

Abstract
Alzheimer's disease (AD) is the most common and irreversible neurodegenerative disorder worldwide. While the precise mechanism behind this rapid progression and multifaceted disease remains unknown, the numerous drawbacks of the available therapies are prevalent, necessitating effective alternative treatment methods. In view of the rising demand for effective AD treatment, numerous reports have shown that tetrahydroisoquinoline (THIQ) is a valuable scaffold in various clinical medicinal molecules and has a promising potential as a therapeutic agent in treating AD due to its significant neuroprotective, anti-inflammatory, and antioxidative properties via several mechanisms that target the altered signaling pathways. Therefore, this review comprehensively outlines the potential application of THIQ derivatives in AD treatment and the challenges in imparting the action of these prospective therapeutic agents. The review emphasizes a number of THIQ derivatives, including Dauricine, jatrorrhizine, 1MeTIQ, and THICAPA, that have been incorporated in AD studies in recent years. Subsequently, a dedicated section of the review briefly discusses the emerging potential benefits of multi-target therapeutics, which lie in their ability to be integrated with alternative therapeutics. Eventually, this review elaborates on the rising challenges and future recommendations for the development of therapeutic drug agents to treat AD effectively. In essence, the valuable research insights of THIQ derivatives presented in this comprehensive review would serve as an integral reference for future studies to develop potent therapeutic drugs for AD research.
Keywords: Alzheimer's disease, Multi-target therapeutics approach, Neuroprotection, Tetrahydroisoquinoline, Therapeutic agent
1. Introduction
Alzheimer's disease (AD) is a type of irreversible neurological disorder that is mainly identified among elderly people globally. Statistically, a staggering 15 million people suffer from AD worldwide, making it the most prevalent neurological condition in the world [1]. The steady increase in AD cases is partly attributed to the aging global population. Dementia, encompassing all causes, has also emerged as the fifth leading cause of global mortality in 2016, accounting for 4.4 % of all deaths [2]. Since then, the number of deaths attributable to dementia has doubled, exhibiting a continuous upward trajectory, partially driven by the consistent population growth and demographic shifts towards an aging population. Despite that this chronic neurological disorder was discovered over a century ago, no permanent cure has been found. The main reason behind the failures in clinical trials is related to the highly complex mechanism of the brain and the limitation among the scientific community to understand the precise mechanism of AD fully.
AD progresses insidiously over time, characterized by a progressive decline in memory, deteriorating cognitive function, and the change in the pathophysiological brain that precedes the emergence of overt clinical manifestations. The multifaceted pathophysiology, including the development of Neurofibrillary Tangles (NFTs) and Amyloid-β (Aβ) plaques, are the two known pathological hallmarks of AD [3]. Researchers have revealed that aberrant Aβ and tau proteins may play a significant role in AD pathogenesis. Generally, it is understood that the amyloid hypothesis of AD describes the neurodegeneration and memory deficits in AD patients due to the buildup of the Aβ plaque in the temporal lobe [1]. Consequently, these aggregates trigger numerous effects, including mitochondrial dysfunction, neuroinflammation, oxidative stress, and calcium imbalances, resulting in the loss of white matter in the brain and neurons.
It is no surprise that Aβ is a normal by-product commonly found in the brain and plasma of all individuals. Nevertheless, the imbalance production of Aβ becomes neurotoxic in several ways, including the formation of pores, enabling ions to leak out, disrupting the cellular calcium balance, and reducing the mitochondria membrane potential [4]. Aβ40 is the dominant Aβ isoform, whereas Aβ42 is the minor and more aggressive isoform aggregating rapidly into amyloid fibrils. These two isoforms of Aβ are heavily enriched in senile plaques [5].
AD can be categorized into two types: familial and sporadic AD. The former, also known as early AD onset, is induced by genetic factors, such as mutations in the genetic coding for Amyloid Precursor Protein (APP), Presenilin 1 (PSEN1), and Presenilin 2 (PSEN2) [6]. Meanwhile, sporadic AD refers to the late AD onset and is affected by several external factors, including dietary intake, stress, and environment. The APP protein is a type-1 integral transmembrane glycoprotein located on chromosome 21 and plays an essential role in AD pathogenesis [7]. Two distinct proteolytic pathways occur in APP: amyloidogenic and non-amyloidogenic pathways. As the APP accumulates, amyloidogenesis takes place at acidic neuronal compartments, such as lysosomes and late endosomes [8]. Note that Aβ peptides are only produced via the amyloidogenic pathway.
In contrast, tauopathy is a neurodegenerative disorder characterized by the accumulation of tau proteins in the brain. In tauopathy, hyperphosphorylated tau protein leads to misfolding, aggregation, and the build-up of NFTs and neuropil threads. These pathological tau aggregates are believed to disrupt normal neuronal functioning, eventually resulting in neuronal death. Since the development of tau pathology is a complex process, researchers are actively exploring this area to elucidate the underlying mechanisms and develop potential treatments to prevent or even reverse this process.
Intense efforts to treat the symptoms and severity of AD are still ongoing. However, clinical trials have shown inconsistent therapeutic effectiveness in inhibiting Aβ and NFTs due to the complex molecular mechanisms of AD [9]. So far, the United States Food and Drug Administration (FDA) has only approved seven AD-related drugs for prescription to relieve the symptoms in AD patients. While AD patients are prescribed a combination of drugs to treat the complications, such medications are often accompanied by numerous side effects. Thus, the research focus is shifting toward the development of alternative therapeutic agents.
Tetrahydroisoquinoline (THIQ) is an organic compound that exists in the form of numerous naturally occurring alkaloids, as shown in Fig. 1. Classified as a secondary amine, this compound is one of the privileged heterocyclic classes obtained from isoquinoline via the hydrogenation process. In nature, THIQ derivatives are found in the brains of primates and rodents, as well as in various foods, such as bananas, cocoa, and milk [10]. It is also naturally present in the human brain in sub-nanomolar concentrations and can easily pass through the blood-brain barrier (BBB) [11].
Fig. 1.

Chemical structure of a naturally occurring THIQ.
This drug class was first reviewed in early 1981 for its pharmacology, chemistry, and biochemistry properties, highlighting its significant effect on the central nervous system [12]. Eventually, THIQ derivatives have been researched for many decades, given that they exhibit a wide range of therapeutic properties, including bronchodilator, anti-convulsant, anti-viral, anti-bacterial, antibiotic, anti-tumor, anti-inflammatory, anti-coagulant, and action on the central nervous system [13,14]. The skeleton structure of the THIQ class was also detected in several clinically effective drugs, including diclofensine, nomifensine, and tubocurarine [15]. This led to the varying synthesis pathways of THIQ analogs for practical use in multiple therapeutic areas.
Over the past decades, THIQ derivatives have been reported for their significant reduction of severity in several AD models. Numerous articles have also been recently published emphasizing the synthesis and development of THIQ derivatives for other disease microenvironments. A number of THIQ derivative patents have been reviewed between 2010 and 2015 for clinical therapeutics of specific diseases, including leishmaniasis, malaria, tuberculosis, Herpes Simplex Virus (HSV), and Human Immunodeficiency Virus (HIV) infections [13]. The unique mechanism of action of these derivatives contributes to their potent biological activities, which can be established as a new class of drugs to treat various diseases.
As far as the author's knowledge is concerned, there is no published review that explores the recent progress in AD research, explicitly using THIQ derivatives. Thus, this paper elucidates a comprehensive overview of the current progress of THIQ derivatives as potential therapeutic agents in AD studies and the challenges toward effective clinical practice. This review provides detailed insights on four key THIQ derivatives, namely, Dau, jatrorrhizine, 1MeTIQ, and THICAPA. This review examines the existing body of research on well-studied THIQ derivatives with potential applications in AD therapy. The alternate potential of multi-target therapeutics will also be briefly discussed. The review also sheds light on the mounting challenges and future recommendations for the smooth development and implementation of THIQ-derived drugs. To the best of the author's knowledge, the present review is the first attempt to compile these derivatives into a holistic perspective on potential AD therapeutics.
2. Tetrahydroisoquinoline derivatives
2.1. Dauricine
Dauricine (Dau) is a dibenzyl THIQ alkaloid extracted from the root of a type of herbal flowering plant known as Menispermum dauricum [16]. The root of M. dauricum, a plant with a rich historical use in traditional Chinese medicine for rheumatic disease treatment, holds great potential for the discovery of new therapeutic agents. The IUPAC name of Dau is 4-[[(1R)-6,7-dimethoxy-2-methyl-3,4-dihydro-1H-isoquinolin-1-yl]methyl]-2-[4-[[(1R)-6,7-dimethoxy-2-methyl-3,4-dihydro-1H-isoquinolin-1-yl]methyl]phenoxy]phenol (molecular formula: C38H44N2O6). It can be chemically classified as an aromatic ether, phenol, and bisbenzyl isoquinoline alkaloid. Dau has been widely used in neurodegenerative research owing to its potent medicinal properties. The following sections describe the therapeutic properties of Dau.
2.1.1. AD treatment
In one study, Liu et al. [17] demonstrated the modulation of Dau in multiple molecular pathways through an in vitro AD disease model. Although the study reported that Dau has no significant cytotoxicity in N2a/APP cells, a substantial loss of viability was reported in Dau-treated N2a/WT cells at higher dosages than 10 μM. This shows that N2a/APP cells have a higher Dau tolerance than N2a/WT cells. The same study also reported that Dau downregulated the amyloidogenic processing pathway in N2a/APP cells. The concentration of Aβ42 in N2a/APP cells was reduced three-fold compared to that in N2a/WT cells. However, the concentration of Aβ40 showed an insignificant reduction, which might be due to the more inclined specific-targeted effect of Dau on minor isoforms of Aβ peptides. In addition, Dau was reported to upregulate sAPPα and downregulate the expression of phosphorylated APP, total APP, Beta-site APP Cleaving Enzyme 1 (BACE1), Presenilin-1 (PSEN1), and sAPPβ. Given that sAPPα essentially prevents the production of Aβ peptides by inhibiting the amyloidogenic pathway, Dau can be an excellent anti-AD agent to induce the upregulation of sAPPα, thus suppressing the production of Aβ peptides.
Another study reported the ability of Dau to inhibit tau pathology in HEK293/Tau and N2a/APP cells [17]. According to the study, Dau significantly reduced the mean phosphorylation levels of glycogen synthase kinase (GSK)-3α and -3β in N2a/APP cells. This finding aligns with a previous study, which revealed Dau modulation in inhibiting tau hyperphosphorylation and bradykinin-induced alteration of calcium homeostasis in N2a cells [18].
2.1.2. Antioxidant properties
Dau exhibits strong antioxidant properties by shielding N2a/APP cells from oxidative stress [17]. The report found that Dau altered the expressions of six protein categories, including metabolite-signaling proteins, oxidative stress-associated proteins, mitochondrial respiration, cytoskeleton-associated proteins, molecular chaperones, endoplasmic reticulum (ER), and stress-associated proteins [17]. The mitochondria are more vulnerable to oxidative stress as it is the main organelle that produces Reactive Oxygen Species (ROS). The oxidative damage immediately leads to direct neuronal apoptosis [19]. The Aβ peptides mediate the Cu2+ toxicity in Aβ-bounded cytochrome C oxidase inhibition, resulting in the hyperphosphorylation of neurons and aggregation of tau proteins [20]. The Cu2+ generates more toxic hydroxyl radicals by directly participating in the Fenton reaction. The study also reported that Dau modified 85 proteins related to cytoskeleton, oxidative stress, mitochondrial function, and unfolded protein response [17]. It was postulated that the antioxidative properties of Dau might have inhibited Ca2+ influx into the mitochondria. However, this study did not further investigate Ca2+ homeostasis in N2a/APP cells. Thus, future research should emphasize the ambiguous role of calcium homeostasis and Dau on pathogenesis related to oxidative stress.
Dau was also reported to reduce worm paralysis in Caenorhabditis elegans strain GMC101 significantly [20]. The high survival rate of worms treated with Dau recorded a lower ROS formation. Since the GMC101 strain progressively expressed Aβ42 under temperature-dependent growth conditions, the treatment process using Dau significantly reduced toxic oligomers. The findings suggest that Dau may promote a potent remedy against oxidative stress.
Nrf2 plays a primary redox-regulated gene and stimulates oxidative stress as it targets Antioxidant Responsive Elements (ARE) in the nucleus, which promotes antioxidant gene expression [21]. Nrf2 has been reported to have dual mechanisms in Parkinson's disease, including antioxidant and anti-inflammatory mechanisms [22]. Concurrently, B-cell lymphoma 2 (Bcl-2) acts directly downstream of Nrf2, which increases the concentration of protein and genes in Bcl-2 [23]. Under normal conditions, the Nrf2/ARE pathway remains inactive. However, the binding and conjugated protein complex formation of Nrf2 and Keap1 will be inhibited under stress-induced environments [24]. This will cause the downregulation of proteasomal degradation and ubiquitin conjugation. In addition, Dau treatment on APPsw was found to regulate the Nrf2/Keap1 pathway [23]. The upregulation of this pathway has been reported for its protective properties against oxidative damage by promoting the transfer and expression of Nrf2 [25]. However, the Nrf2 concentration in the nucleus was inhibited at 1 mΜ Dau compared to a lower concentration of 0.1 mΜ [23]. This suggests that the action of Dau on Nrf2 is concentration-dependent, with higher effectiveness at lower concentrations.
2.1.3. Anti-apoptotic effect
Previously, Wang et al. [23] demonstrated a strong anti-apoptotic effect of Dau when SH-SY5Y cells that overexpress APPsw protein, a Swedish mutant form of the human Aβ precursor, were treated with Cu2+. Another study focusing on Dau also reported that rats injected with Aβ oligomers promoted a high concentration of Cu2+, leading to memory and spatial learning deterioration [26]. Besides, Dau was found to downregulate Bcl-2, which participates in the progression of apoptosis [23]. The Bcl-2 in mitochondria mediates the release of cytochrome C and Bcl-2-related X-protein (Bax), which weakens caspase activation [27]. This suggests that Dau could also exhibit a neuroprotective role by downregulating Bax/Bcl-2 and activating caspase-3 protein from oxidative-stress-induced apoptosis. A previous study also reported that Dau protected the rats from middle cerebral artery occlusion by downregulating Bax/Bcl-2 [28]. The finding shows that the nuclear transport of Dau increases Bcl-2 expression, which is directly involved in the apoptosis of β-cells.
2.1.4. Neuroprotective effect
A study by Li et al. [29] revealed the neuroprotective effect of Dau in cortical neuron culture with hypoglycemia and hypoxia. The brain has been widely reported to have 10,000 times higher extracellular Ca2+ concentration than that of intracellular concentration. The cell membrane plays an essential role in balancing the Ca2+ gradient between the two compartments. Any excessive influx of Ca2+ will lead to cerebral ischemia due to the sustained perturbation in the membrane withstanding Ca2+ that enters through agonist-operated and voltage-dependent channels. The disrupted Ca2+ homeostasis may typically indicate the final mechanism in neurodegenerative diseases.
Past studies have reported that Dau inhibited the voltage-dependent L-type Ca2+ currents (VDDCs) [30]. In addition, VDDC inhibitors can counterbalance the influx of Ca2+ as a result of glucose and oxygen deprivation in cortical neurons [31]. However, the effect of Dau was shorter and weaker than commonly used calcium antagonists [30]. Besides, Dau was found to significantly increase the viability of hypoxia and hypoglycemia neurons [29], owing to the marked reduction in intracellular Ca2+ accumulation. Thus, it was suggested that Dau inhibited Ca2+ influx from the extracellular matrix and efflux to the ER. Apart from that, Dau was shown to suppress the accumulation of intracellular Ca2+ induced by glutamate, an N-methyl-d-aspartic acid receptor (NMDAR) agonist. The suppression of intracellular Ca2+ accumulation might be due to the antioxidative effect of Dau in ameliorating oxidative damage and energy metabolism in mitochondria. Consequently, the deposition of Ca2+ and mitochondrial sequestration were reduced, suppressing intracellular Ca2+ accumulation.
Furthermore, Peng et al. [32] found that Dau provides significant neuroprotection to the brain from secondary brain injury. The administration of Dau to intracerebral hemorrhage (ICH)-induced mice has been shown to reverse the downregulation of glutathione peroxidase 4 (GPX4). The GPX4 is an antioxidant enzyme that plays a vital role in ferroptosis. The enzyme is involved in lipid peroxidation, reduction of phospholipid hydroxide, and over-activation of lipoxygenase, which is often correlated with ICH pathogenesis [33,34]. Dau administration further offers the neuroprotective ability in SH-SY5Y cells induced with RSL3, a ferroptosis inducer. The intracellular oxidative damage and iron accumulation were significantly reduced in vitro [32]. This suggests Dau can inhibit the apoptosis of neurons in neurodegenerative disease progression [35]. Since secondary brain injury is widely seen in late-onset AD patients, the resistance against ferroptosis implies that Dau can be an effective protective agent for the brain.
2.1.5. Neuroinflammation inhibition
Neuroinflammation is another primary mechanism that is widely observed in AD patients. Under normal physiological conditions, immune cells serve as a bridge between peripheral cells and neurons [36]. However, when the aggregated Aβ plaque disrupts the physiological balance of neurons, the astrocytes begin to attack near the plaques, causing the neurons to inflame. To combat the inflammation, neutrophils are recruited and surround the neurons, causing them to release chemotaxis agents and proinflammatory cytokines [37]. In response to the cytokines released, more neutrophils are recruited, which worsens the condition and ultimately triggers ischemic stroke [38].
Pu et al. [39] reported that Dau reduced neutrophil recruitment in mice models, suppressing neuroinflammation. Dau administration to the ischemia-reperfusion (I/R) injury mice model successfully reduced the neutrophil infiltration to the brain [39]. As a result, the chemotaxis factors were downregulated, and the brain infarct size was significantly reduced. However, the reduced infiltration was not due to the change in the permeability of the BBB, suggesting Dau has no effect in recovering BBB but only directly inhibits neutrophil infiltration. This response is possibly due to Dau's downregulation of the chemokine Serpine1 expression at 24 h post-ischemia reperfusion in the ischemic penumbra.
Additionally, Dau was found to inhibit Serpine1 expression, which reversed the infiltration of neutrophils from the brain in the I/R mice model. Serpine1 is a critical chemotaxis factor in neutrophil infiltration in the brain and also a primary inhibitor of tissue-type plasminogen activator that maintains the balance of controlled blood clot degradation and downregulation of fibrinolysis [39]. However, Serpine1 is not merely classified as a senescence biomarker. Serpine1 is essential for sufficient induction of the p53 gene that leads to replicative senescence downstream [40]. Serpine1 might not directly induce AD, but the chemokine can be a potential therapeutic target in combating inflammatory cell migrations in the brain. While the findings show that Dau exhibits anti-inflammatory properties, the precise mechanism is yet to be explored.
2.1.6. Mitigating cognitive decline
A recent study by Xue et al. [41] reported the potential of Dau in mitigating cognitive decline in mice with d-galactose/AlCl3-induced AD, implicating the Ca2+/Calmodulin (CaM) pathway as a potential mechanism of action. CaM is the most important intracellular calcium effector compared to several others [42]. It regulates the activity of APP and BACE1, which are responsible for producing the Aβ plaque, and Ca2+/CAM-dependent kinase II (CaM-KII), which regulates NFT production. Dau treatment on d-galactose/AlCl3 injected mice has shown reduced phosphorylation of tau and CaMKII, as well as the expression of Aβ42, APP, and BACE1 in the cortex and hippocampus. Moreover, the cognitive behavior and memory deficit significantly improved after being treated with 10 mg/kg of Dau. Overall, these findings indicate that Dau exerts potent anti-inflammatory and neuroprotective effects by alleviating both amyloidogenic and tau pathology. Despite a substantial body of research and promising pre-clinical findings, Dau remains in the late pre-clinical phase, highlighting the need for further studies to conclusively establish its druggability properties.
2.2. Jatrorrhizine
Jatrorrhizine is a type of protoberberine derived from plants. It has been widely recognized as a key bioactive moiety in various plant families, including Tinospora sagittate [43], Corydalis yanhusuo [44], Mahonia bealei [45], Coptis Chinensis Franch [46], and Berberis vernae [47]. The IUPAC name of jatrorrhizine is 2,9,10-trimethoxy-5,6-dihydroisoquinolino[2,1-b]isoquinolin-7-ium-3-ol (molecular formula: C20H20NO4) [48]. The diverse therapeutic properties of jatrorrhizine are presented in the following sections.
2.2.1. Beneficial effect on AD
A pronounced reduction in Aβ plaques that formed in the hippocampus and cortex of APP/PSEN1 transgenic mice was reported after being treated with jatrorrhizine [49]. The microscopic observation in the whole brain region of the jatrorrhizine-administered transgenic mice revealed a significant decrease in Aβ plaques compared to the control group. Clear co-localization of active astrocytes was also observed in the brain with a reduced Glial Fibrillary Acidic Protein (GFAP) and expression of diffuse plaques in the hippocampus and cortex. However, the beneficial mechanism of jatrorrhizine in AD is still debatable.
Monoamine oxidase-A (MAO-A) is a mitochondrial membrane-bounded flavoenzyme, which is a vital component in oxidative deamination in the human brain. An increased MAO-A is commonly detected in rodent models with neurodegenerative diseases, stress, and depression. Based on a study by Zhang et al. [50], it was shown that jatrorrhizine recorded the highest inhibition rate of MAO-A ligand (IC50 = 57.73 ± 5.26 μM) compared to ethyl acetate extract. Another study by Kong et al. [51] demonstrated that jatrorrhizine extracted using methanol inhibited MAO-A and MAO-B non-competitively in rat brain mitochondria (IC50 = 4 and 62 mM, respectively). These findings are consistent with the MAO inhibitory effect of N-MeTIQ [52], suggesting that jatrorrhizine can be a suitable MAO inhibitor to alleviate depression-like symptoms in AD patients.
2.2.2. ROS scavenging effect
In general, cells with a higher production of oxidative species exhibit a higher concentration of lactate dehydrogenase (LDH) and malondialdehyde (MDA), which results in cell death [53]. When cell injury occurs, the LDH and MDA permeate through the cells due to the altered cell permeability, which is positively inclined towards cell damage. As the release of LDH and MDA elevates the Mitochondrial Membrane Potential (MMP) concentration in the cytoplasm, nuclear condensation takes place due to the triggered production of cytochrome C [46]. This cascade of the metabolic pathway leads to the production of secondary ROS. Therefore, jatrorrhizine's established antioxidant properties can be potentially utilized to protect cells from oxidative stress.
A study by Luo et al. [53] demonstrated the ROS scavenging effect of jatrorrhizine derived from Coptidis rhizome in rat pheochromocytoma neuronal (PC12) cell lines. When the PC12 cell lines were treated with 200 μM of hydrogen peroxide (H2O2) and 10 μM of jatrorrhizine for 24 h, the cells were unharmed and remained alive. Furthermore, the LDH and MDA levels significantly dropped, while the cytochrome C concentration was elevated compared to cells treated with higher concentrations of H2O2. The expression of HO-1 and superoxide dismutase (SOD) also increased to counteract the overproduction of ROS.
2.2.3. Protection and Maintenance
Jatrorrhizine provides a protective medium for mitochondria to synthesize adenosine triphosphate (ATP) by lowering the MMP induced by H2O2 in the cells. This is essential for preserving healthy cell viability since mitochondria act as a significant powerhouse to maintain the normal physiological function of the cell[19]. Besides, the concentration of caspase-3 was reduced significantly in cells treated with jatrorrhizine. Since caspase-3 is highly present in the cellular apoptosis cascade, the final execution will lead to programmed cell death [54]. In this scenario, jatrorrhizine can regulate the final stage of programmed cell death by acting as an antagonist. This infers that jatrorrhizine exhibits a good antioxidant effect against ROS that are enzymatically produced in intra- and extra-cellular regions.
Moreover, jatrorrhizine also exhibits a protective effect against nerve injury. For instance, Duan and Chen [55] reported that jatrorrhizine alleviated nerve injury induced by Aβ 25–35 in SH-SY5Y cells by mediating through the miR-223-3p/HDAC4 axis. It was reported that histone deacetylase 4 (HDAC4) regulates the survival of neurons by translocating from the cytoplasm to the nucleus. In contrast, jatrorrhizine has been shown to downregulate the overexpression of HDAC4 in Aβ 25–35 treated SH-SY5Y cells. Simultaneously, jatrorrhizine upregulated the expression of miR-223-3p, which was found to be under-expressed in AD cell models. Thus, both of these events alleviated oxidative stress and apoptosis and increased the proliferation rate of Aβ 25–35 treated SH-SY5Y cells.
2.2.4. Regulation of gut microbiota
The gut microbiota plays a vital role in preventing the production of inflammatory cytokines. Any imbalances in the gut microbiota will correlate with a spike in blood cytokines profile, which may lead to brain inflammation, as frequently observed in AD cases [56,57]. Previous studies have also reported that an imbalance of gut microbiota enhances the leakiness of the gut [58], which directly results in neuronal dysfunction [59] and apoptosis [60]. Previously, Wang et al. [49] reported the first-ever study of jatrorrhizine modulation of gut microbiota in a transgenic AD mice model. The gut microbiota balance and memory deficit in the APP/PSEN1 mouse model were recovered following the administration of jatrorrhizine. The study also employed the Morris water maze probe trials and demonstrated that jatrorrhizine improved the spatial memory and learning abilities of APP/PSEN1 AD mice. Furthermore, the exploratory ability of the transgenic mice was significantly enhanced after oral administration of jatrorrhizine for six months. The findings suggest that the anti-oxidative effects of jatrorrhizine might play an essential role in reversing behavioral and memory defects in AD.
The Operational Taxonomic Unit (OTU) of gut microbiota is essential in maintaining the host's health. Various studies have reported that gut microbiota in the intestinal tract, which includes Bifidobacterium and Lactobacillus, produce short-chain fatty acids (SCFAs) that are beneficial for human health. Since the lack of SCFA is highly related to the progression of neurodegenerative disease, improving the alpha diversity of gut microbiota will enhance its abundance [61]. A higher dose of jatrorrhizine results in a higher OTU number in APP/PSEN1 transgenic mice compared to donepezil treatment, which was used as a positive control to evaluate the anti-aging effects on intestinal microbiota. Although donepezil was found to regulate certain bacterial groups, such as Firmicutes, Bacteroidetes, and Tenericutes, the outcome is often accompanied by gastrointestinal side effects, such as vomiting and nausea.
In contrast, jatrorrhizine offers a similar bacterial abundance at recommended doses with potentially fewer side effects [62]. The result suggests that the effectiveness of jatrorrhizine in retaining gut microbiota balance is superior to that of donepezil, an FDA-approved cholinesterase inhibitor for AD. The effectivity of jatrorrhizine as a potent cholinesterase inhibitor is also in line with a study by Jiang et al. [56]. The study reported that jatrorrhizine attached with amino groups at the 3-position exhibited a high inhibition activity for cholinesterase (IC50 = 0.301 μM) compared to donepezil (IC50 = 0.498 μM). This shows that the structural modification of jatrorrhizine effectively enhanced the modulating potency.
Moreover, jatrorrhizine tremendously improved the gut microbiota compared to APP/PSEN1 mice and C57BL/6 mice. Initially, the fecal analysis recorded a very scattered composition in APP/PSEN1 mice and a narrow composition in C57BL/6 mice, which eventually improved the microbiota composition. The jatrorrhizine treatment enriched the beneficial microbiota, including Faecalibaculum, Lactobacillus acidophilus, and Bifidobacterium. Besides, the Firmicutes-to-Bacteroidetes (F/B) ratio decreased in APP/PSEN1 mice after jatrorrhizine administration. The ratio of F/B is a widely used microbiological indicator to measure obesity and has been identified as an independent risk factor for AD progression [63]. Obese individuals were reported to have a higher F/B ratio compared to normal individuals. Hence, the findings from the study showed that jatrorrhizine improved the alpha diversity and reversed the phenotype of mice.
2.2.5. Pharmacokinetics and pharmacodynamics properties
Since different THIQ compounds have different physiochemical properties, jatrorrhizine shows varying in vivo pharmacokinetic properties [64]. For instance, intravenous administration of jatrorrhizine (0.1–3 mg/kg) in rats revealed a biphasic decline in plasma concentration, suggesting a rapid distribution to tissues and potential non-linear pharmacokinetic behavior. This observation was justified by the extensive tissue distribution independent of clearance and half-life but reflected by a dose-dependent increase in the area under the curve (AUC0-∞) [48]. The study on plasma pharmacokinetics also revealed that jatrorrhizine exhibited a greater total exposure (AUC0-∞), higher peak levels (Cmax), and prolonged elimination (T1/2) in insomniac rats, implying potentially increased absorption and bioavailability under pathological conditions [65]. Nevertheless, jatrorrhizine is still in the early pre-clinical phase due to its poor bioavailability and permeability.
Jatrorrhizine interacts with other constituents after administration, which alters its absorptive properties and elimination[48]. The low apparent permeability coefficient of jatrorrhizine (0.23–0.36 × 10−6 cm s−1) in rats after oral administration suggests that P-glycoprotein efflux restricted cellular uptake, which reflects a low bioavailability [66]. The chemical structure of jatrorrhizine is also nearly identical to that of berberine, which exhibits better bioavailability and a safer profile due to its organic acid salt properties and is commonly applied in pharmacological research and clinical usage[64]. Previously, an acute LD50 study in Kunming mice revealed a significantly higher lethal dose of jatrorrhizine (5500 mg/kg) compared to its close relative, berberine (763 mg/kg), underscoring the relatively lower toxicity of jatrorrhizine [67]. Despite its potential application, there are almost no studies on the pharmacokinetics of different jatrorrhizine salts. Thus, it is crucial to study the interaction of jatrorrhizine against different salts to evaluate its physiochemical properties.
2.3. 1MeTIQ
1MeTIQ (IUPAC name: 1-Methyl-1,2,3,4,-tetrahydroisoquinoline; molecular formula: C10H13N) is an endogenous alkaloid found in trace amounts in the mammalian brain, including humans, monkeys, and rodents [68]. It was first identified in the brains of normal rats in 1986 [69], and was subsequently found in foods rich in 2-phenylethylamine, suggesting a potential dietary source and pathway for brain entry [70]. The 1MeTIQ found in the brain exists as a mixture of (R)- and (S)-enantiomers [71]. It is enzymatically synthesized from 2-phenylethylamine and pyruvate by the 1MeTIQ-synthesizing enzyme, a membrane-bound protein localized in the mitochondrial synaptosomal fraction. It is permeable to the BBB and has been found to elicit considerable therapeutic response in the brain.1MeTIQ has shown several promising medicinal benefits, as described in the following sections.
2.3.1. Anti-AD agent
In one study, Kuszczyk et al. [72] demonstrated the neuroprotective effect of 1MeTIQ. The study used primary hippocampal neurons to determine the impact of 1MeTIQ against ROS production and the formation of Aβ peptides. The in vivo study with transgenic AD mouse models validated the synaptotoxic effects of Aβ. They also demonstrated that 1MeTIQ exhibits a low-affinity NMDAR antagonist. The exposure of neurons to Aβ peptides has been linked to a loss of surface NMDAR expression, leading to diminished NMDAR current and impaired activation of the cAMP response element-binding protein [72]. Previously, a toxicity evaluation also showed that 1MeTIQ exhibited no cytotoxicity against primary mammalian neurons even when the cultured cell line was treated with a maximal cut-off concentration of 500 μM [73]. The cytotoxicity findings were further corroborated by the 1MeTIQ's ability to prevent glutamate-induced excitotoxicity and reduce extracellular calcium influx, as demonstrated in studies using 45Ca radioisotope as a tracer.
Excitatory glutamatergic neurotransmission via NMDAR is essential for maintaining neurons’ survival and synaptic plasticity. However, uncontrolled NMDAR activity promotes excitotoxicity and cell mortality, which has been reported in AD. Glutamate-induced excitotoxicity is a key mechanism that leads to chronic neurodegeneration and acute neuronal injuries [74]. In this case, 1MeTIQ is a crucial component in providing neuroprotection by inhibiting the glutamate-induced excitotoxicity mechanism. Previous studies have also demonstrated the neuroprotective and NMDAR antagonist role of 1MeTIQ by inhibiting rotenone-induced injuries [75] and inhibiting morphine tolerances [76].
2.3.2. ROS scavenging properties
The administration of 1MeTIQ on the cerebellar granule cells completely halts the excessive 45Ca2+ influx and NMDAR [73,76], although the mechanism is still under research [76]. This in vitro study highlighted the scavenging properties of 1MeTIQ by inhibiting the Fenton reaction, a method for investigating nonenzymatic generation with hydroxyl radicals. 1MeTIQ was later found to be an uncompetitive NMDAR antagonist that induces tolerance to excitotoxicity. It mimics the same effects as two of the established uncompetitive antagonists of NMDAR, which are MK-801 (0.5 μM) and memantine (5 μM) [77].
In addition, oxidative stress and excitotoxicity occur in a neurodegenerative mechanism caused by complex I inhibitors in the mitochondria of neurons. It is well known that oxidative stress is a critical mechanism in cell apoptosis and aging. On the other hand, excitotoxicity is known for its apoptotic and necrotic properties in complex neurodegenerative pathogenesis. As reported by Michaluk et al. [73], the administration of 1MeTIQ in primary Swiss mouse embryo neocortical tissue showed anti-radical properties, which inhibited glutamate-induced caspase-3 activation. The finding aligns with a study published by Kotake et al. [78], who demonstrated the neurotoxic effect of four dopaminergic neurotoxins in rat mesencephalic neuron cultures. This effect gradually reduced the oxidative stress in the cells.
2.3.3. Synaptic protection
In conjunction with previous evidence, Michaluk et al. [73] proved that 1MeTIQ prevented the loss of Synaptic Density Protein-95 (PSD-95), synaptophysin expression, and NMDAR NR1 subunit. This is the first study to report the inhibition of the NMDAR NR1 subunit by 1MeTIQ, which provides the protective effect of NMDAR antagonists against Aβ [73]. It is also important to note that this is the first-ever study that demonstrated the use of 500 μM 1MeTIQ to inhibit Aβ40-evoked reduction by mimicking the effect of MK-801. Likewise, the study showed the protective effects in synaptic proteins using the established uncompetitive NMDAR antagonist, MK-801, when tested in an in vitro Aβ-induced synaptotoxicity model. This data consistently shows the anti-excitotoxic potential of 1MeTIQ with the previous study, which suggests the inhibition of Aβ production in the synaptic cleft using 1MeTIQ [73]. Consistent with the literature, the results indicated that administering an NMDAR antagonist is a practical synaptoprotective approach against Aβ toxicity [79].
1MeTIQ also showed an inhibitory effect on ROS production in immature hippocampal neurons [72]. The synaptic protein content of Aβ was significantly reduced when the 1MeTIQ-treated dendrites were exposed to exogenous Aβ. However, the surface expression levels of PSD-95 and NR2B subunits of NMDARs were unaltered in the total brain homogenate of APPV717I in AD Tg mice [80]. The PSD-95 and NR2B subunits are vital constituents in developing memory and synaptic plasticity in the brain. Consistent with in vitro observations, the phosphorylated NR2B subunits of NMDAR were reduced, except for the total NR2B expression, which was observed in the hippocampus of hAPP-J20 AD Tg mice. The beneficial impact of 1MeTIQ became more evident at lower concentrations. Michaluk et al. [81] also reported significant inhibition of the MAO-dependent oxidative pathway in Wistar rats by blocking the MAO-A and MAO-B synthesis. Furthermore, administration of 1MeTIQ for 72 h increased the Catechol-O-methyltransferase (COMT)-dependent O-methylation compared to 24 h of administration. The level of dopamine also rose during chronic administration, reflecting the antioxidant and neuroprotective effects of 1MeTIQ.
Overall, 1MeTIQ can be a promising anti-AD agent based on the consistently reported properties of 1MeTIQ, including neuroprotection, synaptoprotection, and ROS inhibition against Aβ toxicity. Thus, the study suggested a new mechanism of 1MeTIQ that provides a neuroprotective effect to induce tolerance of neurons against excitotoxic lesions [78]. However, 1MeTIQ is still in the early phase of pre-clinical study since there are many voids to be filled. A detailed molecular mechanism of 1MetTIQ is still yet to be explored and verified. In this regard, further assessment is needed to unravel the interaction of 1MeTIQ against AD biomarkers and enhance its performance as a potent anti-AD agent.
2.4. THICAPA
Recently, Tan et al. [82] published a new study on a novel THIQ derivative known as THICAPA (IUPAC name: 3-[[(3S)-1,2,3,4-tetrahydroisoquinoline-3 carbonyl]amino] propanoic acid; molecular formula: C13H16N203) [82]. An equivalent term for THICAPA is N-[(3S)-1,2,3,4 tetrahydroisoquinolin-3-ylcarbonyl]-beta-alanine. The chemical structure of THICAPA includes the incorporation of a β-alanine moiety. However, the limited details regarding the source and further characterization of THICAPA were reported by the author.
2.4.1. Neuroprotective effects
Previously, the neuroprotective properties of THICAPA were screened by the Natural Products Depository of RIKEN, Japan. The study utilized two study models: in vitro PC12 cell model and transgenic Drosophila melanogaster expressing human Aβ42 in vivo. The administration of 100 μM THICAPA on PC12 cells treated with 10 μM Aβ42 peptides reported over 92 % cell viability compared to those treated with 10 μM Aβ42 peptides alone (40 %). An increase in the cell proliferation rate was also noted, indicating that THICAPA exhibits an excellent protective effect against Aβ42 induced toxicity due to the presence of beta-alanine in the molecular structure of THICAPA. The high ROS scavenging rate of beta-alanine has been reported to inhibit protein glycation and reduce endothelial cells of immortalized rat brains [83]. In humans, the presence of high beta-alanine serum concentration ameliorated Aβ induced toxicity in the cerebrospinal fluid of AD patients and reduced the risks of dementia [84].
Furthermore, in vivo studies on transgenic Drosophila administered with 100 μM THICAPA showed significant improvement in the eye morphology and life span in both males and females (47.4 % and 69.6 %, respectively). The Drosophila also performed better in the negative geotaxis assay, where the middle-aged phase recorded the fastest climbing speed compared to the early and late phases in both males and females. These findings show that THICAPA plays a protective role in neuronal and non-neuronal cells. THICAPA not only protected the eye tissues from the severe toxicity caused by Aβ42 but also increased the longevity and restored the locomotive functions in Aβ42-expressed Drosophila models. These findings are possibly due to the potential role of THICAPA in inducing compensatory mechanisms, including inflammation regulation, cellular repair processes, hormonal signaling, and immune responses.
2.4.2. Anti-inflammatory properties
Differential expression analysis in THICAPA-treated Drosophila has shown that Imd and Toll pathways, which are essential in innate immunity against infections and inflammations, were downregulated [85]. Drosophila expressing human Aβ42 in photoreceptor cells showed that the Toll pathway was involved in Aβ42-induced neurotoxicity [86]. In contrast, it was claimed that the clumping of Aβ42 was able to protect against microbes and trigger harmful inflammation in the central nervous system [87]. The findings show that THICAPA downregulated genes associated with antimicrobial humoral responses, supporting the theory that THICAPA could possess anti-inflammatory properties. The in vitro and in vivo findings also denote that THICAPA treatment could promote cellular resilience and stimulate cellular defense mechanisms, such as stress response pathways and antioxidant systems.
However, the findings do not demonstrate the direct interaction between THICAPA and Aβ42 peptides. Moreover, the eye assay does not directly confirm the ability of THICAPA to protect neurons against Aβ42-induced toxicity. THICAPA remains in the early stages of pre-clinical development, necessitating further investigation to validate its druggability properties. While THICAPA treatment can downregulate immune response pathways, further in-depth investigations are required to understand the precise action of THICAPA on Aβ42-related neurodegeneration, which are not limited to immune cell infiltration, microglial activation, and neuroinflammation.
2.5. Summary of THIQ derivatives
To wrap it up, this section presents the comprehensive pathway of the THIQ derivatives described in this review, acting through various mechanisms of action within the neuron, as shown in Fig. 2. The review also summarizes the key findings of THIQ derivatives with respect to the experiments and research models incorporated, as listed in Table 1. The comprehensive pathway reveals the diverse potential of the four THIQ derivatives (1MeTIQ, Dau, jatrorrhizine, and THICAPA) in targeting different facets of AD pathology. While all THIQ derivatives share the ability to downregulate Aβ pathology and apoptosis, which implies potential neuroprotective effects, their reported actions are diverse. For instance, Dau exhibits the broadest neuroprotective mechanism, uniquely targeting tau pathology, ER stress, iron homeostasis, GSK-3/BCL-2 signaling, and additionally upregulating Nrf2, Keap1, CAMKII, and GPX4. 1MeTIQ also shares some unique features with Dau, including modulation of NMDAR and calcium overloading and, at the same time, downregulating MAO-A/MAO-B and caspase-3 alongside jatrorrhizine. Moreover, jatrorrhizine stands out by specifically modulating glutamate-induced excitotoxicity, enhancing acetylcholine production, and regulating dopamine levels, potentially mitigating excitotoxicity and neurotransmitter imbalances associated with AD. It also shares the ability to downregulate MAO-A and caspase-3 with 1MeTIQ. Although THICAPA displays a similar ability to downregulate Aβ and apoptosis, it lacks the reported anti-inflammatory and ROS scavenging activity, as observed in the other three THIQ derivatives.
Fig. 2.

Comprehensive pathway of THIQ derivatives through various mechanisms of action within the neuron in AD research. The pink background represents the intracellular environment, and the blue background represents the extracellular compartment of the neuron. The mechanism of the respective THIQ derivatives is labeled with numbers: (1) Dauricine, (2) 1MeTIQ, (3) Jatrorrhizine, and (4) THICAPA. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Table 1.
Different types of THIQ derivatives used in AD studies and key findings.
| THIQ derivatives | Chemical structure | Analytical process | Study model | Findings | Reference |
|---|---|---|---|---|---|
| Dauricine | 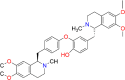 |
|
N2a/APP, N2a/WT |
|
[17] |
| Measurement of phosphorylation level | HEK293/Tau |
|
|||
|
SH-SY5Y/APPsw, Caenorhabditis elegans (GMC101) |
|
[23] | ||
|
Wistar rat primary cortical neuron |
|
[88] | ||
|
Primary microglial cells |
|
[39] | ||
|
57BL/6J (B6) mice |
|
|||
|
SH-SY5Y cell line |
|
[32] | ||
|
C57BL/6 mice |
|
|||
|
Male KM mice |
|
[41] | ||
| Jatrorrhizine | 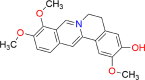 |
|
PC12 cell line |
|
[89] |
|
APP/PSEN1 and C57BL/6 mice |
|
[49] | ||
|
Magnetic beads |
|
[50] | ||
|
Rat brain |
|
[51] | ||
|
SH-SY5Y cell line |
|
[55] | ||
| 1MeTIQ | 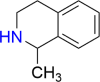 |
|
Primary hippocampal neurons |
|
[77] |
|
Male Wistar rats |
|
[73] | ||
|
Primary neocortical cells |
|
|||
|
Wistar rats |
|
[81] | ||
| THICAPA | 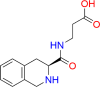 |
|
PC12 cells and Drosophila melanogaster |
|
[82] |
It can be deduced that the four THIQ derivatives discussed in this review have shown good protective effects against all the respective neuron and non-neuronal models. The THIQ derivatives also exhibit strong antioxidant properties and were proven to reduce ROS formation and preserve the cells from oxidative damage. Furthermore, these derivatives showed positive inhibition towards Aβ production and accumulation. While studies on Dau only reported to inhibit tau accumulation, Dau was associated with low permeability in BBB. In contrast, no information was reported regarding BBB permeability for jatrorrhizine, 1MeTIQ, and THICAPA.
Comparatively, there have been more literature studies related to Dau, followed by jatrorrhizine and 1MeTIQ. In contrast, the recent studies focusing on in vitro and in vivo THICAPA research are relatively new in AD research. While the results have been encouraging and good protective effect against human Aβ42, specifically in Drosophila models, in-depth information regarding the druggability, pharmacokinetics, and pharmacodynamics of THICAPA remains scarce. Therefore, future studies should investigate the aforementioned criteria to explore their potential as an alternative therapeutic agent.
3. Emerging potentials of THIQ derivatives via multi-target therapeutic approach
Polypharmacology approaches, exploiting the potential for synergistic effects, offer a promising avenue for developing multifaceted anti-AD formulations with enhanced therapeutic efficacy [90]. However, anti-AD agents targeting only certain biomarkers, genes, or pathways often lead to failures, as AD requires a more sophisticated treatment strategy [91]. Besides, the development of various drugs to treat multiple biological defects is inconvenient and is often accompanied by severe side effects. In view of this, the multi-target therapeutic approach has been widely incorporated in AD treatments to improve overall therapeutic efficiency. Multi-target therapeutic refers to the type of compounds specifically formulated to simultaneously target numerous biological targets, including enzymes, receptors, or other proteins.
Since its successful development two decades ago, the multi-target therapeutic approach has experienced drastic improvement and can be an alternative option to treat AD owing to the heterogeneity of the disease [92]. It is clear from these findings that multi-target therapeutics for AD treatment offer several advantages. Two or more pharmacophore fragments can be combined in multi-target therapeutics to formulate multi-disease-modifying targets to regulate multiple pathological factors simultaneously [93]. The ability of these therapeutics to concurrently modulate multiple pathways at reduced drug dosages while maintaining optimal efficacy is a promising therapeutic strategy. This includes the modulation of Aβ plaque aggregation, NFT, and neurotransmitter imbalance, neuroinflammation, calcium signaling, mitochondrial damage, and oxidative stress [94].
Multi-target therapeutics may also limit the individual side effects of completely blocking specific targets by targeting multiple pathways more comprehensively. This approach can simplify treatment regimens, foster patient adherence, and potentially minimize the side effects associated with polypharmacy. In addition, multi-target therapeutic studies are more efficient and economical than the formulation of various single-target drugs [90]. By addressing the complexity of AD from multiple perspectives, multi-target therapeutics offer a promising approach for developing more effective treatments with good safety profiles for long-term administration. The following sections briefly discuss the current progress on the multi-target therapeutic approach and its potential for combined use in AD treatment.
3.1. Graphene oxide-dauricine (GO@Dau)
Multifunctional drug-loaded nanosystems are a new emerging drug delivery strategy for treating refractory illnesses and complex diseases. As such, graphene oxide (GO) has been chosen as a crucial component in multifunctional drug-loaded nanosystems due to its covalent properties, including high drug-loading concentrations [95]. It has been reported that GO, a graphite derivative, also exhibits excellent biocompatibility, high water solubility, and large surface area compared to other commercial nanosystems in the pharmaceutical industry. The unique physiochemical properties have enabled GO to be used in medical applications, including biosensing [96], bioimaging [97], and other biological applications [98].
In one study, Wang et al. [99] infused GO with Dau (GO@Dau) as a multi-target therapy for AD. In another study, GO was reported to inhibit the Aβ42 production penetrating through Aβ fibrils and disrupt the structures [100]. Li et al. [101] also demonstrated the ability of GO to induce neuronal autophagy and microglia in amyloid plaque clearances. Inspired by the unique biological properties of GO, the study synthesized GO-encapsulated Dau for potential application in AD therapy. This study employed a drug repurposing strategy, utilizing co-incubation of Dau with GO at 37 °C for 48 h to synthesize GO@Dau nanoparticles. Dau and GO were employed for their promising anti-AD properties and versatile non-covalent interactions, respectively. Dau served as the active component, while GO functioned as the drug carrier due to its ability to load therapeutic agents. Eventually, the high biostability and non-covalent interactions of GO have become an added leverage as a potential drug carrier in pharmaceutical modulations [102,103].
Administration of GO@Dau on SH-SY5Y cell lines treated with Aβ42 showed a significant reduction in Aβ concentration compared to the treatment with Dau alone [99]. This indicates that GO@Dau could serve as a more effective anti-Aβ aggregating agent than Dau alone. The in vitro study also showed that GO@Dau inhibited cell apoptosis induced by Aβ due to the significant reduction of MDA and ROS levels, as well as the upregulation of the Nrf2/Keap1 pathway and SOD levels. Consequently, the Nrf2 level increased, and Keap1 decreased in the GO@Dau-treated cells compared to the control group. Concurrently, the Bcl-2/Bax ratio was elevated in GO@Dau-treated cells, which observed the upregulation of Bcl-2 and downregulation of Bax. The p53 and caspase-3 expressions were also downregulated in Aβ42-treated SH-SY5Y cell lines. Conventionally, the downregulated p53, caspase-3, and Bcl-2/Bax are used to deduce the apoptosis levels, where these proteins play a crucial role in apoptosis signal transduction. The finding of this study is consistent with the previous research on the role of Dau in alleviating apoptosis in SH-SY5Y cell lines [23].
Furthermore, GO@Dau administration in the AD mice model was found to significantly improve pathological damage and cognitive decline compared to the administration of Dau alone and the control group [99]. The GO@Dau administration also increased the expression of brain-derived neurotrophic factors and inhibited the recruitment of astrocytes and microglia in the brain triggered by Aβ42, owing to the efficiency of the drug loading rate on GO nanoparticles (6.98 ± 1.20 %). Thus, the substantial loading effect of this nanosystem increased the sustainable release of the drug into the nasal route. Besides, the significant absorption of the drug could be attributed to the presence of a phenyl ring in Dau containing methoxy substituents, which forms non-covalent bonds with GO. However, a detailed focus on the nature of the bonds between GO and Dau is needed to develop an effective drug delivery system.
Notable difficulties in drug development research have been reported, as less than 2 % of the developed drug molecules can pass through the BBB [104]. The barrier, which is composed of glial cells, vascular endothelial cells, and pericytes, filters out potentially harmful substances before passing them to the brain tissue to protect it from possible toxins [105]. Specifically, AD drugs are often reported for their peripheral adverse effect and poor efficacy of the drug as the unique blood-cerebrospinal fluid barrier and BBB block most of the drugs administered in AD patients [106]. Meanwhile, GO infusion in AD treatment has been debated for its limited use as a sensing substance for early detection of mild cognitive impairment, even though the direct pharmacological effects have been studied [107].
Similarly, GO@Dau is also unable to permeate the brain's BBB. Thus, the nasal administration of GO@Dau has been reported as an efficient drug transmission to the brain [99]. The effectiveness of this approach is a step ahead of the administration with Dau alone. The finding was further verified through intra-nasal administration of GO loaded with rhodamine B, which showed that the drug reached different regions of the brain. The effective absorption of GO@Dau by the olfactory neurons to the olfactory bulb by pinocytosis or endocytosis could have enhanced the drug transmission to the different brain regions. Hence, the intra-nasal administration of GO@Dau can be an alternative way for an efficient release of drugs to the brain.
However, the use of THIQ-derived drugs through GO@Dau's multifunctional drug-loaded nanosystem via intra-nasal administration is still a new approach. Besides, the exact mechanism of the nasal transmission route should be further explored to understand the overall treatment outcome. Since intra-nasal administration is non-invasive and can easily reach the brain, GO@Dau can be a potential drug delivery system worth more detailed studies regarding its molecular mechanism, pharmacokinetics, and bioavailability for AD.
3.2. Tetrahydroisoquinoline-benzimidazole
Benzimidazole scaffold is a nitrogen heterocyclic compound found in various bioactive agents and plays an integral role in medicinal and organic chemistry [108]. Some drug candidates incorporating benzimidazole have been developed to treat certain diseases, including the hypertension drug Telmisartan and the anti-viral medicine Maribavir [109]. Previous studies have also demonstrated that benzimidazole compounds exhibited anti-inflammatory properties and served as BACE1 inhibitors. An alternative AD study was conducted by Fang et al. [110] using the concept of Multi-target Directed Ligands (MTDLs) to develop the THIQ-benzimidazole hybrid drug. Referred to as BD3, the hybrid drug showed a positive reaction to AD pathogenesis compared to several other synthesized hybrid compounds. The hybrid BD3 showed good anti-neuroinflammation properties by inhibiting the proinflammatory cytokines in LPS-induced BV2 cells. Moreover, pre-treatment of the LPS-induced BV2 cells using BD3 also significantly reduced the production of inducible nitric oxide synthase (iNOS). Consequently, the proinflammatory cytokines production was inhibited through the suppression of interleukin 1-β (IL-1β) and Tumor Necrosis Factor-α (TNF-α) expressions [110]. The suppressed proinflammatory cytokines followed a dose-dependent manner. Therefore, BD3 can be an excellent anti-neuroinflammatory agent to protect neurons from inflammatory cytokines in AD pathogenesis.
Furthermore, the oxidative stress in glutamate-induced HT22 cells significantly dropped when treated with BD3. The data from the study also reported a substantial increase in cell viability over 91.8 % compared to the HT22 cells treated with glutamate [110]. Apart from that, the hybrid BD3 drug exhibited a robust neuroprotective effect as it reversed the reduction of glutathione (GSH). GSH helps balance the antioxidant's ability for proper brain functioning, making it an essential freeloader of free radicals in the body. Since GSH plays a vital role in scavenging free radicals, reducing GSH production leads to high ROS production and cell death. Remarkably, the study reported that the use of 5 μM hybrid BD3 inhibited ROS production and reversed the GSH depletion by activating the antioxidant mechanism.
In another study, both in vitro and in silico analyses showed that BD3 inhibited the activity of BACE1 (IC50 = 5.70 ± 3.50 μM) by interacting with its catalytic pocket and nitric oxide (IC50 = 5.07 ± 0.54 μM), reducing the formation of Aβ plaque [110]. The results directly correlate with the downregulation of amyloidogenesis and inhibition of Aβ peptides. Concurrently, the drug-profile analysis revealed that the hybrid drug was easily linked to carrier proteins with good absorption in the tissue and high blood-brain transmission. No inhibition of cytochrome P450-2D6 was reported, which is crucial in protecting the liver from hepatotoxicity. However, the water solubility of the drug was lower than the expected value. Thus, it is necessary to optimize pharmacokinetic profiles to formulate a more reliable THIQ-benzimidazole hybrid to increase the bioavailability of the drug.
Overall, the MTDLs combined with THIQ-benzimidazole are worth developing as an anti-AD agent. The current evidence on the formulation of the hybrid drug indicates a potential MTDL candidate for AD therapy. In terms of future prospects, further optimization of multifunctional drug chemistry is required to improve the efficacy and potency of this highly promising drug. A detailed study of molecular mechanisms should also be considered to understand the possible interactions with various biomarkers and pathways. Furthermore, the in silico findings should be verified through in vivo studies to validate the efficiency and possible side effects of the hybrid drug.
3.3. Tetrahydroisoquinoline-oxoisoaporphine
Chen et al. [111] employed a similar strategy by developing a hybrid THIQ-oxoisoaporphine drug as a potential MTDL against Aβ aggregation. Oxoisoaporphine, a naturally occurring compound found in the roots of the Chinese medicinal plant M. dauricum (the same source as Dau), was selected as a hybrid drug candidate due to its diverse range of biological activities. The study departs from utilizing naturally occurring alkaloids and instead focuses on synthesizing oxoisoaporphine derivatives with enhanced biological activities. This was achieved by introducing side chains at various positions (6, 8, 9, 10, and 11) on the 1-azabenzanthrone scaffold. The newly synthesized oxoisoaporphine derivatives demonstrated potent dual activities, inhibiting AChE and reducing Aβ aggregation [112]. These findings suggest their potential as promising candidates as hybrid drugs in combination with THIQ.
The hybrid recorded a relatively high reduction of Aβ42 secretion, up to 37.6 %, when tested on APPsw cells at varying concentrations [111]. A dose-dependent decrease of Aβ42 was observed when the cells were treated with the hybrid at up to 5 μM. The potency of the hybrid was higher in the 8-substitution than in the 11-substitution, indicating that an increase in the side chains reduces the potency of the hybrid against Aβ42 inhibition. Of all the tested hybrids, the 3b and 3c hybrids reported the most substantial Aβ42 inhibition in APPsw cells and SHSY5Y cells. Both hybrids showed no toxic effects when tested up to 5 μM, suggesting that the inhibition of Aβ42 is due to its anti-Aβ properties and not the cytotoxicity.
In addition, the 3b hybrid showed a positive result in inhibiting acetylcholinesterase (AChE) (IC50 = 0.031 ± 0.01 μM), which is known to accelerate the accumulation of amyloid fibrils in the brains of AD patients [113]. The 3b gives a more potent inhibition of AChE than BChE (IC50 = 0.51 ± 0.11 μM) and eight times higher than of tacrine (IC50 = 0.26 ± 0.01 μM). Since the BChE lacks p-aminosalicylic acid (PAS) moiety in its chemical structure compared to AChE, this suggests that the 3b hybrid drug might interact with PAS moiety, leading to a more potent inhibition of AChE. The findings were also in line with those of Aβ42 inhibition.
The 3b and 3c hybrids were also very effective Aβ inhibitors and safe for cell growth, given the marked reduction in the iNOS expression level and insignificant cytotoxicity in BV-2 cells. It is known that the Aβ fibrils induce the production of proinflammatory cytokines and NO in astrocytes and microglia, which contributes to neurodegeneration [114]. Hence, the finding from the study that showed the recovery of nitric concentration in BV-2 microglia and reduced NO production by 65.4 % supports this fact. NO is neurotoxic, as it is endogenously produced from NADPH, l-arginine, and oxygen under proinflammatory stimuli [115]. As a result, the macrophage produces proinflammatory cytokines, including IL-6, IL-1β, and TNF-α, that deteriorate cognitive function and neuron viability [116]. Interestingly, the administration of 15 μM 3b and 3c significantly reduced LPS-induced NO production, with the inhibition by 3b being more substantial than 3c. The results suggest that 3b has a more substantial inhibition effect against proinflammatory cytokines.
A better understanding of the anti-Aβ properties of the 3b hybrid drug was performed in vivo through the worm paralysis assay in C. elegans strain GMC101. This method is an excellent way to study the effects of hybrid drugs on the aggregation of Aβ since the GMC101 strain expresses a full length of the Aβ42 isoform. The worm strain significantly reduced paralysis by half when treated with 5 μM 3b drug compared to the control group. The effect of the hybrid drug is dose-dependent manner. In summary, the study proved the effectiveness of the THIQ-oxoisoaporphine hybrid drug by significantly delaying paralysis in the worm and can be an excellent neuroprotective agent against Aβ42 toxicity. Although the data gives a favorable implication towards Aβ aggregation, they should be interpreted cautiously.
3.4. Tryptophan-tetrahydroisoquinoline
Previously, Lu et al. [11] investigated the development of a tryptophan-THIQ hybrid drug as a multifunctional agent against AD. The essential dietary amino acid, tryptophan, plays a multifaceted role in the brain. It not only serves as a building block for proteins but also contributes to the Aβ peptide regulation under both normal and pathological conditions, including AD [117]. Moreover, tryptophan functions as a precursor for several neuroprotective metabolites, such as kynurenic acid, picolinic acid, and NAD+. Among the identified hit compounds, the study fused tryptophan with THIQ, which exhibited high selectivity and micromolar inhibitory activity against BChE. Thus, the tryptophan-THIQ hybrid has emerged as a promising candidate for potential AD therapy [118].
The tryptophan-THIQ hybrid drug significantly reduced Aβ42 aggregation in SH-SY5Y treated with 50 μM Aβ42 peptides [11]. The Aβ42 was reported to have lower water solubility than Aβ40 but has the highest toxicity, which can trigger neuronal injuries. The synthesized tryptophan-THIQ hybrid exhibited a more comprehensive inhibition range of Aβ42 from 15.5 % to 58.81 %. The inhibition of Aβ42 shows that the hybrid drug could be a good anti-Aβ modulator. The cytotoxicity evaluation on these hybrids also demonstrated good cell viability, suggesting that the inhibition of Aβ42 self-aggregation was due to the potency of the hybrid formulation and not due to cytotoxicity.
The hybrid tryptophan-THIQ was also reported to have low inhibitory potency against AChE and BChE. Among the synthesized hybrids, the 7g(R) was the most potent tryptophan-THIQ hybrid against BChe (IC50 = 0.3 ± 0.1 μM) [11]. The hybrid formulation consisted of a substituted branch and straight alkyl chains and exhibited significant inhibition of AChE and BChE. This shows that tryptophan-THIQ hybrid plays an essential role in cholinesterase inhibition. ACh is widely known for its importance in transporting the neurotransmitters in the hippocampus and cortex region of the brain. A significant reduction of ACh due to an increase in cholinesterase would lead to cell death in the brain [119]. Thus, AChE plays a prominent role in hydrolyzing ACh in the synaptic cleft, while BChE assumes the hydrolyzing role on behalf of AChE at higher concentrations [112]. Despite that BChE is often perceived as a coregulator of Ach due to the damage done by AChE [120], it plays a vital role in the early onset of AD [121] and increases APP metabolism and Aβ40 production [122].
The findings also strongly suggest that the 7g(R) hybrid is a potent cholinesterase inhibitor, especially BChE. Aside from modulating BChE, which counteracts AChE activity loss due to drugs targeting AChE inhibitions, in vivo administration of 7g(R) in a mouse model showed that the hybrid effectively downregulates BChE, which recovers orientation, cognitive functions, and memory. This finding correlates with the cholinergic hypothesis, where the reduction of ACh in AD directly reflects behavioral and cognitive deterioration [123]. The BChE inhibition also aligns with the Aβ fibril inhibition, where BChE eases in vitro generation of amyloid fibril [124].
The 7g(R) hybrid also demonstrates good in vitro permeability. The findings show that the hybrid drug can travel across the BBB at a high membrane permeation (Pe = 10.90 ± 1.99), although the value was not the highest among all other hybrids [11]. Meanwhile, the in vivo study on AD mice reported a good tolerance and rapid recovery after the drug administration via intracerebroventricular injection. Moreover, no passive avoidance impairments were recorded during the post-injection period. The AD mice model also showed improved cognition and behaviors, suggesting the strong neuroprotective effect of the hybrid in preventing injuries induced by amyloid plaques. The study revealed that the hybrid can be used as an anti-AD agent as a cholinesterase inhibitor.
Based on these results, the tryptophan-THIQ hybrid exhibited many significances, especially as an effective BChE and Aβ42 inhibitor. The in vivo and in vitro results also suggest that the hybrid drug provides effective neuroprotection and reverses the injury induced by amyloid plaques. Hence, this multifunctional agent has a higher potential to be developed as an anti-AD agent. However, further in-depth studies should be done focusing on the precise molecular mechanism of this hybrid in AD pathogenesis. Additional structural modifications of the hybrid 7g(R) drug should also be done to improve its potency and efficiency as an anti-AD agent.
3.5. Tetrahydroisoquinoline-cinnamic acid
Generally, cinnamic acid is composed of simple derivatives, including ferulic acid and p-coumaric acid, and is widely found in plants, including vegetables, fruits, whole grains, and honey [125]. Past studies have reported that cinnamic acid exhibits a neuroprotective effect [126], antioxidant [127], and anti-inflammatory properties [128]. When utilized in clinical practice, the administration of this compound alone recorded a low bioavailability, which restricts further development in clinical usages for AD [125]. Regarding this, another MTDL approach was reported by Wang et al. [99] through the synthesis of a novel tetrahydroisoquinoline-cinnamic acid hybrid. In this study, cinnamic acid was fused with benzylpiperidine groups and 1,2,3,4-tetrahydroisoquinoline derivatives to develop the hybrid.
Among all the synthesized hybrids, the 4e hybrid showed the most potent properties in all evaluations. For instance, the 4e hybrid drastically reduced ChE, which is consistent with previous studies reporting the BChE inhibition in THIQ and cinnamic acid [129]. The inhibitory potency of 4e against equine serum BChE was also higher than other derivatives (IC50 = 2.1 ± 0.08 μM). Besides, the hybrid drug restored human BChE activity with donepezil, an FDA-approved cholinesterase inhibitor. The molecular docking study revealed that the 4e hybrid formed multiple interactive sites with BChE, including hydrophobic interaction with some key residues, σ-π interaction with Trp 83 residue of alkylene, two π-π interactions with a THIQ benzene ring, and two π-π interactions with Trp 430 residue of the thiophene ring [99]. This suggests that the 4e hybrid exhibits reversible human BChE inhibitory properties.
Furthermore, the 4e hybrid demonstrated good antioxidant activity in radical oxygen absorbance capacity via the fluorescein assay as well as significant inhibition of the human MAO-B (IC50 = 1.3 ± 0.08). The surge in MAO-B concentration in the brain of AD patients often produces hydroxy radicals, which damage the brain. In the presence of the hydroxyl group in 4e, the drug plays a vital role as an antioxidant by reducing the harmful hydroxy radicals [99]. Although the antioxidant potency of the 4e drug was lower than the other hybrids with secondary amine fragments and organic acid moieties, it exhibits a drastic cell injury reduction in LPS-induced PC12 cells in a dose-dependent manner. The results indicate that the 4e hybrid possesses strong anti-inflammatory properties and is able to protect the neurons from proinflammatory cytokines.
In one study, the administration of 25 μM 4e achieved a prominent decrease in Aβ42 aggregation of 65.2 % compared to curcumin (40.3 % inhibition). Further evaluation through Transmission Electron Microscopy (TEM) revealed progressive deterioration of Aβ oligomers into thin-stranded fibrils when treated with 4e in treated SH-SY5Y culture. The finding is consistent with those in curcumin-treated cell lines [130]. In fact, no prominent cytotoxic effects were observed upon treatment with 50 μM. This shows that the inhibition of the Aβ42 aggregation is due to the anti-Aβ properties of the 4e hybrid and not due to induced cytotoxicity. The 4e hybrid also reported good membrane permeability and obeyed Lipinski's rule of five. Additional assessment of the 4e hybrid found that the hybrid is more stable in artificial gastric than in artificial intestinal fluid, implying that 4e can be practically applied as an orally administered anti-AD drug that can pass through the BBB via passive transport in vivo.
Apart from that, an in vivo study on AlCl3-induced zebrafish reported that 4e was safer to administer with a maximum non-lethal concentration of 8.0 μg/mL. When compared to the donepezil-treated group, the 4e recorded a more significant increase in the distance and speed covered by zebrafish through a dose-dependent manner, where the 4e dosage of up to 0.8 μg/mL showed a better increment. Remarkably, the oral administration of 4e at 1500 mg/kg showed no signs of acute toxicity. The scopolamine-induced memory deterioration of zebrafish also improved significantly after being administered with 4e. Given that AlCl3-induced zebrafish is a well-established AD model [131], the significant improvement following the 4e administration has proven to be a good protective effect against AD.
The in vivo and in vitro studies have provided a wide array of evidence on the potency and efficacy of the THIQ-cinnamic acid hybrid as a potent multi-target anti-AD agent. The fusion of cinnamic acid with THIQ derivative has boosted the bioavailability and effectiveness of the drug. In fact, the hybrid drug reported enhanced performance than commercially available cholinesterase inhibitors and curcumin, which is already an added advantage to developing this drug in the clinical phase further. Additional studies should be carried out to evaluate the efficiency of this hybrid in AD pathogenesis, APP proteolytic pathway, and AD biomarkers.
3.6. Overall Outlook
The five multi-target therapeutics described in this review have demonstrated potent neuroprotective abilities and robust protection against several biological targets involved in AD pathogenesis, both in vitro and in vivo. Table 2 lists the multi-target therapeutics and their respective key biological activities. Accordingly, all these multi-target therapeutics were able to target multiple pathways involved in AD and alleviate the severity of the disease in both neuronal and non-neuronal cells. In fact, multi-target therapeutics provide a better synergistic effect by administering far fewer dosages than the single use of THIQ derivatives. Comparatively, the intra-nasal administration of GO@Dau is considered the most promising multifunctional drug-loaded nanosystem for AD treatment. Although the remaining therapeutic strategies were formulated and tested via oral administration, all multi-target therapeutics described earlier are able to permeate the BBB except for THIQ-oxoisoaporphine. Eventually, these multi-target therapeutics’ safety profiles, pharmacokinetics, and pharmacodynamics should be further studied prior to full implementation.
Table 2.
The list of multi-target therapeutics and the key biological activity.
| Multi-target therapeutics | Chemical structure | Key biological activities | Reference |
|---|---|---|---|
| GO@Dau |  |
|
[99] |
| Tetrahydroisoquinoline-benzimidazole |  |
|
[110] |
| Tetrahydroisoquinoline-oxoisoaporphine |  |
|
[111] |
| Tryptophan-tetrahydroisoquinoline |  |
|
[11] |
| Tetrahydroisoquinoline-cinnamic acid | 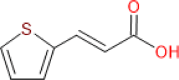 |
|
[130] |
4. Potential challenges
Based on the present review, only a limited number of THIQ derivatives have been explored as potential therapeutic agents for practical use in AD treatment, and most of them are still in the preliminary phase. To the best of the author's knowledge, none of these derivatives has progressed into active clinical trials. While THIQ shows promising potential, several challenges hinder its translation to active clinical trials in AD. These challenges encompass various aspects, including chemistry (efficiency/half-life, limited pharmacological profiles), physical properties (molecular size), and biological factors (inadequacies of research models).
A major obstacle hindering the further development of THIQ drugs lies in the inherent chemical properties of the compounds themselves. Although many studies have reported their positive therapeutic effects, they mostly report on the short-term treatment outcome rather than long-term exposures in vitro. Since an in-depth understanding of long-term drug exposure is beneficial to inhibit the toxic biomarkers and hallmarks of AD over an extended period, it is therefore still necessary to perform long-term and clinical trials to translate these findings into real clinical settings. The THIQ hybrid compounds should also be screened in long-term exposure in vitro to ensure their efficiency in treating AD for an extended duration instead of short-lived effects.
Moreover, there are major data scarcities in published studies that are too complex for other researchers to replicate, such as druggability, pharmacokinetics, pharmacodynamics, stability, and solubility evaluation of the tested compound, while the permeability through the BBB remains ambiguous in most studies published. The positive controls employed in some studies are also often inappropriate, as the compounds/drugs have varying modes of action. Thus, future THIQ studies should precisely define the concrete supporting data for the selected concentrations, chemical properties, modulation of the compounds, and suitable use of controls. This information will serve as a reliable indicator for researchers, industries, and agencies worldwide to understand and translate the findings in different experimental settings effectively.
Another key challenge impeding the further development of THIQ drugs lies in the unfavourable physicochemical properties of these compounds. The size of these compounds also contributes to the high rate of failure in clinical trials. In particular, their large molecular size is unable to penetrate through the BBB. For instance, many THIQ derivatives exceeding 500 Da in molecular weight exhibited low target affinity, suggesting a potential size-related limitation for this class of compounds [132]. Technically, the in vitro concentrations are also too high to be translated into humans. To achieve this goal, studies should design therapeutic compounds with better BBB permeability and effectively reduce the production of biomarkers and toxic hallmarks without causing severe side effects in the human body by considering multiple administration routes (oral, nasal, parenteral, and topical).
The limitations associated with existing research models also deter the advancement of THIQ drugs. Various experimental models that have been employed in most of the pre-clinical testing of THIQ therapeutics to study AD-associated pathology exhibit massive differences with human genetic homology. For example, the findings from vertebrates, such as zebrafish, and invertebrate animals, such as C. elegans and D. melanogaster, do not always mimic the exact genetic homology of humans. Despite the promising results of these model systems, these interventions have yet to record effective clinical impacts in humans. Further concerns exist regarding the reliability of animal models due to the risk of false negative findings, potentially impeding the progress of therapeutic candidates in human trials [133].
Meanwhile, the use of specific types of cells to establish in vitro biocompatibility and toxicity often fails to represent the complex mechanisms of the human body. Compared to an in vitro setting, the body's mechanisms are more complex, which is typically made up of hormones, antibodies, enzymes, peptides, genes, and intricate systems. Furthermore, compounds that were reported to have high biocompatibility often fall short of expectations during clinical trials. In this regard, elucidating the intricate mechanisms of AD pathogenesis is no longer constrained by the limitations of human subject research. Alternatively, a diverse array of advanced methodologies, including novel neuroimaging techniques, patient-derived induced pluripotent stem cells (iPSCs), powerful computational algorithms, and comprehensive "omics" technologies supported by high-throughput platforms and data repositories, now provides unprecedented opportunities to address previously unresolved questions. Nevertheless, the importance of in vitro culture models in AD research should not be overlooked, but the findings should be further corroborated using advanced technologies and comprehensive models.
Although most in vitro studies have attempted to inhibit the excessive accumulation of Aβ, tau, ROS, and the amyloidogenic pathway, the available FDA-approved drugs and drugs under clinical trials that target a similar strategy still fail to halt the progression of AD. Given the multifactorial nature of AD, a comprehensive approach incorporating transdisciplinary factors should be carefully considered. Accumulating evidence from decades of published data has also shown that AD progression is not solely a neuronal issue, as the injuries observed in neurons may be the result of the disease itself. Multiple complex cascades of events occur through a chain reaction throughout the entire body, leading to neuronal injury as the final part. Therefore, future studies utilizing THIQ compounds as therapeutic drugs should encompass a broad range of research models closely resembling the complex human body.
These potential challenges pose considerable economic losses. This avenue arises due to the high cost of clinical trials, coupled with extensive drug development [134]. Research institutes and pharmaceutical companies are facing significant economic challenges. The prospect of repeated failures in AD clinical trials may hinder potential investments in drug development and research in this area. This, in turn, could restrict the discovery of more innovative and comprehensive therapeutic approaches in the long term. To fill the gap between preclinical studies and successful clinical translation in AD, future studies should implement more robust translational approaches.
5. Future recommendations
In line with the current developments in this research field, a number of recommendations for future research should be considered to elevate further the efficacy and effectiveness of THIQ derivatives for AD therapeutics. Since THIQ derivatives are widely available in natural sources, a high throughput screening should be employed in the first place. Several strategies can be employed, such as in vitro screening of ligands of toxic peptides, including Aβ, tau, PSEN, C31, ChE, BACE, MAO, NMDA, and GSK-3β, to identify possible THIQ derivatives that show robust bindings to the subjected ligands.
Researchers can also utilize the growing potential of Artificial Intelligence (AI) in drug screening for AD treatment. The use of AI frameworks integrating multi-omics and human protein-protein interactome data may assist in identifying druggable THIQ targets and high-quality AD Risk Genes (ARGs) associated with variants in genome-wide association studies. Furthermore, AD-associated transcriptional endotypes are reliable metrics for evaluating THIQ therapeutic candidates to modify the disease pathology [135]. This strategy links the gene modules to physiological readouts related to each assigned endotype. For instance, the proportion of cells in the G0-G1, S, and G2-M phases is directly related to the cell cycle endotype, which reflects neuronal re-entry. Therefore, this approach could serve as a means of evaluating each THIQ candidate and validating endotype screening.
Besides, the multifaceted nature of AD disease requires a more effective therapeutic target than in traditional therapeutic approaches. To formulate multi-targets with synergistic therapeutic effects, pharmacophoric motifs of THIQs should be fused, merged, or linked through hybridization against more than one target simultaneously. The research focus should emphasize modifying the chemical structure of THIQ candidates in order to perform better with pharmacodynamics. For example, various THIQ derivatives can be designed and chemically synthesized with methyl substitution on the C-1 position to mitigate cytotoxicity issues. Some specific chemical modifications can also be conducted to synthesize multi-targeted THIQ direct ligands. Additional substituents, such as phenolic and quinoline groups, can be integrated into the THIQ analogs to enhance their antioxidant properties [136].
At the same time, p-I-stilbene (a known Aβ imaging agent) can be merged with N1,N1-dimethyl-N4-(pyridin-2-ylmeth-yl)benzene-1,4-diamine (a known molecule that targets metal−Aβ) and fuse with THIQ analogs to enhance metal chelation, Aβ, and metal−Aβ interactions specifically. A hydroxyl group with oxygen and nitrogen donor atoms from a metal chelator ((8-aminoquinolin-2-yl)methanol) can also enhance the metal-binding properties. Moreover, polar functionalities, such as hydroxyl and amino groups, can be integrated into the THIQ backbone to improve the solubility of the analogs. Thus, future research should ensure that these analogs satisfy Lipinski's rules and logBB for possible drug-likeness and exhibit acceptable penetration levels through the BBB.
Meanwhile, researchers should leverage THIQ drug repurposing and repositioning approaches to expedite the drug development and discovery process [137]. Drug repositioning refers to when a drug is developed for a new indication, whereas repurposing denotes when an existing drug is used for further therapeutic indication. Under such circumstances, researchers can focus on developing effective AD treatment through the repurposing and repositioning of various patented THIQ derivatives, such as N-substituted THIQ derivatives (excellent binding to 5-HT5A receptors), hydroxy group fusion at C-6 position of THIQ ring (good potency against anxiety), and substitution of N-allyl and N-butyl on pyrrolidine ring of THIQ (potent analgesics) [138].
Lastly, future studies are suggested to develop Antibody Drug Conjugates (ADCs) that offer dual action by targeting both the drug and antibody at specific sites, such as AchE, NMDAR, Aβ, tau, and protofibrils. Generally, ADCs are monoclonal antibodies (mAb) that join a linker to a drug (payload). Ideal therapeutic mAbs should seamlessly integrate antigen specificity, a stable systemic linker, and a potent site-cleavable payload targeting DNA, receptors, or other relevant biological targets [139]. For a greater BBB permeability, humanized IgG antibodies with trojan horse technology can be used, as fully humanized mAbs are associated with lower immunogenicity [140].
In terms of the linker selection, cleavable and non-cleavable linkers offer unique benefits. On the one hand, free drugs can be dispensed accurately using cleavable linkers by distinguishing between blood and biological targets, which would be an added advantage in AD therapeutics. On the other hand, non-cleavable linkers, such as thioether or maleimidocaproyl groups with low off-target toxicity, can be utilized as possible linkers in ADCs. Moreover, the payloads should be non-immunogenic, highly soluble, and THIQ derivatives that possess reactive sites to ensure effective linker conjugation in ADCs. These approaches are currently ongoing using numerous established drugs in the market, which opens a promising avenue to employ similar strategies using THIQ derivatives with more significant potential.
6. Conclusion
This review highlights that THIQ is a valuable scaffold in many clinically useful medicinal molecules, particularly in AD studies. The distinct nature of THIQ derivatives not only allows researchers to formulate drugs to combat AD by alleviating mitochondrial dysfunction and neuroinflammation but also regulates oxidative stress, Aβ aggregation, calcium and hormonal imbalance, and genetic factors. The potential modification of the THIQ nucleus at various positions of the ring is also a great benefit, with closer attention being given to selecting the most promising THIQ derivatives to achieve high bioactivities, potency, biological concentration, and specificity under deliberation. This review also explains the emerging prospect of multi-targeted therapeutics as an alternative approach to treating AD. Multi-target therapeutics are superior in terms of safety profile, bioavailability, and pharmacokinetics and are more beneficial compared to the traditional way of combining drugs in AD therapy.
A critical analysis of the multifaceted factors undermining the success of clinical trials has also been extensively presented, with greater emphasis on the underlying challenges inherent to drug discovery. Besides that, the vast majority of drugs in clinical trials have revolved around a single theory of causation of AD, which is a severe flaw, most of the drug formulations were based on AD-related hypotheses, such as the ‘amyloid-cascade hypothesis’ that specifically target β-secretase, γ-secretase, and Aβ that are clearly insufficient to treat or halt the disease progression. The drugs in clinical trials have failed to meet the expectations due to safety concerns, poor BBB permeability, less stability and efficacy, and incorrect drug doses. These failures are also backed by AD's complexity and many ambiguous interacting pathways.
In addressing the growing interest in THIQ-derived drug research, this study offered valid and reasonable future direction for this exciting research area, which could accelerate drug discovery and increase the success rate in treating AD. Most importantly, a rational design of anti-AD agents by chemically fusing THIQ with other potential drugs will significantly boost the drug potency and repair the damages that occur in the multifactorial pathway of AD. The utilization of different modes of drug administration, including nasal, oral, and topical and delivery systems, such as nanosystems, can also enhance penetration of the drugs through the BBB. However, the mechanism of their action needs to be further studied.
In summary, the comprehensive information on THIQ derivatives presented in this review provides a one-stop center for future studies to obtain valuable research insights on the current progress and challenges of THIQ derivatives in AD treatment. Without a doubt, THIQ derivatives have good potential to be developed as disease-modifying biologics and symptom-reducing agents. Therefore, future studies should explore and uncover the molecular mechanism of various THIQ derivatives, which have the potential to be modulated as a potent therapeutic for AD research.
Data availability statement
No data was used for the research described in the article.
Ethics statement
Not applicable.
Funding
This work was supported by the Ministry of Higher Education Malaysia for Transdisciplinary Research Grant Scheme (TRGS) for the project titled “Explicating the novel pathways of THICAPA and POET in different genetic backgrounds of Alzheimer's disease human cell lines” (TRGS/1/2020/USM/02/3/3).
CRediT authorship contribution statement
Danesh Thangeswaran: Writing – review & editing, Writing – original draft, Data curation, Conceptualization. Shaharum Shamsuddin: Visualization, Validation, Supervision, Project administration, Funding acquisition. Venugopal Balakrishnan: Visualization, Validation, Supervision, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Contributor Information
Shaharum Shamsuddin, Email: shaharum1@usm.my.
Venugopal Balakrishnan, Email: venugopal@usm.my.
References
- 1.Behl T., Kaur D., Sehgal A., Singla R.K., Makeen H.A., Albratty M., Alhazmi H.A., Meraya A.M., Bungau S. Therapeutic insights elaborating the potential of retinoids in Alzheimer's disease. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.976799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tahami Monfared A.A., Byrnes M.J., White L.A., Zhang Q. Alzheimer's disease: Epidemiology and clinical progression. Neurol Ther. 2022;11:553–569. doi: 10.1007/s40120-022-00338-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ul Islam B., Zaidi S.K., Kamal M.A., Tabrez S. Exploration of various proteins for the treatment of alzheimer's disease. Curr Drug Metab. 2017;18 doi: 10.2174/1389200218666170203110135. [DOI] [PubMed] [Google Scholar]
- 4.Ly H., Verma N., Sharma S., Kotiya D., Despa S., Abner E.L., Nelson P.T., Jicha G.A., Wilcock D.M., Goldstein L.B., Guerreiro R., Brás J., Hanson A.J., Craft S., Murray A.J., Biessels G.J., Troakes C., Zetterberg H., Hardy J., Lashley T., Aesg, Despa F. The association of circulating amylin with β‐amyloid in familial Alzheimer's disease. Alzheimer's Dementia: Translational Research & Clinical Interventions. 2021;7 doi: 10.1002/trc2.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ul Islam B., Tabrez S. Management of Alzheimer's disease—an insight of the enzymatic and other novel potential targets. Int. J. Biol. Macromol. 2017;97:700–709. doi: 10.1016/j.ijbiomac.2017.01.076. [DOI] [PubMed] [Google Scholar]
- 6.Tsui K.C., Roy J., Chau S.C., Wong K.H., Shi L., Poon C.H., Wang Y., Strekalova T., Aquili L., Chang R.C.-C., Fung M.-L., Song Y.-Q., Lim L.W. Distribution and inter-regional relationship of amyloid-beta plaque deposition in a 5xFAD mouse model of Alzheimer's disease. Front. Aging Neurosci. 2022;14 doi: 10.3389/FNAGI.2022.964336/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan F.H.P., Azzam G. Drosophila melanogaster: Deciphering alzheimer's disease. Malays. J. Med. Sci. 2017;24:6. doi: 10.21315/MJMS2017.24.2.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho Y., Bae H.G., Okun E., Arumugam T.V., Jo D.G. Physiology and pharmacology of amyloid precursor protein. Pharmacol. Ther. 2022;235 doi: 10.1016/J.PHARMTHERA.2022.108122. [DOI] [PubMed] [Google Scholar]
- 9.Pasiek A., Panek D., Malawska B. Exon Publications; 2020. Alzheimer's Disease: Drug Discovery. [DOI] [PubMed] [Google Scholar]
- 10.Hényková E., Kaleta M., Klíčová K., Gonzalez G., Novák O., Strnad M., Kaňovský P. Quantitative Determination of endogenous tetrahydroisoquinolines, potential Parkinson's disease biomarkers, in Mammals. ACS Chem. Neurosci. 2022;13:3230–3246. doi: 10.1021/ACSCHEMNEURO.2C00516/ASSET/IMAGES/LARGE/CN2C00516_0004.JPEG. [DOI] [PubMed] [Google Scholar]
- 11.Lu X., Liu Y., Qin N., Du C., Hu Y., Chen Y., Sun H. Discovery of tryptophan-tetrahydroisoquinoline derivatives as multifunctional agents for treatment of alzheimer's disease. Chin. J. Chem. 2022 doi: 10.1002/cjoc.202200096. [DOI] [Google Scholar]
- 12.Zarranz De Ysern M.E., Ordoñez L.A. Tetrahydroisoquinolines: a review. Prog. Neuro Psychopharmacol. 1981;5:343–355. doi: 10.1016/0364-7722(81)90085-0. [DOI] [PubMed] [Google Scholar]
- 13.Singh I.P., Shah P. Tetrahydroisoquinolines in therapeutics: a patent review (2010-2015) Expert Opin. Ther. Pat. 2017;27:17–36. doi: 10.1080/13543776.2017.1236084. [DOI] [PubMed] [Google Scholar]
- 14.Matada B.S., Pattanashettar R., Yernale N.G. A comprehensive review on the biological interest of quinoline and its derivatives. Bioorg. Med. Chem. 2021;32 doi: 10.1016/j.bmc.2020.115973. [DOI] [PubMed] [Google Scholar]
- 15.Desai N.C., Jadeja K.A., Jadeja D.J., Khedkar V.M., Jha P.C. Design, synthesis, antimicrobial evaluation, and molecular docking study of some 4-thiazolidinone derivatives containing pyridine and quinazoline moiety. Synth. Commun. 2021;51:952–963. doi: 10.1080/00397911.2020.1861302. [DOI] [Google Scholar]
- 16.Xia G.Q., Zhu M.P., Li J.W., Huang H. An alkaloid from Menispermum dauricum, dauricine mediates Ca2+ influx and inhibits NF-κB pathway to protect chondrocytes from IL-1β-induced inflammation and catabolism. J. Ethnopharmacol. 2024;321 doi: 10.1016/J.JEP.2023.117560. [DOI] [PubMed] [Google Scholar]
- 17.Liu P., Chen X., Zhou H., Wang L., Zhang Z., Ren X., Zhu F., Guo Y., Huang X., Liu J., Spencer P.S., Yang X. The isoquinoline alkaloid dauricine targets multiple molecular pathways to ameliorate Alzheimer-like pathological changes in vitro. Oxid. Med. Cell. Longev. 2018;2018 doi: 10.1155/2018/2025914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.L. Wang X., Wang H., Li W., Z X.D., Wang J. Dauricine prevents bradykinin-induced alteration of calcium homeostasis and tau hyperphosphorylation in N2a cells. Prog. Biochem. Biophys. 2005;32:612–617. [Google Scholar]
- 19.ul Islam B., Jabir N.R., Tabrez S. The role of mitochondrial defects and oxidative stress in Alzheimer's disease. J. Drug Target. 2019;27:932–942. doi: 10.1080/1061186X.2019.1584808. [DOI] [PubMed] [Google Scholar]
- 20.Joy T., Rao M.S., Madhyastha S. N-acetyl Cysteine Supplement minimize tau expression and neuronal loss in animal model of alzheimer's disease. Brain Sciences 2018. 2018;8:185. doi: 10.3390/BRAINSCI8100185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zamponi E., Zamponi N., Coskun P., Quassollo G., Lorenzo A., Cannas S.A., Pigino G., Chialvo D.R., Gardiner K., Busciglio J., Helguera P. Nrf2 stabilization prevents critical oxidative damage in Down syndrome cells. Aging Cell. 2018;17 doi: 10.1111/ACEL.12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan G.G., Koch C.M., Connors L.H. Blood Proteomic profiling in Inherited (ATTRm) and Acquired (ATTRwt) forms of Transthyretin-associated Cardiac Amyloidosis. J. Proteome Res. 2017;16:1659–1668. doi: 10.1021/ACS.JPROTEOME.6B00998/SUPPL_FILE/PR6B00998_SI_001.PDF. [DOI] [PubMed] [Google Scholar]
- 23.Wang L., Pu Z., Li M., Wang K., Deng L., Chen W. Antioxidative and antiapoptosis: neuroprotective effects of dauricine in Alzheimer's disease models. Life Sci. 2020;243 doi: 10.1016/j.lfs.2019.117237. [DOI] [PubMed] [Google Scholar]
- 24.Richardson B.G., Jain A.D., Potteti H.R., Lazzara P.R., David B.P., Tamatam C.R., Choma E., Skowron K., Dye K., Siddiqui Z., Wang Y.T., Krunic A., Reddy S.P., Moore T.W. Replacement of a Naphthalene scaffold in Kelch-like ECH-associated protein 1 (KEAP1)/Nuclear factor (Erythroid-derived 2)-like 2 (NRF2) inhibitors. J. Med. Chem. 2018;61:8029–8047. doi: 10.1021/ACS.JMEDCHEM.8B01133/SUPPL_FILE/JM8B01133_SI_002.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bobkov Y.v., Walker W.B., Cattaneo A.M. Altered functional properties of the codling moth Orco mutagenized in the intracellular loop-3. Scientific Reports 2021. 2021;11(1):1–16. doi: 10.1038/s41598-021-83024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behzadfar L., Abdollahi M., Sabzevari O., Hosseini R., Salimi A., Naserzadeh P., Sharifzadeh M., Pourahmad J. Potentiating role of copper on spatial memory deficit induced by beta amyloid and evaluation of mitochondrial function markers in the hippocampus of rats. Metallomics. 2017;9:969–980. doi: 10.1039/C7MT00075H. [DOI] [PubMed] [Google Scholar]
- 27.Jin S.J., Yang Y., Ma L., Ma B.H., Ren L.P., Guo L.C., Wang W.B., Zhang Y.X., Zhao Z.J., Cui M. In vivo and in vitro induction of the apoptotic effects of oxysophoridine on colorectal cancer cells via the Bcl-2/Bax/caspase-3 signaling pathway. Oncol. Lett. 2017;14:8000–8006. doi: 10.3892/OL.2017.7227/HTML. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X.Y., Jiang S.Q., Zhang L., Liu Q.N., Gong P.L. Inhibitory Effect of Dauricine on Inflammatory Process Following Focal Cerebral Ischemia/Reperfusion in Rats. 2012;35:477–486. doi: 10.1142/S0192415X07004990. [DOI] [PubMed] [Google Scholar]
- 29.Li Y.H., Gong P.L. Neuroprotective effect of dauricine in cortical neuron culture exposed to hypoxia and hypoglycemia: Involvement of correcting perturbed calcium homeostasis. Can. J. Physiol. Pharmacol. 2007;85:621–627. doi: 10.1139/Y07-056. [DOI] [PubMed] [Google Scholar]
- 30.Guo D.L., Zhou Z.N., Zeng F.D., Hu C.J. Dauricine inhibited L-type calcium current in single cardiomyocyte of Guinea pig. Zhongguo Yaoli Xuebao. 1997;18:419–421. https://europepmc.org/article/med/10322931 [PubMed] [Google Scholar]
- 31.Pisani A., Calabresi P., Tozzi A., D'Angelo V., Bernardi G. L-type Ca2+ channel Blockers Attenuate Electrical changes and Ca2+ Rise induced by oxygen/glucose deprivation in cortical neurons. Stroke. 1998;29:196–202. doi: 10.1161/01.STR.29.1.196. [DOI] [PubMed] [Google Scholar]
- 32.Peng C., Fu X., Wang K., Chen L., Luo B., Huang N., Luo Y., Chen W. Dauricine alleviated secondary brain injury after intracerebral hemorrhage by upregulating GPX4 expression and inhibiting ferroptosis of nerve cells. Eur. J. Pharmacol. 2022;914 doi: 10.1016/j.ejphar.2021.174461. [DOI] [PubMed] [Google Scholar]
- 33.Cole-Ezea P., Swan D., Shanley D., Hesketh J. Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining oxidative phosphorylation complexes in gut epithelial cells. Free Radic. Biol. Med. 2012;53:488–497. doi: 10.1016/J.FREERADBIOMED.2012.05.029. [DOI] [PubMed] [Google Scholar]
- 34.Alim I., Caulfield J.T., Chen Y., Swarup V., Geschwind D.H., Ivanova E., Seravalli J., Ai Y., Sansing L.H., Emma E.J., Hondal R.J., Mukherjee S., Cave J.W., Sagdullaev B.T., Karuppagounder S.S., Ratan R.R. Selenium Drives a transcriptional Adaptive Program to block ferroptosis and treat stroke. Cell. 2019;177:1262–1279.e25. doi: 10.1016/J.CELL.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 35.Do Van B., Gouel F., Jonneaux A., Timmerman K., Gelé P., Pétrault M., Bastide M., Laloux C., Moreau C., Bordet R., Devos D., Devedjian J.C. Ferroptosis, a newly characterized form of cell death in Parkinson's disease that is regulated by PKC. Neurobiol. Dis. 2016;94:169–178. doi: 10.1016/J.NBD.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 36.Uddin MdS., Kabir MdT., Jalouli M., Rahman MdA., Jeandet P., Behl T., Alexiou A., Albadrani G.M., Abdel-Daim M.M., Perveen A., Ashraf G.M. Neuroinflammatory signaling in the pathogenesis of alzheimer's disease. Curr. Neuropharmacol. 2022;20:126. doi: 10.2174/1570159X19666210826130210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim S.W., Davaanyam D., Seol S.I., Lee H.K., Lee H., Lee J.K. Adenosine triphosphate accumulated following cerebral ischemia induces neutrophil extracellular Trap formation. International Journal of Molecular Sciences 2020. 2020;21:7668. doi: 10.3390/IJMS21207668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-de-Puig I., Miró-Mur F., Ferrer-Ferrer M., Gelpi E., Pedragosa J., Justicia C., Urra X., Chamorro A., Planas A.M. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2014;129(2 129):239–257. doi: 10.1007/S00401-014-1381-0. [DOI] [PubMed] [Google Scholar]
- 39.Pu Z., Bao X., Xia S., Shao P., Xu Y. Serpine1 regulates peripheral neutrophil recruitment and acts as potential target in ischemic stroke. J. Inflamm. Res. 2022;15:2649–2663. doi: 10.2147/JIR.S361072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assem S., Abdelbaki T.N., Mohy-El Dine S.H., Ketat A.F., Abdelmonsif D.A. SERPINE-1 gene methylation and protein as molecular Predictors of Laparoscopic Sleeve Gastrectomy outcome. Obes. Surg. 2020;30(7 30):2620–2630. doi: 10.1007/S11695-020-04533-0. [DOI] [PubMed] [Google Scholar]
- 41.Xue J.S., Li J.Q., Wang C.C., Ma X.H., Dai H., Bin Xu C., Meng X.L. Dauricine alleviates cognitive impairment in Alzheimer's disease mice induced by D-galactose and AlCl3 via the Ca2+/CaM pathway. Toxicol. Appl. Pharmacol. 2023;474 doi: 10.1016/j.taap.2023.116613. [DOI] [PubMed] [Google Scholar]
- 42.O'Day D.H., Eshak K., Myre M.A. Calmodulin binding proteins and alzheimer's disease. J. Alzheim. Dis. 2015;46:553–569. doi: 10.3233/JAD-142772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alami M.M., Shu S., Liu S., Ouyang Z., Zhang Y., Lv M., Sang Y., Gong D., Yang G., Feng S., Mei Z., Xie D.-Y., Wang X. Chromosome-scale genome assembly of Tinospora sagittata (Oliv.) Gagnep. enhances identifying genes involved in the biosynthesis of jatrorrhizine. bioRxiv. 2023;7(20) doi: 10.1101/2023.07.20.549971. [DOI] [Google Scholar]
- 44.Zhang J., He S., Wang J., Wang C., Wu J., Wang W., Li F., Li S., Zhao C., Li F. A review of the traditional Uses, Botany, Phytochemistry, pharmacology, pharmacokinetics, and Toxicology of Corydalis yanhusuo. Nat. Prod. Commun. 2020;15 doi: 10.1177/1934578X20957752/ASSET/IMAGES/LARGE/10.1177_1934578X20957752-FIG9.JPEG. [DOI] [Google Scholar]
- 45.Chen C.Y., Kao C.L., Yeh H.C., Li H.T., Lin R.J. Secondary metabolites of Mahonia bealei. Chem. Nat. Compd. 2022;58:181–184. doi: 10.1007/S10600-022-03629-6/METRICS. [DOI] [Google Scholar]
- 46.Chen H., Fan G., He Y. Species evolution and quality evaluation of four Coptis herbal medicinal materials in Southwest China. 3 Biotech. 2017;7(1 7):1–8. doi: 10.1007/S13205-017-0679-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q., Zhao C., Zhang Y., Du H., Xu T., Xu X., Zhang J., Kuang T., Lai X., Fan G., Zhang Y. 1H NMR-based Metabolomics coupled with molecular docking reveal the anti-Diabetic effects and potential active components of Berberis vernae on type 2 Diabetic rats. Front. Pharmacol. 2020;11:932. doi: 10.3389/FPHAR.2020.00932/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhong F., Chen Y., Chen J., Liao H., Li Y., Ma Y. Jatrorrhizine: a review of sources, pharmacology, pharmacokinetics and toxicity. Front. Pharmacol. 2022;12 doi: 10.3389/fphar.2021.783127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S., Jiang W., Ouyang T., Shen X.Y., Wang F., hua Qu Y., Zhang M., Luo T., Wang H.Q. Jatrorrhizine balances the gut microbiota and reverses learning and memory deficits in APP/PS1 transgenic mice. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-56149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y., Wang Q., Liu R., Zhou H., Crommen J., Moaddel R., Jiang Z., Zhang T. Rapid screening and identification of monoamine oxidase-A inhibitors from Corydalis Rhizome using enzyme-immobilized magnetic beads based method. J. Chromatogr. A. 2019;1592:1–8. doi: 10.1016/j.chroma.2019.01.062. [DOI] [PubMed] [Google Scholar]
- 51.Kong L.D., Cheng C.H., Tan R.X. Monoamine oxidase inhibitors from rhizoma of Coptis chinensis. Planta Med. 2001;67:74–76. doi: 10.1055/s-2001-10874. [DOI] [PubMed] [Google Scholar]
- 52.Patsenka A., Antkiewicz-Michaluk L. Inhibition of rodent brain monoamine oxidase and tyrosine hydroxylase by endogenous compounds - 1,2,3,4-tetrahydro-isoquinoline alkaloids. Pol. J. Pharmacol. 2004;56:727–734. http://www.ncbi.nlm.nih.gov/pubmed/15662085 [PubMed] [Google Scholar]
- 53.Luo T., Zhang H., Zhang W.-W., Huang J.-T., Song E.-L., Chen S.-G., He F., Xu J., Wang H.-Q. Neuroprotective effect of Jatrorrhizine on hydrogen peroxide-induced cell injury and its potential mechanisms in PC12 cells. Neurosci. Lett. 2011;498:227–231. doi: 10.1016/j.neulet.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 54.Eskandari E., Eaves C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. JCB (J. Cell Biol.) 2022;221 doi: 10.1083/JCB.202201159/213213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duan W., Chen X. Jatrorrhizine can improve nerve cell injury induced by Aβ 25-35. Am J Transl Res. 2021;13:4644. [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang C., Li G., Huang P., Liu Z., Zhao B. The gut microbiota and alzheimer's disease. J. Alzheim. Dis. 2017;58:1–15. doi: 10.3233/JAD-161141. [DOI] [PubMed] [Google Scholar]
- 57.Syeda T., Sanchez-Tapia M., Pinedo-Vargas L., Granados O., Cuervo-Zanatta D., Rojas-Santiago E., Díaz-Cintra S., Torres N., Perez-Cruz C. Bioactive food Abates metabolic and synaptic alterations by modulation of gut microbiota in a mouse model of alzheimer's disease. J. Alzheim. Dis. 2018;66:1657–1682. doi: 10.3233/JAD-180556. [DOI] [PubMed] [Google Scholar]
- 58.Pasini E., Corsetti G., Assanelli D., Testa C., Romano C., Dioguardi F.S., Aquilani R. Effects of chronic exercise on gut microbiota and intestinal barrier in human with type 2 diabetes. Minerva Med. 2019;110:3–11. doi: 10.23736/S0026-4806.18.05589-1. [DOI] [PubMed] [Google Scholar]
- 59.Jang S.E., Lim S.M., Jeong J.J., Jang H.M., Lee H.J., Han M.J., Kim D.H. Gastrointestinal inflammation by gut microbiota disturbance induces memory impairment in mice. Mucosal Immunol. 2017;11(2 11):369–379. doi: 10.1038/mi.2017.49. 2018. [DOI] [PubMed] [Google Scholar]
- 60.Hoban A.E., Moloney R.D., Golubeva A.v., McVey Neufeld K.A., O'Sullivan O., Patterson E., Stanton C., Dinan T.G., Clarke G., Cryan J.F. Behavioural and neurochemical consequences of chronic gut microbiota depletion during adulthood in the rat. Neuroscience. 2016;339:463–477. doi: 10.1016/J.NEUROSCIENCE.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 61.Zhao L., Zhang F., Ding X., Wu G., Lam Y.Y., Wang X., Fu H., Xue X., Lu C., Ma J., Yu L., Xu C., Ren Z., Xu Y., Xu S., Shen H., Zhu X., Shi Y., Shen Q., Dong W., Liu R., Ling Y., Zeng Y., Wang X., Zhang Q., Wang J., Wang L., Wu Y., Zeng B., Wei H., Zhang M., Peng Y., Zhang † Chenhong. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359:1151–1156. doi: 10.1126/science.aao5774. www.hmpdacc.org/reference_genomes/finishing [DOI] [PubMed] [Google Scholar]
- 62.Mimica N., Presečki P. Side effects OF approved ANTIDEMENTIVES. Psychiatr. Danub. 2009;21:108–113. [PubMed] [Google Scholar]
- 63.Shen L., Liu L., Ji H.-F. Alzheimer's disease Histological and behavioral Manifestations in transgenic mice correlate with specific gut Microbiome state. J. Alzheim. Dis. 2017;56:385–390. doi: 10.3233/JAD-160884. [DOI] [PubMed] [Google Scholar]
- 64.Cui H.X., Hu Y.N., Li J.W., Yuan K., Guo Y. Preparation and evaluation of Antidiabetic agents of berberine organic acid salts for enhancing the bioavailability. Molecules 2019. 2018;24:103. doi: 10.3390/MOLECULES24010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang X., Chen J., Luo L., He W., Liu G., Gong J., Zeng Y., Xie Z., Liao Q. Comparative brain pharmacokinetic study of Jiaotai pills in normal and insomnic rats using brain microdialysis combinated with LC–MS/MS. Chin Herb Med. 2018;10:206–214. doi: 10.1016/j.chmed.2018.03.006. [DOI] [Google Scholar]
- 66.Cui H.X., Hu Y.N., Li J.W., Yuan K., Guo Y. Preparation and evaluation of Antidiabetic agents of berberine organic acid salts for enhancing the bioavailability. Molecules 2019. 2018;24:103. doi: 10.3390/MOLECULES24010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiu H., Sun S., Ma X., Cui C., Chen G., Liu Z., Li H., Liu M. Jatrorrhizine Hydrochloride Suppresses proliferation, migration, and secretion of Synoviocytes in vitro and Ameliorates rat models of rheumatoid Arthritis in vivo. Int. J. Mol. Sci. 2018;19 doi: 10.3390/IJMS19051514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antkiewicz-Michaluk L., Wąsik A., Michaluk J. 1-Methyl-1,2,3,4-Tetrahydroisoquinoline, an endogenous amine with unexpected mechanism of action: new vistas of therapeutic application. Neurotox. Res. 2014;25:1–12. doi: 10.1007/s12640-013-9402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kohno M., Ohta S., Hirobe M. Tetrahydroisoquinoline and 1-methyl-tetrahydroisoquinoline as novel endogenous amines in rat brain. Biochem. Biophys. Res. Commun. 1986;140:448–454. doi: 10.1016/0006-291X(86)91111-3. [DOI] [PubMed] [Google Scholar]
- 70.Makino Y., Tasaki Y., Ohta S., Hirobe M. Confirmation of the enantiomers of 1-methyl-1,2,3,4-tetrahydroisoquinoline in the mouse brain and foods applying gas chromatography/mass spectrometry with negative ion chemical ionization. Biomed. Environ. Mass Spectrom. 1990;19:415–419. doi: 10.1002/BMS.1200190706. [DOI] [PubMed] [Google Scholar]
- 71.Białoń M., Żarnowska M., Antkiewicz-Michaluk L., Wąsik A. Pro-cognitive effect of 1MeTIQ on recognition memory in the ketamine model of schizophrenia in rats: the behavioural and neurochemical effects. Psychopharmacology (Berl) 2020;237:1577–1593. doi: 10.1007/s00213-020-05484-1/Published. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kuszczyk M.A., Sadowski M.J., Antkiewicz-Michaluk L., Lazarewicz J.W. 1MeTIQ provides protection against Aβ-induced reduction of surface expression of synaptic proteins and inhibits H2O2-induced oxidative stress in primary hippocampal neurons. Neurotox. Res. 2014;25:348–357. doi: 10.1007/s12640-013-9440-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Antkiewicz-Michaluk L., Lazarewicz J.W., Patsenka A., Kajta M., Zieminska E., Salinska E., Wasik A., Golembiowska K., Vetulani J. The mechanism of 1,2,3,4-tetrahydroisoquinolines neuroprotection: the importance of free radicals scavenging properties and inhibition of glutamate-induced excitotoxicity. J. Neurochem. 2006;97:846–856. doi: 10.1111/j.1471-4159.2006.03756.x. [DOI] [PubMed] [Google Scholar]
- 74.Neves D., Salazar I.L., Almeida R.D., Silva R.M. Molecular mechanisms of ischemia and glutamate excitotoxicity. Life Sci. 2023;328 doi: 10.1016/J.LFS.2023.121814. [DOI] [PubMed] [Google Scholar]
- 75.Sasaoka M., Ota T., Kageyama M. Rotenone-induced inner retinal degeneration via presynaptic activation of voltage-dependent sodium and L-type calcium channels in rats. Scientific Reports 2020. 2020;10(1 10):1–16. doi: 10.1038/s41598-020-57638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wąsik A., Antkiewicz-Michaluk L. The mechanism of neuroprotective action of natural compounds. Pharmacol. Rep. 2017;69:851–860. doi: 10.1016/j.pharep.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 77.Kuszczyk M., Słomka M., Antkiewicz-Michaluk L., Saliñska E., Łazarewicz J.W. 1-Methyl-1,2,3,4-tetrahydroisoquinoline and established uncompetitive NMDA receptor antagonists induce tolerance to excitotoxicity. Pharmacol. Rep. 2010;62:1041–1050. doi: 10.1016/S1734-1140(10)70366-2. [DOI] [PubMed] [Google Scholar]
- 78.Kotake Y., Taguchi R., Okuda K., Sekiya Y., Tasaki Y., Hirobe M., Ohta S. Neuroprotective effect of 1-methyl-1,2,3,4-tetrahydroisoquinoline on cultured rat mesencephalic neurons in the presence or absence of various neurotoxins. Brain Res. 2005;1033:143–150. doi: 10.1016/J.BRAINRES.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 79.Companys-Alemany J., Turcu A.L., Schneider M., Müller C.E., Vázquez S., Griñán-Ferré C., Pallàs M. NMDA receptor antagonists reduce amyloid-β deposition by modulating calpain-1 signaling and autophagy, rescuing cognitive impairment in 5XFAD mice. Cell. Mol. Life Sci. 2022;79:1–17. doi: 10.1007/S00018-022-04438-4/FIGURES/8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dewachter I., Filipkowski R.K., Priller C., Ris L., Neyton J., Croes S., Terwel D., Gysemans M., Devijver H., Borghgraef P., Godaux E., Kaczmarek L., Herms J., Van Leuven F. Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol. Aging. 2009;30:241–256. doi: 10.1016/J.NEUROBIOLAGING.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 81.Antkiewicz-Michaluk L., Michaluk J., Mokrosz M., Romanska I., Lorenc-Koci E., Ohta S., Vetulani J. Different action on dopamine catabolic pathways of two endogenous 1,2,3,4-tetrahydroisoquinolines with similar antidopaminergic properties. J. Neurochem. 2001;78:100–108. doi: 10.1046/j.1471-4159.2001.00391.x. [DOI] [PubMed] [Google Scholar]
- 82.Tan F.H.P., Ting A.C.J., Najimudin N., Watanabe N., Shamsuddin S., Zainuddin A., Osada H., Azzam G. 3-[[(3S)-1,2,3,4-Tetrahydroisoquinoline-3-Carbonyl]Amino]Propanoic acid (THICAPA) is protective against Aβ42-induced toxicity in vitro and in an alzheimer's disease Drosophila. J. Gerontol.: Series. 2023;A doi: 10.1093/gerona/glad169. [DOI] [PubMed] [Google Scholar]
- 83.Artioli G.G., Sale C., Jones R.L. Carnosine in health and disease. Eur. J. Sport Sci. 2019;19:30–39. doi: 10.1080/17461391.2018.1444096. [DOI] [PubMed] [Google Scholar]
- 84.Hata J., Ohara T., Katakura Y., Shimizu K., Yamashita S., Yoshida D., Honda T., Hirakawa Y., Shibata M., Sakata S., Kitazono T., Kuhara S., Ninomiya T. Association between serum β-alanine and risk of dementia: the Hisayama study. Am. J. Epidemiol. 2019;188:1637–1645. doi: 10.1093/AJE/KWZ116. [DOI] [PubMed] [Google Scholar]
- 85.Bruno F., Malvaso A., Canterini S., Bruni A.C. Antimicrobial peptides (AMPs) in the pathogenesis of alzheimer's disease: Implications for Diagnosis and treatment. Antibiotics 2022. 2022;11:726. doi: 10.3390/ANTIBIOTICS11060726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deolankar S.C., Najar M.A., Raghu S.V., Prasad T.S.K. Aβ42 expressing Drosophila melanogaster model for alzheimer's disease: Quantitative Proteomics Identifies altered protein dynamics of relevance to neurodegeneration. Https://Home.Liebertpub.Com/Omi. 2022;26:51–63. doi: 10.1089/OMI.2021.0173. [DOI] [PubMed] [Google Scholar]
- 87.Welling M.M., Nabuurs R.J.A., Van Der Weerd L. Potential role of antimicrobial peptides in the early onset of Alzheimer's disease. Alzheimer's Dementia. 2015;11:51–57. doi: 10.1016/J.JALZ.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 88.Li Y.H., Gong P.L. Neuroprotective effects of dauricine against apoptosis induced by transient focal cerebral ischaemia in rats via a mitochondrial pathway. Clin. Exp. Pharmacol. Physiol. 2007;34:177–184. doi: 10.1111/j.1440-1681.2007.04569.x. [DOI] [PubMed] [Google Scholar]
- 89.Luo T., Jiang W., Kong Y., Li S., He F., Xu J., Wang H.-Q. 2012. The Protective Effects of Jatrorrhizine On-Amyloid (25-35)-Induced Neurotoxicity in Rat Cortical Neurons. [DOI] [PubMed] [Google Scholar]
- 90.Proschak E., Stark H., Merk D. Polypharmacology by design: a medicinal Chemist's perspective on Multitargeting compounds. J. Med. Chem. 2019;62:420–444. doi: 10.1021/ACS.JMEDCHEM.8B00760/ASSET/IMAGES/MEDIUM/JM-2018-00760F_0011.GIF. [DOI] [PubMed] [Google Scholar]
- 91.Morsy A., Trippier P.C. Current and emerging pharmacological targets for the treatment of alzheimer's disease. J. Alzheim. Dis. 2019;72:S145–S176. doi: 10.3233/JAD-190744. [DOI] [PubMed] [Google Scholar]
- 92.Jabir N.R., Shakil S., Tabrez S., Khan M.S., Rehman M.T., Ahmed B.A. In silico screening of glycogen synthase kinase-3β targeted ligands against acetylcholinesterase and its probable relevance to Alzheimer's disease. J. Biomol. Struct. Dyn. 2021;39:5083–5092. doi: 10.1080/07391102.2020.1784796. [DOI] [PubMed] [Google Scholar]
- 93.Pasieka A., Panek D., Malawska B. Exon Publications; 2020. Alzheimer's Disease: Drug Discovery. [DOI] [PubMed] [Google Scholar]
- 94.Jabir N.R., Rehman MdT., Tabrez S., Alserihi R.F., AlAjmi M.F., Khan M.S., Husain F.M., Ahmed B.A. Identification of butyrylcholinesterase and monoamine oxidase B targeted ligands and their Putative application in alzheimer's treatment: a computational strategy. Curr. Pharmaceut. Des. 2021;27:2425–2434. doi: 10.2174/1381612827666210226123240. [DOI] [PubMed] [Google Scholar]
- 95.Lopez R.M., White J.R., Truong L., Tanguay R.L. Size- and Oxidation-dependent toxicity of graphene oxide nanomaterials in Embryonic zebrafish. Nanomaterials 2022. 2022;12:1050. doi: 10.3390/NANO12071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abbas Z., Soomro R.A., Kalwar N.H., Tunesi M., Willander M., Karakuş S., Kilislioğlu A. In Situ growth of CuWO4 Nanospheres over graphene oxide for Photoelectrochemical (PEC) Immunosensing of clinical biomarker. Sensors 2020. 2019;20:148. doi: 10.3390/S20010148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sun X., Zebibula A., Dong X., Zhang G., Zhang D., Qian J., He S. Aggregation-induced Emission nanoparticles encapsulated with PEGylated nano graphene oxide and their applications in two-Photon fluorescence bioimaging and Photodynamic therapy in vitro and in vivo. ACS Appl. Mater. Interfaces. 2018;10:25037–25046. doi: 10.1021/ACSAMI.8B05546/SUPPL_FILE/AM8B05546_SI_001.PDF. [DOI] [PubMed] [Google Scholar]
- 98.Buskaran K., Hussein M.Z., Moklas M.A.M., Fakurazi S. Morphological changes and cellular uptake of Functionalized graphene oxide loaded with Protocatechuic acid and folic acid in Hepatocellular Carcinoma cancer cell. International Journal of Molecular Sciences 2020. 2020;21:5874. doi: 10.3390/IJMS21165874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang K., Wang L., Chen L., Peng C., Luo B., Mo J., Chen W. Intranasal administration of dauricine loaded on graphene oxide: multi-target therapy for Alzheimer's disease. Drug Deliv. 2021;28:580–593. doi: 10.1080/10717544.2021.1895909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Z., Ge C., Liu J., Chong Y., Gu Z., Jimenez-Cruz C.A., Chai Z., Zhou R. Destruction of amyloid fibrils by graphene through penetration and extraction of peptides. Nanoscale. 2015;7:18725–18737. doi: 10.1039/C5NR01172H. [DOI] [PubMed] [Google Scholar]
- 101.Li X., Li K., Chu F., Huang J., Yang Z. Graphene oxide enhances β-amyloid clearance by inducing autophagy of microglia and neurons. Chem. Biol. Interact. 2020;325 doi: 10.1016/J.CBI.2020.109126. [DOI] [PubMed] [Google Scholar]
- 102.Maciel E.V.S., Mejía-Carmona K., Jordan-Sinisterra M., da Silva L.F., Vargas Medina D.A., Lanças F.M. The current role of graphene-based nanomaterials in the Sample Preparation Arena. Front. Chem. 2020;8:664. doi: 10.3389/FCHEM.2020.00664/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xia J., Zhu Y., He Z., Wang F., Wu H. Superstrong Noncovalent Interface between Melamine and graphene oxide. ACS Appl. Mater. Interfaces. 2019;11:17068–17078. doi: 10.1021/ACSAMI.9B02971/SUPPL_FILE/AM9B02971_SI_001.PDF. [DOI] [PubMed] [Google Scholar]
- 104.Rohrer J., Lupo N., Bernkop-Schnürch A. Advanced formulations for intranasal delivery of biologics. Int. J. Pharm. 2018;553:8–20. doi: 10.1016/J.IJPHARM.2018.10.029. [DOI] [PubMed] [Google Scholar]
- 105.Sarkar A., Fatima I., Mohammad Sajid Jamal Q., Sayeed U., Khan M. Kalim A., Akhtar S., Amjad Kamal M., Farooqui A., Haris Siddiqui M. Nanoparticles as a carrier system for drug delivery across blood brain barrier. Curr Drug Metab. 2017;18:129–137. doi: 10.2174/1389200218666170113125132. [DOI] [PubMed] [Google Scholar]
- 106.Henrich-Noack P., Nikitovic D., Neagu M., Docea A.O., Engin A.B., Gelperina S., Shtilman M., Mitsias P., Tzanakakis G., Gozes I., Tsatsakis A. The blood–brain barrier and beyond: nano-based neuropharmacology and the role of extracellular matrix. Nanomedicine. 2019;17:359–379. doi: 10.1016/J.NANO.2019.01.016. [DOI] [PubMed] [Google Scholar]
- 107.Zhou J., Meng L., Ye W., Wang Q., Geng S., Sun C. A sensitive detection assay based on signal amplification technology for Alzheimer's disease's early biomarker in exosome. Anal. Chim. Acta. 2018;1022:124–130. doi: 10.1016/J.ACA.2018.03.016. [DOI] [PubMed] [Google Scholar]
- 108.Tahlan S., Kumar S., Narasimhan B. Pharmacological significance of heterocyclic 1H-benzimidazole scaffolds: a review. BMC Chemistry 2019. 2019;13(1):1–21. doi: 10.1186/S13065-019-0625-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dinparast L., Zengin G., Bahadori M.B. Cholinesterases inhibitory activity of 1h-benzimidazole derivatives. Biointerface Res Appl Chem. 2021;11:10739–10745. doi: 10.33263/BRIAC113.1073910745. [DOI] [Google Scholar]
- 110.Fang Y., Zhou H., Gu Q., Xu J. Synthesis and evaluation of tetrahydroisoquinoline-benzimidazole hybrids as multifunctional agents for the treatment of Alzheimer's disease. Eur. J. Med. Chem. 2019;167:133–145. doi: 10.1016/j.ejmech.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 111.Chen Y., Su C., Wang L., Qin J., Wei S., Tang H. Hybrids of oxoisoaporphine–tetrahydroisoquinoline: novel multi-target inhibitors of inflammation and amyloid-β aggregation in Alzheimer's disease. Mol. Divers. 2019;23:709–722. doi: 10.1007/s11030-018-9905-5. [DOI] [PubMed] [Google Scholar]
- 112.Jabir N.R., Khan F.R., Tabrez S. Cholinesterase targeting by polyphenols: a therapeutic approach for the treatment of alzheimer's disease. CNS Neurosci. Ther. 2018;24:753–762. doi: 10.1111/cns.12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pasandideh S., Arasteh A. Evaluation of antioxidant and inhibitory properties of Citrus aurantium L. on the acetylcholinesterase activity and the production of amyloid nano–bio fibrils. Int. J. Biol. Macromol. 2021;182:366–372. doi: 10.1016/J.IJBIOMAC.2021.04.043. [DOI] [PubMed] [Google Scholar]
- 114.Kwon H.S., Koh S.H. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Translational Neurodegeneration 2020. 2020;9(1):1–12. doi: 10.1186/S40035-020-00221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tse J.K.Y. Gut microbiota, nitric oxide, and microglia as Prerequisites for neurodegenerative disorders. ACS Chem. Neurosci. 2017;8:1438–1447. doi: 10.1021/ACSCHEMNEURO.7B00176/ASSET/IMAGES/MEDIUM/CN-2017-001764_0005.GIF. [DOI] [PubMed] [Google Scholar]
- 116.Zeng L., Hu P., Zhang Y., Li M., Zhao Y., Li S., Luo A. Macrophage migration inhibitor factor (MIF): potential role in cognitive impairment disorders. Cytokine Growth Factor Rev. 2024 doi: 10.1016/J.CYTOGFR.2024.03.003. [DOI] [PubMed] [Google Scholar]
- 117.Maitre M., Klein C., Patte-Mensah C., Mensah-Nyagan A.G. Tryptophan metabolites modify brain Aβ peptide degradation: a role in Alzheimer's disease? Prog. Neurobiol. 2020;190 doi: 10.1016/j.pneurobio.2020.101800. [DOI] [PubMed] [Google Scholar]
- 118.Lu X., Yang H., Li Q., Chen Y., Li Q., Zhou Y., Feng F., Liu W., Guo Q., Sun H. Expansion of the scaffold diversity for the development of highly selective butyrylcholinesterase (BChE) inhibitors: discovery of new hits through the pharmacophore model generation, virtual screening and molecular dynamics simulation. Bioorg. Chem. 2019;85:117–127. doi: 10.1016/J.BIOORG.2018.12.023. [DOI] [PubMed] [Google Scholar]
- 119.Ferreira-Vieira T.H., Guimaraes I.M., Silva F.R., Ribeiro F.M. Alzheimer's disease: targeting the cholinergic system. Curr. Neuropharmacol. 2016;14:101–115. doi: 10.2174/1570159X13666150716165726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jasiecki J., Targońska M., Wasąg B. The role of butyrylcholinesterase and iron in the regulation of cholinergic Network and cognitive dysfunction in alzheimer's disease pathogenesis. International Journal of Molecular Sciences 2021. 2021;22:2033. doi: 10.3390/IJMS22042033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jasiecki J., Wasąg B. Butyrylcholinesterase protein Ends in the pathogenesis of alzheimer's disease—could BCHE Genotyping Be helpful in alzheimer's therapy? Biomolecules 2019. 2019;9:592. doi: 10.3390/BIOM9100592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Campos-Peña V., Pichardo-Rojas P., Sánchez-Barbosa T., Ortíz-Islas E., Rodríguez-Pérez C.E., Montes P., Ramos-Palacios G., Silva-Adaya D., Valencia-Quintana R., Cerna-Cortes J.F., Toral-Rios D. Amyloid β, lipid metabolism, basal cholinergic system, and therapeutics in alzheimer's disease. International Journal of Molecular Sciences 2022. 2022;23 doi: 10.3390/IJMS232012092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Knez D., Coquelle N., Pišlar A., Žakelj S., Jukič M., Sova M., Mravljak J., Nachon F., Brazzolotto X., Kos J., Colletier J.P., Gobec S. Multi-target-directed ligands for treating Alzheimer's disease: butyrylcholinesterase inhibitors displaying antioxidant and neuroprotective activities. Eur. J. Med. Chem. 2018;156:598–617. doi: 10.1016/J.EJMECH.2018.07.033. [DOI] [PubMed] [Google Scholar]
- 124.Reid G.A., Darvesh S. Butyrylcholinesterase-knockout reduces brain deposition of fibrillar β-amyloid in an Alzheimer mouse model. Neuroscience. 2015;298:424–435. doi: 10.1016/J.NEUROSCIENCE.2015.04.039. [DOI] [PubMed] [Google Scholar]
- 125.Ruwizhi N., Aderibigbe B.A. Cinnamic acid derivatives and their biological efficacy. International Journal of Molecular Sciences 2020. 2020;21:5712. doi: 10.3390/IJMS21165712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Islam M.A., Saif Khandker S., Alam F., Khalil I., Kamal M.A., Gan S.H. Alzheimer's disease and natural products: future regimens emerging from nature. Curr. Top. Med. Chem. 2017;17:1408–1428. doi: 10.2174/15680266176661701031. [DOI] [PubMed] [Google Scholar]
- 127.Mayer J., Steinbrecher R., Metzsch-Zilligen E., Pfaendner R. Antioxidant activity of Biogenic cinnamic acid derivatives in Polypropylene. Polymers. 2023;15:3621. doi: 10.3390/POLYM15173621/S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Y.P., Brown R.E., Zhang P.C., Zhao Y.T., Ju X.H., Song C. DHA, EPA and their combination at various ratios differently modulated Aβ25-35-induced neurotoxicity in SH-SY5Y cells. Prostaglandins Leukot. Essent. Fatty Acids. 2018;136:85–94. doi: 10.1016/j.plefa.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 129.Lan J.S., Zeng R.F., Jiang X.Y., wei Hou J., Liu Y., Hu Z.H., Li H.X., Li Y., Xie S.S., Ding Y., Zhang T. Design, synthesis and evaluation of novel ferulic acid derivatives as multi-target-directed ligands for the treatment of Alzheimer's disease. Bioorg. Chem. 2020;94 doi: 10.1016/J.BIOORG.2019.103413. [DOI] [PubMed] [Google Scholar]
- 130.Wang K., Shi J., Zhou Y., He Y., Mi J., Yang J., Liu S., Tang X., Liu W., Tan Z., Sang Z. Design, synthesis and evaluation of cinnamic acid hybrids as multi-target-directed agents for the treatment of Alzheimer's disease. Bioorg. Chem. 2021;112 doi: 10.1016/j.bioorg.2021.104879. [DOI] [PubMed] [Google Scholar]
- 131.Sang Z., Wang K., Shi J., Liu W., Cheng X., Zhu G., Wang Y., Zhao Y., Qiao Z., Wu A., Tan Z. The development of advanced structural framework as multi-target-directed ligands for the treatment of Alzheimer's disease. Eur. J. Med. Chem. 2020;192 doi: 10.1016/J.EJMECH.2020.112180. [DOI] [PubMed] [Google Scholar]
- 132.Pardridge W.M. Treatment of alzheimer's disease and blood–brain barrier drug delivery. Pharmaceuticals. 2020;13:1–25. doi: 10.3390/ph13110394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pistollato F., Ohayon E.L., Lam A., Langley G.R., Novak T.J., Pamies D., Perry G., Trushina E., Williams R.S.B., Roher A.E., Hartung T., Harnad S., Barnard N., Morris M.C., Lai M.-C., Merkley R., Chandrasekera P.C. Alzheimer disease research in the 21st century: past and current failures, new perspectives and funding priorities. Oncotarget. 2016;7:38999–39016. doi: 10.18632/oncotarget.9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jain R., Subramanian J., Rathore A.S. A review of therapeutic failures in late-stage clinical trials. Expet Opin. Pharmacother. 2023;24:389–399. doi: 10.1080/14656566.2022.2161366. [DOI] [PubMed] [Google Scholar]
- 135.Caldwell A.B., Liu Q., Zhang C., Schroth G.P., Galasko D.R., Rynearson K.D., Tanzi R.E., Yuan S.H., Wagner S.L., Subramaniam S. Endotype reversal as a novel strategy for screening drugs targeting familial Alzheimer's disease. 2022. [DOI] [PMC free article] [PubMed]
- 136.Hernández-Ayala L.F., Guzmán-López E.G., Galano A. Quinoline derivatives: promising antioxidants with neuroprotective potential. Antioxidants. 2023;12:1853. doi: 10.3390/ANTIOX12101853/S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Athar T., Al Balushi K., Khan S.A. Recent advances on drug development and emerging therapeutic agents for Alzheimer's disease. Mol. Biol. Rep. 2021;48:5629–5645. doi: 10.1007/s11033-021-06512-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Singh I.P., Shah P. Tetrahydroisoquinolines in therapeutics: a patent review (2010-2015) Expert Opin. Ther. Pat. 2017;27:17–36. doi: 10.1080/13543776.2017.1236084. [DOI] [PubMed] [Google Scholar]
- 139.Punyakoti P., Behl T., Sehgal A., Yadav S., Sachdeva M., Anwer M.K., Vargas-De-La-Cruz C., Venkatachalam T., Naqvi M., Verma R., Tuli H.S. Postulating the possible cellular signalling mechanisms of antibody drug conjugates in Alzheimer's disease. Cell. Signal. 2023;102 doi: 10.1016/j.cellsig.2022.110539. [DOI] [PubMed] [Google Scholar]
- 140.Pardridge W.M. Re-engineering therapeutic antibodies for Alzheimer's disease as blood-brain barrier penetrating bi-specific antibodies. Expet Opin. Biol. Ther. 2016;16:1455–1468. doi: 10.1080/14712598.2016.1230195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.