

Abstract
Although all nucleated cells within a multicellular organism contain a complete copy of the genome, cell identity relies on the expression of a specific subset of genes. Therefore, when cells divide they must not only copy their genome to their daughters, but also ensure that the pattern of gene expression present before division is restored. While the carrier of this epigenetic memory has been a topic of much research and debate, post-translational modifications of histone proteins have emerged in the vanguard of candidates. In this paper we examine the mechanisms by which histone post-translational modifications are propagated through DNA replication and cell division, and we critically examine the evidence that they can also act as vectors of epigenetic memory. Finally, we consider ways in which epigenetic memory might be disrupted by interfering with the mechanisms of DNA replication.
Keywords: Epigenetic memory, Gene expression, DNA replication, Histone modifications
Introduction
Histone proteins allow the packaging of several metres of DNA into the eukaryotic nucleus. Eukaryotes possess four core histone proteins, H2A, H2B, H3 and H4. They form an octameric complex, [H2A:H2B]:[H3:H4]2:[H2A:H2B], known as the nucleosome core. The crystal structure of the nucleosome core revealed 146 base pairs of nuclear DNA wrapped around each core particle in 1.65 turns of left-handed superhelix [1]. Nucleosomes are spaced at approximately 200 base pair intervals along the DNA, while the intervening exposed DNA can be bound by one of a number of specialised linker histone proteins (reviewed in [2]). The formation of chromatin by binding of the histone proteins to DNA is the first stage in the processes that compacts DNA 30- to 40-fold into higher order structures within the nucleus (reviewed in [3]).
The need to compact DNA into chromatin is at odds with the requirement for access to the genetic code. Even the formation of nucleosomes results in DNA-binding sites for proteins such as transcription factors becoming occluded as well as acting as a barrier to the key DNA transactions, transcription and replication. Of course, not all DNA sequences need to be accessible all the time. Different cell types exhibit distinct transcriptional programmes that are controlled through the regulation of access to DNA via removing, remodelling or repositioning nucleosomes. Indeed, key to maintaining cell identity is the ability to ‘remember’ which genes are active and which are inactive through DNA replication and mitosis (or occasionally meiosis).
While the restoration of the transcriptional programme following DNA replication could be achieved solely by the maintenance of the signalling environment in which the cell finds itself, there is extensive evidence that cells can also intrinsically maintain gene expression states through cell division. The nature of this ‘memory’, which is not directly encoded by the DNA and hence can be termed ‘epigenetic’ in the sense that it is a heritable trait independent of changes in the underlying DNA sequence [4], has been the subject of intense research. Various vectors of epigenetic memory have been proposed, including DNA methylation, small RNAs, histone tail modifications and histone variants. This review focuses on how histone proteins and particularly post-translational modifications of the histone tails might provide epigenetic memory, and will therefore not deal in detail with DNA methylation and RNA interference-based models, which have been reviewed extensively elsewhere [5, 6].
While the body of the eight histone proteins goes to make up the globular core of the nucleosome and provides the scaffold around which the DNA gyres are wrapped, the N termini of the histone proteins are unstructured and protrude from the core, out past the DNA. The histone proteins, and particularly the N-terminal tails, can be decorated by post-translational modifications including methylation, acetylation and phosphorylation. The effects of these modifications on histone structure are only just beginning to be appreciated, but a great deal of data derived by examination of specific loci and, more recently, through genome-wide studies has shown striking correlations between certain modifications and particular transcriptional states. Thus, transcriptionally active loci are associated with acetylation of H3 at lysine 9 and 14, shortened to H3K9/14ac [7], trimethylation of H3 at lysine 4 (H3K4me3) and acetylation of a number of lysines in the N-terminus of H4. Conversely, repressed loci are found to be enriched for H3K9me2 and me3 or for H3K27me3 (reviewed in [8]).
The array of potential modifications of histones coupled to their association with specific transcriptional states and an increased understanding of the enzymology of mark introduction and reading led to the proposal of a histone code [9, 10]. This concept is linked to the idea that the code can also carry the long-term transcriptional memory necessary for the maintenance of cell identity. However, not all histone modifications have the properties necessary for carrying epigenetic memory, and many are likely to simply mediate short-term signalling functions [11]. Those marks that are candidates for carrying epigenetic memory need to exhibit a number of features, most importantly they must be stable, or at least maintainable, during the cell cycle, and they must be copied onto new chromatin after DNA replication such that they remain associated with the same underlying DNA sequence.
This review will examine the evidence that histone modifications can mediate long-term epigenetic memory in the context of other potential mechanisms and will focus on the mechanisms by which histone modifications are propagated through chromosome replication. Additionally, we discuss how the mechanisms of histone mark propagation might be disturbed by problems encountered by the replication fork and how this can result in instability of epigenetic memory of transcriptional states.
Evidence that histone modifications can be inherited
What evidence is there that histone modifications can act as epigenetic marks, that is, be inherited directly during cell division? General support for this notion has come from the study of epigenetic model systems. In common to each of these is the ability to genetically separate the mechanism of maintaining a gene expression state from the initial process of establishing that state, allowing the components that are required specifically for maintenance of transcriptional states to be identified. In this section we examine four examples from evolutionarily divergent organisms that have all implicated histone modifications as key in epigenetic processes (Fig. 1).
Fig. 1.

Experimental models implicating histone modifications in the propagation of epigenetic states. a A simplified illustration of Polycomb/Trithorax regulation in Drosophila development. In the embryo the expression of Ultrabithorax (UBX) is set by the activity of Hunchback (Hb), which acts as a repressor, and Fushi tarazu (Ftz), which acts as an activator. However, in the adult, neither Hb nor Ftz is present and the embryonic pattern of UBX expression is maintained by binding of the Polycomb and Trithorax complexes. b Position effect variegation in the Drosophila eye. The Drosophila eye in wild type flies has a red colour due to the expression of the gene white. An inversion mutation can place the white gene in proximity to heterochromatin. This can lead, through spreading of the heterochromatin, to stochastic silencing of white and a variegated eye colour phenotype. Suppression of variegation, or Su(var), mutants are identified through reversion of this phenotype. c The S. cerevisiae mating type locus. The mating type is determined by the presence of the a or α mating type cassette within the expressed domain (green), allowing the two genes determining mating type, Mat a/α1 and Mat a/α2, to be expressed. The expressed domain is flanked by silent copies of both a and α mating type cassettes, adjacent to cis-acting silencer elements. Gene conversion can introduce either of the silent cassettes into the expressed cassette to switch mating type. d Live cell imaging to reveal the effect of H3K4 methylation on inheritance of transcriptionally active states in Dictyostelium. The transcription frequency of a reporter locus can be measured directly by observation of RNA production in vivo via binding of the MS2 bacteriophage coat protein to MS2 repeats engineered into the transcript. Cell division gives two daughter cells with good correlation between the transcriptional rates. However, reducing H3K4 methylation by introducing the H3K4A mutation leads to failure of this correlation [38]
Regulation of gene expression by the Polycomb and Trithorax group proteins
During Drosophila development, proteins encoded by pair rule and gap genes switch the expression of crucial genes responsible for patterning segments on or off in different regions of the embryo. Notably, these targets include the highly conserved Hox genes. However, the expression states of Hox genes are then maintained throughout the lifespan of the fly, in the absence of the initial signal (reviewed in [12]). Thus, Hox gene regulation fits the Holliday definition of an epigenetic process [4]. Evidence in favour of a role for histone modifications in this process comes from the study of two classes of mutant, Polycomb and Trithorax, which show defects in maintaining silent and active gene expression states respectively [13, 14]. Both the Polycomb and Trithorax groups of genes include enzymes capable of carrying out histone modifications linked to determination of transcriptional states: the PRC2 Polycomb complex introduces H3K27me3 [15–18] and the Trithorax complexes are involved in trimethylation of lysine 4 of histone H3 [19, 20]. Because the genes that these complexes act at are different in each segment, DNA sequence alone cannot specify their localisation in differentiated cells. Thus the initial targeting of these complexes must be genetically separable from their role in maintaining gene expression states. Instead, the complexes could be retained at specific loci due to epigenetic inheritance of the histone modifications that the complexes themselves introduce (e.g. [21]). Thus, indirectly, the existence and phenotype of the Polycomb and Trithorax group mutants might imply a role for histone modifications in the heritability of gene expression states.
Position effect variegation in Drosophila
The first example of position effect variegation (PEV) involved the stochastic silencing of the white gene when positioned, by a translocation, into pericentromeric chromatin in cells in the developing eye [22, 23]. Silencing of white is established in the imaginal disc, and this silencing is then propagated through to the adult eye. Because the gene, once inactivated, remains inactive for the lifetime of the adult but does not actually become silenced in adult eye cells, initiation and perpetuation of silencing are separable, thus again implicating an epigenetic phenomenon. Multiple mutants have been identified that exhibit defective silencing of a pericentric white allele. Many of these suppressors of variegation, or Su(var), mutations, have been mapped to genes involved in modifying histones (reviewed in [24]): H3K9 methylation and H4K20 methylation, as well as histone deacetylation are all important in PEV.
An important PEV mutant that has given conceptual insights into the role of histone modifications in transcriptional memory is Su(var)2-5, the Drosophila HP1 homologue. HP1 itself lacks the capacity to modify nucleosomes. However, HP1 can bind to the H3K9 methylation mark through its chromodomain [25, 26]. This provides a possible mechanism to explain exactly how the H3K9 methylation modification can lead to transcriptional silencing: HP1 can dimerise, thus potentially allowing it to bind to more than one modified nucleosome simultaneously, contributing to the compaction of chromatin [27]. Moreover, study of HP1 has also given some insight into a possible mechanism for histone modification copying. HP1 can interact with the H3K9 methyltransferase SUV39. This could lead to the recruitment of SUV39 to regions where H3K9me is found, providing a mechanism for spreading of the mark in chromatin and potentially explaining how the modification could be perpetuated through cell division via a “self-recruitment” mechanism.
Mating type switching
Another important system that has given insight into the role chromatin modifications may play in heritable gene expression states is the mating type loci of both the fission yeast Schizosaccharomyces pombe and budding yeast Saccharomyces cerevisiae. Although the initiation of mating type switching differs in the two systems (reviewed in [28, 29]), in both, mating type is determined by the expression of one of two alleles of the mating factor. However, there are also silent copies of both alleles located elsewhere on the chromosome, allowing mating type to ‘switch’ by replacing the expressed allele with the alternative allele. The silent state of these dormant copies is inherited during cell division.
It is important to note that it is more difficult to interpret the yeast mating type switch as epigenetic than it is for the systems from multicellular organisms described above. This is because genes within the silent regions are never expressed in wild type cells, meaning that it is possible that the cis-acting DNA elements, which are required for silencing, could simply re-establish the silent state every time the cell divides. However, there is good evidence from both fission yeast and budding yeast that establishment and maintenance of silencing can be separated genetically, suggesting that some component of the silent state is maintained epigenetically. Simple evidence from S. cerevisiae comes from the phenotype of the sir1 mutant [30]. sir1 mutants exhibit a variegated defect in silencing such that four-fifths of the population show expression of the silent mating type genes and one-fifth keep these genes silent. However, the progeny of cells from the silent or active population show maintenance of the state of their parent over many generations, with rare (1/250) exceptions. This suggests that sir1 mutants are deficient in establishing silencing but not in maintaining it. In fission yeast, where the silent region is larger, evidence of a comparable phenomenon came from the insertion of a reporter gene into the silent region, leading to a variegation effect similar to the sir1 mutant, whereby two states of reporter gene expression (either on or off) were found to be heritable [31, 32]. Furthermore, when cells expressing the reporter were crossed to those not expressing the reporter, the expression state showed Mendelian inheritance such that it co-segregated with the MAT locus [31]. This last experiment dramatically confirms that this is an epigenetic effect and, even more intriguingly, suggests that meiotic inheritance is possible, at least for this system.
Given the epigenetic nature of mating type silencing, the identification of genes involved in histone modification as silencing mutants gives further support to the importance of histone modifications in epigenetic states. Indeed, in S. pombe, homologues of proteins involved in Drosophila PEV have been identified as important in mating type locus silencing, for example Clr4 [33, 34], the S. pombe homologue of the SUV39 H3K9 methyltransferase, and Swi6 [35], the homologue of HP1. Another example is provided by the histone deacetylase Sir2. sir2 mutants in S. cerevisiae are defective in silencing at the mating type locus, and the Drosophila Sir2 homologue is a member of the PRC2 Polycomb complex [36]. These observations suggest deep conservation of a role for histone modification complexes in epigenetic state maintenance. Further, from the extensive study of these relatively simple systems, it has recently become possible to construct detailed computational models of how silencing is maintained [37]. Such studies are useful for understanding the minimal requirements for any transcriptional memory system based on histone modifications and will be discussed in more detail below.
Correlation between transcription and histone modification at the single cell level in Dictyostelium
Though analysis of well-defined genetic model systems is highly suggestive of a role for histone modifications in epigenetic processes, evidence that histone modifications are directly responsible for carrying the information is still lacking. For example, one could imagine a situation where histone modifications act downstream of another factor, itself inherited through cell division, to bring about retention of the transcriptional state. Obtaining more direct evidence that histone modifications are the causal factor inherited through cell division ideally requires following histone modification levels within single cells over multiple cell cycles. Recent work using the slime mould Dictyostelium discoideum has come close to this ideal. Transcription of genes in mother and daughter cells correlates closely, but removing the bulk of H3K4me by replacing one of the H3 genes with an H3K4A mutant led to a reduction in the observed correlation [38]. This suggests that the H3K4me modification is important in the inheritance of transcriptional states through cell division. Future work based on this kind of single cell approach will be invaluable in establishing whether the role of histone modifications is indeed causative. However, the major conceptual barrier to histone modifications being inherited is a mechanistic explanation of how they could survive the process of DNA replication. In order to understand how histone marks might be propagated, it is first necessary to examine the dynamics of histones during replication.
Histone dynamics during DNA replication
A simple consideration of the process of DNA replication leads to the conclusion that when DNA duplicates in S phase, the number of nucleosomes associated with the DNA must also double. Without this, the number of histones per segment of DNA would halve each time the cell divides. Thus, newly synthesised histones also need to be deposited on DNA during DNA replication. At the same time, the nucleosomes ahead of the fork represent a barrier for the unwinding of DNA and therefore need to be removed, at least transiently. Nucleosome incorporation after replication is indeed rapid, and failure to effectively assemble newly synthesised DNA into chromatin behind the replication fork leads to genomic instability and cell death [39]. Crucially though, this incorporation of newly synthesised histones necessarily imposes a challenge to the perpetuation of histone post-translational modifications, because the newly synthesised histones incorporated will not carry the locus-specific information of the parental histones. In order to understand what the final make-up of chromatin might be after replication, in this section we will look at what distinguishes old and new histones, before considering the processes that govern the final distribution of histones after replication.
What distinguishes old and new histones?
Much is known about the modifications that distinguish pre-existing H3 and H4 from de novo H3 and H4 incorporated during replication. Newly synthesised H3.1 and H4 are devoid of methylation marks other than H3K9 monomethylation [40], which is imposed at a very early stage in H3 biosynthesis [41] by an as yet unknown methyltransferase, possibly SETDB1 [40]. It has been proposed that this modification acts as a stepping stone for subsequent further methylation events following incorporation of the H3 into chromatin [40]. Newly synthesised H4 is diacetylated at K5 and K12 in organisms from Tetrahymena [42], in which the equivalent residues are K4 and K11, through to Drosophila and human [40, 43]. This mark is imposed by the histone acetyltransferase HAT1, which associates with the sNASP histone chaperone during the preparation of the H3/H4 dimer for nuclear import [41].
In yeast, newly synthesised H3 is also acetylated at K56 [44] by the HAT Rtt109 [45, 46], and this modification is vital for proper chromatin assembly during S phase [47]. Importantly however, in human cells H3K56ac is found only at very low levels on H3 bound by the histone chaperone Asf1, a pool of H3 that is thought to reflect molecules awaiting loading onto DNA, arguing that this modification is not a general mark of new histones in higher eukaryotes [48].
The modification pattern of newly synthesised histones might be useful for propagation of epigenetic memory as it could allow a distinction between old and new histones to be made. However, to understand how this might work, we first need to consider how parental nucleosomes and newly synthesised nucleosomes come together during replication.
Global dynamics of nucleosomes at the replication fork
Given the requirement not to leave the DNA naked for too long, there are three possible ways in which chromatin might be replicated. First, the histones could be pushed off the end of a linear chromosome. This seems unlikely because there are multiple replication origins on eukaryotic chromosomes and therefore the histones between two origins would be bunched together creating a hideous contortion. A second possibility would be to keep the parental nucleosomes associated with one daughter strand and place the newly synthesised histones on the other strand. Finally, the parental and newly synthesised nucleosomes could be deposited evenly on both daughters, which would have the net effect of placing new and old nucleosomes adjacent to one another (Fig. 2).
Fig. 2.

Management of H3 and H4 during DNA replication. Parental H3/H4 tetramers are displaced ahead of the advancing replicative helicase and are redeposited on the double strand DNA emerging from the polymerases on either leading or lagging strands. Newly synthesised H3/H4 dimers are shown binding first to ASF1 before being handed over to CAF1 bound to PCNA. The stoichiometry of CAF1 at the fork is unclear, but here two CAF1 molecules are shown chaperoning one new dimer each
Several different lines of experimental evidence have converged in favour of the latter possibility. An important technique in elucidating the dynamics of nucleosomes at the replication fork has been electron microscopy of psoralen-crosslinked and deproteinised DNA. Histones protect the DNA from crosslinking and thus, when they are removed by protease and the DNA denatured, a single-strand bubble forms, allowing the visualisation of sites of nucleosome occupancy [49]. This technique demonstrated that nucleosomes were incorporated within about 200 base pairs of the replication fork in SV40 minichromosomes [50]. Furthermore, by inhibiting protein synthesis, it could be shown that nucleosomes still formed on both daughter strands, but with a density of around 50% of the density of nucleosomes ahead of the fork, suggesting that the parental nucleosomes were segregated evenly between the two strands [50]. Parallel elegant biochemical studies employing density gradient centrifugation of radiolabelled histones at cellular replication forks gave rise to broadly similar conclusions [51, 52].
Together, these data suggest that newly synthesised histones are deposited during replication at the same time as parental histones are recycled. Therefore, an important question is whether the old and the new histones can form mixed octamers. The very fact that nucleosomes can be observed on newly synthesised DNA in the absence of protein synthesis implies that mixed octamers are not obligatory; moreover, SV40 chromatin can be replicated with octamers that have been cross-linked together [53]. In vivo, though, data from density labelling of replicating cells suggest that parental H3/H4 tetramers tend to stay together, with a random assortment of newly synthesised and parental H2A/H2B dimers [52]. This makes sense given the assembly pathway of the histone octamer in vitro, where an H3/H4 tetramer is capped by two H2A/H2B dimers [54]. Recently a mass spectrometry study, demonstrating that the vast majority of parental H3.1/H4 remain as tetramers through cell division, has complemented this early work [55]. Additionally, these observations suggest that, if histone proteins and their modifications are to carry epigenetic memory, H3/H4 is a more attractive unit to study than H2A and H2B.
Thus, overall evidence has converged on a simple model whereby, on average, one newly synthesised tetramer will be deposited for every parental H3/H4 tetramer on both strands. Potentially, this allows the H3/H4 tetramer to act as a vector for epigenetic information, raising the important question of which enzymes are needed to control H3/H4 deposition.
Guiding histones: histone chaperones at the replication fork
During chromatin transactions involving histone displacement, specific chaperones are required to prevent unscheduled and potentially disordered reassociation of histones with DNA. These chaperones, which are drawn from diverse protein families, all share in common subunits with acidic regions allowing them to bind to their highly basic client proteins [56].
A large number of histone chaperones have been identified. However, the assembly and disassembly of nucleosomes can take place throughout the cell cycle. Therefore in assessing the function of these chaperones in DNA replication it is important to establish whether they act specifically in S phase, operate entirely outside of DNA replication, or can participate to some degree in both replication-dependent and independent processes. Two chaperones implicated in replication-dependent chromatin assembly are CAF1 and ASF1.
CAF1
The paradigm for a chaperone linked to DNA synthesis is CAF1, discovered due to its ability to support the assembly of SV40 viral DNA into chromatin when used to supplement replication-competent S100 extracts from HeLa cells [57, 58]. Human CAF1 is a trimer consisting of p150, p60 and p48 subunits, which are conserved all the way through to budding yeast [59]. CAF1 was shown to bind histones H3 and H4 in vivo [60], suggesting that it is able to deposit H3 and H4 on newly synthesized DNA, which then nucleates assembly of the complete histone octamer. Deletion of p150 in chicken DT40 cells [61], or its knock-down in human cells [62], results in massive defects in chromatin assembly and rapid lethality, supporting a vital role for CAF1 in chromatin assembly in vivo.
What marks CAF1 out as a DNA synthesis-coupled factor rather than one that is more generally important for chromatin assembly? Conceptually one could imagine two ways in which the activity of a chaperone might be controlled. One would be to ensure regulation such that it only acts at replication forks. Alternatively, the substrate specificity could be such that it only acts on newly synthesised histones in S phase. It turns out that both these mechanisms apply to CAF1.
One simple mechanism regulating CAF1 activity is cell-cycle-regulated phosphorylation, which occurs on the p150 subunit, by the replication-specific kinase CDC7-DBF4 [63]. Phosphorylation in S phase prevents p150 dimerising, thus allowing it to form the active CAF1 heterotrimer and restricting its activity outside of S phase. Perhaps a more important mechanism restricting CAF1 activity to replication is its interaction with PCNA, which means that CAF1 is localised to on-going replication forks [64]. Yeast PCNA mutants that no longer interact with CAF1 are compromised for chromatin assembly [65], suggesting that this interaction is essential for CAF1 function in vivo. However, CAF1 activity is not absolutely restricted to S phase [66, 67], and its interaction with PCNA provides an elegant way in which chromatin assembly can also be linked to DNA repair synthesis, as PCNA loading at sites of repair will be sufficient for CAF1 to be recruited [68].
Pulse labelling studies showed that CAF1 chromatin assembly in vitro can only be carried out using newly synthesised histones [69]. How could this specificity be achieved? Purification of H3.3- or H3.1-bound proteins demonstrated that CAF1 shows preference for the S-phase-specific histone H3.1, rather than the histone variant H3.3, which is expressed throughout the cell cycle [70]. This is intriguing given the modest differences in sequence between H3.3 and H3.1 (reviewed in [71]). The selectivity appears to be so strong that a CAF1-dependent H3.1 deposition ‘signature’ can be detected on newly deposited histones outside of S phase [72], a situation where H3.3 is likely to be in significant excess relative to H3.1. It should be noted however that this experiment utilised an epitope-tagged H3.1 that was not expressed under the endogenous promoter, so was not subject to cell-cycle-dependent regulation; the in vivo H3.3/H3.1 ratio in G1 may be even higher forcing CAF1 to incorporate H3.3.
In yeast the situation is somewhat different. Yeast have two copies of a single H3 gene which is homologous to vertebrate H3.3 [73], and there is therefore no distinction in terms of H3 substrate between replication-dependent and replication-independent nucleosome assembly. In contrast to vertebrate cells yeast mutants lacking CAF1 are viable, though they have defects in chromatin assembly indicated by loss of silencing at the mating type loci and telomeres, which is likely due to the ability of the replication-independent histone chaperone HIRA to substitute for loss of CAF1. Supporting this, a caf1/hira double mutant exhibits much more extensive S-phase trauma and greater silencing defects than either single mutant [74].
Another suggested source of substrate specificity derives from the ability of CAF1 to interact preferentially with acetylated H4 [60]. This would further assist CAF1 in selectively binding newly synthesised histones, which are acetylated on H4 K5/12. Supporting this, CAF1 can be pulled down with acetylated histones [75]. However, the N terminal tails of histone H3 and H4 are dispensable for chromatin assembly in vitro [76], suggesting that if there is an increase in affinity, it is not necessary for CAF1 to work. In yeast however, there is a role for H3K56 acetylation, a mark of newly synthesised histones, in nucleosome assembly that appears to be dependent on CAF1 [47].
Taken together, the evidence suggests that CAF1 is responsible for the deposition of newly synthesised histones H3 and H4, particularly during replication. This would account for the deposition of newly synthesised [H3/H4]2 tetramers, separate from the parental [H3/H4]2 tetramers, implied by the biochemical evidence discussed above. However, this simple model of chromatin assembly during replication must be questioned somewhat by results from the epitope tagging experiments described above. Intriguingly, though chromatin-bound, tagged H3.1 could pull down endogenous H3, suggesting that tagged H3.1 is able to form mixed tetramers, soluble tagged H3.1 (the pool interacting with CAF1) could not pull down endogenous H3. This implies that CAF1 binds an H3/H4 dimer rather than [H3/H4]2 tetramers. Therefore, in order for the activity of CAF1 to support deposition of newly synthesised tetramers either two CAF1 complexes would have to work together, which would be possible as PCNA is trimeric, or another histone chaperone would have to be involved in the deposition process. Important insight into this has come from study of the histone chaperone ASF1, which also seems to be involved in replication-coupled chromatin assembly.
ASF1
ASF1 was initially identified as a locus whose overexpression counteracts silencing in yeast [77]. Evidence for a role in S-phase chromatin assembly came from S. cerevisiae asf1 mutants, which exhibit defects in S-phase progression, and the ability of purified Drosophila ASF1 to stimulate CAF1-mediated nucleosome assembly [78]. The importance of ASF1 in S-phase progression is conserved through to higher organisms: a DT40 mutant lacking ASF1 fails to survive more than two cell cycles after inactivation [79], and in humans, simultaneous knock-down of the two ASF1 genes leads to defects in S-phase progression and chromatin assembly [80].
ASF1 however appears to show some important functional differences to CAF1. First, ASF1 does not appear to be recruited to replication forks in the same way as CAF1. ASF1 has not been shown to interact with PCNA; instead, yeast ASF1 appears to interact with the PCNA loading complex Replication Factor C [81]. Mammalian ASF1 can interact with MCM proteins, probably bridged by H3/H4, as mutant ASF1 protein deficient in histone binding no longer pulls down MCMs [82]. Moreover, the association of ASF1 with unperturbed replication forks appears to be dynamic [48] in contrast to the stable association seen for CAF1.
Another important difference to CAF1 is that ASF1 appears to be involved in replication-independent nucleosome disassembly in yeast, both on a global scale [83] and at specific loci [84]. Human ASF1 knockdown has also been shown to reduce H3.3 deposition in G1. There are also some interesting differences in the phenotypes of the yeast asf1 and caf1 mutants. In particular, asf1 mutants do not show defects in silencing, although asf1/caf1 double mutants show greater silencing defects than either of the single mutants. Overall therefore, these studies suggest that ASF1 is a general histone chaperone, which can contribute in S phase, whereas CAF1 is dedicated to new DNA synthesis. Thus it seems likely that ASF1 and CAF1 cooperate in nucleosome assembly. Below, we consider two classes of model that have been proposed for ASF1 and CAF1 cooperation.
Asf1 as an accessory for de novo nucleosome assembly
The effect of ASF1 on in vitro chromatin assembly, where it appears to be capable of stimulating CAF1 but probably cannot substitute for it [78], supports a view in which ASF1 is an accessory for chromatin assembly during replication. This idea is further supported by the fact that CAF1 and ASF1 interact [85] and are found together in complexes with soluble tagged histone H3.1 [70].
How might such an accessory role for ASF1 be understood? Important insight into this was provided by the crystal structure of ASF1, showing that it interacts with a dimer of H3/H4 [86]. Moreover, the structure showed that the interaction between ASF1 and H3/H4 prevents [H3/H4]2 tetramer formation because the tetramerisation interface is blocked in the complex. This suggests two possibilities for how ASF1 could assist CAF1 in nucleosome assembly. First, as described above, CAF1 would be able to accept histones from ASF1 as it also interacts with dimeric H3/H4. Thus ASF1 would act as a donor in a supply chain to move newly synthesised histones onto chromatin. In this view, two CAF1 complexes would need to collaborate in order to deposit newly synthesised tetramers. A second possibility is that CAF1 and ASF1 could each deposit one dimer to complete an [H3/H4]2 tetramer. This might seem to go against the fact that purified CAF1 alone can mediate nucleosome assembly in vitro [57]. However, CAF1 might be able to function independently of ASF1 in vitro due to the ability of two CAF1 complexes to associate with PCNA, whilst in vivo, perhaps with more limiting CAF1 levels, ASF1 might be able to contribute. This idea might explain the ability of ASF1 to stimulate CAF1 chromatin assembly by increasing the number of dimers available for deposition, without being absolutely essential for the process in vitro.
Asf1 in parental nucleosome management
An alternative but not exclusive possibility for the role of ASF1 derives from a consideration of its role in replication fork progression. Depleting ASF1 from human cells leads to increased replication fork stalling and this correlates to inhibition of unwinding of DNA [82]. This suggests that ASF1 might be helping replication fork progression by assisting in the disassembly of parental nucleosomes ahead of the fork. In support of this idea, overexpressing free H3.1/H4 in order to try and saturate ASF1 with free histones and prevent it from stripping parental histones, seemed to phenocopy Asf1 depletion [82]. This led to the proposal that ASF1 could act both to accept parental H3/H4 from ahead of the fork and supply newly synthesised histones to CAF1.
Since ASF1 can only bind an H3/H4 dimer [86], an important corollary of a role for ASF1 in disassembling nucleosomes ahead of the fork is that parental [H3/H4]2 tetramers must be split, at least transiently, before recombining on the newly synthesised DNA. However, this presents a problem. Since CAF1 also binds dimers, and ASF1 can supply histones to CAF1, there would be nothing to stop CAF1 depositing nucleosomes with an H3/H4 dimer from parental histones and an H3/H4 dimer from newly synthesised nucleosomes to form a mixed tetramer. This runs contrary to the weight of evidence presented above suggesting that parental H3/H4 tetramers stay together during DNA replication. Therefore, the obligate involvement of ASF1 in the displacement of parental histones seems unlikely.
How then might one explain the fork-stalling phenotype observed on ASF1 depletion? One possibility is that tetramer splitting is only required at certain regions of the genome, either due to a compact chromatin structure that is not conducive to replication fork passage, or due to an increased probability of replication stalling due to structured DNA [87]. If replication were to stall such that the synthesis of new DNA was interrupted, parental tetramers removed from DNA ahead of the fork would need to be dissipated into the pool of histones as they could not be deposited. ASF1 might therefore only split parental [H3/H4]2 tetramers under conditions where they cannot be loaded behind the replication fork. Indeed, under conditions of replication stress induced by hydroxyurea treatment, ASF1 does indeed associate to a much greater extent with histones carrying modifications typical of chromatin-bound histones, suggesting that Asf1 is capturing and splitting histones displaced from DNA that cannot be replaced due to interrupted DNA synthesis [48].
Overall therefore, though the possibility of ASF1-mediated parental tetramer-splitting during unperturbed replication is fascinating, there is not yet enough unequivocal data supporting it to outweigh the stable tetramer model. Thus, any model for what happens to histone modifications during cell division must deal with a situation in which H3/H4 tetramers are segregated evenly onto the two daughter strands, and new histones are deposited by CAF1, supplied or supplemented by ASF1, as two H3/H4 dimers. These newly synthesised histones will contain a different complement of post-translational modifications, thus posing a challenge to epigenetic stability. In the next section we will consider models for how histone modifications on parental nucleosomes can be perpetuated despite this challenge.
Mechanisms of histone mark propagation during replication
At the end of a cycle of DNA replication, an even distribution of parental and newly synthesised nucleosomes will be found on both daughter strands. How then can the histone modifications on the parental nucleosomes be introduced onto the newly synthesised nucleosomes to allow the recovery of the original pattern of modifications? We begin with a consideration of the most popular model whereby histone modifications can be perpetuated.
The recruitment-copying model for epigenetic inheritance
A conceptually appealing model to allow parental modifications to be introduced into newly synthesised nucleosomes involves the direct copying of histone modifications onto newly synthesised H3/H4 using adjacent, parental nucleosomes as a template (Fig. 3a). Central to this model is the ability of modified nucleosomes to directly recruit enzyme complexes that are capable of introducing the same modification that mediated the recruitment of the complex. For example, the chromodomain of HP1 can recognise H3K9me2/3 and in turn recruit the histone methyltransferase SUV39H1 [25, 26]. This mechanism potentially allows modification of an adjacent nucleosome and can explain the spreading of heterochromatic domains, as illustrated above with the examples of PEV in Drosophila and silencing of the yeast mating type loci. It could also explain maintenance of heterochromatic regions enriched for H3K9 methylation, because H3K9 methyltransferases would be recruited to regions of H3K9 methylation following replication, where they could introduce the modification onto newly synthesised histones [25, 26].
Fig. 3.
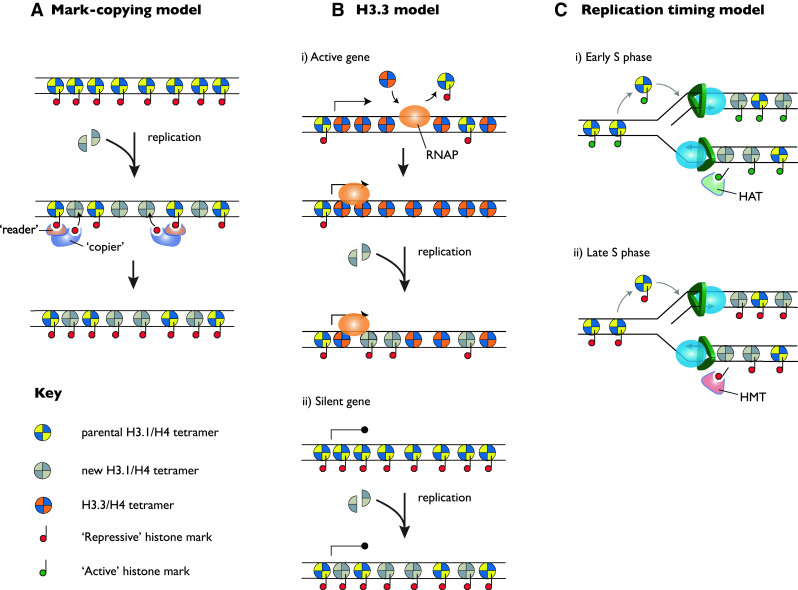
Models for the propagation of histone-based epigenetic memory. a The mark-copying model. For clarity only a single strand is shown. A region containing repressive histones is shown. After replication, approximately 50% of the H3/H4 tetramers are newly synthesised and lack the repressive modifications. These marks can be copied onto the newly synthesised tetramers by the concerted action of a mark-reading enzyme (‘reader’ e.g. HP1) and a mark-copying enzyme (‘copier’ e.g. the histone methyltransferase SUV39H1). This results in reconstitution of the pre-existing chromatin environment. b The H3.3 model. i Passage of RNA polymerase II (RNAP) through an active locus leads to exchange of H3.1 for H3.3. After replication the H3.3 is diluted by incorporation of newly synthesised H3.1, which according to the model behaves in a repressive manner. However, the presence of H3.3 favours recruitment of RNAP and, in turn, leads to replacement of the ‘repressive’ H3.1 with H3.3. ii In a silent locus, the repressive environment means RNAP does not access the DNA for transcription. Thus, no H3.3 is incorporated. During replication, incorporation of H3.1 results in maintenance of this state. c Replication timing model. In this model, active loci (i) are replicated early and this favours histone acetylation, which propagates the active state. Conversely, inactive loci are late replicating and are repressed by lack of acetylation, and incorporation of methyl marks (ii). HAT histone acetyltransferase, HMT histone methyltransferase
Recently this model has also been extended to cover the Polycomb modification H3K27me3. The EZH2 enzyme is solely responsible for introducing H3K27me3 in Drosophila and forms part of the PRC2 complex important in maintenance of Hox gene silencing [15]. Two independent groups showed that the H3K27me3 mark could be recognised by the EED subunit of the PRC2 complex, allowing the potential for EZH2 recruitment to sites where H3K27me3 is found on parental nucleosomes [21, 88]. Furthermore, in accordance with the copying model, EZH2 was found to be recruited to Polycomb repressed regions in S phase. Finally, artificially recruiting EED to a reporter in human cells could recapitulate the observed behaviour of PRC2 target loci including stable transcriptional repression. Together these two studies provide convincing evidence that H3K27me3 could propagate after cell division, giving a potential mechanism for epigenetic inheritance. There are, however, some complicating factors in interpreting these results. One point is that EED does not solely recognise H3K27me3. Indeed, the H3K9me3 modification is actually recognised more strongly [88]. This might suggest that the H3K9 methylation, which is also found at Polycomb repressed sites, might be the causal modification in the reestablishment of the repressive chromatin structure during S phase. Alternatively, a combinatorial effect of two modifications might be a way to install robustness into the system. More challenging for the idea that histone modification self-propagation at Polycomb targets is required for transcriptional repression is the observation that H3K27me3 is not actually found at the Polycomb response elements (PREs) [89]. These DNA sequence elements, which are found upstream of repressed targets in Drosophila and bound in vivo by DNA-binding proteins such as Pipsqueak and Pho, are absolutely required for the maintenance as well as initiation of HOX gene expression patterns. Moreover, Polycomb complexes have been shown to remain bound to these sequences through DNA replication of a plasmid in extracts, leading to the suggestion that the histone modifications are inherited secondary to the Polycomb complexes themselves [90]. Again, however, it is possible that the requirement for PREs may be an example of robustness where both factors together contribute to the reestablishment of the chromatin domain.
Thus far, there is no evidence of a self-recruitment type mechanism for the ‘active’ H3K4me3 mark, because none of the enzymatic activities capable of introducing the mark have been shown to themselves recognise H3K4me3. However, the involvement of this modification in Trithorax gene regulation discussed above is suggestive of a role in epigenetic inheritance. In this regard, the EZH2 studies are important because the EED H3K27me3 recognition domain was not recognisable as a known methyl-histone-binding domain, in contrast to the PhD finger or chromodomain (although, interestingly the structural epitope recognising the modification, an aromatic cage, is superficially similar to the PhD finger) [88]. Therefore, it is possible that self-recruitment mechanisms could apply to other modifications with as yet unknown domains important in recognition. Even so, it is unlikely that every single histone modification will be copied in this self-propagating manner, and ultimately one might imagine a distinction between ‘primary’ marks, which would be genuinely epigenetic, and ‘secondary’ marks whose inheritance would be dependent on that of the primary marks.
This distinction is important in the context of a second restriction on which marks can copy themselves, namely the stability of the modification. After new histones are incorporated in S phase, the modifications on the parental histones must persist for long enough to allow recruitment of their cognate enzymes. A general belief is that the half-life of acetylated histones, which has been measured to be around 15 min [91], is too rapid to allow this modification to act as an epigenetic mark [92]. Recent experiments using SILAC to separate histones incorporated during S phase from those already present in chromatin support the idea that the turnover of methylation is slower in vivo than that of acetylation [93]. Indeed, these studies revealed a fall in the levels of trimethylated H3K27 and H3K9 after S phase, rising gradually before levelling off once pre-S-phase levels are reached after about 6 h, which would fit the recruitment/copying model. In contrast, the levels of acetylation equilibrate much more rapidly, suggesting any step-wise copying would have to occur on a much shorter timescale. How, then, could the pre-S-phase pattern of acetylation be recovered after S phase? Is it possible that histone H3K9/14 acetylation might be a ‘secondary’ epigenetic mark, which could be recruited downstream of a ‘primary’ methylation mark such as H3K4me3? Supporting this type of model is evidence in S. cerevisiae showing that H3K4me3 can recruit the histone acetyltransferase GCN5 through its chromodomain [94]. Moreover, other examples of such secondary histone modification are known: the CW domain in an Arabidopsis H3K36 methyltransferase, ASHH2, recognises H3K4me3 [95]. Given the plethora of known histone modifications, this type of mechanism, whereby a handful of histone modifications are strictly epigenetic with other modifications occurring downstream, would seem to be the most parsimonious way to explain histone modification inheritance.
Although acetylation itself may not have the potential to act as an epigenetic mark, it is worth noting that there is evidence that the recruitment/copying model can apply to unmodified histones as well as modified histones. An important example of this is in S. cerevisiae silencing, where the SIR silencing complex, including the deacetylase SIR2, can be recruited specifically to deacetylated histones [96]. Thus, in this case, the lack of a histone modification on the parental nucleosome would be perpetuated suggesting that deacetylated states might be able to spread even if acetylated states cannot. As a further example of a similar mechanism, the LSD1 corepressor binds to H3 that has been unmodified on H3K4 via its BHC80 subunit [97]. This complex contains the LSD1 demethylase, which can demethylate H3K4me3 [98]. Thus, unmodified H3K4me3 could potentially copy itself during cell division in the same way as deacetylated H3 in S. cerevisiae, allowing a silent state at a promoter to be recovered after S phase.
Recently, the conceptually appealing nature of the mark-copying model has been subject to much more rigorous analysis by the development of computer models that have tested its core assumptions. These models are based on two of the well-understood systems discussed above, namely the S. cerevisiae and S. pombe mating type loci, where there is probably a more complete understanding of the components involved than for any other system. The importance of modelling is to show, first, whether the mechanism proposed can account for the observed stability of the silent state, and second, what the minimum requirements of any epigenetic memory system based on histone modifications are likely to be.
Dodd et al. [37] produced a computer model for the S. pombe mating type switch, discussed above, that used realistic parameters describing enzyme activity and the effect of DNA replication to recapitulate the observed bistability of the transcriptional states of the locus. (Bistability refers to the ability of a system to rest in either of two states, in this case silenced or active.) Importantly, they also found that this bistability was dependent on the ability of a modification to spread more than one nucleosome away, suggesting that restricting the activity of a recruited histone-modifying complex to the adjacent nucleosome is unable to give a robust system.
An interesting set of studies based on the S. cerevisiae mating type switch reached similar conclusions in terms of the importance of mark spreading [99]. They also reached an additional conclusion that was not accessible to the S. pombe study, namely the role of cooperativity in the recruitment-copying model. The reason for this is that S. cerevisiae lacks H3K9 methylation, so that there might in theory be only two modification states: acetylated and deacetylated. This model is able to maintain stable epigenetic states as long as there is cooperativity: an increased probability of recruiting the deacetylase if there are an increased number of deacetylated histone tails within the locus. Intriguingly, in a system with three modification states (acetylation, methylation and no modification), such as is found in the S. pombe mating type locus, recruitment alone without cooperativity is enough to maintain the existing state of the system through multiple cell divisions. Endowing robustness on the system thus provides a possible explanation for some of the observed complexity of the histone code, which will be interesting to follow up with more detailed experiments.
Problems with the mark-copying model
Despite the theoretical and experimental support for the mark-copying model, many authors have questioned whether it can account for epigenetic inheritance, leading to the proposal of alternative mechanisms to explain the propagation of histone modifications through cell division. The objections raised can be grouped together into two general categories.
Robustness of epigenetic states
The first type of problem that has been raised in conjunction with the copying model is its robustness. The central issue is that the 50% segregation of parental histones that occurs on DNA replication is likely to be stochastic. This is unlikely to be a problem for the long domains of modified nucleosomes characteristic of constitutive heterochromatin, but if one were to consider the inheritance of a very short tract of three modified nucleosomes, such as might be envisaged for the extent of the H3K4me3 mark at a transcriptional start site [100], there would be an approximately one in eight chance that none of these would end up on one of the daughter strands and all three would end up on the other. This does not seem to be sufficiently robust for an adequate epigenetic mechanism.
There are three arguments that could be presented to address this issue. First, one could imagine copying from the other strand might occur to restore the original state. However, there is currently no evidence for the copying of nucleosome marks in trans; moreover, it seems difficult to imagine how the precise alignment between sister chromatids that would allow this could be generated. A second possibility is that inherited chromatin domains are much longer than three nucleosomes in length; shorter domains would need then to be reset by some other system. In support of this suggestion it should be noted that whilst H3K4me3 generally marks quite a tight zone around the transcriptional start site of most genes [100], in Trithorax-regulated regions it seems to spread over a much longer distance [89]. Therefore perhaps H3K4me3 is only inherited at Trithorax-regulated genes. Alternatively, crosstalk with another, more widely distributed modification could help to provide robustness in the face of the cases where one modification failed to segregate properly. A third argument is to note that the one in eight chance stated above assumes that there is complete independence between the segregation of parental nucleosomes onto either strand over this length. However, considering an average of 200 bp per nucleosome ahead of the fork, equal segregation would require 600 bp of uncoated double-stranded DNA available behind the fork. In fact, however, nucleosomes are deposited within 200 bp of the fork [50], meaning that the assumption of complete independence over three nucleosomes may not be valid.
Dynamics of nucleosome and histone modification turnover
A second problem with the mark-copying model for chromatin inheritance is a growing view that chromatin is a much more dynamic entity than traditionally thought. This can be subdivided further into the process of introducing and removing modifications on nucleosomal histones and the process of turning over the histones themselves within the nucleosome. The dynamics of histone modification relates to the problem described above for why histone acetylation is generally believed not to be an epigenetic mark, namely that if the turnover of a particular modification is rapid on chromatin, it is not possible for this modification to be used as a template for the restoration of that same modification. The crucial point that takes this further is a consideration of the enzymes that remove modifications from chromatin. Challenging the view that histone methylation is a stable modification, recent work has identified a number of specific histone demethylases [101]. Importantly, histone demethylases are targeted not only against ‘active’ histone modifications such as H3K4me3, but also demethylases that act on H3K27me3 [102] and H3K9me3 [103], which are characteristic of ‘silent’ chromatin. The existence of these enzymes has led to the view that, rather than being a silent, static environment, even regions of silent chromatin are a hub of activity [104]. If this is the case, the lifetime of any particular modification on chromatin might be expected to be too short even for histone methylation to be epigenetic.
Evidence in favour of this dynamic viewpoint has been presented for the H3K4me3 modification by Rando et al. [105] who studied global turnover of H3K4me3 in S. cerevisiae. By synchronizing cells in G1 and releasing them, they could follow the fate of H3K4me3 that had accumulated as a result of arrest over time. Their results show clearly that the H3K4me3 is removed on a timescale that is too rapid to simply represent dilution of the modification by new histone incorporation. Knocking out the yeast H3K4me3-specific demethylase JHD2 led to increased persistence of the modification in chromatin. This was interpreted as evidence against the ability of H3K4me3 to act as an epigenetic mark. However, this interpretation assumes that the activity of the demethylase is constitutive at all loci. Whilst there is some evidence in support of JHD2 activity at specific loci, such as the GAL genes [106], this does not exclude the possibility that JHD2 could be recruited to specific genes in order to actively turn them off after removal of stimuli, for example after alpha factor release. Indeed, the requirement for H3K4 demethylases could suggest the ability of this mark to be propagated independently of the initial signal, at least under some circumstances.
Another source of dynamics in chromatin is the disassembly of nucleosomes outside of S phase. If this is taking place at a rapid rate then this might challenge the idea that histone modifications persist for long enough to act in an epigenetic fashion. In particular, it is expected that rapid nucleosome turnover will occur throughout the gene body during active transcription as RNA polymerase access is blocked by nucleosomes in vitro [107]. This might suggest that modifications found in transcriptionally active chromatin would not exist for long enough to be epigenetic. Interestingly however genome-wide studies of nucleosome turnover in yeast that consider FLAG-tagged histone H3 incorporation outside of S phase showed that, although there is a correlation between RNA pol II ChIP and H3 turnover, it is not particularly strong and there are many highly active genes that show rather low rates of H3 turnover [108]. This suggests that H3 displacement and exchange with non-nucleosomal H3 are not essential for transcriptional activity. This agrees with a recent single-molecule study [109], showing that RNA pol II pushes through a nucleosome by transient unwrapping and rewrapping of the DNA around the nucleosome rather than complete nucleosome disassembly. Therefore, RNA pol II transcription alone would not prevent histone marks on the body of a gene from being epigenetic.
RNA pol II transcription is not the only way that S-phase-independent genome-wide nucleosome disassembly can occur. Chromatin remodelling complexes can catalyse nucleosome turnover outside of S phase and independently of transcription [110]. Outside of S phase, this predominantly involves the incorporation of the histone variant H3.3, which is believed to occur preferentially at active genes and promoters [111]. However, a recent experiment found evidence for a significant rate of genome-wide histone turnover [112]. Using isotope labelling and mass spectrometry in Drosophila, Henikoff and colleagues were able to interrogate the entire genome, including Polycomb/Trithorax-regulated zones. Even in these regions they found an average timescale of nucleosome turnover shorter than the cell cycle, prompting the interpretation that, even in silent regions, modified histones do not persist long enough to template chromatin restoration after S phase.
Importantly however, even in a situation where histones and their modifications are turning over rapidly, it is still possible for an epigenetic mechanism based on mark copying to work. Even if all the regions of the genome turn over their histone modifications and/or nucleosomes at the maximum observed rate, it will be less than the rate that occurs in S phase when effectively 50% of the modified nucleosomes are removed simultaneously within minutes as the replication fork moves through. When nucleosomes or nucleosome modifications are removed stochastically within a tract, therefore, one might imagine that a new, ‘naked’ nucleosome could be ‘clothed’ very easily by virtue of its proximity to its surrounding partners. Additional robustness could be provided by the existence of more than one modification associated with a particular epigenetic state, all of which could help to bring newcomers into line. Indeed, simulations based on the recruitment-copy model explained above can be modified to introduce a term for dynamic nucleosome disassembly, without leading to a failure of bistability [37].
Alternative models of chromatin inheritance
The criticisms presented in the previous section have led some to propose alternative models for histone-dependent inheritance of chromatin states. In this section, two of the most popular of these models will be considered.
H3.3 inheritance model
A model for chromatin inheritance that has been put forward by Henikoff is based on the incorporation of H3.3 into chromatin [113]. The motivation for this model is the lack of concrete evidence that histone modifications, in particular those associated with active genes, can template themselves, coupled with the fact that the dynamics of nucleosome assembly suggest that the persistence of modified nucleosomes may be limited. Three crucial assumptions are necessary for this model. The first is that chromatin with H3.3 is intrinsically more labile and accessible to RNA polymerase. The second is that transcription leads to the deposition of H3.3. The third assumption is that during DNA replication, the default state of newly synthesised histones incorporated into chromatin is most similar to silent chromatin. These three assumptions can be combined into a model (Fig. 3b) which enables the chromatin state of active genes to be perpetuated through cell division in the following way:
A gene that is active during interphase will tend to acquire higher levels of H3.3.
During S phase, H3.3 at an active gene will be segregated evenly between the daughter strands. Thus an active gene will end up with 50% H3.3 and 50% newly synthesised H3.1, but a silent gene will have much less H3.3.
In the following interphase, RNA pol II will try to access the genome for transcription. However, at genes where there is very little H3.3 this access will be at low frequency and therefore the gene will not be transcribed. Only at the genes that have sufficiently high levels of H3.3 remaining will RNA pol II access occur. Thus, it is the H3.3, rather than any modifications carried on the histone, that acts as an epigenetic mark to recall the transcriptional state.
How could this explain histone modification inheritance? The idea is that histone modifications such as H3K4me3 occur downstream of transcription, thus leading to their enrichment at regions where H3.3 is found. This would allow their apparent perpetuation during cell division. By contrast, regions without H3.3 would acquire modifications typical of repressive chromatin, by default. The presumed advantage over the mark-copying model presented above is that transcription could begin very soon after replication of any particular region, suggesting that this mode of inheritance could be compatible with rapid nucleosome turnover.
Although evidence directly supporting a role for H3.3 in transcriptional memory has been provided by Ng and Gurdon, who showed using depletion experiments that H3.3 was essential for epigenetic memory of MyoD transcription in a Xenopus nuclear transfer model [114], there is currently limited evidence in support of any of the three assumptions required for the H3.3 inheritance model to work. First, although there is a broad consensus that H3.3 destabilizes nucleosomes, this seems to depend on the modification status of H3.3 and the composition of the rest of the nucleosome, with the H2Az variant also being important in destabilization [115]. Second, the correlation between H3.3 deposition and transcriptionally active regions is not absolute. H3.3 deposition is also observed to be higher at telomeres, for example, and it is speculated that this is to do with the H3.3-specific chaperone DAXX, partnered by ATRX [116], which may be active at these regions. Moreover, given that RNA pol II transcription does not actually require nucleosome disassembly (see above), the incorporation of H3.3 is unlikely to be due to the act of transcription itself. Without this link some other epigenetic information would be required in order to ensure that H3.3 is perpetuated. In this regard, it is particularly interesting that Ng and Gurdon suggest that the H3.3-dependent memory is independent of transcription, as increased H3.3 levels occur before transcription starts. Further, an H3.3 variant that could not be methylated on K4 failed to preserve transcriptional states, despite being incorporated into chromatin. Thus, their work could be interpreted in favour of a model whereby H3.3 K4 methylation is able to recruit H3.3 insertion and propagation, which is, in essence, simply an extension of the recruitment-copying model.
What about the evidence supporting the third assumption, that the state of newly formed chromatin is most like silenced chromatin? In order for this to be true, newly synthesised histones would have to acquire repressive modifications by default, rather than remaining ‘naïve’ as required for the recruitment/copying model. An important piece of evidence that is consistent with this idea is the association of PCNA with components of heterochromatin formation, which would localise enzymatic activities required for addition of heterochromatic marks to the replication fork, where they can modify newly synthesised histones. For example, PCNA interacts with the DNA methyltransferase DNMT1 [117] and, via DNMT1, the H3K9 methyltransferase G9a [118]. Moreover, CAF1, responsible for binding newly synthesised histones, as well as interacting with PCNA, interacts with the heterochromatin protein HP1 [119].
However, despite the evidence of a molecular link between PCNA and heterochromatin complexes, there is no specific evidence that newly synthesised histones acquire repressive modifications irrespective of their deposition site; it is equally possible therefore that the interactions between PCNA and heterochromatic complexes simply exist to increase the efficiency and speed of heterochromatin reformation after replication by a mark-copying type mechanism. Moreover, there is a significant theoretical challenge to a simple ‘default is silent’ model that comes from the understanding that there is more than one type of silent and active chromatin [120]. In particular, H3K9me3-silenced centromeric heterochromatin is distinct from Polycomb-silenced chromatin as the former is HP1-dependent and latter is not [120]. How would the replication machinery know which silent state to put in during division? On the basis of these problems, we suggest that currently an H3.3-based model is not as well placed to explain epigenetic inheritance as the recruitment-copying model, though of course more evidence in support of the assumptions upon which it is based would change this view.
Replication timing model
A different proposal for how histone modifications may be inherited is based on very early work showing that different regions of the genome are replicated at different times, and that the timing of replication correlates with different levels of chromatin compaction [121]. Generally, regions rich in highly expressed genes, with high levels of histone acetylation, tend to be expressed earlier than chromosomal regions with high levels of repressive chromatin and silent genes [122]. If early replication somehow led to increased deposition of histone acetylation during the replication process itself, whilst late replication led to the increased deposition of repressive histone marks, then changing whether a gene is early or late replicating will change whether it is active or not (Fig. 3c). Since different regions tend to be replicated early or late in different cells this could provide a mechanism to allow duplication of chromatin structure without recourse to direct copying of histone modifications themselves [123].
There are two clear requirements for this theory. The first is to demonstrate that the replication timing changes cause changes in the histone modification spectrum. Proving this is technically challenging although interesting experimental evidence in support of this has come from experiments where reporter genes were injected into early or late S-phase cells [124]. This study revealed that when injected early, the reporter genes became highly acetylated on H3 K9/14, whereas this did not occur when the genes were inserted into late S-phase cells.
The second requirement is that the replication-timing program be inherited independently of histone modifications [125, 126]. Here, the evidence is equivocal. Certain experiments have suggested that replication timing can be functionally separated from gene expression; for example, deletion of the β-globin locus control region followed by its expression in an erythroid background did not alter the replication timing of the β-globin locus despite leading to a loss of expression [127]. However, other experiments have suggested that forced recruitment of histone acetyltransferases to the β-globin locus origin of replication can make replication occur earlier, implying that, in contrast to the requirements of the model, histone modifications may drive replication timing rather than the other way around [128]. Even more confusingly, opposing histone modifications can be associated with early firing origins in the chicken β-globin domain [129]. Therefore there may be many different ways in which origin timing could be regulated, which would argue at least against a very simple form of the model.
A further problem for the model is a lack of explanation for how it might work at a molecular level. Since multiple histone modification complexes interact with PCNA, it is possible that PCNA could somehow coordinate a chromatin replication program such that early replication coincided with histone acetylation and late replication with H3K9 methylation. As mentioned above, PCNA is known to interact with various components of facultative heterochromatin introduction. However, their levels or ability to interact with PCNA would have to be tightly controlled so that the complexes only occur at the end of S phase. This has not been documented. The problem is even more pronounced for early-replicating regions because the interaction of PCNA with histone H3K9/14 acetyltransferases has not yet been shown. These problems make the replication timing theory hard to defend at present.
Taking both theoretical and experimental considerations together, the recruitment/copying model for histone modification inheritance seems at present the best supported of the alternatives that have been put forward. However, there are still significant doubts about the extent to which the model can account for the inheritance of transcriptionally active chromatin, and the relationship between the rate of modification turnover and its inheritance. The model clearly needs to be tested much more rigorously. In the next section, we discuss how recent work on the connection between replication stress and epigenetic inheritance may provide a novel angle to analyse how epigenetic states can be maintained through DNA replication.
Replication stress and epigenetic inheritance
In the previous sections, two important requirements of the mark-copying model, namely mark stability and the ability for marks to recruit histone-modifying complexes, were considered. However, there is another vital requirement, not often mentioned, which relates closely to the mechanism of DNA replication. In order for the information carried by parental nucleosomes to be inherited, it is imperative that they be deposited near their original locations on DNA. In turn, this requirement means that the synthesis of new DNA must be tightly coordinated with the unwinding of DNA ahead of the replication fork. Without this coordination, parental nucleosomes will be displaced but not replaced, and their epigenetic information will be lost.
In this regard, agents that disrupt processive replication might be expected to interfere with the process of histone recycling and thus to a loss of epigenetic information. In particular, uncoupling of the replicative helicase from the synthesis of new DNA will lead to extrusion of long segments of single-stranded DNA, which is a poor substrate for nucleosome assembly [130], and would be expected to lead to an excess of parental histones. Support for this idea comes from experiments mentioned above where the modified histones associated with ASF1 were identified by mass spectrometry [48]. The observation that increased numbers of histones carrying modifications typical of parental histones were bound to ASF1 after replication stress suggests strongly that histones were displaced from the template but not replaced under these conditions (Fig. 4).
Fig. 4.

Buffering of histones by ASF1 resulting from replication arrest. Replication fork arrest following treatment of cells with hydroxyurea leads to uncoupling of the polymerases and helicase due to the helicase running ahead of the deoxynucleotide-depleted DNA polymerases. This results in displacement of parental histones, with their marks, which are split and buffered by a pool of ASF1 associated with the replicative helicase [48]
To test this idea further, one would want to show changes in the level of histone modifications at particular loci as a result of replication stress. DNA-damaging agents and replication fork–stalling agents such as hydroxyurea act stochastically across the genome, so unless very high doses are used, the chance of replication stalling in the same place repeatedly is limited. However, there exist also certain natural sites in DNA that have the capability to stall replication, such as G quadruplex (G4) DNA (reviewed in [87, 131, 132]). In vivo, specific proteins can help the replication fork to deal with difficult to replicate sequences, but in mutants where this ability is compromised, replication stress might lead to repeated aberrant histone recycling around these sites.
We used this idea to look at histone modifications and gene expression changes around sites of G4 DNA in chicken DT40 cells lacking the translesion synthesis polymerase REV1 [133], which has been shown to be required to maintain replication fork progression after DNA damage [134, 135]. Replication forks in cells without REV1 stall more frequently at sites of DNA damage and leave gaps, which are filled in post-replicatively. In addition to its role in DNA damage tolerance, REV1 is required for efficient replication of G4 DNA–containing plasmids [133], thus might be expected to show increased replication fork stalling and uncoupling of the helicase from the synthesis of new DNA at sites where G4 s are found. Both at the G4-containing ρ-globin gene and genome-wide, G4 DNA was associated with loss of gene repression, including loss of the H3K9me2 modification. At the same time, modifications characteristic of newly synthesised histones accumulated, consistent with biased deposition of new histones. This hypothesis was supported further by transplanting a G4 DNA sequence into a locus without one, which led to loss of silencing of the gene in rev1 mutant cells. Thus, these data support a model whereby repeated uncoupling of the helicase and the polymerase at sites of G4 DNA leads to loss of parental histone modifications and loss of the epigenetically maintained state of the gene [133] (Fig. 5).
Fig. 5.

Model for epigenetic instability caused by replication arrest. Leading strand replication is arrested by e.g. a G-quadruplex DNA in REV1-deficient cells [133]. i This leads to uncoupling of leading and lagging strand synthesis and the restart of leading strand replication downstream of the block (ii). Excess parental H3/H4, displaced by the helicase, is buffered by ASF1 as in Fig. 4. The stalling structure is resolved and resulting single strand gap filled in at a later stage, which crucially is remote from a supply of parental histones displaced ahead of the replicative helicase. Thus, chromatinisation of the gap is carried out only with newly synthesised histone lacking any parental modifications. These histones lack repressive modifications and allow RNA PolII access for transcription. The model proposes that loss of modifications is reinforced by the repeated replication arrest at the same structure [133]
The correlation between replication stress at G4 DNA and epigenetic instability is important because it supports a key tenet of the recruitment-copying model, namely that there needs to be precise coupling between the displacement of parental histones ahead of the replication fork and their replacement on newly synthesised DNA. Thus, indirectly, these experiments suggest that histone recycling is indeed important for maintaining gene expression, because interfering with it results in the inability to maintain stable gene expression.
Conclusion
In this article, we have argued that a model of epigenetic memory based on the propagation of histone marks is a viable explanation for how cells maintain gene expression states through cell division. The model is supported by genetic and biochemical evidence from diverse systems, and theoretical evidence from computational simulations. Moreover, we have discussed how recent studies into how defects in replication affect epigenetic inheritance processes can also be interpreted in support of the model. Therefore, though histone post-translational modifications are unlikely to be the sole source of epigenetic information in cells, they are likely to provide at least part of the answer to a question that is of great importance in understanding the development and survival of multicellular organisms: how do different cell types remember what they are supposed to be?
Acknowledgements
We would like to thank members of the lab for discussions and comments on the manuscript. Work in the lab is supported by the Medical Research Council, Association for International Cancer Research and the Fanconi Anemia Research Fund.
Conflict of interest
The authors declare that they have no conflicts of interest.
Contributor Information
Peter Sarkies, Email: psarkies@mrc-lmb.cam.ac.uk.
Julian E. Sale, Phone: +44-1223-252941, Email: jes@mrc-lmb.cam.ac.uk
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Happel N, Doenecke D. Histone H1 and its isoforms: contribution to chromatin structure and function. Gene. 2009;431(1–2):1–12. doi: 10.1016/j.gene.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Bassett A, Cooper S, Wu C, Travers A. The folding and unfolding of eukaryotic chromatin. Curr Opin Genet Dev. 2009;19(2):159–165. doi: 10.1016/j.gde.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 4.Holliday R. The inheritance of epigenetic defects. Science. 1987;238(4824):163–170. doi: 10.1126/science.3310230. [DOI] [PubMed] [Google Scholar]
- 5.Grewal SI, Elgin SC. Transcription and RNA interference in the formation of heterochromatin. Nature. 2007;447(7143):399–406. doi: 10.1038/nature05914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 7.Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol. 2005;12(2):110–112. doi: 10.1038/nsmb0205-110. [DOI] [PubMed] [Google Scholar]
- 8.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Turner BM. Decoding the nucleosome. Cell. 1993;75(1):5–8. [PubMed] [Google Scholar]
- 10.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 11.Turner BM. Defining an epigenetic code. Nat Cell Biol. 2007;9(1):2–6. doi: 10.1038/ncb0107-2. [DOI] [PubMed] [Google Scholar]
- 12.Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–443. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 13.Lewis EB. A gene complex controlling segmentation in Drosophila . Nature. 1978;276(5688):565–570. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 14.Ingham PW. A clonal analysis of the requirement for the trithorax gene in the diversification of segments in Drosophila . J Embryol Exp Morphol. 1985;89:349–365. [PubMed] [Google Scholar]
- 15.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 16.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111(2):185–196. doi: 10.1016/S0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 17.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111(2):197–208. doi: 10.1016/S0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 18.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16(22):2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beisel C, Imhof A, Greene J, Kremmer E, Sauer F. Histone methylation by the Drosophila epigenetic transcriptional regulator Ash1. Nature. 2002;419(6909):857–862. doi: 10.1038/nature01126. [DOI] [PubMed] [Google Scholar]
- 20.Byrd KN, Shearn A. ASH1, a Drosophila trithorax group protein, is required for methylation of lysine 4 residues on histone H3. Proc Natl Acad Sci USA. 2003;100(20):11535–11540. doi: 10.1073/pnas.1933593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen K, Bracken A, Pasini D, Dietrich N, Gehani S, Monrad A, Rappsilber J, Lerdrup M, Helin K. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10(11):1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- 22.Müller HJ. Types of visible variations induced by X-rays in Drosophila. J Genetics. 1930;22:299–334. doi: 10.1007/BF02984195. [DOI] [Google Scholar]
- 23.Schultz J. Variegation in Drosophila and the inert chromosome regions. Proc Natl Acad Sci USA. 1936;22(1):27–33. doi: 10.1073/pnas.22.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fodor BD, Shukeir N, Reuter G, Jenuwein T. Mammalian Su(var) genes in chromatin control. Annu Rev Cell Dev Biol. 2010;26:471–501. doi: 10.1146/annurev.cellbio.042308.113225. [DOI] [PubMed] [Google Scholar]
- 25.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410(6824):120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 26.Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410(6824):116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 27.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292(5514):110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 28.Haber JE. Mating-type gene switching in Saccharomyces cerevisiae . Annu Rev Genet. 1998;32:561–599. doi: 10.1146/annurev.genet.32.1.561. [DOI] [PubMed] [Google Scholar]
- 29.Klar AJ. Lessons learned from studies of fission yeast mating-type switching and silencing. Annu Rev Genet. 2007;41:213–236. doi: 10.1146/annurev.genet.39.073103.094316. [DOI] [PubMed] [Google Scholar]
- 30.Pillus L, Rine J. Epigenetic inheritance of transcriptional states in S. cerevisiae . Cell. 1989;59(4):637–647. doi: 10.1016/0092-8674(89)90009-3. [DOI] [PubMed] [Google Scholar]
- 31.Grewal SI, Klar AJ. Chromosomal inheritance of epigenetic states in fission yeast during mitosis and meiosis. Cell. 1996;86(1):95–101. doi: 10.1016/S0092-8674(00)80080-X. [DOI] [PubMed] [Google Scholar]
- 32.Thon G, Friis T. Epigenetic inheritance of transcriptional silencing and switching competence in fission yeast. Genetics. 1997;145(3):685–696. doi: 10.1093/genetics/145.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thon G, Cohen A, Klar AJ. Three additional linkage groups that repress transcription and meiotic recombination in the mating-type region of Schizosaccharomyces pombe . Genetics. 1994;138(1):29–38. doi: 10.1093/genetics/138.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ekwall K, Ruusala T. Mutations in rik1, clr2, clr3 and clr4 genes asymmetrically derepress the silent mating-type loci in fission yeast. Genetics. 1994;136(1):53–64. doi: 10.1093/genetics/136.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorentz A, Heim L, Schmidt H. The switching gene swi6 affects recombination and gene expression in the mating-type region of Schizosaccharomyces pombe . Mol Gen Genet. 1992;233(3):436–442. doi: 10.1007/BF00265441. [DOI] [PubMed] [Google Scholar]
- 36.Furuyama T, Banerjee R, Breen TR, Harte PJ. SIR2 is required for polycomb silencing and is associated with an E(Z) histone methyltransferase complex. Curr Biol. 2004;14(20):1812–1821. doi: 10.1016/j.cub.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 37.Dodd IB, Micheelsen MA, Sneppen K, Thon G. Theoretical analysis of epigenetic cell memory by nucleosome modification. Cell. 2007;129(4):813–822. doi: 10.1016/j.cell.2007.02.053. [DOI] [PubMed] [Google Scholar]
- 38.Muramoto T, Muller I, Thomas G, Melvin A, Chubb JR. Methylation of H3K4 Is required for inheritance of active transcriptional states. Curr Biol. 2010;20(5):397–406. doi: 10.1016/j.cub.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 39.Ye X, Franco A, Santos H, Nelson D, Kaufman P, Adams P. Defective S phase chromatin assembly causes DNA damage, activation of the S phase checkpoint, and S phase arrest. Mol Cell. 2003;11(2):341–351. doi: 10.1016/S1097-2765(03)00037-6. [DOI] [PubMed] [Google Scholar]
- 40.Loyola A, Bonaldi T, Roche D, Imhof A, Almouzni G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol Cell. 2006;24(2):309–316. doi: 10.1016/j.molcel.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 41.Campos EI, Fillingham J, Li G, Zheng H, Voigt P, Kuo WH, Seepany H, Gao Z, Day LA, Greenblatt JF, Reinberg D. The program for processing newly synthesized histones H3.1 and H4. Nat Struct Mol Biol. 2010;17(11):1343–1351. doi: 10.1038/nsmb.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chicoine LG, Schulman IG, Richman R, Cook RG, Allis CD. Nonrandom utilization of acetylation sites in histones isolated from Tetrahymena. Evidence for functionally distinct H4 acetylation sites. J Biol Chem. 1986;261(3):1071–1076. [PubMed] [Google Scholar]
- 43.Sobel RE, Cook RG, Perry CA, Annunziato AT, Allis CD. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc Natl Acad Sci USA. 1995;92(4):1237–1241. doi: 10.1073/pnas.92.4.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436(7048):294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 45.Driscoll R, Hudson A, Jackson SP. Yeast Rtt109 promotes genome stability by acetylating histone H3 on lysine 56. Science. 2007;315(5812):649–652. doi: 10.1126/science.1135862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider J, Bajwa P, Johnson FC, Bhaumik SR, Shilatifard A. Rtt109 is required for proper H3K56 acetylation: a chromatin mark associated with the elongating RNA polymerase II. J Biol Chem. 2006;281(49):37270–37274. doi: 10.1074/jbc.C600265200. [DOI] [PubMed] [Google Scholar]
- 47.Li Q, Zhou H, Wurtele H, Davies B, Horazdovsky B, Verreault A, Zhang Z. Acetylation of histone H3 lysine 56 regulates replication-coupled nucleosome assembly. Cell. 2008;134(2):244–255. doi: 10.1016/j.cell.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jasencakova Z, Scharf AN, Ask K, Corpet A, Imhof A, Almouzni G, Groth A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol Cell. 2010;37(5):736–743. doi: 10.1016/j.molcel.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 49.Sogo JM, Ness PJ, Widmer RM, Parish RW, Koller T. Psoralen-crosslinking of DNA as a probe for the structure of active nucleolar chromatin. J Mol Biol. 1984;178(4):897–919. doi: 10.1016/0022-2836(84)90318-8. [DOI] [PubMed] [Google Scholar]
- 50.Sogo JM, Stahl H, Koller T, Knippers R. Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J Mol Biol. 1986;189(1):189–204. doi: 10.1016/0022-2836(86)90390-6. [DOI] [PubMed] [Google Scholar]
- 51.Jackson V, Chalkley R. Histone segregation on replicating chromatin. Biochemistry. 1985;24(24):6930–6938. doi: 10.1021/bi00345a027. [DOI] [PubMed] [Google Scholar]
- 52.Jackson V. Deposition of newly synthesized histones: hybrid nucleosomes are not tandemly arranged on daughter DNA strands. Biochemistry. 1988;27(6):2109–2120. doi: 10.1021/bi00406a044. [DOI] [PubMed] [Google Scholar]
- 53.Vestner B, Waldmann T, Gruss C. Histone octamer dissociation is not required for in vitro replication of simian virus 40 minichromosomes. J Biol Chem. 2000;275(11):8190–8195. doi: 10.1074/jbc.275.11.8190. [DOI] [PubMed] [Google Scholar]
- 54.Annunziato AT, Schindler RK, Riggs MG, Seale RL. Association of newly synthesized histones with replicating and nonreplicating regions of chromatin. J Biol Chem. 1982;257(14):8507–8515. [PubMed] [Google Scholar]
- 55.Xu M, Long C, Chen X, Huang C, Chen S, Zhu B. Partitioning of histone H3–H4 tetramers during DNA replication-dependent chromatin assembly. Science. 2010;328(5974):94–98. doi: 10.1126/science.1178994. [DOI] [PubMed] [Google Scholar]
- 56.Park YJ, Luger K. Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol. 2008;18(3):282–289. doi: 10.1016/j.sbi.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith S, Stillman B. Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell. 1989;58(1):15–25. doi: 10.1016/0092-8674(89)90398-X. [DOI] [PubMed] [Google Scholar]
- 58.Stillman B. Chromatin assembly during SV40 DNA replication in vitro. Cell. 1986;45(4):555–565. doi: 10.1016/0092-8674(86)90287-4. [DOI] [PubMed] [Google Scholar]
- 59.Kaufman PD, Kobayashi R, Stillman B. Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 1997;11(3):345–357. doi: 10.1101/gad.11.3.345. [DOI] [PubMed] [Google Scholar]
- 60.Kaufman PD, Kobayashi R, Kessler N, Stillman B. The p150 and p60 subunits of chromatin assembly factor I: a molecular link between newly synthesized histones and DNA replication. Cell. 1995;81(7):1105–1114. doi: 10.1016/S0092-8674(05)80015-7. [DOI] [PubMed] [Google Scholar]
- 61.Takami Y, Ono T, Fukagawa T, Shibahara K, Nakayama T. Essential role of chromatin assembly factor-1-mediated rapid nucleosome assembly for DNA replication and cell division in vertebrate cells. Mol Biol Cell. 2007;18(1):129–141. doi: 10.1091/mbc.E06-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nabatiyan A, Krude T. Silencing of chromatin assembly factor 1 in human cells leads to cell death and loss of chromatin assembly during DNA synthesis. Mol Cell Biol. 2004;24(7):2853–2862. doi: 10.1128/MCB.24.7.2853-2862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerard A, Koundrioukoff S, Ramillon V, Sergere JC, Mailand N, Quivy JP, Almouzni G. The replication kinase Cdc7-Dbf4 promotes the interaction of the p150 subunit of chromatin assembly factor 1 with proliferating cell nuclear antigen. EMBO Rep. 2006;7(8):817–823. doi: 10.1038/sj.embor.7400750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shibahara K, Stillman B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell. 1999;96(4):575–585. doi: 10.1016/S0092-8674(00)80661-3. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Z, Shibahara K, Stillman B. PCNA connects DNA replication to epigenetic inheritance in yeast. Nature. 2000;408(6809):221–225. doi: 10.1038/35041601. [DOI] [PubMed] [Google Scholar]
- 66.Gaillard PH, Martini EM, Kaufman PD, Stillman B, Moustacchi E, Almouzni G. Chromatin assembly coupled to DNA repair: a new role for chromatin assembly factor I. Cell. 1996;86(6):887–896. doi: 10.1016/S0092-8674(00)80164-6. [DOI] [PubMed] [Google Scholar]
- 67.Moggs JG, Grandi P, Quivy JP, Jonsson ZO, Hubscher U, Becker PB, Almouzni G. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol Cell Biol. 2000;20(4):1206–1218. doi: 10.1128/MCB.20.4.1206-1218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Green CM, Almouzni G. Local action of the chromatin assembly factor CAF-1 at sites of nucleotide excision repair in vivo. EMBO J. 2003;22(19):5163–5174. doi: 10.1093/emboj/cdg478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith S, Stillman B. Stepwise assembly of chromatin during DNA replication in vitro. EMBO J. 1991;10(4):971–980. doi: 10.1002/j.1460-2075.1991.tb08031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116(1):51–61. doi: 10.1016/S0092-8674(03)01064-X. [DOI] [PubMed] [Google Scholar]
- 71.Talbert PB, Henikoff S. Histone variants—ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11(4):264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 72.Polo S, Roche D, Almouzni G. New histone incorporation marks sites of UV repair in human cells. Cell. 2006;127(3):481–493. doi: 10.1016/j.cell.2006.08.049. [DOI] [PubMed] [Google Scholar]
- 73.Malik HS, Henikoff S. Phylogenomics of the nucleosome. Nat Struct Biol. 2003;10(11):882–891. doi: 10.1038/nsb996. [DOI] [PubMed] [Google Scholar]
- 74.Kaufman PD, Cohen JL, Osley MA. Hir proteins are required for position-dependent gene silencing in Saccharomyces cerevisiae in the absence of chromatin assembly factor I. Mol Cell Biol. 1998;18(8):4793–4806. doi: 10.1128/mcb.18.8.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Verreault A, Kaufman PD, Kobayashi R, Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell. 1996;87(1):95–104. doi: 10.1016/S0092-8674(00)81326-4. [DOI] [PubMed] [Google Scholar]
- 76.Shibahara K, Verreault A, Stillman B. The N-terminal domains of histones H3 and H4 are not necessary for chromatin assembly factor-1-mediated nucleosome assembly onto replicated DNA in vitro. Proc Natl Acad Sci USA. 2000;97(14):7766–7771. doi: 10.1073/pnas.97.14.7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Le S, Davis C, Konopka JB, Sternglanz R. Two new S-phase-specific genes from Saccharomyces cerevisiae . Yeast. 1997;13(11):1029–1042. doi: 10.1002/(SICI)1097-0061(19970915)13:11<1029::AID-YEA160>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 78.Tyler JK, Adams CR, Chen SR, Kobayashi R, Kamakaka RT, Kadonaga JT. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature. 1999;402(6761):555–560. doi: 10.1038/990147. [DOI] [PubMed] [Google Scholar]
- 79.Sanematsu F, Takami Y, Barman HK, Fukagawa T, Ono T, Shibahara K, Nakayama T. Asf1 is required for viability and chromatin assembly during DNA replication in vertebrate cells. J Biol Chem. 2006;281(19):13817–13827. doi: 10.1074/jbc.M511590200. [DOI] [PubMed] [Google Scholar]
- 80.Groth A, Ray-Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G. Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell. 2005;17(2):301–311. doi: 10.1016/j.molcel.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 81.Franco AA, Lam WM, Burgers PM, Kaufman PD. Histone deposition protein Asf1 maintains DNA replisome integrity and interacts with replication factor C. Genes Dev. 2005;19(11):1365–1375. doi: 10.1101/gad.1305005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Groth A, Corpet A, Cook A, Roche D, Bartek J, Lukas J, Almouzni G. Regulation of replication fork progression through histone supply and demand. Science. 2007;318(5858):1928–1931. doi: 10.1126/science.1148992. [DOI] [PubMed] [Google Scholar]
- 83.Adkins MW, Tyler JK. The histone chaperone Asf1p mediates global chromatin disassembly in vivo. J Biol Chem. 2004;279(50):52069–52074. doi: 10.1074/jbc.M406113200. [DOI] [PubMed] [Google Scholar]
- 84.Korber P, Barbaric S, Luckenbach T, Schmid A, Schermer UJ, Blaschke D, Horz W. The histone chaperone Asf1 increases the rate of histone eviction at the yeast PHO5 and PHO8 promoters. J Biol Chem. 2006;281(9):5539–5545. doi: 10.1074/jbc.M513340200. [DOI] [PubMed] [Google Scholar]
- 85.Tyler JK, Collins KA, Prasad-Sinha J, Amiott E, Bulger M, Harte PJ, Kobayashi R, Kadonaga JT. Interaction between the Drosophila CAF-1 and ASF1 chromatin assembly factors. Mol Cell Biol. 2001;21(19):6574–6584. doi: 10.1128/MCB.21.19.6574-6584.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Natsume R, Eitoku M, Akai Y, Sano N, Horikoshi M, Senda T. Structure and function of the histone chaperone CIA/ASF1 complexed with histones H3 and H4. Nature. 2007;446(7133):338–341. doi: 10.1038/nature05613. [DOI] [PubMed] [Google Scholar]
- 87.Mirkin E, Mirkin S. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71(1):13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Papp B, Muller J. Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 2006;20(15):2041–2054. doi: 10.1101/gad.388706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Francis NJ, Follmer NE, Simon MD, Aghia G, Butler JD. Polycomb proteins remain bound to chromatin and DNA during DNA replication in vitro. Cell. 2009;137(1):110–122. doi: 10.1016/j.cell.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Byvoet P, Shepherd GR, Hardin JM, Noland BJ. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys. 1972;148(2):558–567. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- 92.Jenuwein T, Allis C. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 93.Zee BM, Levin RS, Dimaggio PA, Garcia BA. Global turnover of histone post-translational modifications and variants in human cells. Epigenetics Chromatin. 2010;3(1):22. doi: 10.1186/1756-8935-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pray-Grant MG, Daniel JA, Schieltz D, Yates JR, III, Grant PA. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature. 2005;433(7024):434–438. doi: 10.1038/nature03242. [DOI] [PubMed] [Google Scholar]
- 95.Hoppmann V, Thorstensen T, Kristiansen PE, Veiseth SV, Rahman MA, Finne K, Aalen RB, Aasland R. The CW domain, a new histone recognition module in chromatin proteins. EMBO J. 2011;30(10):1939–1952. doi: 10.1038/emboj.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carmen AA, Milne L, Grunstein M. Acetylation of the yeast histone H4 N terminus regulates its binding to heterochromatin protein SIR3. J Biol Chem. 2002;277(7):4778–4781. doi: 10.1074/jbc.M110532200. [DOI] [PubMed] [Google Scholar]
- 97.Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448(7154):718–722. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 99.David-Rus D, Mukhopadhyay S, Lebowitz JL, Sengupta AM. Inheritance of epigenetic chromatin silencing. J Theor Biol. 2009;258(1):112–120. doi: 10.1016/j.jtbi.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122(4):517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 101.Ng SS, Yue WW, Oppermann U, Klose RJ. Dynamic protein methylation in chromatin biology. Cell Mol Life Sci. 2009;66(3):407–422. doi: 10.1007/s00018-008-8303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449(7163):731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 103.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442(7100):312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 104.Mellor J. Dynamic nucleosomes and gene transcription. Trends Genet. 2006;22(6):320–329. doi: 10.1016/j.tig.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 105.Radman-Livaja M, Liu CL, Friedman N, Schreiber SL, Rando OJ. Replication and active demethylation represent partially overlapping mechanisms for erasure of H3K4me3 in budding yeast. PLoS Genet. 2010;6(2):e1000837. doi: 10.1371/journal.pgen.1000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ingvarsdottir K, Edwards C, Lee MG, Lee JS, Schultz DC, Shilatifard A, Shiekhattar R, Berger SL. Histone H3 K4 demethylation during activation and attenuation of GAL1 transcription in Saccharomyces cerevisiae . Mol Cell Biol. 2007;27(22):7856–7864. doi: 10.1128/MCB.00801-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kireeva ML, Hancock B, Cremona GH, Walter W, Studitsky VM, Kashlev M. Nature of the nucleosomal barrier to RNA polymerase II. Mol Cell. 2005;18(1):97–108. doi: 10.1016/j.molcel.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 108.Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. Dynamics of replication-independent histone turnover in budding yeast. Science. 2007;315(5817):1405–1408. doi: 10.1126/science.1134053. [DOI] [PubMed] [Google Scholar]
- 109.Hodges C, Bintu L, Lubkowska L, Kashlev M, Bustamante C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science. 2009;325(5940):626–628. doi: 10.1126/science.1172926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 111.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet. 2005;37(10):1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 112.Deal RB, Henikoff JG, Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328(5982):1161–1164. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9(1):15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- 114.Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008;10(1):102–109. doi: 10.1038/ncb1674. [DOI] [PubMed] [Google Scholar]
- 115.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21(12):1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA. 2010;107(32):14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science. 1997;277(5334):1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 118.Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20(22):3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Murzina N, Verreault A, Laue E, Stillman B. Heterochromatin dynamics in mouse cells: interaction between chromatin assembly factor 1 and HP1 proteins. Mol Cell. 1999;4(4):529–540. doi: 10.1016/S1097-2765(00)80204-X. [DOI] [PubMed] [Google Scholar]
- 120.Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, van Steensel B. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 2010;143(2):212–224. doi: 10.1016/j.cell.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hand R. Eucaryotic DNA: organization of the genome for replication. Cell. 1978;15(2):317–325. doi: 10.1016/0092-8674(78)90001-6. [DOI] [PubMed] [Google Scholar]
- 122.Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6(10):e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McNairn AJ, Gilbert DM. Epigenomic replication: linking epigenetics to DNA replication. BioEssays News Rev Mol Cell Dev Biol. 2003;25(7):647–656. doi: 10.1002/bies.10305. [DOI] [PubMed] [Google Scholar]
- 124.Lande-Diner L, Zhang J, Cedar H. Shifts in replication timing actively affect histone acetylation during nucleosome reassembly. Mol Cell. 2009;34(6):767–774. doi: 10.1016/j.molcel.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gilbert DM. Replication timing and transcriptional control: beyond cause and effect. Curr Opin Cell Biol. 2002;14(3):377–383. doi: 10.1016/S0955-0674(02)00326-5. [DOI] [PubMed] [Google Scholar]
- 126.Hiratani I, Takebayashi S, Lu J, Gilbert DM. Replication timing and transcriptional control: beyond cause and effect—part II. Curr Opin Genet Dev. 2009;19(2):142–149. doi: 10.1016/j.gde.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Reik A, Telling A, Zitnik G, Cimbora D, Epner E, Groudine M. The locus control region is necessary for gene expression in the human beta-globin locus but not the maintenance of an open chromatin structure in erythroid cells. Mol Cell Biol. 1998;18(10):5992–6000. doi: 10.1128/mcb.18.10.5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goren A, Tabib A, Hecht M, Cedar H. DNA replication timing of the human beta-globin domain is controlled by histone modification at the origin. Genes Dev. 2008;22(10):1319–1324. doi: 10.1101/gad.468308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Prioleau MN, Gendron MC, Hyrien O. Replication of the chicken beta-globin locus: early-firing origins at the 5’ HS4 insulator and the rho- and betaA-globin genes show opposite epigenetic modifications. Mol Cell Biol. 2003;23(10):3536–3549. doi: 10.1128/MCB.23.10.3536-3549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Almouzni G, Mechali M, Wolffe AP. Transcription complex disruption caused by a transition in chromatin structure. Mol Cell Biol. 1991;11(2):655–665. doi: 10.1128/mcb.11.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat Struct Mol Biol. 2006;13(12):1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 132.Lipps HJ, Rhodes D. G-quadruplex structures: in vivo evidence and function. Trends Cell Biol. 2009;19(8):414–422. doi: 10.1016/j.tcb.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 133.Sarkies P, Reams C, Simpson LJ, Sale JE. Epigenetic instability due to defective replication of structured DNA. Mol Cell. 2010;40(5):703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30(4):519–529. doi: 10.1016/j.molcel.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 135.Jansen J, Tsaalbi-Shtylik A, Hendriks G, Gali H, Hendel A, Johansson F, Erixon K, Livneh Z, Mullenders L, Haracska L, de Wind N. Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol. 2009;29(11):3113–3123. doi: 10.1128/MCB.00071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]