

Abstract
Despite the absence of classical tyrosine kinases encrypted in the kinome of Plasmodium falciparum, biochemical analyses have detected significant tyrosine phosphorylation in its cell lysates. Supporting such phosphorylation is critical for parasite development. These observations have thus raised queries regarding the plasmodial enzymes accountable for tyrosine kinase activities in vivo. In the current investigation, immunoblot analysis intriguingly demonstrated that Pfnek3, a plasmodial mitogen-activated protein kinase kinase (MAPKK), displayed both serine/threonine and tyrosine kinase activities in autophosphorylation reactions as well as in phosphorylation of the exogenous myelin basic protein substrate. The results obtained strongly support Pfnek3 as a novel dual-specificity kinase of the malarial parasite, even though it displays a HGDLKSTN motif in the catalytic loop that resembles the consensus HRDLKxxN signature found in the serine/threonine kinases. Notably, its serine/threonine and tyrosine kinase activities were found to be distinctly influenced by Mg2+ and Mn2+ cofactors. Further probing into the regulatory mechanism of Pfnek3 also revealed tyrosine phosphorylation to be a crucial factor that stimulates its kinase activity. Through biocomputational analyses and functional assays, tyrosine residues Y117, Y122, Y172, and Y238 were proposed as phosphorylation sites essential for mediating the catalytic activities of Pfnek3. The discovery of Pfnek3’s dual role in phosphorylation marks its importance in closing the loop for cellular regulation in P. falciparum, which remains elusive to date.
Keywords: Malaria, Plasmodium falciparum, MAPKK, Dual-specificity kinase, Phosphorylation
Introduction
In the eukaryotes, modification of proteins through reversible phosphorylation is an essential mechanism for regulation of cellular processes. This post-translational event is catalyzed by enzymes belonging to the protein kinase superfamily that constitutes approximately 2–3% of the eukaryotic proteome [1, 2]. Eukaryotic protein kinases share extensive sequence and structural homologies within their catalytic domains [1, 3]. Depending on their specificities for the target hydroxyl amino acids, protein kinases are traditionally classified into two major classes: the serine/threonine kinases and the tyrosine kinases [1]. In addition, a smaller class of protein kinases, termed as the dual-specificity kinases, is subsequently discovered with specificity for both the serine/threonine and the tyrosine residues [4]. Through the phosphorylation of specific targets on serine, threonine and/or tyrosine residues, protein kinases play integral roles in a diverse array of cellular events. Thus, their activities have to be tightly regulated. Abnormal functioning of protein kinases has been implicated in a number of major human diseases such as cancer, arthritis, and Alzheimer’s condition [5–7]. Therefore, they have emerged as prime molecular targets for the development of therapeutic intervention against these diseases. In 2001, the Food and Drug Administration approved the first kinase inhibitor, imatinib (Gleevec™), for the treatment of chronic myelogenous leukemia [8]. Its clinical success provided compelling evidence for the effectiveness of kinase-specific inhibitors in the treatment of human diseases.
The growing number of kinase inhibitors for clinical use has raised the interest in whether kinase-targeted therapy can be extended to the treatment of malaria, a parasitic disease that currently causes 800,000 deaths annually [9, 10]. Current efforts to control this disease have been marred by the propensity of Plasmodium falciparum, the most lethal causative agent, to rapidly develop resistance against the front-line artemisinin combination therapies [11]. Therefore, in the context where producing efficacious malarial vaccines remains a great challenge, continued efforts to develop novel antimalarial chemotherapeutics is imperative. One widely adopted strategy for discovering new antimalarial agents is the target-based approach, through which novel targets are first identified followed by the design of parasite-specific inhibitors that act on these targets [12]. The search for promising drug targets from the P. falciparum kinome is relevant, since plasmodial kinases are reportedly divergent in terms of their primary sequences and biochemical properties as compared to their mammalian counterparts [13–15]. More importantly, a number of plasmodial protein kinases have been demonstrated to be essential in the life cycle of P. falciparum, further supporting their potential as “druggable” targets (summarized in [16]).
One of the plasmodial kinases under intense investigation is Pfmap2, a mitogen-activated protein kinase (MAPK) homologue of P. falciparum [17, 18]. It is recognized as an antimalarial drug target following a knock-out study that validated its essentiality in the asexual propagation of P. falciparum [18]. To further probe into the molecular mechanisms by which Pfmap2 is engaged for mediating P. falciparum development, identifying its upstream activating kinases is fundamental. Unfortunately, the P. falciparum kinome lacks clear homologues of the MAPK kinase (MAPKK) and MAPKK kinase (MAPKKK), both of which are components of the conserved MAPK signaling pathway that lead to the activation of MAPKs. On hindsight, this indicates that P. falciparum may possess a MAPK pathway that is distinctively different from the other eukaryotes, including its human host. Hence, other than gaining insights into the mechanistic basis leading to the activation of Pfmap2, identification of its upstream kinases is critical for deciphering the elusive P. falciparum MAPK pathway where novel kinase targets suitable for antimalarial drug discovery can be discerned.
In the absence of traditional MAPKK homologues, distinguishing upstream kinases of Pfmap2 is reliant on the detection and biochemical characterization of plasmodial kinases capable of phosphorylating and activating it. A potential Pfmap2 regulator was first illustrated by Dorin et al. [19], whereby Pfnek1, a kinase belonging to the NIMA-like kinase family, demonstrated the competency to catalyze Pfmap2 phosphorylation in vitro. However, the relevance of this phosphorylation event on the activity of Pfmap2 was not clarified. Contrary to Pfnek1, a second plasmodial NIMA-like kinase, Pfnek3, was subsequently reported to display both the ability to phosphorylate and stimulate Pfmap2 activity [20]. The molecular mechanism by which Pfnek3 activates Pfmap2 was further verified to involve phosphorylation within the latter’s activation motif [21]. As this mode of activation mechanism is highly conserved among the eukaryotes, where the MAPKKs phosphorylate their downstream MAPKs within the activation motif to stimulate them [22], Pfnek3 was consequently recognized as a MAPKK-like enzyme of P. falciparum.
In the current study, further characterization of this unusual MAPK activator is pursued to assess whether it possesses additional properties of a typical MAPKK. Results obtained via Western-blot analysis had intriguingly unveiled the ability of Pfnek3 to autophosphorylate and phosphorylate an exogenous substrate at tyrosine-specific sites, in addition to the serine/threonine residues. Like the serine and threonine phosphorylation illustrated earlier [23], tyrosine phosphorylation was demonstrated to play a crucial role in regulating the kinase activity of Pfnek3. Collectively, the findings of this study indicate that Pfnek3 is a novel dual-specificity kinase of P. falciparum and further highlight the similarities between Pfnek3 and the MAPKKs, which also exert both threonine and tyrosine kinase activities on their MAPK substrates.
Materials and methods
Heterologous expression and purification of recombinant proteins
Escherichia coli BL21-CodonPlus™ cells expressing the glutathione S-transferase (GST)-tagged wild-type Pfnek3, the ∆Pfnek3 and ∆Pfmap2 mutants were obtained previously [20, 21]. They were cultured in 100 ml of Luria–Bertani broth supplemented with ampicillin and chloramphenicol (100 and 40 μg/ml, respectively) at 37°C with continuous agitation until an OD600 of 0.6–0.8 was obtained. They were afterwards induced with 1 mM isopropyl-beta-d-thiogalactopyranoside (Sigma) and incubated at room temperature overnight with shaking. Subsequently, the cells were harvested and lysed by sonication as previously reported [21]. Fusion proteins were purified from soluble cell-free extracts by affinity chromatography using the Microspin GST Purification Module (GE Healthcare) according to the manufacturer’s instructions. Protein concentrations of the purified samples were determined by the Bio-Rad protein assay, using bovine serum albumin (BSA, Sigma) as the reference protein standard. New Pfnek3 site-directed mutants generated in the current study were similarly expressed and purified using the same protocol.
Western blot
Protein samples were routinely boiled for 5 min and separated by either 8 or 15% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE). Resolved proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore), which were then blocked with 2% (w/v) BSA in Tris buffered saline (TBS), following which, the membranes were probed with either the anti-phosphoserine (1:1,000 dilution, Millipore), anti-phosphotyrosine (1:2,000 dilution, Millipore) or anti-phosphothreonine (1:1,000 dilution, Cell Signaling Technology) primary antibodies. A peroxidase-conjugated secondary antibody (1:3,500 dilution, Thermo Fisher Scientific) was then incubated with the membranes. The blots were developed using either the Advance Western Blotting Detection Kit (GE Healthcare) or the ECL Plus Western Blotting Substrate (Thermo Fisher Scientific) and exposed to the CL-XPosure film (Thermo Fisher Scientific). Densitometric quantification of the Western-blot results was performed using ImageJ software [24]. To serve as controls for protein loading, duplicated SDS-PAGE gels were run and stained with Coomassie Blue. Stained gels were visualized using the Bio-Rad Molecular Imager Gel Doc XR system equipped with the Quantity One® software.
Phospho-protein staining
Following in vitro kinase assays, reaction mixtures were subjected to SDS-PAGE using 8% polyacrylamide gels. The gels were stained with Pro-Q® Diamond phosphoprotein gel stain (Invitrogen) according to the manufacturer’s protocol and visualized on a UV transilluminator. Densitometric quantification of the result was performed using ImageJ software [24]. To ensure uniform loading of protein samples for each lane, the gels were subsequently stained with Coomassie Blue.
Tyrosine dephosphorylation of Pfnek3 using protein tyrosine phosphatase 1B
For tyrosine dephosphorylation reactions, a 24 μl assay mixture contained 10 μg of wild-type Pfnek3 and 5 U of protein tyrosine phosphatase 1B (PTP1B, Millipore) in a reaction buffer provided by the manufacturer. The phosphatase was omitted in samples that served as negative controls. Incubations were carried out for 1 h at 30°C before 6 μl of 100 mM sodium orthovanadate was added to the reaction mixture to inhibit the activity of PTP1B (final concentration: 20 mM, NEB). A 12-μl aliquot of the resultant mixture was thereafter subjected to both Western blot and ELISA-based kinase assay for verifying the dephosphorylated status and the kinase activity of the PTP1B-treated Pfnek3.
Generation of Pfnek3 mutants through site-directed mutagenesis
The truncated Pfnek3 gene, encoding a catalytically active form of Pfnek3, was earlier cloned into the GST-encoding pGEX-6P-1 vector [20]. The resultant recombinant expression plasmid was used as the template for site-directed mutagenesis reactions. All GST-tagged Pfnek3 mutants were constructed according to the QuikChange™ site-directed mutagenesis protocol (Stratagene), using mutagenic primers listed in Table 1. The authenticity of the respective Pfnek3 mutants was verified through DNA sequencing to ensure that no spontaneous nucleotide substitution was introduced during the PCR amplification process.
Table 1.
Primers used for generating Pfnek3 site-directed mutants
| Mutation | Sequence of mutagenic primers (5′-3′) |
|---|---|
| Y99F |
F: gtgatgagatatttatatctaaggtatatg R: taccttagatataaatatctcatcactttc |
| Y117F |
F: gatttgaataaatttatgaatgaattatata R: taattcattcataaatttattcaaatcatc |
| Y117D |
F: gatttgaataaagatatgaatgaattatata R: taattcattcatatctttattcaaatcatc |
| Y117E |
F: gatttgaataaagaaatgaatgaattatata R: taattcattcatttctttattcaaatcatc |
| Y122F |
F: tgaatgaattatttataatgaataagttaag R: cttattcattataaataattcattcatatattta |
| Y122D |
F: tgaatgaattagatataatgaataagttaag R: cttattcattatatctaattcattcatatattta |
| Y122E |
F: tgaatgaattagaaataatgaataagttaag R: cttattcattatttctaattcattcatatattta |
| Y172F |
F: aataatgaaatatttacagaaagtgaaata R: ttcactttctgtaaatatttcattatttaatttc |
| Y172D |
F: aataatgaaatagatacagaaagtgaaata R: ttcactttctgtatctatttcattatttaatttc |
| Y172E |
F: aataatgaaatagaaacagaaagtgaaata R: ttcactttctgtttctatttcattatttaatttc |
| Y238F |
F: aattgtttaagttttgaatcaattaaatttaag R: atttaattgattcaaaacttaaacaatttaatg |
| Y238D |
F: aattgtttaagtgatgaatcaattaaatttaag R: atttaattgattcatcacttaaacaatttaatg |
| Y238E |
F: aattgtttaagtgaagaatcaattaaatttaag R: atttaattgattcttcacttaaacaatttaatg |
| Y286F |
F: gaggataaaaattttaaaagctatattatc R: aatatagcttttaaaatttttatcctcaaaaag |
The designations F and R denote the forward and reverse primers, respectively, with the sites of mutagenesis underlined
Protein kinase assays
Kinase assay mixtures of 40 μl aliquots prepared for Western blot comprised of 4 μg of wild-type or mutant Pfnek3 proteins, 500 μM ATP, 10 mM MgCl2/MnCl2 and 50 mM Tris–HCl (pH 7.2). The samples were incubated at 30°C for 30 min before they were boiled for SDS-PAGE and Western-blot analysis. When required, the assay mixtures were supplemented with 10 μg of dephosphorylated myelin basic protein (MBP, Millipore) substrate or the catalytically inactive Pfmap2 mutants [∆Pfmap2 or ∆Pfmap2 (T290A)].
To quantify the kinase activities of the tyrosine dephosphorylated Pfnek3 or its site-directed mutants, an ELISA-based kinase assay was adopted as described in [23]. Briefly, the assays were performed using 96-well microtiter plates (Costar) coated with dephosphorylated MBP. Kinase reaction mix (60 μl) added into each well comprised of 4 μg of recombinant protein, 500 μM ATP, 10 mM MgCl2 and 50 mM Tris–HCl (pH 7.2). The assays were allowed to proceed at 30°C for 30 min. Following which, the serine/threonine and the tyrosine kinase activities of the enzymes were independently assessed, based on the levels of MBP phosphorylation detected using the mixed anti-phosphoserine/threonine (1:500 dilution, Millipore) and the anti-phosphotyrosine (1:1,000 dilution) antibodies, respectively.
Biocomputational analysis
The NetPhos server (http://www.cbs.dtu.dk/services/NetPhos/) was utilized for predicting potential tyrosine phosphorylation sites in Pfnek3 [25]. Using the Swiss-PdbViewer [26], the locality of the predicted phospho-tyrosine sites was determined in the Pfnek3 three-dimensional model generated previously [23].
Results
Recombinant Pfnek3 autophosphorylates on serine, threonine as well as tyrosine residues
Using a generic phosphostain, it was earlier shown that the heterologously expressed Pfnek3 purified from bacterial cells exhibited detectable autophosphorylation [23]. To further examine the autophosphorylation specificity of Pfnek3, Western-blot analysis was performed on the purified fusion kinase using antibodies specific against phospho-serine, phospho-threonine as well as phospho-tyrosine residues. As illustrated in Fig. 1a, the wild-type Pfnek3 was immunoreactive with all three phospho-specific antibodies, indicating that the kinase was autophosphorylated on serine, threonine and tyrosine residues following its expression in bacterial cells. The absence of detectable signals by its catalytically inactive counterpart (∆Pfnek3) supports that phosphorylation of these hydroxyl residues is indeed due to the intrinsic activity of Pfnek3, and not the result of any bacterial kinase phosphorylation.
Fig. 1.

Autophosphorylation characteristics of recombinant Pfnek3. a Recombinant Pfnek3 and its kinase-dead mutant (∆Pfnek3) were transferred to PVDF membrane and probed with antibodies specific against phospho-tyrosine, serine, or threonine (denoted by Anti P-Tyr, Anti P-Ser, and Anti P-Thr, respectively). Coomassie staining of duplicated SDS-PAGE gels indicates similar protein loading for each lane. Representative blots/gel from three independent experiments is shown. b Following protein expression and purification, Pfnek3 was incubated in a kinase assay mix comprising ATP and Mn2+/Mg2+ as shown in the top panel. The mixtures were then subjected to immunoblotting using the specified antibodies. Coomassie staining of duplicated SDS-PAGE gels indicates similar protein loading for each lane. The result shown is representative of three independent experiments. c Densitometric analysis of the results obtained in b. Data is expressed as a percentage of Pfnek3 autophosphorylation in the absence of ATP/Mn2+/Mg2+, with each bar representing the mean autophosphorylation level ± standard deviation from the three independent assays
When the wild-type Pfnek3 was incubated in a kinase assay mix consisting of the ATP phosphate donor and the Mn2+/Mg2+ cofactor prior to Western blotting, a significant increase in its phospho-tyrosine signals was observed (Fig. 1b). Notably, the rise in signal was more evident in the presence of Mn2+ as compared to Mg2+ [294 vs. 211% of untreated Pfnek3 tyrosine autophosphorylation based on densitometric quantification of the Western-blot data (Fig. 1c)]. This indicates that Pfnek3 could undergo further tyrosine autophosphorylation in vitro, with Mn2+ as the preferred metal cofactor. On the other hand, no significant increment was detected for both its phospho-serine and phospho-threonine signals. One plausible explanation could be that the heterologously expressed Pfnek3 has already been substantially autophosphorylated on serine and threonine residues in the bacterial cells. Hence, any further enhancement in the autophosphorylation of serine and threonine residues would not be significantly detected through in vitro experiments.
The ability of Pfnek3 to autophosphorylate on both the serine/threonine and the tyrosine residues suggests its potential as a dual-specificity kinase in P. falciparum. This is intriguing since Pfnek3 is predicted to exert only serine/threonine kinase activity based on its primary sequence homology to the classical serine/threonine-type kinases and thus is not expected to phosphorylate tyrosine residues.
Recombinant Pfnek3 exhibits dual-specificity activity on the exogenous MBP substrate, but phosphorylates its potential in vivo Pfmap2 substrate largely on threonine 290
To provide additional support for Pfnek3 as a dual-specificity kinase, further investigations were pursued to determine if the kinase could likewise phosphorylate substrates on both tyrosine and serine/threonine residues. The in vitro serine/threonine kinase activity of recombinant Pfnek3 has been previously assessed using a panel of generic kinase substrates consisting of MBP, histone H1, casein kinase I and casein kinase II peptide substrates [20]. However, Pfnek3 was found to be competent in phosphorylating only the MBP substrate. In view of this, the MBP substrate is used in the present study to evaluate the tyrosine phosphorylation capacity of Pfnek3. The recombinant kinase was co-incubated with MBP in the presence of ATP and Mg2+/Mn2+ before Western blot was performed to detect for the presence of phospho-tyrosines in the MBP substrate. As observed in Fig. 2a, incubation with Pfnek3 considerably increased the level of phospho-tyrosine signal detected in MBP, providing evidence that Pfnek3 does exert a tyrosine kinase activity towards the exogenous MBP substrate. Furthermore, similar to the phenomenon observed for tyrosine autophosphorylation in Fig. 1b, densitometric analysis of the immunoblot revealed that the MBP phospho-tyrosine signal was 61% higher when the assay mix was supplemented with Mn2+ as compared to Mg2+ (Fig. 2b). This offers additional support that the in vitro tyrosine kinase activity of Pfnek3 is triggered to a greater extent in the presence of the Mn2+ cofactor.
Fig. 2.
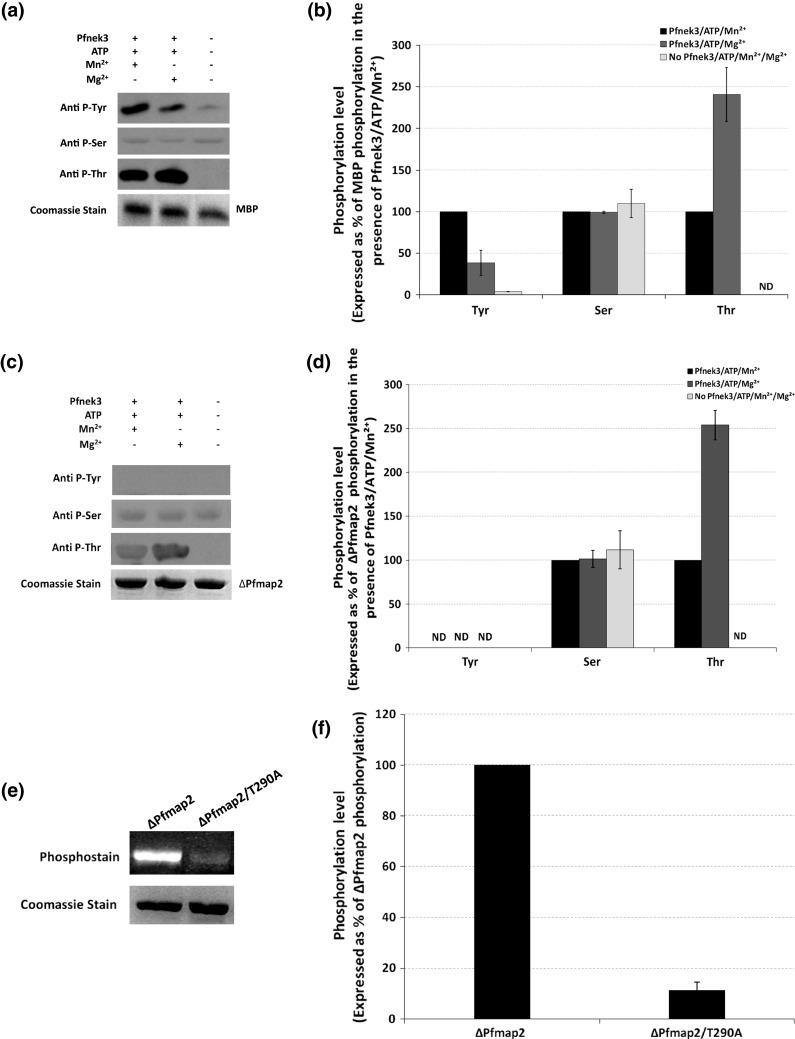
Recombinant Pfnek3 exhibits dual-specificity kinase activity on the exogenous MBP substrate but not Pfmap2. In vitro Pfnek3 kinase assays were performed in the presence of ATP and Mn2+/Mg2+ using a MBP or c ∆Pfmap2 as substrates. The mixtures were resolved by SDS-PAGE and phosphorylation status of the respective substrates was analyzed by Western blot using the indicated phospho-specific antibodies. Coomassie staining of duplicated SDS-PAGE gels indicates similar loading of the respective proteins. Results depicted are representative of three independent experiments. b, d Densitometric quantification of the results obtained in a and c, respectively. For referencing purposes, phosphorylation levels are expressed as percentages of MBP/∆Pfmap2 phosphorylation in the presence of Pfnek3/ATP/Mn2+. Each bar depicts the mean phosphorylation level ± standard deviation from the three independent experiments. e In vitro kinase assays for Pfnek3 were repeated in reaction buffers supplemented with ATP, Mg2+ and either the ∆Pfmap2 or the ∆Pfmap2/T290A substrates. Phosphorylation status of the substrates was subsequently assessed using the Pro-Q® Diamond phosphoprotein gel stain. Coomassie staining of the same gel at the lower panel demonstrates comparable protein loading. A representative result of three independent experiments is illustrated. f Graphical representation of the densitometric analyses for e. Result is expressed as a percentage of ∆Pfmap2 phosphorylation, with each bar representing the mean phosphorylation level ± standard deviation from the three independent assays. ND denotes kinase activity not detected
As the serine/threonine kinase activity of Pfnek3 was previously examined using mixed antibodies specific against both phospho-serines and phospho-threonines, it was unable to clearly distinguish whether phosphorylation had occurred on serine and/or threonine residues. In view of this, Western blot was performed using antibodies specific against either the phospho-serines or the phospho-threonines to further delineate the Pfnek3 phosphorylation specificity on MBP. The results shown in Fig. 2a indicate that Pfnek3 phosphorylates MBP on threonine, but not serine residues. Additionally, in contrast to the trend seen for tyrosine phosphorylation, a stronger phospho-threonine signal was observed when the kinase assay was performed in the presence of Mg2+ instead [141% higher compared to that in the presence of Mn2+ following densitometric quantification of the Western-blot data (Fig. 2b)]. With the competency of Pfnek3 to similarly phosphorylate the exogenous MBP substrate on both the threonine and tyrosine residues, the functionality of Pfnek3 as a dual-specificity kinase is strengthened. In addition, it appears that the dual-specificity activity of the kinase is distinctly influenced by the type of divalent cation present.
Of the two MAPKs (Pfmap1 and Pfmap2) found in the P. falciparum kinome, Pfnek3 was earlier demonstrated to phosphorylate only Pfmap2 in vitro [20]. Hence, other than the exogenous MBP substrate, the dual-specificity kinase activity of Pfnek3 was also evaluated using its plasmodial Pfmap2 substrate in an attempt to gain insights into the functional relevance of its dual-specificity activity in vivo. A catalytically inactive mutant of Pfmap2 (denoted as ∆Pfmap2) was used as the substrate to rule out the effect of its autophosphorylation. In Fig. 2c, Western-blot analysis illustrated that Pfnek3 phosphorylated ∆Pfmap2 at threonine residues. Further densitometric quantification of the immunoblot indicates that the phospho-threonine signal detected in the presence of Mg2+ was 154% more intense as compared to that in Mn2+ (Fig. 2d). This observation coincides with the result obtained from an earlier mass spectrometry study where Pfnek3 was validated to phosphorylate Pfmap2 at the threonine residue, T290 [21]. Apart from threonine residues, Pfnek3 was not observed to phosphorylate ∆Pfmap2 at serine or tyrosine residues. The basal ∆Pfmap2 phosphorylation detected in the phospho-serine blot could be an effect of bacterial kinase phosphorylation when it was heterologously expressed in E. coli. It is believed that Pfnek3 phosphorylates Pfmap2 largely at the validated T290 residue since its phosphorylation was diminished by 89% based on densitometric quantification when the threonine was mutated to a non-phosphorylatable alanine (Fig. 2e, f).
Tyrosine phosphorylation is involved in regulation of the serine/threonine and tyrosine kinase activities of Pfnek3
The detection of Pfnek3 tyrosine autophosphorylation in Fig. 1a has prompted the evaluation of its importance in regulating the catalytic activity of Pfnek3. With this in mind, recombinant Pfnek3 was treated with a tyrosine-specific phosphatase, PTP1B, before its activity was tested in an ELISA-based kinase assay using MBP as the exogenous substrate. In comparison to the untreated Pfnek3, the phosphatase-treated recombinant kinase showed a reduction in its serine/threonine kinase activity by 34%, while its tyrosine kinase activity was lowered by 73% (Fig. 3a). Since prior treatment of Pfnek3 with PTP1B resulted only in the removal of phosphorylation from tyrosine, but not from serine and threonine residues (Fig. 3b), it indicates that the decrease in the activities of Pfnek3 is mainly attributed to the deficiency in tyrosine phosphorylation. Hence, this supports that tyrosine phosphorylation is crucial for the serine/threonine and tyrosine kinase activities of Pfnek3.
Fig. 3.

Effects of tyrosine dephosphorylation on the serine/threonine and tyrosine kinase activities of Pfnek3. Recombinant Pfnek3 was treated with PTP1B as described in the Materials and methods section. In control experiments, PTP1B was omitted. a Aliquots of the mixtures were subjected to ELISA-based kinase assays using MBP as substrate to determine the serine/threonine and tyrosine kinase activities of the PTP1B-treated Pfnek3. Results are expressed as percentage of untreated Pfnek3 activity, with each bar representing the mean activity ± standard deviation from two independent assays. b Phosphorylation status of the PTP1B-treated and control Pfnek3 were verified by Western blot using the indicated antibodies. Coomassie staining of duplicated SDS-PAGE gel illustrates similar loading of the proteins. The results illustrated are representative of two independent experiments
Probing for potential tyrosine phosphorylation sites involved in mediating Pfnek3 kinase activity
Regulation of protein kinase activities can be generally achieved through several mechanisms including phosphorylation by upstream kinases or through autocatalytic events, and/or interaction with regulatory domains or subunits [27, 28]. Among these, phosphorylation within the activation loop, a segment located between the DFG and APE motifs of kinase subdomains VII and VIII, respectively, is a widespread mechanism that controls the function of a wide range of protein kinases [29]. This mode of regulation is similarly adopted by Pfnek3, where previous site-directed mutagenesis and phosphorylation analyses supported S221 and S226 within the activation segment as phospho-activating sites [23]. Other than this, Pfnek3 activity was also demonstrated to rely on phosphorylation of residue T82, located at the N-terminal of the catalytic domain [23]. With the additional observations that Pfnek3 is autophosphorylated on tyrosines residues and that the phosphorylation is essential for its kinase activities (Figs. 1, 3a), determining the specific phospho-tyrosine sites is fundamental to further comprehend the enzymatic regulation of Pfnek3.
However, due to the nature of its amino acid sequence, delineating Pfnek3 phosphorylation sites via mass spectrometry is technically difficult. Despite substantial optimization and troubleshooting, limited protein sequence coverage was constantly obtained, which greatly deterred phospho-site detection [23]. Moreover, as no tyrosine residue is present within the activation loop of Pfnek3, mapping of potential tyrosine phosphorylation sites is further impeded. Hence, a biocomputational approach was undertaken in this study to screen for potential tyrosine phosphorylation sites in Pfnek3. Using the online NetPhos software [25], a total of six candidate phospho-tyrosine sites at Y99, Y117, Y122, Y172, Y238, and Y286 were identified in Pfnek3 (Table 2). With reference to the 3D model of Pfnek3 generated earlier [23], these residues were found to be located on the surface of the protein structure with their hydroxyl side chains projecting outwards (Fig. 4a).
Table 2.
Potential Pfnek3 tyrosine phosphorylation sites predicted by the NetPhos program (http://www.cbs.dtu.dk/services/NetPhos/)
| Tyrosine position | Score | Location in Pfnek3 model |
|---|---|---|
| 99 | 0.981 | Surface |
| 117 | 0.539 | Surface |
| 122 | 0.936 | Surface |
| 172 | 0.959 | Surface |
| 238 | 0.923 | Surface |
| 286 | 0.510 | Surface |
The score represents the probability for each tyrosine residue to undergo phosphorylation. The locality of the residues in the 3D model of Pfnek3 is also indicated
Fig. 4.

Positions of predicted phospho-tyrosine sites in the tertiary model of Pfnek3. a The 3D model of Pfnek3 was generated in an earlier study [23] and viewed using the Swiss-PdbViewer. Location of the predicted phospho-tyrosines are shown and highlighted in red. b Neighboring residues that fall within 4 Å of the predicted phospho-tyrosines were identified and are shown in white. Only residues that form hydrogen bonds (indicated by dotted green lines) with the respective tyrosines are labeled
Mutation of residues Y117, Y122, Y172, and Y238 led to drastic reductions in both Pfnek3 tyrosine phosphorylation and catalytic activities
Based on the hypothesis that phosphorylation occurs at positions Y99, Y117, Y122, Y172, Y238, and Y286, which is in turn essential for stimulating the kinase activity of Pfnek3, site-directed mutagenesis was performed to replace each tyrosine with a non-phosphorylatable phenylalanine. In the event that these tyrosines function as critical phospho-regulatory sites, the mutation would result in the reduction of both the tyrosine phosphorylation of Pfnek3 as well as its catalytic activity.
The GST-tagged mutants (hereafter referred to as Y99F, Y117F, Y122F, Y172F, Y238F, and Y286F) were similarly expressed in E. coli cells as the wild-type kinase and affinity-purified using the glutathione agarose beads. To detect for possible decrease in the tyrosine autophosphorylation of these purified mutants, Western blot was performed using the anti-phosphotyrosine antibody. As illustrated in Fig. 5a, b, both the Y99F and Y286F mutants displayed a comparable level of tyrosine phosphorylation as seen in wild-type kinase. On the contrary, the phospho-tyrosine levels of Y117F, Y122F, Y172F, and Y238F were significantly reduced (29, 51, 27, and 34%, respectively, of wild-type Pfnek3 tyrosine autophosphorylation level based on densitometric analysis of the immunoblot). This supports that tyrosine phosphorylation could originate from residues Y117, Y122, Y172, and Y238, and not from residues Y99 and Y286.
Fig. 5.

Tyrosine phosphorylation levels and catalytic activities of Pfnek3 phenylalanine-substituted mutants. a Purified wild-type Pfnek3 and the indicated site-directed mutants were subjected to Western blot using the phospho-tyrosine antibody. Coomassie staining of duplicated SDS-PAGE gel demonstrates similar protein loading for each lane. The result illustrated is representative of three independent experiments. b Densitometric analysis of the result obtained in a. Data is expressed as a percentage of wild-type Pfnek3 tyrosine autophosphorylation, with each bar representing the mean autophosphorylation level ± standard deviation from the three independent assays. c Via the ELISA-based kinase assay, the in vitro serine/threonine (upper panel) and tyrosine (lower panel) kinase activities of the respective Pfnek3 mutants were determined by their ability to phosphorylate the MBP substrates. Results are presented as a percentage of wild-type Pfnek3 activity, with each bar representing the mean activity ± standard deviation from two independent assays. ND denotes kinase activity not detected
The ELISA-based kinase assays were subsequently conducted to examine the effects of the respective mutations on the serine/threonine and tyrosine kinase activities of Pfnek3. Interestingly, the profile displayed by the mutants for the serine/threonine kinase activity coincided with that for the tyrosine kinase activity (Fig. 5c). It was observed that the Y99F mutant exhibited serine/threonine and tyrosine kinase activities that were similar to the wild-type Pfnek3. On the other hand, the Y117F, Y122F, and Y172F mutants demonstrated significant decrease in both their serine/threonine (5, 26, and 2%, respectively, of wild-type Pfnek3 serine/threonine kinase activity) and tyrosine (13, 26, and 7%, respectively, of wild-type Pfnek3 tyrosine kinase activity) kinase activities. For both the Y238F and Y286F mutants, no significant activity was detected. As the reduction in tyrosine autophosphorylation was concomitant with the decrease in kinase activities for the Y117F, Y122F, Y172F, and Y238F mutants, it supports the notion that phosphorylation does occur at these tyrosine residues and is critical for regulating the catalytic activity of Pfnek3. Nevertheless, structural analyses of the Pfnek3 model revealed that residues Y117 and Y122 each form a hydrogen bond with their neighboring amino acids (Fig. 4b). Therefore, it remains a possibility that the reduction in kinase activity observed for the Y117F and Y122F mutants might be due to the loss of hydrogen bond that could be important for maintaining the structural integrity of the protein.
Based on the results obtained for the Y286F mutant, it is interesting to note that although Y286 is unlikely to be a phosphorylation site, the tyrosine residue was found to be critical for Pfnek3 to phosphorylate the MBP substrate. Considering that autophosphorylation of the Y286F mutant was comparable to that of the wild-type Pfnek3, it suggests that the mutation did not drastically alter the catalytic activity of the kinase. However, the mutant did not exhibit detectable kinase activity using the MBP substrate. A plausible explanation is that the residue Y286 in Pfnek3 is an important binding site residue required for the interaction with the MBP substrate. Therefore, removal of the tyrosine residue via site-directed mutagenesis could have led to a loss of the Pfnek3-MBP interaction and thereby resulted in a lack of phosphorylation in the MBP substrate.
In an attempt to constitutively mimic the effects of phosphorylation at residues Y117, Y122, Y172, and Y238, the tyrosine at these sites were each altered to the negatively charged aspartate or glutamate. The resultant mutants were then assayed for their kinase activities. As depicted in Fig. 6, no significant serine/threonine or tyrosine kinase activity was detected for the Y117D, Y117E, Y172D, Y172E, Y238D, and Y238E phospho-mimetic mutants. In addition, the kinase activities of the Y122D and Y122E mutants did not surpass that of the non-phosphorylatable Y122F mutant. Collectively, it appears that these phospho-mimetic mutations were unable to restore the wild-type activity of Pfnek3. However, it is noteworthy to mention that although aspartate and glutamate are negatively charged amino acids, they may not efficiently mimic the effect of phospho-tyrosines due to the lack of an aromatic ring in their side chains.
Fig. 6.

Catalytic activities of Pfnek3 tyrosine phospho-mimetic mutants. Residues Y117, Y122, Y172, and Y238 were each replaced with either an aspartate or a glutamate to mimic phosphorylation at these residues. Serine/threonine (upper panel) and tyrosine (lower panel) kinase activities of these mutants were analyzed using the ELISA-based kinase assay. Results are presented as a percentage of wild-type Pfnek3 activity, with each bar representing the mean activity ± standard deviation from two independent assays. ND denotes kinase activity not detected
Discussion
The majority of eukaryotic protein kinases are categorized into two broad classes based on whether they phosphorylate serine/threonine or the tyrosine residues. Although the precise basis for conferring amino acid specificity is not entirely clear, serine/threonine and tyrosine kinases can generally be distinguished based on consensus signatures located within the catalytic loop of the kinase domain. The serine/threonine kinases typically contain a HRDLKxxN motif while the tyrosine kinases possess either a HRDLAARN or a HRDLRAAN sequence [1]. However, identification of the third group of protein kinases, the dual-specificity kinases, is less straightforward, as no unique consensus motif has been identified in these kinases. In fact, many of the dual-specificity kinases were initially isolated through phospho-tyrosine antibody screening of cDNA expression libraries designed for detecting novel tyrosine kinases [4]. However, instead of possessing a catalytic domain that was expected to resemble the typical tyrosine kinases, these kinases displayed extensive primary sequence homology to the serine/threonine kinases. Subsequent biochemical analysis revealed their abilities to phosphorylate both the serine/threonine and the tyrosine residues, thus identifying them as dual-specificity kinases. Hence, kinases that are originally predicted as serine/threonine kinases by virtue of their catalytic loop sequences could potentially emerge as dual-specificity kinases.
In the current study, the P. falciparum Pfnek3 was demonstrated via Western-blot analysis to undergo autophosphorylation at not only the serine/threonine but also the tyrosine residues (Fig. 1). Additionally, the kinase could also catalyze phospho-transfer to both groups of hydroxyl amino acids in the exogenous MBP substrate (Fig. 2a, b). These intriguing observations thus point towards a novel dual-specificity kinase activity in Pfnek3. Prior to this study, Pfnek3 was anticipated to phosphorylate merely on serines and/or threonines, since it possesses a catalytic loop (HGDLKSTN) that coincides with the HRDLKxxN consensus element of serine/threonine kinases. Hence, without a more in-depth biochemical examination, the dual-specificity property of Pfnek3 would inevitably be overlooked. These findings thus shed light on another biochemical similarity between Pfnek3 and the classical MAPKK. The latter represents an important dual-specificity kinase in the MAPK signaling pathway essential for activating the downstream MAPK through the concomitant phosphorylation of both threonine and tyrosine residues within the TXY activation site [22].
To assess whether Pfnek3 could exert a similar dual-specificity kinase activity on its Pfmap2 MAPK substrate, its ability to phosphorylate both the serine/threonine and tyrosine residues in Pfmap2 was also investigated. Via mass spectrometry, Pfnek3 was previously verified to phosphorylate Pfmap2 at the threonine residue, T290, within its conserved TSH activation site [21]. This was reflected in the current work where immunoblot analysis demonstrated threonine phosphorylation of Pfmap2 by Pfnek3 (Fig. 2c, d). Other than threonine phosphorylation, no serine or tyrosine phosphorylation was detected. It therefore appears that Pfnek3 exhibits only threonine kinase activity on Pfmap2, and this could occur mainly on residue T290 as phosphorylation of Pfmap2 was largely abrogated when the threonine residue was substituted to an alanine (Fig. 2e, f).
It was also noted that the threonine and tyrosine kinase activities of Pfnek3 were distinctly influenced by the Mg2+ and Mn2+ cofactors (Figs. 1, 2). In the presence of Mn2+, the recombinant Pfnek3 displayed a stronger tyrosine kinase activity in vitro. On the other hand, phosphorylation of threonine residues was more evident in the presence of Mg2+. These findings is in line with the metal cofactor preferences of classical serine/threonine kinases and tyrosine kinases, where Mg2+ and Mn2+ cofactors are favored, respectively [30, 31]. It is plausible that binding of different metal cofactors may induce Pfnek3 to adopt distinct conformations at its active site for promoting the phosphorylation of either serine/threonine or the tyrosine residues.
The catalytic activity of recombinant Pfnek3 has earlier been validated to be influenced by both threonine and serine phosphorylation [23]. In particular, it was demonstrated that phosphorylation of residues T82, S221, and S226 was critical for enhancing the kinase activity of Pfnek3. To complement this finding, results highlighted in Fig. 3 further indicate that tyrosine phosphorylation is also a crucial factor for mediating the kinase activity of Pfnek3. Collectively, the regulatory mechanism involved in controlling Pfnek3 activity seems to become increasingly evident with both serine/threonine and tyrosine phosphorylation playing a pivotal role. Phosphorylation is a major post-translational modification that can modulate protein activity, protein–protein interaction, and protein translocation. Hence, other than acting as molecular switches to regulate its activity, it would be interesting to pursue whether phosphorylation of Pfnek3 can play additional roles in terms of substrate interaction and cellular localization. Efforts have also been made in this study to identify the tyrosine phosphorylation sites that are crucial for modulating Pfnek3 kinase activity. However, as mass spectrometric identification of Pfnek3 tyrosine phosphorylation sites is technically challenging, the current investigation adopted an alternative biocomputational approach for identifying potential tyrosine phosphorylation sites. The NetPhos program predicted a total of six tyrosines that could potentially be phosphorylated (Table 2). The combined data obtained following site-directed mutagenesis and biochemical assays support residues Y117, Y122, Y172, and Y238 as phosphorylation sites crucial for mediating the catalytic activity of Pfnek3 (Fig. 5). Although these tyrosines were phosphorylated based on the autocatalytic activity of recombinant Pfnek3, there remains a possibility for them to be phosphorylated by regulatory kinases in vivo. This would ultimately aid in mapping protein kinase signaling pathways of P. falciparum, which still remains elusive to date. A similar phenomenon has been previously documented for the mammalian ERK2, where bacterially expressed ERK2 was shown to undergo autophosphorylation at the threonine and tyrosine residues within its consensus TEY activation site [32, 33]. However, in vivo, these residues require phosphorylation by the upstream MEK activator to enable ERK2 to achieve its fully activated state [22]. Other than tyrosine phospho-regulatory sites, results obtained from Fig. 5 also suggest that the residue Y286 of Pfnek3 could be an important binding site residue for the interaction with the MBP substrate. This notion, however, requires further investigation and the ability to obtain a crystal structure of the Pfnek3-MBP complex would be advantageous in evaluating the hypothesis.
To summarize, this investigation has provided evidence to support Pfnek3 as a novel dual-specificity kinase of P. falciparum and illustrates an additional biochemical similarity with the MAPKKs. Its serine/threonine and tyrosine kinase activities were also found to be independently stimulated through the binding of distinct metal cofactors. Among all the P. falciparum kinases investigated thus far, Pfmap1, another MAPK homologue, is the only other kinase that reportedly phosphorylates at both the serine/threonine and the tyrosine residues [34]. However, in contrast to Pfnek3, its dual-specificity kinase activity was only restricted to autophosphorylation reactions and whether it exhibits a corresponding activity on its substrates is unclear [34]. An interesting observation highlighted from the study of P. falciparum kinome is that members belonging to the tyrosine kinase family are absent [13]. Despite this, tyrosine phosphorylation was evident throughout the intra-erythrocytic maturation of P. falciparum, and was demonstrated to be essential for parasite survival [35, 36]. This has therefore raised queries regarding the kinases involved in undertaking the role of tyrosine phosphorylation in vivo. The discovery of dual-specificity kinases such as Pfnek3 and Pfmap1 is thus of great biological significance as they could be exploited as key players to perform crucial tyrosine phosphorylation in vivo. More specifically, these kinases may play critical roles in modulating the functions of cellular proteins that require activation by both the serine/threonine and tyrosine phosphorylation. It is also noteworthy to mention that tyrosine-specific phosphatases have been identified in the parasite [37, 38]. Therefore, cellular tyrosine phosphorylation in P. falciparum may be modulated through the cooperative function of these phosphatases and the dual-specificity kinases. Finally, as the dual-specificity property of kinases is difficult to be predicted based on amino acid sequence analysis alone, it is highly likely that additional members of such kinases in P. falciparum could be unveiled through assessment of plasmodial kinases for their tyrosine kinase activities by the biochemical characterization approach.
Ethical standards The authors declare that the experiments comply with the current laws of the country in which they were performed.
Conflict of interest
The authors declare that they have no conflicts of interest.
Abbreviations
- BSA
Bovine serum albumin
- GST
Glutathione S-transferase
- MAPK
Mitogen-activated protein kinase
- MAPKK
Mitogen-activated protein kinase kinase
- MAPKKK
Mitogen-activated protein kinase kinase kinase
- MBP
Myelin basic protein
- PTP1B
Protein tyrosine phosphatase 1B
- PVDF
Polyvinylidene fluoride
- SDS-PAGE
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- TBS
Tris buffered saline
References
- 1.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9(8):576–596. [PubMed] [Google Scholar]
- 2.Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002;27(10):514–520. doi: 10.1016/S0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- 3.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 4.Lindberg RA, Quinn AM, Hunter T. Dual-specificity protein kinases: will any hydroxyl do? Trends Biochem Sci. 1992;17(3):114–119. doi: 10.1016/0968-0004(92)90248-8. [DOI] [PubMed] [Google Scholar]
- 5.Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47(4):409–414. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- 6.Schramek H. MAP kinases: from intracellular signals to physiology and disease. News Physiol Sci. 2002;17:62–67. doi: 10.1152/nips.01365.2001. [DOI] [PubMed] [Google Scholar]
- 7.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1(7):493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 9.World-Health-Organization (2010) World malaria Report 2010
- 10.Doerig C. Protein kinases as targets for anti-parasitic chemotherapy. Biochim Biophys Acta. 2004;1697(1–2):155–168. doi: 10.1016/j.bbapap.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 11.Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, von Seidlein L. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. 2010;8(4):272–280. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 12.Rosenthal PJ. Antimalarial drug discovery: old and new approaches. J Exp Biol. 2003;206(Pt21):3735–3744. doi: 10.1242/jeb.00589. [DOI] [PubMed] [Google Scholar]
- 13.Ward P, Equinet L, Packer J, Doerig C. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics. 2004;5(1):79. doi: 10.1186/1471-2164-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anamika K, Srinivasan N, Krupa A. A genomic perspective of protein kinases in Plasmodium falciparum. Proteins. 2005;58(1):180–189. doi: 10.1002/prot.20278. [DOI] [PubMed] [Google Scholar]
- 15.Doerig C, Meijer L. Antimalarial drug discovery: targeting protein kinases. Expert Opin Ther Targets. 2007;11(3):279–290. doi: 10.1517/14728222.11.3.279. [DOI] [PubMed] [Google Scholar]
- 16.Jirage D, Keenan SM, Waters NC. Exploring novel targets for antimalarial drug discovery: plasmodial protein kinases. Infect Disord Drug Targets. 2010;10(3):134–146. doi: 10.2174/187152610791163381. [DOI] [PubMed] [Google Scholar]
- 17.Dorin D, Alano P, Boccaccio I, Ciceron L, Doerig C, Sulpice R, Parzy D. An atypical mitogen-activated protein kinase (MAPK) homologue expressed in gametocytes of the human malaria parasite Plasmodium falciparum. Identification of a MAPK signature. J Biol Chem. 1999;274(42):29912–29920. doi: 10.1074/jbc.274.42.29912. [DOI] [PubMed] [Google Scholar]
- 18.Dorin-Semblat D, Quashie N, Halbert J, Sicard A, Doerig C, Peat E, Ranford-Cartwright L. Functional characterization of both MAP kinases of the human malaria parasite Plasmodium falciparum by reverse genetics. Mol Microbiol. 2007;65(5):1170–1180. doi: 10.1111/j.1365-2958.2007.05859.x. [DOI] [PubMed] [Google Scholar]
- 19.Dorin D, Le Roch K, Sallicandro P, Alano P, Parzy D, Poullet P, Meijer L, Doerig C. Pfnek-1, a NIMA-related kinase from the human malaria parasite Plasmodium falciparum Biochemical properties and possible involvement in MAPK regulation. Eur J Biochem. 2001;268(9):2600–2608. doi: 10.1046/j.1432-1327.2001.02151.x. [DOI] [PubMed] [Google Scholar]
- 20.Lye YM, Chan M, Sim TS. Pfnek3: an atypical activator of a MAP kinase in Plasmodium falciparum . FEBS Lett. 2006;580(26):6083–6092. doi: 10.1016/j.febslet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Low H, Lye YM, Sim TS. Pfnek3 functions as an atypical MAPKK in Plasmodium falciparum . Biochem Biophys Res Commun. 2007;361(2):439–444. doi: 10.1016/j.bbrc.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Dong C. Regulatory mechanisms of mitogen-activated kinase signaling. Cell Mol Life Sci. 2007;64(21):2771–2789. doi: 10.1007/s00018-007-7012-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Low H, Chua CS, Sim TS. Regulation of Plasmodium falciparum Pfnek3 relies on phosphorylation at its activation loop and at threonine 82. Cell Mol Life Sci. 2009;66(18):3081–3090. doi: 10.1007/s00018-009-0101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abramoff MD, Magalhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11(7):36–42. [Google Scholar]
- 25.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294(5):1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 26.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 27.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15(5):661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 28.Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85(2):149–158. doi: 10.1016/S0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- 29.Adams JA. Activation loop phosphorylation and catalysis in protein kinases: is there functional evidence for the autoinhibitor model? Biochemistry. 2003;42(3):601–607. doi: 10.1021/bi020617o. [DOI] [PubMed] [Google Scholar]
- 30.Swarup G, Dasgupta JD, Garbers DL. Tyrosine-specific protein kinases of normal tissues. Adv Enzym Regul. 1984;22:267–288. doi: 10.1016/0065-2571(84)90018-9. [DOI] [PubMed] [Google Scholar]
- 31.Yuan CJ, Huang CY, Graves DJ. Phosphorylase kinase, a metal ion-dependent dual specificity kinase. J Biol Chem. 1993;268(24):17683–17686. [PubMed] [Google Scholar]
- 32.Seger R, Ahn NG, Boulton TG, Yancopoulos GD, Panayotatos N, Radziejewska E, Ericsson L, Bratlien RL, Cobb MH, Krebs EG. Microtubule-associated protein 2 kinases, ERK1 and ERK2, undergo autophosphorylation on both tyrosine and threonine residues: implications for their mechanism of activation. Proc Natl Acad Sci USA. 1991;88(14):6142–6146. doi: 10.1073/pnas.88.14.6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossomando AJ, Wu J, Michel H, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW. Identification of Tyr-185 as the site of tyrosine autophosphorylation of recombinant mitogen-activated protein kinase p42mapk. Proc Natl Acad Sci USA. 1992;89(13):5779–5783. doi: 10.1073/pnas.89.13.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graeser R, Kury P, Franklin RM, Kappes B. Characterization of a mitogen-activated protein (MAP) kinase from Plasmodium falciparum . Mol Microbiol. 1997;23(1):151–159. doi: 10.1046/j.1365-2958.1997.2071571.x. [DOI] [PubMed] [Google Scholar]
- 35.Sharma A. Protein tyrosine kinase activity in human malaria parasite Plasmodium falciparum . Indian J Exp Biol. 2000;38(12):1222–1226. [PubMed] [Google Scholar]
- 36.Sharma A, Mishra NC. Inhibition of a protein tyrosine kinase activity in Plasmodium falciparum by chloroquine. Indian J Biochem Biophys. 1999;36(5):299–304. [PubMed] [Google Scholar]
- 37.Kumar R, Musiyenko A, Cioffi E, Oldenburg A, Adams B, Bitko V, Krishna SS, Barik S. A zinc-binding dual-specificity YVH1 phosphatase in the malaria parasite, Plasmodium falciparum, and its interaction with the nuclear protein, pescadillo. Mol Biochem Parasitol. 2004;133(2):297–310. doi: 10.1016/j.molbiopara.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 38.Pendyala PR, Ayong L, Eatrides J, Schreiber M, Pham C, Chakrabarti R, Fidock DA, Allen CM, Chakrabarti D. Characterization of a PRL protein tyrosine phosphatase from Plasmodium falciparum . Mol Biochem Parasitol. 2008;158(1):1–10. doi: 10.1016/j.molbiopara.2007.11.006. [DOI] [PubMed] [Google Scholar]