

Abstract
For many proteins, biological function requires the folding of the polypeptide chain into a unique and persistent tertiary structure. This review concerns proteins that adopt a specific tertiary structure to function, but are otherwise partially or completely disordered. The biological cue for protein folding is environmental perturbation or minor post-translational modification. Hence, we term these proteins conditionally disordered. Many of these proteins recognize and bind other molecules, and conditional disorder has been hypothesized to allow for more nuanced control and regulation of binding processes. However, this remains largely unproven. The sequences of conditionally disordered proteins suggest their propensity to fold; yet, under the standard laboratory conditions, they do not do so, which may appear surprising. We argue that the surprise results from the failure to consider the role of the environment in protein structure formation and that conditional disorder arises as a natural consequence of the marginal stability of the folded state.
Keywords: Protein stability, Unstable proteins, Conformational fluctuations, Osmolytes, Coupled binding and folding
Introduction
Christian Anfinsen’s demonstration that Ribonuclease A could be spontaneously refolded in vitro, following chemical denaturation, into a specific and enzymatically active conformation, firmly established protein structure formation as a self-driven process that occurs without external input of energy. This resulted in Anfinsen’s “thermodynamic hypothesis”, namely, that “the three-dimensional structure of the native protein in a given environment (solvent, pH, ionic strength, presence of other components, temperature, etc.) is the one in which the Gibbs free energy of the whole system is a minimum with respect to all degrees of freedom, i.e., that the native conformation is determined by the various interatomic interactions and hence by the amino acid sequence, in a given environment” [1]. For small proteins at least, this is now universally accepted to be true. Once the polypeptide chain has been formed, structure is self-organizing. While Anfinsen worked on stable proteins, which overwhelmingly populate a singular conformational state, the structure of highly dynamic and intrinsically disordered proteins is similarly determined. The amino acid sequence of these proteins, together with the environment, determines the conformational states available to the protein, as well as the pathways and timescales for their interconversion.
Anfinsen’s work was enormously influential, but his carefully worded hypothesis almost immediately underwent radical simplification. In practice, it is frequently reduced to “sequence determines structure”, with the role of the environment neglected. It is certainly true that the conformation of many proteins is only weakly linked to the environment. Proteins may tolerate large changes in temperature and pH without measurable perturbation of their structure, and be quite resistant to chemical denaturation [2]. For relatively stable proteins like this, the simplification is understandable. However, for unstable proteins, like those considered in this review, structure formation itself is strongly linked to the physicochemical environment. If the role of the environment is ignored, then confusion results.
In general, the linkage between protein structure and the environment manifests in a number of ways, some subtle and some dramatic. Haemoglobin and Myoglobin, some of the first proteins to be structurally characterized, undergo pronounced changes in structure as a result of proton and oxygen binding, directly linked to their function in oxygen transport. It is textbook knowledge that their exact structural configuration is linked to pH and the dissolved oxygen concentration. Metamorphic proteins present a more dramatic and more recently discovered example of the environmental dependence of structure formation, being capable of adopting several different tertiary structures dependent on the physicochemical conditions [3–5]. The proteins we consider here are marked by the near complete loss and gain of tertiary structure upon environmental perturbation. This is a feature of all folded proteins, but those we term “conditionally disordered” are demarcated by the occurrence of the structural transitions under near-physiological conditions (Fig. 1). In a biological setting, it is likely that the structure of these proteins is only transiently maintained, forming and possibly dissipating “on demand”. Conditionally disordered proteins thereby force us to recognize the essentiality of both parts of Anfinsen’s hypothesis—structure results from both the amino acid sequence and the environment, a point that has been energetically made by Ben-Naim [6]. To specify the structure of a protein, we need to specify the temperature, pressure, and chemical composition of the solution in which it exists. Based on this observation, we earlier suggested that protein structural domain definition should emphasize the capacity for a sequence to fold, rather than the realization of the folded state in a specific physicochemical environment [7].
Fig. 1.
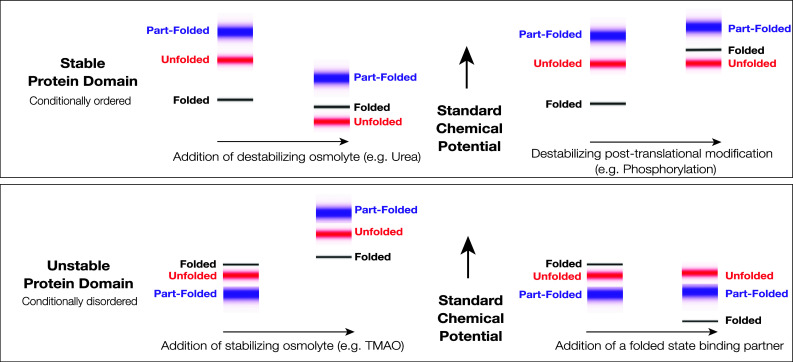
Standard Gibbs energies and protein conformational states. The figure shows schematically the standard chemical potentials (partial molar Gibbs energies) of proteins in the accessible region of the Gibbs energy landscape and their association with protein conformational states. Top A stable protein that folds in two-state fashion under physiological conditions. The low energy states (black) are associated with the very small group of folded state conformations, the higher energy states (red) are associated with the very large group of unfolded state conformations, and the still higher energy states (blue) are associated with the group of partially folded conformations, which may serve as folding transition states. At equilibrium, the folded state conformations are overwhelmingly populated, due to the large difference in the standard chemical potentials of the folded and unfolded states. The folded state is, therefore, the native state. Destabilizing environmental perturbation (e.g., addition of a denaturant like urea) or chemical modification (e.g., phosphorylation) can change the Gibbs energy landscape, allowing the population of the unfolded states. Bottom An unstable, conditionally disordered protein, that has the capacity to fold, but does not do so under physiological conditions. The folded state conformations (black), the unfolded state conformations (red), and the partially folded state conformations (blue) are not well discriminated in the Gibbs energy landscape. Under physiological conditions, the folded state may not be appreciably populated. As depicted here, the partially folded state is the predominant native state. Stabilizing environmental perturbation (e.g., addition of an osmolyte like TMAO or a chemically specific binding partner) can change the Gibbs energy landscape, facilitating partial or complete population of the folded state. In the left panels, the shifts depicted in the figure are consistent in sign with the transfer Gibbs energy measurements carried out by Bolen and coworkers (see [22, 26, 129, 131])
Taxonomy is an artificial business, but for the purposes of this review, it is useful to have an operational definition of a “conditionally disordered” protein. We consider that these proteins have several essential characteristics. (1) They exist in a partially or highly disordered state under the usual conditions for in vitro analysis—in regular physiological buffers, at atmospheric pressure and ambient temperature. (2) They will generally have homologs which are stable and folded under the same conditions, and sequences which are currently not distinguishable from those of folded proteins. This establishes their propensity for structure formation, and discriminates them from intrinsically disordered proteins that cannot fold, which may be characterized by low sequence complexity, and heavily biased amino acid composition [8–11]. (3) Near complete population of the unique and “expected” conformational state can be achieved by environmental perturbation or minor post-translational modification. It is the ability to form tertiary structure, involving non-local sequence contacts, which discriminates these proteins from short linear sequence motifs [12], molecular recognition features (MoRFs) [13, 14], or disordered domains [15].
Environmental perturbations linked to structure formation in conditionally disordered proteins include shifts in temperature or pH, the addition of stabilizing chemical osmolytes, and the addition of chemically specific binding partners. The latter appears of greatest significance for biological function.
In other words, these proteins often participate in particularly extensive coupled folding and binding processes that involve the loss and gain of tertiary structure. The linkage between conformational transitions and binding processes has been well studied from an equilibrium thermodynamic perspective [16–21], and this work provides a theoretical framework for the quantitative treatment of this phenomenon.
However, a characteristic feature of conditionally disordered proteins is that they can often be “force-folded” [22] in the absence of their biological binding partner using stabilizing chemical osmolytes such as trimethylamine-N-oxide (TMAO). Osmolytes are small naturally occurring organic molecules that accumulate intracellularly in response to environmental stresses, such as dehydration [23], and help maintain cellular and molecular organization under these adverse circumstances [24].
The “force folding” of conditionally disordered proteins with co-solutes like TMAO mirrors the much more widely appreciated “force unfolding” of stable proteins with co-solutes like urea (Fig. 1, left). Indeed, the effects of urea and TMAO on protein structure are counteracting and essentially additive [25–27]. While the underlying physical mechanisms are still being investigated [28–32], urea preferentially interacts with the polypeptide backbone, promoting backbone exposure, while TMAO is preferentially excluded, promoting backbone burial. TMAO thereby exerts a general compacting effect on all disordered sequences [27]. However, TMAO will not induce secondary or tertiary structure formation, where no inbuilt structural propensity exists, and it does not specifically guide the structure formation process. Chemical stabilization is, therefore, of enormous practical importance in the study of conditionally disordered proteins, since it can reveal the presence of an accessible folded state within the Gibbs energy landscape of an apparently disordered protein (Fig. 1, left).
Aside from binding, the other principal biological cue for structural reconfiguration of conditionally disordered proteins is post-translational modification [33]. This might include externally catalyzed reactions such as phosphorylation or auto-catalyzed reactions such as ester bond formation. In an strict equilibrium thermodynamic sense, post-translation modification is not an environmental perturbation, since it involves covalent chemical alteration of the polypeptide chain. However, in a biological setting, post-translational modification is a critical regulator of protein structure and function; hence, we have included it in this short review. The effects of post-translational modification are structurally specific, and such modifications might either stabilize or destabilize the folded state of a conditionally disordered protein (Fig. 1, top right).
In the following section, we provide some examples of naturally occurring conditionally disordered proteins and discuss how they respond structurally to environmental perturbation or minor post-translational modifications. While conditionally disordered proteins participate in diverse biological processes, their specific role is almost always molecular recognition. Conditional disorder has been suggested to allow more nuanced control of binding processes; however, this hypothesis remain largely unproven. It may also simply be a manifestation of the marginal stability of the folded state. We consider these issues in the discussion. Regardless of functional advantage, conditional disorder certainly occurs and, in some cases, has become wedded to the regulation of protein function with surprising results.
Examples
Proteins stabilized by chemically specific binding events
The nucleocapsid-binding domains from the RNA-dependent RNA polymerase (RdRp) of rubulaviruses
The paramyxoviruses are a family of respiratory pathogens. Members of this taxon cause measles and mumps, the once familiar diseases of early childhood, as well as numerous other human and animal contagions. The single-stranded RNA genome of these viruses is organized and protected in a protein–RNA complex termed the nucleocapsid [34, 35]. To enable transcription and genome replication, the viral RNA-dependent RNA polymerase (RdRp) must move processively along the viral nucleocapsid. Polymerase translocation and RNA synthesis must be coordinated with the transient displacement of the nucleocapsid protein from the genome (see [36] for review). While there are no detailed molecular models of this process, small (~50 amino acid) helical domains located at the end of the P protein, the non-catalytic subunit of the RdRp, are known to enable processive movement of the RdRp by reversibly binding and releasing the viral nucleocapsid protein. The nucleocapsid-binding domains from the P protein are flexibly tethered to an oligomerizing coiled-coil, and appear to function as the “feet” of the RdRp, enabling its movement along the protein–RNA template.
The interaction between the RdRp foot domain and the nucleocapsid protein was initially characterized for measles virus (genus Morbillivirus). In this case, the RdRp foot domain forms a compact and stable 3-helix bundle, which binds to a linear motif within the intrinsically disordered C-terminus of the nucleocapsid protein [37–39]. As expected for a transiently maintained protein–protein interaction, binding is weak and kinetically rapid. The RdRp–nucleocapsid interactions in Sendai virus (genus Respirovirus) [40, 41] and Hendra virus (genus Henipavirus) [42–44] are similarly configured. However, for mumps virus (genus Rubulavirus), while an X-ray crystal structure of the foot domain (Fig. 2a) revealed the expected 3-helix bundle, the protein exists in solution at low temperature as a compact and dynamic ensemble of partially-formed α-helices—a classical “molten-globule” [45]. This structure largely dissipates at physiological temperatures.
Fig. 2.

Conditionally disordered proteins stabilized by chemically specific binding events. a Nucleocapsid-binding (foot) domain from the mumps virus RdRp (PDB ID: 3BBZ). b B. subtilis RNAse P (PDB ID: 1A6F), with the sulfate ion responsible for structure induction shown in grey. c Third WW domain of human NEDD4-1 (PDB ID: 4N7F). d LEF-1 HMBG domain (PDB ID: 2LEF) bound to DNA. e ET-like domain of human AF9 in complex with AF4 (PDB ID: 2LM0). The ordered part of AF4 (amino acids 761–775) is displayed in grey. f Brinker nuclear repressor (PDB ID: 2GLO) bound to DNA. The program UCSF chimera [130] was used to generate this figure and Fig. 3
This finding prompted a structural survey of the RdRp foot domains from four closely related rubulaviruses [7]. Somewhat surprisingly, with pairwise sequence identities of 30–40%, the rubulavirus foot domains range from stable and folded to almost completely disorganized under the standard laboratory conditions. The mumps virus foot domain sits in the middle of this structural continuum. All but the most disordered domain clearly retain nucleocapsid-binding ability [7]. Characteristically, several of the partially structured foot domains can be “force-folded” by the addition of the stabilizing co-solute TMAO [7, 45]. It seems highly probable, though not yet proven, that the folded state will be transiently adopted in all cases upon nucleocapsid binding. Unlike the other paramyxoviral genera that have been investigated, as described above, the RdRp foot domains of rubulaviruses attach to the structured region of the nucleocapsid protein ([46], unpublished data).
Coupled binding and folding is a recurrent feature of paramyxoviral RdRp–nucleocapsid interactions [38–44] and this has been hypothesized to be a means of moderating the affinity and lifetime of these very transient interactions. Processive movement of the RdRp requires highly specific yet reversible interactions between the feet and template that are strong enough to prevent dissociation, but not so strong as to impede forward motion. For the rubulaviruses, it is hypothesized that the resolution of these competing requirements is frequently achieved through the destabilization of the folded state of the foot domain while still maintaining the ability of this state to form a specific interaction with its binding partner. The energetics of complex formation then subsume both the unfavorable energetic cost of ordering the domain as well as the favorable energetic contribution of binding. Hence, the destabilization of the foot domain serves as a means to precisely tune the affinity and lifetime of the interaction between the polymerase and its template.
However, this idea has not yet been extensively tested. For other virus families in the same order, the RdRp must also traverse a viral nucleocapsid during RNA synthesis, and solutions have evolved that do not involve coupled binding and folding. For example, in the case of rabies viruses [47], the nucleocapsid-binding domains of the phosphoprotein are greatly elaborated [48], appear quite stable, and bind to the structured region of the nucleocapsid protein [49].
Bacillus subtilis RNAse P
RNAse P is an ancient and ubiquitous ribonucleoprotein enzyme responsible for the maturation of transfer RNA through cleavage of 5′ leader sequences present in precursor RNAs [50]. In bacteria, there is a single protein, the RNAse P protein, associated with the single RNA component. The catalytic activity of the enzyme is conferred by the RNA component, which is capable of RNAse activity in isolation under laboratory conditions [51]. However, in the cell, the protein component is essential for transfer RNA maturation. Although not directly involved in catalysis, the RNAse P protein serves to facilitate substrate binding and confer specificity. It may also help maintain the active configuration of the catalytic RNA [52, 53]. The RNAse P protein is ~120 amino acids in size and adopts an α–β sandwich fold (Fig. 2b), which appears to be highly conserved across all bacterial species [54].
A series of studies from the Oas lab [55–61] established that in the absence of the RNA component, the RNAse P protein from B. subtilis exists in a disordered state at low ionic strength, but folds in the presence of small anions such as sulfate and phosphate. In a rigorous and technically sophisticated study, these researchers devised a means to distinguish the folding and binding equilibria, using the osmolyte TMAO to control the extent of chain folding independently of anion binding [55]. They subsequently extended their methods to obtain an integrated kinetic and thermodynamic description of the folding process for this protein [59]. Their results indicated that the protein has a folded state of relatively high Gibbs energy which is not significantly populated unless bound and stabilized by anionic ligands. Furthermore, using TMAO to initiate refolding, they obtained the first estimate for the folding rate constant of a conditionally disordered protein. One finding that emerged from this work is that the RNAse P protein had an unusually slow folding rate in comparison with stable proteins of a similar size. Whether this is a general property of conditionally folded proteins will require studies in other systems using the methods developed by the Oas lab.
The WW domain of human NEDD4-1
WW domains are small (~40 amino acid) protein interaction modules that fold into a triple-stranded antiparallel β-sheet (Fig. 2c), and bind proline-rich peptide motifs [62, 63]. They are named after two conserved tryptophan residues that are central to the domain architecture. WW domains are found in a large number of otherwise unrelated signaling and cytoskeletal proteins. One such protein is NEDD4-1 (neuronal precursor cell-expressed developmentally downregulated 4-1) the prototypical member of a family of ubiquitin–protein ligases [64] that form part of the proteasomal protein degradation system, amongst other functions. Human NEDD4-1 has four centrally located WW domains that are involved in substrate recognition and the regulation of its ubiquitin ligase activity.
A comprehensive NMR study of the third WW domain (WW3) of human NEDD4-1 [65] established that the domain exists in equilibrium between the folded state, competent for peptide binding [66], and a largely unstructured state, characterized by random coil like chemical shifts. Based on global fitting of Carr–Purcell–Meiboom–Gill relaxation dispersion data and circular dichroism data, collected at various temperatures, to a two-state unfolding model, the authors estimate that the unfolded state is approximately 20% populated at physiological temperature, in a phosphate buffered saline solution (pH 6.5). They suggest that peptide recognition is likely to proceed through the conformational selection of the folded state of the WW3 domain, thereby depopulating the unfolded state.
This system differs from the others described in this review, because the folded state remains significantly populated under regular laboratory conditions. There are other examples of proteins exhibiting mixed populations of folded and unfolded states in such circumstances, e.g., the vnd/NK-2 homeodomain [67]. These marginally stable proteins sit on the boundary between order and disorder.
High mobility group box domains
High Mobility Group Box (HMGB) domains are widely distributed in eukaryotic and bacterial genomes [68–70], and generally confer DNA binding function to the protein in which they are embedded. HMGB domains may either recognize specific DNA sequences, or bind to DNA in a sequence-independent fashion. The HMGB domains are formed by three helices, arranged in an “L-shaped” configuration, with the arm created by helices I and II, and the main stem created by helix III and the N-terminal polypeptide. A central hydrophobic core is created at the junction between arm and stem (Fig. 2d). A similar structural motif has recently been identified as binding the stem loop of histone mRNA [71], demonstrating that the L-motif has a broader role in nucleic-acid recognition and remodeling [72]. In addition, the helix–turn–helix structural motif that creates the arm of HMGB domain occurs repeatedly in DNA binding proteins, in a variety of differing structural contexts [73].
The HMGB domains that bind DNA in sequence-independent fashion appear to maintain their structure in the absence of DNA (e.g., the HMG1 A domain [74]). In contrast, some of those that recognize specific DNA sequences are conformationally heterogenous in the unbound state. Human lymphoid enhancer-binding factor-1 (LEF-1) provides an example. LEF-1 is an architectural transcription factor [75, 76] that recognizes a specific nucleotide sequence through an HMGB domain. The LEF-1 HMBG domain introduces a pronounced bend in its cognate DNA sequence [77], facilitating binding of flanking activators, and assembly of a transcriptional complex, sometimes termed the enhanceosome [78]. In the absence of its cognate DNA sequence, the LEF-1 HMGB domain partially destructures. It appears that the α-helical secondary structure is largely retained, based on measurement of protein circular dichroism. However, helix I undergoes slow conformational fluctuations on the microsecond-to-millisecond timescale, such that the backbone NMR resonances are broadened beyond the detection limit [79]. Some HMGB domains found in the SOX family of transcription factors exhibit similar behavior (reviewed in [80]). For the murine SOX-4 HMGB domain, all the three helices are again formed in the absence of DNA, but the failure to observe long-range nuclear Overhauser effects in solution NMR analysis, suggests that the L-motif is flexible, with the position of Helix III varying with respect to the Helix I/II arm. In addition, the N-terminal region of the polypeptide is unstructured and does not pack against Helix III [81]. For the murine SOX-5 HMGB domain, the C-terminus of helix 3 is highly dynamic, but the tertiary structure basically persists in the absence of DNA [82].
While all of these sequence-specific HMGB domains gain conformational mobility in the absence of DNA, they appear to retain most of their helical secondary structure, and the disorder appears to be “segmental” in nature, affecting isolated helices or the relative positioning of helices. It has been speculated that this “floppiness” may be necessary to enable DNA bending [83] or might allow variation of the bending angle in a biological setting [80]. In this regard, it should be noted that some sequence-specific HMGB domains appear to be well structured in the absence of DNA (see [84]).
Transcription factor AF9
Human AF9 is a component of transcriptional elongation complexes and contains an N-terminal YEATS domain, which recognizes chromatin modifications [85, 86]. AF9 and related proteins are associated with the onset of leukemia when chromosomal translocations result in fusion of their coding regions to the MLL (mixed lineage leukemia) gene. At the C-terminus of AF9 there is a domain involved in the recruitment of multiple binding partners. This domain is homologous to the ET domain, a small protein–protein interaction module with a mixed α/β structure, used recurrently in transcriptional and chromatin regulation [87].
The structural basis for binding of the ET-like domain of AF9 to one of its partner proteins, AF4, was investigated by Leach et al. [88]. In the absence of AF4, AF9 is disordered and lacks a significant secondary structure, as evaluated by circular dichroism and NMR spectroscopy. However, in the presence of the relevant linear sequence motif from AF4, a structured ET-like domain results, in which the AF4 sequence forms the third, and exterior strand of the β-sheet (Fig. 2e). Hydrophobic residues from AF4 complete the hydrophobic core of the ET-like domain. As the AF4 peptide is also unstructured in the absence of its binding partner, both elements undergo a coupled folding and binding process in forming this ordered complex. AF4 binds AF9 with high affinity (equilibrium dissociation constant 0.2 nM), as do binding partners Dot1L and BCoR. NMR relaxation measurements show that the bound AF4 retains a significant mobility in the complex, which the authors suggest could lower the activation barrier for exchange of tightly bound partners, something that could be important to facilitate changes in transcription levels in response to varying cellular conditions.
The Brinker nuclear repressor
Brinker is a sequence-specific transcriptional factor that negatively regulates genes involved in Drosophila morphogenesis. When bound to DNA, the Brinker repressor DNA binding domain is comprised of four α-helices, the second and third of which form a classical helix–turn–helix motif which binds in the DNA major groove (Fig. 2f). However, in the absence of DNA, the domain appears to be almost completely unfolded at 25 °C, based on chemical shift dispersion, and the measurement of hetero-nuclear 1H–15N steady-state nuclear Overhauser effect enhancements [89]. Interestingly, a minimal construct encompassing just the DNA binding domain, could be induced to fold at low temperature (5–10 °C), whereas longer constructs, which retained the flanking disordered sequence, could not be stabilized in this fashion. The authors speculate that a particularly high density of positively charged amino acids at the DNA–protein interface leads to conditional disorder in the absence of the bound nucleic acid.
N-terminal regulatory domain of the glucocortocoid receptor
The glucocortocoid receptor (GR) is ubiquitously expressed by all vertebrates, and acts as an intracellular receptor for cortisol and other cholesterol-derived steroid hormones. These have wide-ranging physiological effects, and are central to metabolism and homeostasis. GR is resident in the cytoplasm, and undergoes translocation to the nucleus upon ligand binding, where it can function as either a transcriptional activator or repressor. The N-terminal trans-activation domain (NTD) of GR contains clusters of negatively charged aspartic and glutamic acid side chains, leading to the supposition that this region is disordered—a “negative noodle” as Paul Sigler memorably termed such regions [90]. Indeed, when expressed and purified in isolation, the NTD of GR appears largely unstructured [91, 92]. However, structure is clearly induced in the GR NTD upon interaction with biologically relevant binding partners (e.g., the TATA box binding protein [93]) and also upon stabilization with TMAO [92, 94]. In addition, the induction of structure with TMAO enhances the binding of regulatory molecules to the GR NTD [95]. Based on equilibrium thermodynamic arguments, Hilser and coworkers suggest that TMAO induces stable tertiary structure within the GR NTD [94], but the exact nature of this apparently folded state remains unknown.
Proteins stabilized by post-translational modification
The two-component signaling receiver domain from Erythrobacter litoralis LovR
Bacteria sense and respond to chemical and physical cues in their environment using diverse strategies. One mechanism to accomplish this involves the family of two-component signal transduction systems [96]. The eponymous two components of these systems are a membrane-bound histidine kinase and an intracellular response regulator protein. Typically, the histidine kinase contains an extracellular sensor domain, that will stimulate the kinase activity when bound to its ligand, resulting in phosphorylation of the response regulator and activation of an appropriate intracellular response [97]. Bacterial species may have several dozens or more distinct sensing capabilities encoded in these two-component systems [98].
The phosphorylation site on the response regulator is typically an aspartate residue located on a receiver (REC) domain, which has a flavodoxin-like fold—comprised of five parallel β-strands connected by α-helices (Fig. 3a). In most receiver domains, the phosphorylation site is adjacent to a magnesium-binding site created by the presence of additional acidic side chains. A prevalent model for signaling through the response regulators posits the existence of a conformational equilibrium between a lower energy, signaling-inactive conformation, and a higher energy, signaling-active conformation [99]. Phosphorylation of the receiver domain biases the conformational equilibrium, selectively stabilizing the active conformation, and stimulating the appropriate intracellular response for the lifetime of the phosphorylated state.
Fig. 3.

Conditionally disordered proteins stabilized by post-translational modification. a Two-component signaling receiver domain from E. litoralis LovR (PDB ID: 2MSW). b Structure of 4E-BP2 residues 18–62, uniformly phosphorylated at T37 and T46 (PDB ID: 2MX4). c First IgG-like repeat of the C. perfringens adhesion protein Cpe0147 (PDB ID: 4MKM), with the auto-catalytically generated ester bond in grey
An interesting variation on this mechanism was discovered for the response regulator protein LovR from the marine bacterium E. litoralis [100]. LovR exists in multiple states with different degrees of structure depending on both the binding of the magnesium ion ligand and the protein’s phosphorylation state. In the absence of the magnesium ion, the protein exists in an expanded and protease-sensitive state with limited secondary structure. However, upon the addition of magnesium, the majority of the protein, including the phosphorylation site, adopts a more structured conformation, with much persistent secondary structure and some persistent tertiary structure. Furthermore, upon treatment with the phosphomimetic BeF3 −, the remainder of the protein becomes ordered, adopting a compact and stable structure that resembles other well-characterized receiver domains. Through mutational analysis, the destabilization in the absence of the magnesium ion was attributed to electrostatic repulsion by acidic groups in the vicinity of the phosphorylation site.
The LovR receiver domain, therefore, appears to function mechanistically in a similar fashion to other well-characterized proteins in this class, but the regulation of its activity is accomplished in a novel way. Rather than utilizing a conformational equilibrium between two folded states, in LovR, the signaling-inactive conformation appears to be the partially folded, magnesium-bound state, and the fully folded and presumably signaling-active conformation is only populated, while the protein is phosphorylated.
Eukaryotic translation initiation factor binding protein 4E-BP2
Eukaryotic translation initiation factor eIF4E-binding proteins (4E-BPs) function as a repressors of translation initiation in eukaryotes [101]. eIF4E recognizes and binds the cap structure present at the 5′ end of cellular mRNA, facilitating ribosome attachment. The 4E-BPs bind to eIF4E, preventing assembly of the complete translation apparatus, and thus globally inhibit cap-dependent translation. 4E-BP function is regulated by phosphorylation, with only non-phosphorylated 4E-BP capable of binding eIF4E with high affinity.
In their non-phosphorylated state, 4E-BPs contain a significant complement of secondary structure, but are globally disordered [102]. They interact tightly with eIF4E via a linear YXXXXLΦ motif (where X is any amino acid, and Φ is a hydrophobic amino acid), with other regions of 4E-BP making subsidiary interactions [102]. The YXXXXLΦ motif binds as a helix to eIF4E, mimicking the structural interactions involved in the assembly of the translational apparatus [103, 104]. The mechanism by which phosphorylation relieves this repressive interaction was recently elucidated by a structural study of 4E-BP2, the major neural isoform of 4E-BP.
Phosphorylation was shown to induce the folding of 4E-BP2. The structure of 4E-BP2 residues 18–62, uniformly phosphorylated at T37 and T46, was determined by solution NMR spectroscopy [101], and is built around a four-stranded β-sheet, with a right-handed cross-over connection [105] between the two outer strands (Fig. 3b). Significantly, the YXXXXLΦ motif central to eIF4E binding is largely sequestered in the folded state. The induction of folding appears to be driven by the stabilization of two β-turns within the sheet which contain identical pTPGGT motifs. Although the phosphorylation of 4E-BP2 at positions outside this folded core causes additional repression of eIF4E binding, it is the transition to the folded state, and the concomitant shielding of the canonical YXXXXLΦ binding motif, that appears to be the central mechanism of repression.
Hence, in an intriguing inversion of the usual structure–function paradigm [106], it is the non-phosphorylated and partially structured form of 4E-BP2 that has repressive activity, whereas phosphorylation promotes folding, which deactivates the protein.
Cell-surface adhesion proteins of Gram-positive bacteria
Cpe0147 is an ~220 kDa adhesion protein from the pathogenic bacterium Clostridium perfringens comprising an N-terminal adhesin domain, 11 IgG-like repeats, and a C-terminal peptide motif enabling cell wall anchoring. What results is a long stalk-like molecule, with the functional adhesin domain held some distance from the bacterial cell surface. The IgG-like repeats are very similar, with a minimum pairwise identity of 85%. The structure of the first two repeats of Cpe0147 was determined by X-ray crystallography [107]. Surprisingly, the structure revealed the presence of auto-catalytically generated Thr–Gln ester bond cross-links within the IgG-like repeats, covalently linking the first and last β-strands of the domain at a central location (Fig. 3c). The positioning of the repetitively placed ester bonds appears highly significant, as they create a continuous chain of covalent bonding that extends through the IgG-like repeats, from the cell wall anchor to the adhesin, greatly increasing the tensile strength of the protein along its major axis. Chemically distinct isopeptide bond cross links had been previously identified in pilus components from Gram-positive bacteria [108]. Hence, side chain cross-linking appears to be a quite general strategy for enhancing the stability and mechanical strength of bacterial surface proteins.
For Cpe0147, if residues involved in the creation or catalysis of ester bond formation in the first IgG-like repeat are mutated, the domain unfolds, with both CD spectroscopy and temperature scanning fluorimetry suggesting that the mutant domains contain limited secondary or tertiary structure in the absence of the cross-link [107]. Employing more conservative mutations, which slow, but do not eliminate ester bond formation, subsequent NMR spectroscopic observations have made clear that folding and ester bond formation are tightly coupled (Paul Young, personal communication). Since ester bond formation is auto-catalyzed, it appears that the folded state is at least transiently accessible, prior to the formation of the bond.
It is expected that ester bond formation in Cpe0147 will stabilize the folded state by decreasing the configurational entropy of the unfolded state (and hence increasing its standard chemical potential, refer Fig. 1), analogous to the effects of disulfide bonding [109, 110]. However, it is presently unknown why the rather robust IgG-like domain is so destabilized in the absence of the ester bond cross-link. It is conceivable that this is physical in origin, and related to the burial of the acidic groups required to catalyze bond formation. Alternately, it could be biologically driven by the need to actively transport the nascent molecule across cell membrane. Regardless, it does provide an intriguing and possibly unique example of a protein, whose tertiary structure is regulated by an auto-generated post-translational modification.
Discussion and conclusion
We have highlighted a number of proteins that gain and lose tertiary structure under physiological or near-physiological conditions. Since the biological cue for the disorder–order transition is often the chemically specific binding of another molecule, these proteins provide some of the most extreme examples of coupled folding and binding. The domains involved are structurally diverse, as is the nature of the disordered state in which they natively exist. Where this disordered state retains significant secondary structure, it is usually poorly understood, because of the complexities inherent in the description of conformationally heterogenous proteins, and their investigation using ensemble-averaged experimental data [111–115]. While coupled folding and binding events involving linear sequence motifs are ubiquitous, order/disorder transitions involving the gain and loss of tertiary structure by globular domains appear to be less common. However, there are now enough examples to establish this behavior as part of the normal structure–function repertoire of proteins.
Functional advantage or accident of circumstance?
Since the biological function of conditionally disordered proteins is almost exclusively the binding of other molecules, some of the reasons advanced for the occurrence of conditional disorder mirror those advanced for the utility of coupled folding and binding interactions involving short linear sequence motifs, or longer MoRFs and disordered domains [116–118]. In particular, conditional protein disorder may allow for more nuanced modulation of binding rates and affinities, as suggested for the paramxyoviral RdRp foot domains, and for the ET-like domain of AF9. However, a conditionally disordered protein has more limited structural polymorphism than a MoRF, as its sequence specifies a unique tertiary structure in the appropriate environment. Hence, conditional disorder would not appear to be a likely mechanism for facilitating interactions with highly disparate partners, which is an observed property of MoRFs [119, 120]. For conditionally disordered proteins, any arguments about the necessity of disorder are frequently undercut by the occurrence of fully structured proteins fulfilling the same biological role. The question of whether modest increase in fitness is conferred by conditional disorder is very difficult to test, though potentially addressable through directed evolution.
Irrespective of functional advantage, we hypothesize that the occurrence of conditional disorder may often reflect fundamental physical restraints on the stability of small protein domains. Many of the examples we have cited involve small domains with very simple chain topologies. For example, the RdRP foot domains of rubulaviruses (Fig. 2a) at 50 amino acids in size are among the smallest autonomously folding protein domains known. It is to be expected that conformational fluctuations will become quite significant for the domains of this size [121] and are probably difficult to effectively damp. We have previously suggested that this is the likely reason that conservative sequence variation results in vary large changes in structural and energetic specificity for the foot domains [7]. A similar point could be made about WW domains (~40 amino acids, Fig. 2c), the HMGB domains of architectural transcription factors [~75 amino acids, Fig. 2d), and the Brinker nuclear repressor (~60 amino acids, Fig. 2f)].
In their comprehensive review of the effects of mutation on the internal packing and stability of folded proteins [122], Richards and Lim observed that the large majority of amino acid substitutions in the interior of naturally occurring folded proteins lower protein stability [122]. The key to the generation of a conditionally disordered protein from a stable progenitor is destabilizing mutations which decrease the Gibbs energy gap between the folded state and less structured states, without precluding the possibility of attaining the folded state. Richards and Lim further noted that “It is difficult to find an interior single site (or several site) mutation that completely obliterates the ability of a protein to adopt the basic original fold”. We are not questioning this general conclusion. However, there are likely certain domains, which through a combination of topology and size, are always marginally stable, and are, therefore, amenable to the generation of conditional disorder. For such domains, it is worth noting that many destabilizing mutations would probably generate constitutively disordered proteins that can never fold.
In vitro analysis has shown how readily conditional disorder can be introduced into a binding system. In a particularly striking example, the mutation of one of the conserved Trp residues to Phe in the WW domain of the human YES-associated protein caused almost complete unfolding of the domain. However, the mutant WW domain still binds its proline-rich target peptide, structuring as it does so, with only a threefold reduction in overall binding affinity [123]. This mutation was not observed to occur in any natural WW domain sequence, suggesting that it is probably not tolerated in vivo. The authors suggest that this might be due to the increased sensitivity of the mutant to proteolysis [123]. This makes the important point that the fitness of a protein in a cellular setting reflects not just its ability to perform its primary function, but also its ability to avoid the competing deleterious processes of aggregation and degradation [124]. Notwithstanding, when the primary function of a protein is molecular recognition, protein stability is less important than it is in other contexts, as the loss of structure does not impede binding.
Overall, we suggest that certain protein folds are probably readily destabilized by point mutation, in ways that still allow access to the folded state in the appropriate environment. If the selective pressure for retention of the folded state is relatively weak, as it may be for proteins engaged in molecular recognition, then conditional disorder would naturally emerge. If we accept this hypothesis, then conditional disorder should neither be a surprise, nor interpreted as diagnostic of some special functional need. It is simply part of the repertoire of the things proteins do, ultimately resulting from the weak non-covalent forces which stabilize their tertiary structure. As such, it is a phenomenon that must be accounted for in quantitative modeling of binding processes [16–21], and also one that can be used adaptively by the evolutionary process. The emergence of conditional disorder in the regulation of protein activity provides one such example.
Involvement in regulation of biological activity
Conditionally disordered proteins are characterized by a small or negligible difference in the standard chemical potentials of the folded and unfolded states (Fig. 1). As a consequence, environmental effects which even modestly alter the Gibbs energy of states can substantially alter their relative populations,. In several of the systems, we have reviewed (4E-BP2/4E binding and E. litoralis LovR); protein activity is regulated by redistributing the populations of folded and unfolded states. In both the cases, the cue for the structural transition is phosphorylation. These phenomena can be viewed as an extreme form of allosteric regulation [125–128], with the transitions between structured and unstructured states being driven by a quite local chemical modification.
Regulation of E. litoralis LovR proceeds according to the general framework established for other bacterial response regulators [97], which have structurally similar but not identical active and inactive states, both of which are folded. The difference is that in LovR, the unphosphorylated and inactive folded state has been selectively destabilized, to the level of other unfolded states, presumably through mutation of a stable progenitor.
In contrast, the 4E-BP2/4E binding system provides a remarkable example of our expectations being completely inverted, with activity arising from the unfolded state being regulated by the presence of the folded state. In this system, the unfolded and non-phosphorylated state of 4E-BP2 allows the YXXXXLΦ motif to form productive interactions with elF4E and thereby enable translational initiation. The folded and phosphorylated state of 4E-BP2 partly sequesters this linear sequence motif to prevent initiation. The phosphorylated 4E-BP2 protein is thereby engaging in what might be termed a coupled unfolding and binding process, where association with the interaction partner Elf4E will depopulate the folded state of 4E-BP2.
The kinetic aspects of these systems probably deserve further exploration. While one could construct simple equilibrium models of the regulatory process, involving folded and unfolded states, the structural transitions involved are dramatic, and the timescales of these processes are currently unknown. In addition, these transitions are driven in a cellular setting by protein kinases and phosphatases, creating a situation of considerable complexity.
General conclusion
The widespread occurrence of protein disorder has shown that the protein folding problem is more complex than originally conceived. Functional proteins exhibit varying degrees of structure formation, and protein conformation is often strongly linked to environmental conditions. This review has highlighted proteins that appear to operate according to the traditional paradigm, by adopting a defined tertiary structure. They are not readily differentiated from stable proteins on the basis of sequence. However, their conformational state is strongly linked to the environment, or is dramatically perturbed by post-translational modification of the polypeptide chain. Under the standard laboratory conditions, these proteins will appear disordered, but a structurally specific conformational state will manifest upon environmental perturbation or appropriate chemical modification. In the biological setting, the most common cue for folding will be the binding of a chemically specific partner, while in vitro, folding can generally be promoted with stabilizing chemical osmolytes.
While it remains unclear if conditional disorder offers any pronounced functional advantages for binding processes, available evidence suggests that it arises reasonably readily, particularly for small domains, as a result of the marginal stability of the folded state. Conditionally disordered proteins demonstrate that the modulation of protein structure by the biological environment may go far beyond switching between discrete folded states, and can involve transitions of folded proteins to partially or largely disordered states. For a conditionally disordered protein, activity can potentially be regulated by environmental factors or chemical modifications that act on both folded and unfolded states, since they are barely discriminated in the Gibbs energy landscape. Overall, conditionally disordered proteins serve as a useful reminder that sequence and environment together determine structure, as Anfinsen originally hypothesized. That may not make for a particularly memorable aphorism, but it has the advantage of correctness.
References
- 1.Anfinsen CB, Scheraga HA. Experimental and theoretical aspects of protein folding. Adv Protein Chem. 1975;29:205–300. doi: 10.1016/S0065-3233(08)60413-1. [DOI] [PubMed] [Google Scholar]
- 2.Privalov PL. Stability of proteins: small globular proteins. Adv Protein Chem. 1979;33:167–241. doi: 10.1016/S0065-3233(08)60460-X. [DOI] [PubMed] [Google Scholar]
- 3.Murzin AG. Biochemistry. Metamorphic proteins. Science. 2008;320:1725–1726. doi: 10.1126/science.1158868. [DOI] [PubMed] [Google Scholar]
- 4.Bryan PN, Orban J. Proteins that switch folds. Curr Opin Struct Biol. 2010;20:482–488. doi: 10.1016/j.sbi.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodchild SC, Curmi PMG, Brown LJ. Structural gymnastics of multifunctional metamorphic proteins. Biophys Rev. 2011;3:143–153. doi: 10.1007/s12551-011-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Naim A. The protein folding problem and its solutions. Singapore: World Scientific; 2013. [Google Scholar]
- 7.Yegambaram K, Bulloch EMM, Kingston RL. Protein domain definition should allow for conditional disorder. Protein Sci. 2013;22:1502–1518. doi: 10.1002/pro.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romero P, Obradovic Z, Li X, et al. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Das RK, Pappu RV. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc Natl Acad Sci USA. 2013;110:13392–13397. doi: 10.1073/pnas.1304749110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theillet FX, Kalmar L, Tompa P, Han KH. The alphabet of intrinsic disorder: I. Act like a pro: on the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord Proteins. 2013;1:e24360. doi: 10.4161/idp.24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uversky VN. The alphabet of intrinsic disorder: II. Various roles of glutamic acid in ordered and intrinsically disordered proteins. Intrinsically Disord Proteins. 2013;1:e24684. doi: 10.4161/idp.24684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Roey K, Uyar B, Weatheritt RJ, et al. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem Rev. 2014;114:6733–6778. doi: 10.1021/cr400585q. [DOI] [PubMed] [Google Scholar]
- 13.Oldfield CJ, Cheng Y, Cortese MS, et al. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 14.Mohan A, Oldfield CJ, Radivojac P, et al. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 15.Tompa P, Fuxreiter M, Oldfield CJ, et al. Close encounters of the third kind: disordered domains and the interactions of proteins. BioEssays. 2009;31:328–335. doi: 10.1002/bies.200800151. [DOI] [PubMed] [Google Scholar]
- 16.Wyman J, Gill SJ. Binding and linkage. Mill Valley: University Science Books; 1990. [Google Scholar]
- 17.Freire E. Statistical thermodynamic linkage between conformational and binding equilibria. Adv Protein Chem. 1998;51:255–279. doi: 10.1016/S0065-3233(08)60654-3. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Naim A. Cooperativity and regulation in biochemical processes. USA: Springer; 2001. [Google Scholar]
- 19.Shriver JW, Edmondson SP. Ligand-binding interactions and stability. Methods Mol Biol. 2009;490:135–164. doi: 10.1007/978-1-59745-367-7_6. [DOI] [PubMed] [Google Scholar]
- 20.Hilser VJ, Wrabl JO, Motlagh HN. Structural and energetic basis of allostery. Annu Rev Biophys. 2012;41:585–609. doi: 10.1146/annurev-biophys-050511-102319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermans J, Lentz BR. Equilibria and kinetics of biological macromolecules. New York: Wiley; 2013. [Google Scholar]
- 22.Baskakov I, Bolen DW. Forcing thermodynamically unfolded proteins to fold. J Biol Chem. 1998;273:4831–4834. doi: 10.1074/jbc.273.9.4831. [DOI] [PubMed] [Google Scholar]
- 23.Yancey PH, Clark ME, Hand SC, et al. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 24.Sarkar M, Pielak GJ. An osmolyte mitigates the destabilizing effect of protein crowding. Protein Sci. 2014;23:1161–1164. doi: 10.1002/pro.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holthauzen LMF, Bolen DW. Mixed osmolytes: the degree to which one osmolyte affects the protein stabilizing ability of another. Protein Sci. 2007;16:293–298. doi: 10.1110/ps.062610407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Auton M, Rösgen J, Sinev M, et al. Osmolyte effects on protein stability and solubility: a balancing act between backbone and side-chains. Biophys Chem. 2011;159:90–99. doi: 10.1016/j.bpc.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreon ACM, Moosa MM, Gambin Y, Deniz AA. Counteracting chemical chaperone effects on the single-molecule α-synuclein structural landscape. Proc Natl Acad Sci USA. 2012;109:17826–17831. doi: 10.1073/pnas.1201802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Street TO, Bolen DW, Rose GD. A molecular mechanism for osmolyte-induced protein stability. Proc Natl Acad Sci USA. 2006;103:13997–14002. doi: 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu CY, Pettitt BM, Roesgen J. Osmolyte solutions and protein folding. F1000 Biol Rep. 2009;1:41. doi: 10.3410/B1-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canchi DR, Garcia AE. Cosolvent effects on protein stability. Annu Rev Phys Chem. 2013;64:273–293. doi: 10.1146/annurev-physchem-040412-110156. [DOI] [PubMed] [Google Scholar]
- 31.Candotti M, Esteban-Martín S, Salvatella X, Orozco M. Toward an atomistic description of the urea-denatured state of proteins. Proc Natl Acad Sci USA. 2013;110:5933–5938. doi: 10.1073/pnas.1216589110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma J, Pazos IM, Gai F. Microscopic insights into the protein-stabilizing effect of trimethylamine N-oxide (TMAO) Proc Natl Acad Sci USA. 2014;111:8476–8481. doi: 10.1073/pnas.1403224111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bah A, Forman-Kay JD. Modulation of intrinsically disordered protein function by post-translational modifications. J Biol Chem. 2016;291:6696–6705. doi: 10.1074/jbc.R115.695056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alayyoubi M, Leser GP, Kors CA, Lamb RA. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein–RNA complex. Proc Natl Acad Sci USA. 2015;112:E1792–E1799. doi: 10.1073/pnas.1503941112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruigrok RWH, Crépin T, Kolakofsky D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol. 2011;14:504–510. doi: 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Noton SL, Fearns R. Initiation and regulation of paramyxovirus transcription and replication. Virology. 2015;479–480:545–554. doi: 10.1016/j.virol.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Longhi S, Receveur-Bréchot V, Karlin D, et al. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J Biol Chem. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- 38.Kingston RL, Hamel DJ, Gay LS, et al. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc Natl Acad Sci USA. 2004;101:8301–8306. doi: 10.1073/pnas.0402690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Chu X, Longhi S, et al. Multiscaled exploration of coupled folding and binding of an intrinsically disordered molecular recognition element in measles virus nucleoprotein. Proc Natl Acad Sci USA. 2013;110:E3743–E3752. doi: 10.1073/pnas.1308381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houben K, Marion D, Tarbouriech N, et al. Interaction of the C-terminal domains of sendai virus N and P proteins: comparison of polymerase-nucleocapsid interactions within the paramyxovirus family. J Virol. 2007;81:6807–6816. doi: 10.1128/JVI.00338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider R, Maurin D, Communie G, et al. Visualizing the molecular recognition trajectory of an intrinsically disordered protein using multinuclear relaxation dispersion NMR. J Am Chem Soc. 2015;137:1220–1229. doi: 10.1021/ja511066q. [DOI] [PubMed] [Google Scholar]
- 42.Erales J, Beltrandi M, Roche J, et al. Insights into the Hendra virus NTAIL-XD complex: evidence for a parallel organization of the helical MoRE at the XD surface stabilized by a combination of hydrophobic and polar interactions. Biochim Biophys Acta. 2015;1854:1038–1053. doi: 10.1016/j.bbapap.2015.04.031. [DOI] [PubMed] [Google Scholar]
- 43.Communie G, Habchi J, Yabukarski F, et al. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus. PLoS Pathog. 2013;9:e1003631. doi: 10.1371/journal.ppat.1003631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Habchi J, Blangy S, Mamelli L, et al. Characterization of the interactions between the nucleoprotein and the phosphoprotein of Henipavirus. J Biol Chem. 2011;286:13583–13602. doi: 10.1074/jbc.M111.219857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kingston RL, Gay LS, Baase WS, Matthews BW. Structure of the nucleocapsid-binding domain from the mumps virus polymerase; an example of protein folding induced by crystallization. J Mol Biol. 2008;379:719–731. doi: 10.1016/j.jmb.2007.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kingston RL, Baase WA, Gay LS. Characterization of nucleocapsid binding by the measles virus and mumps virus phosphoproteins. J Virol. 2004;78:10097. doi: 10.1128/JVI.78.16.8630-8640.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivanov I, Yabukarski F, Ruigrok RWH, Jamin M. Structural insights into the rhabdovirus transcription/replication complex. Virus Res. 2011 doi: 10.1016/j.virusres.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 48.Mavrakis M, Mccarthy AA, Roche S, et al. Structure and function of the C-terminal domain of the polymerase cofactor of rabies virus. J Mol Biol. 2004;343:819–831. doi: 10.1016/j.jmb.2004.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ribeiro EA, Leyrat C, Gerard FCA, et al. Binding of rabies virus polymerase cofactor to recombinant circular nucleoprotein-RNA complexes. J Mol Biol. 2009;394:558–575. doi: 10.1016/j.jmb.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 50.Kazantsev AV, Pace NR. Bacterial RNase P: a new view of an ancient enzyme. Nat Rev Microbiol. 2006;4:729–740. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- 51.Kurz JC, Fierke CA. Ribonuclease P: a ribonucleoprotein enzyme. Curr Opin Chem Biol. 2000;4:553–558. doi: 10.1016/S1367-5931(00)00131-9. [DOI] [PubMed] [Google Scholar]
- 52.Crary SM, Niranjanakumari S, Fierke CA. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry. 1998;37:9409–9416. doi: 10.1021/bi980613c. [DOI] [PubMed] [Google Scholar]
- 53.Buck AH, Dalby AB, Poole AW, et al. Protein activation of a ribozyme: the role of bacterial RNase P protein. EMBO J. 2005;24:3360–3368. doi: 10.1038/sj.emboj.7600805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kazantsev AV, Krivenko AA, Harrington DJ, et al. High-resolution structure of RNase P protein from Thermotoga maritima. Proc Natl Acad Sci USA. 2003;100:7497–7502. doi: 10.1073/pnas.0932597100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Henkels CH, Kurz JC, Fierke CA, Oas TG. Linked folding and anion binding of the Bacillus subtilis ribonuclease P protein. Biochemistry. 2001;40:2777–2789. doi: 10.1021/bi002078y. [DOI] [PubMed] [Google Scholar]
- 56.Henkels CH, Oas TG. Thermodynamic characterization of the osmolyte- and ligand-folded states of Bacillus subtilis ribonuclease P protein. Biochemistry. 2005;44:13014–13026. doi: 10.1021/bi0504613. [DOI] [PubMed] [Google Scholar]
- 57.Henkels CH, Oas TG. Ligation-state hydrogen exchange: coupled binding and folding equilibria in ribonuclease P protein. J Am Chem Soc. 2006;128:7772–7781. doi: 10.1021/ja057279+. [DOI] [PubMed] [Google Scholar]
- 58.Henkels CH, Chang Y-C, Chamberlin SI, Oas TG. Dynamics of backbone conformational heterogeneity in Bacillus subtilis ribonuclease P protein. Biochemistry. 2007;46:15062–15075. doi: 10.1021/bi701425n. [DOI] [PubMed] [Google Scholar]
- 59.Chang Y-C, Oas TG. Osmolyte-induced folding of an intrinsically disordered protein: folding mechanism in the absence of ligand. Biochemistry. 2010;49:5086–5096. doi: 10.1021/bi100222h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang Y-C, Franch WR, Oas TG. Probing the folding intermediate of Bacillus subtilis RNase P protein by nuclear magnetic resonance. Biochemistry. 2010;49:9428–9437. doi: 10.1021/bi100287y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mosley PL, Daniels KG, Oas TG. Electrostatic energetics of Bacillus subtilis ribonuclease P protein determined by nuclear magnetic resonance-based histidine pKa measurements. Biochemistry. 2015;54:5379–5388. doi: 10.1021/acs.biochem.5b00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- 63.Macias MJ, Wiesner S, Sudol M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2001;513:30–37. doi: 10.1016/S0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- 64.Yang B, Kumar S. Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ. 2010;17:68–77. doi: 10.1038/cdd.2009.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Panwalkar V, Neudecker P, Schmitz M, et al. The Nedd4-1 WW domain recognizes the PY motif peptide through coupled folding and binding equilibria. Biochemistry. 2016;55:659–674. doi: 10.1021/acs.biochem.5b01028. [DOI] [PubMed] [Google Scholar]
- 66.Bobby R, Medini K, Neudecker P, et al. Structure and dynamics of human Nedd4-1 WW3 in complex with the αENaC PY motif. Biochim Biophys Acta (BBA) Proteins Proteomics. 2013;1834:1632–1641. doi: 10.1016/j.bbapap.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 67.Xiang B, Weiler S, Nirenberg M, Ferretti JA. Structural basis of an embryonically lethal single Ala → Thr mutation in the vnd/NK-2 homeodomain. Proc Natl Acad Sci USA. 1998;95:7412–7416. doi: 10.1073/pnas.95.13.7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Štros M, Launholt D, Grasser KD. The HMG-box: a versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell Mol Life Sci. 2007;64:2590–2606. doi: 10.1007/s00018-007-7162-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baxevanis AD, Landsman D. The HMG-1 box protein family: classification and functional relationships. Nucleic Acids Res. 1995;23:1604–1613. doi: 10.1093/nar/23.9.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grosschedl R, Giese K, Pagel J. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 71.Tan D, Marzluff WF, Dominski Z, Tong L. Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3′hExo ternary complex. Science. 2013;339:318–321. doi: 10.1126/science.1228705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thapar R. Structure-specific nucleic acid recognition by L-motifs and their diverse roles in expression and regulation of the genome. Biochim Biophys Acta. 2015;1849:677–687. doi: 10.1016/j.bbagrm.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rohs R, Jin X, West SM, et al. Origins of specificity in protein-DNA recognition. Annu Rev Biochem. 2010;79:233–269. doi: 10.1146/annurev-biochem-060408-091030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Broadhurst RW, Hardman CH, Thomas JO, Laue ED. Backbone dynamics of the A-domain of HMG1 as studied by 15 N NMR spectroscopy. Biochemistry. 1995;34:16608–16617. doi: 10.1021/bi00051a008. [DOI] [PubMed] [Google Scholar]
- 75.Wolffe AP. Architectural transcription factors. Science. 1994;264:1100–1101. doi: 10.1126/science.8178167. [DOI] [PubMed] [Google Scholar]
- 76.Werner MH, Burley SK. Architectural transcription factors: proteins that remodel DNA. Cell. 1997;88:733–736. doi: 10.1016/S0092-8674(00)81917-0. [DOI] [PubMed] [Google Scholar]
- 77.Love JJ, Li X, Case DA, et al. Structural basis for DNA bending by the architectural transcription factor LEF-1. Nature. 1995;376:791–795. doi: 10.1038/376791a0. [DOI] [PubMed] [Google Scholar]
- 78.Carey M. The enhanceosome and transcriptional synergy. Cell. 1998;92:5–8. doi: 10.1016/S0092-8674(00)80893-4. [DOI] [PubMed] [Google Scholar]
- 79.Love JJ, Li X, Chung J, et al. The LEF-1 high-mobility group domain undergoes a disorder-to-order transition upon formation of a complex with cognate DNA. Biochemistry. 2004;43:8725–8734. doi: 10.1021/bi049591m. [DOI] [PubMed] [Google Scholar]
- 80.Weiss MA. Floppy SOX: mutual induced fit in hmg (high-mobility group) box-DNA recognition. Mol Endocrinol. 2001;15:353–362. doi: 10.1210/mend.15.3.0617. [DOI] [PubMed] [Google Scholar]
- 81.van Houte LP, Chuprina VP, van der Wetering M, et al. Solution structure of the sequence-specific HMG box of the lymphocyte transcriptional activator Sox-4. J Biol Chem. 1995;270:30516–30524. doi: 10.1074/jbc.270.51.30516. [DOI] [PubMed] [Google Scholar]
- 82.Cary PD, Read CM, Davis Ben, et al. Solution structure and backbone dynamics of the DNA-binding domain of mouse Sox-5. Protein Sci. 2001;10:83–98. doi: 10.1110/ps.32801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dyson HJ. Roles of intrinsic disorder in protein–nucleic acid interactions. Mol BioSyst. 2012;8:97–104. doi: 10.1039/C1MB05258F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gao N, Jiang W, Gao H, et al. Structural basis of human transcription factor Sry-related box 17 binding to DNA. Protein Pept Lett. 2013;20:481–488. [PubMed] [Google Scholar]
- 85.Schulze JM, Wang AY, Kobor MS. YEATS domain proteins: a diverse family with many links to chromatin modification and transcription. Biochem Cell Biol. 2009;87:65–75. doi: 10.1139/O08-111. [DOI] [PubMed] [Google Scholar]
- 86.Li Y, Wen H, Xi Y, et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell. 2014;159:558–571. doi: 10.1016/j.cell.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Faure G, Callebaut I. Identification of hidden relationships from the coupling of hydrophobic cluster analysis and domain architecture information. Bioinformatics. 2013;29:1726–1733. doi: 10.1093/bioinformatics/btt271. [DOI] [PubMed] [Google Scholar]
- 88.Leach BI, Kuntimaddi A, Schmidt CR, et al. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 2013;21:176–183. doi: 10.1016/j.str.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cordier F, Hartmann B, Rogowski M, et al. DNA recognition by the brinker repressor—an extreme case of coupling between binding and folding. J Mol Biol. 2006;361:659–672. doi: 10.1016/j.jmb.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 90.Sigler PB. Acid blobs and negative noodles. Nature. 1988;333:210–212. doi: 10.1038/333210a0. [DOI] [PubMed] [Google Scholar]
- 91.Dahlman-Wright K, Baumann H, McEwan IJ, et al. Structural characterization of a minimal functional transactivation domain from the human glucocorticoid receptor. Proc Natl Acad Sci USA. 1995;92:1699–1703. doi: 10.1073/pnas.92.5.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baskakov IV, Kumar R, Srinivasan G, et al. Trimethylamine N-oxide-induced cooperative folding of an intrinsically unfolded transcription-activating fragment of human glucocorticoid receptor. J Biol Chem. 1999;274:10693–10696. doi: 10.1074/jbc.274.16.10693. [DOI] [PubMed] [Google Scholar]
- 93.Kumar R, Volk DE, Li J, et al. TATA box binding protein induces structure in the recombinant glucocorticoid receptor AF1 domain. Proc Natl Acad Sci USA. 2004;101:16425–16430. doi: 10.1073/pnas.0407160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J, Motlagh HN, Chakuroff C, et al. Thermodynamic dissection of the intrinsically disordered N-terminal domain of human glucocorticoid receptor. J Biol Chem. 2012;287:26777–26787. doi: 10.1074/jbc.M112.355651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kumar R, Lee JC, Bolen DW, Thompson EB. The conformation of the glucocorticoid receptor af1/tau1 domain induced by osmolyte binds co-regulatory proteins. J Biol Chem. 2001;276:18146–18152. doi: 10.1074/jbc.M100825200. [DOI] [PubMed] [Google Scholar]
- 96.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 97.Parkinson JS, Kofoid EC. Communication modules in bacterial signaling proteins. Annu Rev Genet. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- 98.Laub MT, Goulian M. Specificity in two-component signal transduction pathways. Annu Rev Genet. 2007;41:121–145. doi: 10.1146/annurev.genet.41.042007.170548. [DOI] [PubMed] [Google Scholar]
- 99.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 100.Ocasio VJ, Corrêa F, Gardner KH. Ligand-induced folding of a two-component signaling receiver domain. Biochemistry. 2015;54:1353–1363. doi: 10.1021/bi501143b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bah A, Vernon RM, Siddiqui Z, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2014;519:106–109. doi: 10.1038/nature13999. [DOI] [PubMed] [Google Scholar]
- 102.Lukhele S, Bah A, Lin H, et al. Interaction of the eukaryotic initiation factor 4E with 4E-BP2 at a dynamic bipartite interface. Structure. 2013;21:2186–2196. doi: 10.1016/j.str.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 103.Marcotrigiano J, Gingras A-C, Sonenberg N, Burley SK. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol Cell. 1999;3:707–716. doi: 10.1016/S1097-2765(01)80003-4. [DOI] [PubMed] [Google Scholar]
- 104.Fukuyo A, In Y, Ishida T, Tomoo K. Structural scaffold for eIF4E binding selectivity of 4E-BP isoforms: crystal structure of eIF4E binding region of 4E-BP2 and its comparison with that of 4E-BP1. J Pept Sci. 2011;17:650–657. doi: 10.1002/psc.1384. [DOI] [PubMed] [Google Scholar]
- 105.Richardson JS. Handedness of crossover connections in beta sheets. Proc Natl Acad Sci USA. 1976;73:2619–2623. doi: 10.1073/pnas.73.8.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bouvignies G, Blackledge M. Structural biology turned on its head. ChemBioChem. 2015;16:1033–1034. doi: 10.1002/cbic.201500101. [DOI] [PubMed] [Google Scholar]
- 107.Kwon H, Squire CJ, Young PG, Baker EN. Autocatalytically generated Thr–Gln ester bond cross-links stabilize the repetitive Ig-domain shaft of a bacterial cell surface adhesin. Proc Natl Acad Sci USA. 2014;111:1367–1372. doi: 10.1073/pnas.1316855111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kang HJ, Middleditch M, Proft T, Baker NA. Isopeptide bonds in bacterial pili and their characterization by X-ray crystallography and mass spectrometry. Biopolymers. 2009;91:1126–1134. doi: 10.1002/bip.21170. [DOI] [PubMed] [Google Scholar]
- 109.Fass D. Disulfide bonding in protein biophysics. Annu Rev Biophys. 2012;41:63–79. doi: 10.1146/annurev-biophys-050511-102321. [DOI] [PubMed] [Google Scholar]
- 110.Flory PJ. Theory of elastic mechanisms in fibrous proteins. J Am Chem Soc. 1956 [Google Scholar]
- 111.Daughdrill GW, Kashtanov S, Stancik A, et al. Understanding the structural ensembles of a highly extended disordered protein. Mol BioSyst. 2012;8:308–319. doi: 10.1039/C1MB05243H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ozenne V, Schneider R, Yao M, et al. Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. J Am Chem Soc. 2012;134:15138–15148. doi: 10.1021/ja306905s. [DOI] [PubMed] [Google Scholar]
- 113.Fenwick RB, Esteban-Martín S, Salvatella X. Understanding biomolecular motion, recognition, and allostery by use of conformational ensembles. Eur Biophys J. 2011;40:1339–1355. doi: 10.1007/s00249-011-0754-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jensen MR, Zweckstetter M, Huang J-R, Blackledge M. Exploring free-energy landscapes of intrinsically disordered proteins at atomic resolution using NMR spectroscopy. Chem Rev. 2014;114:6632–6660. doi: 10.1021/cr400688u. [DOI] [PubMed] [Google Scholar]
- 115.Gibbs EB, Showalter SA. Quantitative biophysical characterization of intrinsically disordered proteins. Biochemistry. 2015;54:1314–1326. doi: 10.1021/bi501460a. [DOI] [PubMed] [Google Scholar]
- 116.Gsponer J, Babu MM. The rules of disorder or why disorder rules. Prog Biophys Mol Biol. 2009;99:94–103. doi: 10.1016/j.pbiomolbio.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 117.Dyson HJ. Expanding the proteome: disordered and alternatively folded proteins. Q Rev Biophys. 2011;44:467–518. doi: 10.1017/S0033583511000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Forman-Kay JD, Mittag T. From sequence and forces to structure, function, and evolution of intrinsically disordered proteins. Structure. 2013;21:1492–1499. doi: 10.1016/j.str.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cumberworth A, Lamour G, Babu MM, Gsponer J. Promiscuity as a functional trait: intrinsically disordered regions as central players of interactomes. Biochem J. 2013;454:361–369. doi: 10.1042/BJ20130545. [DOI] [PubMed] [Google Scholar]
- 120.Oldfield CJ, Meng J, Yang JY, et al. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cooper A. Protein fluctuations and the thermodynamic uncertainty principle. Prog Biophys Mol Biol. 1984;44:181–214. doi: 10.1016/0079-6107(84)90008-7. [DOI] [PubMed] [Google Scholar]
- 122.Richards FM, Lim WA. An analysis of packing in the protein folding problem. Q Rev Biophys. 1993;26:423–498. doi: 10.1017/S0033583500002845. [DOI] [PubMed] [Google Scholar]
- 123.Koepf EK, Petrassi HM, Ratnaswamy G, et al. Characterization of the structure and function of W → F WW domain variants: identification of a natively unfolded protein that folds upon ligand binding. Biochemistry. 1999;38:14338–14351. doi: 10.1021/bi991105l. [DOI] [PubMed] [Google Scholar]
- 124.Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr Opin Struct Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 125.Guarnera E, Berezovsky IN. Allosteric sites: remote control in regulation of protein activity. Curr Opin Struct Biol. 2016;37:1–8. doi: 10.1016/j.sbi.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 126.Nussinov R, Tsai C-J. Allostery without a conformational change? Revisiting the paradigm. Curr Opin Struct Biol. 2015;30:17–24. doi: 10.1016/j.sbi.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 127.Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007 doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu Y, Bolen DW. The peptide backbone plays a dominant role in protein stabilization by naturally occurring osmolytes. Biochemistry. 1995;34:12884–12891. doi: 10.1021/bi00039a051. [DOI] [PubMed] [Google Scholar]
- 130.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 131.Bolen DW. Protein stabilization by naturally occurring osmolytes. Meth Mol Biol. 2001;168:17–36. doi: 10.1385/1-59259-193-0:017. [DOI] [PubMed] [Google Scholar]