

Abstract
Angiogenesis denotes the formation of new blood vessels from pre-existing vasculature. Progression of diseases such as cancer and several ophthalmological disorders may be promoted by excess angiogenesis. Novel therapeutics to inhibit angiogenesis and diagnostic tools for monitoring angiogenesis during therapy, hold great potential for improving treatment of such diseases. We have previously generated so-called biparatopic Affibody constructs with high affinity for the vascular endothelial growth factor receptor-2 (VEGFR2), which recognize two non-overlapping epitopes in the ligand-binding site on the receptor. Affibody molecules have previously been demonstrated suitable for imaging purposes. Their small size also makes them attractive for applications where an alternative route of administration is beneficial, such as topical delivery using eye drops. In this study, we show that decreasing linker length between the two Affibody domains resulted in even slower dissociation from the receptor. The new variants of the biparatopic Affibody bound to VEGFR2-expressing cells, blocked VEGFA binding, and inhibited VEGFA-induced signaling of VEGFR2 over expressing cells. Moreover, the biparatopic Affibody inhibited sprout formation of endothelial cells in an in vitro angiogenesis assay with similar potency as the bivalent monoclonal antibody ramucirumab. This study demonstrates that the biparatopic Affibody constructs show promise for future therapeutic as well as in vivo imaging applications.
Keywords: Affibody molecules, Angiogenesis, Biparatopic affinity protein, Bispecific, VEGF, VEGFR2
Introduction
Angiogenesis is a critical step in many diseases including cancer and ophthalmic diseases, such as neovascular age-related macular degeneration and diabetic retinopathy [1]. One of the major regulators of angiogenesis is the vascular endothelial growth factor receptor 2 (VEGFR2). The receptor is overexpressed on endothelial cells during angiogenesis, and its signaling leads to proliferation, survival, migration, vascular permeability and invasion into surrounding tissue [2]. Signaling by VEGFR2 is initiated by binding of ligands belonging to the vascular endothelial growth factor (VEGF) family. Several structurally and functionally related VEGF polypeptides exist in humans, of which the main regulator of VEGFR2 signaling is VEGFA, although VEGFC and VEGFD are also able to activate this receptor [3]. VEGFA is a homodimeric protein, with two VEGFR2 binding sites [4–6]. Ligand binding induces receptor dimerization and kinase activation, which in turn leads to autophosphorylation of several tyrosines in the intracellular kinase domain of the receptor. Thereby various signaling pathways will be induced, including the mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase-1/2 (ERK1/2) cascade [2, 7].
Inhibition of VEGF–VEGFR2 signaling is an attractive therapeutic strategy for the treatment of conditions such as vascular eye diseases and cancer [8]. Several therapeutics targeting VEGFA are available, such as monoclonal antibodies and alternative scaffold proteins [9, 10], as well as engineered decoy receptors, which sequester the ligand and thereby prevent it from activating the membrane-bound receptor [11]. However, directly targeting the receptor instead of the ligand has the potential advantage of preventing activation by other ligands (e.g., VEGFC and VEGFD). Various therapeutic candidates targeting VEGFR2 have been developed, including small molecule antagonists [12, 13], tyrosine kinase inhibitors [14, 15], monoclonal antibodies including the FDA-approved ramucirumab [16, 17], an adnectin [18], a nanobody-toxin conjugate [19] and antagonistic VEGF variants [20]. Alternative scaffolds or antibody fragments might be advantageous for this application. Fc is not necessarily required for blocking of protein–protein interactions and effector functions such as ADCC and complement activation directed at the endothelial cells might not be desirable. The improved circulation time that is generally provided by Fc can be achieved by other means, for example through association to serum albumin. Although anti-angiogenic therapy has demonstrated positive results in the clinic, and several therapeutic agents have been approved for treatment of various cancers [2, 21], lack of response and development of drug resistance in some patients have been observed [22]. Consequently, biomarkers and diagnostic tools are needed for early stratification of responders versus non-responders and monitoring of the therapeutic progress [23]. In vivo imaging of VEGFR2 could potentially serve this purpose.
Tracers for in vivo imaging should ideally have short circulation time in the blood, high target-affinity and high stability [24, 25]. A type of alternative scaffold protein that has demonstrated excellent properties for imaging purposes is the Affibody scaffold [26]. Affibody molecules have a molecular weight around 6.5 kDa, and a three-helix bundle fold with very fast and reversible folding kinetics [27, 28]. These properties, along with a high solubility and high expression level in bacteria [26] facilitates engineering of multimeric proteins such as bispecific or bivalent constructs [29], which can be valuable for design of therapeutic constructs. The small size, high stability, and high solubility could potentially also be advantageous for delivery using alternative administration routes, such as local application using eye drops.
In a recently published study [30], we selected Affibody molecules binding to human VEGFR2 using a combination of phage display and an in-house developed staphylococcal display method [31–33]. The two most promising candidates demonstrated cross-reactivity to murine VEGFR2, which is a prerequisite to allow pre-clinical studies in mice. Moreover, experiments showed no cross-reactivity to VEGFR1 or VEGFR3, and both Affibody molecules blocked VEGFA from binding to VEGFR2. A surface plasmon resonance (SPR) based competition assay demonstrated that the two Affibody molecules could bind simultaneously to VEGFR2. The results hence indicated that the Affibody molecules targeted two distinct sites that both were located within the VEGF-binding epitope on the receptor. This interesting finding encouraged us to pursue a new strategy for development of a VEGF-blocking agent with potentially even higher affinity. Although the VEGF epitope is relatively large, the results suggested that the two domains interacted at sites that were in close proximity. Our hypothesis was that fusing the domains into one construct would allow for doubling of the binding surface area and achieve a considerable increase in affinity. Formatting the construct as a biparatopic dimer resulted in a dramatic decrease in off-rate compared to the monomers [30]. In addition, the construct was designed to contain an engineered albumin-binding domain ABD [34], intended as a detection- and purification tag as well as a potential means for half-life extension in vivo through interaction with human serum albumin (HSA) [35–37]. The option to express the biparatopic construct with or without ABD would result in adjustable pharmacokinetic properties, allowing for applications requiring either a long circulation time, such as therapy, or fast clearance, such as molecular imaging.
The first aim of this study was to investigate whether the length of the linker, connecting the two Affibody molecules in the biparatopic construct, could be decreased without disturbing simultaneous binding to VEGFR2. For future in vivo imaging purposes, a small size is preferable [24] and minimizing the linker length might also reduce potential issues with proteolytic degradation in serum. A further aim was to evaluate the ability of the biparatopic construct to inhibit cell signaling by blocking VEGFA from binding to VEGFR2. Four different biparatopic constructs were designed with varying linker lengths. The results demonstrated that all constructs were able to bind to VEGFR2-expressing cells, block VEGFA binding, and inhibit VEGFR2 phosphorylation. The smallest construct had the highest affinity and is a promising candidate for further optimization of imaging and therapeutic agents. This construct was also demonstrated to inhibit downstream VEGFR2 signaling, cell proliferation, and angiogenic sprout formation in vitro.
Materials and methods
Design and production of biparatopic VEGFR2-specific Affibody construct with different linker lengths
Genes encoding the following constructs were designed, ordered from DNA2.0 Inc. (Menlo Park, CA, USA), and subcloned in the plasmid pJ411 (DNA2.0 Inc.): (1) ZVEGFR2_Bp1: ZVEGFR2_22-(S4G)-ABD035-(S4G)-ZVEGFR2_40, (2) ZVEGFR2_Bp2: ZVEGFR2_22-(S4G)-ZVEGFR2_40-(S4G)3-ABD035, (3) ZVEGFR2_Bp3: ZVEGFR2_22-(S4G)3-ZVEGFR2_40-(S4G)3-ABD035 and (4) ZVEGFR2_Bp4: ZVEGFR2_22-(S4G)8-ZVEGFR2_40-(S4G)3-ABD035. Plasmids were transformed to E. coli BL21 Star (DE3) cells (Invitrogen, Carlsbad, CA) by heat shock. Colonies were inoculated to TSB medium supplemented with yeast extract (TSB + YE; 30 g/l; Merck, Darmstadt, Germany), and 50 μg/ml kanamycin (Sigma-Aldrich Company Ltd, Dorset, UK) and cultivated overnight at 37 °C and 150 rpm. After 16 h, the cultures were re-inoculated to an OD600 of 0.05–0.1 in TSB + YE medium (Merck) supplemented with 50 μg/ml kanamycin (Sigma-Aldrich). When an OD600 of 0.5–1 was reached, protein expression was induced with isopropyl-beta-d-thiogalactopyranoside (IPTG) (Apollo Scientific Ltd, Stockport, UK), and cultures were incubated for 20 h at 25 °C and 150 rpm. Cells were harvested by centrifugation (2400g, 8 min 4 °C) and proteins were purified using affinity chromatography with HSA immobilized on a Sepharose matrix. The purity and size of the purified proteins were analyzed by SDS-PAGE.
Analysis of binding kinetics using SPR
Surface plasmon resonance (SPR) experiments were performed using a ProteOn XPR36 instrument (Bio-Rad Laboratories, Hercules, CA, USA), using a previously described setup [30]. Briefly, HSA (Sigma-Aldrich) was immobilized on a GLM sensor chip (Bio-Rad Laboratories), and the kinetics of the binding of the Affibody constructs to human or murine VEGFR2 were analyzed by injection of 100 nM Affibody construct, immediately followed by injection of 10, 20, and 40 nM monomeric human or murine VEGFR2 (Sino Biological Inc. Beijing, China). The dissociation constants (and equilibrium constants for ZVEGFR2_Bp2) were obtained from sensorgrams using a monovalent binding equation and non-linear regression. The experiment was performed in duplicates using freshly prepared reagents.
Cell binding analysis
293/KDR cells (Sibtech Inc. Brookfield, CT, USA) were cultivated in Dulbecco’s Modified Eagle Medium (DMEM; Sigma-Aldrich) supplemented with 10 % FBS and 2 mM l-glutamine (Sigma-Aldrich). Cells were washed with PBS and harvested by pipetting using PBSB. HUVECs (Lonza, Basel, Switzerland) were cultivated in Endothelial Growth Basal Medium-2 (EBM-2) (Lonza) supplemented with Endothelial Growth Medium-2 (EGM-2) Bullet Kit (Lonza). Cells were harvested by trypsinization, centrifuged at 220g for 5 min, and pellets were re-suspended in PBS with 1 % BSA (PBSB).
Harvested cells were incubated for 20 min on ice in PBSB with 200 nM of each of the four biparatopic Affibody constructs ZVEGFR2_Bp1, ZVEGFR2_Bp2, ZVEGFR2_Bp3 or ZVEGFR2_Bp4, the biparatopic Affibody construct ZVEGFR2_22-(S4G)4-ABD035-(S4G)4-ZVEGFR2_40, a negative control construct (ZTaq-ABD-ZTaq) [29], or the positive control mouse IgG anti-human VEGFR2 antibody (R&D systems, Minneapolis, MN, USA). Cells were pelleted by centrifugation at 300g for 4 min, and re-suspended in 150 nM HSA (Human serum albumin; Sigma-Aldrich) conjugated to Alexa Fluor 647 (Invitrogen) (or 1 μg/ml Alexa Fluor 488 Rabbit Anti-Mouse IgG (H + L) Antibody (Invitrogen) for the positive control). After incubation for 20 min on ice in the dark, cells were pelleted by centrifugation and re-suspended in PBSB. Cell binding of the biparatopic Affibody constructs was detected by flow-cytometric analysis using a Gallios flow cytometer (Beckman Coulter, Indianapolis, IN, USA).
Evaluation of VEGFA blocking
293/KDR cells (Sibtech Inc.) were cultivated and harvested as described above. Cells were incubated for 15 min on ice with 0.25, 1, 4, 16, 64, 256 or 1024 nM of each of the four Affibody constructs, or 1024 nM of the negative control construct ZTaq-ABD-ZTaq, or without Affibody construct, in PBSB. The cells were pelleted by centrifugation at 300g for 4 min, re-suspended in 50 nM biotinylated VEGFA (Acro Biosystems, Newark, DE, USA), and incubated on ice for 15 min. Thereafter, cells were pelleted by centrifugation, and re-suspended in 1 μg/ml streptavidin-R-Phycoerythrin conjugate (SAPE, Invitrogen). After incubation for 15 min on ice in the dark, cells were pelleted by centrifugation and re-suspended in PBSB. Cell binding of the biotinylated VEGFA was detected by flow-cytometric analysis using a Gallios flow cytometer (Beckman Coulter). Calculation of IC-50 values was performed by non-linear regression to an inhibitor response equation using the GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
VEGFR2 phosphorylation assay
293/KDR cells (Sibtech Inc.) were seeded in Dulbecco’s Modified Eagle Medium (DMEM; Sigma-Aldrich) supplemented with 10 % FSB and 2 mM l-glutamine. 8 h after seeding, cells were shifted to starvation media (DMEM supplemented with 0.5 % FBS), and incubated overnight. Next, 200 or 20 nM of each of the four Affibody constructs, the negative control construct ZTaq-ABD-ZTaq, or PBS (control), was added to the culture dishes. After incubation for 1 h at 37 °C, 1 nM human VEGFA (R&D systems), or PBS, was added, and cells were incubated for 10 min at 37 °C. Thereafter, cells were washed with PBS containing 1 mM sodium orthovanadate (Sigma-Aldrich). Cell lysates were prepared by scraping the cells from the plates and sonication for 1 min in Cell Lysis Buffer (Cell Signaling Technology, Danvers, MA, USA), supplemented with 1 mM sodium orthovanadate (Sigma-Aldrich), 1 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich) and PhosStop (Roche), lysed by sonication and analyzed by ELISA using a PathScan® Phospho-VEGFR-2-Tyr1175 Sandwich ELISA kit (Cell Signaling Technology) in a Tecan Sunrise microplate reader (Tecan Group Ltd., Männedorf, Switzerland), according to the supplier’s recommendations. The same experimental setup was used to determine the IC50 values of ZVEGFR2_Bp2 and Ramucirumab. This was performed using concentrations of 0.5, 1, 3, 10, 20, and 100 nM for the Affibody construct (ZVEGFR2_Bp2) and 0.03, 0.1, 0.7, 3, 15, and 100 nM for Ramucirumab (Cyramza). Calculation of IC-50 values was performed by non-linear regression to an inhibitor response equation using the GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA).
ERK1/ERK2 phosphorylation assay
293/KDR cells (Sibtech Inc.) were seeded in Dulbecco’s Modified Eagle Medium (DMEM; Sigma-Aldrich) supplemented with 10 % FSB and 2 mM l-glutamine. 8 h after seeding, cells were shifted to starvation media (DMEM supplemented with 0.5 % FBS), and incubated overnight. Thereafter, 200 nM ZVEGFR2_Bp2 or PBS (control) was added to the culture dishes. After incubation for 1 h at 37 °C, 10 nM human VEGFA (R&D systems) or PBS was added, and cells were incubated for 10 min at 37 °C. Thereafter, cells were washed with PBS containing 1 mM sodium orthovanadate (Sigma-Aldrich) and 1 μg/ml pepstatin A (Sigma-Aldrich), and 1 mM PMSF (Sigma-Aldrich). Cell lysates were prepared by addition of RIPA Lysis and Extraction buffer (Cell Signaling Technologies), supplemented with 1 mM sodium orthovanadate (Sigma-Aldrich), 1 mM PMSF (Sigma-Aldrich), 1 μg/ml pepstatin A (Sigma-Aldrich), and PhosStop cocktail (Roche), and incubation on ice for 30 min. Thereafter, cell lysates were analyzed by ELISA using a Phospho-ERK1 (T202/Y204)/ERK2 (T185/Y187) DuoSet IC ELISA kit (R&D Systems) in a Tecan Sunrise microplate reader (Tecan Group Ltd.), according to the supplier’s recommendations.
293/KDR cell proliferation assay
293/KDR cells (Sibtech Inc.) were cultivated in Dulbecco’s Modified Eagle Medium (DMEM; Sigma-Aldrich) supplemented with 10 % FSB and 2 mM l-glutamine. 1000 cells/well were seeded in duplicates in 100 μl in 96-well plates in the presence of either 100 nM ZVEGFR2_Bp2 and 10 nM VEGFA (R&D Systems), 10 nM VEGFA only, or no additives (control). After incubation for 72 h, 10 μl Cell Counting Kit-8 (CCK-8; Sigma-Aldrich) was added to the medium and after 5 h and 45 min, the absorbance at 450 nm was measured in a Tecan Sunrise microplate reader (Tecan Group Ltd.). The absorbance at 570 nm was subtracted from the values obtained at 450 nm in order to correct for optical imperfections in the plate.
HUVEC sprouting assay
The HUVEC sprouting assay was performed as described below and as previously described [38]. HUVECs were labeled with PKH67 (Sigma-Aldrich) according to manufacturer instructions, and cells were coated on cytodex3 microcarrier beads (Sigma-Aldrich) for 24 h. Beads were embedded in a fibrin gel [2.5 mg/ml fibrinogen (Sigma-Aldrich) in EBM-2 (Lonza) supplemented with 2 % FBS and 50 mg/ml aprotinin (Sigma-Aldrich)] with fibrinogen solution clotted with 1 U thrombin (Sigma-Aldrich) for 30 min at 37 °C. WI38 cells (25,000 cells/well), in EBM-2 supplemented with 2 % FBS and 50 ng/ml VEGFA (R&D Systems) were then plated on top of the fibrin layer. After 24 h, 50 or 10 nM ZVEGFR2_Bp2, Ramucirumab (ImClone Systems) or ZTaq-ABD-ZTaq was added. Sprouts were imaged 3–4 days later using a Zeiss700 confocal microscope and analysis was performed using Image J.
Results
Design and production of biparatopic VEGFR2-specific Affibody constructs
Previous studies demonstrated that the biparatopic Affibody construct ZVEGFR2_22-(S4G)-ABD035-(S4G)-ZVEGFR2_40 could bind to VEGFR2 with a higher apparent affinity compared to the monomeric ZVEGFR2_22 and ZVEGFR2_40, suggesting simultaneous binding of the two Affibody domains [30]. The results hence indicated that the two epitopes were in relatively close proximity on the receptor (Fig. 1a). To optimize the biparatopic construct and investigate the influence of linker length on VEGFR2-binding and inhibition of VEGFA-stimulated signaling, new heterodimeric constructs based on ZVEGFR2_22 and ZVEGFR2_40 were designed and produced. The genes encoding ZVEGFR2_22 and ZVEGFR2_40 were genetically fused together with an engineered variant of ABD (ABD035 [34]). ABD035 has previously been engineered to femtomolar affinity for human serum albumin and fusions to therapeutic proteins dramatically prolong the serum half-life [35–37]. Four constructs, abbreviated ZVEGFR2_Bp1, ZVEGFR2_Bp2, ZVEGFR2_Bp3, and ZVEGFR2_Bp4, were designed (Fig. 1b), expressed in E. coli and purified by affinity chromatography using HSA-Sepharose. SDS-PAGE confirmed that the proteins were pure and of correct size (data not shown). The successful purification using HSA as ligand during affinity chromatography also demonstrated that the albumin-binding domain was correctly folded and retained the affinity for albumin in the new fusion proteins.
Fig. 1.

Schematic overview of the hypothetical mode of binding and the design of the dimeric constructs. a Schematic outline of VEGF-induced activation of VEGFR2 as well as blocking using respective monomers and the biparatopic construct. VEGFR2 is represented in blue on the cell surface with the VEGF-binding epitope in green. b In ZVEGFR2_Bp1, ABD035 is positioned between the two VEGFR2-binding Affibody molecules, whereas in ZVEGFR2_Bp2, ZVEGFR2_Bp3, and ZVEGFR2_Bp4, it is positioned at the C-terminus. The length and aa composition of the linkers are indicated above each linker and are not in scale. The molecular weights of the constructs are indicated in the figure
Determination of binding kinetics using an SPR-based biosensor assay
Determination of the binding kinetics of the four Affibody constructs to human as well as murine VEGFR2 was performed in an SPR-based assay using a previously described setup [30]. Investigating the interaction using immobilized VEGFR2 and the dimeric Affibody constructs as analyte could result in avidity effects and false interpretation of data. A directed immobilization strategy of the Affibody molecules on the surface was therefore used. Briefly, HSA was immobilized on a sensor chip surface and injection of Affibody constructs was immediately followed by injection of human or murine monomeric VEGFR2. The setup allowed for (1) assessing the ability of the fusion proteins to simultaneously interact with HSA and VEGFR2, and (2) directed immobilization of the Affibody construct on the sensor surface. Although the immobilization of Affibody construct was non-covalent, the femtomolar affinity of ABD035 for HSA [34] results in an extremely slow dissociation rate of the complex [30]. Moreover, monomeric VEGFR2 was used in the assay to avoid potential avidity effects in the interaction between Affibody constructs and VEGFR2. To be able to accurately differentiate between relatively small changes in the interaction, we focused the analysis mainly on the dissociation rate, as it is independent of the concentration of the analyte. The dissociation rate constants (k d) of the four constructs were determined for human and murine VEGFR2 (Fig. 2a–h; Table 1). The dissociation rates for the four constructs were relatively similar, and all were slower than the original construct, ZVEGFR2_22-ABD-ZVEGFR2_40 [30]. The slowest dissociation rate constant (1.8 × 10−5 s−1 for hVEGFR2 and 1.1 × 10−5 s−1 for mVEGFR2) was observed for ZVEGFR2_Bp2, which is even slower than the Fab fragment of the FDA-approved monoclonal antibody ramucirumab [16, 17]. This construct also contained the shortest linker (5 aa) between the two Affibody molecules, and was therefore considered the most promising candidate for further studies. The results also indicated that the binding sites for the two domains are in principle adjacent and might practically be regarded as one continuous epitope. The association rate constant (k a) for ZVEGFR2_Bp2 was determined using the same experimental setup to 7.4 ± 0.1 × 104 M−1 s−1 for hVEGFR2 and 6.5 ± 1.3 × 104 M−1 s−1 for mVEGFR2 and used together with the k d for calculating the equilibrium dissociation constant (K D). The K D was determined to 241 ± 4 pM for human VEGFR2, and 180 ± 36 pM for murine VEGFR2. The SPR results also demonstrated that all four constructs were able to bind to VEGFR2 while simultaneously interacting with HSA, which is promising for future half-life extension purposes in vivo.
Fig. 2.

Determination of kinetics for Affibody molecules binding to VEGFR2. Representative sensorgrams from SPR analysis of ZVEGFR2_Bp1, ZVEGFR2_Bp2, ZVEGFR2_Bp3, and ZVEGFR2_Bp4 binding to human and murine monomeric VEGFR2. Sensorgrams were obtained from a double injection, where a first injection of 100 nM of ZVEGFR2_Bp1, ZVEGFR2_Bp2, ZVEGFR2_Bp3 or ZVEGFR2_Bp4 over immobilized human serum albumin was immediately followed by a second injection of monomeric human or murine VEGFR2 (10, 20, and 40 nM). The sensorgrams show the second injections and the baseline has been normalized to 0 at the start of the second injection. a ZVEGFR2_Bp1 binding human VEGFR2, b ZVEGFR2_Bp2 binding human VEGFR2, c ZVEGFR2_Bp3 binding human VEGFR2, d ZVEGFR2_Bp4 binding human VEGFR2, e ZVEGFR2_Bp1 binding murine VEGFR2, f ZVEGFR2_Bp2 binding murine VEGFR2, g ZVEGFR2_Bp3 binding murine VEGFR2, h ZVEGFR2_Bp4 binding murine VEGFR2. The experiment was performed in duplicates
Table 1.
Dissociation rate constants for the VEGFR2-binding Affibody constructs
| Affibody construct | Mean k d for hVEGFR2 (±SD) (×10−5 s−1) | Mean k d for mVEGFR2 (±SD) (×10−5 s−1) |
|---|---|---|
| ZVEGFR2_Bp1 | 5.3 ± 2.2 | 8.3 ± 4.0 |
| ZVEGFR2_Bp2 | 1.8 ± 1.1 | 1.1 ± 0.4 |
| ZVEGFR2_Bp3 | 2.0 ± 1.0 | 1.3 ± 0.6 |
| ZVEGFR2_Bp4 | 2.9 ± 1.5 | 3.3 ± 1.6 |
Cell binding analysis
A flow-cytometry based assay was performed to investigate the ability of the biparatopic Affibody constructs to bind to VEGFR2 expressed on the surface of mammalian cells. The four dimeric constructs were incubated with either (1) 293/KDR cells, which is a human embryonic kidney (HEK) cell line that has been stably transfected with human recombinant VEGFR2 (also denoted KDR; kinase domain insert, with high expression level), or (2) HUVECs, which are human endothelial VEGFR2-expressing cells (moderate expression level). Fluorescently labeled HSA was used as secondary reagent to achieve signal amplification. The biparatopic Affibody molecule ZVEGFR2_22-(S4G)4-ABD035-(S4G)4-ZVEGFR2_40, which had previously been shown to bind to VEGFR2 with both Affibody molecules simultaneously [30], was included for comparison. The dimeric Affibody construct Z03638-(S4G)4-ABD035-(S4G)4-Z03638, which binds to Taq polymerase [29] (hereafter abbreviated ZTaq-ABD-ZTaq), was included as a negative control. A mouse IgG anti-human VEGFR2 antibody was included as a positive control. Cell binding of the dimeric Affibody constructs was detected using fluorescently labeled HSA (or Alexa Fluor® 488 Rabbit Anti-Mouse IgG Antibody for the positive control). A clear shift in the fluorescent signal was observed for the 293/KDR cells incubated with all four of the dimeric VEGFR2-specific Affibody constructs compared to the negative control (Fig. 3a). As expected, the flow-cytometry results from analysis of the HUVECs resulted in lower signals, with detectable binding above background for ZVEGFR2_Bp1 and ZVEGFR2_Bp2 (Fig. 3b). Please note that different secondary reagents and fluorophores were used for detection of Affibody molecules and the positive control antibody, respectively, and absolute fluorescent signals from the positive control versus the Affibody constructs are hence not comparable. Again, using HSA as a secondary detection reagent confirmed that the fusion proteins were able to bind to the receptor while simultaneously interacting with HSA (Fig. 3a, b).
Fig. 3.

Flow-cytometric analysis of binding of Affibody constructs to VEGFR2-expressing cells and VEGFA blocking. a Binding of the Affibody constructs (ZVEGFR2_Bp1–ZVEGFR2_Bp4) to 293/KDR cells was detected by fluorescently labeled HSA. The biparatopic Affibody molecule ZVEGFR2_22-(S4G)4-ABD035-(S4G)4-ZVEGFR2_40, which had previously been shown to bind to VEGFR2 with both Affibody molecules simultaneously, as well as ZTaq-ABD-ZTaq as negative control were included for comparison. All signals are normalized with the signal from the negative control. Data are represented as mean of duplicates with SD. b Same as in a but for HUVECs. c Evaluation of VEGFA blocking. 293/KDR cells were incubated with varying concentrations of each of the biparatopic Affibody molecules, followed by incubation with biotinylated VEGFA. Binding of VEGFA was detected using fluorescently labeled streptavidin. Background signals from cells treated with secondary reagents are subtracted from all samples. All signals are normalized with the signal from cells only incubated with biotinylated VEGFA and fluorescently labeled streptavidin. Upper dotted line is the signal from cells treated with 1024 nM of negative control Affibody (ZTaq-ABD-ZTaq). The experiment was performed in duplicates. Data are represented as mean of duplicates with SD
Evaluation of VEGFA blocking using flow cytometry
Another flow-cytometry based assay was performed in order to investigate whether the biparatopic Affibody constructs could block VEGFA from binding to VEGFR2 expressed on the surface of mammalian cells. 293/KDR cells were incubated with concentrations varying from 0.25 to 1024 nM of the four VEGFR2-binding dimeric constructs or the negative control construct ZTaq-ABD-ZTaq (1024 nM). After incubation with Affibody constructs, cells were incubated with 50 nM biotinylated VEGFA. Cell binding of the biotinylated VEGFA was detected using streptavidin–phycoerythrin conjugate. A concentration-dependent negative shift in fluorescent signal was observed for the 293/KDR cells incubated with all four of the dimeric VEGFR2-specific Affibody constructs compared to the negative control, indicating blocking of VEGFA binding to VEGFR2 expressed on the surface of 293/KDR cells (Fig. 3c). The estimated IC-50 values obtained from fitting the data to an inhibitor response equation were: 45.1 ± 8 nM for ZVEGFR2_Bp1, 39.5 ± 16 nM for ZVEGFR2_Bp2, 57.6 ± 37 nM for ZVEGFR2_Bp3, and 42.4 ± 8 nM for ZVEGFR2_Bp4. The observed values are consistent with theoretical calculations of IC-50 for the given experiment, using 50 nM VEGFA and assuming a K D of around 400 pM for VEGFA binding to VEGFR2 [39].
Inhibition of VEGFA-induced phosphorylation of VEGFR2
In order to investigate whether the observed ability of the biparatopic Affibody constructs to block VEGFA binding would lead to an inhibition of VEGFR2 phosphorylation, an ELISA-based phosphorylation assay was performed. 293/KDR cells were incubated with 20 or 200 nM of each of the four dimeric constructs or the negative control construct ZTaq-ABD-ZTaq. After incubation with Affibody constructs, VEGFR2 phosphorylation was stimulated by incubation with 1 nM human VEGFA. Cell lysates were prepared and analyzed by ELISA detecting phosphorylation on the tyrosine 1175 in VEGFR2.
A decrease in ELISA signal was observed for all four constructs, demonstrating that all variants could inhibit VEGFR2 phosphorylation (Fig. 4a). Treatment of cells with 200 nM of each Affibody construct in the absence of VEGFA showed no increase in VEGFR2 phosphorylation, indicating that the dimeric Affibody molecules were not agonistic (Fig. 4a). The top candidate ZVEGFR2_Bp2 was subsequently compared to the FDA-approved VEGFR2-specific monoclonal antibody Ramucirumab (ImClone Systems) in a similar assay. The VEGFR2 Tyr1175 phosphorylation levels were determined after incubation with a range of concentrations of ZVEGFR2_Bp2 or Ramucirumab. The obtained IC-50 values were 3.8 ± 1 nM for ZVEGFR2_Bp2 and 0.41 ± 0.01 nM for ramucirumab (Fig. 4b). It should be noted that Ramucirumab is formatted as a full-length bivalent antibody, resulting in avidity effects on the multivalent cell surface.
Fig. 4.

Inhibition of VEGFR2 signaling. a Inhibition of VEGFA-induced VEGFR2 phosphorylation. VEGFR2 phosphorylation level was detected using a Tyr1175 phosphorylation-specific antibody. 293/KDR cells were incubated with 20 nM (purple bars) or 200 nM (green bars) of each of the four biparatopic Affibody molecules prior to VEGFA stimulation. ZTaq-ABD-ZTaq was included as negative control as well as no Affibody. Samples were also treated with 200 nM Affibody constructs without VEGFA stimulation (blue bars). VEGFR2 phosphorylation is on the y-axis and all signals are normalized with signals from cells treated with only VEGFA. Data are represented as mean of duplicates with SD. b Determination of IC-50 value for inhibition of VEGFA-induced VEGFR2 phosphorylation by ZVEGFR2_Bp2 (red) or ramucirumab (green). Solid lines represent fitted equations. Upper dotted line is the signal from cells treated with only VEGFA and lower dotted line is the signal from untreated cells. Data are represented as mean of duplicates with SD. c Inhibition of VEGFA-induced ERK1/ERK2 phosphorylation in 293/KDR cells by ZVEGFR2_Bp2. 293/KDR cells were incubated with 100 nM of ZVEGFR2_Bp2 prior to 10 nM VEGFA stimulation. ERK1/ERK2 phosphorylation is on the y-axis and all signals are normalized with mean signals from cells treated with only VEGFA. Data are represented as mean of duplicates with SD. Data from samples treated with ZVEGFR2_Bp2 + VEGFA and samples treated with only VEGFA was analyzed using an unpaired t test (P = 0.1698)
Inhibition of VEGFA-induced phosphorylation of ERK1/ERK2
The ability of the biparatopic Affibody construct ZVEGFR2_Bp2 to inhibit downstream signaling induced by VEGFR2 was investigated by analysis of the phosphorylation levels of ERK1/ERK2, which are downstream mediators of VEGFR2 signaling. 293/KDR cells were incubated with 200 nM ZVEGFR2_Bp2 or PBS, followed by stimulation with 10 nM VEGFA. Cell lysates were prepared, and the ERK1/ERK2 phosphorylation levels were analyzed by ELISA detecting ERK1 dually phosphorylated at T202/Y204 and ERK2 dually phosphorylated at T185/Y187. Cells treated with VEGFA in the presence of ZVEGFR2_Bp2 showed a decrease in ERK1/ERK2 phosphorylation levels compared to cells treated with VEGFA alone. These results confirmed that the Affibody construct could inhibit downstream VEGFR2 signaling (Fig. 4c).
Inhibition of VEGFA-induced 293/KDR cell proliferation
A cell proliferation assay was performed in order to investigate whether the inhibited VEGFR2 signaling resulted in a decreased proliferation rate of 293/KDR cells. Cells were cultivated in the presence of 10 nM VEGFA and 100 nM ZVEGFR2_Bp2 or PBS as negative control, and the cell count was determined using a CCK8 kit after 72 h. The presence of VEGFA resulted in an increased growth rate (Fig. 5a), which was decreased with the inclusion of ZVEGFR2_Bp2 in the medium (Fig. 5a). Together with the results from the phosphorylation assay, these results suggested that the biparatopic Affibody construct has the ability to inhibit angiogenic effects of VEGFR2-mediated signaling, which is promising for potential therapeutic applications.
Fig. 5.
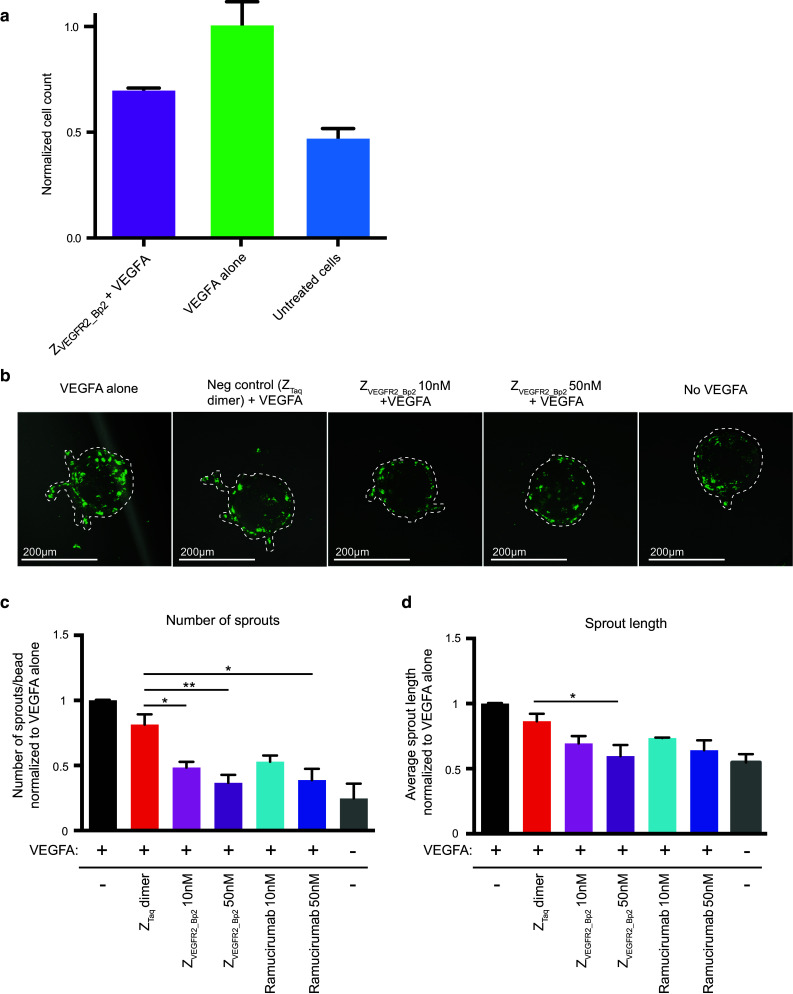
Inhibition of VEGFA-induced 293/KDR proliferation and HUVEC sprout formation in vitro. a Inhibition of VEGFA-induced proliferation of 293/KDR cells. 293/KDR cells were grown in presence of 10 nM VEGFA and 200 nM ZVEGFR2_Bp2. Cells treated with only VEGFA were included as positive control and untreated cells were included as negative control. Cells were grown for 72 h and the relative cell count was determined using CCK8 kit. Relative cell count is on the y-axis and all signals are normalized with mean signals from cells treated with only VEGFA. Data are represented as mean of duplicates with SD. Data from samples treated with ZVEGFR2_Bp2 + VEGFA and samples treated with only VEGFA were analyzed using an unpaired t test (P = 0.0609). b Representative images of a sprouting assay of PKH67 labeled HUVECs (green) after treatment with ZVEGFR2_Bp2. Sprouts treated with only VEGFA were included as positive control, and untreated and ZTaq-ABD-ZTaq treated sprouts were used as a negative control. HUVEC were allowed to sprout for 3–4 days. Sprouts are indicated by hashed lines. c Quantitation of the number of sprouts per bead normalized to VEGFA alone control. d Average sprout length normalized to VEGFA alone control. Data are represented as a mean ± SEM. n = 5 (VEGFA alone, no VEGFA), n = 4 (ZVEGFR2_Bp2 10 nM, ZTaq-ABD-ZTaq), n = 3 (ZVEGFR2_Bp2 50 nM), n = 2 (ramucirumab 10 nM, ramucirumab 50 nM). n = experimental replicates. n > 15 beads imaged per experiment
Inhibition of HUVEC sprout formation in vitro
In order to further investigate the anti-angiogenic effects of the biparatopic Affibody construct, an in vitro angiogenesis assay was performed. HUVECs coated on cytodex3 microcarrier beads were embedded in a fibrin gel and cultivated in the presence of VEGFA and different concentrations of ZVEGFR2_Bp2, Ramucirumab or the negative control construct ZTaq-ABD-ZTaq. Imaging of the sprouts after 3–4 days revealed a substantial decrease in number of sprouts as well as sprout length for cells treated with ZVEGFR2_Bp2 compared to ZTaq-ABD-ZTaq or no Affibody, demonstrating that ZVEGFR2_Bp2 could inhibit sprout formation (Fig. 5b–d). Comparing the results with treatment using the same molar concentrations of Ramucirumab, indicated that the biparatopic Affibody molecule had similar anti-angiogenic effects on endothelial cells in this assay.
Discussion
Angiogenesis is important for the progression of several pathological conditions, including tumor progression and vascular eye disorders [1]. A challenge in angiogenesis-targeted therapy is the preexisting or acquired drug resistance [22]. For ophthalmic diseases, an additional challenge is the delivery of protein therapeutics to the eye, which with the current antibody-based therapeutics requires administration by intraocular injection [40].
In previous work, two different VEGFR2-specific Affibody molecules were generated, and engineered into a biparatopic Affibody construct [30]. The ability of the two Affibody domains in the biparatopic construct to bind simultaneously to VEGFR2 resulted in a dramatic decrease in dissociation rate compared to the monomeric Affibody molecules. The VEGFR2-specific biparatopic Affibody construct can potentially be used for therapeutic as well as in vivo imaging applications. Fusing the small VEGFR2-targeting agent to a 46 aa albumin-binding domain (ABD), allows for shifting the pharmacokinetic behavior to a profile suitable for therapeutic applications, i.e., the small drug obtains a dramatically longer in vivo half-life that is similar to that of serum albumin [35–37], hence resulting in less frequent dosing and higher potency. For topical administration in ocular disorders, the ABD-based strategy would ensure Affibody delivery in an unconjugated mode, resulting in efficient uptake. Once the Affibody reaches the vitreous fluid, however, association to endogenous albumin will decrease the diffusion rate, decrease clearance, and promote prolonged half-life. Since a small size is desirable for in vivo imaging applications [24], as well as for alternative delivery routes such as eye drops, we investigated whether the linker separating the two Affibody domains could be shortened without negatively affecting the VEGFR2 affinity. Four biparatopic constructs were designed, with different linker lengths connecting the two Affibody molecules. Although the ABD will not be part of future constructs intended for in vivo molecular imaging, we included it here to facilitate protein purification, detection of cell binding, and directed immobilization in the biosensor assays. It should be noted that the affibody molecules have been characterized previously without fusions to the albumin-binding domain, demonstrating that the interaction with VEGFR2 is not dependent on the ABD [30]. In three of the constructs (ZVEGFR2_Bp2–ZVEGFR2_Bp4), ABD was positioned at the C-terminal in order to facilitate production without ABD for applications where a short in vivo half-life is preferred. As expected, the improvement in affinity for the new heterodimeric construct compared to the individual monomeric Affibody molecules ZVEGFR2_22 and ZVEGFR2_40 [30] was mainly due to a slower dissociation rate. A decrease in k d value of around 2–3 orders of magnitude was observed for ZVEGFR2_Bp2 compared to the monomers [30]. The new format also demonstrated a fourfold slower dissociation rate compared to the original heterodimeric construct ZVEGFR2_22-ABD-ZVEGFR2_40 [30]. The slowest dissociation rate was observed for ZVEGFR2_Bp2, which had the shortest linker, indicating that the two Affibody binding sites on VEGFR2 are situated in very close proximity and that the binding surface is likely to be practically continuous. However, further structural investigations are needed to determine in detail the exact binding mechanism and epitopes on VEGFR2. Notably, both the biosensor assay and the cell binding experiment demonstrated that the Affibody construct binds to VEGFR2 to the same extent with and without simultaneous binding to albumin, which is essential when using the ABD-based approach for prolonging the circulation time in future therapeutic studies in vivo. Moreover, previous investigations have shown that other ABD-fused therapeutic Affibody molecules retain their targeting capacity in blood, which supports our intended strategy [41].
In order to study the potential of these biparatopic constructs for future therapeutic applications, the ability to block VEGFA binding and inhibit receptor phosphorylation was investigated. The results demonstrated that all four constructs were able to bind to VEGFR2 expressed on 293/KDR cells. The four new constructs were also demonstrated to efficiently block VEGFA binding and thereby inhibit receptor phosphorylation. Due to the slow dissociation rate and the advantages of a shorter linker, ZVEGFR2_Bp2 was considered the top candidate for further analysis. Treatment with ZVEGFR2_Bp2 was shown to decrease the proliferation rate of 293/KDR cells in presence of VEGFA, and inhibit HUVEC sprout formation. These results demonstrate that ZVEGFR2_Bp2 has anti-angiogenic effects comparable with the clinically approved Ramucirumab, which is encouraging for therapeutic strategies with the aim to inhibit VEGFR2 signaling. The molecular weight of ZVEGFR2_Bp2 is around 20 kDa including the ABD, and around 13 kDa without the ABD, which is about half that of an scFv. Importantly, ZVEGFR2_BP2 is a >10-fold smaller protein than a conventional antibody, but with similar affinity as has been reported for Ramucirumab [16, 17]. Therefore, we anticipate that its performance in several aspects will be superior to that of a regular antibody, particularly with regard to: biodistribution, dose response, tissue penetration, clearance in molecular imaging and administration routes. The promising results encourage future preclinical studies to investigate the in vivo performance.
Acknowledgments
This study was financially supported by the Swedish Research Council (621-2012-5236), the Swedish Cancer Society, and the Governmental Agency for Innovation system (2009-00179). EG is supported by Wenner-Gren Stiftelserna.
Footnotes
F. Fleetwood and R. Güler contributed equally.
References
- 1.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 2.Claesson-Welsh L, Welsh M. VEGFA and tumour angiogenesis. J Intern Med. 2013;273(2):114–127. doi: 10.1111/joim.12019. [DOI] [PubMed] [Google Scholar]
- 3.Roskoski R. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007;62(3):179–213. doi: 10.1016/j.critrevonc.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Pötgens AJG, Lubsen NH, van Altena MC, Vermeulen R, Bakker A, Schoenmakers JGG, Ruiter DJ, de Waal RMW. Covalent dimerization of vascular permeability factor vascular endothelial growth factor is essential for its biological activity. J Biol Chem. 1994;269(52):32879–32885. [PubMed] [Google Scholar]
- 5.Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, de Vos AM. Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc Natl Acad Sci USA. 1997;94(14):7192–7197. doi: 10.1073/pnas.94.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, de Vos AM. Crystal structure at 1.7 Å resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell. 1997;91:695–704. doi: 10.1016/S0092-8674(00)80456-0. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001;20(11):2768–2778. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tugues S, Koch S, Gualandi L, Li X, Claesson-Welsh L. Vascular endothelial growth factors and receptors: anti-angiogenic therapy in the treatment of cancer. Mol Aspects Med. 2011;32(2):88–111. doi: 10.1016/j.mam.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57(20):4593–4599. [PubMed] [Google Scholar]
- 10.Mross K, Richly H, Fischer R, Scharr D, Büchert M, Stern A, Gille H, Audoly LP, Scheulen ME. First-in-human phase I study of PRS-050 (Angiocal), an anticalin targeting and antagonizing VEGF-A, in patients with advanced solid tumors. PLoS One. 2013;8(12):1–11. doi: 10.1371/journal.pone.0083232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD, Rudge JS. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99(17):11393–11398. doi: 10.1073/pnas.172398299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polverino A, Coxon A, Starnes C, Diaz Z, DeMelfi T, Wang L, Bready J, Estrada J, Cattley R, Kaufman S, Chen D, Gan Y, Kumar G, Meyer J, Neervannan S, Alva G, Talvenheimo J, Montestruque S, Tasker A, Patel V, Radinsky R, Kendall R. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res. 2006;66(17):8715–8721. doi: 10.1158/0008-5472.CAN-05-4665. [DOI] [PubMed] [Google Scholar]
- 13.Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459–5465. doi: 10.1158/1078-0432.CCR-07-5270. [DOI] [PubMed] [Google Scholar]
- 14.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 15.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. [PubMed] [Google Scholar]
- 16.Lu D, Jimenez X, Zhang H, Bohlen P, Witte L, Zhu Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int J Cancer. 2002;97(3):393–399. doi: 10.1002/ijc.1634. [DOI] [PubMed] [Google Scholar]
- 17.Lu D, Shen J, Vil MD, Zhang H, Jimenez X, Bohlen P, Witte L, Zhu Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J Biol Chem. 2003;278(44):43496–43507. doi: 10.1074/jbc.M307742200. [DOI] [PubMed] [Google Scholar]
- 18.Getmanova EV, Chen Y, Bloom L, Gokemeijer J, Shamah S, Warikoo V, Wang J, Ling V, Sun L. Antagonists to human and mouse vascular endothelial growth factor receptor 2 generated by directed protein evolution in vitro. Chem Biol. 2006;13(5):549–556. doi: 10.1016/j.chembiol.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Behdani M, Zeinali S, Karimipour M, Khanahmad H, Schoonooghe S, Aslemarz A, Seyed N, Moazami-Godarzi R, Baniahmad F, Habibi-Anbouhi M, Hassanzadeh-Ghassabeh G, Muyldermans S. Development of VEGFR2-specific nanobody Pseudomonas exotoxin A conjugated to provide efficient inhibition of tumor cell growth. New Biotechnol. 2013;30(2):205–209. doi: 10.1016/j.nbt.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Siemeister G, Schirner M, Reusch P, Barleon B, Marmé D, Martiny-Baron G. An antagonistic vascular endothelial growth factor (VEGF) variant inhibits VEGF-stimulated receptor autophosphorylation and proliferation of human endothelial cells. Proc Natl Acad Sci USA. 1998;95:4625–4629. doi: 10.1073/pnas.95.8.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waldner MJ, Neurath MF. Targeting the VEGF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):5–13. doi: 10.1517/14728222.2011.641951. [DOI] [PubMed] [Google Scholar]
- 22.Giuliano S, Pages G. Mechanisms of resistance to anti-angiogenesis therapies. Biochimie. 2013;95(6):1110–1119. doi: 10.1016/j.biochi.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Sessa C, Guibal A, Del Conte G, Ruegg C. Biomarkers of angiogenesis for the development of antiangiogenic therapies in oncology: tools or decorations? Nat Clin Pract Oncol. 2008;5(7):378–391. doi: 10.1038/ncponc1150. [DOI] [PubMed] [Google Scholar]
- 24.Olafsen T, Wu AM. Antibody vectors for imaging. Semin Nucl Med. 2010;40(3):167–181. doi: 10.1053/j.semnuclmed.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tolmachev V, Orlova A, Nilsson FY, Feldwisch J, Wennborg A, Abrahmsen L. Affibody molecules: potential for in vivo imaging of molecular targets for cancer therapy. Expert Opin Biol Ther. 2007;7(4):555–568. doi: 10.1517/14712598.7.4.555. [DOI] [PubMed] [Google Scholar]
- 26.Lofblom J, Feldwisch J, Tolmachev V, Carlsson J, Stahl S, Frejd FY. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584(12):2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Myers JK, Oas TG. Preorganized secondary structure as an important determinant of fast protein folding. Nat Struct Biol. 2001;8(6):552–558. doi: 10.1038/88626. [DOI] [PubMed] [Google Scholar]
- 28.Hogbom M, Eklund M, Nygren PA, Nordlund P. Structural basis for recognition by an in vitro evolved affibody. Proc Natl Acad Sci USA. 2003;100(6):3191–3196. doi: 10.1073/pnas.0436100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malm M, Bass T, Gudmundsdotter L, Lord M, Frejd FY, Stahl S, Lofblom J. Engineering of a bispecific affibody molecule towards HER2 and HER3 by addition of an albumin-binding domain allows for affinity purification and in vivo half-life extension. Biotechnol J. 2014;9(9):1215–1222. doi: 10.1002/biot.201400009. [DOI] [PubMed] [Google Scholar]
- 30.Fleetwood F, Klint S, Hanze M, Gunneriusson E, Frejd FY, Stahl S, Lofblom J. Simultaneous targeting of two ligand-binding sites on VEGFR2 using biparatopic Affibody molecules results in dramatically improved affinity. Sci Rep. 2014;4:7518. doi: 10.1038/srep07518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kronqvist N, Lofblom J, Jonsson A, Wernerus H, Stahl S. A novel affinity protein selection system based on staphylococcal cell surface display and flow cytometry. Protein Eng Des Sel. 2008;21(4):247–255. doi: 10.1093/protein/gzm090. [DOI] [PubMed] [Google Scholar]
- 32.Kronqvist N, Malm M, Rockberg J, Hjelm B, Uhlen M, Stahl S, Lofblom J. Staphylococcal surface display in combinatorial protein engineering and epitope mapping of antibodies. Recent Pat Biotechnol. 2010;4(3):171–182. doi: 10.2174/187220810793611536. [DOI] [PubMed] [Google Scholar]
- 33.Fleetwood F, Devoogdt N, Pellis M, Wernery U, Muyldermans S, Stahl S, Lofblom J. Surface display of a single-domain antibody library on Gram-positive bacteria. Cell Mol Life Sci. 2013;70(6):1081–1093. doi: 10.1007/s00018-012-1179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonsson A, Dogan J, Herne N, Abrahmsen L, Nygren PA. Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng Des Sel. 2008;21(8):515–527. doi: 10.1093/protein/gzn028. [DOI] [PubMed] [Google Scholar]
- 35.Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22(6):868–876. doi: 10.1016/j.copbio.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Orlova A, Jonsson A, Rosik D, Lundqvist H, Lindborg M, Abrahmsen L, Ekblad C, Frejd FY, Tolmachev V. Site-specific radiometal labeling and improved biodistribution using ABY-027, a novel HER2-targeting affibody molecule-albumin-binding domain fusion protein. J Nucl Med. 2013;54(6):961–968. doi: 10.2967/jnumed.112.110700. [DOI] [PubMed] [Google Scholar]
- 37.Andersen JT, Pehrson R, Tolmachev V, Daba MB, Abrahmsen L, Ekblad C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J Biol Chem. 2011;286(7):5234–5241. doi: 10.1074/jbc.M110.164848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larrivee B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell. 2012;22(3):489–500. doi: 10.1016/j.devcel.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 40.Davis BM, Normando EM, Guo L, Turner LA, Nizari S, O’Shea P, Moss SE, Somavarapu S, Cordeiro MF. Topical delivery of Avastin to the posterior segment of the eye in vivo using annexin A5-associated liposomes. Small. 2014;10(8):1575–1584. doi: 10.1002/smll.201303433. [DOI] [PubMed] [Google Scholar]
- 41.Tolmachev V, Orlova A, Pehrson R, Galli J, Baastrup B, Andersson K, Sandström M, Rosik D, Carlsson J, Lundqvist H, Wennborg A, Nilsson FY. Radionuclide therapy of HER2-positive microxenografts using a 177Lu-labeled HER2-specific Affibody molecule. Cancer Res. 2007;67(6):2773–2782. doi: 10.1158/0008-5472.CAN-06-1630. [DOI] [PubMed] [Google Scholar]