

Abstract
The genome of a microorganism encodes its potential functions that can be implemented through expressed proteins. It remains elusive how a protein’s selective expression depends on its metabolic essentiality to microbial growth or its ability to claim resources as ecological niches. To reveal a protein’s metabolic or ecological role, we developed a computational pipeline, which pairs metagenomics and metaproteomics data to quantify each protein’s gene-level and protein-level functional redundancy simultaneously. We first illustrated the idea behind the pipeline using simulated data of a consumer-resource model. We then validated it using real data from human and mouse gut microbiome samples. In particular, we analyzed ABC-type transporters and ribosomal proteins, confirming that the metabolic and ecological roles predicted by our pipeline agree well with prior knowledge. Finally, we performed in vitro cultures of a human gut microbiome sample and investigated how oversupplying various sugars involved in ecological niches influences the community structure and protein abundance. The presented results demonstrate the performance of our pipeline in identifying proteins’ metabolic and ecological roles, as well as its potential to help us design nutrient interventions to modulate the human microbiome.
Keywords: metagenomics, metaproteomics, functional redundancy, ecological niche, metabolic essentiality, gut microbiome
Introduction
Metagenomic sequencing has enabled the measurement of the genomic content and functional potential of microbial communities at an unprecedented rate, aiding the understanding of their role in host health [1–3] and biogeochemical cycling [4–6]. Although various computational approaches based on these genomes quantify interactions within microbial communities [7–12] and analyze the functional redundancy (FR) and functional stability of microbial communities [10–12], they focus on potential rather than actual function, as microorganisms only express a subset of genes as proteins [13]. Recent advancements in high-throughput metaproteomics allow us to quantify protein abundances in human gut microbiomes [14], offering insights into gene expression in response to environmental changes when paired with metagenomic data.
From the metabolic perspective, some genes and their encoded proteins are indispensable for cell metabolism under any conditions, as microbial growth halts without these essential functions—aminoacyl-tRNA synthetase [15, 16], ribosomal proteins [17–19], and enzymes involved in glycolysis [20, 21]. From the ecological perspective, gene expression is influenced by ecological selection, with specific proteins indicating which resources a microbe can utilize and defining its ecological niche. For instance, Escherichia coli prefers glucose over lactose due to the repressed expression of lactose-utilizing enzymes, even though it can use both sugars [22, 23]. Such specialization of consuming one resource caused by the selective gene expression may reduce the niche overlap with other species and allow microbial coexistence, as seen with two E. coli strains where one expresses acetyl-coenzyme synthetase (Acs) [24, 25] to consume acetate produced by the other [26–29].
Understanding the selective expression of microbial genes is an outstanding question in microbiology. Does the behavior of selective expression of microbial genes differ between metabolic function (e.g. essential for microbial growth metabolism) and ecological function (e.g. claiming resources as a niche)? To answer this, we developed a computational method to analyze paired metagenomic and metaproteomic [14, 30–33] data, constructing the gene content network (GCN) or protein content network (PCN)—a bipartite graph that connects microbial taxa to their genes or expressed proteins, respectively (Fig. 1A and B). For each gene and its encoding protein, we compared its gene-level (or protein-level) FR, revealing each protein family’s metabolic or ecological role. Our method, validated with several gut microbiome data, accurately predicts that ABC-type transporters are related to ecological niches [34–36], and ribosomal proteins are essential [17–19]. Finally, we performed in vitro culture experiments using human gut microbiome samples to investigate how oversupplying sugars involved in ecological niches influence community structure and protein expression.
Figure 1.
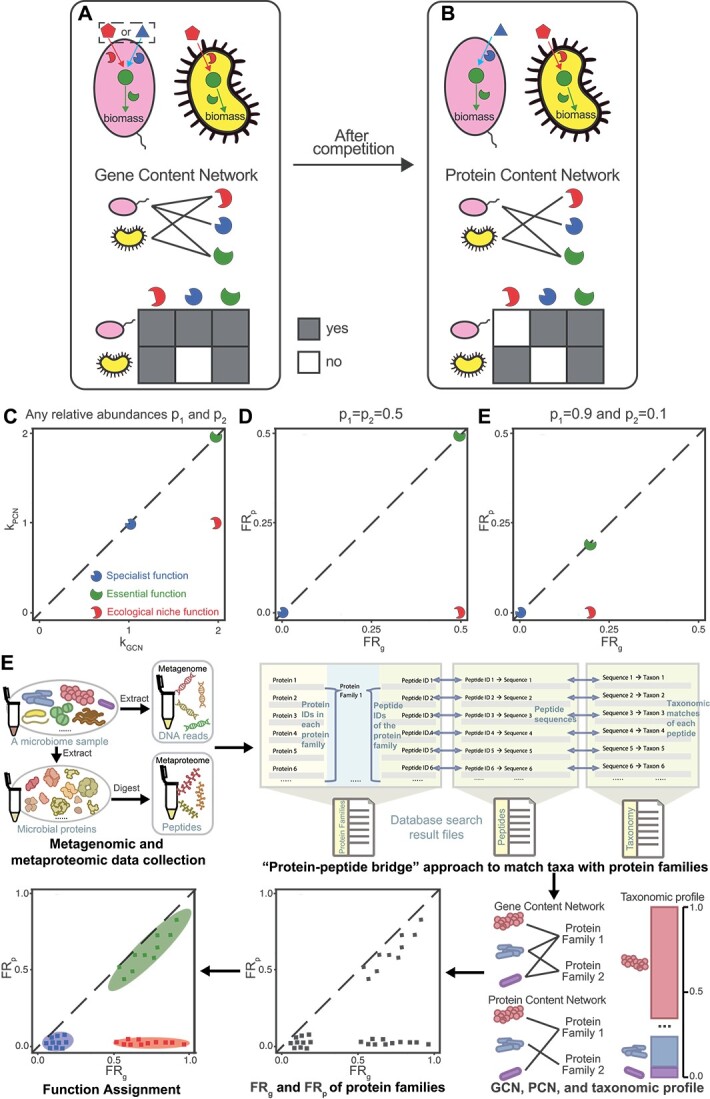
Protein functions involved in determining ecological niches are postulated to have larger discrepancies between the gene-level functional redundancy FRg and protein-level functional redundancy FRp; here we use a hypothetical example with three representative proteins (three broken circles with complementary shapes to their substrates) to demonstrate this point; (A) schematic of the genomic capacity of two microbial taxa (oval vs. indented oval); two resources (pentagon and triangle) are externally supplied to the community; the round-shaped metabolite can be transformed from either resource and further utilized in biomass synthesis; the taxon on the left has the capacity of converting either supplied resource into the metabolite, while the taxon on the right can only convert the pentagon-shaped resource; (B) schematic of expressed proteins for two microbial taxa after their competition in the same community; after the competition, the reduced resource conflict (represented by the taxon on the left choosing the triangle-shaped resource as the sole one to consume) can promote their coexistence; GCN and PCN can be used to capture genomic capacity and expressed protein functions for all taxa; alternatively, this network can be represented as incidence matrices on the bottom (i.e., the presence/absence of edges connecting taxa to proteins); (C and D), the comparison between kGCN and kPCN or between FRg and FRp helps to classify proteins into three protein functional types: specialist function, essential function, and niche function. In the calculation of FRg and FRp, we assume equal abundances of the two species, i.e. 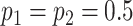 ; (E) the comparison between FRg and FRp when
; (E) the comparison between FRg and FRp when 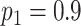 and
and 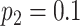 ; (F) the pipeline of assigning the functions (specialist, niche, or essential) to protein families based on the paired metagenomes and metaproteomes; each individual’s gut microbiome sample was subjected to DNA and protein extraction; then a protein-peptide bridge approach can be used for generating the GCN based on the metagenome and PCN based on the metaproteome; when matched metagenomes are available, taxonomic and functional annotations of the metagenomes can be used for PCN generation; based on the generated GCN, PCN, and taxonomic profile, FRg and FRp can be computed and used for the function assignment.
; (F) the pipeline of assigning the functions (specialist, niche, or essential) to protein families based on the paired metagenomes and metaproteomes; each individual’s gut microbiome sample was subjected to DNA and protein extraction; then a protein-peptide bridge approach can be used for generating the GCN based on the metagenome and PCN based on the metaproteome; when matched metagenomes are available, taxonomic and functional annotations of the metagenomes can be used for PCN generation; based on the generated GCN, PCN, and taxonomic profile, FRg and FRp can be computed and used for the function assignment.
Materials and methods
In vitro human gut microbiota culture and metaproteomics
Three healthy individual microbiota samples were collected and biobanked [37]. The frozen microbiome samples were cultured in our optimized culture medium [38] with or without the presence of different sugars in technical triplicates, and were taken at different times for optical density and metaproteomic analyses. For single-strain samples, proteins were extracted with 4% Sodium Dodecyl Sulfate (SDS) 8 M urea buffer in 100 mM Tris–HCl buffer, followed by precipitation and acetone washing. Proteins were digested with trypsin desalted [39] for Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) analysis using an Orbitrap Exploris 480 mass spectrometer. For the cultured microbiomes, an automated process extracted and purified proteins, which were then digested, desalted, and quantified using TMT11plex [40], ensuring mixed representation in labeling to avoid bias. Samples underwent a 2-h LC gradient and were analyzed by mass spectrometry. More details can be found in Supplemental Methods.
Datasets
Metagenomics data from four individual microbiomes were obtained from the previous MetaPro-IQ study [14, 33] (accessible from the National Center for Biotechnology Information (NCBI) sequence read archive under the accession of SRP068619), and the same samples were reanalyzed by an ultra-deep metaproteomics approach [14] via the PRIDE partner repository [41] with the dataset identifier PXD027297. Proteomics dataset of the cultured singles strain samples has been deposited to ProteomeXchange Consortium with the identifier PXD037923. Metaproteomic dataset of the RapidAIM-cultured microbiome samples has been deposited to ProteomeXchange Consortium (identifier PXD037925). The metaproteomic dataset of the mouse gut microbiome comprising 20 gut microbes is derived from a previous study [42] that was deposited to ProteomeXchange Consortium with the dataset identifier PXD009535 and to MassIVE with the dataset identifier MSV000082287.
Database search and data processing
Proteomics database searches used FASTA databases of the individual strains downloaded from NCBI and MaxQuant [43] 1.6.17.0 for analysis, without the label-free quantification. Metaproteomic database searches of cultured microbiome samples were performed using MetaLab V2.2, and the [44] MaxQuant option was used to search the Tandem Mass Tag (TMT) dataset against the integrated gene catalog (IGC) database of the human gut microbiome. The resulting data table was normalized using R package MSstatsTMT [45], and missing values were imputed using R package DreamAI [46]. The “fraction” of each taxon-specific protein is computed by dividing the protein intensity by the sum of the intensities of all proteins assigned to the same taxon. The log2 fold change of each protein is obtained by taking log2 of the ratio between its fraction in the treatment group (with added sugars) and its fraction in the control group (without added sugars).
Statistics
To calculate correlation throughout the study, we used Pearson’s correlation coefficient. All statistical tests were performed using standard numerical and scientific computing libraries in the Python programming language (version 3.7.1) and Jupyter Notebook (version 6.1).
Results
Specialist function, niche function, and essential function
Here, we define three types of functions for protein families that we would like to categorize: (i) “Specialist function”: specialized by only a few taxa and not widely shared within a community. (ii) “Niche function”: arising from ecological competition, widespread among genomes of numerous taxa but selectively expressed under specific ecological conditions. (iii) “Essential function”: metabolically indispensable for and widely shared by many taxa within a microbial community. We emphasize that our definition is not exhaustive; some proteins may display attributes of multiple categories or not align precisely with any single category.
Using a simple hypothetical example of two competing species (Fig. 1A and B), we demonstrated the three function types: (a) the blue protein is a specialist function since it is solely encoded in the pink species’ genome; (b) the red protein belongs to a niche function due to its selective expression by the yellow species even though the protein is encoded in the genomes of both species; (c) the green protein is an essential function because both species need it for biomass synthesis. In coexistence, the pink species specializes in the blue resource, avoiding competition with the yellow species for the red resource.
GCN, PCN, and network degree
We can identify the functional types of proteins in this hypothetical case by comparing the structure of the GCN and PCN (Fig. 1A and B). For example, consider the protein responsible for converting red resource to green metabolite (red broken circle in Fig. 1A and B), its degree in the GCN 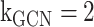 , while its degree in the PCN
, while its degree in the PCN 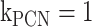 . This degree reduction is due to distinct ecological niches being occupied by two species when they are cocultured. By contrast, the protein responsible for assimilating critical green metabolites (green broken circle in Fig. 1A and B) into biomass does not show a degree reduction (
. This degree reduction is due to distinct ecological niches being occupied by two species when they are cocultured. By contrast, the protein responsible for assimilating critical green metabolites (green broken circle in Fig. 1A and B) into biomass does not show a degree reduction (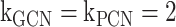 ) because it is essential for microbial growth. Similarly, since the blue protein is only specialized by the pink species, its
) because it is essential for microbial growth. Similarly, since the blue protein is only specialized by the pink species, its 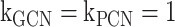 . Thus, three function types occupy different regions in the
. Thus, three function types occupy different regions in the 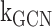 vs.
vs. 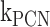 plot (Fig. 1C).
plot (Fig. 1C).
Quantifying gene- and protein-level FR of each gene and its encoded protein
However, the network degree does not consider the significant impact of the microbial taxonomic profile, which provides details about the makeup of a microbial community. This profile is represented by 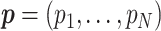 , where
, where 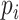 is the relative abundance of taxon-
is the relative abundance of taxon-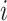 and
and 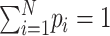 . For a given gene and its encoded protein, we can define its gene-level FR (
. For a given gene and its encoded protein, we can define its gene-level FR (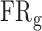 ) and protein-level FR (
) and protein-level FR (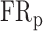 ) within this sample as
) within this sample as
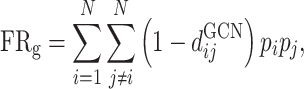 |
(1) |
and
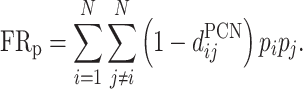 |
(2) |
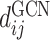 (or
(or 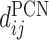 ) is the distance between taxon-
) is the distance between taxon-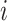 and taxon-
and taxon-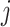 based on their genomic capacity to express this gene (or the presence of the protein). For simplicity, we assume
based on their genomic capacity to express this gene (or the presence of the protein). For simplicity, we assume 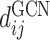 is binary, i.e.
is binary, i.e. 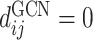 if and only if both taxa share the potential to express the gene, and
if and only if both taxa share the potential to express the gene, and 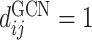 otherwise.
otherwise. 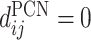 if and only if both taxa have expressed the protein. Here, we define
if and only if both taxa have expressed the protein. Here, we define 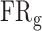 and
and 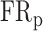 for each protein, different from our previous studies where FR was calculated by including all genes or proteins in a microbial community [12, 14].
for each protein, different from our previous studies where FR was calculated by including all genes or proteins in a microbial community [12, 14].
Comparing 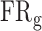 and
and 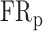 provides deeper insight into proteins’ function types. For the red protein in our hypothetical example,
provides deeper insight into proteins’ function types. For the red protein in our hypothetical example, 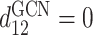 and
and 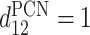 because both species share the potential to express the gene, while only the yellow species have expressed it (Fig. 1A and B). As a result,
because both species share the potential to express the gene, while only the yellow species have expressed it (Fig. 1A and B). As a result, 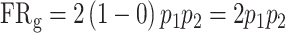 and
and 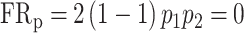 (Fig. 1D and E). Following the same analysis,
(Fig. 1D and E). Following the same analysis, 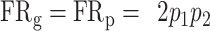 for the green protein, and
for the green protein, and 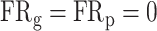 for the blue protein (Fig. 1D and E). Different from composition-independent
for the blue protein (Fig. 1D and E). Different from composition-independent 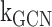 and
and 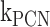 ,
, 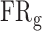 and
and 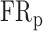 take the microbial composition into account and thus are more ecologically meaningful. Notably, a more uneven abundance distribution would lead to smaller
take the microbial composition into account and thus are more ecologically meaningful. Notably, a more uneven abundance distribution would lead to smaller 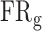 and
and 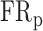 (
(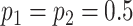 in Fig. 1D;
in Fig. 1D; 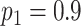 and
and 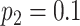 in Fig. 1E). The influence of relative abundances on FR can be mitigated by using the normalized FR: nFR = FR / TD, where TD
in Fig. 1E). The influence of relative abundances on FR can be mitigated by using the normalized FR: nFR = FR / TD, where TD 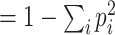 (Supplementary Fig. 1; see Supplementary Information for the definition).
(Supplementary Fig. 1; see Supplementary Information for the definition).
Overview of our computational pipeline
Following the idea of comparing 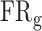 with
with 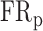 , we developed a computational pipeline to assign the function types (specialist, niche, or essential) to protein families based on the paired metagenome and metaproteome (Fig. 1F). This pipeline starts with DNA sequences from metagenomes and peptides sequences from metaproteomes. Using the “protein-peptide bridge” approach that maps peptides to their taxonomic origins and protein families (i.e. orthologous protein clusters), it generates the GCN, PCN, and taxonomic profile, from which we compute
, we developed a computational pipeline to assign the function types (specialist, niche, or essential) to protein families based on the paired metagenome and metaproteome (Fig. 1F). This pipeline starts with DNA sequences from metagenomes and peptides sequences from metaproteomes. Using the “protein-peptide bridge” approach that maps peptides to their taxonomic origins and protein families (i.e. orthologous protein clusters), it generates the GCN, PCN, and taxonomic profile, from which we compute 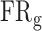 and
and 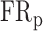 . Details about this approach can be found in the Supplementary Information. Finally, based on the scatterplot of
. Details about this approach can be found in the Supplementary Information. Finally, based on the scatterplot of 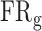 vs.
vs. 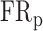 , each protein family is categorized into one of the three function types. Note that the computational pipeline assigns the function type without leveraging the known biological functions. Instead, we validate these assignments against the knowledge about biological functions.
, each protein family is categorized into one of the three function types. Note that the computational pipeline assigns the function type without leveraging the known biological functions. Instead, we validate these assignments against the knowledge about biological functions.
Illustration of our computational pipeline using synthetic data
To illustrate the pipeline’s workflow, we utilized synthetic data generated by a consumer-resource model (CRM). Each niche (or specialist) function is modeled as the consumption of a unique and externally supplied resource (Fig. 2A1), whose loss would make a species unable to consume the corresponding resource (Fig. 2A2 and A3). The loss of an essential function is modeled as reducing a species’ growth rate by 5% (Fig. 2A4).
For each species, each niche (specialist, or essential) function was assigned to the species’ genome with probability 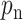 (
(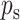 or
or 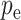 ), respectively (Fig. 2B, left). We set
), respectively (Fig. 2B, left). We set 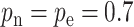 to ensure that we cannot distinguish niche functions from essential functions only based on their
to ensure that we cannot distinguish niche functions from essential functions only based on their 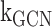 . We set
. We set 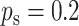 <
< 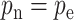 so that specialist functions were assigned to fewer species than niche and essential functions. Species’ actual expressed functions were determined by randomly sampling a subset of its potential functions (Fig. 2B, middle). This behavior of sub-sampling was observed when we cultured single microbial strains in different environments (Supplementary Fig. 21). We simulated community dynamics until reaching a steady state, for which we constructed the PCN of the surviving species (Fig. 2B, right; see Supplementary information for technical details). More technical details of CRM are in Supplementary Information.
so that specialist functions were assigned to fewer species than niche and essential functions. Species’ actual expressed functions were determined by randomly sampling a subset of its potential functions (Fig. 2B, middle). This behavior of sub-sampling was observed when we cultured single microbial strains in different environments (Supplementary Fig. 21). We simulated community dynamics until reaching a steady state, for which we constructed the PCN of the surviving species (Fig. 2B, right; see Supplementary information for technical details). More technical details of CRM are in Supplementary Information.
Figure 2.
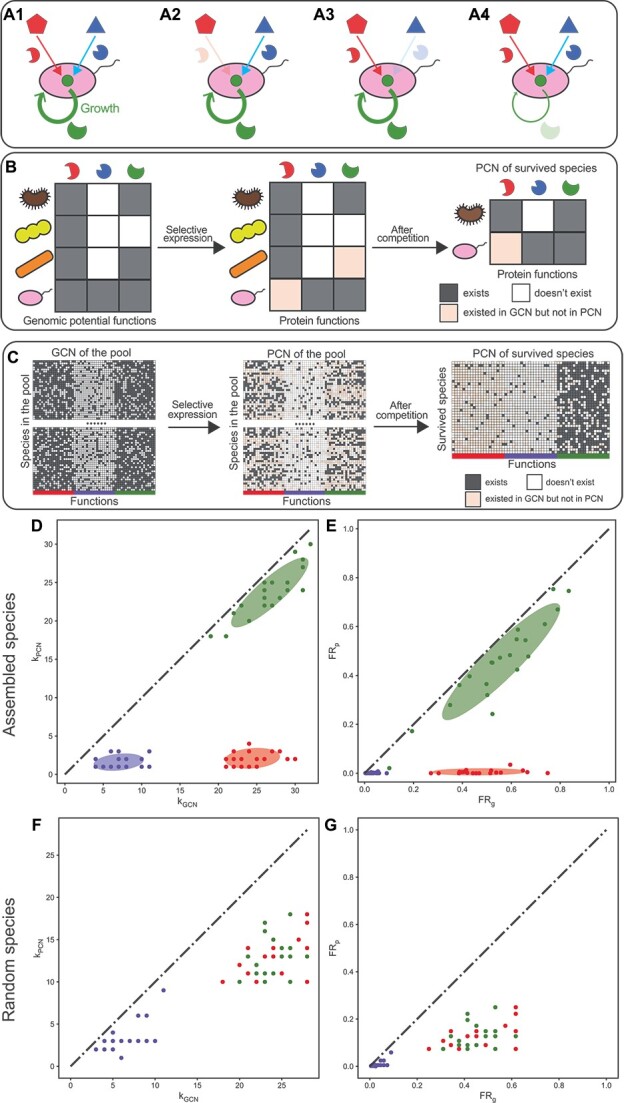
Three protein functional clusters (specialist function, essential function, and niche function) considered in the community assembly model form three distinct clusters when the network degree and FR are compared between the GCN and PCN in model-generated synthetic data; (A1–A4) three types of functions modeled have different ecological and metabolic roles; the niche function (the protein missing in A2) and specialist function (the protein missing in A3) are modeled as abilities to consume externally supplied resources; the role of essential functions (the protein missing in A4) is considered as a reduction in the overall growth rate for each missing essential function; (B) a schematic diagram of the community assembly; species (ovals and indented ovals) with expressed gene functions selected via the sub-sampling of their genomic capacity; then all species are co-cultured together to simulate their ecological competition; (C) a simulation example of the community assembly, and the construction of GCN and PCN for the survived species; (D and E), the comparison of network degree and FR, respectively, based on the GCN and PCN of survived species in the simulation example in panel-C; a Gaussian mixture model with three clusters is used to identify three protein functional clusters; ellipses around clusters cover areas one standard deviation away from their means; (F–G) the comparison of network degree and FR, respectively, based on the GCN and PCN of 35 species randomly selected from the 10 000 species in the initial pool; all points/functions are colored red (niche functions), green (essential functions), and blue (specialist functions) according to their types of functions in the model; kGCN (or kPCN) is the network degree of each function in the GCN (or PCN); FRg (or FRp) is the FR of each function on the gene level (or protein level), respectively.
In our model with 10 000 species and 20 functions for each of the three function types, each species randomly sampled a subset of potential functions (Fig. 2C, left) to express (Fig. 2C, middle). We demonstrated a simulation example with 35 species surviving in the final steady state after the community assembly initialized with 10 000 species (Fig. 2C, right).
We applied the taxonomic profile, GCN, and PCN for the surviving 35 species to our computational pipeline, finding that the three modeled protein function types were correctly classified as three clusters (60 out of 60 were correct) by the Gaussian mixture model in both the comparison of network degree (Fig. 2D) and FR (Fig. 2E). We emphasize that the observed three functional clusters arise from community assembly. When we randomly picked 35 species (same as the number of surviving species) from the initial pool with equal abundances without assembly, niche functions cannot be distinguished from essential functions (Fig. 2F and G). Even when assigning the same randomly picked 35 species with the same abundances of surviving species, we still cannot differentiate these two functional types (Supplementary Fig. 2). Our findings held when varying (1) the number of species and functions or (2) model parameters 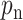 ,
, 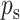 , and
, and 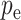 , affirming the method’s robustness in distinguishing function types (Supplementary Figs 3–5).
, affirming the method’s robustness in distinguishing function types (Supplementary Figs 3–5).
Three protein functional clusters observed in human gut microbiomes
Next, we validated our computational pipeline on real data of human mucosal-luminal interface samples previously collected from the ascending colon of four children [14, 33]. Here we focused on the genus level and annotated the identified proteins from metagenomic and metaproteomic data via the clusters of orthologous genes (COGs) database [47, 48]. We chose the genus level due to widely shared peptide sequences across species (Supplementary Fig. 6). We searched metagenomic reads and metaproteomic peptides against the IGC database of the human gut microbiome [49] to generate the GCN and PCN [14] and took the intersected COGs between the two networks. Taxonomic assignment was performed using the “protein-peptide bridge” method as described previously [14]. Our analysis centers on subject HM454, identifying 1542 intersected COGs in both the GCN and PCN and obtaining a taxonomic profile of 85 genera using MetaPhlAn2 [50]. The connectance (i.e. the number of edges divided by the maximal number of possible edges) of the GCN (or PCN) is 0.220 (or 0.049), respectively (Fig. 3A and B). The GCN displayed a higher nestedness (nestedness metric NODF [51]=0.667) than the PCN (NODF = 0.453). More details about data processing and NODF are in Supplementary Information.
Figure 3.

Real data of the human gut microbiome showing three clusters on the plot that compares FRg with FRp; metagenome and metaproteome of subject HM454 mucosal-luminal interface samples [33] were used to construct GCN and PCN, respectively; (A) the GCN shows if a genus owns (or doesn’t own) a COG as its genomic capacity, which is filled (or empty); the GCN matrix is ordered to have decreasing network degrees for both genera and COGs; (B) the PCN shows if a genus expresses (or doesn’t express) a COG as its protein function, which is filled (or empty); the PCN matrix follows the same order as the GCN; (C) differences in network degree for most COGs are large; kGCN is the network degree of each COG in the GCN (i.e. the number of genera owning each COG in the GCN); kPCN is the network degree of each COG in the PCN (i.e. the number of genera owning each COG in the PCN); (D) FRg is larger than FRp for most COGs; three functional clusters are predicted by the Gaussian mixture model with three clusters fitted on synthetic data; the transparent large circles represent centroids of three clusters; (E) the relationship between FRp and network degree of PCN for COGs is not monotonic.
The network degree analysis revealed a general decline from GCN to PCN, with 804 out of 1542 COGs having 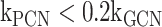 (Fig. 3C; Supplementary Data 1). This decline greatly influences FR but does not fully explain why many COGs have
(Fig. 3C; Supplementary Data 1). This decline greatly influences FR but does not fully explain why many COGs have 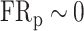 (744 out of 1542 have
(744 out of 1542 have 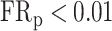 in Fig. 3D) and kPCN nonlinearly correlates with
in Fig. 3D) and kPCN nonlinearly correlates with 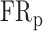 (Fig. 3E). For example, for L-arabinose isomerase (COG2160), its kPCN [7] is fairly close to kGCN [8], but its
(Fig. 3E). For example, for L-arabinose isomerase (COG2160), its kPCN [7] is fairly close to kGCN [8], but its 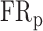 (0.04) is much lower than
(0.04) is much lower than 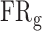 (0.23) since the genus Blautia (relative abundance = 22%) did not express L-arabinose isomerase, even if it has this capacity encoded in its genome.
(0.23) since the genus Blautia (relative abundance = 22%) did not express L-arabinose isomerase, even if it has this capacity encoded in its genome.
Using the Gaussian mixture model fitted on simulated data, we categorized all protein families into three clusters (Fig. 3C and D). Although clusters on real data are not as distinct as on simulated data, the relative positioning of the three clusters (shaded areas in Fig. 3C and D) agrees well with our hypothesis (Fig. 1). The weaker clustering might result from a greater variation in kGCN (or 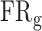 ) for real data (Fig. 3C and D) than that for simulated data (Fig. 2D and E).
) for real data (Fig. 3C and D) than that for simulated data (Fig. 2D and E).
Some COGs have 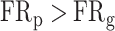 (Fig. 3C and D), contradicting the sub-sampling argument for the gene expression.
(Fig. 3C and D), contradicting the sub-sampling argument for the gene expression. 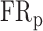 should not exceed
should not exceed 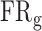 if the PCN was a proper subgraph of the GCN. This contradiction may stem from limitations in metagenomic sequencing and metaproteomic identification depths, as both metagenomics and metaproteomics require sufficient depth to detect genes or proteins, respectively. We tested how the lower detection capability of metaproteomics or metagenomics influences the FR by varying the protein or gene abundance percentile threshold (PAPT or GAPT), which denotes the percentage of most abundant proteins or genes being kept. As PAPT or GAPT decreases, FRp or FRg drops respectively (Supplementary Fig. 15). When GAPT decreases, we observed more proteins with FRp greater than their FRg.
if the PCN was a proper subgraph of the GCN. This contradiction may stem from limitations in metagenomic sequencing and metaproteomic identification depths, as both metagenomics and metaproteomics require sufficient depth to detect genes or proteins, respectively. We tested how the lower detection capability of metaproteomics or metagenomics influences the FR by varying the protein or gene abundance percentile threshold (PAPT or GAPT), which denotes the percentage of most abundant proteins or genes being kept. As PAPT or GAPT decreases, FRp or FRg drops respectively (Supplementary Fig. 15). When GAPT decreases, we observed more proteins with FRp greater than their FRg.
Validating three functional clusters observed in human gut microbiomes
Our computational pipeline accurately assigns functional clusters for protein families, agreeing with their known biological functions. For example, COG0539 (ribosomal protein S1) was assigned as the essential function, which is essential for translational initiation [17–19, 52, 53]. Another example is the assignment of COG1116 (ABC-type nitrate/sulfonate/bicarbonate transport system) [34] as a niche function, whose expression has been shown to be selectively enriched for a few microbial species [54].
The pipeline’s classifications were systematically validated against well-established biological roles of specific protein families: (i) ABC-type transporters are niche proteins due to their connection with ecological metabolic niches [34–36]; (ii) ribosomal proteins are essential proteins because they are indispensable for the microbial growth [55, 56]; (iii) PTS (phosphotransferase system) proteins are specialist proteins because an evolutionary study has shown that various species within the same genus even possess a different set of PTS proteins [57]. To evaluate our pipeline’s performance, we quantified its accuracy in assigning these protein families (ABC-type transporters, ribosomal proteins, or PTS proteins) against the assumed “ground-truth” functions (niche, essential, or specialist functions, respectively).
For HM454, our computational pipeline based on the 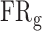 /
/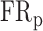 plot correctly categorizes 81 of 122 COGs belonging to ABC-type transporters, ribosomal proteins, or PTS proteins. In comparison, when classifying functions based on the
plot correctly categorizes 81 of 122 COGs belonging to ABC-type transporters, ribosomal proteins, or PTS proteins. In comparison, when classifying functions based on the 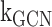 /
/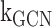 plot, 74 COGs were correctly assigned, slightly worse than that based on the
plot, 74 COGs were correctly assigned, slightly worse than that based on the 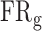 /
/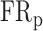 plot. Specifically, 26 of 53 COGs belonging to ABC-type transporters are classified as niche functions. The fraction of ribosomal proteins classified to the cluster of essential functions is 83.0%(=44/53). For the PTS proteins, among the identified 16 COGs, 11 are classified as specialist functions.
plot. Specifically, 26 of 53 COGs belonging to ABC-type transporters are classified as niche functions. The fraction of ribosomal proteins classified to the cluster of essential functions is 83.0%(=44/53). For the PTS proteins, among the identified 16 COGs, 11 are classified as specialist functions.
Alternative clustering and classification methods
Alternatively, we explored the unsupervised K-mean clustering with K = 3, which captured the three representative functional clusters with their positions agreeing with our expectations (Supplementary Fig. 13). We also designed a supervised classifier based on quadratic discriminant analysis (QDA). QDA, trained on ABC-type transporters, PTS proteins, and ribosomal proteins as the niche, specialist, and essential functions, generated clusters closely resembling those from the Gaussian mixture model (Supplementary Fig. 14). For HM454, the K-mean clustering categorizes 48 of these 122 COGs that are ABC-type transporters, ribosomal proteins, or PTS proteins into clusters, respectively, representing niche, essential, or specialist functions (i.e. the accuracy is 39.3%). For the QDA classifier, the accuracy is 59.0% (=72/122). Thus, we select the Gaussian mixture model as the classification method because of its superior accuracy (66.4% = 81/122).
Comparing FRg with FRp identifies ecological niches and metabolic essentiality
We focused on analyzing ABC-type transporters [34–36] and ribosomal proteins [17–19]. ABC-type transporters are energy-requiring transporter proteins that allow microbes to exploit specific niches like glucose uptake [34–36]. For HM454, we indeed found that kGCN for all ABC-type transporters is much larger than their kPCN (Fig. 4A). Similarly, we also found that their 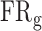 values are much larger than their
values are much larger than their 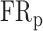 values, classifying many transporter proteins as niche functions (Fig. 4B). Some transporter proteins were classified as specialist functions (blue dots in Fig. 4B) due to the specialization on the gene level, which is carried to the protein level. Some transporter proteins were classified as essential functions (green dots in Fig. 4B). One example is the ABC-type Fe3+/spermidine/putrescine transporter (COG3842), as iron is essential for bacteria to function as a co-factor in iron-containing proteins [58, 59].
values, classifying many transporter proteins as niche functions (Fig. 4B). Some transporter proteins were classified as specialist functions (blue dots in Fig. 4B) due to the specialization on the gene level, which is carried to the protein level. Some transporter proteins were classified as essential functions (green dots in Fig. 4B). One example is the ABC-type Fe3+/spermidine/putrescine transporter (COG3842), as iron is essential for bacteria to function as a co-factor in iron-containing proteins [58, 59].
Figure 4.

Comparison of network degree and FR between the gene and protein level for many protein families including ABC-type transporters and ribosomal proteins from the human gut microbiome; (A) network degrees in GCN are larger than network degrees in PCN for most ABC-type transporter COGs; kGCN (or kPCN) is the network degree of each COG in the GCN (or PCN); (B) FRg is larger than FRp for most ABC-type transporter COGs; (C and D) the distribution of network degrees and functional redundancies (violin plots and boxplots) for ABC-type transporter COGs shows a significantly huge reduction from kGCN to kPCN or from FRg to FRp; (E) network degrees in GCN are comparable with that in PCN for most ribosomal protein COGs; (F) FRg is comparable with FRp for most ribosomal protein COGs; points in scatter plots are colored by the same colors used in Fig. 3d; (G and H) the distribution of network degrees and functional redundancies (violin plots and boxplots) for ribosomal protein COGs shows no significant reduction from kGCN to kPCN or from FRg to FRp; (I) the fraction of assigned specialist, niche, or essential functions based on comparing network degrees kGCN and kPCN for many protein families; (J) the fraction of assigned specialist, niche, or essential functions based on comparing functional redundancies FRg and FRp for many protein families; in all boxplots, the middle white dot is the median, the lower and upper hinges correspond to the first and third quartiles, and the black line ranges from the 1.5 × IQR (where IQR is the interquartile range) below the lower hinge to 1.5 × IQR above the upper hinge; all violin plots are smoothed by a kernel density estimator and 0 is set as the lower bound; all statistical analyses were performed using the two-sided Mann–Whitney-Wilcoxon U test with Bonferroni correction between genomic capacity (GCN) and protein functions (PCN); P-values obtained from the test is divided into five groups: (1) 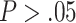 (ns), (2)
(ns), (2) 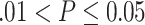 (*), (3)
(*), (3) 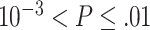 (**), (4)
(**), (4) 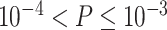 (***), and (5)
(***), and (5) 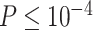 (****); network degree comparison of ABC transporters:
(****); network degree comparison of ABC transporters: 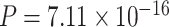 ; network degree comparison of ribosomal proteins: proteins:
; network degree comparison of ribosomal proteins: proteins: 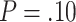 ; redundancy comparison of ABC transporters:
; redundancy comparison of ABC transporters: 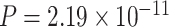 ; redundancy comparison of ribosomal proteins:
; redundancy comparison of ribosomal proteins: 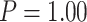 .
.
Ribosomal proteins, critical for protein synthesis and microbial growth [55, 56], showed little variance between kGCN and kPCN (the mean and the standard deviation of the relative difference 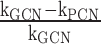 is
is 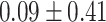 ; Fig. 4E), with most correctly classified as essential (44 out 53 COGs in Fig. 4F). Notably, two ribosomal proteins (L28 and L34), which have been reported as non-essential to microbes such as E. coli [17, 53, 60], were accurately classified as non-essential proteins (red dots in Fig. 4E). Certain specialized ribosomal proteins in microbial genomes continue to be specialized on the protein level and thus were classified as specialist functions.
; Fig. 4E), with most correctly classified as essential (44 out 53 COGs in Fig. 4F). Notably, two ribosomal proteins (L28 and L34), which have been reported as non-essential to microbes such as E. coli [17, 53, 60], were accurately classified as non-essential proteins (red dots in Fig. 4E). Certain specialized ribosomal proteins in microbial genomes continue to be specialized on the protein level and thus were classified as specialist functions.
Alternatively, we looked at the distribution of network degrees (Fig. 4C and G) and FR (Fig. 4D and H). For ABC-type transporters, the distribution of kPCN is close to 0 (median of 2), while the median of kGCN is 25. For ribosomal proteins, the distribution of kPCN (median is 12) is similar to kGCN (median is 14). For ABC-type transporters, the distribution of 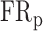 is close to 0 (with a median ~ 0.01), while the median of
is close to 0 (with a median ~ 0.01), while the median of 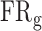 is around 0.30. For ribosomal proteins, the distribution of
is around 0.30. For ribosomal proteins, the distribution of 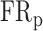 (median ~ 0.20) is similar to the distribution of
(median ~ 0.20) is similar to the distribution of 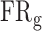 (median ~ 0.21).
(median ~ 0.21).
The similar patterns are also true for the other three individuals (Supplementary Figs 8–10; Supplementary Data 2–4). Variations in the FRg /FRp plot across individuals (Supplementary Figs 8–10) are likely due to differences in gut environments, diets, and microbial composition. Note that HM503 is an outlier due to its lower diversity (36 genera versus the average of 56.7) (Supplementary Fig. 11). Similarly, the Shannon diversity index for HM503 is only 1.41, lower than other individuals (mean and standard deviation are 2.05 and 0.18). The lower diversity in HM503 leads to fewer taxa owning the same function on average, resulting in lower FRg and FRp values.
Extending our analysis to additional protein families, we discovered that proteins linked to glycolysis, RNA polymerase, and the Tricarboxylic Acid cycle (TCA) cycle displayed patterns similar to ribosomal proteins (Fig. 4I and J). By contrast, proteins associated with sugar utilization, glycosyl hydrolase, and aromatic amino acid biosynthesis exhibited patterns more akin to ABC-type transporters. We also analyzed the classification results for the unknown COG functions (with the category “S: Function Unknown”). Out of the 104 unknown COGs in our analysis, the majority are classified as specialist (43/104) or niche functions (40/104), with a smaller portion identified as essential functions (21/104).
We also confirmed our results using the KEGG Orthology (KO) annotation [61–64], which has a lower annotation rate (78%) than COG (92%). For HM454, our computational pipeline categorizes 75 of these 126 KOs that are ABC-type transporters, ribosomal proteins, or PTS proteins into clusters, respectively, representing niche, essential, or specialist functions, similar to the classification accuracy based on the COG. The contrasting difference between ABC-type transporters and ribosomal proteins is well preserved (see Supplementary Fig. 7). Additionally, the distribution of 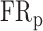 shows a dramatic difference across KO groups (Supplementary Fig. 12). Some ecologically strongly selected KO groups such as ABC transporters have small
shows a dramatic difference across KO groups (Supplementary Fig. 12). Some ecologically strongly selected KO groups such as ABC transporters have small 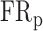 (Supplementary Fig. 12). As a comparison, proteins from aminoacyl-tRNA biosynthesis [15, 16], glycolysis [20, 21], and ribosomes [17–19] have large
(Supplementary Fig. 12). As a comparison, proteins from aminoacyl-tRNA biosynthesis [15, 16], glycolysis [20, 21], and ribosomes [17–19] have large 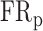 and huge variations within each group (Supplementary Fig. 12).
and huge variations within each group (Supplementary Fig. 12).
Validating our method on the mouse gut microbiome
In testing our method’s feasibility in other microbial communities, we leveraged a metaproteomic dataset from mice gavaged with a synthetic microbiome comprising 20 sequenced bacteria [42]. Since this study lacks paired metagenomes, we used its 16S rRNA gene sequencing data with individual genomes to infer its metagenome. Here we focused on the strain level because peptides of different strains in this simple synthetic gut microbiome can be distinguished. We relied on the comparison between 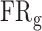 and
and 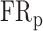 to generate the distribution of functional clusters across many protein families for this dataset (Fig. 5N). The results mirrored those of human gut microbiomes, especially the contrasting patterns between ABC-type transporters and ribosomal proteins (Fig. 5).
to generate the distribution of functional clusters across many protein families for this dataset (Fig. 5N). The results mirrored those of human gut microbiomes, especially the contrasting patterns between ABC-type transporters and ribosomal proteins (Fig. 5).
Figure 5.
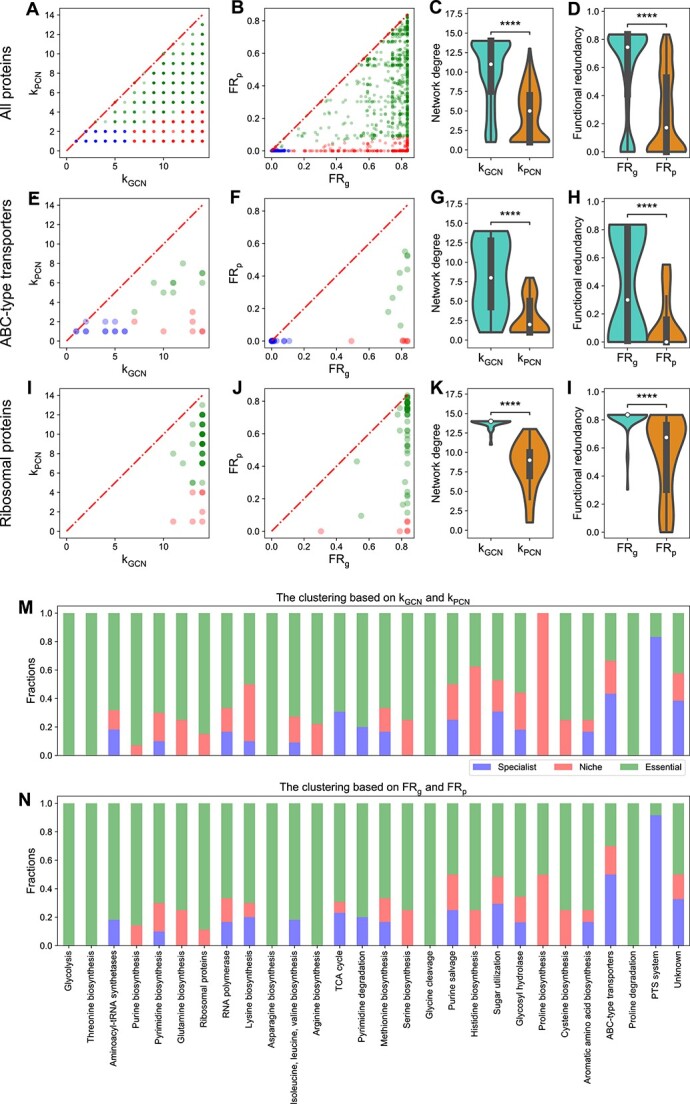
Comparison of network degree and FR between the gene and protein level for many protein families including ABC-type transporters and ribosomal proteins from the synthetic mouse gut microbial community; (A) comparison between network degrees of all COGs in the GCN (kGCN) and network degrees of all COGs in the PCN (kPCN); (B) comparison between functional redundancies of all COGs in the GCN (FRg) and functional redundancies of all COGs in the PCN (FRp); (C and D) the distribution of network degrees and functional redundancies (violin plots and boxplots) for all COGs; (E–H) comparison between kGCN and kPCN, comparison between FRg and FRp, the distribution of network degrees, and the distribution of functional redundancies for ABC-type transporter COGs; (I—L) comparison between kGCN and kPCN, comparison between FRg and FRp, the distribution of network degrees, and the distribution of functional redundancies for ribosomal protein transporter COGs; (M) the fraction of assigned specialist, niche, or essential functions based on comparing network degrees kGCN and kPCN for many protein families; (N) the fraction of assigned specialist, niche, or essential functions based on comparing functional redundancies FRg and FRp for many protein families; in all boxplots, the middle white dot is the median, the lower and upper hinges correspond to the first and third quartiles, and the black line ranges from the 1.5 × IQR (where IQR is the interquartile range) below the lower hinge to 1.5 × IQR above the upper hinge; all violin plots are smoothed by a kernel density estimator and 0 is set as the lower bound; all statistical analyses were performed using the two-sided Mann–Whitney-Wilcoxon U test with Bonferroni correction between genomic capacity (GCN) and protein functions (PCN); P-values obtained from the test are divided into five groups: (1) 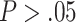 (ns), (2)
(ns), (2) 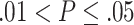 (*), (3)
(*), (3) 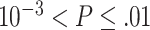 (**), (4)
(**), (4) 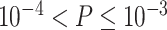 (***), and (5)
(***), and (5) 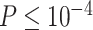 (****).
(****).
Response of community and protein abundance to the introduction of sugars
After identifying niche functions through our computational pipeline, we explored using nutrients associated with niche functions to manipulate the community structure. In ecology, a niche is often defined as an abiotic and biotic factor that supports the survival of species [9, 65–67]. Therefore, niche functions are associated with corresponding limiting resources involved in those functions. For example, COG1879 (ABC-type sugar transporter) is categorized as a niche function due to microbial competition for sugars (Supplementary Fig. 17). Here, we leveraged the in vitro community and studied how expression levels of ATP-type transporters respond to supplied sugars so that a microbial taxon can achieve a better living strategy.
Using the RapidAIM V2.0 approach [68], which replicates the functional profiles of individual gut microbiomes in vitro [38], we cultured three individual human gut microbiota samples and used a semi-automated metaproteomics workflow to observe how taxon-specific proteins respond to the presence of glucose, fructose, and kestose (Fig. 6A). Samples were cultured in technical triplicates, and protein abundances were quantified at 0, 1, 5, 12, and 24 h using 11-plex tandem mass tag (TMT11plex) [40] for a total of 189 samples. We analyzed the Bray–Curtis dissimilarity of metaproteomes over time, finding that more complex sugars induce more pronounced alterations in protein profiles (Supplementary Fig. 16).
Figure 6.
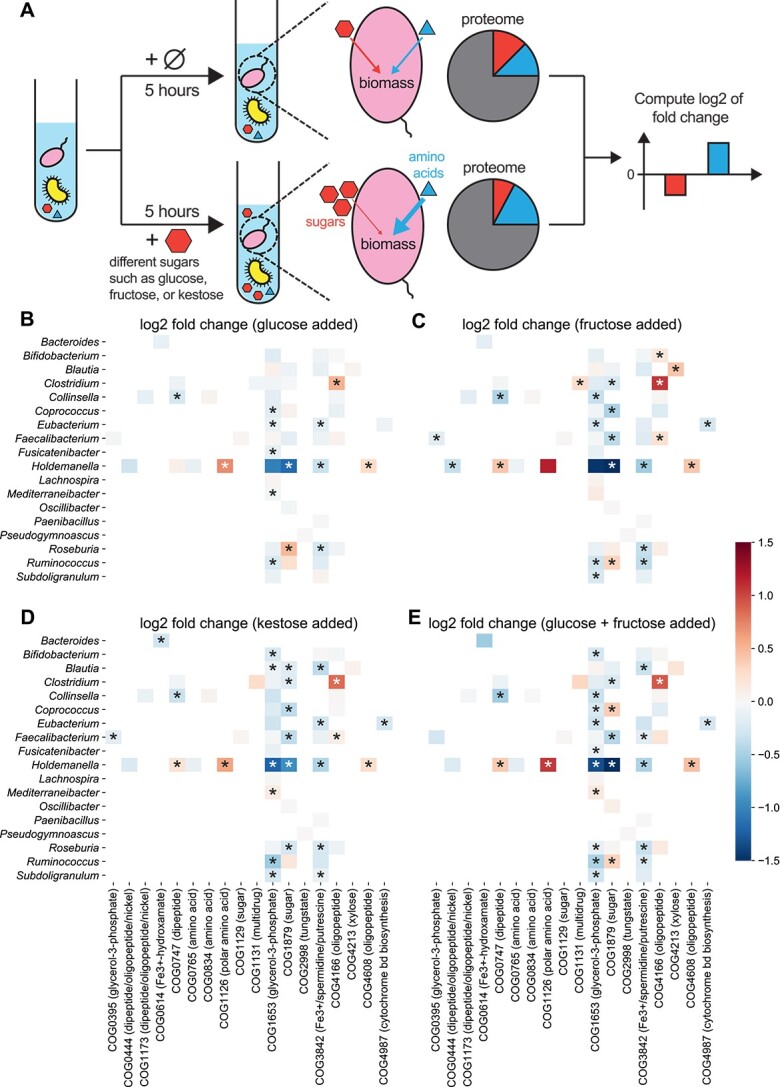
Microbes modify their expression for ABC-type transporters to adapt to added sugars; all heatmaps share the same color bar on the right; (A) schematic of in vitro cultures of a collected human gut microbiome; in the treatment group, one sugar is added to the community; metaproteomic measurements 5 h later were used to compare the intensity of each taxon-specific protein using the log2 fold change of each protein’s fraction (i.e. normalized intensity over each genus) from the treatment group divided by that from the control group; Log2 fold changes of ABC-type transporters were computed 5 h after (B) glucose, (C) fructose, (D) kestose, or (E) glucose and fructose is added; the transported metabolites for each COG are added to the brackets.
To reflect the effect of introduced sugars on protein abundances, we used log2 of fold change in normalized protein abundances/intensities (see Supplemental Methods for details) between the treatment and control group (Fig. 6). We hypothesized that excessive sugars remove the growth limitation on carbon resources, prompting microbes to upregulate other transporters for uptaking more other scarce resources (e.g. nitrogen or amino acids) for better growth (Fig. 6A). We analyzed log2 fold changes of ABC-type transporters 5 h later after sugar introduction (Fig. 6B–D), with most COGs close to zero. Among seven significantly influenced COGs, COG1126 (ABC-type polar amino acid transport system) is the only one that is revealed to be a niche function. Focusing on COG1126, we found that it is specialized by the genus Holdemanella. Holdemanella benefits from upregulating COG1126, as the proportion of Holdemanella proteins significantly increases from 13.5%(± 0.06%) for the control to 15.8%(± 0.08%) with the added glucose (P-value = .04, Mann–Whitney U test applied).
We observed that adding fructose, glucose and fructose, or kestose alters ABC-type transporters’ expression similarly to glucose alone (Fig. 6). The correlation in log2 fold changes of ABC-type transporters between different added sugars is significant (Supplementary Fig. 18). Notably, complex sugars trigger more significant fold changes in microbial protein expression. This pattern persists for metaproteomic measurements 12 and 24 h later, while the fold changes 1 hour later are less significant (Supplementary Fig. 19; P value <.01 for four sugar-adding scenarios, Mann–Whitney U test applied). Additionally, the overwhelmingly positive log2 fold changes of ribosomal proteins (Supplementary Fig. 20; P-value <10−4 for four sugar-adding scenarios, one-sample Wilcoxon test applied) probably imply faster microbial growth when simple sugars are supplied [69, 70].
Discussion
We developed a computational pipeline to classify protein families as specialist, niche, and essential functions by comparing 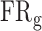 with
with 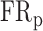 . This approach supplements traditional methods that test metabolic essentiality by gene knockout [17–19] and identify limiting resources by measuring biomass changes upon resource supplies [71–74]. We first illustrated this method on synthetic data and then validated it using real datasets of human and mouse gut microbiomes. We acknowledge a limitation in our validation process—the reliance on limited available literature. Hence, our classification should be seen as a preliminary framework that is open to refinement as the investigation of protein families’ functions improves.
. This approach supplements traditional methods that test metabolic essentiality by gene knockout [17–19] and identify limiting resources by measuring biomass changes upon resource supplies [71–74]. We first illustrated this method on synthetic data and then validated it using real datasets of human and mouse gut microbiomes. We acknowledge a limitation in our validation process—the reliance on limited available literature. Hence, our classification should be seen as a preliminary framework that is open to refinement as the investigation of protein families’ functions improves.
Our findings bridge the gap between the ecological niche theory, which posits that each resource (or niche) can only be occupied by one species for steady-state conditions [67, 75, 76], and the FR revealed by shared functions among microbial genomes [11, 12]. We solved this dilemma by showing niche proteins usually have very small 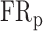 and large
and large 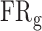 . Additionally, our ecological framework combines genomic capacity and protein functions together by introducing species with sub-sampled functions. The model framework accounts for selective expression due to different environmental conditions [77–79] or evolved strains with distinct metabolic niches [26, 27, 80], reconciling phenotype-focused ecological models with genetic data.
. Additionally, our ecological framework combines genomic capacity and protein functions together by introducing species with sub-sampled functions. The model framework accounts for selective expression due to different environmental conditions [77–79] or evolved strains with distinct metabolic niches [26, 27, 80], reconciling phenotype-focused ecological models with genetic data.
The observed case of FRp > FRg for some COGs could stem from using MetaProIQ, a general microbiome catalog, for metaproteome analysis. Although MetaProIQ facilitates the identification of proteins from various gut microbes, the general search against it may cause anomalies. In contrast, directly searching metaproteomic data against the gene calls from the paired metagenome may deliver more accurate identifications. However, this strategy suffers when the metagenomic sequencing is incomplete, leading to undetected proteins due to missing genes. In our human gut microbiome datasets, limited sequencing depth might cause such issues. For the synthetic mouse gut communities with complete genomes for each microbial strain, we matched metaproteome to microbial genomes and did not encounter any case of FRp > FRg, highlighting the effectiveness of this approach in contexts with complete genomic data.
In this work, we only validated our pipeline on gut-related biomes due to the limited accessibility of paired metagenome-metaproteome in other environments. In nutrient-poor environments, where ribosomal proteins are less expressed [81, 82], essential proteins like ribosomes may be harder to detect, causing potential detection biases. This aspect warrants further investigation to ensure the robustness and applicability of our method in diverse ecological settings. Other technical limitations can also impact clustering accuracy. Smaller ribosomal proteins L28 and L34 could be detected less frequently in metaproteomics, and post-translational modifications may also result in missed cleavage and identification of peptides. Advances such as metaproteomics-assembled proteomes [83] may improve taxon-specific functional annotations and the accuracy of our clustering outcomes.
Supplementary Material
Acknowledgements
We thank Dr Janice Mayne for the help with providing biobanking samples, Dr Zhibin Ning for the help with running mass spectrometry, and Dr Joeselle Serrana for the help with processing the metagenomic data.
Contributor Information
Tong Wang, Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, United States.
Leyuan Li, State Key Laboratory of Medical Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences (Beijing), Beijing Institute of Lifeomics, Beijing 102206, China; School of Pharmaceutical Sciences and Ottawa Institute of Systems Biology, Faculty of Medicine, University of Ottawa, Ottawa, ON K1H8M5, Canada.
Daniel Figeys, School of Pharmaceutical Sciences and Ottawa Institute of Systems Biology, Faculty of Medicine, University of Ottawa, Ottawa, ON K1H8M5, Canada.
Yang-Yu Liu, Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, United States; Center for Artificial Intelligence and Modeling, Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Champaign, IL 61820, United States.
Author contributions
Yang-Yu Liu and Daniel Figeys supervised the study. Tong Wang and Yang-Yu Liu conceived the project. All authors designed the research. Leyuan Li prepared and curated the empirical data as well as performed all wet-lab experiments. Tong Wang analyzed all data and developed the ecological model. Tong Wang wrote the initial manuscript. All authors edited and approved the manuscript.
Conflicts of interest
The authors declare no competing interests. D.F. co-founded MedBiome Inc., a clinical microbiomics company.
Funding
D.F. acknowledges grants from Natural Sciences and Engineering Research Council of Canada (NSERC), and the Government of Canada through Genome Canada and the Ontario Genomics Institute (OGI-114 and OGI-149). D.F. acknowledges a Distinguished Research Chair from the University of Ottawa. Y.-Y.L. is supported by grants R01AI141529, R01HD093761, RF1AG067744, UH3OD023268, U19AI095219, and U01HL089856 from the National Institutes of Health, USA; a pilot grant from the Biology of Trauma Initiative of Broad Institute, USA; and the Office of the Assistant Secretary of Defense for Health Affairs, through the Traumatic Brain Injury and Psychological Health Research Program (Focused Program Award) under award no. (W81XWH-22-S-TBIPH2), endorsed by the Department of Defense, USA.
Data and code availability
All code for simulations used in this manuscript can be found at https://github.com/wt1005203/ecological_niches.
IRB determination/approval statement
The sample collection of human gut microbiomes was approved by the Research Ethics Board of the Children’s Hospital of Eastern Ontario (CHEO), Ottawa, ON, Canada. The written informed consent forms were obtained from their parents.
References
- 1. Tyson GW, Chapman J, Hugenholtz Pet al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004;428:37–43. 10.1038/nature02340 [DOI] [PubMed] [Google Scholar]
- 2. Flint HJ, Scott KP, Louis Pet al. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol 2012;9:577–89. 10.1038/nrgastro.2012.156 [DOI] [PubMed] [Google Scholar]
- 3. Lloyd-Price J, Arze C, Ananthakrishnan ANet al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019;569:655–62. 10.1038/s41586-019-1237-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paerl HW, Pinckney JL. A mini-review of microbial consortia: their roles in aquatic production and biogeochemical cycling. Microb Ecol 1996;31:225–47. 10.1007/BF00171569 [DOI] [PubMed] [Google Scholar]
- 5. Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth’s biogeochemical cycles. Science 2008;320:1034–9. 10.1126/science.1153213 [DOI] [PubMed] [Google Scholar]
- 6. Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016;353:1272–7. 10.1126/science.aaf4507 [DOI] [PubMed] [Google Scholar]
- 7. Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc Natl Acad Sci U S A 2013;110:12804–9. 10.1073/pnas.1300926110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pacheco AR, Moel M, Segrè D. Costless metabolic secretions as drivers of interspecies interactions in microbial ecosystems. Nat Commun 2019;10:103. 10.1038/s41467-018-07946-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fahimipour AK, Gross T. Mapping the bacterial metabolic niche space. Nat Commun 2020;11:4887. 10.1038/s41467-020-18695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Louca S, Jacques SM, Pires APet al. High taxonomic variability despite stable functional structure across microbial communities. Nat Ecol Evol 2016;1:15. 10.1038/s41559-016-0015 [DOI] [PubMed] [Google Scholar]
- 11. Louca S, Polz MF, Mazel Fet al. Function and functional redundancy in microbial systems. Nat Ecol Evol 2018;2:936–43. 10.1038/s41559-018-0519-1 [DOI] [PubMed] [Google Scholar]
- 12. Tian L, Wang XW, Wu AKet al. Deciphering functional redundancy in the human microbiome. Nat Commun 2020;11:6217. 10.1038/s41467-020-19940-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Franzosa EA, Morgan XC, Segata Net al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci U S A 2014;111:E2329–38. 10.1073/pnas.1319284111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li L, Wang T, Ning Zet al. Revealing proteome-level functional redundancy in the human gut microbiome using ultra-deep metaproteomics. Nat Commun 2023;14:3428. 10.1038/s41467-023-39149-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem 2000;69:617–50. 10.1146/annurev.biochem.69.1.617 [DOI] [PubMed] [Google Scholar]
- 16. Parker DJ, Lalanne JB, Kimura Set al. Growth-optimized aminoacyl-tRNA synthetase levels prevent maximal tRNA charging. Cell Syst 2020;11:121–130.e6. 10.1016/j.cels.2020.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shoji S, Dambacher CM, Shajani Zet al. Systematic chromosomal deletion of bacterial ribosomal protein genes. J Mol Biol 2011;413:751–61. 10.1016/j.jmb.2011.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dabbs ER. Mutants lacking individual ribosomal proteins as a tool to investigate ribosomal properties. Biochimie 1991;73:639–45. 10.1016/0300-9084(91)90043-Z [DOI] [PubMed] [Google Scholar]
- 19. Baba T, Ara T, Hasegawa Met al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2006;2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romano AH, Conway T. Evolution of carbohydrate metabolic pathways. Res Microbiol 1996;147:448–55. 10.1016/0923-2508(96)83998-2 [DOI] [PubMed] [Google Scholar]
- 21. Yan Y. Engineering Microbial Metabolism for Chemical Synthesis: Reviews and Perspectives. World Scientific, 2017, Singapore. [Google Scholar]
- 22. Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol 1961;3:318–56. 10.1016/S0022-2836(61)80072-7 [DOI] [PubMed] [Google Scholar]
- 23. Okano H, Hermsen R, Kochanowski Ket al. Regulation underlying hierarchical and simultaneous utilization of carbon substrates by flux sensors in Escherichia coli. Nat Microbiol 2020;5:206–15. 10.1038/s41564-019-0610-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumari S, Beatty CM, Browning DFet al. Regulation of acetyl coenzyme A synthetase in Escherichia coli. J Bacteriol 2000;182:4173–9. 10.1128/JB.182.15.4173-4179.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Starai VJ, Escalante-Semerena JC. Acetyl-coenzyme A synthetase (AMP forming). Cell Mol Life Sci 2004;61:2020–30. 10.1007/s00018-004-3448-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosenzweig RF, Sharp RR, Treves DSet al. Microbial evolution in a simple unstructured environment: genetic differentiation in Escherichia coli. Genetics 1994;137:903–17. 10.1093/genetics/137.4.903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Treves DS, Manning S, Adams J. Repeated evolution of an acetate-crossfeeding polymorphism in long-term populations of Escherichia coli. Mol Biol Evol 1998;15:789–97. 10.1093/oxfordjournals.molbev.a025984 [DOI] [PubMed] [Google Scholar]
- 28. Lin H, Castro NM, Bennett GNet al. Acetyl-CoA synthetase overexpression in Escherichia coli demonstrates more efficient acetate assimilation and lower acetate accumulation: a potential tool in metabolic engineering. Appl Microbiol Biotechnol 2006;71:870–4. 10.1007/s00253-005-0230-4 [DOI] [PubMed] [Google Scholar]
- 29. Kinnersley MA, Holben WE, Rosenzweig F. E Unibus Plurum: genomic analysis of an experimentally evolved polymorphism in Escherichia coli. PLoS Genet 2009;5:1–19.e1000713. 10.1371/journal.pgen.1000713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Penzlin A, Lindner MS, Doellinger Jet al. Pipasic: similarity and expression correction for strain-level identification and quantification in metaproteomics. Bioinformatics 2014;30:i149–56. 10.1093/bioinformatics/btu267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ram RJ, VerBerkmoes NC, Thelen MPet al. Community proteomics of a natural microbial biofilm. Science 2005;308:1915–20. 10.1126/science.1109070 [DOI] [PubMed] [Google Scholar]
- 32. VerBerkmoes NC, Denef VJ, Hettich RLet al. Systems biology: functional analysis of natural microbial consortia using community proteomics. Nat Rev Microbiol 2009;7:196–205. 10.1038/nrmicro2080 [DOI] [PubMed] [Google Scholar]
- 33. Zhang X, Ning Z, Mayne Jet al. MetaPro-IQ: a universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome 2016;4:31. 10.1186/s40168-016-0176-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steinsiek S, Bettenbrock K. Glucose transport in Escherichia coli mutant strains with defects in sugar transport systems. J Bacteriol 2012;194:5897–908. 10.1128/JB.01502-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fath MJ, Kolter R. ABC transporters: bacterial exporters. Microbiol Rev 1993;57:995–1017. 10.1128/mr.57.4.995-1017.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nikaido H. Maltose transport system of Escherichia coli: an ABC-type transporter. FEBS Lett 1994;346:55–8. 10.1016/0014-5793(94)00315-7 [DOI] [PubMed] [Google Scholar]
- 37. Zhang X, Walker K, Mayne Jet al. Evaluating live microbiota biobanking using an ex vivo microbiome assay and metaproteomics. Gut Microbes 2022;14:2035658. 10.1080/19490976.2022.2035658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li L, Abou-Samra E, Ning Zet al. An in vitro model maintaining taxon-specific functional activities of the gut microbiome. Nat Commun 2019;10:4146. 10.1038/s41467-019-12087-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang X, Li L, Mayne Jet al. Assessing the impact of protein extraction methods for human gut metaproteomics. J Proteome 2018;180:120–7. 10.1016/j.jprot.2017.07.001 [DOI] [PubMed] [Google Scholar]
- 40.Creskey M, Li L, Ning Z, et al. An economic and robust TMT labeling approach for high throughput proteomic and metaproteomic analysis. . Proteomics 2023;23:2200116. [DOI] [PubMed]
- 41. Perez-Riverol Y, Bai J, Bandla Cet al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 2022;50:D543–52. 10.1093/nar/gkab1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patnode ML, Beller ZW, Han NDet al. Interspecies competition impacts targeted manipulation of human gut bacteria by fiber-derived glycans. Cell 2019;179:59–73.e13. 10.1016/j.cell.2019.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 2016;11:2301–19. 10.1038/nprot.2016.136 [DOI] [PubMed] [Google Scholar]
- 44. Cheng K, Ning Z, Zhang Xet al. MetaLab 2.0 enables accurate post-translational modifications profiling in metaproteomics. J Am Soc Mass Spectrom 2020;31:1473–82. 10.1021/jasms.0c00083 [DOI] [PubMed] [Google Scholar]
- 45. Huang T, Choi M, Tzouros Met al. MSstatsTMT: statistical detection of differentially abundant proteins in experiments with isobaric labeling and multiple mixtures. Mol Cell Proteomics 2020;19:1706–23. 10.1074/mcp.RA120.002105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma W, Kim S, Chowdhury Set al. DreamAI: algorithm for the imputation of proteomics data 2020.07.21.214205 Preprint at 10.1101/2020.07.21.214205. 2021. [DOI]
- 47. Tatusov RL, Galperin MY, Natale DAet al. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 2000;28:33–6. 10.1093/nar/28.1.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tatusov RL, Fedorova ND, Jackson JDet al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003;4:41. 10.1186/1471-2105-4-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li J, Jia H, Cai Xet al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 2014;32:834–41. 10.1038/nbt.2942 [DOI] [PubMed] [Google Scholar]
- 50. Segata N, Waldron L, Ballarini Aet al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 2012;9:811–4. 10.1038/nmeth.2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Almeida-Neto M, Guimarães P, Guimarães PR Jret al. A consistent metric for nestedness analysis in ecological systems: reconciling concept and measurement. Oikos 2008;117:1227–39. 10.1111/j.0030-1299.2008.16644.x [DOI] [Google Scholar]
- 52. Sørensen MA, Fricke J, Pedersen S. Ribosomal protein S1 is required for translation of most, if not all, natural mRNAs in Escherichia coli in vivo11Edited by D Draper. J Mol Biol 1998;280:561–9. 10.1006/jmbi.1998.1909 [DOI] [PubMed] [Google Scholar]
- 53. Galperin MY, Wolf YI, Garushyants SKet al. Nonessential ribosomal proteins in bacteria and archaea identified using clusters of orthologous genes. J Bacteriol 2021;203:e00058–21. 10.1128/JB.00058-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bakhti SZ, Latifi-Navid S. Oral microbiota and helicobacter pylori in gastric carcinogenesis: what do we know and where next? BMC Microbiol 2021;21:1–15. 10.1186/s12866-021-02130-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cech TR. The ribosome is a ribozyme. Science 2000;289:878–9. 10.1126/science.289.5481.878 [DOI] [PubMed] [Google Scholar]
- 56. Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol 2012;13:355–69. 10.1038/nrm3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Barabote RD, Saier MH. Comparative genomic analyses of the bacterial phosphotransferase system. Microbiol Mol Biol Rev 2005;69:608–34. 10.1128/MMBR.69.4.608-634.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yilmaz B, Li H. Gut microbiota and iron: the crucial actors in health and disease. Pharmaceuticals 2018;11:98. 10.3390/ph11040098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Seyoum Y, Baye K, Humblot C. Iron homeostasis in host and gut bacteria—a complex interrelationship. Gut Microbes 2021;13:1–19. 10.1080/19490976.2021.1874855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bubunenko M, Baker T, Court DL. Essentiality of ribosomal and transcription antitermination proteins analyzed by systematic gene replacement in Escherichia coli. J Bacteriol 2007;189:2844–53. 10.1128/JB.01713-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kanehisa M. A database for post-genome analysis. Trends Genet 1997;13:375–6. 10.1016/S0168-9525(97)01223-7 [DOI] [PubMed] [Google Scholar]
- 62. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30. 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mao X, Cai T, Olyarchuk JGet al. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005;21:3787–93. 10.1093/bioinformatics/bti430 [DOI] [PubMed] [Google Scholar]
- 64. Kanehisa M, Sato Y, Kawashima Met al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44:D457–62. 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. E HG. The Multivariate Niche, Vol. 22. Cold Spring Harbor Symposia on Quantitative Biology, Cold Spring Harbor, New York, 1957, 415–21. [Google Scholar]
- 66. Leibold MA. The niche concept revisited: mechanistic models and community context. Ecology 1995;76:1371–82. 10.2307/1938141 [DOI] [Google Scholar]
- 67. Holt RD. Bringing the Hutchinsonian niche into the 21st century: ecological and evolutionary perspectives. Proc Natl Acad Sci U S A 2009;106:19659–65. 10.1073/pnas.0905137106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li L, Mayne J, Beltran A, Zhang X, Ning Z, Figeys D. RapidAIM 2.0: a high-throughput assay to study functional response of human gut microbiome to xenobiotics. Microbiome Res Rep 2024;3:26. 10.20517/mrr.2023.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. You C, Okano H, Hui Set al. Coordination of bacterial proteome with metabolism by cyclic AMP signalling. Nature 2013;500:301–6. 10.1038/nature12446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Basan M, Hui S, Okano Het al. Overflow metabolism in Escherichia coli results from efficient proteome allocation. Nature 2015;528:99–104. 10.1038/nature15765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tilman D. Resource competition between plankton algae: an experimental and theoretical approach. Ecology 1977;58:338–48. 10.2307/1935608 [DOI] [Google Scholar]
- 72. Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Ann Rev Microbiol 2004;58:611–47. 10.1146/annurev.micro.58.030603.123811 [DOI] [PubMed] [Google Scholar]
- 73. Hibbing ME, Fuqua C, Parsek MRet al. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 2010;8:15–25. 10.1038/nrmicro2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Smith RL, Smith TM. Elements of Ecology. San Francisco: Benjamin Cummings, 2003 [Google Scholar]
- 75. Hutchinson GE. The paradox of the plankton. Am Nat 1961;95:137–45. 10.1086/282171 [DOI] [Google Scholar]
- 76. Marsland R, Cui W, Mehta P. The minimum environmental perturbation principle: a new perspective on niche theory. Am Nat 2020;196:291–305. 10.1086/710093 [DOI] [PubMed] [Google Scholar]
- 77. Cappelletti V, Hauser T, Piazza Iet al. Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ. Cell 2021;184:545–559.e22. 10.1016/j.cell.2020.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schreiber F, Littmann S, Lavik Get al. Phenotypic heterogeneity driven by nutrient limitation promotes growth in fluctuating environments. Nat Microbiol 2016;1:1–7. 10.1038/nmicrobiol.2016.55 [DOI] [PubMed] [Google Scholar]
- 79. Gutierrez-Ríos RM, Freyre-Gonzalez JA, Resendis Oet al. Identification of regulatory network topological units coordinating the genome-wide transcriptional response to glucose in Escherichia coli. BMC Microbiol 2007;7:53. 10.1186/1471-2180-7-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Goyal A, Bittleston LS, Leventhal GEet al. Interactions between strains govern the eco-evolutionary dynamics of microbial communities. Elife 2022;11:e74987. 10.7554/eLife.74987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Basan M, Zhu M, Dai Xet al. Inflating bacterial cells by increased protein synthesis. Mol Syst Biol 2015;11:836. 10.15252/msb.20156178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Serbanescu D, Ojkic N, Banerjee S. Nutrient-dependent trade-offs between ribosomes and division protein synthesis control bacterial cell size and growth. Cell Rep 2020;32:108183. 10.1016/j.celrep.2020.108183 [DOI] [PubMed] [Google Scholar]
- 83. Armengaud J. Metaproteomics to understand how microbiota function: the crystal ball predicts a promising future. Environ Microbiol 2023;25:115–25. 10.1111/1462-2920.16238 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code for simulations used in this manuscript can be found at https://github.com/wt1005203/ecological_niches.