

Abstract
CD24 is a glycosyl-phosphatidylinositol-anchored membrane protein that is frequently over-expressed in a variety of human carcinomas and is correlated with poor prognosis. In cancer cell lines, changes of CD24 expression can alter several cellular properties in vitro and tumor growth in vivo. However, little is known about how CD24 mediates these effects. Here we have analyzed the functional consequences of CD24 knock-down or over-expression in human cancer cell lines. Depletion of CD24 reduced cell proliferation and adhesion, enhanced apoptosis, and regulated the expression of various genes some of which were identified as STAT3 target genes. Loss of CD24 reduced STAT3 and FAK phosphorylation. Diminished STAT3 activity was confirmed by specific reporter assays. We found that reduced STAT3 activity after CD24 knock-down was accompanied by altered Src phosphorylation. Silencing of Src, similar to CD24, targeted the expression of prototype STAT3-regulated genes. Likewise, the over-expression of CD24 augmented Src-Y416 phosphorylation, the recruitment of Src into lipid rafts and the expression of STAT3-dependent target genes. An antibody to CD24 was effective in reducing tumor growth of A549 lung cancer and BxPC3 pancreatic cancer xenografts in mice. Antibody treatment affected the level of Src-phosphorylation in the tumor and altered the expression of STAT3 target genes. Our results provide evidence that CD24 regulates STAT3 and FAK activity and suggest an important role of Src in this process. Finally, the targeting of CD24 by antibodies could represent a novel route for tumor therapy.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1055-9) contains supplementary material, which is available to authorized users.
Keywords: CD24, STAT3, Cancer, Lipid rafts, Signaling
Introduction
CD24 is a highly glycosylated protein with a molecular weight of 30–70 kDa [1] that is linked to the membrane via a glycosyl-phosphatidylinositol (GPI) anchor. In humans, the protein core of CD24 comprises only 31 amino acids with 16 potential O- and N-glycosylation sites showing mucin-like characteristics [2]. CD24 is frequently over-expressed in many human tumors and its expression is associated with a poor prognosis [2–4]. Recently, CD24 has been introduced as a marker for normal and cancer stem cells, although this is controversial and under intensive investigation [5]. The functional role of the CD24 molecule is still undefined.
Initially, CD24 was discovered as a lymphoid differentiation marker in the mouse expressed on hematopoietic cells such as activated T cells, B cells, macrophages, dendritic cells, and neutrophils, but also being present in the developing brain as well as on certain epithelial cells [6]. CD24 knock-out mice were found to be viable but displayed a mild block in bone marrow maturation of B lymphocytes [7]. Interestingly, pre-B cell lines established from CD24 knock-out mice showed reduced α4 integrin-mediated cell binding to activated endothelioma cells as well as fibronectin and this defect was restored by re-expression of CD24 [8]. An impact on integrin-mediated cell binding was also noted in carcinoma cell lines that over-expressed CD24 [9].
In tumor cells, CD24 has been identified as a ligand of P-selectin that supports the rolling of breast carcinoma cells on endothelial cells and adhesion to platelets, and might therefore facilitate the metastatic spread of tumor cells [2, 9]. CD24 expression regulates tumor cell proliferation, adhesion, migration, and invasion and alters gene expression in colon and pancreatic cancer cell lines [10]. More recent studies have also suggested a role of CD24 in mRNA stability in cancer cells [11]. It is unknown how CD24 can regulate such diverse cellular functions.
Due to its GPI anchor, CD24 is exclusively localized in detergent-resistant membrane domains that have been also termed lipid rafts. These membrane microdomains are considered as important platforms for signaling molecules such as Src family tyrosine kinases and G-proteins. GPI-anchored membrane proteins like CD24 reside in opposite leaflets of the same sphingolipid-enriched microdomains [12]. Indeed, a physical interaction of CD24 with members of the Src-kinase family has been observed [13–15]. Two recent reports have suggested that CD24 signals via Src–kinases in human tumors. Baumann et al. [16] found that CD24 interacts and promotes the activity of Src within lipid rafts in breast cancer cells and thereby increases integrin-dependent adhesion. Bretz et al. [17] reported that CD24 knock-down alters cell invasiveness and expression levels of the tissue factor pathway inhibitor-2 (TFPI-2) in an Src-dependent manner. In addition, the over-expression of CD24 augmented contractile forces necessary for cell invasion that was blocked by knock-down of the β1 integrin, in the presence of inhibitors to myosin light chain or Src [18]. It is possible that the CD24-Src association is relevant for other CD24-mediated downstream effects.
In the present study, we used siRNA-mediated knock-down and stable over-expression to investigate the function of CD24 in human carcinoma cell lines. We observed that the knock-down of CD24 affected STAT3 phosphorylation and STAT3-dependent transcriptional activity. MAbs directed against CD24 were found to inhibit growth of human cancer cells in immuno-deficient mice that was accompanied by altered Src phosphorylation and STAT3 target gene expression.
Materials and methods
Cells
The tumor cell lines A549 (lung adenocarcinoma), SKOV3ip (ovarian carcinoma), BxPC3 (pancreatic carcinoma), HS683 and SNB19 (glioblastoma) were either described before (SKOV3ip, [19]) or obtained from the Tumorbank of the German Cancer Research Center. The lung adenocarcinoma cell line A125 stably transfected with CD24 and selected by FACS sorting was described before [18, 20]. SKOV3ip, HS683, and SNB19 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (FBS). A125 and A549 cells were maintained in RPMI 1,640 containing 10 % FBS.
Chemicals and antibodies
The mAb SWA11 to human CD24 was described before [21, 22]. The antibodies against Src, p-Src (Y416, Y527), STAT3 and p-STAT3 (Y705), FAK and p-FAK (Y925, Y397) were obtained from Cell Signaling (Frankfurt, Germany). For immunofluorescence analysis, the p-STAT3 Ab from LifeSpan BioSciences (Seattle, WA) was used. The mAb specific for GAPDH was from Santa Cruz Biotech (Heidelberg, Germany). Secondary antibodies coupled to Peroxidase, Phycoerythrin, Alexa594 or Alexa488 were obtained from Dianova (Hamburg, Germany).
siRNA mediated knock-down
The siRNA for CD24 was described before [22]. Additional CD24-targeting siRNAs were obtained from MWG (Ebersberg, Germany). siRNAs targeting STAT3 and Src as well as GFP-specific siRNA used as a control were also obtained from MWG (Ebersberg, Germany). Sequences for siSTAT3 and siSrc were designed using the “Ambion siRNA target finder” online-tool by Applied Biosystems (Darmstadt, Germany). Transfection of siRNAs was carried out using Oligofectamine™ (Invitrogen, Darmstadt, Germany) following the manufactures protocol.
Quantitative real-time PCR
qPCR was performed as described before [23]. Briefly, total RNA from cells was isolated and transcribed using the ReverseAid H Minus First Strand cDNA Synthesis Kit (Fermentas, St. Leon-Rot, Germany), purified on Microspin G-50 columns (GE Healthcare, Freiburg, Germany) and quantified by a NanoDrop Spectrophotometer (ND-2000; Thermo Scientific). qPCR primers were designed by using the “IDT Primer Quest” online tool and were produced by MWG Eurofines (Ebersberg, Germany). As an internal standard, β-actin was used. The sequences of primers used are available on request.
Luciferase assay
This has been described in detail elsewhere [24]. Briefly, cells were seeded into 96-well plates and subjected to siRNA transfection as described above. The STAT3-specific or control reporter luciferase constructs (Cignal™ STAT3 Reporter Assay Kit, SABioscience, Frederick, MD, USA) were transfected 24 h after siRNA application using SureFECT™ (SABioscience, Frederick, MD, USA) transfection reagent. Cell lysis and luciferase measurement was carried out as described [24]. The experiment was carried out in triplicates.
Apoptosis assay
The apoptosis assay using Annexin-V and propidium iodide (PI) staining has been described elsewhere [25]. An apoptosis detection kit I from BD Pharmingen (Heidelberg, Germany) was used. Cell cycle analysis was carried out after PI staining after knock-down. For data evaluation, the Dean/Jett/Fox modus of cell cycle analysis of the FlowJo software (Ashland, OR) was used.
MTT assay
This assay was described in detail before [26].
Fluorescence-activated cell sorting
This has been described in detail elsewhere [22]. Cells were analyzed with FACS Canto II (Becton–Dickinson, Heidelberg, Germany). For data analysis, FlowJo software (Ashland, OR) was used.
Cell adhesion assay
The cell adhesion assay has been described before [27].
Cell lysis and Western-blot analysis
Cell pellets were solubilized in lysis buffer (250 mM NaCl, 50 mM HEPES, 0.5 % NP-40, 10 % glycerol, 2 mM EDTA, 10 mM NaF, 1 mM Na-orthovanadate, 1 mM PMSF, 10 mg/ml of each leupeptin and aprotinin) for 30 min on ice. Lysates were cleared by centrifugation and boiled with reducing or non-reducing SDS loading buffer. Samples were separated on SDS-PAGE gels and transferred to Immobilon membranes using semi-dry blotting. After blocking with 4 % BSA in Tween-20/TBS, membranes were probed with primary antibodies followed by horseradish peroxidase-conjugated secondary antibody and ECL detection (Amersham-Pharmacia, Freiburg, Germany).
In vivo xenograft tumor models
BxPC3 human pancreatic cancer cells or A549 lung cancer cells (5 × 106 in 100 μl of PBS) were transplanted s.c. into the right flank of Nod/Scid or Scid/beige mice, respectively. SWA11 mAb treatment was initiated when tumors reached a volume of 30–80 mm3. Animals (n = 7–10 per group) received five i.p. injections of SWA11 mAb (IgG2a) at a dose of 10 mg/kg. Control animals received unspecific isotype control IgG2a or PBS. Tumor size was measured externally using a caliper. Data are presented as a relative tumor volume increase from the time of antibody administration. The SKOV3ip therapy model has been described before [19]. Animal experiments were approved by the Baden-Württemberg animal oversight committee (Regierungspräsidium Karlsruhe, Germany).
Immunofluorescence
Double immunofluorescence was performed on 6-μm frozen tumor tissue sections as described elsewhere. Goat anti-mouse IgG conjugated to Alexa 488 or goat anti-rabbit IgG Alexa 594 were used as secondary antibodies. Tissue stainings were examined at 400× magnification using a Leica DMRB microscope. Images were captured using a SPOT Flex digital color camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA) and analyzed with SPOT Advanced version 4.6 software. For the quantification of fluorescent intensities, we selected regions with the maximum of immunofluorescence, i.e., “hot spots”, in both SWA11 mAb-treated tumors and IgG2a controls. We determined the total fluorescence intensity (TFI) of p-Src Y527 and p-Src Y416 staining using the analysis of the images in the single channels and evaluating the integrated density. The ratio between TFI for p-Src Y527 or p-Src Y416 in SWA11 mAb-treated tumors and TFI in IgG2a controls was determined.
Isolation and analysis of membrane lipid rafts
This has been described before [23]. Cells (approx. 1 × 107) were lysed in 10 mM Tris/HCl pH 8.0 containing 1 % Triton X-100, 150 mM NaCl, 1 mM PMSF, 1 μg/ml aprotinin at 4 °C. The lysate was mixed with an equal volume of 85 % sucrose (w/v in TBS containing Boehringer complete™ protease inhibitor cocktail, NaF and NaVO3) and 0.5 ml was transferred to a centrifuge tube. A step gradient was prepared by overlaying with 3 ml of 35 % sucrose in TBS followed by 0.6 ml of 5 % sucrose in TBS as described. The gradient was centrifuged for 18 h at 200,000 × g in a Beckman SW40 rotor. One-ml fractions were collected from the top of the gradient and precipitated with a tenfold volume of acetone. Samples were dissolved in SDS loading buffer. SDS-PAGE, Western blotting, and ECL were done as described above.
Statistical analysis
Data are presented as the mean ± SD. Student’s unpaired t test was used to evaluate the difference between groups. p < 0.05 was considered statistically significant. p values in the figures are indicated as follows: * < 0.05, ** < 0.01, *** < 0.001, n.s. = not significant.
Results
CD24 knock-down affects cell adhesion and proliferation and enhances apoptosis of cancer cell lines
We investigated the effects of transient knock-down of CD24 in four tumor cell lines, i.e., A549 lung carcinoma, SKOV3ip ovarian carcinoma, HS683 and SNB19 glioblastoma cells. The efficacy of CD24 knock-down was verified by Western blot of whole-cell lysates and FACS analysis of cell surface CD24. A reduction of >90 % was observed in SKOV3ip and SNB19 cells whereas the depletion in HS683 and A549 cells was in the range of 60–70 % (Fig. 1a, b).
Fig. 1.
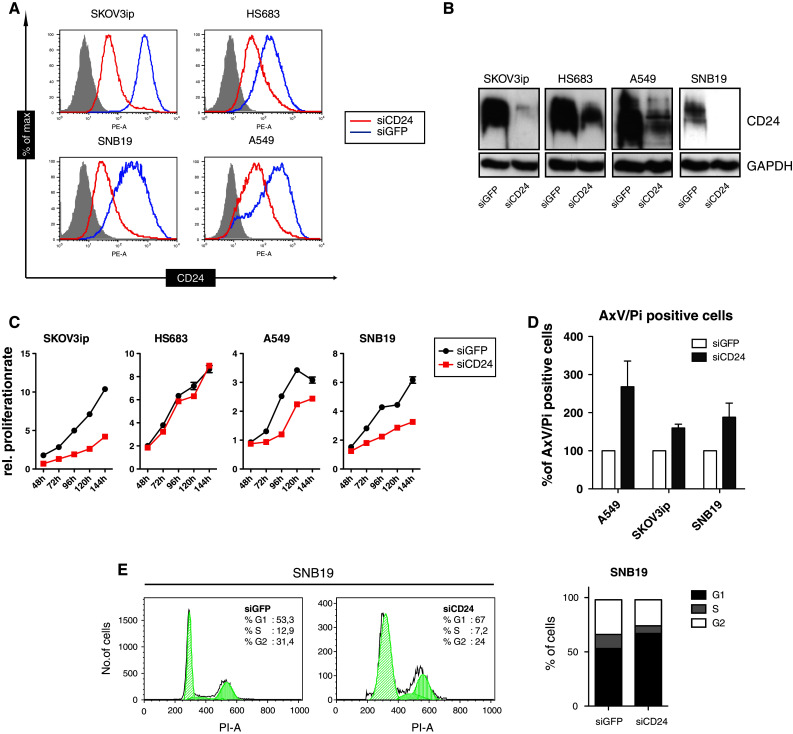
The silencing of CD24 decreases cell proliferation and enhances apoptosis. a The indicated tumor cell lines were treated with siRNA targeting CD24 or GFP as control for 96 h. Cells were stained with antibodies to CD24 (SWA11) followed by PE-conjugated goat anti-mouse IgG and subjected to FACS analysis. b Western-blot analysis of whole-cell lysates using SWA11 and peroxidase-coupled secondary IgG. c MTT proliferation assay of cells after siCD24 knock-down. Proliferation was tested at the indicated time points. d Induction of apoptosis was investigated 72 h after siCD24 transfection using PI- and Annexin-V staining. e Cell cycle analysis on SNB19 cells showing the strongest apoptotic effect was carried out in parallel. Pooled results of n = 3 experiments (d) are shown. Representative data of n = 4 experiments are shown for (a), (b), (c), and (e)
Previous reports have shown that CD24 influences cell proliferation [10, 28]. We found that the depletion of CD24 by siCD24 lead to a profound inhibition of cell proliferation in three out of the four cell lines (Fig. 1c). The low efficacy of CD24 knock-down in HS683 cells may account for this failure. In agreement with previous reports [28], in affected cell lines the loss of proliferation was accompanied by the induction of apoptosis as detected by annexin-V/PI staining (Fig. 1d and Suppl. Fig. 1) and cell cycle analysis showing a growth arrest in the G1 phase (Fig. 1e).
CD24 has been shown to play a role in cell adhesion and invasion [8, 9, 16, 20, 29]. We examined cell adhesion to immobilized ECM components such as laminin and the fibronectin subfragment FN-40, which supports α4β1 integrin-mediated cell adhesion. We observed that CD24 knock-down reduced cell adhesion of HS683 and SNB19 glioblastoma cells to FN-40 but did not affect the binding of A549 and SKOV3ip cells for presently unknown reasons (Fig. 2a). Moreover, the depletion of CD24 diminished the phosphorylation of FAK at the Src-phosphorylation site (FAK Y925) and at the autophosphorylation site (FAK Y397) (Fig. 2b).
Fig. 2.

CD24 knock-down alters cell adhesion and FAK phosphorylation. a The indicated tumor cell lines were subjected to siCD24-mediated knock-down of CD24. Cells were analyzed for cell binding to immobilized substrates FN-40, laminin and BSA for background control. The experiments were carried out in triplicates. b Analysis of FAK phosphorylation in CD24-depleted cells. Cell lysates of the indicated cells were tested for FAK, p-FAK Y925 (Src-dependent) and p-FAK Y397 (autophosphorylation) sites. Representative data of n = 3 experiments are shown
Additional experiments showed that the siCD24-mediated knock-down reduced Matrigel invasion in three of the four cell lines [17]. These results demonstrate that CD24 expression contributes to essential cellular functions of tumor cells such as proliferation, adhesion, and invasion.
CD24 affects gene regulation in human tumor cell lines
In colorectal and pancreatic carcinoma cells, CD24 controls gene expression by an unknown mechanism [10]. To verify this in SKOV3ip cells, we used DNA microarray analysis to identify genes affected by siCD24 treatment. We found that approximately 200 genes were more than 1.5 fold regulated (Suppl. Fig. 2). Based on their background in tumor cell biology, changes in expression levels and significance, we chose four genes (OPG, MMP7, STC-1, and STAT3) for further analysis by qPCR and found them to be either up-regulated (OPG, MMP7, STC-1) or down-regulated (STAT3) by CD24 depletion. Strikingly, we observed that these selected genes were regulated similarly in the other cell lines, i.e., HS683, A549, and SNB19 upon CD24 knock-down (Fig. 3a). Due to its pivotal role in tumor development and progression, we concentrated on the transcription factor STAT3 as a down-regulated gene product. Excluding off-target effects by siCD24, we tested four additional siRNAs (siCD24-1 to 4) for STAT3-regulative properties. All siRNAs were found to deplete CD24 to a variable extent and knock-down was associated with a reduction of STAT3 expression (Fig. 3b). These data suggested that the observed STAT3 regulation was CD24-specific.
Fig. 3.
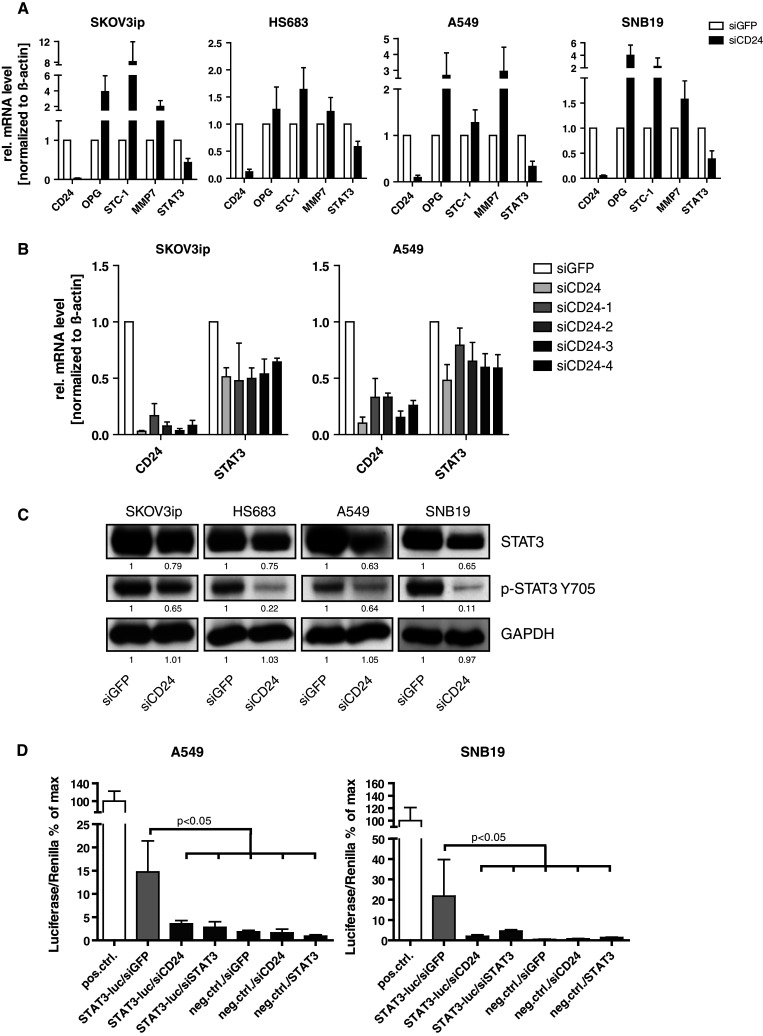
CD24 knock-down mediates gene regulation and reduces STAT3 protein phosphorylation and activity. a Identification of a CD24-dependent target genes signature (OPG, STC-1, MMP-7, and STAT3) by quantitative RT-PCR analysis of mRNAs isolated after siCD24-mediated knock-down. Note that OPG, STC-1, and MMP-7 genes are up-regulated, whereas STAT3 is down-regulated in the indicated cell lines. Pooled results of n = 6 experiments are shown. b Analysis of STAT3 levels after siRNA-mediated knock-down of CD24. Note that all oligonucleotides specific for CD24 reduce STAT3 expression levels. c Western-blot analysis for STAT3 and p-STAT3 Y705 after siCD24-mediated depletion of CD24. d Decreased STAT3 activity after CD24 or STAT3 knock-down (as a positive control) was measured using a STAT3-specific luciferase reporter assay. Pooled results (b) and representative results (c, d) of n = 4 experiments are shown
CD24 regulates STAT3 transcriptional activity
STAT3 was initially described as a key component linking cytokine signaling to transcriptional regulation under physiological conditions, but is now also known to play an important role in cancer [30, 31]. We observed that the knock-down of CD24 significantly decreased the expression and phosphorylation of STAT3 (Y705) in the cell lines analyzed (Fig. 3c).
We investigated whether CD24 depletion could alter STAT3 transcriptional activity. The knock-down of CD24 led to significantly decreased activity in a STAT3-specific luciferase reporter assay and was at a similar level as after STAT3 depletion (Fig. 3d).
In cancer cells, STAT3 signaling is often constitutive and many genes were described to depend on STAT3 activity [30]. To analyze whether CD24 and STAT3 depletion had similar effects, we studied the regulation of Cyclin D1, survivin and MCL-1, which represent well-known target genes of STAT3 in tumor cells [30]. Indeed, the silencing of CD24 or STAT3 caused similar repression of the selected genes in all tumor cell lines (Fig. 4a).
Fig. 4.
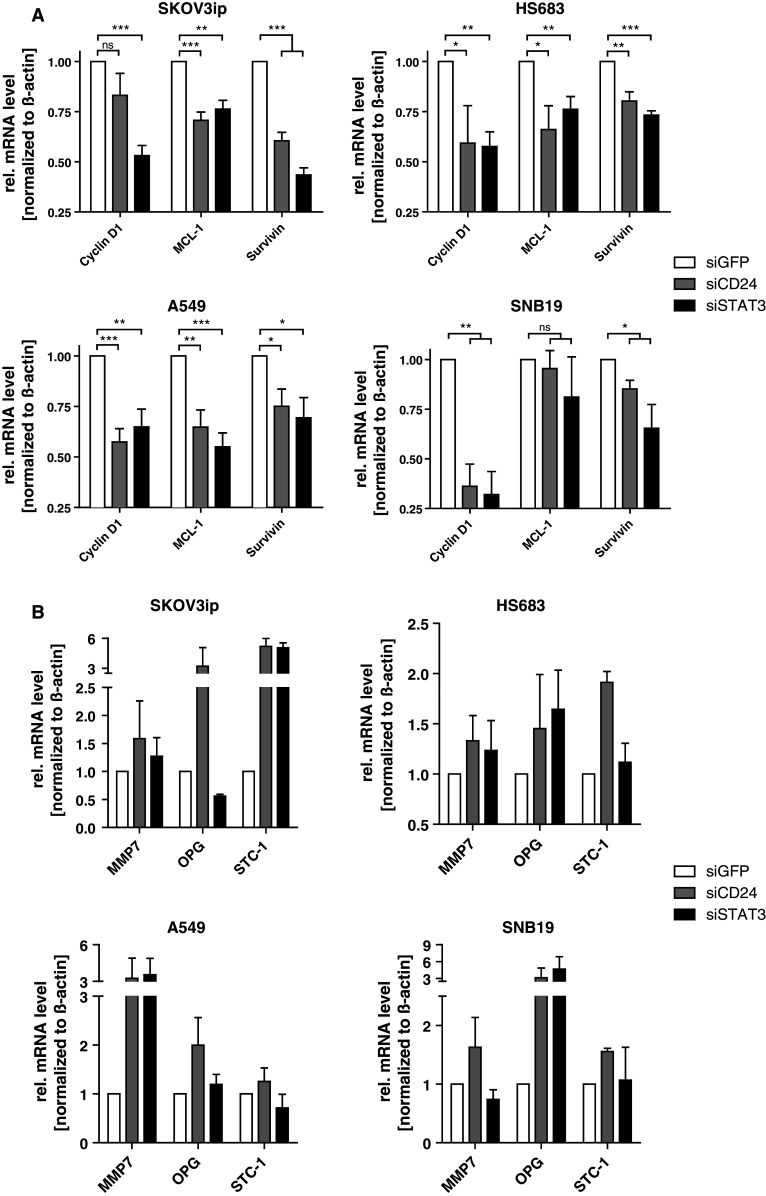
Loss of CD24 affects transcription of classical STAT3 target genes. a Known STAT3 target genes (cyclin D1, MCL-1, survivin) were analyzed by qPCR after CD24 or STAT3 knock-down in indicated tumor cell lines. Relative mRNA levels normalized to β-actin were compared to siGFP control. b Regulation of the CD24-dependent target gene signature expression after STAT3 silencing was determined by qPCR. Relative mRNA levels normalized to β-actin were compared to siGFP control. Pooled results of n = 4 experiments are shown (a, b)
CD24-affected genes are STAT3 target genes
We wondered whether the CD24-dependent genes identified in the initial screening (see Fig. 3a) might also depend on STAT3. To test this hypothesis, we studied the regulation of MMP-7, OPG, and STC-1 after STAT3 depletion. Indeed, several of the genes such as STC-1 in SKOV3ip cells, MMP7 in A549 cells, or OPG in SNB19 cells were regulated by STAT3 knock-down (Fig. 4b). Thus, several of the genes identified by CD24 knock-down appeared to be STAT3 target genes in the respective cell line.
Regulation of STAT3-dependent genes requires Src activity
We recently reported that Src-Y416 phosphorylation (active Src) was significantly diminished after siCD24 knock-down in A549 and SKOV3ip cells [17]. Consistent with a loss of activity, Src Y527 (inactive Src) phosphorylation was increased [17]. Similar results were obtained in the glioblastoma cell lines SNB19 and HS683 (Suppl. Fig. 3).
CD24 and active Src are localized in membrane lipid rafts that can be analyzed after lysis of cells in cold Triton X-100 followed by sucrose density centrifugation. In SKOV3ip and A549 cells, we noticed a loss of Src and active Src Y416 from raft localization after CD24 knock-down. On the contrary, caveolin-1 rafts remained intact (Fig. 5a). These results suggested a link between CD24 and the activation of Src in lipid rafts.
Fig. 5.
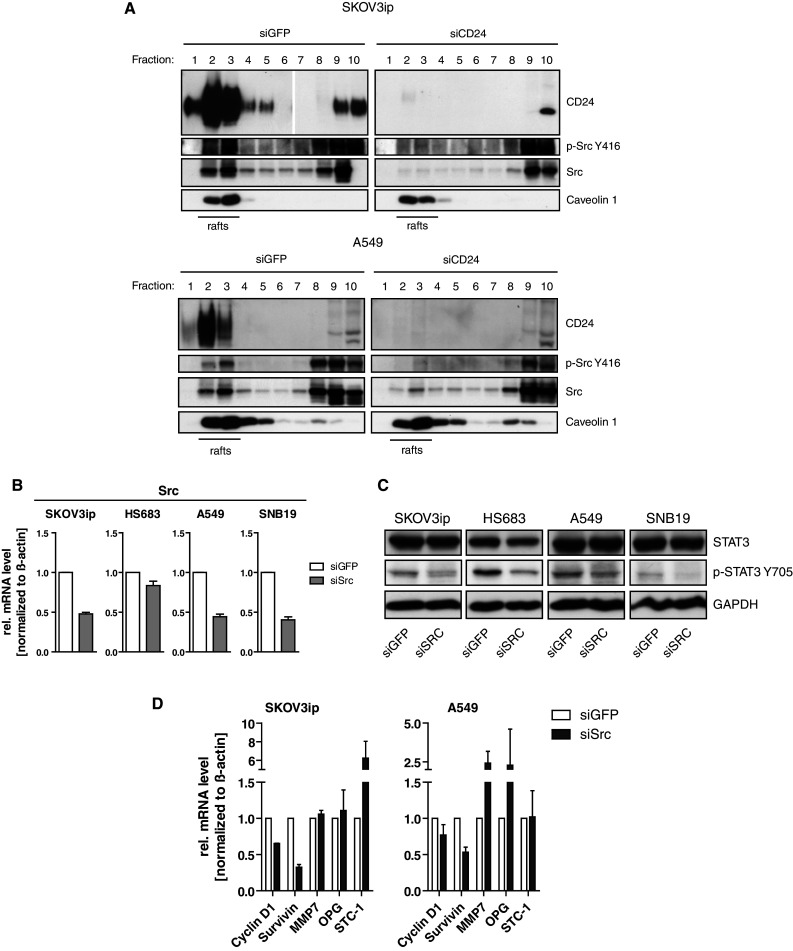
Silencing of Src affects phosphorylation of STAT3 and STAT3 target gene regulation. a Sucrose gradient analysis of lipid rafts after CD24 knock-down. Gradient fractions were probed with the indicated antibodies followed by peroxidase-conjugated secondary antibodies and ECL detection. b The indicated tumor cell lines were subjected to siRNA-mediated knock-down of Src and probed by qPCR for knock-down efficacy. c Phosphorylation of STAT3 after Src knock-down was analyzed by Western blotting. d Expression analysis of STAT3 target genes after specific knock-down of Src. Representative results (a, c) or pooled results (b, d) of at least n = 3 experiments are shown
Since Src can activate STAT3 [32, 33] and FAK [34, 35], we examined whether Src is necessary for STAT3 activity. We depleted Src by specific siRNA and determined silencing efficacy by qPCR in our cell lines (Fig. 5b). The depletion of Src altered the phosphorylation of STAT3 whereas STAT3 levels remained unaffected under these conditions (Fig. 5c). Further analysis indicated that the Src knock-down also affected the expression of STAT3-dependent genes such as Cyclin D1, survivin, MMP-7, OPG, and STC-1 in SKOV3ip and A549 cells (Fig. 5d). Similar results were obtained when cells were treated with the Src-specific inhibitor PP2 (Suppl. Fig. 4). These results suggested that the CD24-Src link played an important role in the regulation of STAT3 transcriptional activity.
CD24 over-expression alters STAT3-dependent genes
To corroborate these findings, we used over-expression of CD24 in A125 lung adenocarcinoma cells. We analyzed A125 cells stably transduced with CD24 showing either high (A125-CD24high) or low (A125-CD24low) expression [18, 20]. The levels of CD24 expression in these cells are depicted in Fig. 6a. In line with the enhanced expression of CD24, we observed a clearly elevated expression of p-STAT3 Y705, p-FAK Y925, and p-Src Y416 (Fig. 6b).
Fig. 6.
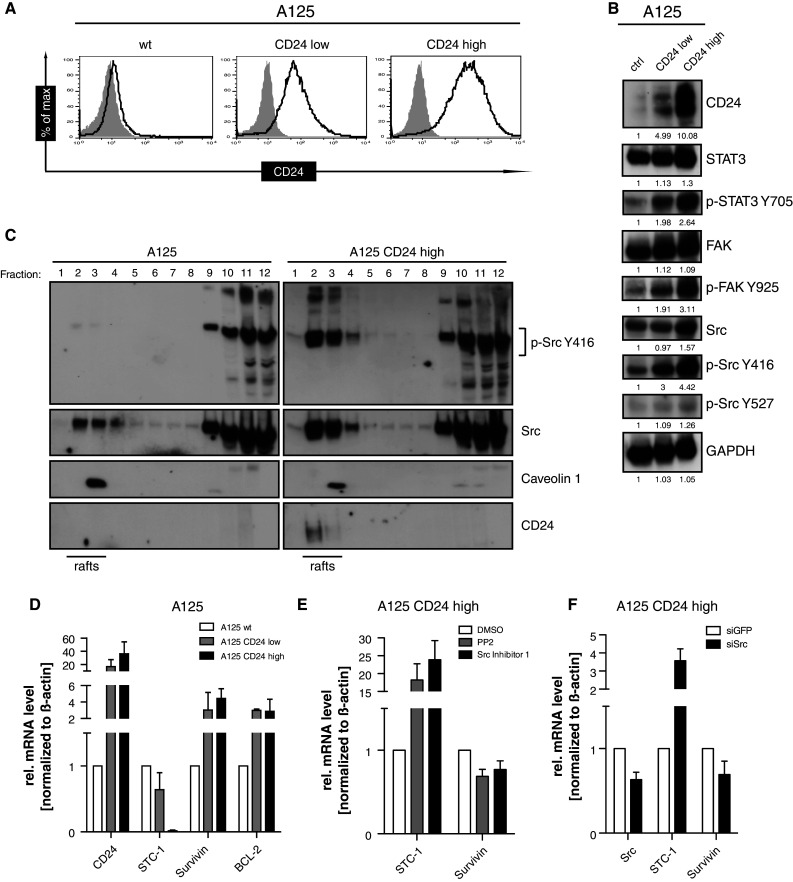
Effect of CD24 over-expression on phosphorylation and gene regulation. a Cytofluorographic analysis for CD24 expression in A125, A125-CD24low, and A125-CD24high-expressing cells. b Western-blot analysis of whole-cell lysates with the indicated phospho-specific antibodies. c Sucrose gradient analysis of lipid rafts as described in the legend to Fig. 5a. d Regulation of selected STAT-3-dependent target genes in the indicated cell lines. e A125 CD24high cells were treated with the inhibitors (20 μM) for overnight and cells were analyzed for the expression of STAT3-dependent genes. f A125 CD24high cells were depleted for Src by siRNA and the effect on the expression of STAT3-dependent genes was analyzed. Representative results of n = 3 analysis are shown in (a, b, c). Pooled results of n = 4 experiments are shown in (d, e, f)
It was shown before that the lipid raft-associated Src family kinase activity is important for cell adhesion and cell cycle progression of breast cancer cells [36]. We analyzed the phosphorylation of Src in lipid rafts of CD24 over-expressing cells. Our observation was a significantly higher expression of p-Src Y416 in A125-CD24high cells compared to A125 control cells. In contrast, the expression and phosphorylation of Src in non-raft fractions was similar and also caveolin-1 rafts were unaltered in amount and distribution. Importantly, in the absence of CD24, we observed a loss of Src from lipid raft localization (Fig. 6c). These findings are consistent with the CD24 knock-down data shown in Fig. 5a and suggest that the expression of CD24 is crucial for the amount and phosphorylation of Src in lipid rafts. This points to an important role for lipid rafts in the regulation of Src and STAT3.
In support of this notion, the expression levels of the STAT3-dependent genes survivin and BCL-2 were significantly up-regulated whereas STC-1 was down-regulated in CD24high expressing cells (Fig. 6d). The CD24-mediated regulation of STAT3 genes required Src activity as the Src inhibitors PP2 or Src inhibitor-1 reverted this expression (Fig. 6e). Similar results were obtained using Src-specific knock-down (Fig. 6f).
MAb directed against CD24 affect tumor growth in vivo
A recent study has shown that CD24 drives lung metastasis formation of bladder cancer and might be a good candidate for mAb-dependent therapy [37]. We analyzed whether the mAb SWA11 directed against CD24 is suitable in preventing tumor growth in xenotransplanted mice. To evaluate the influence of the host immune system, we employed three immunodeficient mice, i.e., Scid/beige (lack T, B, and have non-functional NK cells) for A549 human lung carcinoma, Nod/Scid (lack T and B cells) for BxPC3 pancreatic adenocarcinoma and CD1 mice (lack T cells) for the SKOV3ip ovarian carcinoma model.
In the A549 human lung carcinoma model, mice received five injections of the SWA11 mAb over 25 days. The therapy led to a substantially reduced tumor load. The final tumor volume in SWA11 mAb-treated animals was significantly decreased as compared to the isotype control mAb-treated animals. Similar observations were made in the second therapy model of BxPC3 pancreatic adenocarcinoma cells (Fig. 7a, b). Even more pronounced were the therapeutic effects of the mAb SWA11 application in the SKOV3ip ovarian carcinoma model where the tumor load was decreased by >80 % as compared to the isotype control mAb (Suppl. Fig. 5).
Fig. 7.
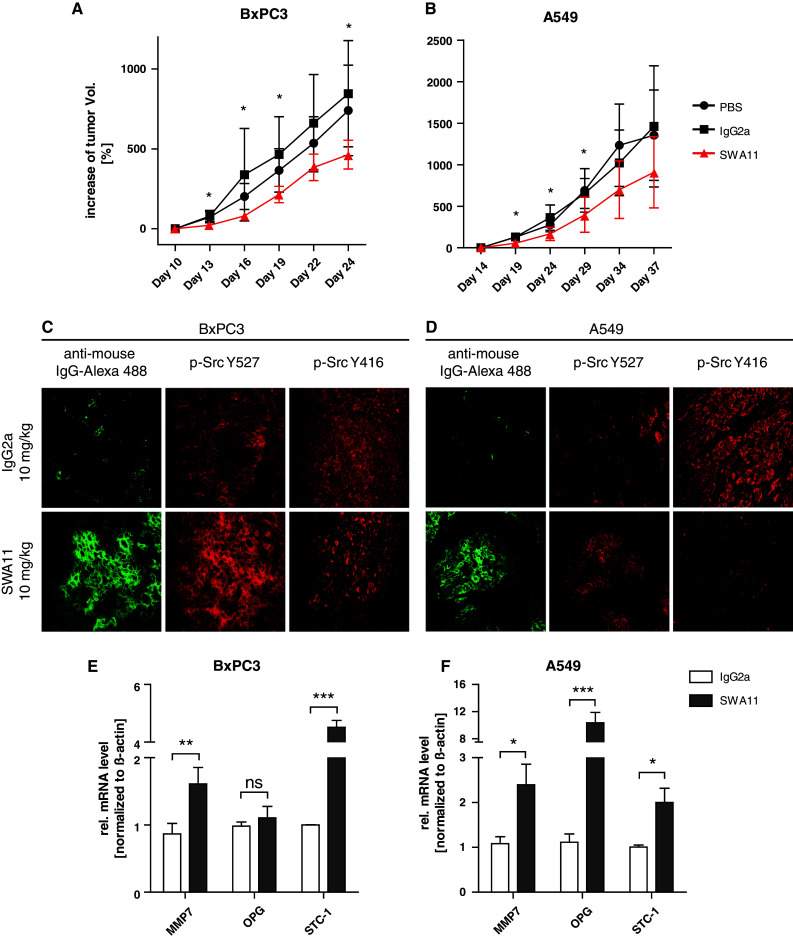
The antibody to CD24 affects phosphorylation of Src in vivo and has a therapeutic effect. a Subcutaneous tumor growth of A549 lung cancer cells in Scid/beige mice treated with either PBS, isotype control IgG2a, or SWA11 mAb. Antibodies (10 mg/kg) were given every 5 days with five injections in total. Tumor growth was measured using a caliper. Animals were killed and the isolated tumors were used for further analysis. b BxPC3 tumor cells were injected s.c. into Nod/Scid mice and treated with SWA11 mAb (10 mg/kg) every 3 days with five injections in total. Animals were killed and the isolated tumors were used for further analysis. c, d Immunofluorescence analysis of Src (Y527 and Y416) in SWA11 versus isotype control-treated animals. Note that p-Src Y416 is decreased and p-Src Y527 staining is increased in tumor areas where mAb SWA11 is bound. e, f) mRNAs were extracted from treated tumors and analyzed by qPCR for indicated STAT3-dependent genes. Pooled results of n = 4 animals per group are shown
To get insights into the effector mechanisms, we analyzed Src phosphorylation in tumor sections using immunofluorescence staining. To confirm that the therapeutic mAb had reached the tumor site, we stained sections with an Alexa488-conjugated pAb to mouse IgG. In both A549 and BxPC3 tumor tissue sections, we observed a decrease in p-Src Y416 (active Src) and an increase in p-Src Y527 staining (inactive Src) in SWA11 mAb-treated tumors compared to isotype control mAb-treated tumors (Fig. 7c, d). The quantification of the total fluorescence intensity (TFI) of p-Src Y527 in “hot spots”, i.e., areas of the tumor with the highest staining intensity, revealed that the ratio between SWA11 mAb-treated and isotype control mAb tumors was 1.26 for A549, 1.25 for BxPC3, and 1.34 for SKOV3ip tumors. Conversely, the values for p-Src Y416 were 0.48 for A549, 0.65 for BxPc-3, and 1.19 for SKOV-3, respectively. In contrast, we did not detect any differences in total Src expression. A similar analysis for p-STAT3 staining revealed a tendency towards reduced staining in SWA11-treated tumors that was however difficult to quantify due to the high basal level of p-STAT3 in the tumor sections (data not shown). Total STAT3 levels did not show any differences (data not shown).
To examine the influence of antibody therapy on STAT3-dependent gene expression, we analyzed tumor tissue by homogenization and extraction of mRNA for qPCR analysis. We observed significant gene-regulation effects evoked in the mAb SWA11-treated tumors compared to the isotype control-treated tumors. This was true for both A549 and BxPC3 mouse tumor models (Fig. 7 e, f) and also for the SKOV3ip model (see suppl. Fig. 5). These results suggest that the siRNA-mediated CD24 depletion in vitro and the antibody treatment in vivo are overlapping in their effects on Src phosphorylation and STAT3 gene expression.
Discussion
Due to its pleiotropic expression, the cellular function of CD24 has raised the interest of researchers in diverse fields such as immunology, tumor biology, and stem cell biology. Using human cancer cells as a model, we investigated how CD24 affects cellular functions. We found that (1) CD24 depletion affects FAK and STAT3 phosphorylation and STAT3-transcriptional activity; (2) CD24 knock-down or over-expression alter the expression of STAT3 dependent genes; (3) the CD24-mediated regulation of STAT3 genes requires Src activity; (4) treatment of mice bearing A459 or BxPC3 tumors with mAb directed against CD24 alters Src activity and STAT3 gene expression in vivo. Our findings are summarized in a model represented in Fig. 8 suggesting a role of CD24 in the regulation of STAT3 and FAK activity via Src that may explain its association with a poor prognosis in cancer.
Fig. 8.
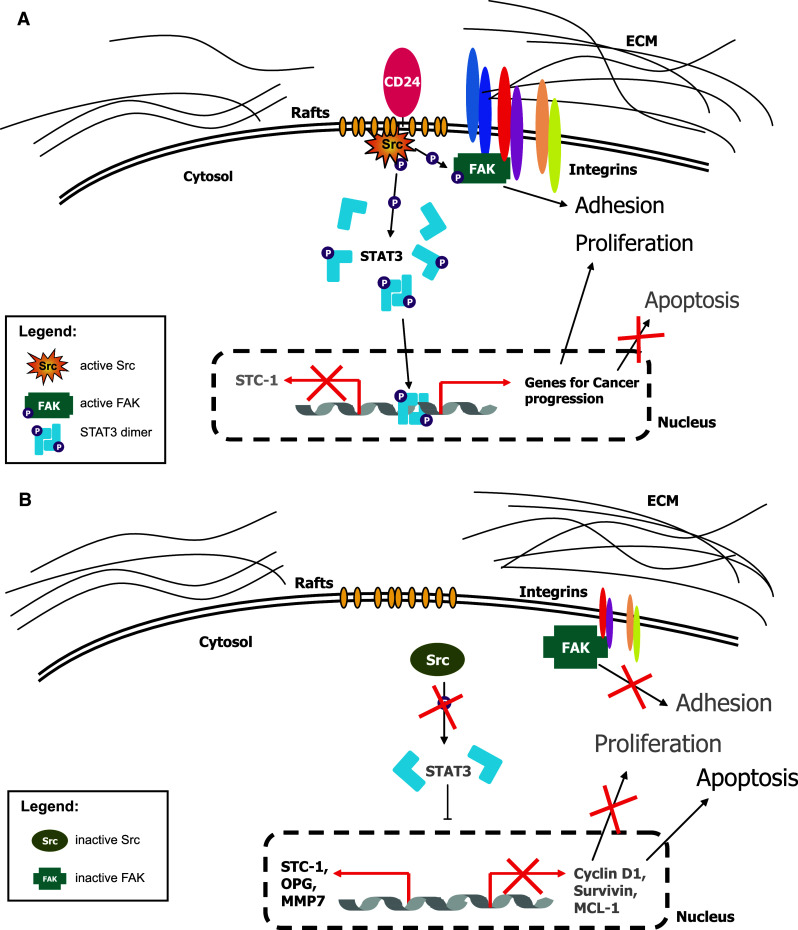
Schematic representation of the functional role of CD24 in tumors. a The presence of CD24 supports the localization and the activity of Src kinase in cholesterol enriched lipid rafts. Active Src is able to activate STAT3 molecules by phosphorylation at tyrosine 705. Activated STAT3 dimerizes and translocates into the nucleus. There, DNA binding of STAT3 dimers results in expression of specific target genes that might influence cellular properties, like survival and proliferation, contributing to oncogenicity of the tumor cell. Additionally, FAK activation might influence integrin-dependent cell adhesion to ECM substrates. b Silencing of CD24 and subsequent depletion from lipid rafts displaces Src kinase from rafts and impairs Src activity. Thereby, decreased STAT3 activation resulting in diminished expression of STAT3 target genes impairs tumor cell proliferation and enhances apoptosis. FAK-regulated adhesion to the ECM is also impaired
We initially observed that the CD24 knock-down affected cell proliferation in three out of four cell lines tested, which was paralleled by the induction of apoptosis. In addition, there was a diminished adhesion towards the α4 integrin substrate FN-40 in two out of four cell lines. These findings are in line with previous publications showing a role for CD24 in cell proliferation, adhesion and invasion [9, 10, 16, 18, 20, 22, 28, 29, 38–40]. Importantly, we observed decreased phosphorylation of FAK Y925 after CD24 knock-down and an enhanced phosphorylation at the same site in A125–CD24high cells. This site in FAK is known to be phosphorylated by Src-kinases and the lack of phosphorylation is associated with impaired adhesion turnover [41]. Thus, these results suggested a role of Src-kinases in CD24-mediated cellular effects.
A recent study by Baumann et al. [16] has come to a similar conclusion. The authors showed that CD24 interacts with Src and augments the kinase activity of Src in breast cancer cells. This occurs within and is dependent upon intact lipid rafts. CD24-augmented Src kinase activity increased the formation of focal adhesion complexes, accelerated phosphorylation of FAK and paxillin and enhanced the integrin-mediated adhesion [16]. Further experiments provided evidence that the Src activity is necessary and sufficient to mediate the effects of CD24 on integrin-dependent adhesion and cell spreading, as well as on invasion [16]. The results presented in our study confirm and extend these observations.
There is evidence that CD24 acts as a “gate-keeper” for lipid raft domains, and thereby influences the activity of other proteins [22]. It was shown before that CD24 recruits β1 integrin to lipid raft domains [20] and down-regulates the activity of the chemokine receptor CXCR4 by expelling it from lipid rafts [22]. In the present paper, we demonstrate that in A125-CD24high cells more Src and p-Src Y416 are present in lipid rafts. Conversely, the knock-down of CD24 in SKOV3ip and A549 cells expelled Src and active Src Y416 from raft localization. Previous studies have shown that the localization of Src family kinases (SFK) is important for its biological activity [36]. It was found that a lipid raft-targeted SFK inhibitory fusion protein inhibited cell adhesion and cell cycle progression of human breast cancer cell lines MCF-7 and MDA-MB-231 whereas the same inhibitor without lipid raft targeting was not effective [36]. This implied an important role for lipid raft associated SFKs. It is presently unclear how CD24 could affect the presence and activation of SFKs in lipid rafts.
Of note, a recent study by Holzer et al. [42] offers a possible explanation for this question. The authors showed that saturated fatty acids (FA) induced Src partitioning into lipid raft membrane domains affecting JNK activation. In comparison to control cells, only in FA- treated cells Src and p-Src Y416 as well as JNK 1/2 were present in lipid rafts [42]. Moreover, it was demonstrated that activation of JNK by FAs was Src dependent and that saturated FAs were preferentially incorporated into lipid rafts [42]. This might be due to their organization of sphingolipids with long and mostly saturated hydrocarbons allowing a tightly packed structure and cholesterols filling the voids between associated sphingolipids [43]. The highly ordered environment of lipid rafts may also allow for the close packing of GPI-anchored proteins, which might support the hypothesis that the GPI anchor is important in signal transduction [44]. Interestingly, the lipid anchor of the phosphoinositol ring in GPI-anchored proteins like CD24 exhibit lipid structures that vary in length, ranging from 14 to 28 carbons and can be either saturated or unsaturated [45]. Due to its structure, it is possible that the GPI anchor of CD24 has a major impact on the recruitment and activity of proteins, especially Src, to membrane rafts. However, this needs to be further investigated.
To identify additional changes induced by CD24, we carried out a genome-wide expression profiling after knock-down in SKOV3ip cells. Focusing on genes relevant in cancer biology (including OPG, MMP7, STC-1 and STAT3) we confirmed their strong regulation by qPCR. The changes in the expression of the selected genes in SKOV3ip cells were also found in the other cell lines investigated. Importantly, STAT3 was down-regulated by CD24 depletion and this was observed with five CD24-specific siRNAs ruling out off-target effects. We also noticed consistently a minor decrease of STAT3 protein expression.
In addition to or as a result of STAT3 reduction, we noticed a significant decrease in phosphorylation at STAT3 Y705 and, subsequently, a loss of transcriptional activity in a STAT3-driven luciferase reporter assay. Typical STAT3-dependent genes such as Cyclin D1, survivin, and MCL-1 were decreased in expression and the CD24 target genes OPG, MMP7, and STC-1 were also identified to be STAT3-dependent. Importantly, the knock-down of either CD24 or Src showed similar changes in the expression of STAT3 target genes.
Over-expression of CD24 in A125 cells, went along with a strong increase of p-STAT3 Y705 and p-Src Y416. It is noteworthy that there was also a minor increase of STAT3 protein upon CD24 over-expression in the latter cells. Nevertheless, the presence of CD24 led to the increased expression of STAT3 target genes, and depletion of Src in CD24 over-expressing cells reverted this gene expression. Thus, our data demonstrate that CD24 affects the transcriptional activity of STAT3 most likely through Src activation and alters the expression of STAT3-dependent genes.
A recent study in hepatocellular carcinoma has shown that CD24 is upregulated in putative tumor-initiating cells that were enriched by their resistance to chemotherapy [46]. The knock-down of CD24 suppressed the stem/progenitor cell characteristics suggesting that CD24 is a functional marker for tumor-initiating cells in the liver. Importantly, the stemness-associated gene NANOG was identified as a downstream target gene of CD24 and the regulation of NANOG expression was through STAT3 phosphorylation at Y705 [46]. Manipulation of CD24 expression consistently altered the activate form of Src (Y416) leading to the conclusion that CD24 likely phosphorylated STAT3 through Src but not JAK. These published results are in support of the presented data.
The classical activation pathway of STAT3 is via upstream kinases such as JAK2 that links STAT3 signaling to cytokine receptors [31]. A recent study has proposed that the STAT3 activation is required for the growth of CD44+ CD24− stem cell-like breast cancer cells [47]. This phenotype appears to be enriched in basal-like breast tumors [48] and was previously proposed to identify breast cancer stem cells [49]. Using primary tumor material and breast cancer cell lines, the authors identified the IL6/JAK2/STAT3 pathway as being crucial for the growth of basal-like breast cancer cells [47]. When analyzing breast tumor tissues sections by triple fluorescence analysis of CD24, CD44 and p-STAT3 it became clear that positive staining for p-STAT3 was not restricted to the CD44+ CD24− cells but resided also in >50 % of CD44+ CD24+ cells [47]. Thus, in the light of our data it is possible that CD24-Src signaling contributed to STAT3 phosphorylation and hence, the IL-6/JAK2 pathway is not the only driver of STAT3 activation.
Another recent study has shown that CD24 is a promising therapeutic target for anti-metastatic therapy of bladder cancer [37]. It was first demonstrated by comparing paired primary and metastatic human bladder cancer samples that CD24 expression was increased in the metastases. Using a model of human urothelial cancer cells transplanted into immuno-compromised mice and therapy with anti-CD24 mAb ALB9, a significantly reduced tumor growth and prolonged survival was observed [37]. Thus, CD24 seems to be an important cellular factor for metastatic progression in bladder cancer and may be promising therapeutic target for anti-metastatic therapy.
In our present work, we confirm and extend these published data by demonstrating that the mAb SWA11 directed against CD24 can inhibit the growth in various xenograft models. We provide evidence by immunohistochemistry that the mAb directed against CD24 reaches the tumor site and decreases p-Src Y416 (active Src) and increases p-Src Y527 (inactive Src) expression. Subsequent analysis of tumor tissue by qPCR revealed that the mAb to CD24 interfered with the expression of STAT3-dependent genes. These results provide first evidence that siRNA to CD24 and/or binding of a CD24-specific mAb can cause similar functional effects that might be beneficial for therapy.
In summary, our results demonstrate that CD24 can affect the Src activity and subsequently the phosphorylation of STAT3 and FAK. It is likely that constitutive STAT3 activation induces cancer-promoting activities involved in cell proliferation, metastasis, or resistance to apoptosis. Our novel insights into CD24 function may help to better understand why CD24 is associated with a poor prognosis in many human cancers.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Suppl. Fig. 1 Dot-blot analysis of apoptosis after siCD24 knock-down using PI- and Annexin-V staining.
Suppl. Fig. 2 List of genes identified after depletion of CD24 by siCD24-dependent knock-down. SiGFP served as a control.
Suppl. Fig. 3 Western-blot analysis of active (Y416) and inactive (Y527) Src in glioblastoma cell lines after CD24 depletion.
Suppl. Fig. 4 Effect of Src inhibition by the specific inhibitor PP2 on the regulation of STAT3-dependent genes.
Suppl. Fig. 5 (A) Intraperitoneal tumor growth of SKOV3ip ovarian cancer cells in CD1 mice treated with either PBS, isotype control, or SWA11 mAb. Antibodies (10 mg/kg) were given twice a week with eight injections in total. Animals were killed and the tumor mass was determined and used for further analysis. (B) MRNAs were extracted from the isolated tumors and analyzed by RT-PCR for the indicated STAT3-dependent genes. Note that p-Src Y527 staining was undetectable both in SWA11- and isotype-control treated groups.
Acknowledgments
This work was supported by a grant from the DKFZ-BSP Alliance to G. M., T.S. and P. A. We thank Helena Kiefel for valuable comments on the manuscript and Volker Kloess for help and technical support.
Abbreviations
- ECM
Extracellular matrix
- FAK
Focal adhesion kinase
- GPI
Glycosyl-phosphatidylinositol
- mAb
Monoclonal antibody
- pAb
Polyclonal antibody
- siCD24
siRNA specific for CD24
- siGFP
siRNA specific for green fluorescent protein (GFP)
- STAT3
Signal transducer and activator of transcription 3
- qPCR
Quantitative real-time PCR
References
- 1.Kay R, Rosten PM, Humphries RK. CD24, a signal transducer modulating B cell activation responses, is a very short peptide with a glycosyl phosphatidylinositol membrane anchor. J Immunol. 1991;147(4):1412–1416. [PubMed] [Google Scholar]
- 2.Kristiansen G, Sammar M, Altevogt P. Tumour biological aspects of CD24, a mucin-like adhesion molecule. J Mol Histol. 2004;35(3):255–262. doi: 10.1023/B:HIJO.0000032357.16261.c5. [DOI] [PubMed] [Google Scholar]
- 3.Kristiansen G, Machado E, Bretz N, Rupp C, Winzer KJ, Konig AK, Moldenhauer G, Marme F, Costa J, Altevogt P. Molecular and clinical dissection of CD24 antibody specificity by a comprehensive comparative analysis. Lab Invest. 2010;90(7):1102–1116. doi: 10.1038/labinvest.2010.70. [DOI] [PubMed] [Google Scholar]
- 4.Lim SC. CD24 and human carcinoma: tumor biological aspects. Biomed Pharmacother. 2005;59(2):S351–S354. doi: 10.1016/S0753-3322(05)80076-9. [DOI] [PubMed] [Google Scholar]
- 5.Woodward WA, Sulman EP. Cancer stem cells: markers or biomarkers? Cancer Metastasis Rev. 2008;27(3):459–470. doi: 10.1007/s10555-008-9130-2. [DOI] [PubMed] [Google Scholar]
- 6.Fang X, Zheng P, Tang J, Liu Y. CD24: from A to Z. Cell Mol Immunol. 2010;7(2):100–103. doi: 10.1038/cmi.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nielsen PJ, Lorenz B, Muller AM, Wenger RH, Brombacher F, Simon M, von der Weid T, Langhorne WJ, Mossmann H, Kohler G. Altered erythrocytes and a leaky block in B-cell development in CD24/HSA-deficient mice. Blood. 1997;89(3):1058–1067. [PubMed] [Google Scholar]
- 8.Hahne M, Wenger RH, Vestweber D, Nielsen PJ. The heat-stable antigen can alter very late antigen 4-mediated adhesion. J Exp Med. 1994;179(4):1391–1395. doi: 10.1084/jem.179.4.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumann P, Cremers N, Kroese F, Orend G, Chiquet-Ehrismann R, Uede T, Yagita H, Sleeman JP. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res. 2005;65(23):10783–10793. doi: 10.1158/0008-5472.CAN-05-0619. [DOI] [PubMed] [Google Scholar]
- 10.Sagiv E, Starr A, Rozovski U, Khosravi R, Altevogt P, Wang T, Arber N. Targeting CD24 for treatment of colorectal and pancreatic cancer by monoclonal antibodies or small interfering RNA. Cancer Res. 2008;68(8):2803–2812. doi: 10.1158/0008-5472.CAN-07-6463. [DOI] [PubMed] [Google Scholar]
- 11.Taniuchi K, Nishimori I, Hollingsworth MA. Intracellular CD24 inhibits cell invasion by posttranscriptional regulation of BART through interaction with G3BP. Cancer Res. 2011;71(3):895–905. doi: 10.1158/0008-5472.CAN-10-2743. [DOI] [PubMed] [Google Scholar]
- 12.Ilangumaran S, Arni S, van Echten-Deckert G, Borisch B, Hoessli DC. Microdomain-dependent regulation of Lck and Fyn protein-tyrosine kinases in T lymphocyte plasma membranes. Mol Biol Cell. 1999;10(4):891–905. doi: 10.1091/mbc.10.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zarn JA, Zimmermann SM, Pass MK, Waibel R, Stahel RA. Association of CD24 with the kinase c-fgr in a small cell lung cancer cell line and with the kinase lyn in an erythroleukemia cell line. Biochem Biophys Res Commun. 1996;225(2):384–391. doi: 10.1006/bbrc.1996.1184. [DOI] [PubMed] [Google Scholar]
- 14.Sammar M, Gulbins E, Hilbert K, Lang F, Altevogt P. Mouse CD24 as a signaling molecule for integrin-mediated cell binding: functional and physical association with src-kinases. Biochem Biophys Res Commun. 1997;234(2):330–334. doi: 10.1006/bbrc.1997.6639. [DOI] [PubMed] [Google Scholar]
- 15.Stefanova I, Horejsi V, Ansotegui IJ, Knapp W, Stockinger H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science. 1991;254(5034):1016–1019. doi: 10.1126/science.1719635. [DOI] [PubMed] [Google Scholar]
- 16.Baumann P, Thiele W, Cremers N, Muppala S, Krachulec J, Diefenbacher M, Kassel O, Mudduluru G, Allgayer H, Frame M, Sleeman JP. CD24 interacts with and promotes the activity of Src within lipid rafts in breast cancer cells, thereby increasing integrin-dependent adhesion. Cell Mol Life Sci. 2012;69(3):435–448. doi: 10.1007/s00018-011-0756-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bretz N, Noske A, Keller S, Erbe-Hofmann N, Schlange T, Salnikov A, Moldenhauer G, Kristiansen G, Altevogt P. CD24 promotes tumor-cell invasion by suppressing tissue factor pathway inhibitor-2 (TFPI-2) in a Src-dependent fashion. Clin Exp Metast. 2012;29(1):27–38. doi: 10.1007/s10585-011-9426-4. [DOI] [PubMed] [Google Scholar]
- 18.Mierke CT, Bretz N, Altevogt P. Contractile forces contribute to increased GPI-anchored receptor CD24 facilitated cancer cell invasion. J Biol Chem. 2011;286(40):34858–34871. doi: 10.1074/jbc.M111.245183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolterink S, Moldenhauer G, Fogel M, Kiefel H, Pfeifer M, Luttgau S, Gouveia R, Costa J, Endell J, Moebius U, Altevogt P. Therapeutic antibodies to human L1CAM: functional characterization and application in a mouse model for ovarian carcinoma. Cancer Res. 2010;70(6):2504–2515. doi: 10.1158/0008-5472.CAN-09-3730. [DOI] [PubMed] [Google Scholar]
- 20.Runz S, Mierke CT, Joumaa S, Behrens J, Fabry B, Altevogt P. CD24 induces localization of beta1 integrin to lipid raft domains. Biochem Biophys Res Commun. 2008;365(1):35–41. doi: 10.1016/j.bbrc.2007.10.139. [DOI] [PubMed] [Google Scholar]
- 21.Jackson D, Waibel R, Weber E, Bell J, Stahel RA. CD24, a signal-transducing molecule expressed on human B cells, is a major surface antigen on small cell lung carcinomas. Cancer Res. 1992;52(19):5264–5270. [PubMed] [Google Scholar]
- 22.Schabath H, Runz S, Joumaa S, Altevogt P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J Cell Sci. 2006;119(Pt 2):314–325. doi: 10.1242/jcs.02741. [DOI] [PubMed] [Google Scholar]
- 23.Riedle S, Kiefel H, Gast D, Bondong S, Wolterink S, Gutwein P, Altevogt P. Nuclear translocation and signalling of L1-CAM in human carcinoma cells requires ADAM10 and presenilin/gamma-secretase activity. Biochem J. 2009;420(3):391–402. doi: 10.1042/BJ20081625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pfeifer M, Schirmer U, Geismann C, Schafer H, Sebens S, Altevogt P. L1CAM expression in endometrial carcinomas is regulated by usage of two different promoter regions. BMC Mol Biol. 2010;11:64. doi: 10.1186/1471-2199-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoeck A, Gast D, Sanderson MP, Issa Y, Gutwein P, Altevogt P. L1-CAM in a membrane-bound or soluble form augments protection from apoptosis in ovarian carcinoma cells. Gynecol Oncol. 2007;104(2):461–469. doi: 10.1016/j.ygyno.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 26.Kiefel H, Bondong S, Erbe-Hoffmann N, Hazin J, Riedle S, Wolf J, Pfeifer M, Arlt A, Schafer H, Muerkoster SS, Altevogt P. L1CAM-integrin interaction induces constitutive NF-kappaB activation in pancreatic adenocarcinoma cells by enhancing IL-1beta expression. Oncogene. 2010;29(34):4766–4778. doi: 10.1038/onc.2010.230. [DOI] [PubMed] [Google Scholar]
- 27.Mechtersheimer S, Gutwein P, Agmon-Levin N, Stoeck A, Oleszewski M, Riedle S, Postina R, Fahrenholz F, Fogel M, Lemmon V, Altevogt P. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J Cell Biol. 2001;155(4):661–673. doi: 10.1083/jcb.200101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith SC, Oxford G, Wu Z, Nitz MD, Conaway M, Frierson HF, Hampton G, Theodorescu D. The metastasis-associated gene CD24 is regulated by Ral GTPase and is a mediator of cell proliferation and survival in human cancer. Cancer Res. 2006;66(4):1917–1922. doi: 10.1158/0008-5472.CAN-05-3855. [DOI] [PubMed] [Google Scholar]
- 29.Senner V, Sturm A, Baur I, Schrell UH, Distel L, Paulus W. CD24 promotes invasion of glioma cells in vivo. J Neuropathol Exp Neurol. 1999;58(8):795–802. doi: 10.1097/00005072-199908000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R, Enkemann S, Jove R, Haura EB. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24(21):3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 31.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao X, Tay A, Guy GR, Tan YH. Activation and association of Stat3 with Src in v-Src-transformed cell lines. Mol Cell Biol. 1996;16(4):1595–1603. doi: 10.1128/mcb.16.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998;18(5):2545–2552. doi: 10.1128/mcb.18.5.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp 125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14(3):1680–1688. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15(2):954–963. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hitosugi T, Sato M, Sasaki K, Umezawa Y. Lipid raft specific knockdown of SRC family kinase activity inhibits cell adhesion and cell cycle progression of breast cancer cells. Cancer Res. 2007;67(17):8139–8148. doi: 10.1158/0008-5472.CAN-06-4539. [DOI] [PubMed] [Google Scholar]
- 37.Overdevest JB, Thomas S, Kristiansen G, Hansel DE, Smith SC, Theodorescu D. CD24 offers a therapeutic target for control of bladder cancer metastasis based on a requirement for lung colonization. Cancer Res. 2011;71(11):3802–3811. doi: 10.1158/0008-5472.CAN-11-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmed MA, Jackson D, Seth R, Robins A, Lobo DN, Tomlinson IP, Ilyas M. CD24 is upregulated in inflammatory bowel disease and stimulates cell motility and colony formation. Inflamm Bowel Dis. 2009;16(5):795–803. doi: 10.1002/ibd.21134. [DOI] [PubMed] [Google Scholar]
- 39.Fukushima T, Tezuka T, Shimomura T, Nakano S, Kataoka H. Silencing of insulin-like growth factor-binding protein-2 in human glioblastoma cells reduces both invasiveness and expression of progression-associated gene CD24. J Biol Chem. 2007;282(25):18634–18644. doi: 10.1074/jbc.M609567200. [DOI] [PubMed] [Google Scholar]
- 40.Wang W, Wang X, Peng L, Deng Q, Liang Y, Qing H, Jiang B. CD24-dependent MAPK pathway activation is required for colorectal cancer cell proliferation. Cancer Sci. 2010;101(1):112–119. doi: 10.1111/j.1349-7006.2009.01370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brunton VG, Avizienyte E, Fincham VJ, Serrels B, Metcalf CA, 3rd, Sawyer TK, Frame MC. Identification of Src-specific phosphorylation site on focal adhesion kinase: dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res. 2005;65(4):1335–1342. doi: 10.1158/0008-5472.CAN-04-1949. [DOI] [PubMed] [Google Scholar]
- 42.Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, Solinas G, Karin M. Saturated fatty acids induce Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147(1):173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rajendran L, Simons K. Lipid rafts and membrane dynamics. J Cell Sci. 2005;118(Pt 6):1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- 44.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 45.Paulick MG, Bertozzi CR. The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry. 2008;47(27):6991–7000. doi: 10.1021/bi8006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9(1):50–63. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 47.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, Wu Z, Gonen M, Mulvey LA, Bessarabova MO, Huh SJ, Silver SJ, Kim SY, Park SY, Lee HE, Anderson KS, Richardson AL, Nikolskaya T, Nikolsky Y, Liu XS, Root DE, Hahn WC, Frank DA, Polyak K. The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24− stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K, Grabau D, Ferno M, Borg A, Hegardt C. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10(3):R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.