

Abstract
Acyl-CoA thioesterase (ACOT) activities are found in prokaryotes and in several compartments of eukaryotes where they hydrolyze a wide range of acyl-CoA substrates and thereby regulate intracellular acyl-CoA/CoA/fatty acid levels. ACOT9 is a mitochondrial ACOT with homologous genes found from bacteria to humans and in this study we have carried out an in-depth kinetic characterization of ACOT9 to determine its possible physiological function. ACOT9 showed unusual kinetic properties with activity peaks for short-, medium-, and saturated long-chain acyl-CoAs with highest V max with propionyl-CoA and (iso) butyryl-CoA while K cat/K m was highest with saturated long-chain acyl-CoAs. Further characterization of the short-chain acyl-CoA activity revealed that ACOT9 also hydrolyzes a number of short-chain acyl-CoAs and short-chain methyl-branched CoA esters that suggest a role for ACOT9 in regulation also of amino acid metabolism. In spite of markedly different K ms, ACOT9 can hydrolyze both short- and long-chain acyl-CoAs simultaneously, indicating that ACOT9 may provide a novel regulatory link between fatty acid and amino acid metabolism in mitochondria. Based on similar acyl-CoA chain-length specificities of recombinant ACOT9 and ACOT activity in mouse brown adipose tissue and kidney mitochondria, we conclude that ACOT9 is the major mitochondrial ACOT hydrolyzing saturated C2-C20-CoA in these tissues. Finally, ACOT9 activity is strongly regulated by NADH and CoA, suggesting that mitochondrial metabolic state regulates the function of ACOT9.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-013-1422-1) contains supplementary material, which is available to authorized users.
Keywords: Acyl-CoA thioesterase, Propionyl-CoA, Amino acid metabolism, Fatty acid metabolism, Mitochondrial metabolism, Organic acids
Introduction
Mitochondrial fatty acid oxidation (β-oxidation) is the principal pathway for oxidation of fatty acids and is responsible for the majority of energy production in mammals. However, peroxisomes play an indispensable role in the β-oxidation and chain shortening of fatty acid derivatives not handled by the mitochondria, including very long chain fatty acids, bile acid intermediates, and branched-chain fatty acids (for reviews see [1–3]). Coenzyme A (CoA) is an obligate co-factor in the β-oxidation of fatty acids and the mitochondria, peroxisomes and cytosol contain separate pools of CoA that need to be regulated. Fatty acids are activated to their CoA esters by acyl-CoA synthetases, coded for by 26 genes in mouse and human with multiple cellular localizations [4, 5]. Intracellular levels of CoA depend on the metabolic state of the cell and a supply of CoA is constantly required to maintain β-oxidation. It can be envisaged that the balance of acyl-CoA/CoA is important to maintain optimal β-oxidation of fatty acids, which would involve acyl-CoA thioesterases (ACOTs) (or acyl-CoA hydrolases, EC3.1.2.2) that catalyze the hydrolysis of acyl-CoAs to release the free fatty acid and CoA (for reviews see [6–9]). To date, 15 ACOTs have been identified and subsequently named ACOT1-15 [9–11]. Structurally, ACOT1-6 belong to the α/β-hydrolase superfamily and ACOT7-15 belong to the hotdog-fold protein superfamily [12–17].
Seven ACOT enzymes have been identified with possible localization in mitochondria in human, rat, and mouse, named ACOT2, ACOT7 (variant 2), ACOT9, ACOT11, and ACOT13-15, which have overlapping substrate specificities and tissue expression. ACOT2 was identified as a mitochondrial ACOT that is mainly active on long-chain acyl-CoAs from C14-CoA to C20-CoA [18–20]. ACOT2 expression is markedly influenced in mouse liver by treatment with peroxisome proliferators and by fasting, and additionally in heart by fasting [20, 21]. ACOT2 was implicated in a futile cycle of fatty acid export from mitochondria together with uncoupling protein 3 (UCP3) in mitochondria [22]. ACOT7 (variant 2) is produced by alternative splicing of the Acot7 coding gene resulting in several isoforms of the enzyme, of which variant 2 contains an N-terminal mitochondrial targeting signal [23, 24]. ACOT7 has broad substrate specificity and hydrolyzes mainly medium and long-chain acyl-CoAs [25]. The cytosolic variant of ACOT7 (ACOT7––variant 1) is highly expressed in macrophages and was shown to hydrolyze arachidonoyl-CoA (C20:4-CoA) and has been implicated in inflammation and in production of prostaglandin D2 and E2 [15]. Interestingly, several fatty acid oxidation disorder patients have been detected with lowered cytosolic and mitochondrial ACOT7 activity in fibroblasts [26] and recently it was shown that ACOT7 plays a crucial role in neuronal fatty acid metabolism [27]. ACOT9 and 10 were initially characterized as MT-ACT48 (variant 1 and 2), and were identified due to co-precipitation with Eps15, a plasma membrane clathrin-coated pit protein required for receptor-mediated endocytosis [28]. MT-ACT48 was identified as a mitochondrial acyl-CoA thioesterase with highest activity with myristoyl-CoA (C14-CoA), but also had detectable activity with hexanoyl-CoA (C6-CoA), decanoyl-CoA (C10-CoA) and stearoyl-CoA (C18:0). However, a functional link or an in vivo interaction between Eps15 and ACOT9 could not be established. The cellular localization of ACOT11 (THEM1) has been unclear, but was recently shown to localize to mitochondria [29]. ACOT11 hydrolyzes a range of acyl-CoAs with a relative preference for longer-chain saturated and unsaturated acyl-CoAs and plays an important role in regulation of energy expenditure [29–32]. ACOT13 (THEM2) is mainly localized to mitochondria [33] and hydrolyzes medium- and long-chain acyl-CoAs [33, 34]. ACOT14 (THEM4), for which the cellular localization is still uncertain, and ACOT15, which is a mitochondrial enzyme, mainly hydrolyze saturated and unsaturated medium- to long-chain acyl-CoAs with only low, or no, activity with short-chain acyl-CoAs [11, 35]. In summary, the mitochondria contain several ACOTs that mainly hydrolyze medium- and long-chain acyl-CoAs, but a mitochondrial short-chain acyl-CoA thioesterase has not yet been identified to date. In the present study, we have characterized the mouse ACOT9 in detail, which revealed that ACOT9 is likely the main short-chain ACOT in mitochondria and has a broad, but yet specific, acyl-CoA specificity. In addition to the activity on short-chain acyl-CoAs, ACOT9 also efficiently hydrolyzes medium- and long-chain acyl-CoAs and a number of CoA esters that are intermediates or products of amino acid metabolism. ACOT9 may thus be classified as a “multi-purpose” ACOT that has the potential to regulate lipid and amino acid metabolism in mitochondria.
Materials and methods
Animals and treatments
C57BL/6 male mice were maintained on a normal chow diet (R36, Lactamin, Vadstena, Sweden) or fasted for 12 h prior to killing. All mice were killed by CO2 asphyxiation and the various organs were removed and immediately frozen in liquid nitrogen and stored at −70 °C until preparation of total RNA and protein extracts. The intestine was taken out, rinsed with ice-cold phosphate buffered saline, and divided into four segments of equal length to map the expression along the small intestine. The epithelium layer was scraped off and transferred into tubes containing Trizol (Invitrogen, Life Technologies) or PBS containing protease inhibitors (Complete, EDTA free, protease inhibitor cocktail tablets, Roche Diagnostics GmbH, Mannheim, Germany) prior to freezing in liquid nitrogen. For isolation of mitochondria, livers, brown adipose tissue (BAT), and kidneys were collected from 12-week-old C57BL/6 mice. The tissues were homogenized in ice-cold medium containing 0.25 M sucrose, 10 mM HEPES, 1 mM EGTA, pH 7.4. The homogenates were centrifuged at 800 × g for 10 min, the pellets were resuspended in homogenization medium and re-centrifuged at 800 × g for 10 min. The combined supernatants were centrifuged at 6,000 × g for 10 min and the pellets were re-suspended in homogenization medium and centrifuged at 6,000 × g for 10 min. The mitochondrial pellets were then re-suspended in 1 ml of homogenization medium and pulse sonicated 6 × 15 s with 10 s intervals at about 5 W using a Sonicator 4000 (Qsonica, LLC, Newton, CT, USA) equipped with a micro-tip. The sonicated samples were centrifuged at 21,000 × g for 40 min and the supernatants were used for thioesterase activity measurements. Ethical permission for all animal experiments was obtained from the Animal Experimental Ethical Committee, Stockholm.
Cloning and expression of mouse ACOT9
The open reading frame of mouse Acot9 was amplified from mouse kidney total RNA using the following primers 5′-CCCTCCTCTAGAATGTCTCAAGGATCCCAATAC-3′ and 5′-CTCCTCTAGACTAGGGCTCCACAGGGTAGTCCTT-3′ (Cybergene AB, Stockholm, Sweden), with added XbaI sites indicated in bold. The Acot9 PCR product was cloned into the XbaI site in the pMal-C2X vector (New England Biolabs, Beverly, MA, USA) to express ACOT9 in fusion with maltose binding protein (MBP). The plasmid was fully sequenced, the correct orientation was verified, and then used to transform BL21 (DES3) pLysS cells (Novagen Inc., Madison, WI, USA). For expression of ACOT9, bacteria were cultured in Luria–Bertani medium at 37 °C, in the presence of 50 μg/ml ampicillin, 34 μg/ml chloramphenicol and 1 g glucose, until an optical density (OD) of ~0.6 at 600 nm was reached. Protein expression was induced by the addition of 0.5 mM isopropyl-1-thio-β-d-galactopyranoside and growth was continued at 37 °C for 3 h. The bacteria were harvested by centrifugation, washed in 10 ml 20 mM Tris–HCl, pH 7.4, centrifuged again, and frozen at −20 °C.
Bacterial pellets were thawed and resuspended in BugBuster protein extraction reagent (Novagen) and Benzonase (Novagen), incubated at room temperature for ~15 min and centrifuged at 25,000 × g for 15 min at +4 °C. The supernatant was then used for purification of the MBP-fusion protein on amylose resin as described by the manufacturer (New England Biolabs) and subsequently used for enzyme activity measurements. Protein concentration was determined using the Bradford assay [36].
Determination of acyl-CoA thioesterase activity
Acyl-CoA thioesterase activity was measured spectrophotometrically at 412 nm with 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB). The medium contained 200 mM KCl, 10 mM HEPES, and 0.05 mM DTNB, pH 7.4. An E 412 = 13,600 M−1cm−1 was used to calculate the activity. Since ACOT9 thioesterase activity was inhibited at substrate concentrations higher than 5–10 μM with acyl-CoAs longer than C12, bovine serum albumin (BSA) was added in a molar ratio of BSA/acyl-CoA of 1:4. The effect of CoA on ACOT9 activity was measured in phosphate buffered saline (with no DTNB added) at 232 nm as the hydrolysis of the thioester bond, using an E 232 = 4,250 M−1cm−1 to calculate the activity. For inhibition studies, the enzyme or the sonicated mitochondrial supernatant was mixed with the inhibitor and the reaction was started by addition of substrate. The commercially available acyl-CoA esters were from Sigma-Aldrich or Avanti Polar Lipids Inc. (Alabaster, AL, USA). 4-Phenylbutyryl-CoA, 2-methylnonanoyl-CoA (2-CH3-C9-CoA) and 4,8,12-methyltridecanoyl-CoA (4,8,12-CH3-C13-CoA) were synthesized from the corresponding fatty acids activated with 1,1-carbonyldiimidazole as described previously [37]. Synthesis of the other CoA-esters has been reported before [37–39]. 4,8-Dimethylnonanoyl-CoA (DMN-CoA, 4,8-CH3-C9-CoA) was a kind gift from Prof. Ronald Wanders, Amsterdam Medical Center. The enzyme kinetics was calculated using GraphPad Prism software.
HPLC analysis was further performed on samples with ACOT9 incubated with C12-CoA and isobutyryl-CoA in 1 ml thioesterase medium described above using 3-hydroxy-3-methylglutaryl-CoA (which is not a substrate for ACOT9) as internal standard. Incubations were terminated by acidification to pH 4, as the activity of ACOT9 is inhibited at this pH. An amount of 500 μl of each incubation was injected onto an HPLC system using a C18-column (Ultrasphere 10 mm × 25 cm) pre-equilibrated with 100 % buffer A (100 mM ammonium phosphate, pH 4). Acyl-CoA esters and CoA were eluted using a linear gradient of 100 % buffer A to 100 % buffer B (buffer A: acetonitrile, 50:50) for 20 min followed by isocratic flow with 100 % buffer B for 14 min. CoA and acyl-CoA were detected by UV-detector at 260 nm and analyzed using Chromelion software (Dionex, Thermo Fisher Scientific). The remaining substrates and formed products were quantitated by peak area integration using the internal standard and standard curves for CoA.
Quantitative real-time polymerase chain reaction (PCR)
Total RNA from various tissues of C57BL/6 mice was extracted after tissue homogenization using the MagMax system (Applied Biosystems, Carlsbad, CA, USA). Total RNA concentration was determined spectrophotometrically using a NanoDrop spectrophotometer (NanoDrop Products, Wilmington, DE, USA) and the quality of RNA was determined using the Experion automated electrophoresis system (Bio-Rad Laboratories, Hercules, CA, USA). Only high-quality RNA was used for further analysis. cDNA was synthesized using High-Capacity RNA-to-cDNA Mastermix (Applied Biosystems) and 0.5 μg total RNA from individual tissue samples or 0.5 μg of total RNA from two pools per tissue containing RNA from 3 animals in each pool (depending on the quality of the RNA). In the case of adrenal, gallbladder and the four intestinal segments, RNA from 4–6 animals was pooled into one sample per tissue (depending on RNA quality). Quantitative real-time PCR was performed in an ABI Prism 7000 sequence detection system, using various gene expression assays (Acot9-Mm01616323_m1, Arbp-Mm00725448_s1 and eukaryotic 18S rRNA, Applied Biosystems). 18S RNA was used as a reference gene for tissue expression assay and Arbp as reference gene for fasting regulation in kidney. All samples were run in triplicate. Data were analyzed using the ABI Prism 7000 SDS software and the relative amounts of each mRNA transcript were calculated using the 2−∆∆Ct method.
Antibody preparation and Western-blot analysis
A peptide, [NH2]CEEEELFKQGELNKS[COOH], was synthesized corresponding to amino acids 225–238 of the mouse ACOT9 protein (with a cysteine added at the N-terminal end for coupling of the peptide). Rabbits were immunized with 500 μg peptide conjugated to keyhole limpet hemocyanin, emulsified in Freund’s complete adjuvant. Booster injections of 250 μg of the peptide-conjugate emulsified in Freund’s incomplete adjuvant were later given and bleeds were carried out according to standard protocols. Antibodies were affinity-purified using a column with the peptide conjugated to Epoxy-activated Sepharose 6B (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Tissues were individually homogenized in 50 mM potassium phosphate buffer, pH 7.4, containing protease inhibitors (Complete, EDTA-free, protease inhibitor cocktail tablets, Roche Diagnostics GmbH), centrifuged for 8 min at 4,000 × g and equal amounts of protein were pooled into tissue pools. Forty μg of each tissue protein pool was separated by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 10 % gels. Western blotting was performed by electrophoretic transfer onto nitrocellulose filters (Nitropure, Micron Separations Inc., Westborough, MA, USA) using a Transblot cell (Bio-Rad). The blots were probed with the ACOT9 antibody followed by horseradish peroxidase-conjugated secondary antibody and visualized by enhanced chemiluminescence (Super Signal West Dura, Thermo Scientific).
Sequence analysis
The mouse ACOT9 protein sequence was used to search various databases at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), TreeFam (http://www.treefam.org/) and Ensembl (http://www.ensembl.org) and complete protein sequences were collected from selected species from bacteria to human. Multiple sequence comparisons were aligned using ClustalW, which was also used to produce the phylogenetic trees, and pairwise sequence alignments were performed with the Lipman–Pearson method.
Results
Genomic characterization of Acot9 and Acot10
ACOT9 and 10 were originally identified as “a single” 48-kDa polypeptide that co-precipitated with GST-Eps15 fusion protein in murine and human cell lysates [28]. Subsequent cDNA cloning and sequence analysis identified two transcripts that were 95 and 96 % identical at nucleotide and protein sequence level, respectively. These two transcripts translate into polypeptides of 439 amino acids, containing a mitochondrial N-terminal targeting signal of 21 amino acids. Homology searches suggested that these polypeptides belong to the acyl-CoA thioesterase family of proteins, and activity measurements of recombinant protein verified acyl-CoA thioesterase activity. Therefore, this protein was named mitochondrial acyl-CoA thioesterase of 48 kDa (MT-ACT48). Based on the more recently revised nomenclature for acyl-CoA thioesterases, these two isoenzymes were renamed ACOT9 and 10. The aim of this study was to characterize ACOT9 in more detail in an attempt to assess the physiological function. A BLAST search using the mouse and human Acot9 nucleotide sequences was carried out against the mouse and human genomic databases to map the genes and the organization of the genes. We identified two genes in the mouse (but not human), as expected, one on the X chromosome and a second gene on chromosome 15. The gene on the X chromosome, which codes for ACOT9, consists of 15 exons and the gene spans about 35 kbp in total. In contrast, the gene on chromosome 15 (coding for ACOT10) lacks introns and was not found in any other mammalian genome. The Acot10 gene was shown to be a recently (after the rat–mouse split) duplicated retrogene showing no evidence of pseudogenization in any of the mouse species investigated [40]. Indeed, Acot10 is expressed at very low levels as there are about 15 times more ESTs for Acot9 than for Acot10 in the mouse EST database (http://www.ncbi.nlm.nih.gov/). Based on the apparently very low expression of ACOT10, we focused the further characterization on ACOT9.
ACOT9 is widely expressed in mouse tissues
Previous Northern-blot analysis showed that Acot9/10 (MT-ACT48) mRNA expression was highest in testis, kidney, skeletal muscle, lung, and heart. In this study, we have used real-time PCR to quantitatively determine the mRNA expression of Acot9 and have also expanded the repertoire of tissues. Our results show that Acot9 has the highest expression in testis, followed by brown adipose tissue (BAT), kidney, adrenal gland, lung, white adipose tissue (WAT), and brain, with the mRNA also expressed in intestinal epithelium, spleen, and thymus (Fig. 1a). Interestingly, the tissue with the lowest expression level of Acot9 was the liver.
Fig. 1.

Mouse Acot9 mRNA and protein is widely expressed. a mRNA expression of Acot9. Total RNA was isolated and pooled into two pools per tissue, with 2–3 animals in each pool, or one tissue pool each for gallbladder, adrenal and the intestinal epithelium segments, with samples from 4–6 animals. Samples were analyzed in triplicate and the relative expression was calculated using the 2−ΔΔCt method, using 18 S as a reference gene. The data are shown as the mean expression in each tissue (±range for the tissues analyzed as two RNA pools) relative to the expression in liver set to 1. BAT brown adipose tissue, WAT white adipose tissue. The intestinal epithelium was divided into four equal segments, S1 (proximal intestine) to S4 (distal intestine). b Western-blot analysis of ACOT9 tissue expression. Forty μg of each tissue pool was separated by 10 % SDS/PAGE, blotted and probed with an ACOT9 peptide antibody. The first lane contains the molecular mass standard with the 50- and 70-kDa bands indicated and the band corresponding to ACOT9 indicated by an arrow. BAT brown adipose tissue, WAT white adipose tissue, Int epi prox and dist correspond to the S1 and S4 segments shown in a. Skel. mu (Gast) gastrocnemius skeletal muscle, Skel. mu (Abd) abdominis skeletal muscle
Western-blot analysis was carried out on various mouse tissue lysates using a peptide antibody generated to mouse ACOT9. A major band of about 48 kDa was detected in WAT, kidney, BAT, testis, and brain with weaker expression in lung, heart, and spleen (Fig. 1b). Higher molecular mass bands were seen in heart, skeletal muscle, and weakly in kidney and liver. Control experiments showed that excess recombinant ACOT9 competed out antibody binding to the 48-kDa band while the bands of higher molecular masses, being most prominent in heart and skeletal muscle, remained detectable at similar levels in the absence or presence of the recombinant protein, suggesting that only the 48-kDa band corresponds to ACOT9 (data not shown). The Western-blot data largely confirmed the mRNA data except for testis, skeletal muscle, and intestine, where the protein levels were lower than expected based on the mRNA expression.
Mouse ACOT9 is a “multi-purpose” acyl-CoA thioesterase
We expressed recombinant ACOT9 to characterize its catalytic activity in more detail to provide evidence as to its putative physiological function. Using the spectrophotometric method, we determined K m and V max that was used to calculate K cat/K m for a large variety of acyl-CoA esters, as summarized in Table 1. Comparison of the V max values for straight-chain saturated acyl-CoAs of 2–20 carbon atoms surprisingly revealed three peaks of activity; a very distinct peak with propionyl- and butyryl-CoA (C3- and C4-CoA), a drop in activity with valeryl-CoA (C5-CoA) and hexanoyl-CoA (C6-CoA), a second “peak” with C8-CoA, a clear drop with C9-CoA (nonanoyl-CoA), and a broad peak at C10-C14-CoA followed by a concomitant drop in activity with acyl-CoAs of ≥C16-CoA (palmitoyl-CoA). The drop in V max values with ≥C16-CoA were also accompanied by striking drops in K ms, which “maintained” relatively high K cat/K m values, at least for C16-18-CoAs. The activity with unsaturated acyl-CoAs was much lower than with the corresponding saturated acyl-CoAs, both as judged from the V max values as well as K cat/K m in spite of low K m values.
Table 1.
Kinetic characterization of mouse ACOT9 with various acyl-CoA esters
| V max μmol/min/mg | K m μM | K cat s−1 | K cat/K m M−1 s−1 | |
|---|---|---|---|---|
| Cn-CoA | ||||
| C2 | 27.0 ± 7.3 | 31.3 ± 6.1 | 21.6 ± 5.8 | 7.2 × 105 ± 3.3 × 105 |
| C3 | 49.7 ± 15.5 | 35.4 ± 10.8 | 45.3 ± 6.6 | 12.9 × 105 ± 3.0 × 105 |
| C4 | 45.1 ± 9.5 | 27.3 ± 10.5 | 36.1 ± 7.6 | 13.7 × 105 ± 2.5 × 105 |
| C5 | 16.6 ± 7.6 | 15.1 ± 1.7 | 13.3 ± 6.1 | 9.1 × 105 ± 5.1 × 105 |
| C6 | 21.1 ± 5.9 | 24.0 ± 5.2 | 16.9 ± 4.8 | 7.4 × 105 ± 3.6 × 105 |
| C8 | 34.6 ± 13.5 | 17.4 ± 3.8 | 27.7 ± 10.8 | 15.9 × 105 ± 6.4 × 105 |
| C9 | 8.8 ± 1.8 | 32.3 ± 13.2 | 7.0 ± 1.4 | 2.3 × 105 ± 0.5 × 105 |
| C10 | 38.1 ± 6.8 | 19.9 ± 7.4 | 30.5 ± 5.5 | 17.1 × 105 ± 9.1 × 105 |
| C12 | 26.8 ± 9.2 | 8.9 ± 2.7 | 21.4 ± 7.4 | 25.9 × 105 ± 11.5 × 105 |
| C14 | 26.5 ± 7.3 | 7.8 ± 2.0 | 21.2 ± 5.8 | 29.4 × 105 ± 13.6 × 105 |
| C16 | 8.4 ± 2.0 | 3.1 ± 1.3 | 6.7 ± 1.6 | 24.5 × 105 ± 15.4 × 105 |
| C18 | 5.2 ± 0.9 | 1.8 ± 0.9 | 4.1 ± 0.7 | 25.8 × 105 ± 9.1 × 105 |
| C20 | 1.7 ± 0.5 | 1.8 ± 1.5 | 1.3 ± 0.4 | 10.3 × 105 ± 6.6 × 105 |
| C16:1 | 2.0 ± 0.3 | 1.2 ± 0.4 | 1.6 ± 0.3 | 12.8 × 105 ± 1.6 × 105 |
| C18:1 | 1.2 ± 0.3 | 1.9 ± 0.2 | 0.9 ± 0.2 | 4.8 × 105 ± 0.5 × 105 |
| C18:2 | 1.0 ± 0.8 | 2.9 ± 0.2 | 1.2 ± 0.6 | 4.0 × 105 ± 2.0 × 105 |
| C18:3 | 1.7 ± 0.4 | 6.0 ± 7.4 | 1.4 ± 0.3 | 10.3 × 105 ± 13.1 × 105 |
| C20:4 | N.D | N.D | N.D | N.D |
| C22:6 | N.D | N.D | N.D | N.D |
| 4,8-CH3-C9 (DMN) | 12.4 ± 1.5 | 7.2 ± 3.7 | 9.9 ± 1.2 | 16.3 × 105 ± 10.1 × 105 |
| 3-CH3-C9 | 2.4 ± 0.4 | 5.5 ± 0.5 | 1.9 ± 0.3 | 3.6 × 105 ± 0.9 × 105 |
| 4-CH3-C9 | 5.2 ± 1.3 | 7.0 ± 0.0 | 4.2 ± 1.1 | 6.0 × 105 ± 1.6 × 105 |
| 4,8,12-CH3-C13 | 3.8 ± 3.0 | 5.6 ± 4.2 | 3.0 ± 2.4 | 5.2 × 105 ± 0.4 × 105 |
| 2-CH3-C16 | 1.0 ± 0.3 | 4.9 ± 5.1 | 0.8 ± 0.3 | 4.1 × 105 ± 4.8 × 105 |
| 3,7-CH3-C8:1 | 0.6 ± 0.4 | 6.5 ± 5.4 | 0.4 ± 0.3 | 1.3 × 105 ± 1.6 × 105 |
| 2-CH3-C6 | 0.9 ± 0.2 | 8.2 ± 2.1 | 0.7 ± 0.2 | 0.9 × 105 ± 0.5 × 105 |
| 3-CH3-C4 | 11.8 ± 2.5 | 30.8 ± 4.5 | 9.5 ± 2.0 | 3.2 × 105 ± 1.1 × 105 |
| 4-phenyl-C4 | 1.3 ± 0.9 | 11.0 ± 6.0 | 1.0 ± 0.7 | 0.9 × 105 ± 0.2 × 105 |
| X-CoA | ||||
| Glutaryl | 12.1 ± 0.8 | 29.3 ± 6.3 | 9.7 ± 0.6 | 3.4 × 105 ± 0.5 × 105 |
| Isovaleryl (3-CH3-C4) | 15.0 ± 0.1 | 14.2 ± 5.5 | 12.0 ± 0.1 | 9.2 × 105 ± 3.6 × 105 |
| Acetoacetyl | 17.1 ± 3.3 | 19.6 ± 10.9 | 13.7 ± 2.6 | 8.3 × 105 ± 4.3 × 105 |
| Isobutyryl (2-CH3-C3) | 31.9 ± 7.9 | 30.8 ± 8.0 | 25.5 ± 6.3 | 8.9 × 105 ± 4.4 × 105 |
| 3-OH-butyryl | 3.3 ± 0.1 | 46.8 ± 26.2 | 2.6 ± 0.1 | 0.7 × 105 ± 0.4 × 105 |
| Crotonyl | 7.9 ± 0.3 | 33.5 ± 4.2 | 6.3 ± 0.3 | 1.9 × 105 ± 0.2 × 105 |
| Malonyl | 1.3 ± 1.0 | 4.0 ± 2.6 | 1.0 ± 0.8 | 2.4 × 105 ± 0.3 × 105 |
| Methylmalonyl | 11.9 ± 0.9 | 25.2 ± 15.5 | 9.5 ± 0.7 | 4.6 × 105 ± 2.5 × 105 |
| Phenylacetyl | 18.1 ± 8.9 | 16.4 ± 4.6 | 14.5 ± 7.2 | 8.5 × 105 ± 2.0 × 105 |
| Succinyl | 13.3 ± 4.1 | 26.6 ± 11.8 | 10.6 ± 3.3 | 4.1 × 105 ± 0.6 × 105 |
| 3-OH-methyl-glutaryl | N.D. | N.D. | N.D. | N.D. |
| Methylcrotonyl | N.D. | N.D. | N.D. | N.D. |
| Benzoyl | N.D. | N.D. | N.D. | N.D. |
| 2-propyl-C5 | N.D. | N.D. | N.D. | N.D. |
| 2-ethyl-C6 | N.D. | N.D. | N.D. | N.D. |
Enzyme kinetics were calculated using GraphPad Prism software and are presented in the table as respective unit ± SD from 2–5 experiments
N.D. not detected, meaning activity too low to allow kinetic analysis, DMN dimethylnonanoyl-CoA
The drop in activity with nonanoyl-CoA is striking and was consistent with different enzyme preparations. Nonanoic acid is a rare fatty acid and likely is a very rare physiological substrate, however, 4,8-CH3-C9-CoA (4,8-dimethylnonanoyl-CoA, DMN-CoA for short) is the end product of the peroxisomal β-oxidation of pristanic acid that is transferred to mitochondria for further β-oxidation. We therefore tested whether DMN-CoA is a substrate for ACOT9 and indeed, as seen in Table 1, Supplementary Fig. 1, V max is higher and K m is lower resulting in K cat/K m being several-fold higher with DMN-CoA than with C9-CoA, suggesting an “adaptation of function” of ACOT9 to this substrate. This finding led us to test various other methyl-branched acyl-CoA esters and other CoA derivatives. Of the methyl-branched acyl-CoAs, the highest V max was found with isobutyryl-CoA (2-CH3-C3-CoA) followed by isovaleryl-CoA (3-CH3-C4-CoA), but the activity with longer methyl-branched acyl-CoAs was much lower in spite of low K ms. While ACOT9 readily accepts isomers of short-chain acyl-CoAs (e.g., isobutyryl-CoA and isovaleryl-CoA), methyl-branched long-chain acyl-CoAs (e.g., 4,8,12-CH3-C13-CoA and 2-CH3-C16-CoA) are much poorer substrates. “Bulkier” branched short-chain acyl-CoAs (4-phenyl-C4-CoA, 2-propyl-C5-CoA and 2-ethyl-C6-CoA) were poor substrates or not hydrolyzed at all.
We further tested all commercially available acyl-CoA intermediates or products of amino acid metabolism, e.g., shorter CoA esters of dicarboxylic acids (malonyl-, succinyl-, glutaryl- and methylmalonyl-CoA), acetoacetyl-CoA, crotonyl-CoA, 3-methylcrotonyl-CoA, 3-hydroxy-3-methylglutaryl-CoA, phenylacetyl-CoA and benzoyl-CoA, many of which are intermediates/products of amino acid metabolism. Of these, acetoacetyl-CoA, succinyl-CoA, glutaryl-CoA, phenylacetyl-CoA (in contrast to 4-phenyl-C4-CoA) and methylmalonyl-CoA were substrates for ACOT9. Taken together, ACOT9 shows broad acyl-CoA substrate specificity with a clear preference for hydrophobic short-, medium-, and long-chain saturated acyl-CoAs with some activity also with short-chain dicarboxylic CoA esters.
ACOT9 catalyzes the simultaneous hydrolysis of short-chain and long-chain acyl-CoA
The broad acyl-chain specificity of ACOT9 raises questions as to its physiological function, in particular when comparing the K cat/K m values, which indicate that ACOT9 would preferentially hydrolyze C12-C18-CoAs in comparison to C3-C4-CoAs. To test this, we compared the activity of ACOT9 with C12-CoA (one of the best long-chain substrates) and the activity with isobutyryl-CoA (one of the best short-chain substrates) to the activity with mixtures of the two CoA esters at a fixed concentration of C12-CoA (at 7 μM, corresponding to the K m for C12-CoA) and varying concentrations of isobutyryl-CoA (Fig. 2a). The V max and K m with C12-CoA was 18.7 ± 5.0 μmol/min/mg protein and 8.5 ± 1.7 μM, respectively, and 31.9 ± 7.9 μmol/min/mg protein and 30.8 ± 5.7 μM with isobutyryl-CoA. Surprisingly, the activity of ACOT9 in the presence of 7 μM C12-CoA and increasing concentration of isobutyryl-CoA was additive up to 10 μM isobutyryl-CoA, and remained higher than the activity of either substrate alone at any given substrate concentration. However, the V versus [S] curve with the mixture of the two substrates showed a biphasic saturation profile, with V max1 = 13.7 ± 4.3 μmol/min/mg and V max2 = 32.0 ± 12.1 μmol/min/mg and K m1 = 0.8 ± 0.3 μM and K m2 = 16.7 ± 1.5 μM. The K cat/K m for the “high-affinity activity” is about sixfold higher compared to C12-CoA alone and nearly 20-fold higher compared to isobutyryl-CoA alone, suggesting that ACOT9 becomes catalytically more efficient in the presence of the two substrates. The spectrophotometric assay measures the release of CoA and does not distinguish between the two substrates and we therefore repeated incubations with the two substrates (at 5 μM each in the same medium as for the spectrophotometric assay) and analyzed the disappearance of the two substrates and appearance of CoA using a HPLC-based method. As shown in Fig. 2b, which shows the consumption of the substrates over time from 0 to 40 s, both C12-CoA and isobutyryl-CoA decrease in parallel, indicating that the enzyme does not prefer any substrate over the other. Taken together, these data suggest that ACOT9 is a short- and long-chain ACOT that hydrolyzes these substrates simultaneously with increased efficiency in spite of the markedly different V max, K m, and K cat/K m values for these substrates.
Fig. 2.

ACOT9 is a more efficient thioesterase in the presence of both short-chain and long-chain acyl-CoAs. a Spectrophotometric measurement of ACOT9 activity with two substrates. ACOT9 activity was measured with various concentrations of isobutyryl-CoA (grey circles with the Michaelis–Menten curve fit in light grey), C12-CoA (black circles with the Michaelis-Mentens curve fit in black) and the activity with different concentrations of isobutyryl-CoA at a fixed concentration of 7 μM C12-CoA (grey squares, with two Michaelis–Mentens curve fits, one for the lower concentrations of isobutyryl-CoA and one for the higher). b HPLC analysis of ACOT9 activity with two substrates. A mixture of 5 μM of isobutyryl-CoA and 5 μM C12-CoA were incubated together with 3-hydroxy-3-methylglutaryl-CoA with ACOT9, and the reaction was terminated by acidification at different time points (0–40 s) and analyzed by HPLC. The figure shows five merged HPLC chromatograms representing different time points. The peak at 17 min corresponds to isobutyryl-CoA and the peak at 27 min corresponds to C12-CoA. The large peak at 18 min is DTNB that was added to the reaction to prevent possible inhibition by released CoA during the reaction
ACOT9 is the major acyl-CoA thioesterase in brown adipose tissue and kidney mitochondria, but is absent in liver mitochondria
The broad acyl-chain length specificity of recombinant ACOT9 with short-, medium-, and long-chain activity resembles the ACOT activity previously reported for hamster and rat BAT mitochondria [41, 42]. Apart from testis, ACOT9 is most strongly expressed in BAT and kidney and the lowest expression was found in liver. We therefore isolated mitochondria from these three tissues and measured the thioesterase activity with C2-C18 acyl-CoAs. ACOT activity in both BAT and kidney mitochondria showed distinct peaks of activity with C3-CoA and broad peaks with longer-chain acyl-CoAs, similar to the activity of recombinant ACOT9 (Fig. 3a). In contrast, isolated liver mitochondria lacked the peak of activity with C3-CoA and rather appear to harbor a medium-chain ACOT activity. These data indicate that ACOT9 is responsible for the majority of the activity in BAT and kidney mitochondria while liver mitochondria lack ACOT9, in line with the very low/absent expression levels at mRNA and protein level in liver (Fig. 1). To lend further support to this notion, we sought to utilize an inhibitor of ACOT9 activity to apply to the mitochondrial activity. Myristoyl-CoA is one of the best substrates for ACOT9 (as judged from highest K cat/K m) and we therefore tested a commercially available myristoyl-CoA thioether (which is not hydrolyzed by ACOTs) on the activity of recombinant ACOT9 with C12-CoA (being the longest acyl-CoA ester used for activity measurements without the requirement for addition of BSA). Indeed, the C14-CoA thioether is a potent inhibitor of ACOT9 when measured with C12-CoA (IC50 ≈ 2.5 μM) and even more potent when measured with C3-CoA (IC50 ≈ 0.5-1.0 μM) (Fig. 3b). We have previously tested the effect of the C14-CoA ether on ACOT1 (a representative Type-I ACOT), which showed that this inhibitor had no effect on ACOT1 activity (data not shown). We next employed the inhibitor on the activity in soluble extracts of isolated mitochondria and indeed the activity with the entire range of CoA-esters was substantially decreased with the short-chain activity peak being almost completely abolished in BAT and kidney mitochondria with minor effect in liver mitochondria. Interestingly, a distinct peak of activity with C10-CoA remained in kidney, and also to a lower extent in liver and BAT, suggesting that these tissues contain a novel, yet uncharacterized, medium-chain mitochondrial ACOT activity. Although we cannot exclude the possibility that the C14-CoA ether inhibits other type-II ACOTs, the data taken together suggest that ACOT9 is responsible for the majority of the short-chain activity and possibly also the medium- to long-chain activity in BAT and kidney mitochondria.
Fig. 3.
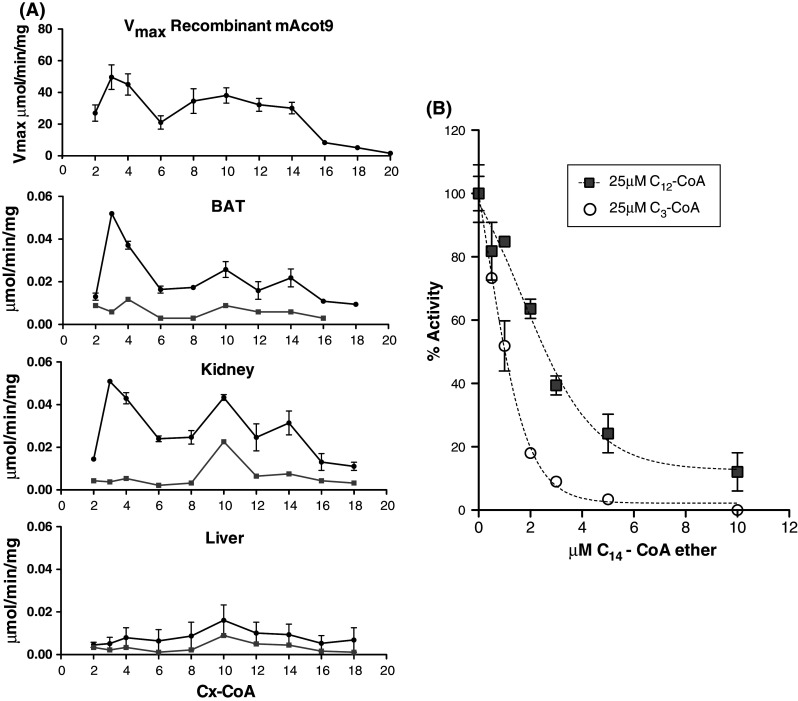
Acyl-CoA chain-length specificity of recombinant ACOT9 and thioesterase activity in isolated mitochondria. a Thioesterase activity of ACOT9 was measured spectrophotometrically at 412 nm with various concentrations of saturated acyl-CoAs and the V max values were calculated and plotted in the upper panel (mean ± SEM, n = 2–5). Mitochondria were isolated from BAT, kidney, and liver, sonicated and centrifuged, and thioesterase activity was measured in the supernatants as described under the "Materials and methods" section. The black circles show the activity with 50 μM of acyl-CoA esters ranging from C2 to C12-CoA, 25 μM C14-CoA and 10 μM C16-C18-CoA (data are mean ± SEM of two independent experiments). Note the very similar activity pattern for recombinant ACOT9 and BAT mitochondria. The grey squares show the remaining thioesterase activity after 2 min pre-incubation with 20 μM C14-CoA thioether with mitochondrial extracts. b C14-CoA thioether is a potent inhibitor of ACOT9 activity. Recombinant ACOT9 was pre-incubated with various concentrations of C14-CoA thioether (0–10 μM) and the activity was measured at fixed concentrations of C3-CoA (50 μM) (open circles) and C12-CoA (25 μM) (grey squares) using the spectrophotometric assay at 412 nm (mean ± SEM with two different enzyme preparations)
Regulation of ACOT9 and mitochondrial activity by NADH and CoA
A striking feature of the BAT thioesterase activity in both hamster and rat BAT mitochondria was the pronounced inhibition by NADH [41, 42]. Based on the dual chain-length preference of ACOT9, we tested the effect of NADH on ACOT9 activity with C3-CoA and C14-CoA (Fig. 4a). NADH inhibited the activity with C3-CoA with an IC50 ≈ 300 μM and the activity with C14-CoA with an IC50 ≈ 500 μM. The effect of 400 μM NADH was then tested on C3-CoA and C14-CoA thioesterase activity in BAT, kidney, and liver mitochondria, which inhibited C3-CoA thioesterase activity by about 65 and 75 % in BAT and kidney mitochondria, respectively, while NADH had no effect on the activity in liver mitochondria (Fig. 4b). The same concentration of NADH inhibited the activity of recombinant ACOT9 by about 65 %, supporting the hypothesis that ACOT9 is the main propionyl-CoA (C3-CoA) thioesterase in BAT and kidney mitochondria. The activity with C14-CoA was less sensitive to NADH; the inhibition was about 35 and 45 % in BAT and kidney mitochondria, respectively, compared to about 40 % inhibition with recombinant ACOT9. Again, NADH was without effect in liver mitochondria, in line with the lack of detectable expression of ACOT9 in this tissue.
Fig. 4.
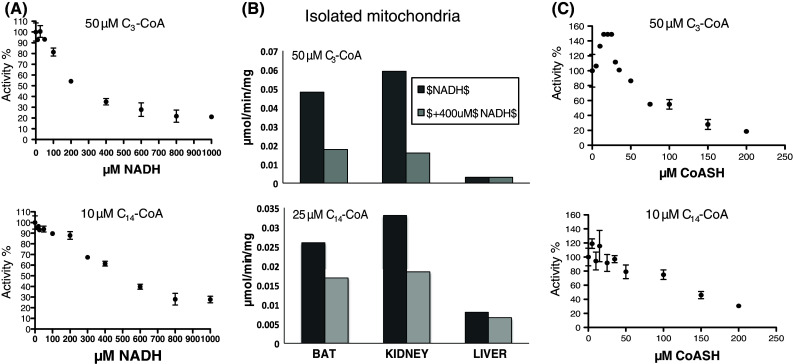
ACOT9 activity is regulated by NADH and CoA. a Recombinant ACOT9 was incubated at fixed concentrations of C3-CoA (50 μM) or C14-CoA (10 μM) and varying concentrations of NADH (0–1,000 μM). The activity was measured using the standard spectrophotometric assay at 412 nm and the graph shows one representative experiment out of three for C3-CoA (upper panel) and one out of two experiments for C14-CoA (lower panel). The data are shown as the % remaining activity in the presence of NADH (mean ± SEM of 1–4 measurements). b Thioesterase activity was measured with soluble extracts of mitochondria isolated from BAT, kidney, and liver with 50 μM C3-CoA or 25 μM C14-CoA in the presence (light grey bars) or absence (dark grey bars) of 400 μM NADH. c Recombinant ACOT9 was incubated at fixed concentrations of C3-CoA or (50 μM) and C14-CoA (10 μM) in the presence of varying concentrations of CoA (0–200 μM) and activity was measured using the UV spectrophotometric assay. One representative experiment out of three is shown for C3-CoA and one out of two for C14-CoA. The values represent mean ± SEM of 2–5 measurements
Based on the apparent regulation of ACOT9 activity by nucleotides, we also tested the effect of CoA, which inhibited the activity with C3-CoA with IC50 ≈ 100 μM and the activity with C14-CoA with IC50 ≈ 150 μM (Fig. 4c). However, the effect of CoA was biphasic in that low concentrations of CoA (up to ≈ 25 μM) stimulated the activity with C3-CoA but not the activity with C14-CoA activity.
Fasting regulates Acot9 mRNA expression in mouse kidney
The finding that ACOT9 is a “multi-purpose” ACOT having high activity with short-chain CoA esters (propionyl-CoA and intermediates in amino acid metabolism) led us to examine the regulation of this enzyme by conditions that affect the metabolic status of the cell and especially mitochondrial metabolism. Fasting condition results in an increase in mitochondrial β-oxidation, and we therefore determined the regulation of Acot9 mRNA in kidney and abdominis and gastrocnemius skeletal muscle following 12-h fasting. While there was no clear effect of fasting in muscle on Acot9 mRNA levels, mRNA was downregulated by ~40 % in kidney (data not shown).
ACOT9 homologs are found from bacteria to man
In a recent classification, thioesterases were grouped into 23 families based on sequence conservation, tertiary structure superposition, and mechanisms [13]. ACOT9 falls into group TE7, which are found in eukaryotes and bacteria. We therefore performed a “global” BLAST search to search for ACOT9 homologs, which showed that ACOT9 orthologous genes are found from bacteria to man (see Supplementary Fig. 1, which shows a phylogenetic tree based on ACOT9). In most species, only one gene exists, but in e.g., the nematode (Caenorhabditis elegans) and zebra fish (Danio rerio), there are three and two genes, respectively, showing homology to mouse ACOT9. Homologous genes also exist in bacteria, fungi, plants, insects, other invertebrates such as tunicates (Ciona intestinalis), and non-mammalian vertebrates as well as mammals. In some mammals, e.g., Chinese hamster (Cricetulus griseus), dog (Canis lupus), guinea pig (Cavia porcellus), and primates, the Acot9 gene is alternatively spliced at exon 3 to produce two variants of 448 and 439 amino acids, respectively. At least in human, both isoforms are expressed in most tissues (Agnes Hedenström, Veronika Tillander and Stefan Alexson, unpublished results). Interestingly, in some insects and fungi, the protein sequence ends in –SKL, which is the consensus peroxisomal type-1 targeting sequence, suggesting that ACOT9 is mitochondrial and/or peroxisomal in these species (Supplementary Fig. 2).
Identification of active site and catalytic amino acid residues
In order to identify the catalytic residues of ACOT9, we aligned the sequence of mouse ACOT9 with the sequences of mouse ACOT7, ACOT11-13, and the ACOT9-related protein from the archaea bacteria Haloquadratum walsbyi (Supplementary Fig. 3). The catalytic machinery of ACOT7 was identified as Asn-24, located in the N-terminal thioesterase domain, and Asp-213 located in the C-terminal thioesterase domain, which confers active site I in ACOT7 while active site II (Glu-39 and Thr-198) is not active, however both domains are required for activity [15]. The sequence alignment identified active site II as the putative active site in ACOT9 (Asp-120 and Asn-305) as well as in ACOT11 and 12. In contrast, ACOT13 and some bacterial proteins (e.g., H. walsbyi) only contain one thioesterase domain, which upon dimerization probably produces two active sites per dimer.
We next aligned mouse ACOT9 to homologs from phylogenetically diverse species including bacterial proteins that also contain two thioesterase domains (e.g., Desulfobulbus propionicus) (Supplementary Fig. 4). The corresponding catalytic Asn and Asp residues are conserved in all the analyzed putative ACOT9 sequences and in all proteins they confer active site II. The Hotdog fold thioesterase proteins form dimers and although only one active site is apparently functional, it is likely that both thioesterase domains are required for activity.
Discussion
The sequence identity of mouse ACOT9 and 10 is 96 % at protein level and a previous brief kinetic characterization of ACOT9/10 suggested that they function mainly as long-chain ACOTs [28]. Based on the higher expression of ACOT9 compared to 10, we have here characterized ACOT9 kinetically and examined tissue expression to identify its putative physiological function. This in-depth characterization showed that ACOT9 is active on a much broader range of CoA-esters than previously anticipated, in particular a distinct activity with short-chain (C2-C5) acyl-CoAs was found. Further characterization identified also short-chain methyl-branched acyl-CoAs (isobutyryl-CoA and isovaleryl-CoA) and several intermediates in amino acid metabolism as good substrates while hydroxylated short-chain acyl-CoAs (e.g., 3-hydroxybutyryl-CoA) are poor substrates. These data indicate that ACOT9 may regulate both fatty acid metabolism and amino acid metabolism in mitochondria.
ACOT9 is likely the main short- and medium-chain ACOT enzyme in BAT and kidney mitochondria
The high activity with propionyl-CoA and our findings that NADH and CoA are potent inhibitors of ACOT9 suggested to us that ACOT9 may be the enzyme responsible for the previously described NADH-sensitive short-chain ACOT activity in BAT mitochondria [41, 42] and probably also a previously partially purified mitochondrial thioesterase from pig heart [43]. It was proposed by Alexson and Nedergaard [41] that the short- and medium-chain NADH-inhibited activities in hamster BAT mitochondria were due to separate enzymes based on the acyl-CoA chain-length specificity and difference in NADH sensitivity, however, our data suggest that these activities apparently are catalyzed by a single enzyme. The very similar acyl-CoA chain-length specificity in mitochondria isolated from BAT and kidney to the specificity of ACOT9 indicates that ACOT9 is the main mitochondrial short- and medium-chain ACOT in these tissues. To obtain further support for this notion, we therefore sought to take advantage of the inhibitory effect of NADH and the very potent inhibitor C14-CoA thioether (but not CoA since the UV-based spectrophotometric method is not suitable for crude protein samples giving high UV absorbance) to compare the effects on ACOT9 activity with the effects on mitochondrial ACOT activity. Indeed, NADH inhibited the activity with recombinant ACOT9 and in mitochondrial extracts from BAT to kidney, but not liver, to a similar extent. In an attempt to further characterize the mitochondrial activity, we measured ACOT activity with CoA esters ranging from C2-C18 in mitochondria from BAT, kidney, and liver in the absence or presence of the C14-CoA thioether. These measurements showed that the ACOT activity in BAT and kidney, but not in liver mitochondria, closely resembled the V max pattern of recombinant ACOT9 and that the addition of the C14-CoA thioether strongly inhibited the mitochondrial ACOT activity in BAT and kidney mitochondria but not in liver mitochondria. The only prominent residual activity with the thioether was a peak with C10-CoA in kidney mitochondria, which is also evident (but less pronounced) in BAT and liver mitochondria. Although being circumstantial evidence, taken together these data suggest that ACOT9 is the major ACOT that hydrolyzes saturated C2-C18 acyl-CoAs in BAT and kidney and that ACOT9 is hardly detectable in liver mitochondria. The data also suggest that mitochondria contain a rather specific medium-chain ACOT that has not yet been identified at protein or gene level.
The short-chain acyl-CoA thioesterase activity of ACOT9 suggests regulatory functions in amino acid metabolism and the Krebs cycle
The short-chain thioesterase activity peak is very distinct and suggests a function for ACOT9 in metabolism/formation of e.g., acetate, acetoacetate, propionate, (iso)butyrate, and isovalerate. This is supported also by the fact that ACOT9 homologs are found in several of the (fermentative) bacteria (e.g., Pelobacter propionicus [44], Pelobacter carbonolicus [45], Desulfobulbus propionicus [46], and Geobacter metallireducens [47]) identified in our BLAST search using the mouse ACOT9 protein sequence as template (Supplementary Fig. 2), which utilize or produce acetate, propionate, butyrate, and isobutyrate, suggesting an evolutionary conserved function of ACOT9 in metabolism of short-chain fatty acids. Further substrate characterization showed that ACOT9 also hydrolyzes a number of other short-chain CoA esters produced by amino acid metabolism. A summary of the acyl-CoA esters involved in amino acid metabolism that were tested in this study is highlighted in a metabolic scheme in Fig. 5. The only mammalian short-chain ACOTs identified so far is ACOT12, a cytosolic/peroxisomal acetyl-CoA thioesterase [48, 49], whose physiological function is still unclear, and 3-hydroxyisobutyryl-CoA hydrolase (HIBCH) [50] that has important functions in valine metabolism and 3-hydroxyisobutyric aciduria [50–52]. HIBCH hydrolyzes only 3-hydroxyisobutyryl-CoA and 3-hydroxypropionyl-CoA, with <0.1 % activity with isobutyryl-CoA and propionyl-CoA [53]. Although we did not test activity of ACOT9 with 3-hydroxyisobutyryl-CoA or 3-hydroxypropionyl-CoA (not being available to us), we anticipate that both are poor substrates since ACOT9 activity with 3-hydroxbutyryl-CoA is less than 10 % compared to the activity with butyryl-CoA. Further, indirect, support for a role in amino acid metabolism stems from the tissue expression of ACOT9; the mRNA and protein expression is broad but lowest expression was found in liver, intestine, and skeletal muscle, similar to the expression/activity of BCAT (branched-chain aminotransferase, catalyzing the first step in the metabolism of valine, leucine, and isoleucine) in various rat and canine tissues [54, 55].
Fig. 5.
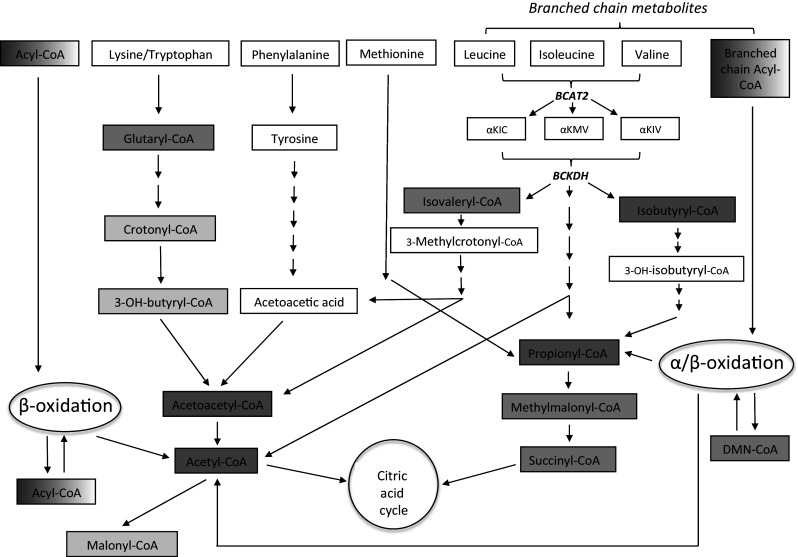
ACOT9 potentially regulates fatty acid and amino acid metabolism in mitochondria. Hypothetical scheme of fatty acid and amino acid metabolism pathways in which ACOT9 may be involved. Products and intermediates as CoA esters that were tested and found to be substrates for ACOT9 are colored in grey, where a darker shade indicates higher activity. Color: Darkest grey = V max > 15 μmol/min/mg, Darker grey = V max 10–15 μmol/min/mg, Light grey = V max < 10 μmol/min/mg. Boxes with graded shade intensity of grey symbolize groups of substrates that include both good and poor substrates for ACOT9. BCAT branched-chain aminotransferase, BCKDH branched-chain ketoacid dehydrogense, αKIV α-ketoisovaleric acid, αKMV α-keto-β-methylvaleric acid, αKIC α-ketoisocaproic acid, DMN-CoA dimethylnonanoyl-CoA. The activity with 3-methylcrotonyl-CoA was too low to allow kinetic analysis. 3-Hydroxyisobutyryl-CoA was not tested as a substrate for ACOT9 but a specific mitochondrial thioesterase (HIBCH) that hydrolyzes 3-hydroxyisobutyryl-CoA and also 3-hydroxypropionyl-CoA has been identified in mammals [50]
Short-chain organic acids increase in blood and/or urine in a number of pathological conditions, for example propionic acid, butyric acid, isobutyric acid, isovaleric acid, dicarboxylic acids, and metabolites of these acids (e.g., 3-hydroxy derivatives), together with carnitine esters and glycine conjugates and other organic acids [56, 57]. Of the short-chain substrates produced in pathological conditions, propionyl-CoA, butyryl-CoA, isobutyryl-CoA, and isovaleryl-CoA are the best substrates for ACOT9. ACOT9 is therefore a strong candidate to produce several of these short-chain acids that are found under normal and pathological conditions. Propionyl-CoA is derived from many sources, including catabolism of essential amino acids (isoleucine, valine, threonine, and methionine) in mitochondria, oxidation of odd chain-length fatty acids and methyl-branched chain fatty acids in peroxisomes and mitochondria and the β-oxidation of bile acid intermediates in peroxisomes [58]). The propionyl-CoA produced in peroxisomes can be transported to the mitochondria as carnitine esters (formed by carnitine acetyltransferase, CRAT, which is localized both in peroxisomes and mitochondria) via the carnitine/acylcarnitine carrier (CACT) and once inside the mitochondria, propionyl-CoA is re-formed via the reverse action of CRAT [59]. Propionyl-CoA can subsequently be converted to succinyl-CoA by the action of three specialized enzymes, propionyl-CoA carboxylase (PCCA), methylmalonyl-CoA epimerase, and methylmalonyl-CoA mutase, and the produced succinyl-CoA can enter the Krebs cycle. Propionic acidemia is an inborn error of amino acid metabolism due to a defect in PCCA that catalyzes the biotin-dependent conversion of propionyl-CoA to methylmalonyl-CoA. These patients have elevated propionylcarnitine and 3-hydroxypropionate, and the presence of methylcitrate, tiglylglycine, and propionylglycine that are not normally found in the urine. The toxic effects of propionyl-CoA accumulation are likely due to excretion of propionylcarnitine, which leads to carnitine deficiency and in turn CoA deficiency, and in addition propionyl-CoA is converted to methylcitrate, which likely inhibits the Krebs cycle [60]. Under these conditions, NADH levels may be reduced and in combination with CoA sequestration, ACOT9 activity would be increased but not sufficiently to maintain carnitine levels. Tiglylglycine and propionylglycine are formed in liver and kidney by the activity of glycine N-acyltransferase (GLYAT), which is a mitochondrial enzyme expressed only in liver and kidney [61] that serves as a salvage pathway to excrete excess tiglic and propionic acid. The accumulation of propionyl-CoA in the mitochondria also leads to inhibition of a number of enzymes including the pyruvate dehydrogenase complex (PDHc) and α-ketoglutarate dehydrogenase complex [62, 63]. Interestingly, the acyl-CoA chain-length specificity of PDHc inhibition is reciprocal to the acyl-CoA specificity of ACOT9, i.e., strong inhibition with C2-C4-CoA, weakest inhibition with C6-CoA, intermediate with C8-CoA, and strong inhibition with C10-C16-CoA, and it was also shown that C8-CoA inhibits mitochondrial complex III [62]. Based on the substrate specificity of ACOT9, it is plausible that ACOT9 may function to counteract these effects to maintain efficient mitochondrial metabolism.
ACOT9 may have a regulatory function in the mitochondrial metabolism of methyl-branched fatty acids
A striking feature of the acyl-CoA specificity of ACOT9 (and in isolated BAT mitochondria, data not shown) was the sharp drop in activity with nonanoyl-CoA (C9-CoA, see supplementary Fig. 1), which is in contrast to the hamster and rat BAT mitochondrial activity. The kinetic characterization with nonanoyl-CoA compared to the activity with DMN-CoA revealed that V max was higher and K m was lower with DMN-CoA, resulting in 3–4-fold higher K cat/K m. The K m values were lower also with 3-methylnonanoyl-CoA and 4-methylnonanoyl-CoA, resulting in higher K cat/K m also for these substrates compared to nonanoyl-CoA, suggesting that increased hydrophobicity increases the affinity for these substrates. DMN-CoA is a physiological substrate (unlike nonanoyl-CoA) since it is the end product of β-oxidation of pristanic acid in peroxisomes, which can be transferred to mitochondria either as the carnitine ester or as the free acid. It is conceivable that the rat and hamster enzymes also hydrolyze DMN-CoA (although this was never tested) and that the activity with methyl-branched medium-chain substrates is a propensity of the medium-chain ACOT9 activity.
ACOT9 hydrolyzes short- and long-chain acyl-CoAs simultaneously, functional and structural implications
As discussed above, the kinetic characterization of ACOT9 showed that C3-CoA, C4-CoA, and isobutyryl-CoA were the best short-chain substrates while C12-CoA and C14-CoA were the best long-chain substrates, based on V max and K cat/K m values. However, the K cat/K m was about threefold higher with the longer acyl-CoAs, which suggest that ACOT9 would function mainly as a long-chain ACOT. We tested this hypothesis by measuring the activity in the presence of both a short- and long-chain CoA ester and surprisingly the activity was additive, suggesting that ACOT9 hydrolyzes short-chain and long-chain acyl-CoAs simultaneously at similar rates. This was further verified by HPLC analysis of the disappearance of the two substrates. Closer analysis of the Michaelis–Menten kinetics with two substrates showed a biphasic saturation profile indicative of high- and low-affinity activities/sites, which raises questions as to the structure and mechanism of catalytic action of ACOT9. A biphasic saturation curve in which V max2 > V max1 and K m2 >> K m1 is compatible with a two-enzyme model or, as in this case with recombinant ACOT9 a two-site model [64]. The ACOT9 polypeptide contains two thioesterase domains and two putative active sites. However, our sequence analysis showed that only one site (site II) is apparently active and therefore a dimeric structure is required to form the active enzyme. ACOT9 most likely forms a trimer of dimers similar to ACOT7 [15], and a hexameric structure of ACO9 is supported by the estimated molecular mass by size-exclusion chromatography of the partially purified pig heart enzyme of ≈300 kDa [43] and the rat BAT mitochondrial NADH-sensitive activity of >240 kDa [42] (assuming that these activities correspond to ACOT9). This implies that most likely the active site contains two, or possibly three, substrate binding sites, or pockets, to accommodate short-, medium-, and long-chain acyl-CoAs, respectively. In addition, whether the “inactive” site can bind acyl-CoA and thereby regulate the activity is still an open question. Of interest are also the findings that NADH, CoA, and the non-hydrolyzable myristoyl-CoA thioether inhibits the short- and long-chain ACOT activities with different sensitivities and that low concentrations of CoA and NADH did not inhibit, or even slightly stimulated, the activity with C3-CoA. We did not further characterize the type of inhibition by CoA and NADH due to the unusual Michaelis–Menten kinetics, but most likely NADH mediates allosteric regulation while inhibition by CoA may at least in part be competitive since the CoA-moiety is probably involved in substrate binding. CoA was shown to induce dramatic structural changes in a hexameric hotdog domain-containing short-chain ACOT from Bacillus halodurans [65]. Binding of CoA, which negatively regulates the activity, caused dramatic changes at both the dimer and trimer-of-dimer interfaces and in the C-terminal helices, which play a role in regulating the activity and it was concluded that these conformational changes are likely conserved from bacteria through to humans. Thus, CoA provides a direct inhibitory feedback and also likely acts to sense intra-mitochondrial CoA levels. The inhibition by CoA and NADH within physiological concentrations suggests that ACOT9 activity is tightly regulated and may provide a fine-tuning regulation of lipid and amino acid metabolism in mitochondria. Many, if not most, of the characteristics and proposed functions of mouse ACOT9 can be translated to the human ACOT9 enzyme, which share the kinetic properties of the mouse enzyme (Agnes Hedenström, Veronika Tillander, and Stefan Alexson, unpublished results).
Electronic supplementary material
Below is the link to the electronic supplementary material.
4,8-Dimethylnonanoyl-CoA is a substrate for ACOT9. 4,8-Dimethylnonanoyl-CoA (DMN-CoA) is formed in peroxisomes via α- and subsequent β-oxidation of phytanic acid. DMN-CoA is a substrate either for carnitine octanoyltransferase (CROT) or ACOT8, a thioesterase with very broad acyl-CoA specificity [66, 67]. DMN can be transferred to mitochondria either as the non-esterified acid or as a carnitine ester, where it can be metabolized to propionyl-CoA and acetyl-CoA via mitochondrial β-oxidation (indicated in the upper right panel). The upper left panel shows a plot of ACOT9 V max and K m values with nonanoyl-CoA being clearly distinguishable from the other acyl-CoA esters in the curve. The lower left panel shows a graph of K cat/K m values including the K cat/K m for DMN-CoA. The lower right panels show the Michaelis–Menten kinetics of ACOT9 with nonanoyl-CoA (C9-CoA) and DMN-CoA (one representative experiment out of two). Note that V max is higher and K m is lower with DMN-CoA, resulting in a 3–4fold higher K cat/K m (PPTX 226 kb)
Phylogenetic tree of ACOT9 homologs from selected species ranging from bacteria to primates. The mouse ACOT9 protein sequence was used as a template to search various databases and full-length protein sequences were aligned using ClustalW. While some species contain more than one ACOT9 homologue, e.g. C. elegans and Danio rerio, some mammalian species including human and other primates contain one gene that is alternatively spliced (3rd exon coding for nine amino acids) resulting for proteins of 439 and 448 amino acids, indicated as -1 and -2 respectively. *Indicates the presence of putative C-terminal peroxisomal Type-1 targeting signal, with its amino acid sequence being added after the name (PPTX 358 kb)
Identification of the putative catalytic residues and active sites I and II in ACOT9. The protein sequences of mouse ACOT7, ACOT9, ACOT11, ACOT12 and ACOT13 were aligned together with a homologous thioesterase from H. walsbyi (Haloquadratum walsbyi) using ClustalW. The upper panel (a cladogram) shows the relationship of H. walsbyi thioesterase, ACOT7, ACOT9, ACOT11, ACOT12 and ACOT13 despite low sequence similarity. It should be noted that ClustalW uses the ‘neighbor-joining method’, which rather represent the data in the form of an additive tree and therefore provides a branching pattern rather than relative time or relative amount of amino acid changes (evolutionary changes). The lower panel shows the alignment of the corresponding amino acid sequences that highlights (boxed) the catalytic residues of active site I (Asn-36 and Asp-225, which are crucial for activity) and II (Glu-51 and Thr-210) of ACOT7 as identified in [15]. In ACOT9, ACOT11 and ACOT12 only site II is active (corresponding Asp and Asn residues). ACOT13 and the H. walsbyi thioesterase contain only one thioesterase domain with one active site (Asn and Asp) that upon dimerization of the proteins results in two active sites (Asn/Asp and Asn/Asp respectively) per dimer in these proteins (PPTX 436 kb)
The catalytic residues of the putative active site II (Asp and Asn) are conserved in a variety of ACOT9 homologs. A selection of ACOT9 homologs from various species from archaea bacteria to human were aligned using ClustalW. Note that Asp and Asn of active site II are conserved in all ACOT9 proteins containing two thioesterase domains. The archaea bacterium Haloquadratum walsbyi (H. walsbyi) is included as a bacterial homologue containing only one thioesterase domain and Desulfobulbus propionicus (D. propionicus) and Geobacter uraniireducens (G. uraniireducens) are included as examples of bacterial ACOT9 homologs containing two thioesterase domains. M. musculus; Mus musculus, C. elegans; Caenorhabditis elegans, C. intestinalis; Ciona intestinalis, H. sapiens, Homo sapiens, I. scapularis; Ixodes scapularis, M. gallopavo; Meleagris gallopavo, N. vectensis; Nematostella vectensis, R. communis, Ricinus communis, T. stipitatus; Talaromyces stipitatus, V. carteri; Volvox carteri (PPTX 812 kb)
Acknowledgments
The authors are grateful to Prof. Ingemar Björkhem and Dr. Maura Heverin for supplying the C57BL/6 male mice for tissue mitochondrial preparations and to Prof. Ronald Wanders for the kind gift of DMN-CoA. We also want to thank docent Ulrika von Döbeln for valuable discussions. This study was supported by the Swedish Research Council, NordForsk under the Nordic Centre of Excellence Programme in Food, Nutrition and Health (Project 070010) “MitoHealth”, the FP6 European Union Project “Peroxisome” (LSHG-CT-2004-512018), Carl Trygger Foundation, Professor Nanna Svartz Fond Åke Wibergs stiftelse and by a grant from the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen (FWO G.0721.10 N).
Conflict of interests
All authors declare no conflicts of interest.
References
- 1.Van Veldhoven PP. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res. 2010;51:2863–2895. doi: 10.1194/jlr.R005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wanders RJ, Ruiter JP, IJ L, Waterham HR, Houten SM. The enzymology of mitochondrial fatty acid β-oxidation and its application to follow-up analysis of positive neonatal screening results. J Inherit Metab Dis. 2010;33:479–494. doi: 10.1007/s10545-010-9104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 4.Aas M. Organ and subcellular distribution of fatty acid activating enzymes in the rat. Biochim Biophys Acta. 1971;231:32–47. doi: 10.1016/0005-2760(71)90253-0. [DOI] [PubMed] [Google Scholar]
- 5.Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res. 2007;48:2736–2750. doi: 10.1194/jlr.M700378-JLR200. [DOI] [PubMed] [Google Scholar]
- 6.Hunt MC, Alexson SE. The role Acyl-CoA thioesterases play in mediating intracellular lipid metabolism. Prog Lipid Res. 2002;41:99–130. doi: 10.1016/S0163-7827(01)00017-0. [DOI] [PubMed] [Google Scholar]
- 7.Hunt MC, Alexson SE. Novel functions of acyl-CoA thioesterases and acyltransferases as auxiliary enzymes in peroxisomal lipid metabolism. Prog Lipid Res. 2008;47:405–421. doi: 10.1016/j.plipres.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Hunt MC, Siponen MI, Alexson SE. The emerging role of acyl-CoA thioesterases and acyltransferases in regulating peroxisomal lipid metabolism. Biochim Biophys Acta. 2012;1822:1397–1410. doi: 10.1016/j.bbadis.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Kirkby B, Roman N, Kobe B, Kellie S, Forwood JK. Functional and structural properties of mammalian acyl-coenzyme A thioesterases. Prog Lipid Res. 2010;49:366–377. doi: 10.1016/j.plipres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Hunt MC, Yamada J, Maltais LJ, Wright MW, Podesta EJ, et al. A revised nomenclature for mammalian acyl-CoA thioesterases/hydrolases. J Lipid Res. 2005;46:2029–2032. doi: 10.1194/jlr.E500003-JLR200. [DOI] [PubMed] [Google Scholar]
- 11.Zhuravleva E, Gut H, Hynx D, Marcellin D, Bleck CK, et al. Acyl-CoA thioesterase Them5/Acot15 is involved in cardiolipin remodeling and fatty liver development. Mol Cell Biol. 2012;32:2685–2697. doi: 10.1128/MCB.00312-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brocker C, Carpenter C, Nebert DW, Vasiliou V. Evolutionary divergence and functions of the human acyl-CoA thioesterase gene (ACOT) family. Hum Genom. 2010;4:411–420. doi: 10.1186/1479-7364-4-6-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cantu DC, Chen Y, Reilly PJ. Thioesterases: a new perspective based on their primary and tertiary structures. Prot Sci. 2010;19:1281–1295. doi: 10.1002/pro.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dillon SC, Bateman A. The hotdog fold: wrapping up a superfamily of thioesterases and dehydratases. BMC Bioinfo. 2004;5:109. doi: 10.1186/1471-2105-5-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forwood JK, Thakur AS, Guncar G, Marfori M, Mouradov D, et al. Structural basis for recruitment of tandem hotdog domains in acyl-CoA thioesterase 7 and its role in inflammation. Proc Natl Acad Sci USA. 2007;104:10382–10387. doi: 10.1073/pnas.0700974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandel CR, Tweel B, Tong L. Crystal structure of human mitochondrial acyl-CoA thioesterase (ACOT2) Biochem Biophys Res Commun. 2009;385:630–633. doi: 10.1016/j.bbrc.2009.05.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pidugu LS, Maity K, Ramaswamy K, Surolia N, Suguna K. Analysis of proteins with the ‘hot dog’ fold: prediction of function and identification of catalytic residues of hypothetical proteins. BMC Struct Biol. 2009;9:37. doi: 10.1186/1472-6807-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Svensson LT, Alexson SEH, Hiltunen JK. Very long chain and long-chain acyl-CoA thioesterases in rat liver mitochondria. Identification, purification, characterization, and induction by peroxisome proliferators. J Biol Chem. 1995;270:12177–12183. doi: 10.1074/jbc.270.20.12177. [DOI] [PubMed] [Google Scholar]
- 19.Hunt MC, Rautanen A, Westin MAK, Svensson LT, Alexson SEH. Analysis of mouse and human acyl-CoA thioesterase (ACOT) gene clusters shows that convergent functional evolution results in a reduced number of peroxisomal ACOTs. FASEB J. 2006;20:1855–1864. doi: 10.1096/fj.06-6042com. [DOI] [PubMed] [Google Scholar]
- 20.Svensson LT, Engberg ST, Aoyama T, Usuda N, Alexson SEH, et al. Molecular cloning and characterization of a mitochondrial peroxisome proliferator-induced acyl-CoA thioesterase from rat liver. Biochem J. 1998;329:601–608. doi: 10.1042/bj3290601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunt MC, Lindquist PJG, Peters JM, Gonzalez FJ, Diczfalusy U, et al. Involvement of the peroxisome proliferator-activated receptor alpha (PPARα) in regulation of long-chain acyl-CoA thioesterases. J Lipid Res. 2000;41:814–823. [PubMed] [Google Scholar]
- 22.Himms-Hagen J, Harper ME. Physiological role of UCP3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: an hypothesis. Exp Biol Med (Maywood) 2001;226:78–84. doi: 10.1177/153537020122600204. [DOI] [PubMed] [Google Scholar]
- 23.Hunt MC, Greene S, Hultenby K, Svensson LT, Engberg S, et al. Alternative exon usage selectively determines both tissue distribution and subcellular localization of the acyl-CoA thioesterase 7 gene products. Cell Mol Life Sci. 2007;64:1558–1570. doi: 10.1007/s00018-007-7062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamada J, Kuramochi Y, Takagi M, Watanabe T, Suga T. Human brain acyl-CoA hydrolase isoforms encoded by a single gene. Biochem Biophys Res Comm. 2002;299:49–56. doi: 10.1016/S0006-291X(02)02587-1. [DOI] [PubMed] [Google Scholar]
- 25.Kuramochi Y, Takagi-Sakuma M, Kitahara M, Emori R, Asaba Y, et al. Characterization of mouse homolog of brain acyl-CoA hydrolase: molecular cloning and neuronal localization. Mol Brain Res. 2002;98:81–92. doi: 10.1016/S0169-328X(01)00323-0. [DOI] [PubMed] [Google Scholar]
- 26.Hunt MC, Ruiter J, Mooyer P, van Roermund CWT, Ofman R, et al. Identification of fatty acid oxidation disorder patients with lowered acyl-CoA thioesterase activity in human skin fibroblasts. Eur J Clin Invest. 2005;35:38–46. doi: 10.1111/j.1365-2362.2005.01447.x. [DOI] [PubMed] [Google Scholar]
- 27.Ellis JM, Wong GW, Wolfgang MJ. Acyl coenzyme a thioesterase 7 regulates neuronal fatty acid metabolism to prevent neurotoxicity. Mol Cell Biol. 2013;33:1869–1882. doi: 10.1128/MCB.01548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poupon V, Begue B, Gagnon J, Dautry-Varsat A, Cerf-Bensussan N, et al. Molecular cloning and characterization of MT-ACT48, a novel mitochondrial acyl-CoA thioesterase. J Biol Chem. 1999;274:19188–19194. doi: 10.1074/jbc.274.27.19188. [DOI] [PubMed] [Google Scholar]
- 29.Chen D, Latham J, Zhao H, Bisoffi M, Farelli J, et al. Human brown fat inducible thioesterase variant 2 cellular localization and catalytic function. Biochemistry. 2012;51:6990–6999. doi: 10.1021/bi3008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen DE. New players on the metabolic stage: how do you like them acots? Adipocyte. 2013;2:3–6. doi: 10.4161/adip.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han S, Cohen DE. Functional characterization of thioesterase superfamily member 1/Acyl-CoA thioesterase 11: implications for metabolic regulation. J Lipid Res. 2012;53:2620–2631. doi: 10.1194/jlr.M029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Li Y, Niepel MW, Kawano Y, Han S, et al. Targeted deletion of thioesterase superfamily member 1 promotes energy expenditure and protects against obesity and insulin resistance. Proc Natl Acad Sci USA. 2012;109:5417–5422. doi: 10.1073/pnas.1116011109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J, Kang HW, Cohen DE. Thioesterase superfamily member 2 (Them2)/acyl-CoA thioesterase 13 (Acot13): a homotetrameric hotdog fold thioesterase with selectivity for long-chain fatty acyl-CoAs. Biochem J. 2009;421:311–322. doi: 10.1042/BJ20090039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao J, Xu H, Zhao H, Gong W, Dunaway-Mariano D. The mechanisms of human hotdog-fold thioesterase 2 (hTHEM2) substrate recognition and catalysis illuminated by a structure and function based analysis. Biochemistry. 2009;48:1293–1304. doi: 10.1021/bi801879z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H, Martin BM, Bisoffi M, Dunaway-Mariano D. The Akt C-terminal modulator protein is an acyl-CoA thioesterase of the hotdog-fold family. Biochemistry. 2009;48:5507–5509. doi: 10.1021/bi900710w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein/dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 37.Foulon V, Asselberghs S, Geens W, Mannaerts GP, Casteels M, et al. Further studies on the substrate spectrum of phytanoyl-CoA hydroxylase: implications for refsum disease? J Lipid Res. 2003;44:2349–2355. doi: 10.1194/jlr.M300230-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Van Veldhoven PP, Croes K, Asselberghs S, Herdewijn P, Mannaerts GP. Peroxisomal beta-oxidation of 2-methyl-branched acyl-CoA esters: stereospecific recognition of the 2S-methyl compounds by trihydroxycoprostanoyl-CoA oxidase and pristanoyl-CoA oxidase. FEBS Lett. 1996;388:80–84. doi: 10.1016/0014-5793(96)00508-X. [DOI] [PubMed] [Google Scholar]
- 39.Van Veldhoven PP, Vanhove G, Assselberghs S, Eyssen HJ, Mannaerts GP. Substrate specificities of rat liver peroxisomal acyl-CoA oxidases: palmitoyl-CoA oxidase (inducible acyl-CoA oxidase), pristanoyl-CoA oxidase (non-inducible acyl-CoA oxidase), and trihydroxycoprostanoyl-CoA oxidase. J Biol Chem. 1992;267:20065–20074. [PubMed] [Google Scholar]
- 40.Gayral P, Caminade P, Boursot P, Galtier N. The evolutionary fate of recently duplicated retrogenes in mice. J Evol Biol. 2007;20:617–626. doi: 10.1111/j.1420-9101.2006.01245.x. [DOI] [PubMed] [Google Scholar]
- 41.Alexson SE, Nedergaard J. A novel type of short- and medium-chain acyl-CoA hydrolases in brown adipose tissue mitochondria. J Biol Chem. 1988;263:13564–13571. [PubMed] [Google Scholar]
- 42.Alexson SE, Svensson LT, Nedergaard J. NADH-sensitive propionyl-CoA hydrolase in brown-adipose-tissue mitochondria of the rat. Biochim Biophys Acta. 1989;1005:13–19. doi: 10.1016/0005-2760(89)90025-8. [DOI] [PubMed] [Google Scholar]
- 43.Lee KY, Schulz H. Isolation, properties, and regulation of a mitochondrial acyl coenzyme A thioesterase from pig heart. J Biol Chem. 1979;254:4516–4523. [PubMed] [Google Scholar]
- 44.Wagener S, Schink B. Fermentative degradation of nonionic surfactants and polyethylene glycol by enrichment cultures and by pure cultures of homoacetogenic and propionate-forming bacteria. Appl Environ Microbiol. 1988;54:561–565. doi: 10.1128/aem.54.2.561-565.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lovley DR, Phillips EJ, Lonergan DJ, Widman PK. Fe(III) and S0 reduction by Pelobacter carbinolicus . Appl Environ Microbiol. 1995;61:2132–2138. doi: 10.1128/aem.61.6.2132-2138.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kremer DR, Hansen TA. Pathway of propionate degradation in Desulfobulbus propionicus . FEMS Microbiol Lett. 1988;49:273–277. doi: 10.1111/j.1574-6968.1988.tb02729.x. [DOI] [Google Scholar]
- 47.Aklujkar M, Krushkal J, DiBartolo G, Lapidus A, Land ML, et al. The genome sequence of Geobacter metallireducens: features of metabolism, physiology and regulation common and dissimilar to Geobacter sulfurreducens . BMC Microbiol. 2009;9:109. doi: 10.1186/1471-2180-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prass RL, Isohashi F, Utter MF. Purification and characterization of an extramitochondrial acetyl coenzyme A hydrolase from rat liver. J Biol Chem. 1980;255:5215–5223. [PubMed] [Google Scholar]
- 49.Suematsu N, Okamoto K, Shibata K, Nakanishi Y, Isohashi F. Molecular cloning and functional expression of rat liver cytosolic acetyl-CoA hydrolase. Eur J Biochem. 2001;268:2700–2709. doi: 10.1046/j.1432-1327.2001.02162.x. [DOI] [PubMed] [Google Scholar]
- 50.Hawes JW, Jaskiewicz J, Shimomura Y, Huang B, Bunting J, et al. Primary structure and tissue-specific expression of human β-hydroxyisobutyryl-coenzyme A hydrolase. J Biol Chem. 1996;271:26430–26434. doi: 10.1074/jbc.271.42.26430. [DOI] [PubMed] [Google Scholar]
- 51.Loupatty FJ, Clayton PT, Ruiter JP, Ofman R, Ijlst L, et al. Mutations in the gene encoding 3-hydroxyisobutyryl-CoA hydrolase results in progressive infantile neurodegeneration. Am J Hum Genet. 2007;80:195–199. doi: 10.1086/510725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loupatty FJ, van der Steen A, Ijlst L, Ruiter JP, Ofman R, et al. Clinical, biochemical, and molecular findings in three patients with 3-hydroxyisobutyric aciduria. Mol Genet Metab. 2006;87:243–248. doi: 10.1016/j.ymgme.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 53.Shimomura Y, Murakami T, Nakai N, Huang B, Hawes JW, et al. 3-hydroxyisobutyryl-CoA hydrolase. Meth Enz. 2000;324:229–240. doi: 10.1016/S0076-6879(00)24235-3. [DOI] [PubMed] [Google Scholar]
- 54.Hutson SM, Wallin R, Hall TR. Identification of mitochondrial branched-chain aminotransferase and its isoforms in rat tissues. J Biol Chem. 1992;267:15681–15686. [PubMed] [Google Scholar]
- 55.Ooiwa T, Goto H, Tsukamoto Y, Hayakawa T, Sugiyama S, et al. Regulation of valine catabolism in canine tissues: tissue distributions of branched-chain aminotransferase and 2-oxo acid dehydrogenase complex, methacrylyl-CoA hydratase and 3-hydroxyisobutyryl-CoA hydrolase. Biochim Biophys Acta. 1995;1243:216–220. doi: 10.1016/0304-4165(94)00061-2. [DOI] [PubMed] [Google Scholar]
- 56.Colombo S, Casati A, Capocasa T, Cornero G, Gallioli G, et al. Treatment of severe acidemia in a young woman affected by diabetic ketoacidosis. Minerva Anestesiol. 1997;63:379–382. [PubMed] [Google Scholar]
- 57.Seashore MR. The organic acidemias: an overview. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. Gene reviews. Washington: Seattle; 1993. [Google Scholar]
- 58.Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 59.Jakobs BS, Wanders RJA. Fatty acid β-oxidation in peroxisomes and mitochondria: the first, unequivocal evidence for the involvement of carnitine in shuttling propionyl-CoA from peroxisomes to mitochondria. Biochem Biophys Res Commun. 1995;213:1035–1041. doi: 10.1006/bbrc.1995.2232. [DOI] [PubMed] [Google Scholar]
- 60.Cheema-Dhadli S, Leznoff CC, Halperin ML. Effect of 2-methylcitrate on citrate metabolism: implications for the management of patients with propionic acidemia and methylmalonic aciduria. Pediatr Res. 1975;9:905–908. doi: 10.1203/00006450-197512000-00008. [DOI] [PubMed] [Google Scholar]
- 61.Matsuo M, Terai K, Kameda N, Matsumoto A, Kurokawa Y, et al. Designation of enzyme activity of glycine-N-acyltransferase family genes and depression of glycine-N-acyltransferase in human hepatocellular carcinoma. Biochem Biophys Res Commun. 2012;420:901–906. doi: 10.1016/j.bbrc.2012.03.099. [DOI] [PubMed] [Google Scholar]
- 62.Sauer SW, Okun JG, Hoffmann GF, Koelker S, Morath MA. Impact of short- and medium-chain organic acids, acylcarnitines, and acyl-CoAs on mitochondrial energy metabolism. Biochim Biophys Acta. 2008;1777:1276–1282. doi: 10.1016/j.bbabio.2008.05.447. [DOI] [PubMed] [Google Scholar]
- 63.Schwab MA, Sauer SW, Okun JG, Nijtmans LG, Rodenburg RJ, et al. Secondary mitochondrial dysfunction in propionic aciduria: a pathogenic role for endogenous mitochondrial toxins. Biochem J. 2006;398:107–112. doi: 10.1042/BJ20060221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Korzekwa KR, Krishnamachary N, Shou M, Ogai A, Parise RA, et al. Evaluation of atypical cytochrome P450 kinetics with two-substrate models: evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. Biochemistry. 1998;37:4137–4147. doi: 10.1021/bi9715627. [DOI] [PubMed] [Google Scholar]
- 65.Marfori M, Kobe B, Forwood JK. Ligand-induced conformational changes within a hexameric Acyl-CoA thioesterase. J Biol Chem. 2011;286:35643–35649. doi: 10.1074/jbc.M111.225953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hunt MC, Solaas K, Kase BF, Alexson SE. Characterization of an acyl-coA thioesterase that functions as a major regulator of peroxisomal lipid metabolism. J Biol Chem. 2002;277:1128–1138. doi: 10.1074/jbc.M106458200. [DOI] [PubMed] [Google Scholar]
- 67.Ofman R, el Mrabet L, Dacremont G, Spijer D, Wanders RJ. Demonstration of dimethylnonanoyl-CoA thioesterase activity in rat liver peroxisomes followed by purification and molecular cloning of the thioesterase involved. Biochem Biophys Res Commun. 2002;290:629–634. doi: 10.1006/bbrc.2001.6245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
4,8-Dimethylnonanoyl-CoA is a substrate for ACOT9. 4,8-Dimethylnonanoyl-CoA (DMN-CoA) is formed in peroxisomes via α- and subsequent β-oxidation of phytanic acid. DMN-CoA is a substrate either for carnitine octanoyltransferase (CROT) or ACOT8, a thioesterase with very broad acyl-CoA specificity [66, 67]. DMN can be transferred to mitochondria either as the non-esterified acid or as a carnitine ester, where it can be metabolized to propionyl-CoA and acetyl-CoA via mitochondrial β-oxidation (indicated in the upper right panel). The upper left panel shows a plot of ACOT9 V max and K m values with nonanoyl-CoA being clearly distinguishable from the other acyl-CoA esters in the curve. The lower left panel shows a graph of K cat/K m values including the K cat/K m for DMN-CoA. The lower right panels show the Michaelis–Menten kinetics of ACOT9 with nonanoyl-CoA (C9-CoA) and DMN-CoA (one representative experiment out of two). Note that V max is higher and K m is lower with DMN-CoA, resulting in a 3–4fold higher K cat/K m (PPTX 226 kb)
Phylogenetic tree of ACOT9 homologs from selected species ranging from bacteria to primates. The mouse ACOT9 protein sequence was used as a template to search various databases and full-length protein sequences were aligned using ClustalW. While some species contain more than one ACOT9 homologue, e.g. C. elegans and Danio rerio, some mammalian species including human and other primates contain one gene that is alternatively spliced (3rd exon coding for nine amino acids) resulting for proteins of 439 and 448 amino acids, indicated as -1 and -2 respectively. *Indicates the presence of putative C-terminal peroxisomal Type-1 targeting signal, with its amino acid sequence being added after the name (PPTX 358 kb)
Identification of the putative catalytic residues and active sites I and II in ACOT9. The protein sequences of mouse ACOT7, ACOT9, ACOT11, ACOT12 and ACOT13 were aligned together with a homologous thioesterase from H. walsbyi (Haloquadratum walsbyi) using ClustalW. The upper panel (a cladogram) shows the relationship of H. walsbyi thioesterase, ACOT7, ACOT9, ACOT11, ACOT12 and ACOT13 despite low sequence similarity. It should be noted that ClustalW uses the ‘neighbor-joining method’, which rather represent the data in the form of an additive tree and therefore provides a branching pattern rather than relative time or relative amount of amino acid changes (evolutionary changes). The lower panel shows the alignment of the corresponding amino acid sequences that highlights (boxed) the catalytic residues of active site I (Asn-36 and Asp-225, which are crucial for activity) and II (Glu-51 and Thr-210) of ACOT7 as identified in [15]. In ACOT9, ACOT11 and ACOT12 only site II is active (corresponding Asp and Asn residues). ACOT13 and the H. walsbyi thioesterase contain only one thioesterase domain with one active site (Asn and Asp) that upon dimerization of the proteins results in two active sites (Asn/Asp and Asn/Asp respectively) per dimer in these proteins (PPTX 436 kb)
The catalytic residues of the putative active site II (Asp and Asn) are conserved in a variety of ACOT9 homologs. A selection of ACOT9 homologs from various species from archaea bacteria to human were aligned using ClustalW. Note that Asp and Asn of active site II are conserved in all ACOT9 proteins containing two thioesterase domains. The archaea bacterium Haloquadratum walsbyi (H. walsbyi) is included as a bacterial homologue containing only one thioesterase domain and Desulfobulbus propionicus (D. propionicus) and Geobacter uraniireducens (G. uraniireducens) are included as examples of bacterial ACOT9 homologs containing two thioesterase domains. M. musculus; Mus musculus, C. elegans; Caenorhabditis elegans, C. intestinalis; Ciona intestinalis, H. sapiens, Homo sapiens, I. scapularis; Ixodes scapularis, M. gallopavo; Meleagris gallopavo, N. vectensis; Nematostella vectensis, R. communis, Ricinus communis, T. stipitatus; Talaromyces stipitatus, V. carteri; Volvox carteri (PPTX 812 kb)