

Abstract
Increased demands on the protein folding capacity of the endoplasmic reticulum (ER) trigger the unfolded protein response (UPR). Comprised of a tripartite signaling system, the UPR regulates translation and gene transcription to manifest pro-adaptive and, if necessary, pro-apoptotic outcomes. The three UPR pathways, initiated by activating transcription factor 6, inositol requiring enzyme 1, and protein kinase RNA-activated-like ER kinase (PERK), direct distinct downstream signaling events. However, it is becoming increasingly clear that interplay between the cascades is vital in shaping the UPR. In particular, recent discoveries have revealed that PERK-dependent signals mediate both inter- and intra-pathway regulation within the UPR, underscoring the critical role of the PERK pathway in the cellular response to ER stress.
Keywords: Endoplasmic reticulum, Unfolded protein response, ATF6α, ATF4, XBP1, PERK
Introduction
The endoplasmic reticulum (ER), an elaborate network of flattened sac-like and tubular structures, serves as a factory for protein and lipid production, a specialized protein folding compartment and the major depot for intracellular Ca2+ [1, 2]. Hence, cells are exquisitely sensitive to physiologic conditions that either perturb the normal ER environment or that increase demand for synthesis and maturation of secretory pathway proteins. In general, ER stress is considered to occur when the load of client proteins exceeds the folding capacity of the ER. Without appropriate remedy, such stress can be detrimental to the ER, the entire secretory pathway, and the cell as a whole. Physiologic ER stress occurs in a variety of normal processes such as when B lymphocytes differentiate into antibody-secreting plasma cells [3] and pancreatic β cells engage in episodic, high-rate insulin synthesis [4]. Similarly, a number of patho-physiologic situations can stress the ER including the infection of cells by viruses that hijack the secretory apparatus for viral glycoprotein synthesis and replication [5], and the hypoxia and/or glucose deprivation experienced by cancer cells when the available vasculature fails to meet the metabolic needs of growing tumors [6]. Pharmacologic agents such as tunicamycin, an inhibitor of N-linked glycosylation, and thapsigargin, an inhibitor of the ER-Ca2+ ATPase, grossly disrupt protein folding in the ER, thus potently inducing ER stress. Using such ER poisons as experimental tools, investigators elucidated the molecular details of a multi-faceted cellular response to ER stress, collectively termed the unfolded protein response (UPR) [7] (Fig. 1). Here, the basics of pro-adaptive and pro-apoptotic UPR signaling are reviewed and special emphasis is placed on recent discoveries providing new insight into how the PERK pathway intersects with the entire UPR to influence the fate of cells under ER stress.
Fig. 1.

Conceptual framework for the cellular response to ER stress. An increased workload on the machinery for synthesis, folding, maturation, and transport of secretory pathway proteins renders ER stress, thereby activating the UPR. To adapt to increased demands on the ER, mammalian cells engage translational regulation to reduce the flow of client proteins into the ER and enhance transcription of genes encoding proteins that expand ER capacity. If the pro-adaptive outcomes are insufficient to restore ER homeostasis, the UPR elicits pro-apoptotic mechanisms to promote cell death
Pro-adaptive UPR signaling
When faced with ER stress, cells utilize the UPR to address the problem on multiple fronts (Fig. 1). The UPR can slow the flow of nascent polypeptides into the ER lumen, enhance the ER machinery needed for protein folding and assembly, augment the system for disposal of mis-folded client proteins, and coordinate expansion of the ER compartment. The mammalian UPR is composed of three signaling pathways that are separately initiated by the ER transmembrane proteins activating transcription factor 6 (ATF6) [8], inositol requiring enzyme 1 (IRE1, first identified in yeast) [9, 10], and protein kinase RNA-activated (PKR)-like ER kinase (PERK) [11, 12], each of which possesses an ER lumenal domain that senses stress. The transmembrane regions of IRE1 and PERK detect alterations in the lipid composition of the ER membrane [13, 14]. Each of the three primary UPR signaling molecules contains a cytosolic domain that propagates downstream events (Fig. 2) directed toward balancing load with capacity in the ER, thereby restoring ER homeostasis [15].
Fig. 2.
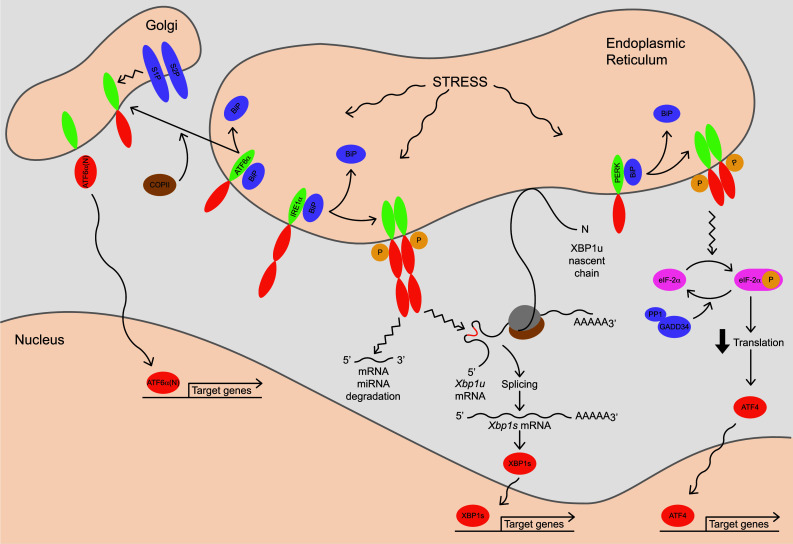
Signaling pathways of the mammalian UPR. Conditions that increase demand on the ER induce ‘stress’ and trigger the UPR. Upon release from the chaperone BiP, an exposed Golgi-localization signal and interaction with the COPII complex allows ATF6α to be ferried to the Golgi apparatus where it undergoes regulated intramembrane proteolysis by S1P and S2P. Liberated from the membrane, its cytosolic domain, ATF6α(N), moves into the nucleus where it functions as a bZIP transcription factor to up-regulate expression of genes via the ERSE promoter motif. ATF6α primarily targets genes involved in various ER quality control processes such as protein folding and ERAD. The IRE1α proteins oligomerize when BiP releases, activating their C-terminal endoribonuclease domains that execute site-specific cleavage of Xbp1 mRNA at two sites. Splicing of the resulting fragments in the cytoplasm yields a transcript with an altered reading frame encoding XBP1s, a bZIP transcription factor that acts on the ERSE and UPRE promoter motifs to up-regulate genes involved throughout the secretory pathway. Translation of Xbp1 mRNA prior to UPR-directed splicing yields XBP1u, a bZIP factor which lacks a transactivation domain. A hydrophobic region near the C-terminus of nascent XBP1u associates with the ER membrane. This interaction facilitates IRE1α-initiated splicing as it positions ribosome-engaged Xbp1 mRNA near the ER membrane. Activated IRE1α also cleaves and degrades select mRNA and miRNA. Upon release of BiP, the PERK proteins oligomerize and phosphorylate the translation initiation factor eIF-2α, effectively reducing translation. Translation of ATF4 increases when global protein synthesis decreases. ATF4 acts on the CARE promoter motif to induce a variety of targets including genes involved in cellular redox homeostasis, amino acid metabolism, protein synthesis and apoptosis. The GADD34–PP1 complex de-phosphorylates eIF-2α, allowing translation to resume
The ATF6 pathway
The ubiquitously expressed ATF6 protein localizes to the ER with a type II transmembrane topology [8]. In the ‘resting’ state, the ER lumenal domain of ATF6 is bound to immunoglobulin binding protein (BiP), an abundant, soluble ER-resident chaperone. Under conditions of ER stress, ATF6 dissociates from BiP, unmasking a Golgi-localization signal in the ATF6 lumenal domain which facilitates its transport to the Golgi [16, 17] via coat protein II-coated vesicles [18]. Reduction of intra- and inter-disulfide bonds in ATF6 oligomers [19], the interaction of its lumenal domain with thrombospondin proteins in the ER [20] and its under-glycosylation and subsequent release from the ER chaperone calreticulin [21] all appear to govern the transit of ATF6 to the Golgi. Upon arrival in the Golgi, ATF6 undergoes regulated intramembrane proteolysis by the site-1 (S1P) and site-2 (S2P) proteases [22], a process that liberates its cytosolic domain from the membrane. This ATF6 fragment, termed ATF6(N), migrates to the nucleus and functions as a basic leucine zipper (bZIP) transcription factor. In the presence of nuclear factor Y, also known as CCAAT-box binding factor, ATF6(N) binds directly to the cis-acting ER stress response element (ERSE; CCAAT-N9-CCACG) [23] found within the promoter regions of many ER stress-responsive genes [24, 25]. Many ATF6-regulated genes encode ER-resident proteins that participate in ER quality control processes. For example, ATF6 targets include molecular chaperones (such as BiP), folding enzymes (such as the protein disulfide isomerase-like protein ERp72), and components of the ER-associated degradation (ERAD) system (such as degradation in ER protein 3, Derlin-3) for disposal of mis-folded proteins [26, 27].
Cells express two isoforms of ATF6, α and β, both of which are functional and responsive to ER stress [8, 28, 29]. Single deletion of either Atf6α or Atf6β in mice does not disrupt normal development [30, 31]. However, combined deletion of these genes results in embryonic lethality [31], suggesting that ATF6α and ATF6β can compensate for each other, at least during embryonic development. Interestingly, studies of Atf6α −/− and Atf6β −/− mouse embryo fibroblasts (MEFs) in vitro indicate that only ATF6α is essential for induction of UPR target genes and cell survival during ER stress [26, 30, 31]. In addition, ATF6α can drive lipid biosynthesis and ER biogenesis [32, 33], and ATF6α has been linked to the physiology of multiple cell types including hepatocytes [30, 34–36], dopaminergic neurons [37], skeletal muscle cells [38], pancreatic β cells [39], and dormant tumor cells [40]. It is important to point out that several other ATF6-like proteins have been identified, including OASIS, LUMAN, BBF2H7, CREBH, and CREB3L4 (Tisp40). These ER transmembrane, bZIP transcription factors appear to mediate specialized UPR functions in specific organs and cell types as they exhibit differences in activating signals, tissue distribution, and response element binding [41]. In this article, further discussion of the ATF6 pathway will center on ATF6α.
The IRE1 pathway
The IRE1 protein contains a serine/threonine kinase module and an endoribonuclease (RNase) domain in its cytoplasmic region [9, 10]. Like ATF6, the ER lumenal domain of inactive IRE1 is bound to BiP and then dissociates when ER stress occurs. In yeast, IRE1 is activated by the binding of unfolded proteins with a groove in its ER lumenal domain [42–44], and the IRE1–BiP interaction fosters appropriate IRE1 triggering [45]. However, in mammalian cells, IRE1 activation does not appear to rely on its association with unfolded proteins [46, 47]. In yeast and mammalian cells, oligomerization and clustering of IRE1 in the ER membrane correlates with onset of IRE1 autophosphorylation and RNase activity [44, 48–50].
One function for the IRE1 RNase is to instigate the demise of various RNA substrates. This mechanism, referred to as regulated IRE1-dependent decay (RIDD), was first shown to target mRNA molecules encoding signal-sequence bearing proteins destined for the secretory pathway [51–53]. Thus, RIDD provides one way for the UPR to stem the flow of nascent polypeptides into the stressed ER [54–56]. Equally important, the activated IRE1 RNase excises a 26-nucleotide intron from X-box binding protein 1 (Xbp1) mRNA, initiating a novel spliceosome-independent, cytosolic splicing mechanism that alters the reading frame of the transcript [57–61]. Without modification by IRE1-dependent splicing, the unspliced (u) Xbp1 transcript encodes XBP1u, a bZIP factor unable to transactivate genes. In sharp contrast, the spliced (s) transcript yields XBP1s, a bZIP factor with a carboxyl (C)-terminal transactivation domain [58, 59, 61]. As the nascent XBP1u polypeptide is synthesized, a 26-amino acid (AA) peptide at its C-terminus mediates a translational pause, consequently stabilizing the mRNA-ribosome-nascent chain complex. This brief suspension in XBP1u synthesis provides sufficient time for a slightly upstream hydrophobic region that has already emerged from the ribosome to associate with the ER membrane, thereby facilitating IRE1-dependent generation of Xbp1s transcripts as ribosome-engaged Xbp1u mRNA is brought into the vicinity of ER-localized IRE1 [62, 63].
XBP1s, like ATF6α(N), can regulate gene targets via the ERSE [61]. Additionally, XBP1s activates ER stress-responsive genes via interaction with a promoter motif termed the UPR element (UPRE; TGACGTGG/A) [64, 65], both as XBP1s homodimers and as XBP1s/ATF6α(N) heterodimers [31]. Exerting considerable influence on cellular secretory capacity, XBP1s regulates ER biogenesis [66–68] and enhances expression of genes involved throughout the exocytic pathway. These include gene products that facilitate the entry of nascent polypeptides into the ER (such as SRP54, a component of the signal recognition particle), protein folding and assembly in the ER (such as ERdj4, a co-chaperone and DnaJ/Hsp40 homolog), and vesicular transport (such as SEC23b, an ER-Golgi transport protein) [26, 67, 69, 70].
Xbp1 is required for proper development of the liver and is therefore essential for embryogenesis [71]. Studies employing tissue-specific Xbp1 deletion have revealed a vital role for this factor in a number of specialized secretory cell types such as pancreatic acinar cells [66], salivary gland cells [66], antibody-secreting plasma cells [72, 73], and plasmacytoid dendritic cells [74], all of which possess a highly developed ER network. Of the two IRE1 isoforms, IRE1α is ubiquitously expressed [9], while expression of IRE1β has been identified specifically in gut and bronchial epithelium where it optimizes mucin production [75, 76]. Similar to XBP1, deletion of IRE1α causes embryonic lethality due to liver dysfunction [77]. Interestingly, targeted expression of IRE1α in the placenta rescues this defect [78], allowing for the birth of IRE1α-deficient mice that exhibit several mild, but measurable, phenotypes including hypoinsulinemia, hyperglycemia, and reduced antibody levels [79]. For the purposes of this review, further discussion of the IRE1 pathway will focus on IRE1α.
The PERK pathway
The ubiquitously expressed PERK protein utilizes its cytoplasmic serine/threonine kinase domain to signal downstream events [11, 12]. Analogous to the ATF6 and IRE1 proteins, PERK activation correlates with the release of its ER lumenal domain from BiP [16]. One substrate for PERK is nuclear erythroid 2-related factor, a transcription factor involved in cellular redox homeostasis [80, 81]. However, the most heavily studied and well-characterized PERK target is a translation initiation factor, eukaryotic initiation factor-2 (eIF-2) [82]. Normally, eIF-2 binds guanosine triphosphate (GTP) and brings the initiator Met-tRNAMeti to assembling translational machinery. Phosphorylation of serine 51 on the α subunit of eIF-2 disables the ability of eIF-2B to promote conversion of inactive eIF-2-guanosine diphosphate to active eIF-2-GTP. Consequently, the supply of translation initiation complexes rapidly shrinks [83]. Thus, PERK-mediated phosphorylation of eIF-2α effectively curtails cellular protein synthesis, including the production of nascent polypeptides that would otherwise enter an ER environment unfavorable for folding and maturation [82].
Translational attenuation, however, must be transient if the cell is to efficiently translate new transcripts encoding ER folding assistants and ERAD components that can resolve the problem of mis-folded proteins in the ER. To this end, ATF4 is selectively synthesized through a novel mechanism of translational shunting when the level of phosphorylated eIF-2α (eIF-2α~P) is high and translation initiation complexes are scarce. The ATF4 mRNA contains two small, upstream open reading frames that help direct or shunt the ribosomes to initiate translation at the correct start site for production of ATF4 protein [84, 85]. ATF4 activates gene transcription by binding to CCAAT-enhancer binding protein-activating transcription factor (C/EBP-ATF) response elements (CARE; TGATGXAAX), which consist of half-sites for C/EBP and ATF family members [86, 87]. These CARE motifs are often referred to AA response elements in situations of protein or AA deprivation [88]. A key target for ATF4 is the gene encoding growth arrest and DNA damage-inducible 34 (GADD34) [89], a binding partner for type 1 protein phosphatase (PP1) [90]. GADD34 promotes de-phosphorylation of eIF-2α~P by PP1, allowing for translational recovery [89, 91]. ATF4 also up-regulates expression of many other genes involved in diverse processes including AA metabolism, redox control, protein folding, and autophagy [92–94].
It is important to note that additional stress conditions, besides ER stress, activate distinct eIF-2α kinases, resulting in translational inhibition and induction of ATF4. These eIF-2α kinases include general control nonrepressed 2, activated by nutrient deprivation; heme-regulated inhibitor, stimulated by heme deficiency and oxidative stress; and PKR, turned on by double-stranded RNA in virally-infected cells [82, 83]. With distinct cellular stress responses converging on the eIF-2α~P/ATF4 pathway, this signaling node is considered as the integrated stress response [93, 94].
The PERK pathway provides a means for governance of translation according to the status of the ER, and this is crucial for survival of cells subjected to pharmacologically-induced ER stress in vitro [95]. Moreover, studies of gene-targeted mice revealed that PERK is essential for proper development and function of certain cell types, including specialized secretory cells in the pancreas and skeletal system [96–98]. In humans, loss of PERK causes Wolcott–Rallison syndrome, a disorder involving dysfunction of the exocrine pancreas and liver, neonatal diabetes, skeletal anomalies, and growth retardation [99, 100].
Pro-apoptotic UPR signaling
Despite the concerted efforts of the UPR to adapt to increased demands on the ER, certain ER stress states are incompatible with cell survival. Indeed, under conditions that severely and/or chronically disrupt the ER environment, the UPR can facilitate the execution of stressed cells. Central to ER stress-induced cell death is the mitochondrial apoptosis pathway, a process controlled by the balance of various anti- and pro-apoptotic members of the B cell lymphoma-2 (BCL-2) protein family. These proteins are grouped according to the presence of BCL-2 homology (BH) domains, with pro-apoptotic members harboring BH1, BH2, and BH3 domains or only the BH3 domain [101]. Conformational changes activate the pro-apoptotic BCL-2 family members, BCL-2 associated X protein (BAX) and BCL-2-antagonist or killer (BAK), leading to permeabilization of the outer mitochondrial membrane. Subsequent release of various mitochondrial proteins, such as cytochrome c, triggers caspase activation and apoptosis [102]. A number of UPR-mediated events have been linked to the mitochondrial apoptosis mechanism, and these have been the subject of several recent comprehensive reviews [103–106]. Here, a few examples of such connections are highlighted to further set the stage for how crosstalk within the UPR can affect cell fate during ER stress.
While XBP1s is certainly a pro-adaptive/pro-survival factor, IRE1α signaling can also elicit pro-apoptotic outcomes. For example, the interaction of activated IRE1α with tumor necrosis factor receptor-associated factor 2 leads to activation of apoptosis signal-regulating kinase 1 and its target JUN N-terminal kinase (JNK) [107–109]. JNK-mediated phosphorylation reduces the activity of anti-apoptotic BCL-2 family members, but enhances the function of pro-apoptotic members [110]. Thus, the IRE1α-JNK connection, although incompletely understood, may provide a mechanism whereby the UPR modulates the relative amounts of key anti- and pro-apoptotic factors to promote cell death. More recently, evidence has emerged that IRE1α-directed RIDD couples ER stress to apoptosis by selective cleavage of microribonucleic acids (miRNAs). A subset of miRNAs (miR-17, -34a, -96, -125b) that limit translation of caspase-2 (CASP2) are degraded by IRE1α, allowing for increased synthesis of this pro-apoptotic caspase [111]. In turn, CASP2 cleaves and activates the BH3-only protein BID (BH3-interacting domain death agonist), a critical factor in driving BAX/BAK-dependent apoptosis [112, 113]. IRE1α-mediated decay of miR-17 has also been implicated in stabilizing mRNA encoding thioredoxin-interacting protein (TXNIP), a pro-oxidant protein that promotes activation of the NOD-like receptor protein 3 inflammasome and the apoptotic death of pancreatic β cells under ER stress [114, 115].
A growing body of evidence indicates that IRE1α functions as part of a multi-protein signaling platform, referred to as the ‘UPRosome’, which includes various BCL-2 family members that modulate its activity [116, 117]. For example, the association of BAX and BAK with the cytosolic domain of IRE1α regulates the initiation and duration of IRE1α activity [118]. This checkpoint in IRE1α signaling is calibrated by the levels of the ER transmembrane protein BI-1 (BAX inhibitor), an ER transmembrane protein that antagonizes BAX [119], and bi-functional apoptosis regulator, an ER-associated E3 ubiquitin ligase that promotes BI-1 degradation [120]. In addition, recent work indicates that the BH3-only proteins BCL-2-interacting mediator of cell death (BIM) and p53 up-regulated modulator of apoptosis also interact with IRE1α and sustain its activity as ER stress proceeds [121]. Diminishment of IRE1α activity, concomitant with ongoing PERK signaling, during prolonged ER stress may be pivotal in shifting the UPR toward a pro-apoptotic outcome [122, 123]. Therefore, the apoptosis-unrelated functions of certain BCL-2 family members in managing IRE1α activity may, in fact, affect cell fate determination when the UPR is engaged.
Downstream of PERK, reduced synthesis of secretory pathway cargo and ATF4-induced expression of pro-adaptive genes favors cell survival during ER stress. However, ATF4 also up-regulates expression of C/EBP homologous protein (CHOP, also known as GADD153), a multi-tasking, pro-apoptotic transcription factor [86, 93, 124, 125]. Acting on an ERSE in the CHOP promoter, ATF6α may contribute to transcriptional induction of this factor in the UPR [23]. CHOP collaborates with ATF4 to activate expression of GADD34 and, under conditions of unmitigated ER stress, GADD34-mediated recovery of translation likely exacerbates problems within the ER, leading to cell death [126]. Along these same lines, very recent data indicate that ATF4 and CHOP work together in up-regulating transcription of a large cohort of genes involved in protein synthesis including aminoacyl-tRNA synthetases and translation initiation factors [127]. In so doing, ATF4 and CHOP promote protein synthesis, and this can lead to oxidative stress and cell death if conditions in the ER remain unfavorable for proper protein folding [127]. These findings further illustrate how the delicate balance between repression and restoration of protein synthesis downstream of PERK signaling influences cell fate in the UPR.
Notably, CHOP has been implicated in the down-regulation of BCL-2 [128], an anti-apoptotic factor that sequesters BH3-only proteins that are required for BAX/BAK-dependent apoptosis [101]. In parallel, CHOP contributes to increased expression of pro-apoptotic BH3-only proteins such as BIM [129]. ER oxidase 1α (ERO1α), another transcriptional target of CHOP, facilitates disulfide bond formation in newly synthesized polypeptides in the ER lumen [126]. During chronic ER stress, however, ERO1α may promote a hyper-oxidizing ER environment, leading to activation of the ER calcium-release receptor inositol tris–phosphate receptor 1 and apoptosis via calcium-sensing calmodulin-dependent protein kinase II [130]. CHOP has been implicated in regulating expression of other apoptosis-associated proteins including death receptor-5 [131], tribbles-related protein 3 [132], and the ATF5 transcription factor [133], although the contributions of these players to the UPR are not fully defined. Clearly, when ER stress conditions persist, CHOP can affect multiple mechanisms to tilt the balance toward apoptosis.
Regulatory crosstalk in the UPR: much work for PERK
Given the complexity of the ER stress response and its myriad downstream effects, it is not surprising that a variety of regulatory interactions occur within and among the three UPR pathways. For example, while ATF6α(N) and XBP1s are responsible for up-regulating distinct sets of ER stress-responsive genes [26, 31], ATF6α(N)/XBP1s heterodimers induce a separate batch of targets, many of which are involved in ERAD [31]. In contrast, XBP1u can partner with and accelerate the cytosolic degradation of both XBP1s and ATF6α(N) [134–136]. Other potential, albeit incompletely understood, regulatory relationships include negative regulation of ATF6α activation by the ER stress-inducible proteins nucleobindin-1 [137] and Wolfram syndrome 1 [138], enhancement of PERK signaling by a cytosolic splice variant of BiP [139], and inhibition of PERK activity by p58 inhibitor of protein kinase [140, 141], an ER co-chaperone up-regulated by ATF6α(N) and XBP1s [31, 142]. A series of recent publications have reported mechanisms by which PERK-dependent signals can modulate both pro-adaptive and pro-apoptotic UPR outcomes, and these are discussed in detail below.
Cranking-up the ATF6α pathway: PERK holds the keys
Studies of ER stress-responsive gene expression in MEFs lacking PERK [30, 93] and in MEFs expressing a non-phosphorylatable form of eIF-2α (eIF-2αSer51Ala) [97] uncovered the initial clues of a relationship between the PERK and ATF6α pathways. Surprisingly, transcriptional induction of ER chaperones such as BiP, well characterized as ATF6α gene targets, was found to be defective in these cell types deficient in PERK-dependent signaling. ATF4 does not bind to the cis-acting ERSE vital for transcriptional activation of such genes [124], suggesting that another event downstream of PERK might be necessary for the activation and/or function of ATF6α.
Adachi and colleagues [26] provided the first direct evidence linking PERK to ATF6α activation. These investigators demonstrated that ER stress-induced cleavage of full-length ATF6α to generate the active ATF6α(N) transcription factor is weak and not sustained in PERK-deficient MEFs. Extending these studies, Teske et al. [143] showed that ATF6α activation is defective in MEFs expressing non-phosphorylatable eIF-2αSer51Ala and in MEFs lacking ATF4. Furthermore, they evaluated the ATF6α pathway in the livers of wild-type and liver-specific PERK knockout mice after intraperitoneal injection of the ER stress-inducing agent tunicamycin. In this situation of ER stress in vivo, the generation of ATF6α(N) and induction of ATF6α target genes were found to be severely attenuated in PERK-deficient liver tissue [143]. Therefore, an intact PERK/eIF-2α~P/ATF4 pathway is necessary for successful activation of the ATF6α pathway in vitro, and this appears to hold true for in vivo models of ER stress.
While the mechanism(s) that couples PERK-mediated signaling to ATF6α activation is not fully defined, experiments in the Teske et al. [143] study revealed a major role for ATF4 in this process. First, ATF4 promotes increased transcription of Atf6α during ER stress, thereby helping to replenish full-length ATF6α as it is being cleaved to yield ATF6α(N). Next, ATF4 is required for proper movement of ATF6α from the ER to the Golgi during ER stress. Finally, in keeping with its role in ATF6α trafficking, ATF4 is essential for maximal expression of certain genes involved in ER-Golgi transport that are transcriptionally activated in a PERK-dependent manner during the UPR. Together, these data indicate that the PERK/eIF-2α~P/ATF4 cascade ‘cranks-up’ the ATF6α pathway by driving ATF6α synthesis and supplying the machinery to ferry it to the Golgi for proteolytic activation (Fig. 3). Knowledge of this regulatory circuit provides further insight into the phenotypic characteristics of both PERK-deficient cells and mice. Whether reliance on the PERK pathway is a feature for all physiologic modes of ATF6α activation awaits further study.
Fig. 3.
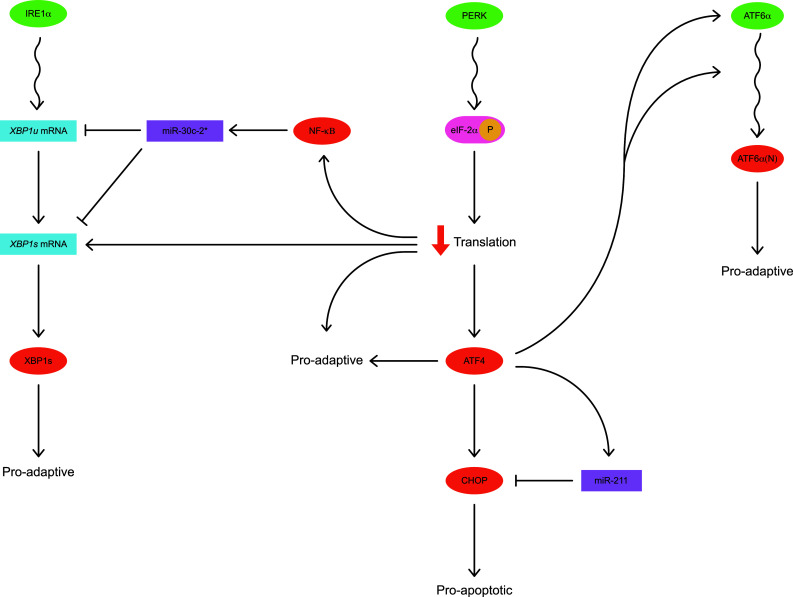
PERK-dependent signals mediate inter- and intra-pathway regulatory crosstalk within the UPR. Translational repression, downstream of PERK, is pro-adaptive as it reduces entry of new client proteins into the stressed ER. In parallel, translational repression leads to induction of ATF4, a transcriptional activator that up-regulates expression of the pro-apoptotic transcription factor CHOP. ATF4 induces expression of miR-211 which negatively regulates CHOP expression in the early UPR. Thus, the ATF4/miR-211/CHOP relationship is an example of intra-pathway regulation within the PERK cascade of the UPR. Furthermore, ATF4 up-regulates expression of ATF6α and promotes trafficking of ATF6α to the Golgi apparatus where it is activated, yielding the pro-adaptive transcription factor ATF6α(N). Hence, ATF4 mediates inter-pathway regulatory crosstalk between the PERK and ATF6α pathways of the UPR. Finally, PERK-dependent translational inhibition leads to stabilization of Xbp1s mRNA, thereby maximizing synthesis of the pro-adaptive transcription factor XBP1s. PERK signaling, via a mechanism involving NF-κB, increases expression miR-30c-2* which can negatively regulate Xbp1 mRNA. Therefore, PERK-dependent signals converge on Xbp1 mRNA, mediating inter-pathway regulatory crosstalk between the PERK and IRE1α pathways of the UPR
Optimizing XBP1s levels: PERK makes it happen
Similar to the initial discovery of the PERK–ATF6α connection, studies of PERK-deficient MEFs [58] and MEFs expressing the non-phosphorylatable eIF-2αSer51Ala [144] provided the first hints that the PERK/eIF-2α pathway governs expression of Xbp1. Calfon and colleagues [58] showed that PERK knockout MEFs fail to up-regulate Xbp1 mRNA during ER stress, but the underlying mechanism was unclear. While ATF6α(N) can induce Xbp1 transcription [59, 61], ATF6α is not essential for induction of Xbp1 mRNA in the UPR [26, 31]. Thus, the inability of PERK-deficient MEFs to up-regulate Xbp1 mRNA cannot be easily attributed to a lack of ATF6α activation in these cells. The Hatzoglou laboratory demonstrated that accumulation of XBP1s protein in the UPR is at least partially dependent on eIF-2α~P [144], suggesting an event(s) downstream of PERK/eIF-2α~P has a positive impact on the generation, stability, and/or translation of Xbp1s mRNA.
To investigate this phenomenon further, the Hatzoglou group examined the relationship between Xbp1 mRNA levels and translational control during the UPR. Using MEFs and thapsigargin-induced ER stress as an experimental system, these investigators tied PERK/eIF-2α~P-mediated translational control to the regulation of Xbp1s mRNA turnover [145]. Specifically, Xbp1s mRNA is stabilized during early (initial 3 h of thapsigargin treatment), but not late (≥7 h of thapsigargin treatment), ER stress. Stabilization of Xbp1s mRNA requires eIF-2α~P and the corresponding inhibition of protein synthesis. Interestingly, for Xbp1s mRNA half-life to increase during the UPR, these transcripts must be derived by IRE1α-mediated cytoplasmic splicing of Xbp1u mRNA. The authors proposed that cytoplasmic splicing somehow ‘marks’ Xbp1s transcripts for stabilization in the early UPR when protein synthesis is repressed [145]. Such a mechanism would allow the cell to stockpile Xbp1s mRNA for translation when repression is eventually lifted, thus ensuring maximal levels of XBP1s protein are achieved. These data spawn a number of intriguing questions. For example, what is the molecular composition of the proposed ‘marks’ on Xbp1s mRNA bestowed by the cytoplasmic splicing process, and does this putative modification shield these transcripts from a degradative mechanism? Also, is turnover of Xbp1s mRNA similarly regulated in physiologic processes involving XBP1s, but in which the PERK/eIF-2α pathway is not engaged, such as the development of antibody-secreting plasma cells [146, 147] and Toll-like receptor-mediated activation of macrophages [148]?
Making matters more complicated is recent work linking a miRNA up-regulated downstream of PERK to negative regulation of Xbp1 expression in the UPR. Employing computational tools and bioinformatics analyses, Byrd and colleagues [149] identified miR-30c-2* (also known as miR-30c-2-3p) as having potential regulatory activity toward a target site in the Xbp1 mRNA 3′ untranslated region. These investigators provided evidence that miR-30c-2* has the capacity to limit the level of Xbp1 mRNA and XBP1s protein, thereby tempering the magnitude of XBP1s-mediated gene transcription in the UPR. Intriguingly, expression of miR-30c-2* increases during ER stress in a PERK-dependent fashion via a mechanism involving NF-κB [149], a transcription factor activated under many conditions including PERK-dependent translational inhibition [150, 151]. Whether miR-30c-2* activity participates in the intricate mechanisms controlling Xbp1 mRNA half-life remains to be determined, as does its potential significance in physiologic processes involving XBP1s. While many questions are unanswered, the emerging story is that PERK/eIF-2α~P-mediated translational control intersects with the IRE1α–XBP1 pathway by fostering robust expression of XBP1s, as well as fine-tuning the amount of this pro-adaptive transcription factor as cells cope with ER stress (Fig. 3).
Timing CHOP expression: PERK keeps the clock
Among the many puzzles in UPR research are the ways cells appropriately coordinate pro-survival and pro-apoptotic signals during ER stress. For example, certain PERK-dependent events, such as translational control and induction of ATF4 target genes that maintain redox homeostasis, clearly promote adaptation and survival, whereas ATF4-driven CHOP expression favors apoptosis. It follows that balancing these distinct signaling outcomes would be fundamental in determining cell fate depending on the severity and duration of a given ER stress condition.
Shedding new light on this aspect of UPR control, Chitnis and colleagues [152] recently identified an ER stress-responsive miRNA that negatively regulates expression of CHOP [encoded by DNA damage-inducible transcript 3 (Ddit3)]. Using MEFs and thapsigargin treatment as the primary model of ER stress, these investigators showed that induction of miR-211 is dependent on PERK/eIF-2α~P-mediated translational inhibition and ATF4. Acting through two sites in the proximal Ddit3 promoter, miR-211 increases histone methylation and impedes Ddit3 transcription, thus limiting synthesis of pro-apoptotic CHOP. Importantly, specific suppression of miR-211 hastened CHOP-dependent apoptosis of cells under ER stress, indicating that Ddit3 is a significant target for miR-211 [152]. Therefore, the PERK/eIF-2α~P/ATF4 pathway mediates both positive and negative regulation of CHOP expression (Fig. 3). This seemingly paradoxical arrangement appears to make sense within the timing of the UPR. After increasing during the initial 5 h of thapsigargin-induced stress, miR-211 levels decrease thereafter, and this corresponds with accumulation of CHOP [152]. Based on these data, the authors proposed that PERK/ATF4-dependent miR-211 induction in the early UPR guards against a premature buildup of CHOP, thereby allowing sufficient time for restoration of ER homeostasis before initiation of a terminal apoptotic outcome. Sustained ER stress, however, leads to diminishment of miR-211, consequently ensuring maximal CHOP production and tilting the balance toward apoptosis. Defining the mechanism by which ATF4 and potentially other factors mediate differential expression of miR-211 in early and late ER stress is of obvious interest as this may represent a decisive ‘switch’ in cell fate determination in the UPR. Likewise, it will be important to evaluate the possible function of miR-211 during physiologic settings of UPR activity such as the tumor microenvironment [6]. In this regard, the Chitnis and colleagues study [152] included evidence that elevated levels of miR-211 in mouse mammary tumors correlate with increased PERK expression and reduced CHOP mRNA. Certainly, the miR-211/CHOP connection, an exciting example of intra-pathway regulation within the PERK branch of the UPR, is ripe for further analysis.
Concluding remarks
Approximately 15 years ago, the PERK protein was identified and characterized [11, 12], providing a molecular explanation for the drastic attenuation of global protein synthesis known to occur when cells encounter ER stress [153, 154]. Since then, a number of excellent studies have yielded a wealth of information regarding how this ER-localized kinase functions through its substrate eIF-2α and downstream effector ATF4 as cells respond to conditions that challenge the ER environment. With effects extending well beyond its immediate impact on translation, the PERK pathway influences the entire UPR as it facilitates ATF6α activation, optimizes XBP1s synthesis, and coordinates temporal expression of CHOP. Thus, the PERK pathway interconnects with both the ATF6α and IRE1α pathways and modulates itself to affect pro-adaptive and pro-apoptotic outcomes at multiple points in the complex circuitry of the UPR.
Acknowledgments
The author apologizes to any investigators whose work in the UPR field was not cited due to the scope of this article. The author thanks Jason P. Clark (University of South Alabama) for expert technical assistance with artwork. J.W.B. was supported by a Grant from the US National Institutes of Health (GM061970).
References
- 1.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 2.Fagone P, Jackowski S. Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res. 2009;50(Suppl):S311–S316. doi: 10.1194/jlr.R800049-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewer JW, Hendershot LM. Building an antibody factory: a job for the unfolded protein response. Nat Immunol. 2005;6:23–29. doi: 10.1038/ni1149. [DOI] [PubMed] [Google Scholar]
- 4.Volchuk A, Ron D. The endoplasmic reticulum stress response in the pancreatic β-cell. Diabetes Obes Metab. 2010;12(Suppl 2):48–57. doi: 10.1111/j.1463-1326.2010.01271.x. [DOI] [PubMed] [Google Scholar]
- 5.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 6.Wang G, Yang Z-Q, Zhang K. Endoplasmic reticulum stress response in cancer: molecular mechanism and therapeutic potential. Am J Transl Res. 2010;2:65–74. [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 8.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5717. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 12.Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 α-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18:7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell. 2011;22:3520–3532. doi: 10.1091/mbc.E11-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. 2013;110:4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 16.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 17.Shen J, Chen X, Hendershot LM, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 18.Schindler AJ, Schekman R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci USA. 2009;106:17775–17780. doi: 10.1073/pnas.0910342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. 2007;27:1027–1043. doi: 10.1128/MCB.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, Lynch KA, Okada T, Aronow BJ, Osinska H, Prywes R, Lorenz JN, Mori K, Lawler J, Robbins J, Molkentin JD. A thrombospondin-dependent pathway for a protective ER stress response. Cell. 2012;149:1257–1268. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong M, Luo S, Baumeister P, Huang JM, Gogia RK, Li M, Lee AS. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. J Biol Chem. 2004;279:11354–11363. doi: 10.1074/jbc.M309804200. [DOI] [PubMed] [Google Scholar]
- 22.Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy B, Lee AS. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999;27:1437–1443. doi: 10.1093/nar/27.6.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 26.Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33:75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- 27.Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366:585–594. doi: 10.1042/BJ20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haze K, Okada T, Yoshida H, Yanagi H, Yura T, Negishi M, Mori K. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem J. 2001;355:19–28. doi: 10.1042/0264-6021:3550019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thuerauf DJ, Marcinko M, Belmont PJ, Glembotski CC. Effects of the isoform-specific characteristics of ATF6α and ATF6β on endoplasmic reticulum stress response gene expression and cell viability. J Biol Chem. 2007;282:22865–22878. doi: 10.1074/jbc.M701213200. [DOI] [PubMed] [Google Scholar]
- 30.Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 32.Bommiasamy H, Back SH, Fagone P, Lee K, Meshinchi S, Vink E, Sriburi R, Frank M, Jackowski S, Kaufman RJ, Brewer JW. ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci. 2009;122:1626–1636. doi: 10.1242/jcs.045625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maiuolo J, Bulotta S, Verderio C, Benfante R, Borgese N. Selective activation of the transcription factor ATF6 mediates endoplasmic reticulum proliferation triggered by a membrane protein. Proc Natl Acad Sci USA. 2011;108:7832–7837. doi: 10.1073/pnas.1101379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cinaroglu A, Gao C, Imrie D, Sadler KC. Activating transcription factor 6 plays protective and pathological roles in steatosis due to endoplasmic reticulum stress in zebrafish. Hepatology. 2011;54:495–508. doi: 10.1002/hep.24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. doi: 10.1038/nature08111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, Mori K. Induction of liver steatosis and lipid droplet formation in ATF6α-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21:2975–2986. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem. 2011;286:7947–7957. doi: 10.1074/jbc.M110.156430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Bostrom P, Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, Kaufman RJ, Spiegelman BM. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab. 2011;13:160–169. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teodoro T, Odisho T, Sidorova E, Volchuk A. Pancreatic β-cells depend on basal expression of active ATF6α-p50 for cell survival even under non-stress conditions. Am J Physiol Cell Physiol. 2012;302:C992–C1003. doi: 10.1152/ajpcell.00160.2011. [DOI] [PubMed] [Google Scholar]
- 40.Schewe DM, Aguirre-Ghiso JA. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. 2008;105:10519–10524. doi: 10.1073/pnas.0800939105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kondo S, Saito A, Asada R, Kanemoto S, Imaizumi K. Physiological unfolded protein response regulated by OASIS family members, transmembrane bZIP transcription factors. IUBMB Life. 2011;63:233–239. doi: 10.1002/iub.433. [DOI] [PubMed] [Google Scholar]
- 42.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pincus D, Chevalier MW, Aragon T, van Anken E, Vidal SE, El-Samad H, Walter P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. Plos Biol. 2010;8:e1000415. doi: 10.1371/journal.pbio.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. 2009;315:2496–2504. doi: 10.1016/j.yexcr.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci USA. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Korennykh AV, Behrman SL, Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci USA. 2010;107:16113–16118. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han D, Lerner AG, Vande Walle L, Upton J-P, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009;186:323–331. doi: 10.1083/jcb.200903014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 54.Iqbal J, Dai K, Seimon T, Jungreis R, Oyadomari M, Kuriakose G, Ron D, Tabas I, Hussain MM. IRE1β inhibits chylomicron production by selectively degrading MTP mRNA. Cell Metab. 2008;7:445–455. doi: 10.1016/j.cmet.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee A-H, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci USA. 2011;108:8885–8890. doi: 10.1073/pnas.1105564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lipson KL, Ghosh R, Urano F. The role of IRE1α in the degradation of insulin mRNA in pancreatic beta-cells. PLoS ONE. 2008;3:e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Back SH, Lee K, Vink E, Kaufman RJ. Cytoplasmic IRE1α-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J Biol Chem. 2006;281:18691–18706. doi: 10.1074/jbc.M602030200. [DOI] [PubMed] [Google Scholar]
- 58.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 59.Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uemura A, Oku M, Mori K, Yoshida H. Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J Cell Sci. 2009;122:2877–2886. doi: 10.1242/jcs.040584. [DOI] [PubMed] [Google Scholar]
- 61.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 62.Yanagitani K, Imagawa Y, Iwawaki T, Hosoda A, Saito M, Kimata Y, Kohno K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol Cell. 2009;34:191–200. doi: 10.1016/j.molcel.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 63.Yanagitani K, Kimata Y, Kadokura H, Kohno K. Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science. 2011;331:586–589. doi: 10.1126/science.1197142. [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. 2003;4:265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 66.Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, Hurt EM, Petroulakis E, Sonenberg N, Yewdell JW, Calame K, Glimcher LH, Staudt LM. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 68.Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sriburi R, Bommiasamy H, Buldak GL, Robbins GR, Frank M, Jackowski S, Brewer JW. Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J Biol Chem. 2007;282:7024–7034. doi: 10.1074/jbc.M609490200. [DOI] [PubMed] [Google Scholar]
- 71.Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–157. [PMC free article] [PubMed] [Google Scholar]
- 72.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 73.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 74.Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204:2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martino MB, Jones L, Brighton B, Ehre C, Abdulah L, Davis CW, Ron D, O’Neal WK, Ribeiro CM. The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol. 2013;6:639–654. doi: 10.1038/mi.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsuru A, Fujimoto N, Takahashi S, Saito M, Nakamura D, Iwano M, Iwawaki T, Kadokura H, Ron D, Kohno K. Negative feedback by IRE1β optimizes mucin production in goblet cells. Proc Natl Acad Sci USA. 2013;110:2864–2869. doi: 10.1073/pnas.1212484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1α is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1α in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iwawaki T, Akai R, Kohno K. IRE1 disruption causes histological abnormality of exocrine tissues, increase of blood glucose level, and decrease of serum immunoglobulin level. PLoS ONE. 2010;5:e13052. doi: 10.1371/journal.pone.0013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem. 2004;279:20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 81.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9:2357–2371. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 83.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- 87.Wolfgang CD, Chen BP, Martindale JL, Holbrook NJ, Hai T. gadd153/Chop10, a potential target gene of the transcriptional repressor ATF3. Mol Cell Biol. 1997;17:6700–6707. doi: 10.1128/mcb.17.11.6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kilberg MS, Balasubramanian M, Fu L, Shan J. The transcription factor network associated with the amino acid response in mammalian cells. Adv Nutr. 2012;3:295–306. doi: 10.3945/an.112.001891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–34873. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- 90.Connor JH, Weiser DC, Li S, Hallenbeck JM, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol Cell Biol. 2001;21:6841–6850. doi: 10.1128/MCB.21.20.6841-6850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ameri K, Harris AL. Activating transcription factor 4. Int J Biochem Cell Biol. 2008;40:14–21. doi: 10.1016/j.biocel.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 93.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 94.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 95.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 96.Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in Perk −/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 97.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 98.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2α kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-β kinase 3, is mutated in patients with Wolcott–Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 100.Senee V, Vattem KM, Delepine M, Rainbow LA, Haton C, Lecoq A, Shaw NJ, Robert J–J, Rooman R, Diatloff-Zito C, Michaud JL, Bin-Abbas B, Taha D, Zabel B, Franceschini P, Topaloglu AK, Lathrop GM, Barrett TG, Nicolino M, Wek RC, Julier C. Wolcott–Rallison syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53:1876–1883. doi: 10.2337/diabetes.53.7.1876. [DOI] [PubMed] [Google Scholar]
- 101.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 102.Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 103.Jager R, Bertrand MJ, Gorman AM, Vandenabeele P, Samali A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol Cell. 2012;104:259–270. doi: 10.1111/boc.201100055. [DOI] [PubMed] [Google Scholar]
- 104.Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis. 2013;18:537–546. doi: 10.1007/s10495-013-0818-6. [DOI] [PubMed] [Google Scholar]
- 105.Rodriguez D, Rojas-Rivera D, Hetz C. Integrating stress signals at the endoplasmic reticulum: the BCL-2 protein family rheostat. Biochim Biophys Acta. 2011;1813:564–574. doi: 10.1016/j.bbamcr.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 106.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim I, Shu C-W, Xu W, Shiau C-W, Grant D, Vasile S, Cosford NDP, Reed JC. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J Biol Chem. 2009;284:1593–1603. doi: 10.1074/jbc.M807308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 110.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, Papa FR, Oakes SA. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gu H, Chen X, Gao G, Dong H. Caspase-2 functions upstream of mitochondria in endoplasmic reticulum stress-induced apoptosis by bortezomib in human myeloma cells. Mol Cancer Ther. 2008;7:2298–2307. doi: 10.1158/1535-7163.MCT-08-0186. [DOI] [PubMed] [Google Scholar]
- 113.Upton J-P, Austgen K, Nishino M, Coakley KM, Hagen A, Han D, Papa FR, Oakes SA. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cell Biol. 2008;28:3943–3951. doi: 10.1128/MCB.00013-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lerner AG, Upton JP, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, Greiner D, Kaufman RJ, Bortell R, Urano F. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1α interactome. Mol Cell. 2009;35:551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36:329–337. doi: 10.1016/j.tibs.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 118.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 119.Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, Reed JC, Glimcher LH, Hetz C. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Mol Cell. 2009;33:679–691. doi: 10.1016/j.molcel.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rong J, Chen L, Toth JI, Tcherpakov M, Petroski MD, Reed JC. Bifunctional apoptosis regulator (BAR), an endoplasmic reticulum (ER)-associated E3 ubiquitin ligase, modulates BI-1 protein stability and function in ER Stress. J Biol Chem. 2011;286:1453–1463. doi: 10.1074/jbc.M110.175232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rodriguez DA, Zamorano S, Lisbona F, Rojas-Rivera D, Urra H, Cubillos-Ruiz JR, Armisen R, Henriquez DR, Cheng EH, Letek M, Vaisar T, Irrazabal T, Gonzalez-Billault C, Letai A, Pimentel-Muinos FX, Kroemer G, Hetz C. BH3-only proteins are part of a regulatory network that control the sustained signalling of the unfolded protein response sensor IRE1. EMBO J. 2012;31:2322–2335. doi: 10.1038/emboj.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lin JH, Li H, Zhang Y, Ron D, Walter P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE. 2009;4:e4170. doi: 10.1371/journal.pone.0004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 125.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Han J, Back SH, Hur J, Lin Y-H, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA, Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 130.Li G, Mongillo M, Chin K-T, Harding H, Ron D, Marks AR, Tabas I. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol. 2009;186:783–792. doi: 10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yamaguchi H, Wang H-G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279:45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 132.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Teske BF, Fusakio ME, Zhou D, Shan J, McClintick JN, Kilberg MS, Wek RC. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell. 2013;15:2477–2490. doi: 10.1091/mbc.E13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tirosh B, Iwakoshi NN, Glimcher LH, Ploegh HL. Rapid turnover of unspliced Xbp-1 as a factor that modulates the unfolded protein response. J Biol Chem. 2006;281:5852–5860. doi: 10.1074/jbc.M509061200. [DOI] [PubMed] [Google Scholar]
- 135.Yoshida H, Oku M, Suzuki M, Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol. 2006;172:565–575. doi: 10.1083/jcb.200508145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yoshida H, Uemura A, Mori K. pXBP1(U), a negative regulator of the unfolded protein response activator pXBP1(S), targets ATF6 but not ATF4 in proteasome-mediated degradation. Cell Struct Funct. 2009;34:1–10. doi: 10.1247/csf.06028. [DOI] [PubMed] [Google Scholar]
- 137.Tsukumo Y, Tomida A, Kitahara O, Nakamura Y, Asada S, Mori K, Tsuruo T. Nucleobindin 1 controls the unfolded protein response by inhibiting ATF6 activation. J Biol Chem. 2007;282:29264–29272. doi: 10.1074/jbc.M705038200. [DOI] [PubMed] [Google Scholar]
- 138.Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, Hayashi E, Ishihara H, Oka Y, Permutt MA, Urano F. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J Clin Invest. 2010;120:744–755. doi: 10.1172/JCI39678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ni M, Zhou H, Wey S, Baumeister P, Lee AS (2009) Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS ONE [Electronic Resource] 4:e6868 [DOI] [PMC free article] [PubMed]
- 140.van Huizen R, Martindale JL, Gorospe M, Holbrook NJ. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2α signaling. J Biol Chem. 2003;278:15558–15564. doi: 10.1074/jbc.M212074200. [DOI] [PubMed] [Google Scholar]
- 141.Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, Katze MG. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc Natl Acad Sci USA. 2002;99:15920–15925. doi: 10.1073/pnas.252341799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, Harding HP, Goodman AG, Harant H, Garrison JL, Taunton J, Katze MG, Ron D. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126:727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 143.Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011;22:4390–4405. doi: 10.1091/mbc.E11-06-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang CC, Li Y, Lopez AB, Chiang C-M, Kaufman RJ, Snider MD, Hatzoglou M. Temporal regulation of Cat-1 (cationic amino acid transporter-1) gene transcription during endoplasmic reticulum stress. Biochem J. 2010;429:215–224. doi: 10.1042/BJ20100286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Majumder M, Huang C, Snider MD, Komar AA, Tanaka J, Kaufman RJ, Krokowski D, Hatzoglou M. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol Cell Biol. 2012;32:992–1003. doi: 10.1128/MCB.06665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gass JN, Jiang H-Y, Wek RC, Brewer JW. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol Immunol. 2008;45:1035–1043. doi: 10.1016/j.molimm.2007.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ma Y, Shimizu Y, Mann MJ, Jin Y, Hendershot LM. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones. 2010;15:281–293. doi: 10.1007/s12192-009-0142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Martinon F, Chen X, Lee A-HL, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Byrd AE, Aragon IV, Brewer JW. MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J Cell Biol. 2012;196:689–698. doi: 10.1083/jcb.201201077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the α subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chitnis NS, Pytel D, Bobrovnikova-Marjon E, Pant D, Zheng H, Maas NL, Frederick B, Kushner JA, Chodosh LA, Koumenis C, Fuchs SY, Diehl JA. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol Cell. 2012;48:353–364. doi: 10.1016/j.molcel.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Prostko CR, Brostrom MA, Brostrom CO. Reversible phosphorylation of eukaryotic initiation factor 2 α in response to endoplasmic reticular signaling. Mol Cell Biochem. 1993;127–128:255–265. doi: 10.1007/BF01076776. [DOI] [PubMed] [Google Scholar]
- 154.Wong WL, Brostrom MA, Kuznetsov G, Gmitter-Yellen D, Brostrom CO. Inhibition of protein synthesis and early protein processing by thapsigargin in cultured cells. Biochem J. 1993;289:71–79. doi: 10.1042/bj2890071. [DOI] [PMC free article] [PubMed] [Google Scholar]