

Abstract
Upon massive DNA damage cells fail to undergo productive DNA repair and trigger the cell death response. Resistance to cell death is linked to cellular transformation and carcinogenesis as well as radio- and chemoresistance, making the underlying signaling pathways a promising target for therapeutic intervention. Diverse DNA damage-induced cell death pathways are operative in mammalian cells and finally culminate in the induction of programmed cell death via activation of apoptosis or necroptosis. These signaling routes affect nuclear, mitochondria- and plasma membrane-associated key molecules to activate the apoptotic or necroptotic response. In this review, we highlight the main signaling pathways, molecular players and mechanisms guiding the DNA damage-induced cell death response.
Keywords: Adriamycin, ATM, ATR, Axin, c-Abl, BAX, Cancer therapy, Caspase 2, Chk1, Chk2, Doxorubicin, FOXO3, HIPK2, Hippo pathway, Ionizing radiation, Yap1, MLKL, p53, p53 Serine 46, p73, PML, PIDD, Pin1, PUMA, RIPK1, RIPK3, SUMO, XAF1
Introduction
The cells of an organism are constantly exposed to DNA damage caused by various intrinsic and extrinsic sources. Metabolic by-products such as reactive oxygen species (ROS) stemming from the mitochondrial respiratory chain are presumably the most frequent intrinsic source of DNA damage. Naturally occurring DNA damage counts for up to 200,000 lesions per cell per day [1]. Extrinsic or environmental sources of DNA damage include ultraviolet (UV) light, ionizing radiation (IR) and exposure to genotoxic agents such as chemotherapeutic drugs. For example, UV light from the sun can induce up to 100,000 DNA lesions per cell per day (mainly pyrimidine dimers and 6–4 photoproducts) [2, 3]. Given the constant challenge of DNA lesions, cells had to evolve a molecular toolbox to appropriately handle the damage. This toolbox has been coined DNA damage response (DDR) and is a complex signaling network composed of versatile sensing, signal transduction and execution systems. Mildly damaged cells may repair the DNA lesions and recover. However, if the damage is irreparable, cells can ultimately trigger the cell death response to eliminate damaged, potentially threatening cells. To do so, mammalian cells have evolved a number of signaling pathways and mechanisms to engage the DNA damage-induced cell death response. Before we will turn to molecular players, pathways and mechanisms of the DNA damage-induced cell death response, we need to set the stage and to briefly discuss the physiological and pathophysiological impact of the DDR, which is linked to balanced or rather unbalanced DNA damage-induced cell death.
Physiology/pathophysiology of the DNA damage response: cancer, aging, immunodeficiency and neurodegeneration
In the light of the high numbers of DNA lesions which accumulate in a cell per day [3], it is not surprising that the deregulation of the DDR is linked to pathophysiology and disease. Indeed, an impaired DDR is associated with a number of clinically relevant diseases including premature aging, tissue dysfunction, immunodeficiency, neurodegenerative disorders and cancer formation (Fig. 1) [4–6]. In this context a well-balanced cell death response system is of fundamental importance. Whereas neurodegeneration and immunodeficiency are linked to an increased, unscheduled death of neurons or lymphocytes, respectively, cancer has been interconnected to a defective cell death response and to survival of DNA-damaged cells that display genomic instability.
Fig. 1.

Role of the DNA damage response in physiology and pathophysiology. The cells in our body are constantly damaged by various intrinsic and extrinsic sources. To repair the caused DNA damage, the cells have evolved a sophisticated repair mechanism termed the DNA damage response (DDR). Proper DNA repair is essential for genome stability, prevention of transformation and tumor suppression. This physiological operating DDR is opposed by a defective DNA damage response. If the DDR is not functioning correctly, the DNA damage cannot be resolved and leads to severe phenotypes, such as cancer formation, neurodegenerative diseases and premature aging
Mutations or genetic deletions of central regulators of the DDR, such as the DNA damage checkpoint kinases ATM and ATR, cause complex disorders and disease syndromes in mice and man [4]. Many of these syndromes display phenotypes of premature aging, neurological abnormalities, growth defects, immunological defects and increased cancer susceptibility—highlighting the significance of the DDR in physiology and pathophysiology [4, 7]. As a consequence, tissue-specific modulation of the DDR appears to be a promising approach for the treatment of these diseases, which are a major problem in our aging societies.
In general, the DDR lies at the heart of cancer biology. This is illustrated by a major dichotomy: on the one hand, DNA damage is the main driving force towards genomic instability and thus plays a causative role in carcinogenesis; on the other hand, targeted local (radiotherapy) or systemic (chemotherapy) induction of DNA damage is currently the most widely exploited and effective therapeutic principle in cancer treatment. Concerning development of cancer, it has been demonstrated that the DDR is inactivated in early pre-cancerous lesions, illustrating a critical barrier function in counteracting cellular transformation and the development of neoplastic lesions [8]. If DNA damage is not correctly repaired or stays unrepaired, the resulting DNA scars can harbor mutations or may lead to chromosomal aberrations including deletions and insertions, which can foster genomic instability, cellular transformation and ultimately cancer development.
In regard to cancer treatment, a large number of chemotherapeutic drugs used as first-line therapeutics, such as Adriamycin/Doxorubicin, Etoposide, Camptothecin, Bleomycin and Cisplatin, are well-known inducers of DNA damage and potently activate the DDR [6]. The desired biological response upon administration of these DNA-damaging therapies is the activation of apoptosis and the destruction of the cancer cells. However, sustained induction of the apoptotic response during therapy is frequently limited by cancer cell resistance to DNA-damaging therapy. In addition, adverse toxicity to healthy tissue is observed with numerous chemotherapeutic drugs. For example, major collateral damage includes nephrotoxicity and neurotoxicity for platinum anticancer drugs and cardiotoxicity for Doxorubicin/Adriamycin [9, 10]. To optimize cancer treatment options and to overcome cancer cell resistance and toxic side effects, it is of clinical relevance to understand the reasons underlying both cancer cell resistance to therapy and tissue-specific toxicity. For this purpose, detailed and in-depth knowledge of the signaling pathways, the molecules involved, potential cross-talk between the different pathways and the underlying molecular mechanisms of the DNA damage-induced cell death response is mandatory.
In this review, we aim at providing an overview on the most prominent pathways, molecular players and mechanisms guiding the DNA damage-induced cell death response. For a more detailed introduction into the DNA damage response and DNA repair, we recommend some recent reviews on this topic [11–13].
The DNA damage response
To set the stage for explaining and discussing the pathways and mechanisms cells use to activate the cell death response after DNA damage, we first need to provide a short introduction into the DDR and the principles of how cells sense and respond to DNA damage.
The cells of our body are continuously confronted with a large number of DNA lesions, which are generated by intrinsic (e.g., reactive oxygen species) and extrinsic sources (e.g., by ionizing radiation, ultraviolet radiation). In addition to DNA damage-induced lesions, the cells have to deal with spontaneous lesions, such as AP (apurinic/apyrimidinic) sites or the deamination of bases. To keep our genome stable and secure cellular homeostasis, it is essential for the cells to counteract DNA damage by activating the DDR which finally coordinates cell fate decision making. The DDR in damaged somatic cells can lead to cellular responses like cell cycle arrest, DNA repair, cellular senescence or apoptosis (Fig. 2). For adult stem cells, an additional cell fate option has been observed, which is the activation of a terminal differentiation program [14]. While the cellular senescence response permanently arrests the cell cycle of the damaged cell and thus blocks proliferation, the cell death response facilitates destruction and removal of the damaged cell. Since senescent cells are not passive but actively secrete pro-inflammatory cytokines, and since cellular senescence can be overrun [15], senescent cells pose a constant source of danger to the organism. Therefore, induction of cell death appears to be the safest response to finally inactivate severely damaged cells.
Fig. 2.

DNA damage-induced cell fate decisions. DNA damage is directly linked to the fate of a cell. It is essential for the proper function and survival of an organism to counteract and clear the caused damage. Therefore, the DNA damage response (DDR) lies at the center of the cell fate decision making process. In the course of the DDR, the DNA damage is sensed and its severity classified. The DDR distinguishes between repairable and non-repairable damage and coordinates different cellular outcomes like transient cell cycle arrest and DNA repair, cell death, senescence or differentiation
The cellular DDR is a hierarchically organized response system in the shape of a kinase cascade which can be divided into three different steps: (a) DNA damage sensing, (b) DNA damage signaling and (c) activation of an appropriate cellular response.
DNA damage is sensed by specialized, multimeric sensor protein complexes, which are directly recruited to the site of the DNA lesion. Since DNA damage comes in different flavors, such as DNA double-strand breaks (DSBs), single-strand breaks (SSBs) or DNA adducts by base alkylation, various sensor complexes have evolved to facilitate the recognition of these qualitative different lesions (Fig. 3). All these sensor complexes recruit and activate master DNA damage-responsive Ser/Thr kinases, which belong to the family of PI3 (phosphatidylinositide 3-kinases)-like kinases and are placed at the apex of the DNA damage signaling cascade. DSBs are sensed by the heterotrimeric Mre11/Rad50/NBS1 (MRN) complex [16]. The MRN complex is recruited to DSBs and serves as an activation platform for the DNA damage checkpoint kinase ATM (Ataxia telangiectasia mutated). The inactive ATM dimers get recruited to DSBs, undergo auto-phosphorylation at Ser residue 1981 and convert to active monomers [7]. In turn, ATM phosphorylates histone variant H2AX at Ser residue 139 (γH2AX) in the proximal region to the DNA break. Phosphorylation of H2AX allows the DNA damage signal to spread along the chromatin. γH2AX directly binds DNA damage mediators such as MDC1 (Mediator of DNA damage Checkpoint protein 1) and NBS1 (also termed Nibrin) which further enhance ATM binding and increase H2AX phosphorylation in a positive feedback loop. Activated ATM phosphorylates a huge set of cellular substrates and thereby spreads the damage signal into numerous cellular pathways and processes—shifting the damaged cell into an alarm mode [17].
Fig. 3.

DNA damage recognition and DNA damage checkpoint kinase activation. Different types of DNA damage can be caused after genotoxic stress, such as a DNA double-strand breaks (DSBs), b single-strand breaks (SSBs) or c DNA adducts by base alkylation. Dependent on the type, the DNA damage is recognized by different sensor protein complexes that initiate the activation of the DNA damage response and recruit the apical kinases ATM, ATR and DNA-PKcs. These proteins are all members of the PI3K-like protein kinase family. The Mre11-Rad50-Nbs1 (MRN) sensor complex and the Ku70-Ku80 heterodimer recognize double-strand breaks and recruit the apical kinases ATM or DNA-PKcs. Single-strand breaks are recognized by the RPA–ATRIP complex, which recruits the checkpoint kinase ATR. First, RPA binds ATRIP and the 9-1-1 checkpoint clamp is loaded at the 5′ primer junction. Finally, TOPBP1 is recruited and activates ATR. ATR can also be recruited to sites of DNA adducts by base alkylation. A direct interaction between ATR–ATRIP and MutS–MutL can promote the activation of ATR independently of RPA and single-stranded DNA breaks
ATM plays a major role in coordinating DNA repair and it has been shown that ATM is directly involved in DSB repair by homologous recombination (HR) [18]. ATM phosphorylates and removes its substrate protein KAP1 (KRAB-associated protein-1) from the heterochromatin environment of the DNA lesion and enables the recruitment of HR proteins such as MRE11 and CtIP (CtBP-interacting protein) [18, 19]. In turn, 3′ single-stranded nucleotide overhangs are created, RPA (replication protein A) is loaded and the RAD51 nucleofilament is formed [18]. ATM is not only involved in the initiation of HR, but also at later time points after the formation of the RAD51 nucleofilament [20]. Thus, ATM plays a major role in DNA repair by facilitating HR, a highly accurate way to repair DSBs that uses the intact sister chromatid as a template. For readers interested in a more detailed description of ATM in DNA repair coordination, we recommend a number of recent reviews [11, 12].
DSBs also activate another DNA damage-responsive kinase, the catalytic subunit (cs) of DNA-PK (DNA-activated protein kinase). DNA-PKCS is recruited to sites of DNA breaks by the Ku70/Ku80 sensor complex, which binds free DNA ends, and is subsequently activated. DNA-PK plays a critical role in coordinating a highly efficient and fast mechanism to repair DSBs, the so-called non-homologous end-joining (NHEJ) pathway. In contrast to the HR pathway, NHEJ is more error-prone since the ends of the DNA breaks are directly ligated without the need for a homologous template. More details on the function of DNA-PK can be obtained in a recent review [21].
When cells encounter SSBs or replication errors, a third kinase belonging to the same family as ATM and DNA-PK is activated, termed ATR (ATM and Rad3-related). ATR activation includes site-specific autophosphorylation and requires the adaptor protein ATRIP, which recruits ATR to single-stranded DNA (ssDNA) [22]. First, the ssDNA is detected by RPA (replication protein A). In a second step, ATRIP directly binds to RPA-coated ssDNA and consequently recruits ATR to sites of DNA damage. However, the localization of the ATR–ATRIP complex to sites of DNA damage is not sufficient to fully activate ATR. The kinase activity of ATR has to be activated by TopBP1 (topoisomerase II binding protein 1). Subsequently, active ATR triggers a cell cycle checkpoint by phosphorylation and activation of Chk1 (Checkpoint kinase 1). This results in an arrest of the cells at the G2/M phase by inhibiting the CyclinB-CDK1 activity via the Cdc25 phosphatase [22]. In consequence, the arrested cells have time to repair the damage.
Although ATR is mainly activated in response to replication stress and single-strand breaks, it gets also activated in response to IR damage, which predominantly induces DSBs [23]. Thus, IR activates both ATM and ATR, and activation of ATM occurs irrespective of the cell cycle phase, whereas ATR is mainly activated in S and G2 phase [23]. Cross-talk between both kinases occurs during 5′-end resection, and ATM is required for the recruitment of ATR- to RPA-coated single-stranded overhangs [23, 24]. In the classical model, the apical master kinases ATM and ATR act on different types of DNA breaks: ATM is activated in response to double-strand breaks, whereas ATR acts in response to single-strand breaks. However, several studies demonstrate that there is cross-talk between the pathways and that both ATM and ATR are involved in repair of DSBs.
Taken together, different types of DNA damage get sensed by specific sensor complexes and result in the activation of the DDR master kinases ATM, DNA-PK and ATR, respectively. The signal is transduced further downstream by a kinase cascade. Activation of ATM results in the activation of the downstream kinase Chk2 (checkpoint protein 2), and ATR results in the activation of Chk1 (checkpoint protein 1). In turn, these kinases stabilize effector proteins like p53 or Cdc25 by phosphorylation. Finally, target genes which coordinate cell cycle arrest, DNA repair, apoptosis and senescence are activated.
DNA damage-induced programmed cell death: apoptosis vs. necroptosis
Apoptosis
In general, cells can trigger apoptosis and necroptosis in response to DNA damage, but apoptosis appears to be the prevailing form of cell death. Apoptotic cell death can not only occur during normal developmental and maintenance processes of cell populations, but also as a cellular response to DNA damage. As already mentioned, the pathway is strongly regulated. The key molecules of the apoptotic pathway will be described in the following sections.
A characteristic of apoptotic signaling pathways is the activation of caspases. Caspases are aspartate-specific cysteine proteases with mainly two functions: the processing and activation of pro-inflammatory cytokines and the cleavage of various proteins during apoptosis. Caspases which are involved in apoptosis can be divided into initiator caspases (Caspase-2, -8, -9 and -10) and effector caspases (Caspase-3, -6 and -7). Within a cell, all caspases are present as inactive zymogens. The procaspases are activated through proteolytic cleavage after a specific internal aspartate residue. The initiator caspases are activated through autocatalytic interchain cleavage at high molecular weight adaptor complexes such as the apoptosome, the death-inducing signaling complex (DISC) or the PIDDosome [25–27].
Mitochondria play a critical role in apoptosis execution and intrinsic stimuli such as metabolic, replicative or genotoxic stress result in the induction of the intrinsic, mitochondrial apoptotic pathway. The key initiators of the intrinsic pathway belong to the B cell lymphoma-2 (BCL-2) family of proteins such as PUMA, BAX and NOXA. Members of the BCL-2 family of proteins contain one to four BCL-2 homology (BH) domains and are able to regulate the release of mitochondrial cytochrome c. There are three subfamilies: the anti-apoptotic BCL-2 subfamily, the pro-apoptotic BAX-like subfamily and the BH3-only subfamily. The members of the pro-apoptotic BAX-like subfamily contain three BH domains (BH1, BH2 and BH3) and promote apoptosis by the formation of pores in the mitochondrial outer membrane. BAX, BAK and BOK belong to the aforementioned subfamily. The pro-apoptotic BH3-only subfamily is more diverse and just contains one BH3 domain. It has eight members: BID, BAM, BIM, BIK, BMF, NOXA, PUMA and HRK. The most prominent members are NOXA, PUMA and BID, which are transcriptionally upregulated by p53 following DNA damage [28, 29].
Exposure to stress results in the induction of BH3-only proteins which in turn neutralize the pro-survival proteins of the BCL-2 subfamily. The BH3-only proteins can bind to the pro-survival BCL-2 family members via their BH3 domains with high affinity. Individual BH3-only proteins have varying affinities for different pro-survival proteins. Whereas BIM, tBID (the cleaved, active form of BID) and PUMA can bind to all pro-survival proteins, BAD and NOXA are more selective. BAD only binds to BCL-2, BCL-XL and BCL-W with high affinity, and NOXA only to MCL1 and A1. Efficient apoptosis requires the neutralization of all pro-survival BCL-2 family members within a cell.
The activation of the pro-apoptotic BAX and BAK is still not fully understood. The ‘direct activation’ model suggests that certain BH3-only proteins such as BIM, tBID and PUMA bind to BAX and BAK transiently, thereby triggering their conformational change and subsequent oligomerization. In contrast to this ‘hit and run’ model, the ‘indirect activation’ model suggests that the BH3-only proteins simply bind to the pro-survival BCL-2 members and prevent them from inactivating BAX or BAK. Activated BAX and/or BAK oligomerize and form pores in the mitochondrial outer membrane which leads to its permeabilization. The mitochondrial outer membrane permeabilization (MOMP) enables the release of cytochrome c and the pro-apoptotic SMAC/DIABLO from the mitochondrial intermembrane space into the cytosol. Release of cytochrome c from mitochondria after MOMP triggers in concert with APAF-1, Caspase-9 and ATP the formation of a macromolecular complex, the so-called apoptosome. The initiator Caspase-9 gets activated, which in turn cleaves, processes and thereby activates the executioner caspases: Caspase-3, -6 and -7 [30, 31]. In addition, SMAC/DIABLO, which is released from the inner mitochondrial membrane space, contributes to Caspase-9 activation by inhibiting IAPs (inhibitors of apoptosis), which function as Caspase-9 antagonists [30].
In contrast to the intrinsic apoptotic pathway, the extrinsic pathway is receptor mediated and involves death receptors that belong to the tumor necrosis factor receptor (TNFR) superfamily [32]. Members of the TNF receptor family are characterized by cysteine-rich extracellular domains. The death receptors additionally share a cytoplasmic ‘death domain’ (DD) which is important for the transmission of the death signal from the cell surface to the intracellular signaling pathways. The best known death receptors are CD95 (APO1/Fas), TNF receptor 1 (TNF-R1, CD120a), TNF-R2 (CD120b), TNF-related apoptosis-inducing ligand-receptor 1 (TRAIL-R1/DR4) and TRAIL-R2 (DR5), DR3 (TRAMP/APO3/WSL-1/LARD) or DR6. After binding of death ligands such as CD95L (APO1L/FASL) or APO2L/TRAIL, the death receptors assemble a so-called death-inducing signaling complex (DISC). Ligand binding results in receptor trimerization and clustering of the cytoplasmic death domains. The trimeric receptors serve as a docking platform and recruit adaptor molecules such as Fas-associated protein with death domains (FADD) for CD95 or TNF receptor-associated protein with death domains (TRADD) for TNF-R1/TNF-R2. The assembled complex provides a scaffold for the recruitment of the procaspases-8 or -10. The procaspases are recruited to the activated receptor to form the DISC. The procaspases oligomerize at the DISC and are activated by self-cleavage. Subsequently, Caspase-8 or Caspase-10 activates downstream effector caspases such as Caspase-3. Caspase-3 in turn activates Caspases-6 and -7 to initiate apoptosis. Members of this receptor family, in particular TNF-R1, do not exclusively transmit an apoptotic signal, but also inflammatory ones through activation of the transcription factors NF-κB and AP-1. Interestingly, under specific conditions death receptors can also activate programmed necrosis, termed necroptosis [33].
Necroptosis
Necroptosis is a form of regulated necrosis. Its morphological phenotype resembles that of necrosis and is characterized by plasma membrane rupture, cell swelling and spill-out of the cellular content in the extracellular space [31, 34, 35]. The term necroptosis (a mixture of necrosis and apoptosis) has been coined by Vandenabeele et al., to differentiate between the process of unregulated necrosis and the ordered, programmed form of necrosis [36]. In contrast to apoptosis, necroptotic cell death is characterized by the lack of caspase activation and the requirement of RIPK1 and RIPK3 (receptor-interacting protein kinase 1 and 3) [37, 38]. Necroptosis can be activated by death receptor ligands, such as TNF-α, the death ligand FasL or Apo2L/TRAIL [37, 39]. To trigger necroptosis in human cells under laboratory conditions, caspase activation, and hence apoptosis—needs to be blocked. Accordingly, one needs to apply an inhibitor of apoptosis (IAP) antagonist (SMAC/DIABLO mimetic) and the pan-caspase inhibitor zVAD to block the apoptotic death receptor pathway [38].
Since the classical necroptosis model system is the ubiquitously expressed cytokine receptor TNFR1 (TNF receptor 1), the necroptotic pathway will be exemplarily explained for that receptor (Fig. 4). Upon stimulation of the TNFR1 by TNF, a receptor-bound macromolecular complex is formed, termed complex I. TRADD (TNF receptor-associated death domain) gets recruited to TNFR1 via its death domain (DD), and in turn recruits RIPK1, cIAP1, cIAP2 (cellular inhibitor of apoptosis protein 1, 2) and TRAF2 and TRAF5 (TNF receptor-associated factor 2 and 5). In a further step, RIPK1 is poly-ubiquitinated by the E3 ligases of the cIAP family [40]. This post-translational modification of RIPK1 allows for the recruitment of TAK1 (transforming growth factor-β-activated kinase 1) in complex with TAB 2 or TAB 3 (TAK1 binding protein 2 or 3), which leads to the assembly of the IKK (inhibitor of NF-κB kinase) complex and subsequent activation of the NF-κB pathway [41]. NF-κB finally triggers the expression of pro-inflammatory cytokines. Consecutively, the Lys63-linked polyubiquitination is removed from RIPK1 via CYLD (cylindromatosis) and complex I is destabilized [42]. RIPK1 can dissociate from the complex and interact with TRADD, FADD (Fas-associated death domain), pro-caspase-8 and FLIP (FLICE-like inhibitory protein) to form the so-called complex II [43]. The long isoforms of FLIP, FLIPL, and pro-caspase-8 form a heterodimeric caspase which cleaves RIPK1, RIPK3 and CYLD. This leads to necroptosis inhibition and cell death via the apoptotic pathway. A similar complex can also form in the absence of cIAPs. When cIAPs are blocked, RIPK1 does not get poly-ubiquitinated and NF-κB is activated by a non-canonical pathway via the upregulation of NIK (NF-κB-inducing kinase) [44, 45]. In the absence of cIAPS, a complex II without TRADD can form. This complex is called Ripoptosome and consists of RIPK1, RIP3, FADD, FLIPL and pro-Caspase-8 [43]. As before, RIPK1 and RIPK3 get inactivated and apoptosis is induced by caspase-8. However, if Caspase-8 is inhibited and kept inactive, the RHIM domains (RIP homotypic interaction motif) of RIPK1 and RIPK3 associate in a microfilament-like structures termed necrosome [36]. Subsequently, programmed cell death by necroptosis is initiated via the trans- and auto-phosphorylation of RIPK1 and RIPK3 and the recruitment of MLKL (mixed lineage kinase domain-like) [38].
Fig. 4.

DNA damage-induced necroptosis signaling. The necroptotic response is induced by death receptors. The classical necroptosis death receptor is TNFR1 which gets stimulated by TNF and forms a large macromolecular complex with TRADD, RIPK1, cIAP2, cIAP5, TRAF2 and TRAF5. This complex is also referred to as complex I. The essential necroptotic protein RIPK1 gets poly-ubiquitinated by cIAPs and leads to TAK1–TAB 2–TAB 3 complex formation. This leads to the assembly of IKK and canonical NF-κB activation. In a further step, RIPK1 gets de-ubiquitinated and complex I gets destabilized. Subsequently, RIPK1 can form a complex together with TRADD, FADD, cFLIP and pro-caspase-8 (complex II). RIPK1 gets de-activated and apoptosis is induced via caspase-8. In the absence of cIAPs, RIPK1 cannot get de-ubiquitinated. The absence of cIAPS leads to a non-canonical activation of the NF-κB pathway via NIK. Together with FADD, cFLIP and pro-caspase-8, RIPK1 can still form a complex II. This complex is called Ripoptosome and leads to apoptosis via activation of caspase-8. However, if caspase-8 is blocked, RIPK1 and RIPK3 interact and associate in microfilament-like structures, the so-called necrosome. RIPK1 and RIPK3 get activated via trans- and auto-phosphorylation, which finally leads to MLKL recruitment and necroptosis. A RIPK3-dependent necroptosis pathway is also activated in response to treatment with DNA-damaging chemotherapeutic drugs (as Doxorubicin). The exact mechanisms of signal transduction involved are currently unclear
The process of necroptosis relies on the activation of the Ser/Thr protein kinase RIPK3, which phosphorylates the pseudokinase MLKL at several residues (human: Thr357 and Ser358; mouse: Ser345, Ser347, Thr349) and thereby facilitates its plasma membrane targeting, membrane rupture, cell swelling and death [46–48]. In contrast to apoptosis, mitochondria do not contribute to the execution of necroptosis. The necroptotic activity of RIPK3 can be activated by three different upstream activators—either by the aforementioned RIPK1, by DAI (DNA-dependent activator of interferon) or TRIF (toll-like receptor-interacting factor). All of these proteins contain the RHIM domain [38, 49, 50]. Necroptosis induces a strong inflammatory response due to the spill-out of the cell content, whereas apoptosis usually does not. The cell content of apoptotic cells is kept engulfed by the plasma membrane in so-called apoptotic bodies, which are cleared by phagocytosis [31, 51]. Mice with knockout in RIPK3 or MLKL are viable and develop normally, but show defects in clearing certain viral infections. This is in line with the view that necroptosis is not relevant during development but is critical for pathogen defense.
Interestingly, necroptosis is also induced upon treatment with chemotherapeutic drugs [52]. How cells decide between both modes of cell death in response to DNA damage remains largely unclear. However, the essential necroptosis activator RIPK3 appears to be frequently lost in cancer cell [52], which prevents necroptosis induction. We will discuss this interesting finding in the section on "DNA damage-induced necroptosis" latter in our review.SS
Molecular players and pathways of the DNA damage-induced cell death response
In principle cells can respond to DNA damage using different strategies including DNA repair, induction of cellular senescence or cell death. Both apoptosis and necroptosis are ultimate cellular decisions, which result in the physical loss of the damaged cells. Because of the irreversibility of these processes, it is reasonable that such cellular decisions are tightly controlled to prevent a premature activation, which could lead to loss of homeostasis, tissue damage or even organ failure and death. On the other hand, the decision needs to be that robust to avoid frequent bypass of damaged cells, which would increase the risk of genomic instability and cancer. In the following section we will describe the main pathways and mechanisms that have evolved to switch on the cell death response upon DNA damage. Cells have the choice between different signaling pathways to induce the cell death program in response to severe DNA damage. The cellular origin (cell specificity) plays at least partially a role in the decision which pathway is finally selected. However, it still remains unclear whether these pathways act in parallel and to which extent cross-talk occurs between them.
In response to DNA damage a set of apoptotic signaling routes can be stimulated, including the ATM/ATR–HIPK2–p53, the ATM–PIDD–Caspase-2, the ATM-c-ABL–Yap1–p73 and c-Abl–HIPK2–p53 signaling axes. In addition, DNA damage has also been linked to the RIPK3–MLKL pathway of necroptosis. In the following we will discuss the most prominent signaling pathways in more detail. For readers interested in more details of how specific DNA lesions and repair mechanisms are coupled to the DNA damage response and cell death machinery we recommend a recent review [53].
The tumor suppressor p53
The most prominent regulator of DNA damage-induced cell death is presumably p53, which is linked to numerous cell death pathways (Figs. 5, 6). It is a well-known tumor suppressor frequently mutated or deleted in human cancers [54, 55]. p53 is activated in response to a wide range of cellular stress, including DNA damage—and acts as transcription factor, which binds to a broad set of target gene promoters (for detailed review see [56]). p53 binds to promoter regions of its target genes via so-called response elements (REs). However, the affinity of p53 for its REs differs between target genes. Interestingly, it has been found that the transactivation potential of p53 is higher towards cell cycle arrest genes than towards pro-apoptotic genes [57]. p53 target genes that are involved in cell cycle arrest contain robust REs, which are frequently evolutionary conserved [58]. In contrast, p53 REs of pro-apoptotic genes like BAX, CD95/Fas and p53AIP1 are less conserved across species and even appear to be primate specific, indicating that the p53 response has substantially changed during evolution [59]. Comparison between p53 REs of cell cycle arrest and pro-apoptotic genes by a computational approach revealed that REs of cell cycle arrest genes are exposed on the nucleosomal surface whereas many sites of pro-apoptotic genes are found buried inside the nucleosome [60]. This suggests that REs of cell cycle arrest genes are conserved and easily accessible for p53, while most REs of pro-apoptotic target genes are less conserved and less accessible for p53 binding.
Fig. 5.
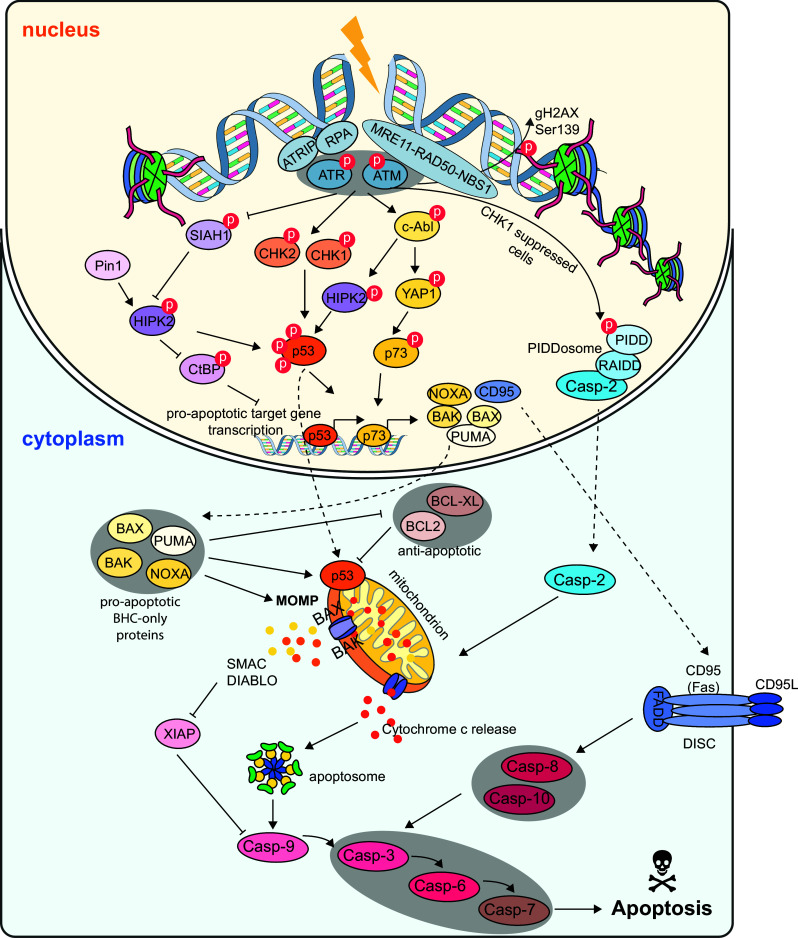
DNA damage-induced apoptosis signaling pathways. After genotoxic stress, the DNA damage response gets activated. Cells can sense sites of DNA breaks by sensor protein complexes (Mre11–Rad50–Nbs1 and RPA–ATRIP) which recruit and activate the checkpoint kinases ATM and ATR. The apical kinases get activated by phosphorylation and the signal is transduced further downstream by a kinase cascade including Chk1, Chk2, HIPK2 and cAbl-YAP1. These kinases stabilize the tumor suppressors p53 and p73 by phosphorylation. Finally, p53/p73 target genes which coordinate DNA repair, apoptosis and senescence are transcribed. Amongst these target genes are the pro-apoptotic BHC-only proteins BAX, BAK, PUMA and NOXA. They get activated and translocated from the nucleus to the cytoplasm. Once in the cytoplasm, the pro-apoptotic BHC-only proteins inhibit the anti-apoptotic family members BCL2 and BCL-XL. Furthermore, BAX and BAK oligomerize at the mitochondrial outer membrane and form pores which lead to mitochondrial outer membrane permeabilization (MOMP) and subsequent cytochrome c and SMAC/DIABLO release. These events trigger the formation of the so-called apoptosome, a platform for the activation of pro-caspase 9. Caspase-9 in turn activates the executioner caspases Caspase 3, 6 and 7 which finally leads to apoptosis. Another platform for caspase activation can be directly activated by the checkpoint kinase ATM. ATM phosphorylates PIDD which can bind to RAIDD and pro-caspase-2 to form the so-called PIDDosome. The activated caspase-2 subsequently signals to the mitochondrion. The mitochondrial pathways leading to apoptosis are also termed the intrinsic pathway. A third possibility for caspase activation is via the extrinsic, receptor-mediated pathway. After the binding of death ligands to the respective death receptors (e.g., CD95/Fas or TNFR), the so-called death-inducing signaling complex (DISC) assembles. The assembled complex provides a platform for the activation of pro-Caspase-8 or -10. They get activated by self-cleavage and finally activate the effector caspases to induce apoptosis
Fig. 6.

p53 Serine 46 phosphorylation: the deadly phosphorylation mark on p53. Upon DNA damage, a kinase cascade is activated which modifies p53 via phosphorylation of several serine residues. These post-translational modifications finally lead to p53-dependent pro-apoptotic target gene transcription. P53 is modified by the checkpoint kinases ATM, ATR and HIPK2. The apical kinases ATM and ATR are recruited to sites of DNA breaks and get activated via phosphorylation. Both ATM and ATR can directly phosphorylate p53 at Serine residue 15 and indirectly via Chk2 and Chk1 at Serine residue 20. After DNA damage, a third checkpoint kinase is activated, HIPK2. HIPK2 phosphorylates p53 at Serine residue 46 exclusively after severe, non-repairable damage. The post-translational modification of Serine 46 thus serves as an pro-apoptotic mark on p53. Subsequently, the pro-apoptotic p53 target genes BAX, PUMA, NOXA and p53AIP1 are transcribed. These pro-apoptotic factors finally activate the cell death pathway via the mitochondrial, intrinsic pathway. However, p53 can also act in a transcription-independent mode. HIPK2-mediated phosphorylation of Serine residue 46 initiates the binding of Pin1 to p53. Binding of Pin1 leads to the targeting of p53 to the mitochondrion, BAX activation and mitochondrial outer membrane permeabilization (MOMP). These events finally trigger the apoptosis response. p53 can activate apoptosis both in an indirect, transcription-mediated manner and directly in the cytosol at the mitochondrial membrane
The target genes which are regulated by p53 guide cell fate decision-making towards different responses including cell cycle arrest and DNA repair, cell death, autophagy and cellular senescence [61, 62]. How p53 selects between its different target genes is still not fully understood, although there is good evidence that posttranslational modifications play a key role. Beside its function as transcription factor in the cell nucleus, p53 can also act in the cytoplasm, independently of its transcriptional function—and directly stimulate the mitochondrial cell death route by activating the pro-apoptotic proteins BAK and BAX. It targets BAX to the outer mitochondrial membrane, leading to MOMP, cytochrome c release and apoptosis induction [63–65]. The cytoplasmic role of p53 appears to be in particular important in cells which show high sensitivity to p53-induced apoptosis such as lymphocytes.
DNA damage-induced activation of p53 is regulated at the level of its post-translational modifications. Normal proliferating cells have only very little p53 due to its proteasomal degradation, which is facilitated by complex formation with different E3 ubiquitin ligases, the most prominent being Mdm2, which tags p53 with ubiquitin and thereby targets it for degradation [66]. The DNA damage-responsive kinases ATM and ATR and their downstream mediators Chk2 and Chk1 phosphorylate p53 at Ser15 and Ser20, respectively [67]. This leads to dissociation of Mdm2 and results in p53 stabilization and enables the binding to its target gene promoters.
Besides its well-documented function in apoptosis induction, p53 has also been reported to initiate necrosis in response to oxidative stress by H2O2 treatment. P53 has been shown to accumulate in mitochondria and trigger the opening of the mitochondrial permeability transition pore (PTP), a water and solute channel placed at the contact points of the inner and outer mitochondrial membrane [68, 69]—by interaction with its essential regulator, the prolyl isomerase Cyclophilin D [68, 69]. Whether this activity of p53 is also regulated by its post-translational modifications remains to be studied. In the past years, the p53 pathway received a lot of attention and numerous seminal findings paved the way to our current understanding on the mechanisms by which p53 is activated/inactivated, and how it selects its different target gene sets upon genotoxic stress. To guide the DNA damage response towards cell death in damaged tumor cells, it remains of fundamental importance to reveal molecular insight in the mechanisms by which p53 discriminates between its apoptotic and non-apoptotic target genes.
p53 Ser46 phosphorylation: the deadly phospho-mark on p53
A central finding for our current understanding of the DNA damage-induced p53 response was the identification of the p53 Serine 46 phosphorylation site and its specific linkage to pro-apoptotic p53 target gene expression [70]. Remarkably, p53 Ser46 phosphorylation is strictly linked to lethal, non-repairable DNA damage and can be stimulated by different treatments such as IR, UV and by DNA-damaging chemotherapeutic drugs such as Cisplatin and Adriamycin/Doxorubicin [70–76]. p53 phosphorylation at Ser46 is found in conjunction with Ser15 and Ser20 phosphorylation of p53, since the kinases phosphorylating Ser15 (ATM, ATR) and Ser20 (Chk1, Chk2) are activated also in response to lethal damage (Fig. 6). p53 Ser46 phosphorylation apparently controls a number of apoptotic target genes including p53AIP1, p53DINP1, PUMA and Noxa [70, 77–79].
Which kinases phosphorylate p53 at the Ser46 residue? HIPK2 has been identified as the first p53 Ser46 kinase [71, 72]. Later additional kinases including DYRK2, ATM, PKCδ and AMPK were linked to p53 Ser46 phosphorylation under genotoxic and energetic stress conditions [74, 80–82]. HIPK2 is the best understood p53 Ser46 kinase and mediates this phosphorylation in response to a variety of DNA damage-inducing stimuli, including the aforementioned UV, IR, Cisplatin and Doxorubicin/Adriamycin—to trigger apoptotic target gene expression and apoptosis [83]. Along these lines, defective p53 Ser46 phosphorylation was found to account for apoptosis resistance in oral squamous cell carcinoma cells [78]. Ser46 phosphorylation of p53 is also a recognition signal for the phospho-specific prolyl isomerase Pin1, which mediates changes in p53 conformation and plays an important role in assisting transactivation of apoptotic p53 target genes [84–87].
Interestingly, p53 Ser46 phosphorylation has also been linked to the neurotoxicity of mutant huntingtin protein, which causes the fatal neurodegenerative disorder Huntington disease [88]. Inhibition of HIPK2, ATM and PKCδ prevented the apoptotic effects of mutant huntingtin in neuronal cells, indicating a pathophysiological function of this deadly phospho-mark [88].
Recent reports demonstrated that Ser46-phosphorylated p53 also acts in a transcription independent fashion in the cytosol upon DNA damage (Fig. 6). HIPK2-mediated p53 Ser46 phosphorylation initiates phosphorylation-dependent binding of the cis/trans isomerase Pin1, which potentiates mitochondrial targeting of p53 and apoptosis [89]. Moreover, Pin1-mediated isomerization of phospho-Ser46 p53 provokes BAX activation and targeting into the mitochondrial outer membrane leading to MOMP and transcription-independent apoptosis induction [90].
Regulation of the p53 Ser46 kinase HIPK2
HIPK2 is a nuclear body-associated enzyme and an important regulator of stress signaling and the DNA damage response [91–96]. The kinase is largely regulated by posttranslational modifications including SUMOylation, phosphorylation, acetylation and ubiquitination [91, 97–100]. Unstressed cells keep HIPK2 inactive through interaction and targeted proteolysis by the E3 ubiquitin ligases Siah-1, Siah-2 and WSB-1 [92, 101–103]. After DNA damage, ATM and ATR kinases phosphorylate Siah-1 and dissociate the HIPK2-Siah1 complex and thereby mediate HIPK2 stabilization (Fig. 5) [101]. Also Zyxin, a protein known to regulate cell migration, mediates HIPK2 stabilization after Doxorubicin treatment, presumably through its interaction with Siah-1 and Siah-2 [104]. Specific activation of HIPK2 in response to DNA damage requires site-specific autophosphorylation at Ser880/Thr882 [76]. This phospho-mark is recognized by the Pin1 isomerase, which changes the conformation of HIPK2 and profoundly contributes to its stabilization [76]. The apoptotic function of activated HIPK2 can be further boosted by caspase-mediated cleavage, which removes a C terminal autoinhibitory domain and stimulates its kinase activity [105].
During cellular recovery from DNA damage, HIPK2 is rapidly degraded by Siah-1 [101]. Interference with HIPK2 degradation by depletion of Siah-1 results in the induction of apoptosis in response to a sublethal UV dose. Interestingly, no increase in p53 Ser46 phosphorylation is detected under these conditions, but instead increased HIPK2-mediated CtBP degradation [101]. CtBP has been identified as an HIPK2 phospho-target, which is targeted for degradation upon Ser322 phosphorylation and triggers a p53-independent cell death route via HIPK2 upon UV damage [106].
HIPK2 is also a target of oncogenic signaling. For instance, the cell growth-inducing non-receptor Tyr kinase c-Src phosphorylates HIPK2 at numerous Tyr residues and targets HIPK2 into the cytoplasm resulting in inhibition of Doxorubicin-induced p53 Ser46 phosphorylation and apoptosis [107]. In addition, the E6 protein of human papillomavirus 23 (HPV23), a cutaneous papilloma-inducing virus, interacts with HIPK2 and interferes with p53 Ser46 phosphorylation [108]. This anti-apoptotic mechanism might be important for the proliferation-inducing capacity of this papillomavirus. Taken together, the activation of the p53 Ser46 kinase HIPK2 is tightly regulated and governed by the DNA damage checkpoint kinases ATM and ATR.
The c-Abl–HIPK2–p53 axis
Recently, HIPK2 was shown to be regulated by tyrosine phosphorylation through the non-receptor tyrosine kinase c-Abl (Fig. 5) [109]. It is already well established that c-Abl is involved in apoptotic DNA damage signaling to p73; however, any contribution of c-Abl in the p53 response remained largely unclear. It has been shown that c-Abl and HIPK2 form a complex in vitro and in vivo, which results in phosphorylation of HIPK2 at a number of Tyr residues, including Tyr360 next to its activation loop. Interestingly, the c-Abl–HIPK2 interaction is already present in the absence of DNA damage, suggesting a role for c-Abl in balancing HIPK2 steady-state levels. c-Abl increases the stability of HIPK2 by interfering with its degradation by Siah-1 [101] and finally leading to HIPK2-mediated p53 Ser46 phosphorylation and apoptosis. Pharmacological inhibition of c-Abl with STI571 (Imatinib) resulted in a profound reduction of HIPK2 stabilization and p53 Ser46 phosphorylation after IR and UV treatment [109], indicating that c-Abl is critical to activate the apoptotic function of endogenous HIPK2. To which extent the effects of c-Abl are controlled by c-Abl-mediated Tyr phosphorylation remains to be revealed in the future.
The ATM/ATR–HIPK2–p53 axis
Checkpoint kinase ATM, and also ATR, plays an essential role in DNA damage-induced HIPK2 activation (Fig. 5). In undamaged cells, HIPK2 is an unstable protein due to constant ubiquitination by the E3 ubiquitin ligase Siah-1 and consequent proteasomal degradation. In response to DNA damage-inducing treatments (IR, UV, chemotherapeutic drugs) HIPK2 protein levels increase in an ATM- and ATR-dependent fashion due to improved protein stability [73, 101]. Mechanistically, ATM and ATR can phosphorylate Siah-1 at Ser19 and thereby weaken the HIPK2–Siah-1 complex, resulting in HIPK2 stabilization. As soon as the ATM/ATR signaling is silenced (for example, by the ATM/ATR-inhibitor caffeine), HIPK2 gets rapidly destabilized, demonstrating the critical role of ATM/ATR activity. Similar, upon recovery from weak DNA damage, HIPK2 levels drop rapidly, which is in line with terminated ATM/ATR checkpoint signaling after successful DNA repair [101]. In addition, the Pin1 isomerase is also essential for HIPK2 stabilization, since it specifically binds to the activated, autophosphorylated HIPK2 isoform and mediates its stabilization through introducing a conformational change [76]. Of note, HIPK2 stabilization can be found after sublethal and lethal DNA damage likewise, but Ser46 phosphorylation occurs exclusively after lethal damage when the ATM/ATR checkpoints stay activated [101]. This argues that critical factors for the assembly of a functional HIPK2–Ser46 kinase complex are not available or excluded from the complex after sublethal damage. One such factor might be p53DINP1/TP53INP1, which is induced upon high-dose DNA damage and interacts with p53 and HIPK2 in PML-NBs [110]. However, the important question whether p53DINP1/TP53INP1 plays a direct role in HIPK2-mediated p53 Ser46 phosphorylation remains to be answered.
The HIPK2–Axin–p53 axis
Beside its well-established role in Wnt/β-catenin signaling, Axin was shown to play a role in the regulation of p53 Ser46 phosphorylation and cell death upon UV damage, as well. Axin forms a complex with p53 as well as HIPK2, and overexpressed Axin stimulates p53 Ser46 phosphorylation [111]. Somewhat unexpected, the endogenous HIPK2–Axin–p53 complex was found in the absence of any DNA damage, and it remains to be addressed how this complex may be altered in response to UV damage, when p53 Ser46 phosphorylation is evident. This suggests that the p53–Axin–HIPK2 complex is already pre-assembled in cells and presumably requires an activation signal upon UV damage to assist in HIPK2-controlled p53 phosphorylation. The later point remains to be tested. In addition, Daxx, which also binds HIPK2 [112], interacts with the p53–Axin–HIPK2 complex and takes part in p53 Ser46 phosphorylation and apoptosis activation upon UV treatment [113]. Daxx was also found to be associated with Axin in the absence of any UV treatment and the important question whether UV damage has an impact on this complex remains to be clarified. Finally, it was proposed that the HIPK2–Axin–p53 complex is kept silent by the E3 ubiquitin ligase Pirh2 in a ligase-independent adaptor mechanism by competing with HIPK2 in the complex. Upon UV or Doxorubicin treatment Tip60 is targeted in an ATM/ATR-dependent fashion and removes Pirh2 from the Axin complex and allows for the incorporation of HIPK2 and thereby p53 Ser46 phosphorylation [114]. The underlying molecular mechanisms for the exchange of Pirh2 and Tip60 and the subsequent recruitment of HIPK2 into the Axin complex are currently unclear. Likewise, it will be interesting to determine the physiological significance of the pre-formed HIPK2–Axin–p53 complexes observed in cells [111].
The ATM–Chk2–FOXO3a–p53 axis
Forkhead box-O (FOXO) transcription factors fulfill important functions in cell proliferation, cell differentiation, development and cell survival. One of the most intriguing roles of the FOXO members is probably their function in aging and life span regulation. The FOXO transcription factors are either regulated by post-translational modifications or protein–protein interactions. In a recent study, FOXO3 has been shown to be directly involved in apoptosis. It has been demonstrated that FOXO3 shuttles from the cytoplasm to the nucleus after DNA damage and gets recruited to sites of DNA breaks. At the so-called DNA damage foci, FOXO3 co-localizes with the DNA damage marker protein γH2AX, the activated kinase ATM and phosphorylated p53. Furthermore, it has been shown that FOXO3 forms a complex with ATM and regulates the autophosphorylation of ATM at Ser1981 and its activation upon DNA damage [82, 83]. FOXO3 interacts with the ATM–Chk2–p53 complex, which is essential for DNA damage-induced apoptosis induction. It is suggested that FOXO3 regulates the chromatin retention of the ATM–Chk2–p53 complex through a mechanism that is currently unknown [115]. It also remains unclear how FOXO3 itself gets recruited to the sites of DNA breaks, and whether the DNA binding domain of FOXO3 may be involved in its role at DNA damage foci—or whether this represents a transcription and DNA binding-independent FOXO3 function.
The p53–XAF1–HIPK2 axis
The X-linked inhibitor of apoptosis (XIAP)-associated factor 1 (XAF1) is involved in apoptosis signaling by both antagonizing the anti-Caspase activity and the anti-apoptotic activity of XIAP [116, 117]. In a more recent report, XAF1 has been directly linked to p53 and the apoptotic switch of p53 signaling. XAF1 has been shown to be a direct transcriptional target of p53 by chromatin immunoprecipitation assay [118]. Wild-type p53 downregulates XAF1 on both mRNA and protein level. In contrast, overexpression of XAF1 leads to the activation of wild-type p53, its nuclear accumulation and its increased transcriptional activity towards pro-apoptotic target genes—suggesting for a p53–XAF1 feedback loop. In a further report, the underlying mechanism has been deciphered in detail. XAF1 has been found to be a competitor of the E3 ubiquitin ligase MDM2 in binding to p53 [119]. However, XAF1 not only disrupts the p53–MDM2 regulatory feedback loop, but also the HIPK2-targeting function of Siah-2 which results in stabilization of HIPK2. In turn, stabilized HIPK2 promotes p53 phosphorylation at Ser46 upon DNA damage and shifts the cell fate towards apoptotic cell death [119]. Moreover, in the same study XAF1 has been described to activate the zinc finger protein 313 (ZNF313) leading to ubiquitination and degradation of the cell cycle regulator p21, and thus further shifting the cell fate towards apoptosis [119]. Together, XAF1 promotes the genotoxic stress-induced cell death response through numerous activities.
p53Ser46–AREG–miR-15
As mentioned before, the affinity for p53 for its target genes changes upon genotoxic stress, and especially the phosphorylation of p53 at Ser46 by HIPK2 or the DYRK2 kinase after a lethal dose of DNA damage shifts cell fate towards apoptosis. In a recent report, it has been shown that the post-translational modification of p53 specifically changes its target gene affinity and indirectly affects microRNA biogenesis [120]. Taira et al. could show that p53 phosphorylated at Ser46 selectively binds to the promoter region of amphiregulin (AREG) and induces its expression upon DNA damage. Consequently, AREG shuttles to the nucleus under DNA damage conditions and interacts with the DEAD-box RNA helicase p68 (DDX5). DDX5 is one of the components of the Drosha complex which is important for microRNA processing. The group could demonstrate that AREG specifically regulates the processing of pri-miR-15 and the production of miR-15 [120]. In previous studies, it was found that pre-miR-15 is increased after DNA damage and targets the anti-apoptotic BCL-2, thus promoting the apoptotic response [121, 122].
This study could show that p53 indirectly regulates miRNA production in response to DNA damage via regulation of the Drosha complex by AREG. However, there are still open questions and it is not clear, how AREG localizes into the nucleus. Moreover, miR-15 is not the only microRNA which gets regulated by p53 in response to DNA damage. Other microRNAs, such as miR-34a, get transactivated by p53 and are involved in apoptosis, as well [123, 124]. Furthermore, other key players of the DDR have been shown to be involved in microRNA biogenesis, such as ATM which phosphorylates KH-type splicing regulatory protein (KSRP), enhancing its interactions with pri-miRNAs [125]. There is evidence that microRNA metabolism is dysregulated in tumors, and that microRNA levels are decreased [126, 127]. Thus, the regulation of microRNA processing seems to be a critical step for DNA damage-induced apoptosis and tumor inhibition. For a more detailed overview on the growing cell death regulatory microRNA network some review papers to this topic are recommended [128, 129].
p53, PML nuclear bodies (PML-NBs) and DNA damage-induced cell death
PML nuclear bodies (PML-NBs), are multi-protein domains responsive to different kinds of stress including DNA damage [130]. PML-NBs play an important role in the DNA damage-induced cell death response by p53 and p73. Upon DNA damage, p53 is recruited along with HIPK2 by a specific PML isoform to PML-NBs and HIPK2 mediates p53 Ser46 phosphorylation in association with PML nuclear bodies (PML-NBs) [71, 72, 95]. Numerous additional p53 and p73 regulating enzymes including CBP, Chk2, ATM, ATR are found at PML-NBs under specific conditions [131–133] and also p53 acetylation was linked to PML-NB-associated CBP/p300 [134, 135]. Since there are numerous reviews on the role of PML-NBs in apoptosis regulation, we recommend these to those readers interested in the role of PML-NBs in apoptosis and DDR regulation in more detail [83, 131, 132, 136].
Although it is now known for quite a while that p53 regulators meet at PML-NBs and regulate p53 function, it remained until recently unclear if cross-talk between p53 regulators takes place at PML-NBs, and whether this shapes cell fate decision-making upon DNA damage. Indeed, cross-talk between the p53 activator HIPK2 and the p53-inhibiting deacetylase SIRT1 upon Adriamycin/Doxorubicin treatment was observed [137]. HIPK2 forms a complex with SIRT1 upon DNA damage and specifically phosphorylates SIRT1 at Ser682 upon lethal DNA damage, but not in response to cytostatic DNA damage. In addition, Ser682 phosphorylated SIRT1 co-localizes with HIPK2 at PML-NBs, and SIRT1 Ser682 phosphorylation upon DNA damage requires PML expression, as depletion of PML by RNA interference inhibits SIRT1 Ser682 phosphorylation. Mechanistically, Ser682 phosphorylation dissociates AROS, an activator of SIRT1, from SIRT1 and inhibits SIRT1 activity, which is reflected by increased p53 acetylation and apoptotic target gene expression [137]. Thus, SIRT1 Ser682 is, beyond p53 Ser46, a second HIPK2-mediated phospho-site linked to lethal DNA damage, and based on cross-talk of SIRT1 and HIPK2 at PML-NBs. It will be interesting to see in the future whether additional cross-talk between p53 regulatory enzymes takes place at PML-NBs.
The tumor suppressor p73
A further key molecule controlling DNA damage-induced cell death is the transcription factor p73, a protein showing a very similar modular organization to p53. Comparable to p53, p73 is an unstable protein which is expressed in a variety of isoforms, with p73α being the most widely studied [138]. In unstressed cells, p73 is degraded by the proteasome, which is facilitated by interaction with the E3 ubiquitin ligase Itch [139–141]. In response to genotoxic stress, the destabilization of Itch mediates the accumulation of p73 upon numerous kinds of genotoxic stress stimuli (IR, UV, Doxorubicin, Cisplatin) [138, 141]. p73 transactivates many apoptotic target genes which are also known to be targeted by p53—including PUMA and Noxa, and induces apoptosis through inducing MOMP [139, 140, 142]. Similar to p53, p73 activity is regulated through association with PML-NBs [143]. For a more detailed overview on p73 function and regulation, we recommend a number of recent reviews [131, 144, 145]. In the following, we will discuss the most prominent DNA damage-induced cell death pathways involving p73.
The ATM–cAbl–YAP1–p73 axis
The apoptotic activity of p73 in response to cisplatin-induced and IR-mediated DNA damage was found to be regulated by interaction with and phosphorylation by c-Abl kinase (Fig. 5) [146, 147]. In absence of DNA damage, c-Abl is retained in the cytoplasm through interaction with 14-3-3ζ protein. Upon Doxorubicin treatment, JNK (c-Jun N-terminal kinase) phosphorylates 14-3-3ζ, and thereby triggers the release and nuclear translocation of c-Abl [148]. In the cell nucleus the non-receptor tyrosine kinase c-Abl is activated upon DNA damage directly by the ATM checkpoint kinase, which phosphorylates c-Abl at Ser465 [149, 150]. Posttranslational modifications—in particular phosphorylation at Tyr99 by c-Abl and acetylation by the acetyltransferase p300—activate the cell death-inducing activity of p73 [151]. Accordingly, both c-Abl and ATM were found to cooperate in response to DNA damage during nervous system development [152]. c-Abl also phosphorylates a key co-activator protein of p73, YAP1 (Yes-Associated Protein 1). Yap1 is phosphorylated at Tyr357 after IR and Cisplatin treatment, which guides its co-activator function away from the proliferation-driving TEAD transcription factors towards p73 to potentiate the transcription of apoptotic target genes [153]. In addition, YAP1 can also protect p73 from Itch-mediated proteolysis and potentiates p73 activity independent of Tyr357 phosphorylation [154]. Taken together, c-Abl exerts important functions in DNA damage-induced apoptosis through controlling p73 activity.
The ATM–NEMO/IKKγ–RIPK1 axis
Following DNA damage, ATM can activate the NF-κB pathway to promote cell survival [155] [156, 157]. However, in response to severe damage, ATM and NEMO/IKK-γ (NF-κB essential modulator)-dependent NF-κB activation can lead to caspase-8 activation and subsequent apoptosis by the extrinsic pathway [158].
For NF-κB activation following genotoxic stress, post-translational modification of NEMO/IKK-γ is required. NEMO is sumoylated at lysine residues 277 and 309 by the SUMO E3 ligase PIASy/PIAS4 (protein inhibitor of activated STAT y) and phosphorylated at Serine 85 by activated ATM [157, 159–161]. Both post-translational modifications are a prerequisite for the monoubiquitination of NEMO at lysine residues 277 and 309 by cellular inhibitor of apoptosis-1 (cIAP1) [162]. Whether the sumoylation and subsequent ubiquitination can co-exist is unclear. The proposed mechanism involves a transient sumoylation followed by the phosphorylation and monoubiquitination of NEMO. The ubiquitination promotes the nuclear export of NEMO to facilitate downstream signal transduction and the activation of the NF-κB pathway. Interestingly NEMO can distribute a small proportion of ATM to the cytoplasm [161], where it activates an additional kinase, TAK1 (transforming growth factor β-activated kinase-1), which has been shown to be required for NF-κB activation after DNA damage [162, 163]. TAK1 requires further activation either by polyubiquitination of ELKS (a protein rich in glutamate, leucine, lysine, and serine) by auto-ubiquitination of TRAF6 (TNFR-associated factor 6) or by RIPK1 ubiquitination [164–166].
A recent report [158] has revealed that DNA damage signaling via the ATM–NEMO–RIPK1 signaling axis serves as a cell fate switch and drive cells into apoptosis. NF-κB signaling is both activated after low and high doses of DNA damage and leads to a TNFα–TNFR1 signaling loop. After a sustained lethal dose of DNA damage, this feed-forward loop is further triggered and excessive production of TNFα leads to a second wave of NF-κB activity and the phosphorylation of RIPK1 [158]. In turn, the formation of a NEMO–RIPK1 complex stimulates JNK (c-Jun N-terminal kinase) phosphorylation, which leads to the production of the pro-inflammatory cytokine IL-8. These events finally lead to recruitment of FADD to the complex. FADD serves as a docking for the initiator caspase-8. Caspase-8 gets activated at this complex and triggers apoptosis via the extrinsic pathway. The RIPK1–NEMO–FADD–caspase-8 complex is similar to the aforementioned Ripoptosome.
The piddosome: a nuclear caspase 2 activation platform
One of the characteristics of the apoptotic process is the sequential activation of caspases. It has been shown that the initiator caspases get activated at large macromolecular scaffold structures, such as the apoptosome, which activates caspase-9, or the DISC (death-induced signaling complex), which activates caspases-8 and 10. A third caspase-activating platform is the PIDDosome [167]. Remarkably, the PIDDosome is localized to the cell nucleus. The PIDDosome is a high molecular weight complex and consists of PIDD (p53-induced death domain protein), RAIDD (receptor-interacting protein-associated Ich-1/CED homologous protein with DD) and the initiator caspase-2 (Fig. 5) [167]. Caspase-2 is a tumor suppressor and is important for p53-independent, mitochondrial apoptosis signaling, and siRNA-mediated knockdown of caspase-2 inhibits the induction of apoptosis by various DNA damaging drugs [168, 169]. Similar effects have been observed with MEFs from caspase-2 deficient mice [170, 171]. Loss of caspase-2 also leads to an acceleration of tumor onset in a mouse lymphoma model. However, the effects of caspase-2 on tumorigenesis seem to be independent of its activation at the PIDDosome and its pro-apoptotic activity [172]. Interestingly, caspase-2 KO MEFs escape cellular senescence in culture and show signs of high genomic instability such as the accumulation of micronuclei [173].
PIDD has been suggested as a molecular switch between cell survival and cell death following DNA damage. PIDD signals towards apoptosis in connection with RAIDD and caspase-2, whereas binding to RIPK1 leads to NF-κB signaling and cell survival [174]. The pro-apoptotic PIDDosome assembly upon DNA damage is initiated via phosphorylation of the PIDD death domain (DD) by the checkpoint kinase ATM in Chk1-inhibited cells. The post-translational modification allows for recruitment of RAIDD and complex assembly [175]. However, there is evidence from knockout mouse models that DNA damage-induced caspase-2 activation does also function in RAIDD−/− and PIDD−/− cells, demonstrating that the PIDDosome is not required for DNA damage-induced caspase-2 activation in vivo [172, 176]. Indeed, loss of Caspase-2, but not Pidd1, reduced the cell death response after γ-irradiation in mouse embryonic fibroblasts [176]. In a more recent study, the same group investigated the effect of Raidd in a mouse lymphoma model. It was known before that loss of Caspase-2 in the Eµ-Myc mouse lymphoma model leads to an acceleration of tumor onset, whereas loss of Pidd1 delays tumor onset. Interestingly, Raidd deficiency showed no modulatory effect on the development of radiation-induced thymic lymphomas, demonstrating that the PIDDosome scaffold protein RAIDD is dispensable for Caspase-2 activation in this tumor model [172]. What in detail accounts for these conflicting results in mice and in human cancer cells remains to be determined in the future. Taken together, the PIDDosome is a versatile protein complex consisting of different signaling molecules, but exclusively assembly of PIDD–RAIDD–caspase-2 complex initiates cell death signaling after DNA damage [26].
DNA damage-induced necroptosis
Treatment with DNA-damaging chemotherapeutic drugs does not solely engage the apoptotic response but can also activate necroptosis. Two recent studies provide evidence that the expression of the essential necroptosis activator RIPK3 is lost in numerous cancer cells including hepatocellular carcinoma, breast carcinoma, glioblastoma and melanoma [52, 177]. Interestingly, in the hematopoietic cell lines investigated only 20 % showed a loss of RIPK3 expression, in contrast to broad loss of RIPK3 in about 80 % of non-hematopoietic cell lines [52]. In virtually all cells with lost RIPK3 expression, the addition of an inhibitor of DNA methylation reconstituted the expression of RIPK3 mRNA and protein, demonstrating that RIPK3 is a target of epigenetic silencing [52]. Remarkably, DNA hypomethylation using 5-azacytidine (5-AD) specifically sensitized RIPK3-silenced cells, but not the RIPK3-expressing counterparts, to chemotherapeutic drugs including Doxorubicin, Etoposide, Cisplatin or Camptothecin and facilitated MLKL phosphorylation and necroptotic cell death (Fig. 4) [52]. The authors also confirmed this effect in vivo in a mouse xenograft model. In addition, using immunohistochemistry, the authors demonstrate that RIPK3 is downregulated in numerous breast cancer specimens [52]. However, the fascinating question by which molecular mechanisms these diverse DNA-damaging (Etoposide, Doxorubicin) and mitosis-arresting drugs (Taxanes) engage the necrosome remains to be elucidated in the future. Interestingly, since prolonged mitotic arrest has been shown to trigger a telomere-derived DNA damage response [178], it appears possible that DNA damage checkpoint kinases might be involved in sending a damage signal to the necrosome. In summary, necroptosis appears to be suppressed in numerous tumor cell lines and in breast cancer due to lost RIPK3 expression. Reconstitution of RIPK3 expression sensitizes these cells to necroptotic cell death upon treatment with various chemotherapeutic drugs, making the necrosome an attractive target in cancer treatment.
Perspective
In the past years major progress has been made to our understanding of the underlying mechanisms of DNA damage-induced cell death responses. Numerous signaling pathways, molecules and mechanisms involved have been identified and studied. Despite of this gain of knowledge, there are still essential questions to be answered in the future to reveal a more detailed understanding.
For example, how do cells decide between the utilization of these different cell death signaling routes? The simplest explanation would be that key molecules of a given cell death route are expressed in a cell type-specific fashion or get lost in cancer cells. However, this apparently applies only to a limited number of key players such as p53 and RIPK3, which are lost or inactivated to a significant extent in cancer cells. Thus, it may be a promising strategy to identify the cell death pathways, which in principle remain functional in cancer cells under the given settings, to guide and assist full activation of these pathways by the means of suited drugs.
The development of more efficient and safer anticancer drugs is one of the major goals in cancer research. Up to now, first-line therapy for many cancer entities is classical DNA damage-inducing therapies, such as IR and chemotherapeutic drugs as Cisplatin, Doxorubicin or Etoposide. The harmed cancer cells react by activating the DDR, ideally culminating in the removal of the tumor cells by apoptosis. Limitations arise from acquired resistance of cancer cells to the administered drug and from toxic side effects on specific tissues of the patient treated. Over the last years, drugs based on small molecule inhibitors and on hydrocarbon-stapled peptides have been designed to specifically target central players of the DDR including p53, Chk1 and Chk2 [179, 180]. First results appear promising although on-target side effects need to be further reduced. It will be interesting to see how these drugs perform in combination with DNA-damaging treatments.
A further important question requiring more scientific attention is if and how the different death signaling cascades do cross-talk with each other. Since for instance ATM is at the apex of many DNA damage-induced cell death signaling pathways, it remains to be studied if and how these pathways branch downstream of ATM and may converge again at central nodes downstream. The identification of the key molecules mediating cross-talk, branching and signal integration would provide promising targets for the design of small molecules and/or hydrocarbon-stapled peptides to potentiate or to limit the activity/function of these signaling hubs.
How many different cell death routes are operative in a given (cancer) cell (line) and which routes are the most broadly engaged ones in response to DNA damaging drug treatment or radiation therapy? Such knowledge will be essential to define new target proteins in the respective pathways and to design and develop pharmacological molecules. With the knowledge of the relevant death pathways operative in the given cancer cell or cancer tissue, combinations of drugs and inhibitors that potentiate this pathways could be designed and analyzed for their efficacy in cancer cell killing.
Results from death receptor signaling studies indicate that caspase activation during apoptosis inhibits the activation of necroptosis. Do these findings also apply to the DNA damage-induced signaling pathways? This could mean that DNA damage-induced apoptosis and necroptosis are mutually exclusive in a DNA-damaged cancer cells. Then, it would be likely that cancer cells showing only limited caspase activation, which is insufficient to trigger apoptosis, might suppress necroptotic death and thus mediate cancer cell survival. If this is true, we need to consider novel strategies in cancer cell killing, such as transient inhibition of caspases by caspase inhibitors to re-activate the necroptotic machinery.
Finally, much of our current knowledge on DNA damage-induced cell death routes is obtained from the use of cancer cell lines. This knowledge needs to be transferred to organismic model systems to understand the in vivo relevance of these pathways and mechanisms in cancer cell killing.
Taken together, despite the accumulating wealth of knowledge, we still need to fill major knowledge gaps in order to enable a targeted manipulation of the DNA damage-induced cell death response in cancer.
Acknowledgments
We thank Tilman Polonio-Vallon for help with the figure design and preparation and for discussion. This work has been supported by the SFB 1036 “Cellular surveillance and damage control” funded by the Deutsche Forschungsgemeinschaft (DFG). We also want to apologize to all authors of important publication which could not be mentioned here due to focus and space limitation.
Abbreviations
- ATM
Ataxia-telangiectasia mutated
- ATR
ATM and Rad3-related
- Chk1
Checkpoint kinase 1
- Chk2
Checkpoint kinase 2
- DDR
DNA damage response
- DSB
Double-strand break
- γH2AX
H2AX phosphorylated at Ser139
- HIPK2
Homeodomain-interacting protein kinase 2
- IR
Ionizing radiation
- MLKL
Mixed-lineage kinase like
- MOMP
Mitochondrial outer membrane permeabilization
- PIDD
p53-induced death domain containing protein
- PML
Promyelocytic leukemia
- RIPK3
Receptor-interacting protein kinase 3
- Siah-1
Seven in absentia homologue 1
- SSB
Single-strand break
- UV
Ultraviolet
- Yap1
Yes-associated protein 1
References
- 1.Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proc Natl Acad Sci USA. 2000;97:686–691. doi: 10.1073/pnas.97.2.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. DNA damage, aging, and cancer. New Engl J Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 3.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5:772–776. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klingseisen A, Jackson AP. Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev. 2011;25:2011–2024. doi: 10.1101/gad.169037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- 8.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nature Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 10.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Disc. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 11.Panier S, Durocher D. Push back to respond better: regulatory inhibition of the DNA double-strand break response. Nat Rev Mol Cell Biol. 2013;14:661–672. doi: 10.1038/nrm3659. [DOI] [PubMed] [Google Scholar]
- 12.Jackson SP, Durocher D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell. 2013;49:795–807. doi: 10.1016/j.molcel.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Jeggo PA, Lobrich M. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem J. 2015;471:1–11. doi: 10.1042/BJ20150582. [DOI] [PubMed] [Google Scholar]
- 14.Oh J, Lee YD, Wagers AJ. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nature Med. 2014;20:870–880. doi: 10.1038/nm.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burton DG, Faragher RG. Cellular senescence: from growth arrest to immunogenic conversion. Age. 2015;37:27. doi: 10.1007/s11357-015-9764-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, 3rd, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/S0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 18.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Lobrich M, Jeggo PA. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 19.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 20.Bakr A, Oing C, Kocher S, Borgmann K, Dornreiter I, Petersen C, Dikomey E, Mansour WY. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nuc Acid Res. 2015;43:3154–3166. doi: 10.1093/nar/gkv160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis AJ, Chen BP, Chen DJ. DNA-PK: a dynamic enzyme in a versatile DSB repair pathway. DNA Repair. 2014;17:21–29. doi: 10.1016/j.dnarep.2014.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor Perspect Biol. 2013;5:1–14. doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 24.Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, Lopez E, Fernandez-Capetillo O. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J Exp Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 26.Janssens S, Tinel A. The PIDDosome, DNA-damage-induced apoptosis and beyond. Cell Death Differ. 2012;19:13–20. doi: 10.1038/cdd.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar S. Caspase 2 in apoptosis, the DNA damage response and tumour suppression: enigma no more? Nat Rev Cancer. 2009;9:897–903. doi: 10.1038/nrc2745. [DOI] [PubMed] [Google Scholar]
- 28.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 29.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 30.Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 31.Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–344. doi: 10.1038/nrm3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 33.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 34.Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nature Immunol. 2015;16:689–697. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 35.Linkermann A, Green DR. Necroptosis. New Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 37.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 38.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 39.Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 41.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 42.Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE. 2013;8:e76841. doi: 10.1371/journal.pone.0076841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nature Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 44.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, Zobel K, Dynek JN, Elliott LO, Wallweber HJ, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 45.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nature Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, et al. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2015;23:76–88. doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 49.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Scie USA. 2011;108:20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 52.Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–725. doi: 10.1038/cr.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332:237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 54.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 56.Vousden KH, Prives C. Blinded by the Light: the Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 57.Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements. J Mol Biol. 2005;348:589–596. doi: 10.1016/j.jmb.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 58.Horvath MM, Wang X, Resnick MA, Bell DA. Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS Genet. 2007;3:e127. doi: 10.1371/journal.pgen.0030127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jegga AG, Inga A, Menendez D, Aronow BJ, Resnick MA. Functional evolution of the p53 regulatory network through its target response elements. Proc Natl Acad Sci USA. 2008;105:944–949. doi: 10.1073/pnas.0704694105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cui F, Zhurkin VB. Rotational positioning of nucleosomes facilitates selective binding of p53 to response elements associated with cell cycle arrest. Nuc Acid Res. 2014;42:836–847. doi: 10.1093/nar/gkt943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 62.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 63.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 64.Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- 65.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/S1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 66.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 68.Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 70.Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/S0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 71.Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H, Schmitz ML. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol. 2002;4:1–10. doi: 10.1038/ncb715. [DOI] [PubMed] [Google Scholar]
- 72.D’Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 2002;4:11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 73.Dauth I, Kruger J, Hofmann TG. Homeodomain-interacting protein kinase 2 is the ionizing radiation-activated p53 serine 46 kinase and is regulated by ATM. Cancer Res. 2007;67:2274–2279. doi: 10.1158/0008-5472.CAN-06-2884. [DOI] [PubMed] [Google Scholar]
- 74.Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell. 2007;25:725–738. doi: 10.1016/j.molcel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 75.Di Stefano V, Rinaldo C, Sacchi A, Soddu S, D’Orazi G. Homeodomain-interacting protein kinase-2 activity and p53 phosphorylation are critical events for cisplatin-mediated apoptosis. Exp Cell Res. 2004;293:311–320. doi: 10.1016/j.yexcr.2003.09.032. [DOI] [PubMed] [Google Scholar]
- 76.Bitomsky N, Conrad E, Moritz C, Polonio-Vallon T, Sombroek D, Schultheiss K, Glas C, Greiner V, Herbel C, Mantovani F, et al. Autophosphorylation and Pin1 binding coordinate DNA damage-induced HIPK2 activation and cell death. Proc Natl Acad Sci USA. 2013;110:E4203–E4212. doi: 10.1073/pnas.1310001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, Monden M, Nakamura Y. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell. 2001;8:85–94. doi: 10.1016/S1097-2765(01)00284-2. [DOI] [PubMed] [Google Scholar]
- 78.Ichwan SJ, Yamada S, Sumrejkanchanakij P, Ibrahim-Auerkari E, Eto K, Ikeda MA. Defect in serine 46 phosphorylation of p53 contributes to acquisition of p53 resistance in oral squamous cell carcinoma cells. Oncogene. 2006;25:1216–1224. doi: 10.1038/sj.onc.1209158. [DOI] [PubMed] [Google Scholar]
- 79.Li X, Dumont P, Della Pietra A, Shetler C, Murphy ME. The codon 47 polymorphism in p53 is functionally significant. J Biol Chem. 2005;280:24245–24251. doi: 10.1074/jbc.M414637200. [DOI] [PubMed] [Google Scholar]
- 80.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 81.Kodama M, Otsubo C, Hirota T, Yokota J, Enari M, Taya Y. Requirement of ATM for rapid p53 phosphorylation at Ser46 without Ser/Thr-Gln sequences. Mol Cell Biol. 2010;30:1620–1633. doi: 10.1128/MCB.00810-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshida K, Liu H, Miki Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J Biol Chem. 2006;281:5734–5740. doi: 10.1074/jbc.M512074200. [DOI] [PubMed] [Google Scholar]
- 83.Bitomsky N, Hofmann TG. Apoptosis and autophagy: regulation of apoptosis by DNA damage signalling—roles of p53, p73 and HIPK2. FEBS J. 2009;276:6074–6083. doi: 10.1111/j.1742-4658.2009.07331.x. [DOI] [PubMed] [Google Scholar]
- 84.Polonio-Vallon T, Kruger D, Hofmann TG. ShaPINg cell fate upon DNA damage: role of Pin1 isomerase in DNA damage-induced cell death and repair. Front Oncol. 2014;4:148. doi: 10.3389/fonc.2014.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, Del Sal G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature. 2002;419:853–857. doi: 10.1038/nature01120. [DOI] [PubMed] [Google Scholar]
- 86.Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf GM, Gu L, Tang X, Lu KP, Xiao ZX. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–853. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 87.Wulf GM, Liou YC, Ryo A, Lee SW, Lu KP. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J Biol Chem. 2002;277:47976–47979. doi: 10.1074/jbc.C200538200. [DOI] [PubMed] [Google Scholar]
- 88.Grison A, Mantovani F, Comel A, Agostoni E, Gustincich S, Persichetti F, Del Sal G. Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc Natl Acad Sci USA. 2011;108:17979–17984. doi: 10.1073/pnas.1106198108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sorrentino G, Mioni M, Giorgi C, Ruggeri N, Pinton P, Moll U, Mantovani F, Del Sal G. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ. 2013;20:198–208. doi: 10.1038/cdd.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Follis AV, Llambi F, Merritt P, Chipuk JE, Green DR, Kriwacki RW. Pin1-induced proline isomerization in cytosolic p53 mediates BAX activation and apoptosis. Mol Cell. 2015;59:677–684. doi: 10.1016/j.molcel.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hofmann TG, Glas C, Bitomsky N. HIPK2: a tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. BioEssays. 2013;35:55–64. doi: 10.1002/bies.201200060. [DOI] [PubMed] [Google Scholar]
- 92.Sombroek D, Hofmann TG. How cells switch HIPK2 on and off. Cell Death Differ. 2009;16:187–194. doi: 10.1038/cdd.2008.154. [DOI] [PubMed] [Google Scholar]
- 93.Saul VV, Schmitz ML. Posttranslational modifications regulate HIPK2, a driver of proliferative diseases. J Mol Med. 2013;91:1051–1058. doi: 10.1007/s00109-013-1042-0. [DOI] [PubMed] [Google Scholar]
- 94.D’Orazi G, Rinaldo C, Soddu S. Updates on HIPK2: a resourceful oncosuppressor for clearing cancer. J Exp Clin Cancer Res. 2012;31:63. doi: 10.1186/1756-9966-31-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moller A, Sirma H, Hofmann TG, Rueffer S, Klimczak E, Droge W, Will H, Schmitz ML. PML is required for homeodomain-interacting protein kinase 2 (HIPK2)-mediated p53 phosphorylation and cell cycle arrest but is dispensable for the formation of HIPK domains. Cancer Res. 2003;63:4310–4314. [PubMed] [Google Scholar]
- 96.Moller A, Sirma H, Hofmann TG, Staege H, Gresko E, Ludi KS, Klimczak E, Droge W, Will H, Schmitz ML. Sp100 is important for the stimulatory effect of homeodomain-interacting protein kinase-2 on p53-dependent gene expression. Oncogene. 2003;22:8731–8737. doi: 10.1038/sj.onc.1207079. [DOI] [PubMed] [Google Scholar]
- 97.Siepi F, Gatti V, Camerini S, Crescenzi M, Soddu S. HIPK2 catalytic activity and subcellular localization are regulated by activation-loop Y354 autophosphorylation. Biochim Biophys Acta. 2013;1833:1443–1453. doi: 10.1016/j.bbamcr.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Saul VV, de la Vega L, Milanovic M, Kruger M, Braun T, Fritz-Wolf K, Becker K, Schmitz ML. HIPK2 kinase activity depends on cis-autophosphorylation of its activation loop. J Mol Cell Biol. 2013;5:27–38. doi: 10.1093/jmcb/mjs053. [DOI] [PubMed] [Google Scholar]
- 99.de la Vega L, Grishina I, Moreno R, Kruger M, Braun T, Schmitz ML. A redox-regulated SUMO/acetylation switch of HIPK2 controls the survival threshold to oxidative stress. Mol Cell. 2012;46:472–483. doi: 10.1016/j.molcel.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 100.Hofmann TG, Jaffray E, Stollberg N, Hay RT, Will H. Regulation of homeodomain-interacting protein kinase 2 (HIPK2) effector function through dynamic small ubiquitin-related modifier-1 (SUMO-1) modification. J Biol Chem. 2005;280:29224–29232. doi: 10.1074/jbc.M503921200. [DOI] [PubMed] [Google Scholar]
- 101.Winter M, Sombroek D, Dauth I, Moehlenbrink J, Scheuermann K, Crone J, Hofmann TG. Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nat Cell Biol. 2008;10:812–824. doi: 10.1038/ncb1743. [DOI] [PubMed] [Google Scholar]
- 102.Choi DW, Seo YM, Kim EA, Sung KS, Ahn JW, Park SJ, Lee SR, Choi CY. Ubiquitination and degradation of homeodomain-interacting protein kinase 2 by WD40 repeat/SOCS box protein WSB-1. J Biol Chem. 2008;283:4682–4689. doi: 10.1074/jbc.M708873200. [DOI] [PubMed] [Google Scholar]
- 103.Kim SY, Choi DW, Kim EA, Choi CY. Stabilization of HIPK2 by escape from proteasomal degradation mediated by the E3 ubiquitin ligase Siah1. Cancer Lett. 2009;279:177–184. doi: 10.1016/j.canlet.2009.01.036. [DOI] [PubMed] [Google Scholar]
- 104.Crone J, Glas C, Schultheiss K, Moehlenbrink J, Krieghoff-Henning E, Hofmann TG. Zyxin is a critical regulator of the apoptotic HIPK2-p53 signaling axis. Cancer Res. 2011;71:2350–2359. doi: 10.1158/0008-5472.CAN-10-3486. [DOI] [PubMed] [Google Scholar]
- 105.Gresko E, Roscic A, Ritterhoff S, Vichalkovski A, Del Sal G, Schmitz ML. Autoregulatory control of the p53 response by caspase-mediated processing of HIPK2. EMBO J. 2006;25:1883–1894. doi: 10.1038/sj.emboj.7601077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Q, Yoshimatsu Y, Hildebrand J, Frisch SM, Goodman RH. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell. 2003;115:177–186. doi: 10.1016/S0092-8674(03)00802-X. [DOI] [PubMed] [Google Scholar]
- 107.Polonio-Vallon T, Kirkpatrick J, Krijgsveld J, Hofmann TG. Src kinase modulates the apoptotic p53 pathway by altering HIPK2 localization. Cell Cycle. 2014;13:115–125. doi: 10.4161/cc.26857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Muschik D, Braspenning-Wesch I, Stockfleth E, Rosl F, Hofmann TG, Nindl I. Cutaneous HPV23 E6 prevents p53 phosphorylation through interaction with HIPK2. PLoS ONE. 2011;6:e27655. doi: 10.1371/journal.pone.0027655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reuven N, Adler J, Porat Z, Polonio-Vallon T, Hofmann TG, Shaul Y. The tyrosine kinase c-Abl promotes homeodomain-interacting protein kinase 2 (HIPK2) accumulation and activation in response to DNA damage. J Biol Chem. 2015;290:16478–16488. doi: 10.1074/jbc.M114.628982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tomasini R, Samir AA, Carrier A, Isnardon D, Cecchinelli B, Soddu S, Malissen B, Dagorn JC, Iovanna JL, Dusetti NJ. TP53INP1 s and homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity. J Biol Chem. 2003;278:37722–37729. doi: 10.1074/jbc.M301979200. [DOI] [PubMed] [Google Scholar]
- 111.Rui Y, Xu Z, Lin S, Li Q, Rui H, Luo W, Zhou HM, Cheung PY, Wu Z, Ye Z, et al. Axin stimulates p53 functions by activation of HIPK2 kinase through multimeric complex formation. EMBO J. 2004;23:4583–4594. doi: 10.1038/sj.emboj.7600475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hofmann TG, Stollberg N, Schmitz ML, Will H. HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 2003;63:8271–8277. [PubMed] [Google Scholar]
- 113.Li Q, Wang X, Wu X, Rui Y, Liu W, Wang J, Wang X, Liou YC, Ye Z, Lin SC. Daxx cooperates with the Axin/HIPK2/p53 complex to induce cell death. Cancer Res. 2007;67:66–74. doi: 10.1158/0008-5472.CAN-06-1671. [DOI] [PubMed] [Google Scholar]
- 114.Li Q, Lin S, Wang X, Lian G, Lu Z, Guo H, Ruan K, Wang Y, Ye Z, Han J, et al. Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nat Cell Biol. 2009;11:1128–1134. doi: 10.1038/ncb1927. [DOI] [PubMed] [Google Scholar]
- 115.Chung YM, Park SH, Tsai WB, Wang SY, Ikeda MA, Berek JS, Chen DJ, Hu MC. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat Comm. 2012;3:1000. doi: 10.1038/ncomms2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fong WG, Liston P, Rajcan-Separovic E, St Jean M, Craig C, Korneluk RG. Expression and genetic analysis of XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics. 2000;70:113–122. doi: 10.1006/geno.2000.6364. [DOI] [PubMed] [Google Scholar]
- 117.Liston P, Fong WG, Kelly NL, Toji S, Miyazaki T, Conte D, Tamai K, Craig CG, McBurney MW, Korneluk RG. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat Cell Biol. 2001;3:128–133. doi: 10.1038/35055027. [DOI] [PubMed] [Google Scholar]
- 118.Zou B, Chim CS, Pang R, Zeng H, Dai Y, Zhang R, Lam CS, Tan VP, Hung IF, Lan HY, et al. XIAP-associated factor 1 (XAF1), a novel target of p53, enhances p53-mediated apoptosis via post-translational modification. Mol Carcinogen. 2012;51:422–432. doi: 10.1002/mc.20807. [DOI] [PubMed] [Google Scholar]
- 119.Lee MG, Han J, Jeong SI, Her NG, Lee JH, Ha TK, Kang MJ, Ryu BK, Chi SG. XAF1 directs apoptotic switch of p53 signaling through activation of HIPK2 and ZNF313. Proc Natl Acad Sci USA. 2014;111:15532–15537. doi: 10.1073/pnas.1411746111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taira N, Yamaguchi T, Kimura J, Lu ZG, Fukuda S, Higashiyama S, Ono M, Yoshida K. Induction of amphiregulin by p53 promotes apoptosis via control of microRNA biogenesis in response to DNA damage. Proc Natl Acad Sci USA. 2014;111:717–722. doi: 10.1073/pnas.1313675111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 122.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 125.Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41:371–383. doi: 10.1016/j.molcel.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 127.Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.He L, He X, Lowe SW, Hannon GJ. MicroRNAs join the p53 network-another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–5974. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- 130.Hofmann TG, Will H. Body language: the function of PML nuclear bodies in apoptosis regulation. Cell Death Differ. 2003;10:1290–1299. doi: 10.1038/sj.cdd.4401313. [DOI] [PubMed] [Google Scholar]
- 131.Krieghoff-Henning E, Hofmann TG. Role of nuclear bodies in apoptosis signalling. Biochim Biophys Acta. 2008;1783:2185–2194. doi: 10.1016/j.bbamcr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 132.Bernardi R, Papa A, Pandolfi PP. Regulation of apoptosis by PML and the PML-NBs. Oncogene. 2008;27:6299–6312. doi: 10.1038/onc.2008.305. [DOI] [PubMed] [Google Scholar]
- 133.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–170. doi: 10.1016/S0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 134.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35021000. [DOI] [PubMed] [Google Scholar]
- 135.Gostissa M, Hofmann TG, Will H, Del Sal G. Regulation of p53 functions: let’s meet at the nuclear bodies. Curr Opin Cell Biol. 2003;15:351–357. doi: 10.1016/S0955-0674(03)00038-3. [DOI] [PubMed] [Google Scholar]
- 136.Dellaire G, Bazett-Jones DP. Beyond repair foci: subnuclear domains and the cellular response to DNA damage. Cell Cycle. 2007;6:1864–1872. doi: 10.4161/cc.6.15.4560. [DOI] [PubMed] [Google Scholar]
- 137.Conrad E, Polonio-Vallon T, Meister M, Matt S, Bitomsky N, Herbel C, Liebl M, Greiner V, Kriznik B, Schumacher S, et al. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016;23:110–122. doi: 10.1038/cdd.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Melino G. p73, the “assistant” guardian of the genome? Ann N Y Acad Sci. 2003;1010:9–15. doi: 10.1196/annals.1299.002. [DOI] [PubMed] [Google Scholar]
- 139.Conforti F, Sayan AE, Sreekumar R, Sayan BS. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012;3:e285. doi: 10.1038/cddis.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Allocati N, Di Ilio C, De Laurenzi V. p63/p73 in the control of cell cycle and cell death. Exp Cell Res. 2012;318:1285–1290. doi: 10.1016/j.yexcr.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 141.Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–848. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Flinterman M, Guelen L, Ezzati-Nik S, Killick R, Melino G, Tominaga K, Mymryk JS, Gaken J, Tavassoli M. E1A activates transcription of p73 and Noxa to induce apoptosis. J Biol Chem. 2005;280:5945–5959. doi: 10.1074/jbc.M406661200. [DOI] [PubMed] [Google Scholar]
- 143.Bernassola F, Salomoni P, Oberst A, Di Como CJ, Pagano M, Melino G, Pandolfi PP. Ubiquitin-dependent degradation of p73 is inhibited by PML. J Exp Med. 2004;199:1545–1557. doi: 10.1084/jem.20031943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tomasini R, Mak TW, Melino G. The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol. 2008;18:244–252. doi: 10.1016/j.tcb.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 145.Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harbor Perspect Biol. 2010;2:a004887. doi: 10.1101/cshperspect.a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399:809–813. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 147.Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Jr, Levrero M, Wang JY. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 148.Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat Cell Biol. 2005;7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- 149.Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, Barlow C, Baltimore D, Wynshaw-Boris A, Kastan MB, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387:516–519. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 150.Shafman T, Khanna KK, Kedar P, Spring K, Kozlov S, Yen T, Hobson K, Gatei M, Zhang N, Watters D, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387:520–523. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 151.Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, Fontemaggi G, Fanciulli M, Schiltz L, Blandino G, et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol Cell. 2002;9:175–186. doi: 10.1016/S1097-2765(02)00431-8. [DOI] [PubMed] [Google Scholar]
- 152.Miller HL, Lee Y, Zhao J, Chong MJ, McKinnon PJ. Atm and c-Abl cooperate in the response to genotoxic stress during nervous system development. Dev Brain Res. 2003;145:31–38. doi: 10.1016/S0165-3806(03)00192-5. [DOI] [PubMed] [Google Scholar]
- 153.Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell. 2008;29:350–361. doi: 10.1016/j.molcel.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 154.Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007;14:743–751. doi: 10.1038/sj.cdd.4402063. [DOI] [PubMed] [Google Scholar]
- 155.Piret B, Schoonbroodt S, Piette J. The ATM protein is required for sustained activation of NF-kappaB following DNA damage. Oncogene. 1999;18:2261–2271. doi: 10.1038/sj.onc.1202541. [DOI] [PubMed] [Google Scholar]
- 156.Li N, Banin S, Ouyang H, Li GC, Courtois G, Shiloh Y, Karin M, Rotman G. ATM is required for IkappaB kinase (IKKk) activation in response to DNA double strand breaks. J Biol Chem. 2001;276:8898–8903. doi: 10.1074/jbc.M009809200. [DOI] [PubMed] [Google Scholar]
- 157.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell. 2003;115:565–576. doi: 10.1016/S0092-8674(03)00895-X. [DOI] [PubMed] [Google Scholar]
- 158.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 159.Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell. 2009;36:365–378. doi: 10.1016/j.molcel.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 160.Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat Cell Biol. 2006;8:986–993. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- 161.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 162.Jin HS, Lee DH, Kim DH, Chung JH, Lee SJ, Lee TH. cIAP1, cIAP2, and XIAP act cooperatively via nonredundant pathways to regulate genotoxic stress-induced nuclear factor-kappaB activation. Cancer Res. 2009;69:1782–1791. doi: 10.1158/0008-5472.CAN-08-2256. [DOI] [PubMed] [Google Scholar]
- 163.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nature Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 164.Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol Cell. 2010;40:75–86. doi: 10.1016/j.molcel.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol Cell. 2010;40:63–74. doi: 10.1016/j.molcel.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 166.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S S, Kelliher MA. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-κB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol. 2011;31:2774–2786. doi: 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–846. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 168.Lassus P, Opitz-Araya X, Lazebnik Y. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science. 2002;297:1352–1354. doi: 10.1126/science.1074721. [DOI] [PubMed] [Google Scholar]
- 169.Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;277:29803–29809. doi: 10.1074/jbc.M204185200. [DOI] [PubMed] [Google Scholar]
- 170.Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, et al. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ho LH, Taylor R, Dorstyn L, Cakouros D, Bouillet P, Kumar S. A tumor suppressor function for caspase-2. Proc Natl Acad Sci USA. 2009;106:5336–5341. doi: 10.1073/pnas.0811928106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Peintner L, Dorstyn L, Kumar S, Aneichyk T, Villunger A, Manzl C. The tumor-modulatory effects of Caspase-2 and Pidd1 do not require the scaffold protein Raidd. Cell Death Differ. 2015;22:1803–1811. doi: 10.1038/cdd.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S, Kumar S. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 2012;19:1288–1298. doi: 10.1038/cdd.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-κB activation in response to DNA damage. Cell. 2005;123:1079–1092. doi: 10.1016/j.cell.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 175.Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, Look AT, Wang J, Bouchier-Hayes L, Sidi S. PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol Cell. 2012;47:681–693. doi: 10.1016/j.molcel.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Manzl C, Fava LL, Krumschnabel G, Peintner L, Tanzer MC, Soratroi C, Bock FJ, Schuler F, Luef B, Geley S, et al. Death of p53-defective cells triggered by forced mitotic entry in the presence of DNA damage is not uniquely dependent on Caspase-2 or the PIDDosome. Cell Death Dis. 2013;4:e942. doi: 10.1038/cddis.2013.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J, Gough PJ, Feoktistova M, Leverkus M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015;6:e1884. doi: 10.1038/cddis.2015.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387–394. doi: 10.1038/nsmb.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Disc. 2014;13:217–236. doi: 10.1038/nrd4288. [DOI] [PubMed] [Google Scholar]
- 180.O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]