

Abstract
Given that novel anticancer therapies have different toxicity profiles and mechanisms of action, it is important to reconsider the current approaches for dose selection. In an effort to move away from considering the maximum tolerated dose (MTD) as the optimal dose, the Food and Drug Administration Project Optimus points to the need of incorporating long-term toxicity evaluation, given that many of these novel agents lead to late-onset or cumulative toxicities and there are no guidelines on how to handle them. Numerous methods have been proposed to handle late-onset toxicities in dose-finding clinical trials. A summary and comparison of these methods is provided. Moreover, using PI3K inhibitors as a case study, we show how late-onset toxicity can be integrated into the dose optimization strategy using current available approaches. We illustrate a re-design of this trial to compare the approach to those that only consider early toxicity outcomes and disregard late-onset toxicities. We also provide proposals going forward for dose optimization in early development of novel anticancer agents with considerations for late-onset toxicities.
Keywords: Dose optimization, Dose-finding, Oncology, Toxicity efficacy designs, Delayed Toxicities, Cumulative Toxicities
Introduction and background
The release of the recent Project Optimus guidelines from the Food and Drug Administration (FDA) calls for the evaluation of the approach that has been used for dose recommendations for novel oncology drugs.1 Many of the methods that have been proposed and that we continue to use today focus on the estimation of the maximum tolerated dose (MTD) based on the traditional chemotherapy treatment paradigm. With different toxicity profiles and mechanisms of action, the MTD may not be the optimal dose for novel anticancer therapies, such as targeted therapies and immunotherapies. Thus, it is important to reconsider the way we have been selecting doses along with the assumptions that are made in the process. This is particularly important given that post-approval dose-reduction considerations have happened for several drugs in oncology over the past years.2 While for chemotherapies, the assumption has been that most toxicities occur shortly after treatment initiation, this is not always true for targeted therapies and immunotherapies which have been associated with late-onset toxicities as well as cumulative toxicities.
The FDA Dose Optimization Guidelines do not explicitly provide guidelines for handling late-onset or cumulative toxicities. The definition of the dose limiting toxicity (DLT) evaluation window is a very important consideration in the context of dose-finding given that the tolerability of an optimal dose is defined relative to the specified window. To ensure that the majority of toxicities are captured, it is important to select a toxicity evaluation window that encompasses most of the toxicities. However, having long windows can threaten the ability to complete the studies in an adequate amount of time by substantially increasing trial duration given that dose-finding methods for cancer therapies are adaptive and sequential to ensure patient safety. We, and others, have evaluated the timing of toxicities using completed phase 1 and 2 clinical trials as case examples3–5 and have shown that targeted therapies can often lead to toxicities even 3 to 5 cycles after treatment initiation. Similarly, immunotherapies have also been shown to be associated with toxicity onset months after treatment initiation.6,7 Thus, selecting optimal doses based on toxicities from the first cycle, as has traditionally been done for chemotherapy dose-finding clinical trials, could lead to the recommendation of doses associated with higher than desired toxicities in the long run.
In this paper, we first present a motivating example of a drug that was approved and later was discovered to be not well tolerated in later clinical trials. This drug would have benefited from a longer DLT observation window and the use of a design that allows for the inclusion of late-onset toxicities. Afterwards, we present a systematic review of dose-finding methods in the setting of late-onset toxicity, re-design the motivating example using two of these designs, and perform a simulation study to illustrate the potential impact of not including late-onset toxicities in dose recommendations. Furthermore, Project Optimus particularly points out the violation regarding the assumption that increased toxicity leads to increased efficacy and the need to move away from the MTD paradigm and incorporate efficacy to optimize dose selection to ensure that we recommend an efficacious dose with minimum toxicity. Thus, in the last section, we discuss the best path forward for performing dose-finding and optimization in the drug development of novel anticancer treatments using current available approaches to ensure long-term tolerability.
Motivating example
In April 2022, the FDA published a briefing that it had prepared for the oncologic drugs advisory committee for Phosphatidylinositol 3-Kinase (PI3K) Inhibitors in hematologic malignancies which was the impetus behind the need for dose optimization.8 In the briefing, the drug development of PI3K inhibitors is discussed in light of their toxicities, the concerning effects on overall survival, and the inadequate optimization of the doses used, which have led to the recommendation of dose optimization studies after approval.
While several PI3K inhibitors exist, for simplicity we will focus on idelalisib which was approved by the FDA in 2014. Using idelalisib as an example of failure in the dose-finding process of new anticancer therapies, we review the studies that were used for its dose selection process. Three dose-finding trials of idelalisib monotherapy were done in patients with different hematologic malignancies: 64 patients with relapsed indolent non-Hodgkin lymphoma (iNHL),9 40 patients with relapsed/refractory chronic lymphocytic leukemia (CLL),10 and 54 patients with relapsed/mantle cell leukemia (CLL).11 These trials were done concurrently with similar designs evaluating eight doses of idelalisib (150 mg and 300 mg QD (28 days), 150 mg BID (21 days), and 50mg, 100 mg BID, 150 mg, 200 mg, 350 mg BID (28 days). All of them estimated the maximum tolerated dose (MTD) using similar definitions of DLT with a DLT window of up to day 28 even though the drug was to be administered for 48 weeks. Thus, the recommended dose was the MTD based solely on acute severe toxicity defined over the first 28 days of treatment. All three papers reported no DLT and indicated recommending the dose of 150mg BID based on clinical response, as well as pharmacokinetic and pharmacodynamic data. Unfortunately, the papers did not report adverse events and dose modifications by dose level, so the rate of late-onset toxicities at the recommended dose of 150mg BID was unclear. However, the FDA briefing indicated dose modifications even in later cycles and lower doses such as 100mg BID which also showed objective response. Idelalisib monotherapy was granted accelerated approval for patients with relapsed follicular B-cell non-Hodgkin lymphoma or small lymphocytic leukemia after at least 2 prior systemic therapies based on overall response rate in a single arm trial. It was also granted approval as a combination therapy with rituximab based on a randomized controlled trial which showed improvement in progression free survival in patients with relapsed chronic lymphocytic leukemia (CHL). However, the FDA briefing indicated that three later randomized controlled clinical trials showed increased risk of death, toxicity, and drug modifications for patients, leading to black box warnings in the USA. Boxed warnings, or black box warnings, refer to the most stringent safety-related warning that the Food and Drug Administration (FDA) can assign to an approved drug or medical device. It presents in the form of printed bold text within a black box on the package and prescription information of a drug or medical device.12 Thus currently, it is well known that PI3K inhibitors are associated with increased risk of adverse events and late-onset toxicities which can occur up to 6 months after initiation of treatment.13,14 The question is would a more tolerable dose of idelalisib have been selected with the inclusion of more cycles for the definition of DLT.
Methods for late-onset toxicity
Over the past decades, various methods have been proposed to handle late-onset toxicities in dose-finding trials while allowing for continuous accrual. To identify these methods, we conducted a systematic search of the literature using PubMed. The search terms were: ((”dose finding”[Title/Abstract]) OR (”dose-finding”[Title/Abstract]) OR (”phase I”[Title/Abstract]) OR (”phase 1”[Title/Abstract])) AND ((”late-onset toxicities”[Title/Abstract]) OR (”late onset toxicities”[Title/Abstract]) OR (”delayed toxicities”[Title/Abstract]) OR (”delayed toxicity”[Title/Abstract]) OR (”continuous patient enrollment”[Title/Abstract]) OR (”continuous accrual”[Title/Abstract]) OR (”Time-to-Event”[Title/Abstract]) OR (”Time to Event”[Title/Abstract]) OR (”Time to event”[Title/Abstract]) OR (”Time-to-event”[Title/Abstract])) with publication date filtered from 1 January 2000 to 30 September 2023. This yielded a total of 170 publications which were assessed for inclusion by two independent reviewers. In case of disagreement, the article was discussed and consensus was reached. After excluding clinical trial reports, reviews, and statistical publications related to phase I/II designs or phase I designs including other endpoints, modeling multiple treatment cycles, or drugs combination, a total of 18 publications were selected. Thirteen of these were model-based dose-finding designs and five were model-assisted designs. These are summarized in Table 1 along with the available corresponding software.
Table 1.
Summary of phase I dose-finding designs for late-onset toxicities. TITE: Time to Event; CRM: Continual Reassessment Method; PRT: Predicted Risk of Toxicity; EM: Expectation Maximization; DA: Data Augmentation; EWOC: Escalation with Overdose Control; IR: Isotonic Regression; CDP: Conaway, Dunbar, and Peddada; BOIN: Bayesian Optimal Interval; POD-i: Probability of Decision interval; CFO: Calibration Free Odds.
| Authors | Type of design | Method for pending data | Available software |
|---|---|---|---|
|
| |||
| Model-Based | |||
| Cheung & Chappell (2000) | TITE-CRM | Weighting scheme | R package dfcrm, R shiny TITE-CRM |
| Braun (2006) | Generalized TITE-CRM | Weighting scheme | None listed |
| Bekele et al. (2008) | PRT | Model predicted risk of toxicity | Windows software mdanderson.org |
| Mauguen et al. (2011) | TITE-EWOC | Weighting scheme | None listed |
| Yuan & Yin (2011) | EM-CRM | EM algorithm | None listed |
| Liu et al. (2013) | DA-CRM | Data augmentation process | Windows software mdanderson.org |
| Yin et al. (2013) | Fractional CRM | Kaplan-Meier estimator | R shiny fCRM, Github fCRM |
| Tighiouart et al. (2014) | EWOC-Time to toxicity | Cox proportional hazards model | Web tool EWOC |
| Zheng et al. 2016 | Bayesian logistic model | Weighting scheme | None listed |
| Chapple et al. (2019) | TITE-IR | Weighting scheme | R package titeir |
| Andrillon et al. (2020) | Survival-CRM | Survival exponential model | Github SurvivalCRM |
| Zhu et al. (2021) | Rolling-CRM | Truncated piecewise exponential model | R package crmPack |
| Wages et al. (2023) | TITE-CDP | Weighting scheme | R shiny TITE-CDP |
| Model-Assisted | |||
|
| |||
| Yuan et al. (2018) | TITE-BOIN | Bayesian data imputation | R shiny TITE-BOIN |
| Lin & Yuan (2020) | TITE-Keyboard | Weighting scheme | R shiny TITE-KEYBOARD |
| Zhou et al. (2021) | Late onset BOIN | Poisson process | None listed |
| Xu & Lin (2022) | POD-i3+3 | Weighting scheme | R Shiny podi3 |
| Jin & Yin (2023) | TITE-CFO | Imputation potential outcomes | Github CFO |
Many of these designs aim to recommend a single dose based on a binary toxicity endpoint over a prolonged DLT observation window. They mostly rely on weighted binomial likelihoods or imputation and data augmentation for missing data to handle partially observed follow-up during dose escalation. Other approaches model toxicity as time-to-event endpoints to handle late onset toxicities, some accounting for the cumulative effect of each consecutive treatment cycle,15–18 others considering the prolonged toxicity window as a whole.19,20 In contrast to designs using binary toxicity outcomes, designs modeling toxicity as time-to-event endpoints account for the timing of the toxicities in the dose selection process. Most of these methods have available software and can be readily applied to design dose-finding clinical trials. We refer the interested reader to Barnett et al’s recent work for a simulation-based comparison of late-onset toxicities designs.21
Re-designing the idelalisib dose-finding clinical trials
The dose-finding data for idelalisib suggest the need to define DLT for a longer duration to ensure the recommendation of a tolerable dose. To do so, we can apply the two most commonly used dose-finding designs presented in the previous section to select doses with acceptable toxicity over a prolonged observation window, TITE-CRM and TITE-BOIN.22,23 The TITE-CRM is an extension of the CRM design which is a model-based dose-finding design.22,24 It relies on modeling the dose-toxicity relationship after each cohort of patients to estimate the MTD based on all available data and assign this dose to the next cohort of patients. The TITE-CRM incorporates incomplete observations in the MTD estimation process during the trial by weighting them according to the available fraction of follow-up time. Therefore, it allows for continuous accrual and is ideal in the setting of late-onset toxicities. Similarly, TITE-BOIN is an extension of the model-assisted BOIN design23,25 which weights incomplete observations for MTD computations and dose assignments during the trial. BOIN designs do not rely on a dose-toxicity model for dose escalation decisions but rather assign doses based on the probability of DLT at the current dose level. The estimation of the MTD at the end of the trial is based on an isotonic regression using the complete data.
To illustrate the use of TITE-CRM and TITE-BOIN designs in the setting of the idelalisib motivating example, we extended the DLT window to 16-week based on the FDA briefing regarding delayed effects of the drug. We compared these results to those using the initial setting of the idelalisib trials, with a 4-week DLT window under various scenarios. A sample size of 60 patients was used based on the original dose-finding trials. A reduced sample size of 30 patients was also evaluated, in line with usual practice in phase I trials. For the application of the designs, we assumed a target DLT rate of 25% using cohorts of 3 patients. In the case of the TITE-CRM, scaled doses were selected using the method by Lee and Cheung assuming dose level 3 was the anticipated MTD, in line with 150mg BID being the recommended dose from the initial idelalisib trials, and ensuring that the methods will eventually select a dose that yields between 20% and 30% risk of DLT.26
The assumed true scenario for the occurrences of toxicity was obtained based on the data from the idelalisib trials in the FDA briefing. While the dose-finding trials had 8 dose levels with different schedules, we limited the re-design of this trial to 5 dose levels with the twice a day schedule. The cumulative incidence of toxicity at 16 weeks from treatment initiation for the 5 dose levels was (0.10, 0.25, 0.45, 0.60, 0.75), with dose level 2 being the true MTD. Based on these probability of toxicities at 16 weeks, we further derived the 4-week cumulative incidences of DLT, assuming either a constant risk of DLT over time (i.e. constant hazard) or a decreasing risk of DLT over time (i.e. decreasing hazard). As illustrated in Figure 2 for dose level 2, which is the true MTD at 16 weeks, a constant risk corresponds to a quarter of DLT events occurring by 4 weeks, while decreasing risks of DLT, corresponds to either 59% or 78% of toxicity events occurring by week 4. Table 2 displays the corresponding probabilities of DLT for each of the dose levels at 4 and 16 weeks under the different risk scenarios. In all cases, as expected, considering a shorter DLT window (i.e., 4 weeks instead of 16), reduces the proportion of events which can be observed with a shorter observation window. Under the scenario that 78% of DLTs are observed by 4 weeks, the MTD is dose level 2. However, under the scenario of constant risk over time, the MTD shifts up to dose level 5. We did not consider any scenarios in which the risk increases over time given that we would expect worse performance. Moreover, we assumed an accrual rate of 2 patients per month in line with the accrual rate for the idelalisib phase 1 trials.
Figure 2.
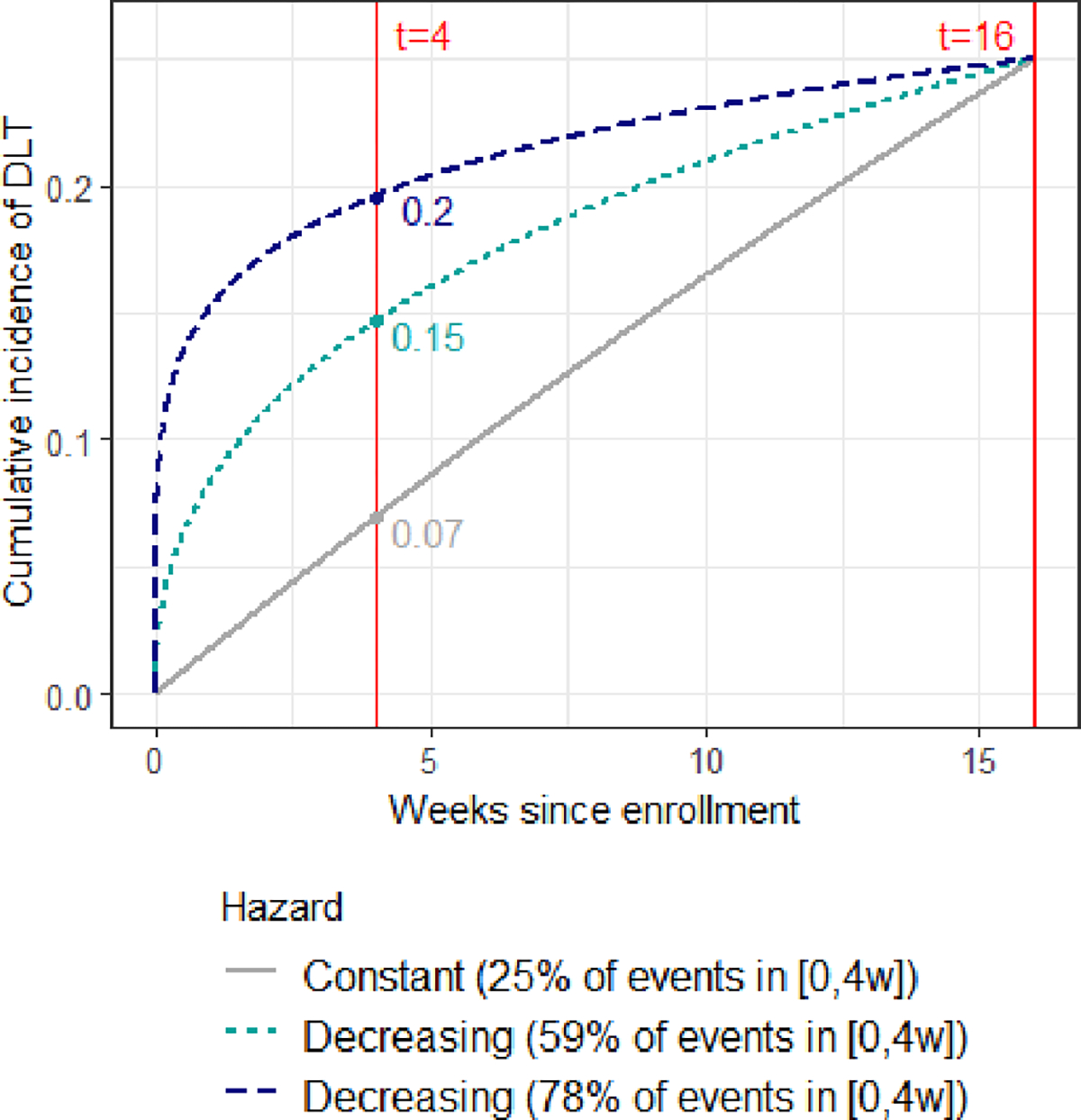
Cumulative incidence of dose limiting toxicities (DLT) in the simulation scenario for dose level 2, within a 16-week follow-up window. Cumulative incidence is plotted according to the shape of DLT hazard, given a common value of 25% at week 16. t refers to the time since enrollment, in weeks.
Table 2.
Simulations scenarios following the idelalisib example: cumulative incidence of DLT at the end of the observation window (Obs. window) by dose level (D1 to D5), and by length of window. Maximum tolerated dose is given in bold.
| Cumulative incidence of DLT at the end of window | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Obs. window | Hazard | D1 | D2 | D3 | D4 | D5 |
|
| ||||||
| 16 weeks | Constant (100% of toxicity events in [0,16w]) | 0.10 | 0.25 | 0.45 | 0.60 | 0.75 |
| 4 weeks | Decreasing (78% of toxicity events in [0,4w]) | 0.077 | 0.196 | 0.364 | 0.501 | 0.650 |
| 4 weeks | Decreasing (59% of toxicity events in [0,4w]) | 0.056 | 0.147 | 0.281 | 0.396 | 0.534 |
| 4 weeks | Constant (25% of toxicity events in [0,4w]) | 0.026 | 0.069 | 0.139 | 0.205 | 0.293 |
Figure 1 represents an example simulated trial using the TITE-CRM design based on a sample size of 30 patients with the above specifications and either a 16-week or a 4-week DLT window with constant risk over time. The trial started by assigning the first cohort of patients to the lowest dose level and escalated to dose level 2 for the second cohort. With a 4-week observation window, no DLTs were observed, and the third cohort of patients was assigned to dose level 3. However, with a longer observation window of 16-week, we observed that patient 4 experienced toxicity leading to a de-escalation for the next cohort of patients. At the end of the trial with a 4-week window, 80% of the patients were assigned to a dose higher than dose level 2, while when a longer toxicity observation window was considered all patients were treated at dose level 2 or below. Moreover, the recommended dose was dose level 4 based solely on a 4-week toxicity assessment and dose level 2 based on a 16-week toxicity assessment. It took about 64 weeks to complete the entire trial with the 4-week observation window, whereas an additional 12 weeks would have been needed if the 16-week windows had been used.
Figure 1.
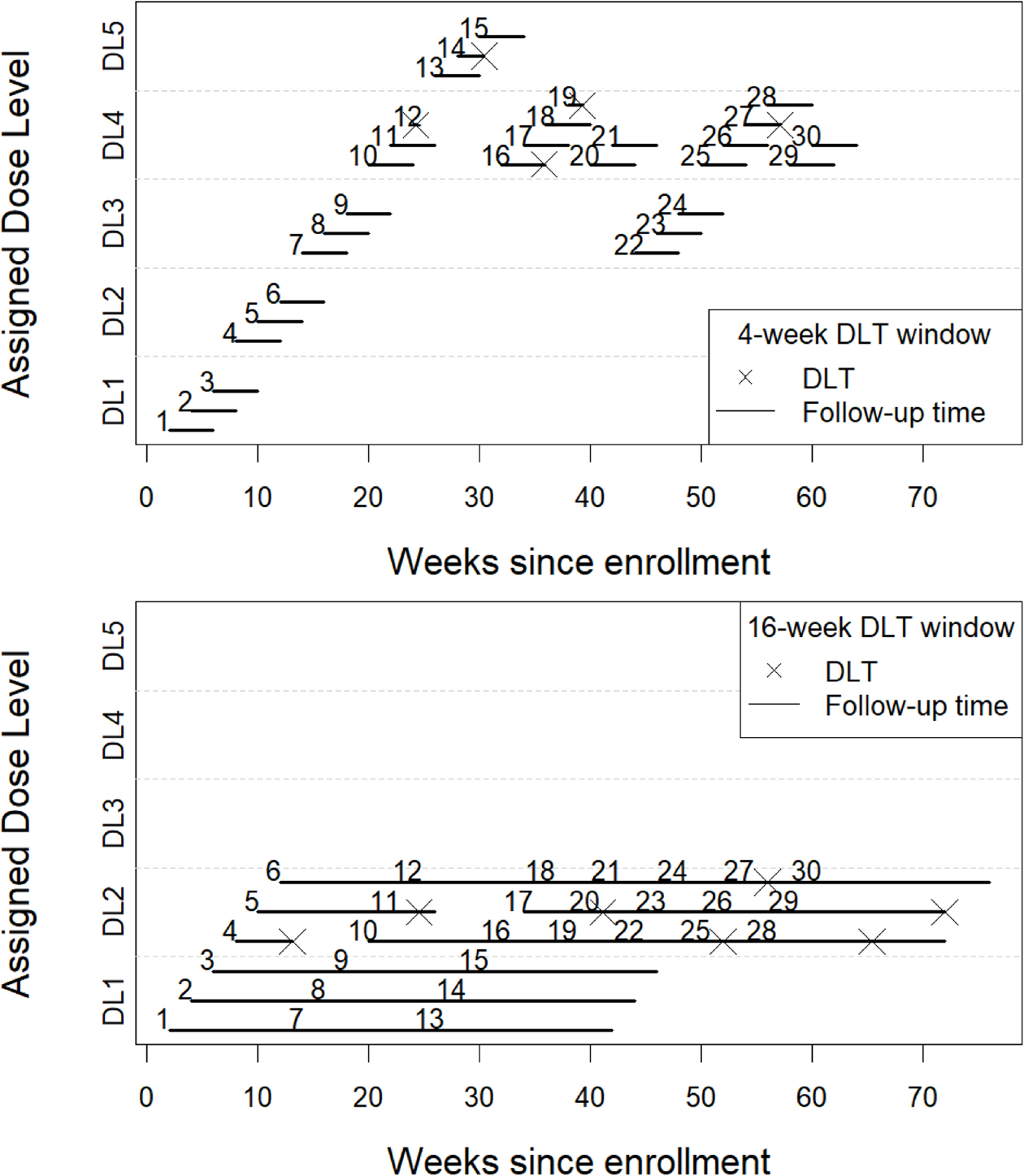
Trial data from an illustrative simulated example using a TITE-CRM with a 16-week observation window for toxicity (plot on top) and 4-week observation window (plot on bottom), a total sample size of 30 patients, an accrual rate of 2 patients per month. DL: dose level, DLT: Dose-limiting toxicity
Results over 10,000 simulated trials comparing DLT windows of 16 week versus 4 weeks with various scenarios of risk over time are reported in Table 3. Considering a 16-week observation window, the probability of correctly selecting dose level 2 as the MTD was 82% with the TITE-CRM and the TITE-BOIN with an average trial duration of 136 weeks and 161 weeks, respectively. However, if we only took into account toxicities in the first 4-weeks of follow-up, as was done in the real trials for idelalisib, and assume a constant hazard of DLT (corresponding to 25% of DLTs happening by week 4), we found that the designs mostly recommended higher dose levels, selecting dose level 4 47% and dose level 5 45% using the TITE-CRM. This is expected given that the true probabilities of DLTs are much lower for any given dose level at week 4 compared to week 16, as illustrated in Table 2 and Figure 2. When almost 80% of the toxicities occur before 4 weeks (very early toxicity), the TITE-CRM recommended dose level 2 in 65% of cases and dose level 3 in 34% of cases. Moreover, when 60% of toxicities occur before 4 weeks, the designs recommended dose level 3 most often. In terms of trial duration, with an accrual rate of 2 patients per month and a 16-week DLT assessment window (i.e., 8 patients expected per observation window), the trial duration was extended by 12 weeks compared to having a 4-week observation window. It is worth noting that using dose-finding designs that required complete follow-up, such as the CRM or BOIN, at 4 weeks results in similar performances in selecting the MTD, but almost double the trial duration (data not shown). This is because they require complete observations in current patients, before each dose assignment. While the original trials used between 40–60 patients, we found that with a reduced sample size of 30 patients the TITE-CRM and TITE-BOIN designs with a 16-week window resulted in 66% and 71% correct selection of dose level 2 as the MTD, with an average trial duration of 76 and 100 weeks, respectively (Supplemental material). This is comparable to, if not more efficient than, using designs that require complete follow-up (e.g. CRM, EWOC, BOIN or 3+3) with 60 patients and a 4-week window.
Table 3.
Simulations results for the idelalisib example: percent of recommendations of each dose level (D1 to D5), estimated over 10000 simulated trials, with a 4-week or 16-week observation window for toxicity, a total sample size of 60 patients, an accrual rate of 2 patients per month.
| Hazard | D1 | D2 | D3 | D4 | D5 | Duration | |
|---|---|---|---|---|---|---|---|
|
| |||||||
| 16-week obs. window (8 patients per obs.window) | |||||||
|
| |||||||
| Constant | True Probability of Toxicity | 0.10 | 0.25 | 0.45 | 0.60 | 0.75 | |
| % recommended by TITE-CRM | 10 | 82 | 8 | 0 | 0 | 136 weeks | |
| % recommended by TITE-BOIN | 12 | 82 | 6 | 0 | 0 | 161 weeks | |
|
| |||||||
| 4 week obs. window (2 patients per obs.window) | |||||||
|
| |||||||
| Decreasing | True Probability of Toxicity | 0.077 | 0.196 | 0.364 | 0.501 | 0.650 | |
| % recommended by TITE-CRM | 1 | 65 | 34 | 0 | 0 | 122 weeks | |
| % recommended by TITE-BOIN | 3 | 69 | 27 | 0 | 0 | 131 weeks | |
| Decreasing | True Probability of Toxicity | 0.056 | 0.147 | 0.281 | 0.396 | 0.534 | |
| % recommended by TITE-CRM | 0 | 24 | 68 | 8 | 0 | 122 weeks | |
| % recommended by TITE-BOIN | 0 | 32 | 60 | 8 | 0 | 131 weeks | |
| Constant | True Probability of Toxicity | 0.026 | 0.069 | 0.139 | 0.205 | 0.293 | |
| % recommended by TITE-CRM | 0 | 0 | 7 | 47 | 45 | 122 weeks | |
| % recommended by TITE-BOIN | 0 | 1 | 12 | 44 | 43 | 131 weeks | |
Proposals for dose optimization and considerations going forward
Given ethical concerns, dose assignments for dose-finding designs are done sequentially to ensure safety of lower doses before proceeding to higher doses. Thus, it is imperative to identify a safe set of doses first before evaluating the efficacy of the doses using a randomized design. It is also worth noting that, generally, patients enrolled in dose-finding trials are at an advanced stage of disease which may preclude the possibility for the assessment of long-term toxicities. For example, over half of trials in the Bortezomib case study by Lee et al. had median treatment follow-up of one or two cycles.3 While designs for late-onset toxicities could be used to estimate optimal doses, we can only guarantee that the dose is tolerable for the follow-up duration of the dose-finding trial and not necessarily for extended administration because of the short-follow-up. Moreover, the patient population for dose-finding designs and later stages of development are not always the same. Phase 1 trials in oncology often enroll all solid tumors while in later stages the patient population resembles more the intended population for treatment. Given all these considerations, we propose a departure from the traditional naming of phase 1 and phase 2 trials to naming the trials based on their objectives as dose-finding trials followed by dose-optimization trials. The goal of the dose-finding trial would be to identify a tolerable set of doses and the goal of the dose-optimization trial would be to evaluate efficacy and further evaluate late-onset or delayed toxicity to identify an optimal dose for the comparative trial. In Table 4, we summarize our proposal for dose optimization in the setting of late-onset toxicities.
Table 4.
Proposal for dose optimization in the setting of late-onset toxicities.
| Designs | Population | Toxicity window | |
|---|---|---|---|
|
| |||
| Dose-finding clinical trial | Toxicity only | General | 3–6 cycles |
| Toxicity and efficacy | Intended | 3–6 cycles | |
|
| |||
| Dose optimization clinical trial | Dose ranging | Intended | Entire treatment period |
| Selection designs | |||
If late-onset toxicities are expected, dose-finding trials should expand the window beyond the first cycle to ensure the inclusion of late-onset toxicities in the identification of a tolerable set of doses. The length of the toxicity window should be chosen depending on the toxicity profile of the drug and expected time to progression. Given the advanced stage of the patient population and the sequential nature of the designs, windows of 3–6 cycles should be considered. Windows longer than 6 cycles may lead to longer than desired dose-finding clinical trials. The population in phase I trials may often be general, particularly for solid tumors. Thus, toxicity only designs are preferred given that efficacy may depend on the population of interest. While these designs generally recommend a single estimate of the MTD or Recommended Phase 2 Dose (RP2D), it may be appropriate to also select one or two doses below the MTD as the set of tolerable doses for the dose optimization trial to more formally evaluate efficacy and delayed toxicities. We can also use designs that include other sources of toxicity information to define the MTD with late-onset toxicities. A few examples of these are designs incorporating pharmacokinetic information,27 toxicity information at multiple treatment cycles,28–31 toxicity grade32 or patient-reported toxicity information.33
Furthermore, when planning a dose-finding design, if the population is the intended population, toxicity and efficacy designs can be used to formally recommend dose(s) that maximize efficacy with acceptable toxicity; many of these designs allow the recommendation of more than one dose for further evaluation of efficacy and toxicity. While these designs have often been proposed as phase 1/2 designs given that they include efficacy as an endpoint, their aim remains dose-finding and their sample sizes are still reasonable. Thus, it is appropriate to use these designs for estimating an optimal set of doses for further evaluation in the dose optimization trial. The selection of design depends on the desired efficacy outcome and the suspected dose-efficacy relationship. Efficacy outcomes can be activity data such as pharmacodynamic data or more formal clinical efficacy endpoints such as response. In some cases, a plateau or U-shape relationship is suspected on the dose-efficacy relationship, instead of a strictly increasing relationship. Many of the approaches discussed above have been extended to incorporate efficacy endpoints and we provide some examples of designs in the Supplemental Material. Designs have been proposed for a binary (e.g., response) or time to event (e.g., progression-free survival or time to efficacy) efficacy outcome.34–39 Proposals are numerous and this is not an exhaustive list. The proposals differ in their definition of the optimal dose(s) which depend on the design’s objective: e.g., maximizing the efficacy criterion while satisfying a toxicity constraint,20,34–36,40,41 or optimizing an efficacy–toxicity trade-off or utility measure.38,42–45
Given the small sample sizes and short follow-up periods in a dose-finding trial, it is important to continue the evaluation of efficacy and longer-term tolerability in the dose optimization trial using selection designs and dose ranging designs in the intended patient population. Given that the dose-finding designs have provided a tolerable set of doses, patients can be randomized to the selected doses. It is worth noting that the analysis and reporting of adverse event data in phase 1 and 2 trials have been inconsistent and limited.46,47 More comprehensive methods for the analysis of adverse events and guidelines for reporting would improve our understanding of treatment tolerability to better design future phase 3 trials by selecting more tolerable doses, proposing strategies to handle treatment related adverse events, and optimizing the eligibility criteria. Various methods and data visualizations approaches have been proposed to better understand treatment tolerability and the timing of adverse events.4,48–51 While many of these methods have been illustrated in the context of phase 3 trials they can also be applied in randomized phase 2 before proceeding to phase 3 registration trials. This is particularly recommended for dose-optimization trials when the dose-finding trials used to identify the recommended phase 2 doses have limited follow-up information.
Supplementary Material
Acknowledgements
We thank the Reviewer and Associate Editor of the journal for their comments and suggestions that greatly improved the manuscript. We also thank Prof. Yin Yuan for kindly providing the R code for the TITE-BOIN design, which was used to perform simulations in this work.
References
- 1.Optimizing the dosage of human prescription drugs and biological products for the treatment of oncologic diseases: Draft guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/optimizing-dosage-human-prescription-drugs-and-biological-products-treatment-oncologic-diseases. Accessed: 2023-06-01. [Google Scholar]
- 2.Shah M, Rahman A, Theoret MR et al. The drug-dosing conundrum in oncology — when less is more. New England Journal of Medicine 2021; 385(16): 1445–1447. DOI: 10.1056/NEJMp2109826. [DOI] [PubMed] [Google Scholar]
- 3.Lee SM, Backenroth D, Cheung YKK et al. Case example of dose optimization using data from bortezomib dose-finding clinical trials. Journal of Clinical Oncology 2016; 34(12): 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SM, Zhang Y, Minasian LM et al. Using delayed toxicities to re-evaluate tolerability in phase 2 trials: a case example using bortezomib. Cancer Investigation 2017; 35(7): 484–489. [DOI] [PubMed] [Google Scholar]
- 5.Postel-Vinay S, Gomez-Roca C, Molife LR et al. Phase I trials of molecularly targeted agents: should we pay more attention to late toxicities. Journal of Clinical Oncology 2011; 29(13): 1728–1735. [DOI] [PubMed] [Google Scholar]
- 6.Postel-Vinay S, Aspeslagh S, Lanoy E et al. Challenges of phase 1 clinical trials evaluating immune checkpoint-targeted antibodies. Annals of Oncology 2016; 27(2): 214–224. [DOI] [PubMed] [Google Scholar]
- 7.Puzanov I, Diab A, Abdallah K et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunotherapy Cancer 2017; 5(1): 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phosphatidylinositol 3-kinase (PI3K) inhibitors in hematologic malignancies. https://fda.report/media/157762/ODAC-20220421-Backgrounder-FDA.pdf. Accessed: 2023-06-01. [Google Scholar]
- 9.Flinn IW, Kahl BS, Leonard JP et al. Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-δ, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood, The Journal of the American Society of Hematology 2014; 123(22): 3406–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kahl BS, Spurgeon SE, Furman RR et al. A phase 1 study of the pi3kδ inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (mcl). Blood, The Journal of the American Society of Hematology 2014; 123(22): 3398–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown JR, Byrd JC, Coutre SE et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood, The Journal of the American Society of Hematology 2014; 123(22): 3390–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warnings and precautions, contraindications, and boxed warning sections of labeling for human prescription drug and biological products — content and format, FDA Guidance document. https://www.fda.gov/media/71866/download. Accessed: 2023-10-24. [Google Scholar]
- 13.Coutre S, Barrientos J, Brown J et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma 2015; 56(10): 2779–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanlon A and Brander D. Managing toxicities of phosphatidylinositol-3-kinase (PI3K) inhibitors. Hematology Am Soc Hematol Educ Program 2020; 2020(1): 346–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braun TM, Yuan Z and Thall PF. Determining a maximum-tolerated schedule of a cytotoxic agent. Biometrics 2005; 61(2): 335–343. [DOI] [PubMed] [Google Scholar]
- 16.Braun TM, Thall PF, Nguyen H et al. Simultaneously optimizing dose and schedule of a new cytotoxic agent. Clinical Trials 2007; 4(2): 113–124. [DOI] [PubMed] [Google Scholar]
- 17.Liu CA and Braun TM. Parametric non-mixture cure models for schedule finding of therapeutic agents. Journal of the Royal Statistical Society Series C: Applied Statistics 2009; 58(2): 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J and Braun TM. A phase I Bayesian adaptive design to simultaneously optimize dose and schedule assignments both between and within patients. Journal of the American Statistical Association 2013; 108(503): 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tighiouart M, Liu Y and Rogatko A. Escalation with overdose control using time to toxicity for cancer phase i clinical trials. PloS one 2014; 9(3): e93070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andrillon A, Chevret S, Lee SM et al. Surv-CRM-12: A Bayesian phase I/II survival CRM for right-censored toxicity endpoints with competing disease progression. Statistics in Medicine 2022; 41(29): 5753–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnett H, Boix O, Kontos D et al. Dose finding studies for therapies with late-onset toxicities: A comparison study of designs. Statistics in Medicine 2022; 41(30): 5767–5788. DOI: 10.1002/sim.9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung YK and Chappell R. Sequential designs for phase i clinical trials with late-onset toxicities. Biometrics 2000; 56(4): 1177–1182. [DOI] [PubMed] [Google Scholar]
- 23.Yuan Y, Lin R, Li D et al. Time-to-event Bayesian optimal interval design to accelerate phase i trials. Clinical Cancer Research 2018; 24(20): 4921–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Quigley J, Pepe M and Fisher L. Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics 1990;: 33–48. [PubMed] [Google Scholar]
- 25.Liu S and Yuan Y. Bayesian optimal interval designs for phase I clinical trials. Journal of the Royal Statistical Society: Series C: Applied Statistics 2015; 64(3): 507–523. [Google Scholar]
- 26.Lee SM and Cheung YK. Model calibration in the continual reassessment method. Clinical Trials 2009; 6(3): 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunhan BK, Weber S, Seroutou A et al. ¨ Phase I dose-escalation oncology trials with sequential multiple schedules. BMC Medical Research Methodology 2021; 21: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wages NA, O’Quigley J and Conaway MR. Phase I design for completely or partially ordered treatment schedules. Statistics in Medicine 2014; 33(4): 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandes LL, Taylor JM and Murray S. Adaptive phase I clinical trial design using Markov models for conditional probability of toxicity. Journal of biopharmaceutical statistics 2016; 26(3): 475–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyu J, Curran E and Ji Y. Bayesian adaptive design for finding the maximum tolerated sequence of doses in multicycle dose-finding clinical trials. JCO Precision Oncology 2018; 2: 1–19. [DOI] [PubMed] [Google Scholar]
- 31.Ursino M, Biard L and Chevret S. DICE: A Bayesian model for early dose finding in phase I trials with multiple treatment courses. Biometrical Journal 2022; 64(8): 1486–1497. [DOI] [PubMed] [Google Scholar]
- 32.Lee SM, Ursino M, Cheung YK et al. Dose-finding designs for cumulative toxicities using multiple constraints. Biostatistics 2019; 20(1): 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrillon A, Biard L and Lee SM. Incorporating patient-reported outcomes in dose-finding clinical trials with continuous patient enrollment. Journal of biopharmaceutical statistics 2023; 0(0): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riviere MK, Yuan Y, Jourdan JH et al. Phase I/II dose-finding design for molecularly targeted agent: plateau determination using adaptive randomization. Statistical methods in medical research 2018; 27(2): 466–479. [DOI] [PubMed] [Google Scholar]
- 35.Yan D, Tait C, Wages NA et al. Generalization of the time-to-event continual reassessment method to bivariate outcomes. Journal of Biopharmaceutical Statistics 2019; 29(4): 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeda K, Morita S and Taguri M. TITE-BOIN-ET: time-to-event Bayesian optimal interval design to accelerate dose-finding based on both efficacy and toxicity outcomes. Pharmaceutical Statistics 2020; 19(3): 335–349. [DOI] [PubMed] [Google Scholar]
- 37.Altzerinakou MA and Paoletti X. An adaptive design for the identification of the optimal dose using joint modeling of continuous repeated biomarker measurements and time-to-toxicity in phase I/II clinical trials in oncology. Statistical methods in medical research 2020; 29(2): 508–521. [DOI] [PubMed] [Google Scholar]
- 38.Koopmeiners JS and Modiano J. A Bayesian adaptive phase I–II clinical trial for evaluating efficacy and toxicity with delayed outcomes. Clinical Trials 2014; 11(1): 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hobbs BP, Thall PF and Lin SH. Bayesian group sequential clinical trial design using total toxicity burden and progression-free survival. Journal of the Royal Statistical Society Series C Applied Statistics 2016; 65(5): 273–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan Y and Yin G. Bayesian dose finding by jointly modelling toxicity and efficacy as time-to-event outcomes. Journal of the Royal Statistical Society: Series C (Applied Statistics) 2009; 58(5): 719–736. [Google Scholar]
- 41.Biard L, Lee SM and Cheng B. Seamless phase I/II design for novel anticancer agents with competing disease progression. Statistics in medicine 2021; 40(21): 4568–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin IH, Liu S, Thall PF et al. Using data augmentation to facilitate conduct of phase I–II clinical trials with delayed outcomes. Journal of the American Statistical Association 2014; 109(506): 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu S and Johnson VE. A robust Bayesian dose-finding design for phase I/II clinical trials. Biostatistics 2016; 17(2): 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Cao S, Zhang C et al. A Bayesian adaptive phase I/II clinical trial design with late-onset competing risk outcomes. Biometrics 2021; 77(3): 796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y, Lin R, Lee JJ et al. Tite-boin12: A Bayesian phase I/II trial design to find the optimal biological dose with late-onset toxicity and efficacy. Statistics in medicine 2022; 41(11): 1918–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips R, Hazell L, Sauzet O et al. Analysis and reporting of adverse events in randomised controlled trials: a review. BMJ Open 2019; 9: e024537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yap C, Bedding A and de Bono J et al. The need for reporting guidelines for early phase dose-finding trials: Dose-finding consort extension. Nat Med 2022; 28: 6–7. [DOI] [PubMed] [Google Scholar]
- 48.Francis KE, Gebski V, Lord SJ et al. Reporting the trajectories of adverse events over the entire treatment course in patients with recurrent platinum-sensitive ovarian cancer treated with platinum-based combination chemotherapy regimens: A graphical approach to trial adverse event reporting. European Journal of Cancer 2021; 148: 251–259. [DOI] [PubMed] [Google Scholar]
- 49.Stegherr R, Schmoor C, Lubbert M et al. ¨ Estimating and comparing adverse event probabilities in the presence of varying follow-up times and competing events. Pharmaceutical Statistics 2021; 20(6): 1125–1146. [DOI] [PubMed] [Google Scholar]
- 50.Stegherr R, Schmoor C and Beyersmann Jea. Survival analysis for adverse events with varying follow-up times (savvy)-estimation of adverse event risks. Trials 2021; 22(1): 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SM, Fan W, Wang A et al. Novel approaches for dynamic visualization of adverse event data in oncology clinical trials: A case study using immunotherapy trial S1400-I (SWOG). JCO Clinical Cancer Informatics 2023; 7: e2200165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.