

Abstract
Members of the ADP-ribosylation factor (Arf) family of small GTPases are implicated in vesicle traffic in the secretory pathway, although their precise function remains unclear. We generated a series of 23 clustered charge-to-alanine mutations in the Arf1 protein of Saccharomyces cerevisiae to determine the portions of this protein important for its function in cells. These mutants display a number of phenotypes, including conditional lethality at high or low temperature, defects in glycosylation of invertase, dominant lethality, fluoride sensitivity, and synthetic lethality with the arf2 null mutation. All mutations were mapped onto the available crystal structures for Arf1p: Arf1p bound to GDP, to GTP, and complexed with the regulatory proteins ArfGEF and ArfGAP. From this systematic structure-function analysis we demonstrate that all essential mutations studied map to one hemisphere of the protein and provide strong evidence in support of the proposed ArfGEF contact site on Arf1p but minimal evidence in support of the proposed ArfGAP-binding site. In addition, we describe the isolation of a spatially distant intragenic suppressor of a dominant lethal mutation in the guanine nucleotide-binding region of Arf1p.
INTRODUCTION
ADP-ribosylation factors (Arfs) are small monomeric GTP-binding proteins that comprise a subfamily of the Ras-related superfamily. Arfs were originally identified as necessary in vitro cofactors for cholera toxin-mediated ADP-ribosylation of Gsα (Kahn and Gilman, 1986). However, subsequent work has determined an important, although poorly defined, in vivo role for Arfs in the secretory pathway. The best-studied function of Arfs is in the formation of COPI-coated vesicles from the Golgi (reviewed in Wieland and Harter, 1999). Arfs have also been implicated in the formation of a number of different vesicle coats and in many different steps in secretion (Balch et al., 1992; Boman et al., 1992; Lenhard et al., 1992; Stamnes and Rothman, 1993; Traub et al., 1993; Letourneur et al., 1994; West et al., 1997; Ooi et al., 1998; Hirst et al., 1999). Indeed, mutations of ARF1 in yeast result in pleiotropic defects in the processing of secreted proteins, as well as aberrant morphology of intracellular organelles (Stearns et al., 1990b; Gaynor et al., 1998; Yahara et al., 2001).
Arf proteins are highly conserved and ubiquitous proteins in eukaryotes. Human and yeast Arf1 proteins are 78% identical, and the human ARF1 gene can complement a yeast arf1 mutation (Kahn et al., 1991). Thus, studies of the function of Arf1 in yeast should provide insights that can be generally applied to eukaryotic cells. Saccharomyces cerevisiae has three ARF genes. ARF1 and ARF2 encode proteins that are 96% identical at the amino acid level, form an essential pair, and seem to be functionally homologous (Stearns et al., 1990a). Mutations of ARF1 are reported to result in a variety of phenotypes, including cold sensitivity, slow growth, pleiotropic defects in secretion, and fluoride sensitivity (Stearns et al., 1990a,b; Gaynor et al., 1998; Yahara et al., 2001). Deletion of ARF2 results in no observable phenotype, likely due to low expression of this gene relative to ARF1 (Stearns et al., 1990b). ARF3 does not compensate for the loss of ARF1 and ARF2 and based on sequence comparison with human Arfs, which are subdivided into three classes, represents a separate subclass of Arf protein.
Monomeric GTPases cycle between GDP-bound and GTP-bound states, which are accompanied by large conformational changes, primarily in the so-called “switch” regions (reviewed in Bourne et al., 1991). This cycle is modulated by proteins termed guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). In general, GTP binding is thought to promote binding of the GTPase to target or “effector” proteins, whereas hydrolysis of GTP results in dissociation of these interactions. Such conditional binding to target proteins allows GTPases to function as molecular switches in a number of cellular processes, including secretion, nuclear transport, and signaling. With Arf, the GTPase cycle is proposed to regulate the formation and disassembly of coated vesicles as well as the selection of vesicle cargo (Goldberg, 1999; reviewed in Wieland and Harter, 1999). For Arf proteins, the coupling of protein binding to membrane association is thought to occur via extrusion of the unique myristoylated N terminus upon GTP binding (Antonny et al., 1997).
In this study we aimed to determine the regions of the yeast protein Arf1 that are important for its function in cells. Previous mutagenesis studies have provided valuable information about particular residues such as those involved in nucleotide binding and N-myristoylation and crystallographic studies have identified binding sites for ArfGEF and ArfGAP (Kahn et al. 1995; Goldberg, 1998, 1999; Yahara et al., 2001). However, large portions of Arf1p, including those determined by crystallography to bind other proteins, have not been tested directly for their function in cells. In this study we have used a systematic clustered charge-to-alanine mutagenesis approach (Bass et al., 1991; Bennett et al., 1991; Gibbs and Zoller, 1991) to generate mutations widely distributed on the surface of Arf1p in yeast. This approach is unbiased by expectations based on structure or sequence homology to other GTPases and has been used successfully in the related Rho-family GTPase CDC42 to discover novel phenotypes and to identify regions essential for cellular function (Kozminski et al., 2000). This approach has allowed us to determine directly the importance in vivo of regions of Arf1p implicated by crystallography and biochemistry to be important for such functions as binding to membrane or to other proteins as well as regions for which no prior functional information was available.
MATERIALS AND METHODS
Media and Genetic Manipulations
Yeast media for growth and sporulation are as described previously (Guthrie and Fink, 1991), with the exception that YPD was always supplemented with 50 mg/l adenine sulfate and 20 mg/l tryptophan. In addition, yeast nitrogen base medium was supplemented with 100 mg/l leucine, as needed. Permissive temperature for all experiments was 25°C. Diploids were sporulated by diluting overnight cultures 50-fold into sporulation medium. Asci were digested with Zymolyase. Fluoride-containing medium was made by dissolving solid NaF (Mallinckrodt, Paris, KY) in warm YP agar medium to a final concentration of 30, 40, or 50 mM NaF before the addition of glucose, adenine sulfate, and tryptophan. Yeast were tested for growth on various media by inoculating individual yeast colonies into liquid medium, growing over 2 nights, serially diluting into medium in 96-well plates, and pipetting an aliquot of each dilution onto agar plates. In general, strains were grown in yeast nitrogen base drop-out medium lacking only the appropriate amino acids. Yeast transformations were done by electroporation as described previously (Becker and Guarente, 1991).
In Vitro Mutagenesis of ARF1
Methods were modified from those of Kunkel et al. (1987) and Sambrook et al. (1998) and were described in detail previously (Kunkel et al., 1987; Sambrook et al., 1989; Miller et al., 1996). Briefly, an oligonucleotide bearing the desired mutation was annealed to a single-stranded ARF1 plasmid template containing a high frequency of misincorporated uracils. The second strand of DNA was synthesized in vitro and the resulting plasmid was transformed into an Escherichia coli strain that repaired the misincorporated uracils in the “wild-type” DNA strand by using the mutant oligonucleotide DNA as a template, resulting in incorporation of the desired mutation into ARF1. Table 1 shows a summary of Ala scan mutations made. The correct sequence of each mutation was verified by restriction digestion followed by sequence analysis with an automated sequencing machine. For two alleles, arf1-107 and -108, the designed mutation did not introduce or delete any restriction sites. For these two alleles, the TaqMan assay (Livak et al., 1995) was used to distinguish wild-type from mutant plasmids. TaqMan assay conditions were as described previously (Ranade et al., 2001).
Table 1.
Summary of ARF1 ala-scan mutations
| arf1 Allele | Amino acid substitutions | DNA sequence changesa | Diagnostic restriction sites |
|---|---|---|---|
| 101 | G2A | GGT | Gain HaeIII |
| GCC | |||
| 102 | K7A | AAG | Lose DdeI |
| GCA | |||
| 103 | K16, E17, R19 | AAAGAAATGCGT | Gain BbvI |
| GCAGCAATGGCT | |||
| 104 | D26 | GAT | Gain HpaII |
| GCC | |||
| 105 | K30 | AAG | Gain BanI |
| GCC | |||
| 106 | K36, K38, K41 | AAGTTGAAATTGGGTGAA | Gain BsgI |
| GCCTTGGCATTGGGTGCA | |||
| 107 | E54 | GAA | Noneb |
| GCC | |||
| 108 | K59 | AAG | Noneb |
| GCA | |||
| 109 | D67 | GAT | Gain HaeIII |
| GCC | |||
| 110 | D72, R73, R75 | GACAGAATTAGA | Lose BglII |
| GCCGCAATTGCA | |||
| 111 | R79, H80 | AGACAC | Lose BsmA1 |
| GCAGCC | |||
| 112 | R83, E86 | AGAAACACTGAA | Gain PstI |
| GCAAACACTGCA | |||
| 113 | D93, D96, R97 | GATTCTAACGATAGA | Gain SfaNI |
| GCTTCTAACGCTGCA | |||
| 114 | R99 | CGT | Lose BstUI |
| GCT | |||
| 115 | E102, R104, E105 | GAAGCTAGAGAA | Gain Pst1 |
| GCTGCTGCAGCA | |||
| 116 | R109 | AGA | Gain NlaIII |
| GCT | |||
| 117 | E113, D114, E115 | GAAGATGAATTGAGA | Gain PvuII |
| R117 | GCAGCTGCATTGGCA | ||
| 118 | K127, D129, E132 | AAGCAAGATTTGCCAGAA | Gain HindIII |
| GCTCAAGCTTTGCCAGCA | |||
| 119 | E138, E141, K142 | GAAATCACTGAAAAA | Gain PstI |
| GCAATCACTGCAGCA | |||
| 120 | H146, R149, R151 | CATTCTATTAGAAACCGT | Gain SfaNI |
| GCATCTATTGCAAACGCT | |||
| 121 | E164 | GAA | Gain BspMI |
| GCA | |||
| 122 | E168, E171 | GAAGGTTTGGAA | Gain Bgl I |
| GCCGGTTTGGCA | |||
| 123 | K178 | AAA | Gain HaeIII |
| GCC |
Mutant sequence is below the corresponding wild-type sequence. Altered codons are in bold; altered nucleotides are underlined.
No restriction site changes were introduced by the mutation. TaqMan (see MATERIALS AND METHODS) was used to identify mutants.
Strains and Plasmids
Strains used in this study are listed in Table 2. Plasmids used are listed in Table 3. A detailed list of yeast strains made in this study can be downloaded from the Web supplement. All deletion strains were made by double-fusion polymerase chain reaction (Amberg et al., 1995). To make strain DBY9166, primers that remove the entire ARF1 coding sequence as well as 15 base pairs (bp) upstream and 8 bp downstream were used (see the Web supplement for sequences of all primers used to construct gene deletions). Correct insertion of the arf1::HIS3 deletion construct at the ARF1 locus was confirmed by polymerase chain reaction with primers that lie outside of the ends of the ARF1 deletion construct. The arf1::HIS3 deletion construct was transformed into the diploid made by crossing YPH102 and YPH250 to generate the heterozygous mutant diploid DBY9163. DBY9163 was sporulated and dissected, and a His+ Trp+ spore colony was selected as DBY9166. DBY9169 was constructed using primers that remove the ARF2 open reading frame plus 35 bp upstream and 26 bp downstream. The arf2::HIS3 deletion construct was transformed into a diploid made by crossing YPH102 and YPH250 to generate DBY9168. DBY9168 was dissected to generate DBY9169. The control strain DBY9556, which contains LEU2 integrated downstream of ARF1 and oriented in the same direction as ARF1, was made by transforming the NheI-PstI fragment of pRB2927 into DBY9163. A transformant in which ARF1-LEU2 had integrated at the arf1::HIS3 locus (a Leu+ His− transformant, DBY9172) was selected and dissected to generate DBY9556.
Table 2.
Yeast strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| YPH250 | MATa ura3-52 lys2-801 ade2-101 trp1-Δ1 his3-Δ200 leu2-Δ1 | Sikorski and Hieter, 1989 |
| YPH102 | MATα ura3-52 lys2-801 ade2-101 his3-Δ200 leu2-Δ1 | Sikorski and Hieter, 1989 |
| CKY59 | MATa sec18-1 ura3-52 his4-619 | C. Kaiser |
| DBY9163 | MATa/α arf1∷HIS3/ARF1 trp1-Δ1/TRP1 ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 | This study |
| DBY9165 | MATα ura3-52 lys2-801 ade2-101 leu2-Δ1 his2-Δ200 | This study |
| DBY9166 | MATa arf1∷HIS3 ura3-52 lys2-801 ade2-101 leu2-Δ1 his2-Δ200 | This study |
| DBY9167 | MATa/α arf1∷HIS3/ARF1 ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 | This study |
| DBY9168 | MATa/α arf2∷HIS3/ARF2 trp1-Δ1/TRP1 ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 | This study |
| DBY9169 | MATα arf2∷HIS3 ura3-52 lys2-801 ade2-101 leu2-Δ1 his2-Δ200 | This study |
| DBY9172 | MATa/α ARF1-LEU2/ARF1 trp1-Δ1/TRP1 ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 | This study |
| DBY9176–9178 | MATa/α arf1-101-LEU2/ARF1 ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 | This study |
| DBY9180–9254 | Like DBY9176 & 9178 but with arf1 Ala-scan alleles (two isolates of two independently derived diploids for each) | This study |
| DBY9175 | MATα ARF1-LEU2 ura3-52 lys2-801 ade2-101 leu2-Δ1 his2-Δ200 | This study |
| DBY9556 | MATa ARF1-LEU2 ura3-52 lys2-801 ade2-101 leu2-Δ1 his2-Δ200 | This study |
| DBY9255–9334 | Like DBY9175 but with arf1 Ala-scan alleles (four each, two of each mating type) | This study |
| DBY9521 | MATa/α ARF1-LEU2/ARF1 arf2∷HIS3/ARF2 ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 | This study |
| DBY9335–9374 | Like DBY9521 but with arf1 Ala-scan alleles (two isolates of each) | This study |
| DBY9522 | MATα ARF1-LEU2 arf2∷HIS3 ura3-52 lys2-801 ade2-101 leu2-Δ1 his2-Δ200 | This study |
| DBY9375–9426 | Like DBY9522 but with arf1 Ala-scan alleles (two of each, one of each mating type) | This study |
| DBY9524–DBY9526 | MATa/alpha (arf1-124)-LEU2/ARF1 ura3-52/ura3-52 lys2-801/lys2-801 ade2-101/ade2-101 his3-Δ200/his3-Δ200 leu2-Δ1/leu2-Δ1 | This study |
| DBY9534–DBY9539 | Like DBY9175 but with arf1 Ala-scan allele arf1-124 (three sets of independent isolates from dissection of DBY9524–9526, each with one of each mating type) | This study |
Table 3.
Plasmids used in this study
| Plasmid | Description | Source |
|---|---|---|
| pUC119 | pUC19 with M13 origin of replication | Sambrook et al., 1989 |
| pJJ283a | LEU2 in pUC18 | Jones and Prakash, 1990 |
| pRB1291 | ARF1 in pUC19 | This laboratory |
| PRB1297 | ARF1 in Ycp50 | Stearns et al., 1990 |
| pRB2925 | ARF1 (EcoRI-PstI) from pRB1291 in pUC119 | This study |
| pRB2927 | ARF1-LEU2 in pUC119. BamHI-XbaI (LEU2) of pJJ283 Klenow-filled and inserted into XbaI site in pEC1 just downstream of ARF1. LEU2 and ARF1 are oriented in the same direction. | This study |
| pRB2816–2861 | pRB2927 mutagenized by site-directed mutagenesis to give two isolates of each Ala-scan mutation arf1-101 through arf1-123 | This study |
PJJ283 replaces PJJ252.
Mutant alleles of ARF1 were constructed by in vitro mutagenesis of pRB2927 (see “In Vitro Mutagenesis of ARF1”). The mutant plasmids were digested with NheI and PstI (except for alleles arf1-112, -115, and -119, which contain PstI sites; these were digested with NheI and SphI), and the restriction digestion mixture was transformed into DBY9167 to generate the heterozygous diploid strains DBY9180–9254. Two independent diploids were made for each mutation (with the exception of arf1-119) from independently generated plasmids. These diploids were sporulated and dissected to generate DBY9255–9334. These haploid strains were mated to DBY9169 to generate DBY9335–9374. These strains were dissected, and Leu+ His+ spore colonies were selected to generate DBY9375–9426. The arf1-LEU2 ARF2 (DBY9255–9334) and arf1-LEU2 arf2Δ (DBY9375–9426) haploid strains generated from each of the independent mutant diploid parental strains (DBY9180–9254) were compared with each other for growth at 25°C and all but three of the strains were also tested in duplicate for growth at 11 and 37°C and on fluoride-containing medium to verify that the independent mutants had concordant phenotypes.
Oligonucleotides
Oligonucleotides used are described in the Web supplement. Primers were obtained from Genset (La Jolla, CA) and probes for the TaqMan assay (see below) were purchased from Applied Biosystems (Foster City, CA).
Invertase Westerns
Preparation of protein samples for Western analysis was performed essentially as described previously (Kaiser et al., 1987; Stearns et al., 1990b). Individual yeast colonies were inoculated into 25 ml of YPD medium containing 5% glucose and grown overnight at 25°C. Cultures were diluted back to OD600 0.1 and grown until OD600 0.2–0.45. Cultures were centrifuged and resuspended in YPD containing 0.1% glucose to an OD600 (calculated) of ∼8–12. Then 250 μL of this suspension was added to each of two flasks containing 26 ml of prewarmed 0.1% glucose medium and grown 3 h with shaking at 25 or 37°C. For a subset of strains, cultures were also incubated at 11°C. A 1-ml aliquot of each culture was then taken for measurement of the OD600. The remaining 25 ml was transferred to 50-ml conical tubes containing ice and 40 μl of 1 M sodium azide. Cells were centrifuged, transferred to a Sarstedt tube, centrifuged again to completely remove the supernatant, and quick-frozen in an ethanol/dry ice bath. Protein samples were prepared by adding 1 μl of sample buffer (80 mM Tris pH 6.8, 2% SDS, 0.01% bromphenol blue, 0.1 M dithiothreitol, 10% glycerol, 2 mM phenylmethylsulfonyl fluoride) per 0.02 OD600 unit and 0.1 g of glass beads, vortexing 2 min, and then heating at 95°C for 10 min with occasional vortexing. The samples were transferred to a fresh tube, centrifuged once more to remove any remaining cellular debris, and the supernatant was transferred to a fresh tube and stored at −20°C. A 25-μl aliquot of each sample was run per lane in a 7.5% polyacrylamide gel. Protein was transferred to nitrocellulose membrane (MSI, Westborough, MA) overnight at 250 mA. Nitrocellulose membranes were probed with anti-invertase antibody (Preuss et al., 1991) at 1:2000 and horseradish peroxidase-conjugated protein A at 1:5000 (Cappell, West Chester, PA) and chemiluminescent detection was performed using ECL reagents (Amersham Biosciences UK, Little Chalfont, Buckinghamshire, United Kingdom).
Molecular Modeling
The published structures of human Arf1p bound to GTP (Goldberg, 1998), to GDP (Amor et al., 1994), to ArfGEF (Goldberg, 1998), and to ArfGAP (Goldberg, 1999) were used for modeling Arf1p mutations. The “A” monomer of the Arf1-GDP dimer (protein database code IHUR) (Amor et al., 1994) was used for figures of the monomer. It should be noted that an N-terminal truncation Arf1 protein (Δ1–17) was used to obtain the crystal structures of Arf1-GTP, Arf1p bound to ArfGEF, and Arf1p bound to ArfGAP. By using the program Swiss-Pdb Viewer (Guex and Peitsch, 1997), the root mean square difference between the Cα atoms of human and yeast Arf1p (structure generously provided by Y. Wang and D. Ringe, Brandeis, University) bound to GDP was determined to be 1.39 Å. Because of this high structural similarity between the yeast and human proteins, “yeast” Arf1p models were generated from the human Arf1p structures by changing residues that differ between the two proteins to the appropriate yeast residues without adjusting the backbones of the human Arf1 proteins. Using the program O (Copyright 1990 by Alwyn Jones), each of the yeast Arf1p models was aligned along the Cα atoms against Arf1p in the Arf1p-ArfGAP structure to visualize these structures in the same orientation (Figure 4 and Web supplement). The root mean square differences for each of these alignments were as follows: Arf1p-ArfGAP vs. Arf1p-GDP, 0.831 Å; Arf1p-ArfGAP vs. Arf1p-ArfGEF, 1.073 Å; and Arf1p-ArfGAP vs. Arf1p-GTP, 1.211 Å. All figures shown herein were generated using RASMOL (Sayle and Milner-White, 1995). Interactive views of each of the “yeast” Arf1p protein models are available at the Botstein laboratory Web site at http://genome-www.stanford.edu/Arf1/.
Figure 4.

Localization of lethal alleles on the crystal structure of Arf1p. Dominant lethal alleles are shown in red, recessive-lethal alleles in orange, and alleles that are synthetically lethal with arf2Δ in yellow. All lethal alleles are numbered. Alleles that do not produce a lethal phenotype are colored gray. Nucleotide is shown in dark blue, and magnesium ion in light blue. (A) Arf1p bound to GDP. (B) Arf1p bound to the GTP-analog GppNHp. The switch 1 and switch 2 regions are circled. Each structure on the right is rotated 180o along the vertical axis from the figure on the left.
RESULTS
Mutations of ARF1 were made by changing all charged residues within a window of six amino acids to alanine. Two exceptions were made, however, for amino acids for which specific structural or biochemical data had already been obtained. D26 has been mutated previously (Kahn et al., 1995), so this residue was mutated separately from K30. E54 was shown by crystallography to ligate magnesium in the Arf-GDP conformation, so this residue was mutated separately from K59 (Amor et al., 1994). Most charged residues of Arf1p are clustered with other charged residues. However, the few charged residues that are isolated in the protein were also mutated to alanine in this study. In addition, a single noncharged residue, the site of N-myristoylation (G2), was also mutated to alanine. Yeast Arf1p is 78% identical and 89% similar to human Arf1p at the amino acid level. All but nine of the 45 charged residues of yeast Arf1p are identical in human Arf1p. Of the nine, six represent conservative changes and three represent nonconservative changes (Figure 1).
Figure 1.

Overview of ARF1 Ala-scan mutants. The amino acid sequence of S. cerevisiae Arf1p is shown in large print. Charged residues are shown in blue and numbered (normal font). All charged residues bracketed by a black bar were mutated simultaneously in a single allele (numbered, italics). A single uncharged residue (G2, the site of N-myristoylation) was also mutated to alanine. Residues of human Arf1p that differ from yeast Arf1p are shown in small letters below the yeast Arf1p sequence. Conservative changes are in gray, nonconservative changes in black.
Mutant arf1 alleles were constructed by site-directed mutagenesis and integrated at the chromosomal ARF1 locus by homologous recombination to generate heterozygous arf1/ARF1 diploids (see MATERIALS AND METHODS). Each mutant was made twice independently and verified by restriction digestion and sequence analysis. Two independent haploid isolates of each mutation, in both the ARF2 and arf2Δ genetic backgrounds, were tested for phenotype to ensure that the phenotypes were concordant. Testing for phenotypic concordance decreased the likelihood that any second-site mutations would contribute to the observed phenotype. To track the presence of the mutant ARF1 alleles, the LEU2 gene was integrated downstream of the ARF1 locus. Integration of LEU2 downstream of the wild-type ARF1 gene did not result in any phenotypic difference from the parental wild-type strain with respect to any of the phenotypes assayed in this study (our unpublished data). Therefore, any phenotypes associated with the mutant ARF1 genes marked by LEU2 can be ascribed to the specific mutation in ARF1 and not to the presence of the LEU2 gene downstream of ARF1. In all assays of phenotype, an appropriate ARF1-LEU2 control strain was used.
Phenotypes of arf1 Mutants
Each mutant was analyzed in three different genetic backgrounds: heterozygous mutant diploid (arf1/ARF1, ARF2/ARF2), haploid with wild-type ARF2 (arf1 ARF2), and haploid in the absence of any wild-type Arf1 or Arf2 protein (arf1 arf2Δ). The resulting growth phenotypes are summarized in Figure 2. Lethal mutations were recovered in each genetic background analyzed. Two mutations (arf1-109 and arf1-118) conferred a dominant lethal phenotype. This is inferred from the finding that heterozygous mutant diploid strains could not be recovered for these mutations. One mutation, arf1-113, was incompletely dominant lethal, a phenotype that has not been previously described for an arf1 mutation. The heterozygous mutant diploid grows less well than the wild-type diploid, and the haploid (arf1-113 ARF2) is lethal. Seven alleles were synthetically lethal with the arf2 null mutation, and in this respect have the same phenotype as an arf1 null mutation. This result is consistent with previously published phenotypes for two mutations, arf1[G2A] (this study, arf1-101), which removes the myristoylation site of Arf1p, and arf1[D26A] (this study, arf1-104), which results in decreased affinity for GTP (Kahn et al., 1995).
Figure 2.

Summary of phenotypes of arf1 ala-scan mutants. Growth phenotypes at three temperatures (11, 25, and 37°C) and on NaF-containing medium, as well as invertase glycosylation phenotypes (SEC) are summarized for each arf1 ala-scan allele in three different genetic backgrounds: heterozygous diploid (arf1/ARF1, ARF2/ARF2), haploid (arf1 ARF2), and haploid with ARF2 deleted (arf1 arf2Δ). Mutations that are inviable in a particular genetic background are colored black, those with no observed phenotype are colored white, and those with a phenotype are represented with colors along a gradient from red (severe phenotype) to yellow (mild phenotype).
Each of the mutants was also tested for growth at high, low, and permissive temperature (11, 25, and 37°C). Several alleles confer slow growth at one or more temperatures analyzed, much like the null mutant, which is slow growing at all three temperatures. The arf1Δ mutation is reported to be cold sensitive (Stearns et al. 1990a). In this study arf1Δ was found to be slow growing but viable at 11°C. In contrast, several of the mutant alleles generated in this study display a conditional lethal phenotype at high and/or low temperature. The alleles arf1-105 and arf1-114 are both cold and temperature sensitive, whereas arf1-107 and arf1-110 are temperature sensitive only. These alleles are all cold and/or temperature sensitive in the wild-type ARF2 background, and thus have a more severe phenotype than the arf1 null mutant. This suggests that these mutations do not result simply in loss of function, but rather confer aberrant function to the protein.
The arf1Δ mutant is unable to grow in medium containing fluoride ion (Stearns et al., 1990a). Several mutations isolated in this study also have a fluoride-sensitive growth defect. Three alleles, arf1-101, -104, and -105, have fluoride-sensitive growth defects comparable with that of the null mutation, whereas alleles arf1-102, -103, -106, -110, and -114 have a milder defect. The fluoride-sensitive growth defect of arf1-103 is apparent only in the arf2Δ background.
Mutations in ARF1 have been shown to result in defects in the processing of a number of secreted proteins, including invertase (Stearns et al., 1990b; Gaynor et al., 1998; Yahara et al., 2001). This enzyme, which hydrolyzes sucrose, is encoded by the yeast SUC2 gene. A cytoplasmic form of invertase is constitutively expressed, whereas the secreted form of invertase is produced only in response to glucose limitation (Carlson and Botstein, 1982). The secreted form of invertase is core glycosylated in the endoplasmic reticulum and further glycosylated in the Golgi apparatus. In wild-type cells, visualization of invertase by Western analysis resolves the cytoplasmic form as well as the heterogeneously glycosylated secreted invertase that runs as a smear between 100 and 140 kDa (Esmon et al., 1981). The arf1 null mutant shows an invertase processing defect that is characterized by a downward shift in the molecular weight range of glycosylated invertase (Stearns et al., 1990b). This underglycosylation of invertase does not represent a block in secretion of the protein, however, because invertase is secreted to the cell surface at rates that have been estimated to be “comparable to wild-type” or “4.5-fold slower than the wild-type transport half-time of one minute,” respectively (Stearns et al., 1990b; Gaynor et al., 1998).
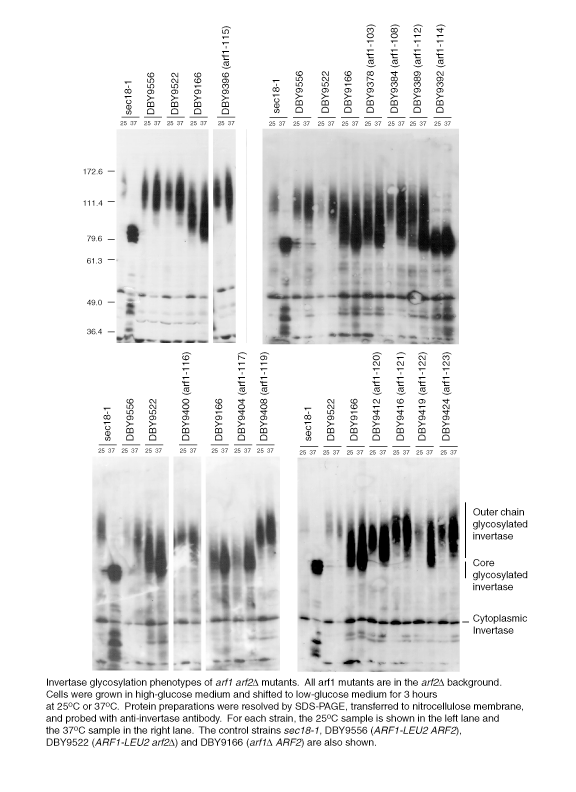
The invertase glycosylation phenotype was determined for all mutant alleles in the ARF2 and arf2Δ genetic backgrounds at both 25 and 37°C. Examples of invertase glycosylation phenotypes for some arf1 ARF2 mutants are shown in Figure 3A (see Web supplement for complete Western analysis data) and the phenotypes for all mutants are summarized in Figure 2. In Figure 3A, the glycosylation defect of a temperature-sensitive allele of SEC18 is shown for comparison. SEC18 encodes the yeast NSF protein, and the sec18-1 allele shows a complete endoplasmic reticulum (ER) block at high temperature and thus only the core-glycosylated form is visible (Esmon et al., 1981). Invertase glycosylation defects represent the most commonly observed phenotype of the arf1 mutant alleles. Of the 20 viable haploid arf1 mutant strains, 14 show some degree of invertase glycosylation defect in the ARF2 and/or arf2Δ genetic backgrounds. Two alleles (arf1-105 and -114) have a more severe invertase glycosylation defect than the null mutant, as defined by a lower molecular weight range of glycosylated invertase. It is interesting to note that although the molecular weight range of invertase glycosylation is very low compared with wild type for the arf1-105 mutant in particular, it is clearly distinguishable from the defect of sec18-1. Thus, arf1-105 would seem not to result in a complete ER block. The remaining alleles that have an invertase processing defect show a phenotype that is either comparable with or less severe than that of the arf1 null mutant.
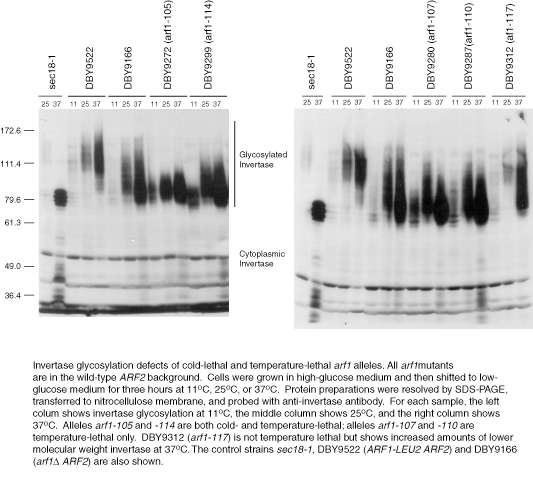
Figure 3.

Examples of invertase glycosylation defects of arf1 ala-scan mutants. Synthesis of the secreted form of invertase was induced by growth in low-glucose medium at various temperatures and detected with anti-invertase antibody. (A) Wild-type, arf1Δ, sec18-1, or arf1 ala-scan mutants cultures were incubated at 25°C (left column in each pair of lanes grouped by a black bar) and 37°C (right column). (B) Same as in A except cultures were grown at 11°C (left column of each set of lanes grouped by a black bar), 25°C (middle), and 37°C (right).
The effect of high and low temperature on cold-sensitive (CS) and temperature-sensitive (TS) alleles (CS/TS: arf1-105, arf1-114; TS arf1-107, and arf1-110) of ARF1 was also analyzed. At 11°C, the invertase glycosylation defect of arf1-114, in particular, is more severe than at permissive temperature, whereas that of arf1-105 is roughly comparable with that at the permissive temperature (Figure 3B). In contrast, the severity of the glycosylation defect of these mutants or of the temperature lethal alleles arf1-107 and arf1-110 does not seem to be appreciably greater at high temperature vs. permissive temperature (Figure 3A). As with both the wild-type and arf2Δ strains, there is more glycosylated invertase present in the 37°C samples and a slight downward shift in its molecular weight. However, these changes do not correlate with temperature sensitivity for the null or wild-type strains. Thus, with the exception of arf1-114, the conditional lethality of these strains does not seem to be correlated with the severity of the invertase glycosylation defect at high or low temperature and suggests that the lethality of these alleles is due to perturbation of some other cellular function.
This set of mutations also allowed us to address the question of whether the various phenotypes of the arf1 null mutation can be dissected apart from each other. As shown in Figure 2, most mutants have overlapping phenotypes and there is no clear distinction between mutants with certain phenotypes and those with other phenotypes. However, one mutation, arf1-107, is notable in that it has an invertase secretion defect comparable with the null mutation and yet exhibits no fluoride sensitivity. This result suggests that at least the phenotypes of fluoride sensitivity and defective invertase glycosylation may result from perturbation of distinct molecular functions of Arf1p.
Structure-Function Relationships
The amino acids mutated in this study were mapped onto the available crystal structures for Arf1 protein: Arf1p bound to GTP, Arf1p bound to GDP, Arf1p complexed with ArfGEF, and Arf1p complexed with ArfGAP (see MATERIALS AND METHODS) (Amor et al., 1994; Goldberg, 1998, 1999). Figure 4 shows the distribution of all lethal mutations (see Web supplement for distribution of alleles with other phenotypes). All mutations with dominant growth defects (dominant lethal, arf1-109 and -118; or incompletely dominant lethal, arf1-113) map near the nucleotide-binding site. Residue D67 (mutated in arf1-109) ligates magnesium via a water molecule in the GTP conformation of Arf and is conserved in all GTPases (Bourne et al., 1991; Goldberg, 1998). Arf1-118 contains two mutations (K127A and D129A) within the so-called guanine specificity region (consensus sequence NKxD), which is highly conserved in small GTPases (Bourne et al., 1991). Mutant allele arf1-113 has an incompletely dominant phenotype. Analysis of the crystal structure of Arf1p reveals that residue D93 of arf1-113 forms a salt bridge with K127 of the guanine nucleotide specificity region. Therefore, D93 may be important for maintaining the precise positioning of K127, and the phenotypic effects of the arf1-113 allele are likely to be secondary to perturbation of this interaction.
Mutations that are synthetically lethal with the arf2 null mutation are shown in yellow in Figure 4. One explanation for the phenotype of synthetic lethality with arf2Δ is that these residues represent essential regions of class I Arf protein (combined Arf1p and Arf2p) and that some amount of wild-type protein, normally supplied by Arf2p, is required for viability. It is notable that all of the mutations that are synthetically lethal with arf2Δ, as well as those with dominant effects on viability discussed above, map to one hemisphere of Arf1p (Figure 4).
The distribution of all mutant alleles (lethal and nonlethal) on the structures of Arf1p bound to GDP and to GTP is also shown in Figure 4. The arf1 ala-scan mutations are relatively evenly distributed over the surface of the protein, with the exception of a patch of hydrophobic residues, which includes the Switch1 domain. Also, most mutated residues are indeed on the surface of the protein, as expected for mutations made by a clustered charge-to-alanine approach. The few residues not on the surface of the protein are involved in nucleotide (D26 and K30) or metal (E54 and D67) binding.
Mutations in Proposed ArfGEF- and ArfGAP-binding Regions of Arf1p
All known ArfGEF proteins contact Arf1p via a conserved so-called SEC7 domain of the GEF (reviewed in Cherfils and Chardin, 1999; Jackson and Casanova, 2000). The contact surface of the SEC7 domain consists of a hydrophobic groove and a hydrophilic loop termed the FG loop. Residues primarily in the switch 1 and switch 2 regions of Arf1p contact the hydrophobic groove of SEC7, and the FG loop of SEC7 inserts into the nucleotide-binding site of Arf1p (Goldberg, 1998). Three residues of Arf1p that bind the GEF were mutated in this study. R73, mutated in the arf1-110 allele, is one of the switch 2 residues that contacts the GEF, and K30 (mutated in arf1-105) and R99 (arf1-114) contact the FG loop (Figure 5). These latter two mutations are notable in that they are related both structurally and phenotypically: they are the only mutations isolated in this study with cold-sensitive and invertase glycosylation phenotypes more severe than the null mutant. This concordance of phenotype and structure suggests that these phenotypes are due to perturbations in the interaction between Arf1p and ArfGEF.
Figure 5.

Cold-sensitive mutations of Arf1p bind the catalytic loop of ArfGEF. (A) Residues of Arf1p (white) that contact ArfGEF (gray) are shown in yellow and purple. Those residues of Arf1p that when mutated confer a conditional lethal phenotype at low temperature are shown in purple. The catalytic loop (FG loop) of ArfGEF is shown in blue. Residues of Arf1p that contact ArfGEF and that were mutated in this study are underlined. (B) Detailed view of the interaction between Arf1p and the catalytic loop of ArfGEF. All color designations are as in A.
The crystal structure of Arf1p bound to GDP and to the minimal catalytic domain of ArfGAP has been solved (Goldberg, 1999). The interface between Arf1p and ArfGAP consists of switch 2 and helix α3 of Arf1p and one β strand and two α helices of ArfGAP. All but one of the residues that lie in the ArfGAP-binding site of Arf1p were mutated in this study (Figure 6). Mutations of residues in the ArfGAP-binding region do not produce a single common phenotype. Mutations in the more N-terminal portion of the binding site (arf1-110 and arf1-111) are synthetically lethal with the arf2 null, whereas mutations in the C-terminal portion of the binding site (arf1-115, arf1-116, and arf1-117) have no phenotype in the ARF2 background. Of these three alleles, only arf1-117 shows any phenotype, a defect in invertase glycosylation, even in the arf2Δ background.
Figure 6.

Allele numbers are shown in parentheses. Localization of residues of Arf1p that contact ArfGAP. Residues of Arf1p that contact ArfGAP (yellow) are labeled and shown in green. Those Arf1p residues in this binding site that were mutated in this study are underlined. The only residue of Arf1p which contacts ArfGap and which was not mutated in this study is 106 (E106 in human Af1p, V106 in yeast. Other residues included in these mutations (arf1-110, -111, -115, -116, and -117) but that do not contact ArfGAP residues are shown in gray. The zinc ion in the zinc-finger motif of ArfGAP is shown in magenta, GDP is shown in dark blue, and magnesium ion in light blue. (B) Same as in A, except that the complex has been rotated 90o clockwise along the vertical axis.
Identification of an Intragenic Suppressor of a Dominant Lethal Mutation
In the process of sequencing all ARF1 mutant plasmids to verify the correct sequence for each mutation, an arf1 mutant carrying two separate mutations, corresponding to two oligonucleotide-directed changes, was discovered. This mutant (designated arf1-124) carried the mutation corresponding to arf1-118 (K127A, D129A, and E132A) as well as that corresponding to arf1-117 (E113A, D114A, E115A, and R117A). To verify the phenotype of this serendipitously discovered double mutant, the double mutation was remade de novo and analyzed in three independent isolates. Interestingly, although the mutant arf1-118 alone is dominant lethal, the double mutant was found to be viable, both as a diploid and as a haploid in the wild-type ARF2 background. Thus, arf1-117 is an intragenic suppressor of arf1-118 dominant lethality. As discussed above, the mutation arf1-118 disrupts residues in the guanine specificity region. As shown in Figure 7, arf1-117 is physically separated on the surface of the protein from the arf1-118 mutation and lies in the crystallographically determined ArfGAP-binding region of Arf1p
Figure 7.

Mutation arf1-117 suppresses the dominant lethality of arf1-118 and maps to the ArfGAP-binding domain of Arf1p. Arf1p (white) residues mutated in arf1-118 are shown in red and those in arf1-117 are in green. Arf1GAP is shown in yellow and the zinc ion in the zinc-finger motif is shown in magenta. GDP bound to Arf1p is shown in dark blue and magnesium ion in light blue. The structure on the right is rotated 180o along the vertical axis from the figure on the left.
DISCUSSION
We report herein the first systematic mutagenesis of the small GTPase Arf1p. By relating the phenotypes of these mutants to the available crystal structures of Arf1p we have been able to identify a number of regions of the protein important for its function in cells.
Mutations in N Terminus of Arf1p
The myristoylated N terminus of Arf proteins is unique to this subclass of Ras-related monomeric GTPase proteins. It is thought to be extruded from the protein upon GTP binding and to be critical for association of Arf proteins with cellular membranes (Antonny et al., 1997). The structure of the N terminus may also influence nucleotide binding and hydrolysis (Amor et al., 2001). In this study we confirm the finding that mutation of the myristoylation site results in a null-like phenotype with respect to growth and fluoride sensitivity (Kahn et al., 1995), and extend this observation to include a null-like defect in invertase glycosylation, an assay of secretory function. Thus, for all phenotypes assayed thus far, myristoylation of Arf1p seems to be essential for the function of Arf1p in cells. In addition, positively charged residues in the N terminus have been postulated to be important for membrane binding by participating in electrostatic interactions with the lipid bilayer (Amor et al., 1994; Antonny et al., 1997). The mutation arf1-102 (K7A) results in a phenotype very similar to that of the myristoylation site mutation. Although K7 is an essential residue, it, and the N terminus in general, is poorly conserved, implying that the precise position of positively charged residues in the N terminus is not important for these interactions. Alternatively, K7 lies just adjacent to the consensus sequence for N-myristoyl transferase (Met-Gly-X4-Ser/Thr), which catalyzes the transfer of myristoyl-CoA to protein, and either lysine or arginine is preferred at this position (Resh, 1999). Thus, K7 may contribute to efficient myristoylation of Arf1p and different Arf isotypes may be myristoylated with different efficiencies. Mutation of Arf1p residues K16, E17, and R19 (arf1-103), in contrast, results only in mild phenotypes, suggesting that their contribution to membrane binding of Arf1p is modest.
Mutations in Proposed ArfGEF-binding Site of Arf1p
Our results strongly support the proposed binding site for ArfGEF on Arf1p determined by crystallography (Goldberg, 1998). Only two mutations isolated in this study result in cold-sensitive and invertase glycosylation defects more severe than those of the arf1 null and these mutations cluster in a region of Arf1p that contacts the catalytic (FG) loop of ArfGEF. Nucleotide exchange on Arf1p is proposed to involve a ternary complex of Arf, nucleotide, and GEF in which the positioning of a critical glutamine (“glutamine finger”) of the GEF in the Arf active site results in steric and hydrostatic repulsion of the nucleotide and Mg2+ and thus promotes nucleotide release (Mossessova et al., 1998; Goldberg, 1999). Arf1p residue K30 (arf1-105) contacts E97 of ArfGEF, the glutamine finger. Previous biochemical studies have demonstrated that E97 is critical for catalytic activity and that mutation of this residue results in the aberrant formation of a stable but catalytically inert complex between Arf1p and ArfGEF and inhibits nucleotide exchange on Arf1p by wild-type ArfGEF (Béraud-Dufour et al., 1998; Betz et al., 1998; Cherfils et al., 1998; Mossessova et al., 1998). Additional functional data also suggest that interactions between Arf and the FG loop of ArfGEF are indeed important in vivo. Mutation of ArfGEF E97 to lysine results in the emb30 mutant of Arabidopsis (Shevell et al., 1994), and in yeast, the ArfGETs GEA1 and GEA2 were identified as high-copy suppressors of the semidominant cold-sensitive mutations arf2-G29A and arf2-T31N, which lie to either side of K30 (arf1-105) (Peyroche et al., 1996). The binding partner of Arf1 R99 (arf1-114), SEC7 Q99, is also important for catalytic activity, because mutation of this residue results in a 90% loss of exchange activity relative to wild-type protein (Mossessova et al., 1998). We hypothesize that the primary role of Arf1p R99 is to stabilize the FG loop of ArfGEF for interaction of ArfGEF E97 and Arf1p K30. Residue R99 is completely conserved in all Arf proteins but is not generally conserved in other small GTPases, further suggesting its importance for the specific interaction between Arf and ArfGEF proteins. Based on this cumulative evidence, we propose the biochemically testable hypothesis that the severe cold-sensitive and invertase glycosylation phenotypes of arf1-105 and arf1114 are due to a dominant negative mechanism whereby ArfGEF protein is sequestered in a catalytically inactive complex with the mutant Arf1p proteins.
Mutations in Proposed ArfGAP-binding Site of Arf1p
In contrast to the findings relative to the ArfGEF-binding site, most mutations in the proposed ArfGAP-binding region result in surprisingly mild phenotypes. Mutations that alter the interaction between Arf1p and ArfGAP, which is required for GTP hydrolysis, might have been expected to produce phenotypes that resemble a GTPase-deficient Arf1p mutant (dominant lethality) or ArfGAP null mutations (for example, fluoride, cold, temperature sensitivity, and defective invertase glycosylation) (Kahn et al., 1995; Zhang et al., 1998; Blader et al., 1999; Poon et al., 1999). Mutation arf1-110 does have a temperature-sensitive phenotype more severe than the null mutant as well as phenotypes (for example, fluoride sensitivity) similar to those of ArfGAP null mutants. However, this mutation contains one residue in the proposed ArfGAP-binding site and one residue in the proposed ArfGEF-binding site, complicating interpretation of these phenotypes. The mutations arf1-110 and arf1-111 are lethal in the arf2Δ background, indicating their functional importance, but arf1- 115, arf1-116, and arf1-117 have either no phenotype or a very mild phenotype. The absence of phenotypes for mutations arf1-115 and arf1-116 is surprising, given that helix α3 of Arf1 (residues 100–112) is shown by crystallography to be a key recognition site for ArfGAP (Goldberg, 1999).
These discrepancies between expected and observed phenotypes could be explained if none of these individual mutant alleles alone significantly impair binding to ArfGAP. Alternatively, they may indicate that residues implicated by crystallographic studies to interact with ArfGAP are not critical for the mechanism of GTP hydrolysis. Indeed, the precise mechanism by which ArfGAP catalyzes GTP hydrolysis on Arf proteins remains controversial (Goldberg, 1999; Mandiyan et al., 1999; Szafer et al., 2000). The structure of Arf1p complexed with ArfGAP was solved for the GDP-bound form of Arf1p, the product of the reaction, so no direct information about the role of particular residues in the mechanism of GTP hydrolysis is available. Thus, our results might suggest that other residues, not identified in this structure, are important for GTP hydrolysis. As another alternative, a model has been proposed whereby GTP hydrolysis on Arf includes a tripartite complex of Arf1p, ArfGAP, and coatomer (Goldberg, 1999). Thus, our results could also indicate that the relative contribution of ArfGAP to GTP hydrolyis in this tripartite complex is modest.
The model proposed by Goldberg differs significantly from the mechanism of GTP hydrolysis on other small GTPases such as Ras and Rho in which an arginine residue essential for hydrolysis (an “arginine finger”) is supplied to the nucleotide binding site in trans by the GAP protein (reviewed in Scheffzek et al., 1998). The contribution of an arginine finger by ArfGAP is apparently incompatible with the structure of Arf1p bound to ArfGAP because ArfGAP binds far from the nucleotide-binding site. Goldberg's model is based on the finding that the addition of coatomer to Arf1p and ArfGAP enhances GTP hydrolysis on Arf1p several orders of magnitude and it is suggested that coatomer may contribute an arginine finger residue. However, this model has been challenged by the finding that under certain conditions lipid can also accelerate the rate of GTP hydrolysis and that the addition of coatomer does not further increase this rate (Szafer et al., 2000). Also, the binding site for ArfGAP on Arf1p seems to be incompatible with the crystal structure of another ArfGAP, PAPβ. The structure of PAPβ cannot be fitted to Arf1p along the binding site determined for ArfGAP because in this position, significant overlap occurs between Arf1p and the ankyrin repeats of PAPβ. Finally, some evidence that ArfGAP may indeed provide an arginine finger is provided by the finding that a single arginine residue conserved in ArfGAPs, located near the zinc-finger domain, is necessary for GTP hydrolysis on Arf1p (Mandiyan et al., 1999). Importantly, in this study we have generated several novel mutations in the putative ArfGAP-binding region of Arf1p that will allow the importance of these residues in ArfGAP-mediated GTP hydrolysis to be determined directly.
Isolation of an Intragenic Suppressing Mutation
In this study we also report the discovery of an intragenic suppressor of a dominant lethal mutation. Although mutation arf1-118 is dominant lethal, a double mutation consisting of arf1-118 and arf1-117 (arf1-124) is viable. The dominant lethality of arf1-118 implies an aberrant function of this protein, for example, increased binding with other proteins. We propose that residues in arf1-117 form the binding site for some protein that binds to Arf1p when it is in the conformation conferred by the arf1-118 mutation, and that disruption of this binding site abolishes this dominant negative interaction. Residues in arf1-117 do not alter the residues mutated in arf1-118 directly because the residues of these two mutations are located far apart in the Arf1p crystal structure (Figure 7). Furthermore, although destabilization of the double mutant cannot be ruled out, the mutation arf1-117 alone clearly does not significantly decrease the stability of Arf1p because this mutation has a very mild phenotype on its own (Figure 2). Mutation arf1-118 is in the so-called guanine nucleotide specificity region, mutations of which have been shown by studies in multiple small GTPase family members to result in decreased affinity of protein for nucleotide primarily as a result of an increased nucleotide dissociation rate (Sigal et al., 1986; Walter et al., 1986). This, in turn, has been proposed to result in one or both of the following effects in cells: a shift toward the GTP-bound conformation and interaction with effector proteins or a shift toward nucleotide-free protein and subsequent sequestration of GEFs (Feig et al., 1986; Ziman et al., 1991; Jones et al., 1995; Schmidt et al., 1996; Cool et al., 1999). However, in two-hybrid studies, Arf1-N126I (a mutation in the guanine specificity region) not only binds effector proteins coatomer subunits β and ε but also the GAP proteins Gcs1 and Glo3 (Eugster et al., 2000). This, combined with the observation that arf1-117 maps to the crystallographically determined ArfGAP-binding site (Goldberg, 1999), suggests that the lethality of arf1-118 may be due to aberrant binding to ArfGAP.
To conclude, we performed a systematic structure-function analysis of the surface of the Arf1 protein in yeast. In this study we found that all Arf1p functions seem to require myristoylation of the N terminus, strong support for the location of the proposed ArfGEF interaction site (Goldberg, 1998) and minimal support for the proposed ArfGAP binding site (Goldberg, 1999). These mutations provide the tools for continued genetic and biochemical studies aimed at understanding the precise function of Arf1p in cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yi Wang, Dagmar Ringe, and Jonathan Goldberg for providing crystallographic data. E.C. thanks Bryan Sutton and Mark Breidenbach for assistance with aligning crystal structures; Kirk Anders for critical reading of the manuscript and assistance with molecular modeling and web design; Jonathan Binkley, Kristy Richards, Koustbh Ranade, and Tracy Ferea for helpful discussions; and Katja Schwartz for technical assistance. This work is supported by National Institutes of Health grant GM-46406 (to D.B.) and by a Medical Scientist Training Grant (to E.C.).
Footnotes
Online version of this article contains video and supplementary dataset materials. Online version available at www.molbiolcell.org.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.02–01–0007. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.02–01–0007.
REFERENCES
- Amberg D, Botstein D, Beasley E. Precise gene disruption in Saccharomyces cerevisiae by double fusion polymerase chain reaction. Yeast. 1995;11:1275–1280. doi: 10.1002/yea.320111307. [DOI] [PubMed] [Google Scholar]
- Amor C, Harrison D, Kahn R, Ringe D. Structure of the human ADP-ribosylation factor 1 complexed with GDP. Nature. 1994;372:704–708. doi: 10.1038/372704a0. [DOI] [PubMed] [Google Scholar]
- Amor J, Horton J, Zhu X, Wang Y, Sullards C, Ringe D, Cheng X, Kahn R. Structures of yeast ARF2 and ARL1. Distinct roles for the N terminus in the structure and function of ARF family GTPases. J Biol Chem. 2001;276:42477–42484. doi: 10.1074/jbc.M106660200. [DOI] [PubMed] [Google Scholar]
- Antonny B, Beraud-Dufour S, Chardin P, Chabre M. N-Terminal hydrophobic residues of the G-protein ADP-ribosylation factor-1 insert into membrane phospholipids upon GDP to GTP exchange. Biochemistry. 1997;36:4675–4684. doi: 10.1021/bi962252b. [DOI] [PubMed] [Google Scholar]
- Balch WE, Kahn RA, Schwaninger R. ADP-ribosylation factor is required for vesicular trafficking between the endoplasmic reticulum and the cis-Golgi compartment. J Biol Chem. 1992;267:13053–13061. [PubMed] [Google Scholar]
- Bass S, Mulkerrin M, Wells J. A systematic mutational analysis of hormone-binding determinants in the human growth hormone receptor. Proc Natl Acad Sci USA. 1991;88:4498–4502. doi: 10.1073/pnas.88.10.4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D, Guarente L. High-efficiency transformation of yeast by electroporation. Methods Enzymol. 1991;194:182–187. doi: 10.1016/0076-6879(91)94015-5. [DOI] [PubMed] [Google Scholar]
- Bennett W, Paoni N, Keyt B, Botstein D, Jones A, Presta L, Wurm F, Zoller M. High resolution analysis of functional determinants on human tissue-type plasminogen activator. J Biol Chem. 1991;266:5191–5201. [PubMed] [Google Scholar]
- Béraud-Dufour S, Robineau S, Chardin P, Paris S, Chabre M, Cherfils J, Antonny B. A glutamic finger in the guanine nucleotide exchange factor ARNO displaces Mg2+ and the beta-phosphate to destabilize GDP on ARF1. EMBO J. 1998;17:3651–3659. doi: 10.1093/emboj/17.13.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz S, Schnuchel A, Wang H, Olejniczak E, Meadows R, Lipsky B, Harris E, Staunton D, Fesik S. Sec7 domain and its interaction with the GTPase ADP ribosylation factor 1. Proc Natl Acad Sci USA. 1998;95:7909–7914. doi: 10.1073/pnas.95.14.7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blader I, Cope M, Jackson T, Profit A, Greenwood A, Drubin D, Prestwich G, Theibert A. GCS1, an Arf guanosine triphosphatase-activating protein in Saccharomyces cerevisiae, is required for normal actin cytoskeletal organization in vivo and stimulates actin polymerization in vitro. Mol Biol Cell. 1999;10:581–596. doi: 10.1091/mbc.10.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman AL, Taylor TC, Melançon P, Wilson KL. A role for ADP-ribosylation factor in nuclear vesicle dynamics. Nature. 1992;358:512–514. doi: 10.1038/358512a0. [DOI] [PubMed] [Google Scholar]
- Bourne H, Sanders D, McCormick F. The GTPase superfamily: conserved structure and molecular mechanism. Nature. 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- Carlson M, Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell. 1982;28:145–154. doi: 10.1016/0092-8674(82)90384-1. [DOI] [PubMed] [Google Scholar]
- Cherfils J, Chardin P. GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem Sci. 1999;24:306–311. doi: 10.1016/s0968-0004(99)01429-2. [DOI] [PubMed] [Google Scholar]
- Cherfils J, Menetrey J, Mathieu M, Bras GL, Robineau S, Beraud-Dufour S, Antonny B, Chardin P. Structure of the Sec7 domain of the Arf exchange factor ARNO. Nature. 1998;392:101–105. doi: 10.1038/32210. [DOI] [PubMed] [Google Scholar]
- Cool RH, Schmidt G, Lenzen CU, Prinz H, Vogt D, Wittinghofer A. The Ras mutant D119N is both dominant negative and activated. Mol Cell Biol. 1999;19:6297–6305. doi: 10.1128/mcb.19.9.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmon B, Novick P, Scheckman R. Compartmentalized assembly of oligosaccharides on exported glycoproteins in yeast. Cell. 1981;25:451–460. doi: 10.1016/0092-8674(81)90063-5. [DOI] [PubMed] [Google Scholar]
- Eugster A, Frigerio G, Dale M, Duden R. COPI domains required for coatomer integrity, and novel interactions with ARF and ARF-GAP. EMBO J. 2000;19:3905–3917. doi: 10.1093/emboj/19.15.3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA, Pan B-T, Roberts TM, Cooper GM. Isolation of ras GTP-binding mutants using an in situ colony-binding assay. Proc Natl Acad Sci USA. 1986;83:4607–4611. doi: 10.1073/pnas.83.13.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynor E, Chen C, Emr S, Graham T. ARF is required for maintenance of yeast Golgi and endosome structure and function. Mol Biol Cell. 1998;9:653–670. doi: 10.1091/mbc.9.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs C, Zoller M. Rational scanning mutagenesis of a protein kinase identifies functional regions involved in catalysis and substrate interactions. J Biol Chem. 1991;266:8923–8931. [PubMed] [Google Scholar]
- Goldberg J. Structural basis for activation of ARF GTPase: mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell. 1998;95:237–248. doi: 10.1016/s0092-8674(00)81754-7. [DOI] [PubMed] [Google Scholar]
- Goldberg J. Structural and functional analysis of the ARF1-ARFGAP complex reveals a role for coatomer in GTP hydrolysis. Cell. 1999;96:893–902. doi: 10.1016/s0092-8674(00)80598-x. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch M. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. In: Guide to Yeast Genetics and Molecular Biology. Abelson JN, Simon MI, editors. Vol. 194. San Diego: Academic Press; 1991. [Google Scholar]
- Hirst J, Bright NA, Rous B, Robinson MS. Characterization of a fourth adaptor-related protein complex. Mol Biol Cell. 1999;10:2787–2802. doi: 10.1091/mbc.10.8.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CL, Casanova JE. Turning on ARF. the Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol. 2000;10:60–67. doi: 10.1016/s0962-8924(99)01699-2. [DOI] [PubMed] [Google Scholar]
- Jones S, Litt RJ, Richardson CJ, Segev N. Requirement of nucleotide exchange factor for Ypt1 GTPase mediated protein transport. J Cell Biol. 1995;130:1051–1061. doi: 10.1083/jcb.130.5.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J, Prakash L. Yeast Saccharomyces cerevisiae selectable markers in pUC18 polylinkers. Yeast. 1990;6:363–366. doi: 10.1002/yea.320060502. [DOI] [PubMed] [Google Scholar]
- Kahn RA, Clark J, Rulka C, Stearns T, Zhang C-J, Randazzo PA, Terui T, Cavenagh M. Mutational analysis of Saccharomyces cerevisiae ARF1. J Biol Chem. 1995;270:143–150. doi: 10.1074/jbc.270.1.143. [DOI] [PubMed] [Google Scholar]
- Kahn RA, Gilman AG. The protein cofactor necessary for ADP-ribosylation of Gs by cholera toxin is itself a GTP binding protein. J Biol Chem. 1986;261:7906–7911. [PubMed] [Google Scholar]
- Kahn R, Kern F, Clark J, Gelmann E, Rulka C. Human ADP-ribosylation factors. A functionally conserved family of GTP-binding proteins. J Biol Chem. 1991;266:2606–2614. [PubMed] [Google Scholar]
- Kaiser C, Preuss D, Grisafi P, Botstein D. Many random sequences functionally replace the secretion signal sequence of yeast invertase. Science. 1987;235:312–317. doi: 10.1126/science.3541205. [DOI] [PubMed] [Google Scholar]
- Kozminski K, Chen A, Rodal A, Drubin D. Functions and functional domains of the GTPase Cdc42p. Mol Biol Cell. 2000;11:339–354. doi: 10.1091/mbc.11.1.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel T, Roberts J, Zakour R. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Lenhard JM, Kahn RA, Stahl PD. Evidence for ADP-ribosylation factor (ARF) as a Regulator of in vitro endosome-endosome fusion. J Biol Chem. 1992;267:13047–13052. [PubMed] [Google Scholar]
- Letourneur F, Gaynor E, Hennecke S, Demoliere C, Duden R, Emr S, Riezman H, Cosson P. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Livak K, Marmaro J, Todd J. Towards fully automated genome-wide polymorphism screening. Nat Genet. 1995;9:341–342. doi: 10.1038/ng0495-341. [DOI] [PubMed] [Google Scholar]
- Mandiyan V, Andreev J, Schlessinger J, Hubbard S. Crystal structure of the ARF-GAP domain and ankyrin repeats of PYK2-associated protein β. EMBO J. 1999;18:6890–6898. doi: 10.1093/emboj/18.24.6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Doyle T, Bobkova E, Botstein D, Reisler E. Mutational analysis of the role of hydrophobic residues in the 338–348 helix on actin in actomyosin interactions. Biochemistry. 1996;35:3670–3676. doi: 10.1021/bi952645v. [DOI] [PubMed] [Google Scholar]
- Mossessova E, Gulbis J, Goldberg J. Structure of the guanine nucleotide exchange factor Sec7 domain of human Arno and analysis of the interaction with ARF GTPase. Cell. 1998;92:415–423. doi: 10.1016/s0092-8674(00)80933-2. [DOI] [PubMed] [Google Scholar]
- Ooi CE, Dell'Angelica EC, Bonifacino JS. ADP-ribosylation factor 1(ARF1) regulates recruitment of the AP-3 adaptor complex to membranes. J Cell Biol. 1998;142:391–402. doi: 10.1083/jcb.142.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyoche A, Paris S, Jackson CL. Nucleotide exchange on ARF mediated by Gea1 protein. Nature. 1996;384:479–481. doi: 10.1038/384479a0. [DOI] [PubMed] [Google Scholar]
- Poon PP, Cassel D, Spang A, Rotman M, Pick E, Singer RA, Johnston GC. Retrograde transport from the yeast Golgi is mediated by tow ARF GAP proteins with overlapping function. EMBO J. 1999;18:555–564. doi: 10.1093/emboj/18.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss D, Mulholland J, Kaiser C, Orlean P, Albright C, Rose M, Robbins P, Botstein D. Structure of the yeast endoplasmic reticulum: localization of ER proteins using immunofluorescence and immunoelectron microscopy. Yeast. 1991;7:891–911. doi: 10.1002/yea.320070902. [DOI] [PubMed] [Google Scholar]
- Ranade K, et al. High-throughput genotyping with single nucleotide polymorphisms. Genome Res. 2001;11:1262–1268. doi: 10.1101/gr.157801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochem Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch E, Maniatis T. Molecular Cloning: A Laboratory Manual, 2 ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Sayle R, Milner-White E. RASMOL: biomolecular graphics for all. Trends Biochem Sci. 1995;20:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR, Wittinghoffer A. GTPase-activating proteins: helping hands to complement an active site. Trends Biochem Sci. 1998;23:257–262. doi: 10.1016/s0968-0004(98)01224-9. [DOI] [PubMed] [Google Scholar]
- Schmidt G, Lenzen C, Simon I, Deuter R, Cool RH, Goody RS, Wittinghofer A. Biochemical and biological consequences of changing the specificity of p21ras from guanosine to xanthine nucleotides. Oncogene. 1996;12:87–96. [PubMed] [Google Scholar]
- Shevell D, Leu E, Gillmor C, Xia G, Feldmann K, Chua N. EMB30 is essential for normal cell division, cell expansion, and cell adhesion in Arabidopsis and encodes a protein that has similarity to SEC7. Cell. 1994;77:1051–1062. doi: 10.1016/0092-8674(94)90444-8. [DOI] [PubMed] [Google Scholar]
- Sigal IS, Gibbs JB, D'Alonzo JS, Temeles GL, Wolanski BS, Socher SH, Scolnick EM. Mutant ras-encoded proteins with altered nucleotide binding exert dominant biological effects. Proc Natl Acad Sci USA. 1986;83:952–956. doi: 10.1073/pnas.83.4.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnes MA, Rothman JE. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell. 1993;73:999–1005. doi: 10.1016/0092-8674(93)90277-w. [DOI] [PubMed] [Google Scholar]
- Stearns T, Kahn R, Botstein D, Hoyt M. ADP ribosylation factor is an essential protein in Saccharomyces cerevisiae and is encoded by two genes. Mol Cell Biol. 1990a;10:6690–6699. doi: 10.1128/mcb.10.12.6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns T, Willingham M, Botstein D, Kahn R. ADP-ribosylation factor is functionally and physically associated with the Golgi complex. Proc Natl Acad Sci USA. 1990b;87:1238–1242. doi: 10.1073/pnas.87.3.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafer E, Pick E, Rotman M, Zuck S, Huber I, Cassel D. Role of coatomer and phospholipids in GTPase activating protein-dependent hydrolysis of GTP by ADP-ribosylation factor-1. J Biol Chem. 2000;275:23615–23619. doi: 10.1074/jbc.M003171200. [DOI] [PubMed] [Google Scholar]
- Traub LM, Ostrom JA, Kornfeld S. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J Cell Biol. 1993;123:561–573. doi: 10.1083/jcb.123.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M, Clark SG, Levinson AD. The oncogenic activation of human p21ras by a novel mechanism. Science. 1986;233:649–652. doi: 10.1126/science.3487832. [DOI] [PubMed] [Google Scholar]
- West MA, Bright NA, Robinson MS. A role of ADP-ribosylation factor and phospholipase D in adaptor recruitment. J Cell Biol. 1997;138:1239–1254. doi: 10.1083/jcb.138.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland F, Harter C. Mechanisms of vesicle formation: insights from the COP system. Curr Opin Cell Biol. 1999;11:440–446. doi: 10.1016/s0955-0674(99)80063-5. [DOI] [PubMed] [Google Scholar]
- Yahara N, Ueda T, Sato K, Nakano A. Multiple roles of Arf1 GTPase in the yeast exocytic and endocytic pathways. Mol Biol Cell. 2001;12:221–238. doi: 10.1091/mbc.12.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-J, Cavenagh MM, Kahn RA. A family of Arf effectors defined as suppressors of the loss of Arf function in the yeast Saccharomyces cerevisiae. J Biol Chem. 1998;273:19792–19796. doi: 10.1074/jbc.273.31.19792. [DOI] [PubMed] [Google Scholar]
- Ziman M, O'Brien J, Ouellette L, Church W, Johnson D. Mutational analysis of CDC42Sc, a Saccharomyces cerevisiae gene that encodes a putative GTP-binding protein involved in the control of cell polarity. Mol Cell Biol. 1991;11:3537–3544. doi: 10.1128/mcb.11.7.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}