

Abstract
Guanylate cyclase activating protein 1 (GCAP1) is a neuronal Ca2+ sensor (NCS) that regulates the activation of rod outer segment guanylate cyclases (ROS-GCs) in photoreceptors. In this study, we investigated the Ca2+-induced effects on the conformation and the thermal stability of four GCAP1 variants associated with hereditary human cone dystrophies. Ca2+ binding stabilized the conformation of all the GCAP1 variants independent of myristoylation. The myristoylated wild-type GCAP1 was found to have the highest Ca2+ affinity and thermal stability, whereas all the mutants showed decreased Ca2+ affinity and significantly lower thermal stability in both apo and Ca2+-loaded forms. No apparent cooperativity of Ca2+ binding was detected for any variant. Finally, the nonmyristoylated mutants were still capable of activating ROS-GC1, but the measured cyclase activity was shifted toward high, nonphysiological Ca2+ concentrations. Thus, we conclude that distorted Ca2+-sensor properties could lead to cone dysfunction.
Keywords: Neuronal calcium sensor, GCAP, Photoreceptor, Cone dystrophy
Introduction
The concentration of cytoplasmic Ca2+ in photoreceptor cells changes dynamically upon light exposure, ranging from 500 to 600 nM in dark-adapted cells to 50–100 nM in the light.
Guanylate cyclase-activating proteins (GCAPs) are expressed in retinal rod and cone cells and function as neuronal calcium sensors (NCS) [1] that detect changes in Ca2+ concentration and modulate the activity of membrane-bound photoreceptor-specific guanylate cyclases (ROS-GCs) in a Ca2+-dependent manner [2–4]. Ca2+/GCAP-dependent regulation of ROS-GC activity forms a powerful feedback mechanism that sets the overall photoresponse and is highly involved in adaptation phenomena [5, 6]. Among GCAPs, GCAP1 shows stronger immunoreactivity in cones than in rods [7] and was found to be capable of restoring recovery of rod and cone photoresponses in the absence of GCAP2 [3, 8]. However, recent studies showed that the kinetics of recovery from light flashes is slowed down in mice lacking GCAP2, suggesting a role for GCAP2 in adjusting the operating range of rods to higher light intensities [9].
In a very recent work, three novel (i.e., E89K, D100E, G159V) [10] and one previously identified (i.e., L151F) [11, 12] mutations in the gene encoding GCAP1 were found in patients suffering from different forms of cone dystrophies, and the corresponding heterologously expressed proteins were investigated in their capability of activating ROS-GC1 [10]. The disease-associated GCAP1 variants showed as a common hallmark the persistent stimulation of ROS-GC1 at physiological calcium [10]. Moreover, the GCAP1 conformational change upon Ca2+ binding was monitored for each variant by fluorescence spectroscopy and gel electrophoresis, highlighting differences between the wild-type (WT) and the mutated forms [10].
In this study, we investigate whether the persistent stimulation of ROS-GC1 at physiological Ca2+ of disease-associated GCAP1 mutants is related to altered Ca2+ affinity and/or altered Ca2+-induced response of the protein conformation and stability. We thus present a biophysical and functional characterization of four disease-associated GCAP1 mutants and compare their structural stability with that of the WT in the presence and in the absence of Ca2+. We further investigate the influence of the myristoyl group on stability, conformation and function in these GCAP1 variants. The equilibrium Ca2+-binding constants were measured for each GCAP1 variant, and our data are consistent with non-cooperative sequential binding that is strongly influenced both by myristoylation and by the point mutations.
Materials and methods
Building the homology model of human myristoylated GCAP1
The homology model of human myristoylated (myr) GCAP1 was built as follows. The FASTA sequence of human GCAP1 was retrieved from the ExPASy Proteomics Server (http://www.expasy.ch) and used as an input for the Modeller web server for protein structure modeling (ModWeb server: http://modbase.compbio.ucsf.edu/ModWeb20-html/modweb.html). Twenty-six models were retained out of 213 calculated by the server, which were ranked according to pre-defined indexes based on the reliability index and a composite score calculated with respect to the X-ray structure of chicken GCAP1 [13] (PDB entry: 2R2I) used as a template (sequence identity: 85.00%). The models were checked with the Protein Health module in the QUANTA2006 package (http://www.accelrys.com), and the highest score model passed all the quality checks in terms of main and side chain conformations and atomic contacts. Superimposition of the modeled human GCAP1 with the template (chicken GCAP1) led to a 0.34 Å Cα-root mean-square deviation, indicating high structural similarity in particular in the EF-hand loop regions. The three Ca2+ ions and the myristoyl chain were manually placed, followed by rotamer adjustment of the surrounding side chains. The resulting three-dimensional (3D) model is shown in Fig. 1.
Fig. 1.

Three-dimensional homology model of human myrGCAP1. a Cartoon representation of the myrGCAP1 overall fold. EF1 is colored in yellow, EF2 in green, EF3 in orange and EF4 in light blue. N- and C-terminals are colored in pale grey and pale cyan, respectively, and the myristoyl group is represented by grey spheres. Calcium ions are represented by red spheres, while residue targets of cone dystrophy-associated mutations are labeled and shown by dark blue sticks. b Details of the EF3-EF4 domain shown from a different angle. Some key hydrophobic residues (see “Discussion”) are shown by green sticks and labeled; all the other structural features are represented in transparency with the same colors as in a
Expression and purification of GCAP1 variants
Wild-type and mutants of GCAP1 in their nonmyristoylated and myristoylated forms were obtained as described previously [10]. In brief, by employing the same vectors pET21-bGCAP1 and pET21-bGCAP1-D6S as described in [10], the GCAP1 variants were obtained by overexpression in BL21 E. coli cells. The expressed GCAP1 forms were extracted from the insoluble fraction of the inclusion bodies by homogenization in 6 M guanidine hydrochloride and dialyzed twice against 3 l of Tris buffer (20 mM Tris–HCl, 150 mM NaCl, 1 mM DTT pH 7.4), and insoluble fractions were removed by centrifugation (100,000×g for 30 min). The supernatant was concentrated to 5 ml, and the GCAP variants were purified in the presence of 1 mM EDTA and 1 mM EGTA by a combination of size exclusion and ion-exchange chromatography using a Superdex75 column and a UnoQ-column, respectively (BioRad Life Science Group, Hercules, CA) [14]. The degree of myristoylation was analyzed by reversed-phase high-performance liquid chromatography [15].
Protein sample preparation and circular dichroism spectroscopy
For CD spectroscopy and UV monitored Ca2+ titrations in the presence of chelator, proteins were dissolved in a decalcified Tris-buffer (5 mM Tris-HCl, 150 mM KCl, pH 7.5). Decalcification was achieved by insertion of a five-times boiled dialysis tube (3,500 MW cutoff, 18 mm width, 11.5 mm diameter, 1 ml/cm capacity) filled with Chelex-100 resin (Bio-Rad) in the plastic bottle containing the buffer, and left on a rocker for few days. Adjustment of the pH following the decalcification process was performed with particular care (inserting the pH electrode in a withdrawn fraction) to avoid calcium contamination of the buffer. For each preparation, MilliQ water was employed (Millipore Corporation).
All circular dichromism (CD) experiments were carried out using a JASCO J-815 spectropolarimeter (JASCO, Easton, MD) with a Peltier type thermostated cell holder. Far-UV CD spectra (200–250 nm) were recorded at 37°C with a scan rate of 20 nm/min, a band width of 4 nm and a digital integration time of 16 s. Four spectra were accumulated and averaged for each sample. The spectrum of the buffer alone was subtracted from the spectrum of each protein sample. The protein concentration was 19–25 μM in 0.1-cm cuvettes. The spectra of the apo-GCAP1 forms were first recorded in the presence of 100 μM EDTA (Fig. 2, black dashed line). CaCl2 was then added to a final concentration of 200 μM, and the spectra of the Ca2+-loaded forms of the GCAP1 variants were recorded (Fig. 2, black solid line).
Fig. 2.
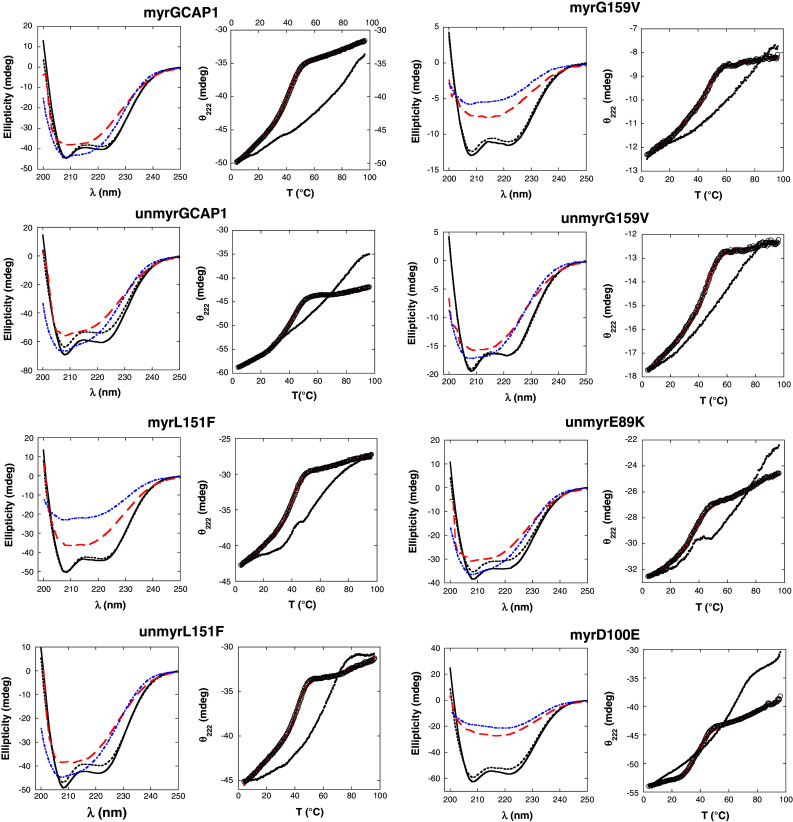
Data from CD spectroscopy performed on GCAP1 variants. For each GCAP1 variant, the panel on the left reports the far UV-CD spectra recorded in the 200–250 nm range for the apo form at T = 37°C (black dashed line), the Ca2+ form at T = 37°C (solid black line), the Ca2+ form after thermal denaturation (T = 96°C, red dashed line) and the Ca2+ form after partial renaturation (T = 4°C, blue dashed line). In the right panels, the thermal unfolding curves are reported for each variant in terms of CD signal at 222 nm as a function of temperature in the 4–96°C range. Data points are reported in the absence (open circles) and in the presence (filled circles) of saturating Ca2+. For the apo cases, computer-fitted curves to apo-protein data by the two-state model (Eq. 1) are shown by solid red lines
The thermal denaturation of all the GCAP1 variants in both their apo- and Ca2+-loaded forms was monitored between 4 and 96°C using the same conditions and concentration range as for the far-UV spectra. The ellipticity at 222 nm (θ222) was recorded with a scan rate of 1°C/min and a response time of 16 s using 0.1 cm quartz cuvettes with a Teflon stopper. For the Ca2+-loaded form of each variant, a 250–200 nm CD spectrum was recorded 5–30 min after reaching the maximum unfolding temperature (T = 96°C) (red dashed lines in Fig. 2). The denatured samples were then cooled to 4°C to check for reversibility, and 5–30 min after reaching the minimum temperature another CD spectrum was recorded (blue dashed line in Fig. 2).
Analysis of thermal denaturation data
The analysis of thermal denaturation curves was performed for each variant assuming a two-state transition. If equilibrium were reached between a fully folded and a fully unfolded state, the θ222 as a function of the temperature could be described quantitatively by accounting for the fraction of folded and unfolded protein. However, in our experiments no complete unfolding was achieved for any variant and in any condition, as is clearly shown by the CD signals that remain negative even at 96°C. The “unfolded” state indeed refers to the denatured state observed in our experiments, for which residual structure is clearly present. Hence, the theoretical fitting curve was used as an ideal approximation for assessing the apparent transition temperature (T t):
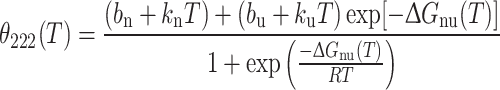 |
1 |
where n indicates the native, folded state and u the (ideal) unfolded state; b is the baseline value and k the slope; T is the temperature and ΔG nu is the Gibbs free energy for the folded–unfolded transition and is given by:
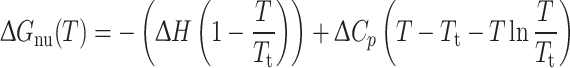 |
2 |
where ΔH and ΔC p respectively indicate the change in enthalpy and heat capacity upon denaturation at constant pressure. We point out that the only parameter obtained from the fitting procedure that was actually employed in comparison between the GCAP1 variants was the apparent transition temperature T t, since the lack of a real equilibrium between the native and fully unfolded state in our experiments made a detailed thermodynamic characterization of the process unfeasible.
While the thermal denaturation curves of the GCAP1 apo-forms were in every case fitted well by the two-state unfolding curve (Fig. 2, right columns panels, full red lines), the Ca2+-loaded forms showed in general a much more gradual transition, indicating a denaturation process of low cooperativity, and within the tested temperature range a quantitative fitting was not always possible. The experimental setting in fact limits the study to temperatures <100°C. Table 1 reports the fitted or the estimated values of T t for each GCAP1 variant.
Table 1.
Results from far-UV CD spectroscopy, thermal denaturation and Ca2+-binding experiments with GCAP1 variants
| Relative Δθa222 | Tapot (°C)b | TCat (°C)b | logK c1 | logK c2 | logK c3 | log(K 1 K 2)/2 | IC50 (μM)d | |
|---|---|---|---|---|---|---|---|---|
| myrGCAP1 | 0.047 | 50.1 ± 1.5 | >70 | 8.4 ± 0.4 | 7.51 ± 0.07 | 5.9 ± 0.4 | 7.9 ± 0.3 | 0.8 ± 0.2e |
| nonmyrGCAP1 | 0.12 | 42.7 ± 0.2 | >60 | 7.8 ± 0.5 | 6.7 ± 0.3 | 3.3 ± 3.0 | 7.2 ± 0.4 | 2.2 ± 0.8f |
| myrG159V | 0.044 | 48.4 ± 0.2 | >60 | 6.7 ± 0.2 | 4.7 ± 0.5 | 3.3 ± 1.9 | 5.6 ± 0.2 | 14.5 ± 1.9e |
| nonmyrG159V | 0.006 | 47.1 ± 0.2 | 63 ± 8 | 6.7 ± 0.3 | 5.81 ± 0.16 | 3.6 ± 1.7 | 6.2 ± 0.2 | 40.3 ± 1.0 |
| myrL151F | 0.023 | 43.0 ± 0.3 | 63 ± 3 | 7.3 ± 0.2 | 4.0 ± 1.6 | 5.1 ± 0.7 | 5.6 ± 0.8 | 39 ± 5 |
| nonmyrL151F | 0.075 | 43.5 ± 0.3 | 65.3 ± 0.7 | 6.9 ± 0.9 | 6.6 ± 0.2 | 5.0 ± 0.3 | 6.8 ± 0.5 | 576 ± 131 |
| myrD100E | 0.073 | 37.5 ± 0.2 | 66.1 ± 0.6 | 6.37 ± 0.13 | 5.0 ± 0.5 | 5.1 ± 0.3 | 5.6 ± 0.3 | 21.9 ± 1.3e |
| nonmyrE89K | 0.12 | 36.9 ± 0.3 | 74.4 ± 0.7 | 7.0 ± 0.2 | 6.4 ± 0.5 | 5.0 ± 0.2 | 6.6 ± 0.3 | 40 ± 9 |
aMeasured at T = 37°C
bTt represents the transition temperature according to the two-state native-unfolded transition model (see Eqs. 1 and 2); apo refers to the calcium-free form, whereas Ca to the calcium-loaded form
cK1, K2 and K3 are the three macroscopic equilibrium constants for calcium binding to GCAP1 variants
dIC50 is the calcium concentration at which the GC activity is half maximal
eData from ref. [10]
fData from ref. [14]
Determination of Ca2+-binding constants using a chromophoric chelator
Calcium binding to GCAP1 variants was monitored by an accurate titration method described previously [16–18], which is based on the competition for Ca2+ between the protein and a chromophoric chelator whose absorbance changes upon Ca2+ binding. The precision of the method is very high for the determination of the equilibrium binding constants, especially when a set of proteins, i.e., different mutants, are compared [16, 17]. Approximately equal amounts (10–50 μM) of chelator and protein are titrated with Ca2+, and the binding of the cation to the chelator is monitored by UV absorbance at 263 nm, allowing the extraction of the binding constants by computer fitting the absorbance as a function of the total Ca2+ concentration. In detail, a Ca2+-free solution of the 5,5′Br2-BAPTA chelator was prepared by dissolving the chelator in the decalcified Tris-buffer described above, and the exact chelator concentration C Q was spectroscopically obtained by measuring the absorbance at 239.5 nm after addition of saturating Ca2+. It was found to range between 19 and 21 μM. The initial concentration of Ca2+ in the chelator solution [Ca Q] was also spectroscopically determined to be between 1.6 and 3 μM for all samples. For each titration experiment, lyophilized proteins were dissolved in the decalcified chelator solution to obtain a similar concentration to that of the chelator. The precise concentration of each protein stock Cp was determined by amino acid analysis following acid hydrolysis (Biomedical Center, Uppsala, Sweden). In each experiment, 1 ml of chelator-protein solution was inserted in a 1-cm cuvette, and the absorbance at 263 nm (λmax for the Ca2+-free chelator) was monitored upon titration with fixed amounts (2.5 μl) of 3 mM CaCl2. Titrations were performed until no significant change in the absorbance was detected. Two or three replications of each titration experiment were performed, and the data (Fig. 3) were fitted using a Newton-Raphson direct least-square fitting procedure implemented in the CaLigator software [16]. In brief, the model used for the fitting allows determining the macroscopic binding constants, the response signals being dependent only on the stoichiometric number of bound Ca2+. Therefore, since GCAP1 has three binding sites, the involved equilibria concerning the protein are:
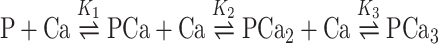 |
3 |
where P denotes the protein (GCAP1 variant), and the K′s are the macroscopic equilibrium constants. The analysis of the equilibria including the one with the chelator leads to the following equation for the total Ca2+:
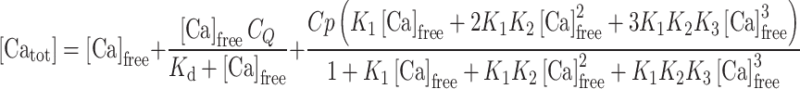 |
4 |
where K d is the dissociation constant for the chelator-Ca2+ complex, which is 2.3 μM for 5,5′Br2-BAPTA in Tris 2 mM, 150 mM KCl, at pH 7.5 [17]. The absorbance at each titration point is calculated as:
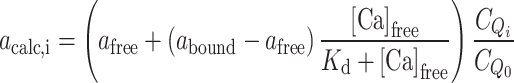 |
5 |
where a
free is the absorbance when no Ca2+ is added, a
bound is the final absorbance in saturating conditions, when no more Ca2+ is added and 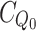 and
and 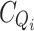 are the total chelator concentrations at the start and at the titration point i, respectively.
are the total chelator concentrations at the start and at the titration point i, respectively.
Fig. 3.

Ca2+-titration curves for the eight variants of GCAP1. In the upper left panel, the experimental points for myrGCAP1 (empty circles) are shown together with the optimal curve by computer fitting (see “Materials and methods”) and the theoretical (simulated) curve for a protein that binds Ca2+ several orders of magnitude lower than the chelator. The curves include the effects of dilution upon titration. In the other panels, empty circles and grey curves refer to myrGCAP1, reported for comparison, while filled circles and black curves refer to one representative set of titration data obtained for that variant. The axes have been normalized in the following way: 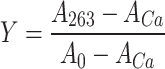 , where A263 is the absorbance read at 263 nm, ACa is the fitted absorbance at saturating Ca2+, and A0 is the fitted absorbance in the absence of Ca2+ additions,
, where A263 is the absorbance read at 263 nm, ACa is the fitted absorbance at saturating Ca2+, and A0 is the fitted absorbance in the absence of Ca2+ additions, 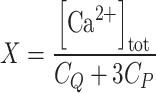 , where [Ca2+]tot is the total Ca2+ concentration (μM) after each addition, C
Q is the total concentration of the chelator (μM), C
p the total concentration of the protein (μM), and 3 is the number of Ca2+-binding sites per protein variant
, where [Ca2+]tot is the total Ca2+ concentration (μM) after each addition, C
Q is the total concentration of the chelator (μM), C
p the total concentration of the protein (μM), and 3 is the number of Ca2+-binding sites per protein variant
During the fitting procedure, [Ca]free in Eq. 5 is replaced by a numerical solution of Eq. 4. The sum of the squares of residuals χ2 is obtained by summing over all points in the titration:
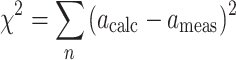 |
6 |
and the variable parameters are iterated in a separate procedure until an optimal fit, i.e., minimum χ2, is obtained. The fitting procedure is rendered automatically by the software CaLigator [16].
All the Ca2+-binding experiments were performed at room temperature (T ~ 21°C), which ensures thermal stability for all the variants analyzed in this study.
Guanylate cyclase assay
Guanylate cyclase activity was measured by using bovine rod outer segment membranes, of which ca. 96% of the cyclase activity originates from ROS-GC1 [19]. Purified GCAP1 forms were reconstituted with washed ROS membranes devoid of intrinsic GCAP activity as described before [20]. The assay buffer was slightly modified to yield free Mg2+ of 1 mM. All incubation steps were performed under very dim red light, and incubation time was 5 min. Synthesis of cGMP was quantified by high performance liquid chromatography as described [20].
Results
Cone dystrophy-associated mutations in myrGCAP1: a structural perspective
Based on the available high-resolution structure of Ca2+-bound myrGCAP1 from chicken [13], we built a reliable homology model of human myrGCAP1 to focus on the regions targeted by three novel and one known mutation recently found in patients suffering from different forms of cone dystrophy [10]. The model is shown in Fig. 1, where the EF-hand motifs (EF1: pseudo EF hand, in yellow; EF2, EF3 and EF4: Ca2+-binding EF hands, in green, orange and blue, respectively) are shown together with the N-(grey) and C-terminal (pale cyan) helices, with the myristoyl group shown by grey spheres and the residues affected by point mutations shown by sticks, namely E89, G159, D100 and L151. A visual inspection of the homology model revealed a perturbation of helical structures in the EF-hand domains, as well as changes in Ca2+ coordination and in surface charges (for a detailed discussion of structural aspects, see below). Since it is known that protein surface charges can greatly affect the thermodynamics of Ca2+ binding, even if not directly involved in the metal coordination [21], and that they may also significantly influence the overall protein stability [22], we investigated the influences of the point mutations in GCAP1 on conformation and stability.
Far UV-CD spectroscopy of myrGCAP1 variants: effects of Ca2+-binding
Far-UV CD spectra were recorded for all the GCAP1 variants in the absence and presence of Ca2+, and the results are shown in Fig. 2. In the absence of Ca2+ all the GCAP1 variants clearly showed the typical CD spectrum of α-helical proteins, indicating that the mutations do not appreciably perturb the protein secondary structure. Moreover, the absence of the myristoyl moiety did not affect the shape of the spectra, indicating that the secondary structure was in no case significantly perturbed by the acyl modification (Fig. 2).
Similarly to what has been previously observed for WT GCAP2 [23], addition of Ca2+ leads to an increase of the CD signal for all the variants (Fig. 2). Depending on the GCAP1 variant, the change in ellipticity at 222 nm ranges from no change (0.6%) to 12% (Table 1). The detected changes are similar to or lower than the 14% increase observed for myrGCAP2 upon Ca2+ binding [23].
Effects of Ca2+ and myristoylation on the stability of GCAP1 variants
Thermal denaturation was investigated in the 4–96°C range for all the GCAP1 variants by monitoring the ellipticity at 222 nm, in the absence and presence of Ca2+. The raw data are shown in Fig. 2, and the values of the transition temperature Tt obtained by fitting a two-state transition model to these data (see “Materials and methods”) are reported in Table 1.
When denaturation occurred in the absence of Ca2+, a clear transition between two structural states could be observed, allowing for an accurate fitting (Fig. 2). However, addition of Ca2+ shifted the transition to higher temperatures, and the denatured state in some cases was not detectable (Fig. 2), thus making the estimate of T t only approximate. The stabilizing effect of Ca2+ was apparent for all the GCAP1 variants as the transition toward denatured states always occurred at higher temperatures compared to the apo-forms (Fig. 2).
Wild-type myrGCAP1 showed the highest thermal stability in both the apo (T t = 50.1 ± 1.5°C) and Ca2+-loaded forms (T t > 70°C) compared to all other variants. These estimates are consistent with the very recent differential scanning calorimetry (DSC) experiments by Lim et al. [24], who measured a transition temperature of 52°C for apo-WT myrGCAP1 and of 103°C for the Ca2+-bound form. A slightly lower transition temperature (T t = 42.7 ± 0.2°C) was instead obtained in this study for the apo-nonmyrGCAP1 compared to the value obtained by DSC measurements (T t = 47°C, [24]). Nevertheless, in both experiments the stabilizing effect of the myristoyl group on the WT structure of GCAP1 was confirmed. However, the stabilizing effect of myristoylation was not observed for the mutants G159V and L151F for which we detected only minor differences in T t, compared to nonmyristoylated forms both in the absence and in the presence of Ca2+ (Table 1). Apparently, the destabilizing effect of the mutations was not compensated by the presence of the myristoyl group.
Among all tested GCAP1 variants, myrD100E (T t = 37.5 ± 0.2°C) and nonmyrE89K (T t = 36.9 ± 0.3°C) were found to be the most unstable in the absence of Ca2+. When saturating Ca2+ was added, all GCAP1 variants became more stable than their apo-forms as shown by a shift to higher T ts that largely exceeded 15°C in most cases (Table 1). Particularly impressive is the Ca2+-induced stabilization of nonmyrE89K that exhibited a native-like stability and a difference in T t between the apo and the Ca2+-bound form of >37°C.
We did not observe reversible unfolding for any of the GCAP1 samples similarly to recently reported DSC experiments [24]. In order to assess the protein secondary structure after denaturation, we recorded the far UV-CD spectrum for all the Ca2+-loaded variants at 96°C (Fig. 2, red dashed lines). While some secondary structure was clearly retained, the spectra demonstrated a loss of α-helical content when compared to the ones recorded at 37°C. In an attempt to follow protein refolding, the samples were cooled back to 4°C (Fig. 2; blue dashed lines). With respect to the spectra at 96°C, some CD signal is recovered for most of the variants, indicative of a partial recovery of secondary structure. However, the spectral shape is still far from the native, purely α-helical one. Renaturation curves were incompatible with the two-state transition model and did not overlap with the corresponding denaturation curves (results not shown).
Ca2+ binding to GCAP1 variants
Competition with the 5,5′Br2-BAPTA chelator was monitored spectroscopically to follow the binding of Ca2+ ions to GCAP1. The titration curves obtained for all the variants are shown in Fig. 3. Overall, Ca2+ binding to GCAP1 appeared to be sequential and to occur with no apparent cooperativity. All the variants were found to bind Ca2+ to some extent, as is clearly shown by comparing to the simulated curve for a case where the Ca2+ affinity is several orders of magnitude lower for the proteins than for the chelator (Fig. 3, upper panel). All data sets were reasonably fitted by a model in which the protein has three Ca2+-binding sites (see “Materials and methods”), and the value of the binding constants could hence be obtained by computer fitting (Table 1). The working conditions under which the Ca2+ binding experiments were performed ensure robust and reliable determinations. In fact, the experiments were performed at protein concentrations significantly lower than 100 μM, which is the lowest concentration at which GCAP1 dimerization was observed [24]. On the other hand, preliminary studies showed that some mutations may change the monomer/dimer equilibrium, but do not cause aggregation of the proteins under these conditions (manuscript in preparation).
Our results are consistent with myrGCAP1 having two high affinity Ca2+ binding sites (represented by K 1 and K 2 in Table 1) and one site with much lower affinity (K 3 in Table 1). Indeed, the detected affinity for the third binding site was in most cases similar to or even below the experimental detection limit using 5,5′Br2-BAPTA (logK chelator = 5.6, K Dchel = 2.3 μM). The method measures macroscopic binding constants, and for a case with two sites with identical high affinity sites and one site with much lower affinity, logK 1 will be 0.6 higher than logK 2 [25]. A larger difference is seen for all variants except one, suggesting that the two high affinity sites vary somewhat in affinity, although the error limits for the individual logKs are generally larger than for their product, which can be determined with considerably higher accuracy. Hence, we report also as an apparent affinity the average of the logarithms of the first two macroscopic binding constants ((logK 1 + logK 2)/2). Given the much lower affinity for the third site, this apparent affinity is not affected by the poorly accurate information on the low affinity binding site (i.e., logK 3).
Among the GCAP1 variants, the WT myr form showed the highest affinity for Ca2+ (Table 1), followed by the nonmyr form, which is observed to bind Ca2+ with a ~5-fold lower affinity. Such a difference concerns both high-affinity binding sites, as the first macroscopic dissociation constant (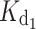 ) increased from 4 nM for the myr form to 16 nM for the nonmyr form, while
) increased from 4 nM for the myr form to 16 nM for the nonmyr form, while 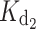 increased from 31 nM for the myr form to 200 nM. All the GCAP1 mutants associated with cone dystrophy showed impaired apparent Ca2+ affinity, which for some variants (myrG159V, myrD100E, myr L151F) is even two log units lower, i.e., 100-fold lower affinity than WT (Table 1). The most apparent effect on
increased from 31 nM for the myr form to 200 nM. All the GCAP1 mutants associated with cone dystrophy showed impaired apparent Ca2+ affinity, which for some variants (myrG159V, myrD100E, myr L151F) is even two log units lower, i.e., 100-fold lower affinity than WT (Table 1). The most apparent effect on 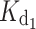 was observed for the myrD100E mutant (
was observed for the myrD100E mutant (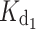 = 426 nM), whereas
= 426 nM), whereas 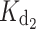 is particularly affected by the myrG159V (
is particularly affected by the myrG159V (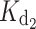 = 20 μM) and myrD100E (
= 20 μM) and myrD100E (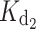 = 10 μM) mutations.
= 10 μM) mutations.
Myristoylation affected Ca2+ binding to all variants, in some cases enhancing the affinity (WT GCAP1), whereas in some others decreasing it (G159V, L151F).
GCAP1-dependent regulation of GC activity
In order to investigate the functionality of GCAP1 variants and their Ca2+-dependent activation of photoreceptor GC, we performed GC activity assays for the cases that had not been previously analyzed (Fig. 4). These include various mutant forms recently found to be associated with cone dystrophy, i.e., nonmyrG159V, myrL151F, nonmyrL151F and nonmyrE189K. Table 1 compares the data of the present with the previous study in terms of IC50, i.e., the Ca2+-concentration at which the GC activity is half maximal. The tested variants showed IC50 values significantly shifted to high Ca2+ concentrations compared to the native myrGCAP1 (Table 1, Fig. 4). In particular, nonmyrG159V, myrL151F and nonmyr E89K showed a ~50-fold shift in Ca2+ sensitivity, whereas nonmyrL151F showed an impressive ~720-fold shift.
Fig. 4.

Regulation of ROS-GC1 by nonmyrGCAP1 variants. Washed bovine rod outer segment membranes containing ROS-GC1 were reconstituted with 10 μM nonmyrGCAP1 (filled circle), nonmyrL151F (open circle), nonmyrE89K (filled inverted triangle) and nonmyrG159V (open triangle) at varying Ca2+ concentrations. Guanylate cyclase activity of nonmyrGCAP1 was measured as in ref. [19] and that of the mutants as described in the “Materials and methods.” Activity was calculated as nmol cGMP/min/mg rhodopsin and is expressed as normalized activity
Discussion
Gathering detailed biochemical and biophysical information on the molecular determinants of disease-associated states is essential in order to be able to quantitatively describe phototransduction events in normal and altered conditions, and hence to design effective interventions [6]. In this study we attempted to characterize the effects of Ca2+ binding to some GCAP1 variants associated with human cone dystrophies and to investigate the role of myristoylation in both protein stability and GCAP1-mediated activation of GC. To date, ten mutations have been found in the gene encoding GCAP1 that are related to autosomal dominant cone dystrophy (adCD), autosomal dominant rod-cone dystrophy (adCRD) and macular dystrophy (adMD), which have been characterized in previous works [12, 26–32]. Among these mutants, four were recently investigated in a combined clinical and molecular characterization [10]. The same variants of GCAP1 have been further analyzed here in order to highlight common disease-associated hallmarks from a biophysical and functional perspective.
Interestingly, all four mutations analyzed in this study, which are involved in cone dystrophies, are located in the EF3-EF4 domain of GCAP1 (Fig. 1). This region is known to undergo major conformational changes upon Ca2+ binding [33], and we followed the structural effects of Ca2+ binding by CD spectroscopy. The results showed some common features shared by WT and mutated GCAP1s (Fig. 2). The apo form of each variant exhibited a typical α-helical pattern, indicative of a well-defined secondary structure. Addition of saturating Ca2+ increased the intensity of the CD signal, as it was quantified by positive Δθ222s (Table 1), without altering the shape of the spectrum. This behavior suggests that in the absence of Ca2+, all GCAP1 variants form a flexible molten globule state, with fully native secondary structure content but lack of stable tertiary structure. Differently from the interpretation of enhanced helical content proposed for GCAP2 [23], we suggest that the increase observed in the CD signal upon addition of Ca2+ is a consequence of the protein transition to a more rigid and compact state. The same behavior was observed for other Ca2+-binding proteins, including GCAP2 [34]. Furthermore, this interpretation is compatible with very recent two-dimensional NMR data [24] that show a typical molten-globule spectrum for the apo form and the clear appearance of characteristic EF-hand peaks upon Ca2+ saturation. CD spectra also suggest that the presence of the myristoyl group affected the flexibility of the protein in a manner that depended on the point mutation.
The idea that Ca2+-loaded forms are more stable than the respective apo forms was further confirmed by thermal denaturation studies (Fig. 2; Table 1). In every case, but to a different extent depending on the GCAP1 variant, the transition to a less folded state is shifted toward higher temperatures in the presence of Ca2+. This result is in agreement with recent DSC observations by Lim. et al. [24] on WT GCAP1 being stabilized by metal binding. Likewise, we also noticed that myristoylation at the N-terminus had a stabilizing effect on the WT form both in the presence and in the absence of Ca2+ (Table 1). In addition, we proved that the effect of acylation on protein stability nevertheless depends on the GCAP1 variant, as both G159V and L151F showed virtually unaltered stabilities independent of myristoylation (Table 1). Interestingly, the change in the transition temperature upon addition of Ca2+, quantitatively determined for most of the variants (Table 1), correlated well with the change in CD signal detected at 222 nm (correlation coefficient R = 0.92; see Table 1). This seems to further support our interpretation that the increased signal upon Ca2+ saturation is due to GCAP1 gaining a more compact and overall stable conformation rather than to undergoing a change in its secondary structure as postulated before for GCAP2 [23].
In the present study, we measured for WT myrGCAP1 a slightly higher affinity for Ca2+ compared to that recently measured by Lim et al. [24] by isothermal titration calorimetry (ITC). The ITC data of Lim et al. [24] are compatible with two nanomolar affinity binding sites (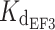 = 80 nM and
= 80 nM and 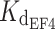 = 200 nM) and one micromolar affinity site (
= 200 nM) and one micromolar affinity site (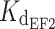 = 0.9 μM) when measured in the absence of Mg2+. When the constants measured in the present study for WT myrGCAP1 are given as macroscopic K
ds, the values are
= 0.9 μM) when measured in the absence of Mg2+. When the constants measured in the present study for WT myrGCAP1 are given as macroscopic K
ds, the values are 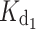 = 4 nM,
= 4 nM, 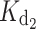 = 31 nM and
= 31 nM and 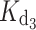 = 1.2 μM. The fact that we measured macroscopic binding constants as well as differences in the running buffer and the method employed to measure the initial protein concentration could account for the slight divergence with respect to ITC determinations. One of the advantages offered by the chelator method employed in this study is its proven precision when comparing between protein sets (e.g., WT and mutants) [16, 17]. Hence, the differences in apparent affinities for the GCAP1 variants listed in Table 1 are expected to be highly reliable.
= 1.2 μM. The fact that we measured macroscopic binding constants as well as differences in the running buffer and the method employed to measure the initial protein concentration could account for the slight divergence with respect to ITC determinations. One of the advantages offered by the chelator method employed in this study is its proven precision when comparing between protein sets (e.g., WT and mutants) [16, 17]. Hence, the differences in apparent affinities for the GCAP1 variants listed in Table 1 are expected to be highly reliable.
The impact of disease-related mutations was further analyzed by referring to the modeled protein structure of myr human GCAP1. Thus, we restrict this aspect of our discussion to the myristoylated GCAP1 mutants. When the analysis is limited to the myristoylated forms, the impaired ability of binding Ca2+ observed experimentally for all the cone dystrophy-associated mutants for WT GCAP1 could be partially explained by analyzing the modeled protein structure (Fig. 1B). The 3D structure of the Ca2+-loaded myristoylated form of chicken GCAP1 was resolved recently by X-ray crystallography at 2.0 Å resolution (PDB entry: 2R2I). In contrast to recoverin (Rec), which upon Ca2+-binding exposes the acyl group to the solvent, GCAP1 keeps the myristoyl group buried inside the protein milieu, hence providing evidence for no Ca2+-mediated switching [13]. This result confirms previous surface plasmon resonance studies that highlighted an influence of the myristoyl group on the Ca2+-dependent regulation of ROS-GC1 by GCAP1, but no Rec-like switching for GCAP1 and GCAP2 [14]. A structural comparison between myrGCAP1, nonmyrGCAP2 and nonmyrGCAP3 [35] reveals that the overall architecture and conformation of the Ca2+-binding EF hands are conserved independent of the acylation, but the structure of the N- and C-terminal helices is severely affected in the nonmyristoylated proteins [13]. In detail, the absence of the myristoyl group was found to cause a severe destabilization of the C-terminal helix in nonmyrGCAP2, which could not be resolved by NMR spectroscopy, as well a considerably increased flexibility of the N-terminal region [34]. Overall, these structural findings suggest that the N-terminal myristoylation of GCAPs is important for maintaining the physiological conformation of the N- and C-terminal helices, which is finally required for functional interaction with ROS-GC1 [14, 36].
The analysis of protein structure further reveals some specific features of the sites that are targeted by mutagenesis in the cone-dystrophy conditions. In this respect, G159 was found to be disease-associated when mutated into V [10]. The native G159 lies at the border between the two helices making up the EF4 motif (Fig. 1b), and a mutation to V is expected to perturb sterically the tight interaction with I143 in the facing helix. This perturbation might propagate and eventually affect the Ca2+-binding process. L151 is tightly packed in the core between EF3 and EF4 (Fig. 1b), in a hydrophobic cleft formed by F156, F140 and again I143. The observed mutation to F is likely to modify the hydrophobic interactions and finally destabilize the whole EF3-EF4 domain as F140 would need to drastically change its conformation in order to accommodate the new side chain. The perturbation would likely affect EF4 wholly, with a possible repercussion on the Ca2+ binding in its loop. D100 is directly involved in Ca2+ coordination in the loop of EF3 (Fig. 1). While conserving the net charge, mutation to E is expected to affect the Ca2+ binding. The site has evolved to accommodate D, and the larger E may be sterically disfavored, leading to suboptimal Ca2+ coordination. Finally, E89 is totally solvent-exposed (Fig. 1a) and is not involved in significant short-range electrostatic interactions. The closest intra-helical interaction is with R172, which is stabilized by a stronger coulombic interaction with D168; such interaction is in fact expected to be weak (E89-R172 distance = 13 Å). The effect on Ca2+ binding, structure and stability is therefore expected to be limited and exerted through purely electrostatic effects.
The spectroscopic titration method to follow the competition with a chelator also allows very accurate detection of cooperativity mechanisms in Ca2+-binding proteins (see, for example, [17, 18, 37, 38]). The unique advantage of this technique is that, different from other approaches that are based on chemical modifications of the protein, it allows detecting and quantifying cooperativity mechanisms without perturbing the system. The shape of the titration curve indeed contains the information on interaction between binding sites and hence on potential cooperativity. It has been demonstrated that positive cooperativity and sequential binding result in S-shaped curves of opposite curvature [37], hence making a clear distinction between the two mechanisms possible. In none of the cases tested here did we observed apparent cooperativity for Ca2+ binding to GCAP1 (Fig. 3), but rather our data are consistent with sequential binding. This is in contrast with what has been very recently reported by Lim et al. [24], who observed positive cooperativity in Ca2+ binding between EF3 and EF4, differently from a previous work in which the overall Ca2+-binding isotherm of GCAP1 obtained by analysis of fluorescent chelator binding showed no evidence of cooperativity [39]. The observed difference might be due to the mutants employed in the study to disable specific binding sites, which may perturb the system and affect the measured change in the free energy of Ca2+ binding [24]. Double mutants that selectively impair Ca2+ binding to EF2, EF3 or EF4 could have unpredictable structural and thermodynamic effects on the other EF hands and affect their Ca2+ affinity, as the present study clearly shows. Therefore, while the Ca2+-mediated activation process of ROS-GC1 by GCAP1 definitively shows the features of cooperativity in Ca2+ binding [40], our data strongly suggest that metal binding to GCAP1 alone is not cooperative.
Surprisingly, we could not find any significant correlation between the apparent Ca2+ affinity and IC50 values for the set of mutants studied here. No correlation was found also when focusing on individual binding constants, even when the set of previously analyzed mutants (see ref. [10]) was included in the analysis. The interpretation of this finding is not trivial, as the information contained in the IC50 values summarizes a rather complex series of events. As discussed and suggested previously [24, 39, 41], the activation step of ROS-GC1 by GCAP1 would include: (1) dissociation of Ca2+ from GCAP1 while it is bound to ROS-GC1; (2) an induced conformational change in GCAP1 that is transferred to the catalytic domain of ROS-GC1; (3) a rearrangement of the dimer interface in ROS-GC1 leading to an accelerated cGMP synthesis. The concerted action of these sequential steps is mirrored in the cooperativity of ROS-GC1 activation [40], but Ca2+ binding to isolated GCAP1 occurs without cooperativity. Furthermore, GCAP1 might display different Ca2+ affinities when GCAP1 is bound to ROS-GC1. This phenomenon is not uncommon and has been observed with calmodulin and other Ca2+-sensor proteins and their corresponding targets [42]. These issues, however, deserve future investigation.
Finally, the minor stabilizing effect of myristoylation in WT GCAP1 (Fig. 2; Table 1) is in agreement with previous studies [13] as well as with the effect of acylation on the control of the Ca2+ sensitivity [14, 15, 43]. These results suggest an allosteric mechanism of communication between the Ca2+-binding sites and the myristoyl moiety without a switch mechanism [14].
In conclusion, the results of this study support the idea that disease-associated forms of GCAP1 are less stable and have lower affinity for Ca2+ than the native form. Although they can be still functional, they would require a much higher Ca2+ concentration to inhibit the cyclase, a phenomenon that is even more apparent for the nonmyristoylated forms. Hence, in vivo, these forms are expected to maintain ROS-GC1 in a state of constitutive activity, eventually driving the photoreceptor to degeneration.
Acknowledgments
This work was supported in part by a FEBS Short-Term Fellowship, by a Research Fellowship grant by the Alexander von Humboldt Foundation (both to DDO), by ProRetina Deutschland e.V. (Pro-Re/Prom-Stip.Koch/Behnen.1) and by the Swedish Research Council, VR (to SL). Research support by the Blanceflor Boncompagni-Ludovisi Foundation (Stockholm, Sweden) is also gratefully acknowledged.
Contributor Information
Sara Linse, Phone: +46-46-2228246, FAX: +46-46-2224543, Email: sara.linse@biochemstry.lu.se.
Karl-Wilhelm Koch, Phone: +49-441-7983640, FAX: +49-441-798193640, Email: karl.w.koch@uni-oldenburg.de.
References
- 1.Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat Rev Neurosci. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch KW, Duda T, Sharma RK. Photoreceptor specific guanylate cyclases in vertebrate phototransduction. Mol Cell Biochem. 2002;230:97–106. doi: 10.1023/A:1014209711793. [DOI] [PubMed] [Google Scholar]
- 3.Mendez A, Burns ME, Sokal I, Dizhoor AM, Baehr W, Palczewski K, Baylor DA, Chen J. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci USA. 2001;98:9948–9953. doi: 10.1073/pnas.171308998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palczewski K, Polans AS, Baehr W, Ames JB. Ca(2+)-binding proteins in the retina: structure, function, and the etiology of human visual diseases. Bioessays. 2000;22:337–350. doi: 10.1002/(SICI)1521-1878(200004)22:4<337::AID-BIES4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 5.Burns ME, Arshavsky VY. Beyond counting photons: trials and trends in vertebrate visual transduction. Neuron. 2005;48:387–401. doi: 10.1016/j.neuron.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Dell’Orco D, Schmidt H, Mariani S, Fanelli F. Network-level analysis of light adaptation in rod cells under normal and altered conditions. Mol Biosyst. 2009;5:1232–1246. doi: 10.1039/b908123b. [DOI] [PubMed] [Google Scholar]
- 7.Cuenca N, Lopez S, Howes K, Kolb H. The localization of guanylyl cyclase-activating proteins in the mammalian retina. Invest Ophthalmol Vis Sci. 1998;39:1243–1250. [PubMed] [Google Scholar]
- 8.Pennesi ME, Howes KA, Baehr W, Wu SM. Guanylate cyclase-activating protein (GCAP) 1 rescues cone recovery kinetics in GCAP1/GCAP2 knockout mice. Proc Natl Acad Sci USA. 2003;100:6783–6788. doi: 10.1073/pnas.1130102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makino CL, Peshenko IV, Wen XH, Olshevskaya EV, Barrett R, Dizhoor AM. A role for GCAP2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J Biol Chem. 2008;283:29135–29143. doi: 10.1074/jbc.M804445200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitiratschky VB, Behnen P, Kellner U, Heckenlively JR, Zrenner E, Jagle H, Kohl S, Wissinger B, Koch KW. Mutations in the GUCA1A gene involved in hereditary cone dystrophies impair calcium-mediated regulation of guanylate cyclase. Hum Mutat. 2009;30:E782–E796. doi: 10.1002/humu.21055. [DOI] [PubMed] [Google Scholar]
- 11.Jiang L, Katz BJ, Yang Z, Zhao Y, Faulkner N, Hu J, Baird J, Baehr W, Creel DJ, Zhang K. Autosomal dominant cone dystrophy caused by a novel mutation in the GCAP1 gene (GUCA1A) Mol Vis. 2005;11:143–151. [PubMed] [Google Scholar]
- 12.Sokal I, Dupps WJ, Grassi MA, Brown J, Jr, Affatigato LM, Roychowdhury N, Yang L, Filipek S, Palczewski K, Stone EM, Baehr W. A novel GCAP1 missense mutation (L151F) in a large family with autosomal dominant cone-rod dystrophy (adCORD) Invest Ophthalmol Vis Sci. 2005;46:1124–1132. doi: 10.1167/iovs.04-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stephen R, Bereta G, Golczak M, Palczewski K, Sousa MC. Stabilizing function for myristoyl group revealed by the crystal structure of a neuronal calcium sensor, guanylate cyclase-activating protein 1. Structure. 2007;15:1392–1402. doi: 10.1016/j.str.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang JY, Koch KW. Calcium- and myristoyl-dependent properties of guanylate cyclase-activating protein-1 and protein-2. Biochemistry. 2002;41:13021–13028. doi: 10.1021/bi026618y. [DOI] [PubMed] [Google Scholar]
- 15.Hwang JY, Koch KW. The myristoylation of the neuronal Ca2+-sensors guanylate cyclase-activating protein 1 and 2. Biochim Biophys Acta. 2002;1600:111–117. doi: 10.1016/s1570-9639(02)00451-x. [DOI] [PubMed] [Google Scholar]
- 16.Andre I, Linse S. Measurement of Ca2+-binding constants of proteins and presentation of the CaLigator software. Anal Biochem. 2002;305:195–205. doi: 10.1006/abio.2002.5661. [DOI] [PubMed] [Google Scholar]
- 17.Linse S. Calcium binding to proteins studied via competition with chromophoric chelators. In: Vogel HJ, editor. Calcium-binding protein protocols: volume 2: methods and techniques. Totowa: Humana Press; 2002. pp. 15–24. [DOI] [PubMed] [Google Scholar]
- 18.Linse S, Johansson C, Brodin P, Grundstrom T, Drakenberg T, Forsen S. Electrostatic contributions to the binding of Ca2+ in calbindin D9k. Biochemistry. 1991;30:154–162. doi: 10.1021/bi00215a023. [DOI] [PubMed] [Google Scholar]
- 19.Helten A, Saftel W, Koch KW. Expression level and activity profile of membrane bound guanylate cyclase type 2 in rod outer segments. J Neurochem. 2007;103:1439–1446. doi: 10.1111/j.1471-4159.2007.04923.x. [DOI] [PubMed] [Google Scholar]
- 20.Helten A, Koch KW. Guanylate cyclase-based signaling in photoreceptors and retina. In: Fliesler SJ, Kisselev OG, editors. Signal transduction in the retina. Boca Raton: Taylor & Francis Group, LLC; 2008. pp. 121–164. [Google Scholar]
- 21.Linse S, Brodin P, Johansson C, Thulin E, Grundstrom T, Forsen S. The role of protein surface charges in ion binding. Nature. 1988;335:651–652. doi: 10.1038/335651a0. [DOI] [PubMed] [Google Scholar]
- 22.Dell’Orco D, Xue WF, Thulin E, Linse S. Electrostatic contributions to the kinetics and thermodynamics of protein assembly. Biophys J. 2005;88:1991–2002. doi: 10.1529/biophysj.104.049189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes RE, Brzovic PS, Dizhoor AM, Klevit RE, Hurley JB. Ca2+-dependent conformational changes in bovine GCAP-2. Protein Sci. 1998;7:2675–2680. doi: 10.1002/pro.5560071222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim S, Peshenko I, Dizhoor A, Ames JB. Effects of Ca2+, Mg2+, and myristoylation on guanylyl cyclase activating protein 1 structure and stability. Biochemistry. 2009;48:850–862. doi: 10.1021/bi801897p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber G. Energetics of ligand binding to proteins. Adv Protein Chem. 1975;29:1–83. doi: 10.1016/S0065-3233(08)60410-6. [DOI] [PubMed] [Google Scholar]
- 26.Dizhoor AM, Boikov SG, Olshevskaya EV. Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1. Possible role in causing human autosomal dominant cone degeneration. J Biol Chem. 1998;273:17311–17314. doi: 10.1074/jbc.273.28.17311. [DOI] [PubMed] [Google Scholar]
- 27.Nishiguchi KM, Sokal I, Yang L, Roychowdhury N, Palczewski K, Berson EL, Dryja TP, Baehr W. A novel mutation (I143NT) in guanylate cyclase-activating protein 1 (GCAP1) associated with autosomal dominant cone degeneration. Invest Ophthalmol Vis Sci. 2004;45:3863–3870. doi: 10.1167/iovs.04-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olshevskaya EV, Calvert PD, Woodruff ML, Savchenko AB, Makino CL, Ho YS, Fain GL, Dizhoor AM. The Y99C mutation in guanylyl cyclase-activating protein 1 increases intracellular Ca2+ and causes photoreceptor degeneration in transgenic mice. J Neurosci. 2004;24:6078–6085. doi: 10.1523/JNEUROSCI.0963-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Payne AM, Downes SM, Bessant DA, Taylor R, Holder GE, Warren MJ, Bird AC, Bhattacharya SS. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7:273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]
- 30.Sokal I, Li N, Surgucheva I, Warren MJ, Payne AM, Bhattacharya SS, Baehr W, Palczewski K. GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol Cell. 1998;2:129–133. doi: 10.1016/S1097-2765(00)80121-5. [DOI] [PubMed] [Google Scholar]
- 31.Wilkie SE, Li Y, Deery EC, Newbold RJ, Garibaldi D, Bateman JB, Zhang H, Lin W, Zack DJ, Bhattacharya SS, Warren MJ, Hunt DM, Zhang K. Identification and functional consequences of a new mutation (E155G) in the gene for GCAP1 that causes autosomal dominant cone dystrophy. Am J Hum Genet. 2001;69:471–480. doi: 10.1086/323265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodruff ML, Olshevskaya EV, Savchenko AB, Peshenko IV, Barrett R, Bush RA, Sieving PA, Fain GL, Dizhoor AM. Constitutive excitation by Gly90Asp rhodopsin rescues rods from degeneration caused by elevated production of cGMP in the dark. J Neurosci. 2007;27:8805–8815. doi: 10.1523/JNEUROSCI.2751-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sokal I, Otto-Bruc AE, Surgucheva I, Verlinde CL, Wang CK, Baehr W, Palczewski K. Conformational changes in guanylyl cyclase-activating protein 1 (GCAP1) and its tryptophan mutants as a function of calcium concentration. J Biol Chem. 1999;274:19829–19837. doi: 10.1074/jbc.274.28.19829. [DOI] [PubMed] [Google Scholar]
- 34.Ames JB, Dizhoor AM, Ikura M, Palczewski K, Stryer L. Three-dimensional structure of guanylyl cyclase activating protein-2, a calcium-sensitive modulator of photoreceptor guanylyl cyclases. J Biol Chem. 1999;274:19329–19337. doi: 10.1074/jbc.274.27.19329. [DOI] [PubMed] [Google Scholar]
- 35.Stephen R, Palczewski K, Sousa MC. The crystal structure of GCAP3 suggests molecular mechanism of GCAP-linked cone dystrophies. J Mol Biol. 2006;359:266–275. doi: 10.1016/j.jmb.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otto-Bruc A, Buczylko J, Surgucheva I, Subbaraya I, Rudnicka-Nawrot M, Crabb JW, Arendt A, Hargrave PA, Baehr W, Palczewski K. Functional reconstitution of photoreceptor guanylate cyclase with native and mutant forms of guanylate cyclase-activating protein 1. Biochemistry. 1997;36:4295–4302. doi: 10.1021/bi963000d. [DOI] [PubMed] [Google Scholar]
- 37.Linse S, Helmersson A, Forsen S. Calcium binding to calmodulin and its globular domains. J Biol Chem. 1991;266:8050–8054. [PubMed] [Google Scholar]
- 38.Linse S, Thulin E, Sellers P. Disulfide bonds in homo- and heterodimers of EF-hand subdomains of calbindin D9k: stability, calcium binding, and NMR studies. Protein Sci. 1993;2:985–1000. doi: 10.1002/pro.5560020612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peshenko IV, Dizhoor AM. Ca2+ and Mg2+ binding properties of GCAP-1. Evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J Biol Chem. 2006;281:23830–23841. doi: 10.1074/jbc.M600257200. [DOI] [PubMed] [Google Scholar]
- 40.Koch KW, Stryer L. Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature. 1988;334:64–66. doi: 10.1038/334064a0. [DOI] [PubMed] [Google Scholar]
- 41.Koch KW. Target recognition of guanylate cyclase by guanylate cyclase-activating proteins. Adv Exp Med Biol. 2002;514:349–360. doi: 10.1007/978-1-4615-0121-3_21. [DOI] [PubMed] [Google Scholar]
- 42.Vetter SW, Leclerc E. Novel aspects of calmodulin target recognition and activation. Eur J Biochem. 2003;270:404–414. doi: 10.1046/j.1432-1033.2003.03414.x. [DOI] [PubMed] [Google Scholar]
- 43.Hwang JY, Lange C, Helten A, Hoppner-Heitmann D, Duda T, Sharma RK, Koch KW. Regulatory modes of rod outer segment membrane guanylate cyclase differ in catalytic efficiency and Ca(2+)-sensitivity. Eur J Biochem. 2003;270:3814–3821. doi: 10.1046/j.1432-1033.2003.03770.x. [DOI] [PubMed] [Google Scholar]