residue and G*, A*, T*, C* phosphorothioat DNA bases.
Transfections and co-immunoprecipitations
2.5 × 106 HeLa cells were seeded in 9 cm plates and transfected on the next day (at approx. 90 % confluency) with 3 µg 32P-labeled HCV reporter RNA (40––60 pmol) and 3 µg of active miRNA (i.e., 6 µg of miR duplex = 400 pmol duplex) using Lipofectamine 2000. The RNA amounts per cell and the reporter RNA/microRNA ratios correspond to the conditions found previously to be optimal for analysis of miR-122 action on HCV reporter RNAs in cells [27, 32]. Cells were harvested after 6 h incubation [23, 54], since HCV reporter RNA expression in HeLa cells was found to be maximal after 6 h [22]. For input RNA controls, total RNA was isolated prior to co-IP from 10 % of the cell extract. The residual cell lysates were used for the co-IP essentially according to the protocol of Beitzinger et al. [54] for 3 h using the Ago2 monoclonal antibody 11A9 [55] and Protein G beads (New England Biolabs, NEB) [23, 54]. A mouse anti-Flag M2 antibody (Sigma-Aldrich) was used for controls. After the co-IP and four washing steps, 10 % of the beads were taken for western blots. Anti-Ago2 western blots were performed as described [23, 55]. From the remaining beads, co-IP RNA was isolated [23, 54]. Radiolabeled input RNAs and RNAs recovered by co-IP were purified by proteinase K digestion, phenol/chloroform extraction and ethanol precipitation. RNAs were separated by gel electrophoresis on 7 M urea/6 % polyacrylamide gels and analyzed using a PerkinElmer Cyclone plus phosphorimager.
To calculate the miRNA binding strength from the phosphorimager scans, first the background binding intensity values were subtracted from all binding intensity values of RNAs. Then, each Ago co-IP readout was normalized to the corresponding amount of input RNA, and finally these normalized Ago-co-IP binding intensity values were divided by Flag-co-IP binding intensity values to obtain relative binding intensities. For each target RNA, means and standard deviations were calculated from biological replicates.
Immunochemistry, western blots and translation assays
One day before transfection coverslips were heated in pure Ethanol and covered for 30 min with 0.1 mg/ml Poly-l-Lysin (30,000–70,000). Two days after transfection of HCV full-length Jc1 genomes, cells were washed with PBS and fixed with 4 % paraformaldehyde for 10 min. Cells were washed again 3× with ice cold PBS, permeabilized with cold acetone for 10 min at −20 °C and again washed. Then, cells were incubated with 1 % BSA, 22.5 mg/ml glycine in PBST for 10 min. For staining, cells were incubated with a 1:400 dilution of Anti-HCV NS3 antibody (8 G-2, abcam) in 1 % BSA for 1.5 h at room temperature. Cells were washed 3× with PBS and then incubated with a 1:200 dilution of the secondary antibody (goat-anti-mouse IgG1, Alexa Fluor® 488 conjugate) for 1 h at 37 °C in the dark. Cells were again washed 3× with PBS, incubated with 1 ml DAPI-solution (D1306, Thermo) (5 mg/ml) 1:2000 in Glycin-PBS for 5 min, washed again and mounted on object plates with 13 µl Moviol with DABCO. Western blots were performed as described Conrad et al. [23] using the Anti-HCV NS3 antibody, a HCV Core antigen EA antibody (QE210796, Life Technologies), an anti-GAPDH mAb (clone GAPDH-71.1; Sigma-Aldrich) and as secondary antibody a goat-anti-mouse IgG HOR (Life Technologies). Translation assays were performed as described [23].
Total RNA preparation and quantification of HCV RNA by RT-qPCR
At 3 days posttransfection cells were washed thoroughly with PBS and lysed by Trizol reagent (Ambion). After collection of lysates, total RNA was extracted with chloroform and precipitated with isopropanol in the presence of GlycoBlue reagent. Next, total RNA was resuspended in RNase-free water; DNaseI treatment was conducted with 1 U of DNaseI, and the RNA was purified again using an RNA Clean-up Kit (Life Technologies). Finally, total RNA samples were eluted in equivalent amounts of RNase-free water. cDNA was produced using the qScript Flex cDNA Kit (Quanta Biosciences) according to the manufacturer’s instructions. A reverse transcriptase with RNase H activity revealed important for the quantification of different RNA amounts. For the quantitative PCR, the prepared cDNA was used with the PerfeCTa SYBR Green FastMix (Quanta Biosciences) according to the manufacturer’s instructions; all measurements were conducted using Eppendorf Mastercycler ep realplex2.
RNA structure predictions and calculation of single strand probabilities
The RNA sequences specified in Supplementary Figure S6 were used to predict RNA secondary structures using the Vienna RNAfold WebServer (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) [56, 57]. Basic options were set to “avoid isolated base pairs”, advanced folding options to “no dangling end energies” with the Turner model, 2004 [58]. An equivalent example RNAfold command line call would be “RNAfold -p -d0—noLP < RNA_5B.2a.fa > RNA_5B.2a.out”. The Vienna RNA structure prediction dot-bracket output is shown in Supplementary Figure S6. For visualization of predicted RNA structures, RNA sequences and Vienna dot-bracket outputs were loaded into VARNAv3-92 (http://varna.lri.fr) [59]. Base pair probability (bpp) values were loaded into VARNAv3-92 from the Vienna dot plot EPS file output, and the color code was set to Vienna style. The resulting predicted RNA structures are shown in Supplementary Figures S7–S16, details are shown in Figs. 3, 4 and 5.
For calculating the single strand probability (ssp) values for miRNA binding sites as a measure for target site accessibility, we used the Vienna output EPS files for “MFE structure drawing encoding base-pair probabilities”. This Vienna minimum free energy (MFE) output returns base pair probability (bpp) values for bases predicted to pair, but single strand probabilities (ssp = 1 − bpp) for bases predicted to be unpaired. Therefore, we directly used the ssp values for each unpaired base, but calculated the ssp values for those bases predicted to pair as (ssp = 1 − bpp) from the Vienna MFE output. Then, mean ssp values for each miRNA target site (or parts of it) were calculated by dividing the sum of base ssp values by the number of the respective bases. For calculating the mean ssp values for miR-122 target sites including the presence or absence of an additional A residue directly downstream of the seed target site (which binds into a pocket of the Ago2 protein [14]), the ssp of this A residue (if present) was included in the calculation of mean ssps.
For plotting the binding strength of miR-122-Ago2 complexes scanned from experiments over target site accessibility (Supplementary Figures 16 and 17), mean (AVERAGE) values were calculated from the binding strength of different experiments with the same RNA. From these binding strength values, also standard deviations (SD; STABW.N) were calculated and indicated by error bars above and below mean values. For correlation, Excel returns the coefficient of determination (RSQ). The square root of RSQ yields the correlation coefficient R shown in the diagrams in Supplementary Figures 16 and 17.
For plotting target site average single strand probability over sequence window position (Fig. 6), a window of 200 nts was moved in steps of three nucleotides over the sequence containing the 5B.3 target site, and the predicted average ssp of the site was plotted over the position of the upstream edge of the window. Then the ensemble of all possible foldings was calculated with the function “RNAfold -p” of the Vienna RNAfold WebServer. Nucleotide single strand probabilities were then calculated using “relplot.pl -u”of the Vienna RNAfold WebServer. From these, the average miRNA target site single strand probability of the miRNA target site was calculated and plotted over the nucleotide position at the window’s left edge.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, Germany (DFG; IRTG 1384, MA-5082/1, SFB 1021, NI-604/2-2) and the Carl-Zeiss-Stiftung. We thank Volker Lohmann for providing plasmid pFK\I389/NS3-39/Hygubi/5.1, Charles M. Rice for plasmid pFL-J6-JFH-1, Thomas Pietschmann and Ralf Bartenschlager for the Jc1 clone, and Yann Ponty for programming the VARNAv3-92 update including Vienna style handling.
Footnotes
G. K. Gerresheim and N. Dünnes contributed equally to this work.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 3.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schnall-Levin M, Zhao Y, Perrimon N, Berger B. Conserved microRNA targeting in Drosophila is as widespread in coding regions as in 3′UTRs. Proc Natl Acad Sci USA. 2010;107:15751–15756. doi: 10.1073/pnas.1006172107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orom UA, Nielsen FC, Lund AH. microRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 8.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 9.Eichhorn SW, Guo H, McGeary SE, Rodriguez-Mias RA, Shin C, Baek D, Hsu SH, Ghoshal K, Villen J, Bartel DP. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol Cell. 2014;56:104–115. doi: 10.1016/j.molcel.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brümmer A, Hausser J. microRNA binding sites in the coding region of mRNAs: extending the repertoire of post-transcriptional gene regulation. Bioessays News Rev Mol Cell Dev Biol. 2014;36:617–626. doi: 10.1002/bies.201300104. [DOI] [PubMed] [Google Scholar]
- 11.Petri S, Dueck A, Lehmann G, Putz N, Rüdel S, Kremmer E, Meister G. Increased siRNA duplex stability correlates with reduced off-target and elevated on-target effects. RNA. 2011;17:737–749. doi: 10.1261/rna.2348111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature. 2008;456:921–926. doi: 10.1038/nature07666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jinek M, Doudna JA. A three-dimensional view of the molecular machinery of RNA interference. Nature. 2009;457:405–412. doi: 10.1038/nature07755. [DOI] [PubMed] [Google Scholar]
- 14.Schirle NT, Sheu-Gruttadauria J, MacRae IJ. Structural basis for microRNA targeting. Science. 2014;346:608–613. doi: 10.1126/science.1258040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science. 2005;309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 16.Chang J, Nicolas E, Marks D, Sander C, Lerro A, Buendia MA, Xu C, Mason WS, Moloshok T, Bort R, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. doi: 10.4161/rna.1.2.1066. [DOI] [PubMed] [Google Scholar]
- 17.Jopling C. Liver-specific microRNA-122: biogenesis and function. RNA Biol. 2012;9:137–142. doi: 10.4161/rna.18827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohmann V. Hepatitis C virus RNA replication. Curr Top Microbiol Immunol. 2013;369:167–198. doi: 10.1007/978-3-642-27340-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moradpour D, Penin F. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol. 2013;369:113–142. doi: 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
- 20.Niepmann M. Hepatitis C virus RNA translation. Curr Top Microbiol Immunol. 2013;369:143–166. doi: 10.1007/978-3-642-27340-7_6. [DOI] [PubMed] [Google Scholar]
- 21.Ito T, Tahara SM, Lai MM. The 3′-untranslated region of hepatitis C virus RNA enhances translation from an internal ribosomal entry site. J Virol. 1998;72:8789–8796. doi: 10.1128/jvi.72.11.8789-8796.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song Y, Friebe P, Tzima E, Jünemann C, Bartenschlager R, Niepmann M. The hepatitis C virus RNA 3′-untranslated region strongly enhances translation directed by the internal ribosome entry site. J Virol. 2006;80:11579–11588. doi: 10.1128/JVI.00675-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Conrad KD, Giering F, Erfurth C, Neumann A, Fehr C, Meister G, Niepmann M. MicroRNA-122 dependent binding of Ago2 protein to hepatitis C virus RNA is associated with enhanced RNA stability and translation stimulation. PLoS One. 2013;8:e56272. doi: 10.1371/journal.pone.0056272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimakami T, Yamane D, Jangra RK, Kempf BJ, Spaniel C, Barton DJ, Lemon SM. Stabilization of hepatitis C virus RNA by an Ago2-miR-122 complex. Proc Natl Acad Sci USA. 2012;109:941–946. doi: 10.1073/pnas.1112263109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimakami T, Yamane D, Welsch C, Hensley L, Jangra RK, Lemon SM. Base pairing between hepatitis C virus RNA and MicroRNA 122 3′ of its seed sequence is essential for genome stabilization and production of infectious virus. J Virol. 2012;86:7372–7383. doi: 10.1128/JVI.00513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci USA. 2013;110:1881–1886. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henke JI, Goergen D, Zheng J, Song Y, Schüttler CG, Fehr C, Jünemann C, Niepmann M. microRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J. 2008;27:3300–3310. doi: 10.1038/emboj.2008.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niepmann M. Activation of hepatitis C virus translation by a liver-specific microRNA. Cell Cycle. 2009;8:1473–1477. doi: 10.4161/cc.8.10.8349. [DOI] [PubMed] [Google Scholar]
- 29.Jangra RK, Yi M, Lemon SM. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J Virol. 2010;84:6615–6625. doi: 10.1128/JVI.00417-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts AP, Lewis AP, Jopling CL. miR-122 activates hepatitis C virus translation by a specialized mechanism requiring particular RNA components. Nucleic Acids Res. 2011;39:7716–7729. doi: 10.1093/nar/gkr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson JA, Zhang C, Huys A, Richardson CD. Human Ago2 is required for efficient microRNA 122 regulation of hepatitis C virus RNA accumulation and translation. J Virol. 2011;85:2342–2350. doi: 10.1128/JVI.02046-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goergen D, Niepmann M. Stimulation of hepatitis C Virus RNA translation by microRNA-122 occurs under different conditions in vivo and in vitro. Virus Res. 2012;167:343–352. doi: 10.1016/j.virusres.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 33.Fehr C, Conrad DK, Niepmann M. Differential stimulation of hepatitis C Virus RNA translation by microRNA-122 in different cell cycle phases. Cell Cycle. 2012;11:277–285. doi: 10.4161/cc.11.2.18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mauger DM, Golden M, Yamane D, Williford S, Lemon SM, Martin DP, Weeks KM. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc Natl Acad Sci USA. 2015;112:3692–3697. doi: 10.1073/pnas.1416266112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fricke M, Dünnes N, Zayas M, Bartenschlager R, Niepmann M, Marz M. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA. 2015;21:1219–1232. doi: 10.1261/rna.049338.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 37.Hofacker IL. How microRNAs choose their targets. Nat Genet. 2007;39:1191–1192. doi: 10.1038/ng1007-1191. [DOI] [PubMed] [Google Scholar]
- 38.Obernosterer G, Tafer H, Martinez J. Target site effects in the RNA interference and microRNA pathways. Biochem Soc Trans. 2008;36:1216–1219. doi: 10.1042/BST0361216. [DOI] [PubMed] [Google Scholar]
- 39.Marin RM, Vanicek J. Efficient use of accessibility in microRNA target prediction. Nucleic Acids Res. 2011;39:19–29. doi: 10.1093/nar/gkq768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C, Mallick B, Long D, Rennie WA, Wolenc A, Carmack CS, Ding Y. CLIP-based prediction of mammalian microRNA binding sites. Nucleic Acids Res. 2013;41:e138. doi: 10.1093/nar/gkt435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luna JM, Scheel TK, Danino T, Shaw KS, Mele A, Fak JJ, Nishiuchi E, Takacs CN, Catanese MT, de Jong YP, et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell. 2015;160:1099–1110. doi: 10.1016/j.cell.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nasheri N, Singaravelu R, Goodmurphy M, Lyn RK, Pezacki JP. Competing roles of microRNA-122 recognition elements in hepatitis C virus RNA. Virology. 2011;410:336–344. doi: 10.1016/j.virol.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 43.Wu L, Belasco JG. Micro-RNA regulation of the mammalian lin-28 gene during neuronal differentiation of embryonal carcinoma cells. Mol Cell Biol. 2005;25:9198–9208. doi: 10.1128/MCB.25.21.9198-9208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nielsen CB, Shomron N, Sandberg R, Hornstein E, Kitzman J, Burge CB. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA. 2007;13:1894–1910. doi: 10.1261/rna.768207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mortimer SA, Doudna JA. Unconventional miR-122 binding stabilizes the HCV genome by forming a trimolecular RNA structure. Nucleic Acids Res. 2013;41:4230–4240. doi: 10.1093/nar/gkt075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Long D, Lee R, Williams P, Chan CY, Ambros V, Ding Y. Potent effect of target structure on microRNA function. Nat Struct Mol Biol. 2007;14:287–294. doi: 10.1038/nsmb1226. [DOI] [PubMed] [Google Scholar]
- 47.Marin RM, Voellmy F, von Erlach T, Vanicek J. Analysis of the accessibility of CLIP bound sites reveals that nucleation of the miRNA:mRNA pairing occurs preferentially at the 3′-end of the seed match. RNA. 2012;18:1760–1770. doi: 10.1261/rna.033282.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khorshid M, Hausser J, Zavolan M, van Nimwegen E. A biophysical miRNA–mRNA interaction model infers canonical and noncanonical targets. Nat Methods. 2013;10:253–255. doi: 10.1038/nmeth.2341. [DOI] [PubMed] [Google Scholar]
- 49.Frese M, Barth K, Kaul A, Lohmann V, Schwarzle V, Bartenschlager R. Hepatitis C virus RNA replication is resistant to tumour necrosis factor-alpha. J Gen Virol. 2003;84:1253–1259. doi: 10.1099/vir.0.18997-0. [DOI] [PubMed] [Google Scholar]
- 50.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 51.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 52.Kato T, Date T, Miyamoto M, Furusaka A, Tokushige K, Mizokami M, Wakita T. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology. 2003;125:1808–1817. doi: 10.1053/j.gastro.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 53.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci USA. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beitzinger M, Peters L, Zhu JY, Kremmer E, Meister G. Identification of human microRNA targets from isolated argonaute protein complexes. RNA Biol. 2007;4:76–84. doi: 10.4161/rna.4.2.4640. [DOI] [PubMed] [Google Scholar]
- 55.Rüdel S, Flatley A, Weinmann L, Kremmer E, Meister G. A multifunctional human Argonaute2-specific monoclonal antibody. RNA. 2008;14:1244–1253. doi: 10.1261/rna.973808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lorenz R, Bernhart SH, Zu Siederdissen CH, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA Package 2.0. Algorithms for Molecular Biology: AMB 6,26 [DOI] [PMC free article] [PubMed]
- 58.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Darty K, Denise A, Ponty Y. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.