

Abstract
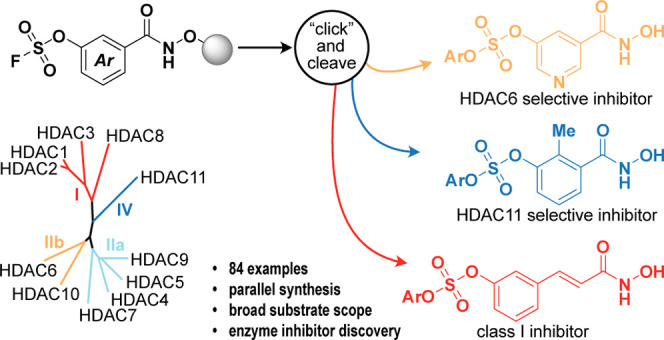
Multistep synthesis performed on solid support is a powerful means to generate small-molecule libraries for the discovery of chemical probes to dissect biological mechanisms as well as for drug discovery. Therefore, expansion of the collection of robust chemical transformations amenable to solid-phase synthesis is desirable for achieving chemically diverse libraries for biological testing. Here, we show that sulfur(VI) fluoride exchange (SuFEx) chemistry, exemplified by pairing phenols with aryl fluorosulfates, can be used for the solid-phase synthesis of biologically active compounds. As a case study, we designed and synthesized a library of 84 hydroxamic acid-containing small molecules, providing a rich source of inhibitors with diverse selectivity profiles across the human histone deacetylase enzyme family. Among other discoveries, we identified a scaffold that furnished inhibitors of HDAC11 with exquisite selectivity in vitro and a selective inhibitor of HDAC6 that was shown to affect the acetylation of α-tubulin over histone sites H3K18, H3K27, as well as SMC3 in cultured cells. Our results encourage the further use of SuFEx chemistry for the synthesis of diverse small-molecule libraries and provide insight for future design of selective HDAC inhibitors.
Keywords: SuFEx, library synthesis, solid-phase synthesis, HDAC inhibitors, epigenetics
Introduction
Drug discovery campaigns often rely on the generation and screening of collections of chemical compounds during both hit identification and lead optimization.1 The development of solid-phase synthesis technology, originally for peptide synthesis,2 enabled the chemical synthesis of libraries of peptides3,4 and led to the development of combinatorial chemistry.5−8 The synthesis of compound libraries rapidly evolved to include nonoligomeric chemotypes9−24 and attention has since been dedicated to the importance of the structural diversity of the compound collections for success in identifying biologically relevant ligands.25−27 Still, most small-molecule compound libraries are generated by using a limited set of chemical transformations: amide coupling, aromatic nucleophilic substitution, reductive amination, and transition-metal-catalyzed cross-coupling reactions.28 These choices may, at least in part, reflect the requirements that reactions used for library synthesis need to be reliable, high-yielding, and have broad functional group tolerance. As such, sulfur(VI) fluoride exchange (SuFEx) chemistry, which has found considerable use in covalent chemical probe design,29−35 has been widely developed36−38 and included as a viable addition to this toolbox,39−41 since it was coined in 2014.42 While compounds that contain SuFEx “war-heads” have been prepared on solid support,43−45 the SuFEx reaction has only been performed on solid phase for polymer synthesis previously.46 In this work, we showcase the extension of SuFEx chemistry, for the generation of biologically active small molecules, to solid-phase synthesis, which enables the rapid generation and handling of large compound libraries.
Results and Discussion
As a case study, we chose to investigate the discovery of histone deacetylase (HDAC) inhibitors with novel selectivity profiles by combining a selection of resin-bound SuFEx hubs with a range of phenols in a parallel synthesis format. Histone deacetylases are therapeutically relevant drug targets,47,48 and inhibitors that bind to the active sites of these enzymes generally contain a zinc-binding group, a linker that mimics the side chain of a lysine residue, and a capping group that interacts with the surface of the enzyme.49 We therefore designed a strategy that could be readily extended to a number of different aryl fluorosulfate building blocks. First, these aryl fluorosulfates were prepared in a two-chamber reactor that allows for the generation of sulfuryl fluoride in one chamber and consumption of the gas in the other chamber (II; Scheme 1A).50−52 The resulting building blocks can then be converted to their corresponding acid chlorides (III, Scheme 1B) and coupled to a solid-supported hydroxylamine to give resin-bound hydroxamic acids, which have previously been applied for the discovery of HDAC inhibitors.53−58 The resin-bound hydroxamates prepared in this study (IV) displayed the aryl fluorosulfate SuFEx hubs for functionalization with a panel of phenols, to give diverse hydroxamic acid-containing compounds (V) upon cleavage from the resin (Scheme 1B).
Scheme 1. (A) Synthesis of Aryl Fluorosulfates by Utilizing a Two-Chamber System for the Formation and Consumption of SO2F2. (B) Strategy for On-Resin, Accelerated SuFEx Chemistry to Generate Compound Arrays.
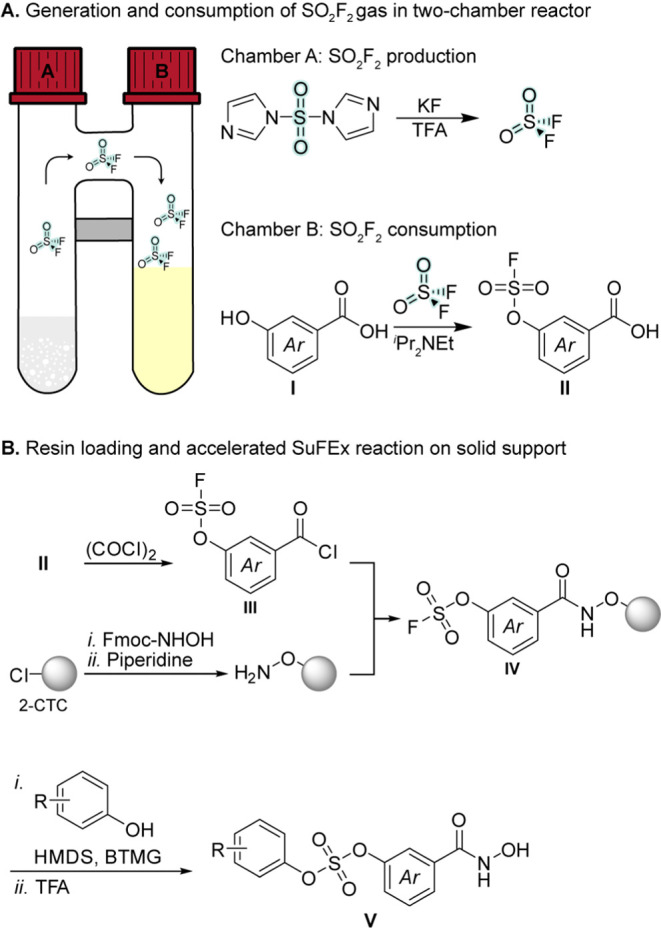
The aryl fluorosulfate SuFEx functionality was strategically chosen due to its latent reactivity profile to enable the manipulations required for preparing and handling the resins. Importantly, we then envisioned that the recently developed conditions for accelerated SuFEx chemistry59 could be adapted to catalyze the functionalization of our resins (IV) [using 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG; “Barton’s base”) and hexamethyldisilazane (HMDS)].
Our initial attempts to use acetonitrile as the solvent, as originally reported by Moses and co-workers,59 led to poor swelling of our polystyrene-based resins. Further, the heating required for product formation caused rapid evaporation of the solvent, prohibiting extended reaction times (Table 1, entry 2). Polystyrene-based resins have excellent swelling properties in dichloromethane, but low conversion was also observed in this solvent at room temperature (entries 3 and 4). We therefore investigated N,N-dimethylformamide (DMF) and found that elevated temperature (65 °C) for 2 h gave acceptable conversion (entries 5–8), while extended reaction time (16 h) at this temperature gave full conversion (entry 9).
Table 1. Screening of Reaction Conditions for the On-Resin SuFEx Reactiona,b.

| entry | solvent | phenol | time (h) | temp. (°C) | conv. (%)c |
|---|---|---|---|---|---|
| 1 | MeCN | PhOH | 2 | 20 | 4 |
| 2 | MeCNd | PhOH | 2 | 65 | 52 |
| 3 | CH2Cl2 | PhOH | 0.5 | 20 | 12 |
| 4 | CH2Cl2 | PhOH | 2 | 20 | 9 |
| 5 | DMF | PhOH | 0.5 | 20 | 9 |
| 6 | DMF | PhOH | 2 | 20 | 47 |
| 7 | DMF | PhOH | 0.5 | 65 | 19 |
| 8 | DMF | PhOH | 2 | 65 | 75 |
| 9 | DMF | PhOH | 16 | 65 | >99 |
| 10 | DMFe | p-CF3-PhOH | 16 | 65 | >99 |
| 11 | DMFe | o-iPr-PhOH | 16 | 65 | >99 |
Monitored by HPLC after cleavage from resin.
Conditions: phenol (5.0 equiv), HMDS (5.0 equiv), and BTMG (1.0 equiv) relative to the resin loading (2.6 μmol scale), solvent (200 μL).
Conversion given as AUCproduct × (AUCproduct + AUCunreacted ArOSO2F)−1 measured by HPLC at 215 nm.
Solvent evaporated during the reaction time.
20 μmol scale.
These optimized conditions also gave full conversion when using a sterically hindered or an electron-deficient phenol (entries 10 and 11). For the cleavage step, we found that dilute trifluoroacetic acid (TFA) was superior to the milder 1,1,1,3,3,3-hexafluoro-isopropyl alcohol, and the resulting crude chromatograms revealed minor impurities from the catalysts. The on-resin functionalization thus appears to reduce the degree of Lossen rearrangement, which is a known side reaction between hydroxamic acids and aryl fluorosulfates.45 Final compounds were still purified by preparative high-performance liquid chromatography (HPLC) using a short gradient to give purities >95%.
For the first-generation compound series, we chose scaffold A in combination with a selection of 30 phenols/hydroxy-aryl compounds (R–OH with R = 1–30; Figure 1A). Final products A1–A28 were all successfully prepared and only the reactions with pyrimidin-2-ol (29) and 5-hydroxyindole (30) were unproductive according to LC-mass spectrometry (LC-MS) analysis of the cleaved crude material. The potencies of the initial compound series were then evaluated against a selection of human zinc-dependent HDACs (HDAC1–11). We selected HDACs 1, 6, 8, and 11 as targets for inhibition to include representatives of subclasses I, IIb, and IV. Class IIa enzymes were not included in the initial screen because this class of isozymes has generally required hydroxamic acid-containing inhibitors with substantial steric congestion or nonchelating chemotypes for potent inhibition.60−63 The HDAC6 was considered a particularly interesting target due to its recent status as an orphan drug target by the US Food and Drug Administration. Also, HDAC8 plays a potential role in several cancers, including T-cell lymphoma,64 and HDAC11 is of interest due to the under representation of tool compounds available to study its biological function.65
Figure 1.

(A) Structures of compounds A1–A30 (for compound synthesis, see Supporting Figures S1 and S2); NP, no product formed. (B) Structures of scaffolds B–F for the second-generation compound series. (C) Heat maps representing Ki values of selected first-generation members and the full second-generation series against HDAC1, HDAC6, HDAC8, and HDAC11. The Ki values are calculated using the Cheng–Prusoff equation, from IC50 values determined by at least two individual end point assays, performed in duplicate. See Supporting Figures S4 and S9 for full dose–response curves and Supporting Tables S1 for IC50 and calculated Ki values. NI, no inhibition as determined by <50% inhibition at 5 μM inhibitor; ND, not determined as compound was left out of the series.
All compounds A1–A28 exhibited limited potencies against HDACs 1 and 11 but generally inhibited HDACs 6 and 8 (see Supporting Figure S3 for a heat map and Supporting Table S1 for Ki values). Potent inhibition of both HDACs 6 and 8 were observed, for example, for compounds A22 (Ki = 17 nM against HDAC6) and A17 (Ki = 22 nM against HDAC8), respectively. However, modest selectivity below 10-fold was observed between the inhibition of the two enzymes.
In the next iteration of the synthesis and biochemical evaluation, we focused on altering the structure of the scaffold to give series B–F (Figure 1B). We selected phenols to represent ones from potent HDAC6 and 8 inhibitors (A1, A2, and A23), from compounds that inhibited HDAC11 (A3, A11, A12, A19, and A21), and finally from nonselective inhibitors (A4, A8, A9, and A24) (Figure 1C). Combining these phenols with the five scaffolds provided series B–F, incorporating substituents in the 2- and 5-positions of the scaffold (B–D) and including a meta-substituted pyridine ring (E). The series F was based on 3-hydroxy-cinnamic acid, resembling the clinically approved, nonselective HDAC inhibitor belinostat (Beleodaq).48 When testing these series against the same selection of enzymes as selected in the initial screen, the series B compounds showed a general decrease in potency against HDACs 6 and 8, while some potency was retained against HDAC11 (Figure 1C). Substituting the methyl group in B for a fluorine atom in C resulted in retained potency against HDAC6 compared to the series A, but with improved selectivity due to lower potencies observed against HDAC8. For example, compound C1 exhibited 20-fold selectivity for HDAC6 over HDAC8, while compound A1 was equipotent against these two enzymes (Supporting Table S1). In series D, where a methyl group was introduced in the 2-position of the scaffold, a striking loss of activity was observed against both HDAC6 and 8. On the other hand, a general increase in potency against HDAC11 was recorded and compounds D3, D9, and D21 inhibited this enzyme with Ki values in the 150–190 nM range (Supporting Table S1). Thus, these compounds exhibited excellent isozyme selectivity with no inhibition of the other tested HDACs at concentrations up to 5 μM. Interestingly, a recent study from Wang and co-workers also reported HDAC11-selective inhibitor with an ortho-substituted benzohydroxamic acid,66 suggesting that this motif may offer a general avenue for the future development of optimized inhibitors of HDAC11.
The pyridine-containing series (E), like series C, generally furnished inhibitors with selectivity for HDAC6, but with decreased selectivity over other isozymes (Figure 1C). Compound E24 was identified as the most potent compound of the library with a Ki value of 14 nM against HDAC6 and ∼20-fold selectivity over HDAC8 (Supporting Table S1). The series F was devoid of HDAC11 inhibitors and instead provided several compounds with potency against all of the remaining three HDACs (1, 6, and 8), consistent with the activities of the known cinnamic hydroxamate, belinostat.48
To investigate the selectivity of selected inhibitors across a broader selection of HDAC isozymes, we picked eight compounds with different selectivity profiles in the initial screen (A10, C1, D21, E3, E23, E24, F2, and F3) (Supporting Figures S10 and S11 and Table S2). This analysis revealed inhibitors with selectivity for HDAC6 (C1, E3, E23, and E24) (Figure 2A,B), HDAC8 (A10), and HDAC11 (D21) (Figure 2C). Further, scaffold F furnished nonselective ones (F2 and F3) that target all enzymes except HDAC11 and class IIa isozymes, which have been reported to have noncatalytic function and are thus generally poorly targeted by hydroxamic acids.67,68
Figure 2.
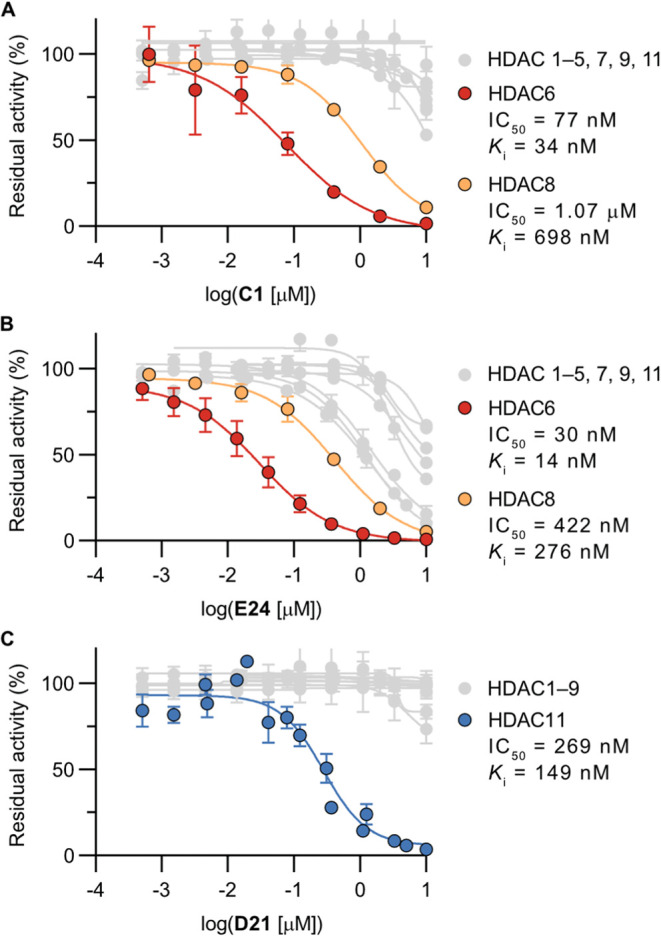
Selectivity of selected compounds against HDACs 1–9 and 11. (A) IC50 curves for C1. (B) IC50 curves for compound E24. (C) IC50 curves for compound D21. The Ki values are calculated from IC50 values determined by at least two individual end point assays performed in duplicate by using the Cheng–Prusoff equation. See Supporting Figures S10 and S11 and Table S2 for additional data.
Although the hydrolysis rates of bis-aryl sulfate diesters under alkaline conditions have been investigated in detail,69,70 we evaluated the stability of the selected hit compounds (C1, D21, E24) under the applied assay conditions, as well as in the presence of glutathione. All experiments revealed half-lives of the compounds above 20 h, confirming that the compounds were stable during the performed assays (Supporting Figure S12).
Next, we performed preincubation dose–response experiments for compounds C1, D21, and E24 against their target enzyme in vitro, because examples of small-molecule hydroxamic acid-containing HDAC inhibitors have been shown to exhibit tight-binding kinetics.71 For the selected compounds, we did not observe any decrease in IC50 value upon preincubation, showing no indication of slow, tight-binding kinetics or covalent inhibition (Supporting Figure S13).
With our novel series of inhibitors, exhibiting a wide range of selectivity profiles against recombinant enzymes in biochemical assays in vitro, we were interested in demonstrating target engagement and determining the selectivity in cells. For this purpose, we treated human embryonic kidney (HEK)293T cells with compounds C1 or E24 for 5 h, and subsequent cell lysis and Western blot analysis revealed significant upregulation of the acetylation of the cytosolic HDAC6 target α-tubulin but not the histones (H3K18 or H3K27) or the HDAC8 target SMC3 compared to DMSO control (Figures 3 and S14).
Figure 3.
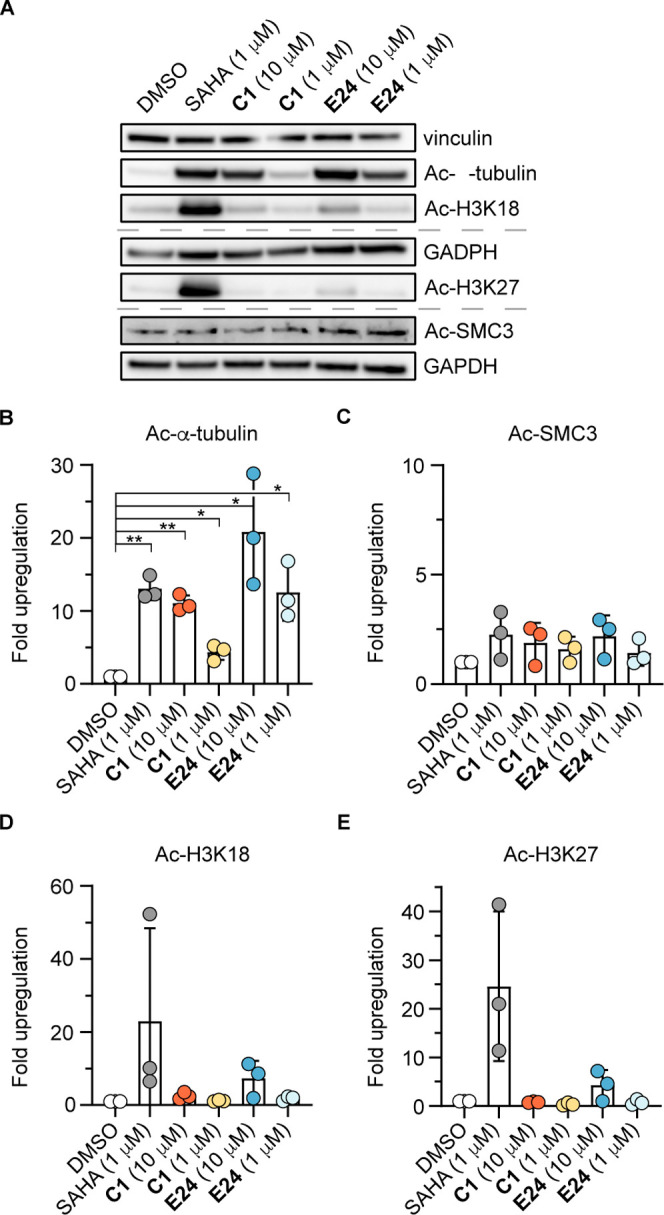
Inhibition of HDACs in cells. (A) Representative Western blots of lysine acetylation (α-tubulin, H3K18, H3K27, and SMC3) upon 5 h treatment with compounds C1 or E24 (1 or 10 μM), SAHA (1 μM), or DMSO. (B–E) Quantification of acetylation, normalized to vinculin or GAPDH loading control, and relative to DMSO (data represent mean ± SD, n = 3 biologically independent samples). See Supporting Figure S14 for full blots and replicates.
We also performed Western blot-based cellular thermal shift assays (CETSA),72,73 which we have previously used for investigating inhibitors targeting sirtuins (NAD+-dependent class III HDACs).74−77 Because HDAC11 has very low expression levels in most cell lines, we were not able to produce a viable melting curve for this enzyme. Thus, we tested the most potent HDAC6 inhibitor of our collection (E24). With HDAC8 being the closest targeted isozyme by compound E24 (Figure 2B), we also evaluated the thermal shift of this enzyme. In HEK293T cells, we observed a thermal shift of HDAC6 of ∼2° relative to DMSO control, while no shift was observed for HDAC8 (Supporting Figures S15 and S16). These data are also in agreement with the in vitro assays.
Conclusions
In summary, we have developed a strategy for using SuFEx click chemistry on a solid support to generate arrays of biologically active compounds. In this case study, we successfully applied the aryl fluorosulfate functionality as a hub for diversification during the solid-phase synthesis of potential HDAC inhibitors. By parallel synthesis, we prepared a library of 84 diverse compounds that furnished potent HDAC inhibitors with a broad range of selectivity profiles. Thus, the present work demonstrates SuFEx chemistry as an addition to the existing arsenal of reactions for the diversification of small-molecule libraries on the solid phase. The bis-aryl sulfate diester motif itself has found limited use in medicinal chemistry previously,78 perhaps in part due to a lack of efficient methods for its formation prior to the development of the accelerated SuFEx conditions59 also applied herein. However, the lower number of hydrogen bond donors compared to sulfonamides, sulfamates, or sulfamides, which are accessible by SuFEx chemistry using anilines and amines, may be an attractive feature of the sulfate diesters.
These results argue for the future application of additional SuFEx reactions on solid support, and we further envision that our data should serve as encouragement for similar expansion of the chemical transformations applied for the synthesis of DNA encoded libraries.
The biochemical evaluation of the library against recombinant HDAC enzymes furnished a structure–activity relationship (SAR) study, which identified key features of importance for the targeting of individual HDAC isozymes. Particularly, we successfully identified novel isozyme selective inhibitors against HDACs 6 and 11, of which the former mentioned was confirmed to inhibit the desired target with selectivity in HEK293T cells.
Methods
General Procedure 1: Aryl Fluorosulfate Formation52
Chamber A of a two-chamber reactor COware gas reactor; Sigma #STW1 (1 mmol) or #STW5 (5 mmol) was charged with 1,1′-sulfonyldiimidazole (1.5 equiv) and potassium fluoride (4.0 equiv). Chamber B was charged with the desired phenol (1 equiv) followed by H2O–MeCN (1:1, 4 mL) and iPr2NEt (3.0 equiv). The two chambers were sealed and TFA (0.5 mL per mmol phenol) was added by injection through the septum of chamber A. The reaction was stirred for 16 h at room temperature before the caps were removed and the reaction was stirred for another 15 min to ensure that all sulfuryl fluoride was vented out of the fume hood. Next, the content of chamber B was transferred to a round-bottom flask and volatiles were removed under reduced pressure. The residue was acidified to pH 2 with aqueous HCl (1 M), extracted twice with CH2Cl2, dried over Na2SO4, filtered, and concentrated under reduced pressure to give the crude residue.
General Procedure 2: Formation of Acid Chloride and Resin Functionalization
Aryl fluorosulfate-modified carboxylic acid (1 equiv) was dissolved in anhydrous CH2Cl2 (10 mL/mmol). The solution was stirred in an ice bath, and oxalyl chloride (2 equiv) was added followed by DMF (two drops). The reaction was allowed to warm up to room temperature and stirred for 2 h before volatiles were removed under reduced pressure to give the crude acid chloride, which was used immediately in the next step (General Procedure 3: Loading of 2-Chlorotrityl Chloride Resin and Formation of Aryl Fluorosulfate-Functionalized Resin Section).
General Procedure 3: Loading of 2-Chlorotrityl Chloride Resin and Formation of Aryl Fluorosulfate-Functionalized Resin
2-Chlorotrityl chloride polystyrene resin was allowed to swell in anhydrous CH2Cl2 in a fritted syringe for 15 min. After the solvent was removed, N-Fmoc-hydroxylamine (1.5 equiv related to the theoretical loading from the vendor) and iPr2NEt (5 equiv) in anhydrous CH2Cl2–DMF (10:1, 11 mL/g resin) were added, and the mixture was agitated for 2 h at room temperature. The resin was then drained and capped by incubation with CH2Cl2–MeOH–iPr2NEt (17:1:2, 10 mL/g resin, 2 × 15 min) and then washed with CH2Cl2 (3 × 1 min), DMF (2 × 1 min), and CH2Cl2 (2 × 1 min). The loading was determined spectrophotometrically, quantifying the amount of released fluorene upon cleavage of the Fmoc group from a small sample of dried resin.79 Loading was typically determined to be 0.5–0.6 mmol/g for different batches of resin. Fmoc deprotection was achieved with DMF–piperidine (4:1, v/v, 9 mL; 2 min; then repeated for 15 min), followed by washing with DMF (5 × 1 min) and CH2Cl2 (3 × 1 min). The hydroxylamine functionalized resin was then incubated with the corresponding aryl fluorosulfate-modified acid chloride (1.5 equiv) and iPr2NEt (6.0 equiv) in anhydrous CH2Cl2 (0.2 M of acid chloride) for 90 min at room temperature, followed by washing with DMF (3 × 1 min), MeOH (3 × 1 min), and CH2Cl2 (3 × 1 min), before the resin was drained and dried under reduced pressure. It should be noted that we did not measure the efficiency of the acid chloride reaction with the resin, which may have affected the isolated yields of the final compounds.
General Procedure 4: On-Resin Sulfur(VI) Fluoride Exchange (SuFEx) Reaction
The aryl fluorosulfate-functionalized resin was placed in a fritted syringe and swelled in anhydrous DMF (0.25 mL/10 μmol of resin). After 5–10 min, the resin was drained by suction, indicated phenol (5 equiv), BTMG (1 equiv), and HMDS (5 equiv) in anhydrous DMF (0.025 mL/μmol resin) were added, and the mixture was agitated for 16 h at 65 °C. The resin was then washed with DMF (5 × 1 min) and CH2Cl2 (5 × 1 min). The hydroxamic acid-containing compound was released from the solid support by CH2Cl2–TFA (9:1, 0.5 mL, 2 × 15 min). Volatiles were removed under a stream of nitrogen, and the crude residue was purified by preparative HPLC.
Optimization of On-Resin SuFEx Reaction
Resin A (2.6 μmol) was swelled in anhydrous solvent (200 μL) and reacted with phenol (5 equiv), BTMG (1 equiv), and HMDS (5 equiv) in the desired solvent (200 μL) for the indicated time and temperature. After the reaction, the resin was drained and washed with DMF (5 × 1 min) and CH2Cl2 (5 × 1 min) before the product was cleaved with CH2Cl2–TFA (9:1, 200 μL, 2 × 15 min). Volatiles were removed under a stream of nitrogen, and the crude residue was dissolved in acetonitrile and analyzed by ultrahigh-performance liquid chromatography. Conversion was estimated as the ratio between the area under the curve (AUC) of the product and the released unreacted aryl fluorosulfate in the UV chromatogram at 215 nm (eq 1)
| 1 |
Acknowledgments
The authors thank Iben Jensen and Huy T. Nguyen for technical assistance. They also thank the Erasmus+ Student Mobility Programme for support to L.G. This work was supported by the Independent Research Fund Denmark–Medical Sciences (0134-00435B; C.A.O.), the Independent Research Fund Denmark–Technical and Production Sciences (0136-00421A; C.A.O.), and the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (grant agreement number: 725172–SIRFUNCT, C.A.O.)
Glossary
Abbreviations
- 2-CTC
2-chlorotrityl chloride
- AUC
area under the curve
- BTMG
2-tert-butyl-1,1,3,3-tetramethylguanidine
- CETSA
cellular thermal shift assay
- DMF
N,N-dimethylformamide
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HMDS
hexamethyldisilazane
- HDAC
histone deacetylase
- HPLC
high-performance liquid chromatography
- HEK
human embryonic kidney
- LC-MS
liquid chromatography–mass spectrometry
- NMR
nuclear magnetic resonance
- SuFEx
sulfur(VI) fluoride exchange
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.4c00042.
Synthesis of building blocks and final compounds; biochemical assay data; biochemical data behind the heat maps; experimental methods; chemical synthesis and compound characterization data; and copies of HPLC traces, 1H, 13C, and 19F NMR spectra (PDF)
Author Present Address
† Lundbeck Pharma A/S, Ottiliavej 9, DK-2500 Valby, Denmark
Author Contributions
‡ T.N.H. and D.D. contributed equally to this work. CRediT: Tobias N. Hansen conceptualization, data curation, formal analysis, investigation, visualization, writing-original draft, writing-review & editing; Daniela Dankova conceptualization, data curation, formal analysis, investigation, writing-review & editing; Michael Bæk conceptualization, investigation, writing-review & editing; Linda Grlas investigation; Christian A. Olsen conceptualization, project administration, supervision, visualization, writing-original draft, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Gerry C. J.; Schreiber S. L. Chemical probes and drug leads from advances in synthetic planning and methodology. Nat. Rev. Drug Discovery 2018, 17, 333–352. 10.1038/nrd.2018.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield R. B. Solid Phase Peptide Synthesis. 1. Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Geysen H. M.; Meloen R. H.; Barteling S. J. Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 3998–4002. 10.1073/pnas.81.13.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghten R. A. General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 5131–5135. 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furka Á.; Sebestyen F.; Asgedom M.; Dibo G. General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res. 1991, 37, 487–493. 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- Houghten R. A.; Pinilla C.; Blondelle S. E.; Appel J. R.; Dooley C. T.; Cuervo J. H. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991, 354, 84–86. 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- Lam K. S.; Salmon S. E.; Hersh E. M.; Hruby V. J.; Kazmierski W. M.; Knapp R. J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- Lam K. S.; Lebl M.; Krchnak V. The ″One-Bead-One-Compound″ Combinatorial Library Method. Chem. Rev. 1997, 97, 411–448. 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- Thompson L. A.; Ellman J. A. Synthesis and Applications of Small Molecule Libraries. Chem. Rev. 1996, 96, 555–600. 10.1021/cr9402081. [DOI] [PubMed] [Google Scholar]
- Watson C. Polymer-Supported Synthesis of Non-Oligomeric Natural Products. Angew. Chem., Int. Ed. 1999, 38, 1903–1908. . [DOI] [PubMed] [Google Scholar]
- Breinbauer R.; Vetter I. R.; Waldmann H. From Protein Domains to Drug Candidates—Natural Products as Guiding Principles in the Design and Synthesis of Compound Libraries. Angew. Chem., Int. Ed. 2002, 41, 2878–2890. . [DOI] [PubMed] [Google Scholar]
- Dolle R. E.; Bourdonnec B. L.; Worm K.; Morales G. A.; Thomas C. J.; Zhang W. Comprehensive survey of chemical libraries for drug discovery and chemical biology: 2009. J. Comb. Chem. 2010, 12, 765–806. 10.1021/cc100128w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle R. E. Comprehensive survey of combinatorial library synthesis: 1999. J. Comb. Chem. 2000, 2, 383–433. 10.1021/cc000055x. [DOI] [PubMed] [Google Scholar]
- Dolle R. E. Comprehensive survey of combinatorial library synthesis: 2000. J. Comb. Chem. 2001, 3, 477–517. 10.1021/cc010049g. [DOI] [PubMed] [Google Scholar]
- Dolle R. E. Comprehensive survey of combinatorial library synthesis: 2001. J. Comb. Chem. 2002, 4, 369–418. 10.1021/cc020039v. [DOI] [PubMed] [Google Scholar]
- Dolle R. E. Comprehensive survey of combinatorial library synthesis: 2002. J. Comb. Chem. 2003, 5, 693–753. 10.1021/cc0340224. [DOI] [PubMed] [Google Scholar]
- Tan D. S.; Foley M. A.; Stockwell B. R.; Shair M. D.; Schreiber S. L. Synthesis and Preliminary Evaluation of a Library of Polycyclic Small Molecules for Use in Chemical Genetic Assays. J. Am. Chem. Soc. 1999, 121, 9073–9087. 10.1021/ja992144n. [DOI] [Google Scholar]
- Nicolaou K. C.; Pfefferkorn J. A.; Barluenga S.; Mitchell H. J.; Roecker A. J.; Cao G. Q. Natural product-like combinatorial libraries based on privileged structures. 3. The ″libraries from libraries″ principle for diversity enhancement of benzopyran libraries. J. Am. Chem. Soc. 2000, 122, 9968–9976. 10.1021/ja0020355. [DOI] [Google Scholar]
- Nicolaou K. C.; Pfefferkorn J. A.; Mitchell H. J.; Roecker A. J.; Barluenga S.; Cao G. Q.; Affleck R. L.; Lillig J. E. Natural product-like combinatorial libraries based on privileged structures. 2. Construction of a 10 000-membered benzopyran library by directed split-and-pool chemistry using NanoKans and optical encoding. J. Am. Chem. Soc. 2000, 122, 9954–9967. 10.1021/ja002034c. [DOI] [Google Scholar]
- Nicolaou K. C.; Pfefferkorn J. A.; Roecker A. J.; Cao G. Q.; Barluenga S.; Mitchell H. J. Natural product-like combinatorial libraries based on privileged structures. 1. General principles and solid-phase synthesis of benzopyrans. J. Am. Chem. Soc. 2000, 122, 9939–9953. 10.1021/ja002033k. [DOI] [Google Scholar]
- Fenniri H.; Ding L.; Ribbe A. E.; Zyrianov Y. Barcoded resins: a new concept for polymer-supported combinatorial library self-deconvolution. J. Am. Chem. Soc. 2001, 123, 8151–8152. 10.1021/ja016375h. [DOI] [PubMed] [Google Scholar]
- Tallarico J. A.; Depew K. M.; Pelish H. E.; Westwood N. J.; Lindsley C. W.; Shair M. D.; Schreiber S. L.; Foley M. A. An alkylsilyl-tethered, high-capacity solid support amenable to diversity-oriented synthesis for one-bead, one-stock solution chemical genetics. J. Comb. Chem. 2001, 3, 312–318. 10.1021/cc000107i. [DOI] [PubMed] [Google Scholar]
- Brohm D.; Philippe N.; Metzger S.; Bhargava A.; Muller O.; Lieb F.; Waldmann H. Solid-phase synthesis of dysidiolide-derived protein phosphatase inhibitors. J. Am. Chem. Soc. 2002, 124, 13171–13178. 10.1021/ja027609f. [DOI] [PubMed] [Google Scholar]
- Umarye J. D.; Lessmann T.; Garcia A. B.; Mamane V.; Sommer S.; Waldmann H. Biology-oriented synthesis of stereochemically diverse natural-product-derived compound collections by iterative allylations on a solid support. Chem. - Eur. J. 2007, 13, 3305–3319. 10.1002/chem.200601698. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- Burke M. D.; Berger E. M.; Schreiber S. L. Generating diverse skeletons of small molecules combinatorially. Science 2003, 302, 613–618. 10.1126/science.1089946. [DOI] [PubMed] [Google Scholar]
- Dobson C. M. Chemical space and biology. Nature 2004, 432, 824–828. 10.1038/nature03192. [DOI] [PubMed] [Google Scholar]
- Brown D. G.; Boström J. Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?. J. Med. Chem. 2016, 59, 4443–4458. 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- Grimster N. P.; Connelly S.; Baranczak A.; Dong J.; Krasnova L. B.; Sharpless K. B.; Powers E. T.; Wilson I. A.; Kelly J. W. Aromatic sulfonyl fluorides covalently kinetically stabilize transthyretin to prevent amyloidogenesis while affording a fluorescent conjugate. J. Am. Chem. Soc. 2013, 135, 5656–5668. 10.1021/ja311729d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranczak A.; Liu Y.; Connelly S.; Du W. G.; Greiner E. R.; Genereux J. C.; Wiseman R. L.; Eisele Y. S.; Bradbury N. C.; Dong J.; Noodleman L.; Sharpless K. B.; Wilson I. A.; Encalada S. E.; Kelly J. W. A fluorogenic aryl fluorosulfate for intraorganellar transthyretin imaging in living cells and in Caenorhabditis elegans. J. Am. Chem. Soc. 2015, 137, 7404–7414. 10.1021/jacs.5b03042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Dong J.; Plate L.; Mortenson D. E.; Brighty G. J.; Li S.; Liu Y.; Galmozzi A.; Lee P. S.; Hulce J. J.; Cravatt B. F.; Saez E.; Powers E. T.; Wilson I. A.; Sharpless K. B.; Kelly J. W. Arylfluorosulfates Inactivate Intracellular Lipid Binding Protein(s) through Chemoselective SuFEx Reaction with a Binding Site Tyr Residue. J. Am. Chem. Soc. 2016, 138, 7353–7364. 10.1021/jacs.6b02960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q.; Ouyang X.; Wan X.; Gajiwala K. S.; Kath J. C.; Jones L. H.; Burlingame A. L.; Taunton J. Broad-Spectrum Kinase Profiling in Live Cells with Lysine-Targeted Sulfonyl Fluoride Probes. J. Am. Chem. Soc. 2017, 139, 680–685. 10.1021/jacs.6b08536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. H. Emerging Utility of Fluorosulfate Chemical Probes. ACS Med. Chem. Lett. 2018, 9, 584–586. 10.1021/acsmedchemlett.8b00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Gago P.; Olsen C. A. Arylfluorosulfate-Based Electrophiles for Covalent Protein Labeling: A New Addition to the Arsenal. Angew. Chem., Int. Ed. 2019, 58, 957–966. 10.1002/anie.201806037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Jones L. H. Covalent drug discovery using sulfur(VI) fluoride exchange warheads. Expert Opin. Drug Discovery 2023, 18, 725–735. 10.1080/17460441.2023.2218642. [DOI] [PubMed] [Google Scholar]
- Barrow A. S.; Smedley C. J.; Zheng Q.; Li S.; Dong J.; Moses J. E. The growing applications of SuFEx click chemistry. Chem. Soc. Rev. 2019, 48, 4731–4758. 10.1039/C8CS00960K. [DOI] [PubMed] [Google Scholar]
- Zeng D. M.; Deng W. P.; Jiang X. F. Linkage Chemistry of S(VI) Fluorides. Chem. - Eur. J. 2023, 29, e202300536 10.1002/chem.202300536. [DOI] [PubMed] [Google Scholar]
- Zeng D. M.; Deng W. P.; Jiang X. F. Advances in the construction of diverse SuFEx linkers. Natl. Sci. Rev. 2023, 10, nwad123 10.1093/nsr/nwad123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S.; Zheng Q. H.; Woehl J. L.; Solania A.; Chen E.; Dillon N.; Hull M. V.; Kotaniguchi M.; Cappiello J. R.; Kitamura S.; Nizet V.; Sharpless K. B.; Wolan D. W. Sulfur(VI) Fluoride Exchange (SuFEx)-Enabled High-Throughput Medicinal Chemistry. J. Am. Chem. Soc. 2020, 142, 10899–10904. 10.1021/jacs.9b13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnar-Wortzel L.; Bishop T. R.; Kitamura S.; Milosevich N.; Asiaban J. N.; Zhang X.; Zheng Q.; Chen E.; Ramos A. R.; Ackerman C. J.; Hampton E. N.; Chatterjee A. K.; Young T. S.; Hull M. V.; Sharpless K. B.; Cravatt B. F.; Wolan D. W.; Erb M. A. Chemical Inhibition of ENL/AF9 YEATS Domains in Acute Leukemia. ACS Cent. Sci. 2021, 7, 815–830. 10.1021/acscentsci.0c01550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Li G.; Smedley C. J.; Giel M. C.; Kitamura S.; Woehl J. L.; Bianco G.; Forli S.; Homer J. A.; Cappiello J. R.; Wolan D. W.; Moses J. E.; Sharpless K. B. Diversity oriented clicking delivers beta-substituted alkenyl sulfonyl fluorides as covalent human neutrophil elastase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2208540119 10.1073/pnas.2208540119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J.; Krasnova L.; Finn M. G.; Sharpless K. B. Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew. Chem., Int. Ed. 2014, 53, 9430–9448. 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- Faucher F. F.; Abegg D.; Ipock P.; Adibekian A.; Lovell S.; Bogyo M. Solid Phase Synthesis of Fluorosulfate Containing Macrocycles for Chemoproteomic Workflows. Isr. J. Chem. 2023, 63, e202300020 10.1002/ijch.202300020. [DOI] [Google Scholar]
- Chen W.; Dong J.; Li S.; Liu Y.; Wang Y.; Yoon L.; Wu P.; Sharpless K. B.; Kelly J. W. Synthesis of Sulfotyrosine-Containing Peptides by Incorporating Fluorosulfated Tyrosine Using an Fmoc-Based Solid-Phase Strategy. Angew. Chem., Int. Ed. 2016, 55, 1835–1838. 10.1002/anie.201509016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Liu X.; Zhou M.; Xia C.; Lyu Y.; Peng Q.; Soni C.; Zhou Z.; Su Q.; Wu Y.; Weerapana E.; Gao J.; Chatterjee A.; Lin C.; Niu J. Fluorosulfate as a Latent Sulfate in Peptides and Proteins. J. Am. Chem. Soc. 2023, 145, 20189–20195. 10.1021/jacs.3c07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.; Flynn J. P.; Niu J. Facile Synthesis of Sequence-Regulated Synthetic Polymers Using Orthogonal SuFEx and CuAAC Click Reactions. Angew. Chem., Int. Ed. 2018, 57, 16194–16199. 10.1002/anie.201811051. [DOI] [PubMed] [Google Scholar]
- Falkenberg K. J.; Johnstone R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discovery 2014, 13, 673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- Ho T. C. S.; Chan A. H. Y.; Ganesan A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- Jung M.; Hoffmann K.; Brosch G.; Loidl P. Analogues of trichostatin A and trapoxin B as histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 1997, 7, 1655–1658. 10.1016/S0960-894X(97)00284-9. [DOI] [Google Scholar]
- Friis S. D.; Lindhardt A. T.; Skrydstrup T. The Development and Application of Two-Chamber Reactors and Carbon Monoxide Precursors for Safe Carbonylation Reactions. Acc. Chem. Res. 2016, 49, 594–605. 10.1021/acs.accounts.5b00471. [DOI] [PubMed] [Google Scholar]
- Veryser C.; Demaerel J.; Nas V. B.; Gilles P.; De Borggraeve W. M. Ex Situ Generation of Sulfuryl Fluoride for the Synthesis of Aryl Fluorosulfates. Org. Lett. 2017, 19, 5244–5247. 10.1021/acs.orglett.7b02522. [DOI] [PubMed] [Google Scholar]
- Bolding J. E.; Martin-Gago P.; Rajabi N.; Gamon L. F.; Hansen T. N.; Bartling C. R. O.; Strømgaard K.; Davies M. J.; Olsen C. A. Aryl Fluorosulfate Based Inhibitors That Covalently Target the SIRT5 Lysine Deacylase. Angew. Chem., Int. Ed. 2022, 61, e202204565 10.1002/anie.202204565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn M. P.; Kahnberg P.; Boyle G. M.; Hansford K. A.; Hans D.; Martyn A. C.; Parsons P. G.; Fairlie D. P. Antiproliferative and phenotype-transforming antitumor agents derived from cysteine. J. Med. Chem. 2004, 47, 2984–2994. 10.1021/jm030222i. [DOI] [PubMed] [Google Scholar]
- Kahnberg P.; Lucke A. J.; Glenn M. P.; Boyle G. M.; Tyndall J. D. A.; Parsons P. G.; Fairlie D. P. Design, synthesis, potency, and cytoselectivity of anticancer agents derived by parallel synthesis from α-aminosuberic acid. J. Med. Chem. 2006, 49, 7611–7622. 10.1021/jm050214x. [DOI] [PubMed] [Google Scholar]
- Montero A.; Beierle J. M.; Olsen C. A.; Ghadiri M. R. Design, synthesis, biological evaluation, and structural characterization of potent histone deacetylase inhibitors based on cyclic alpha/beta-tetrapeptide architectures. J. Am. Chem. Soc. 2009, 131, 3033–3041. 10.1021/ja809508f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen R.; Le Quement S. T.; Nielsen T. E. Synthesis of a natural product-like compound collection through oxidative cleavage and cyclization of linear peptides. Angew. Chem., Int. Ed. 2014, 53, 11778–11782. 10.1002/anie.201405747. [DOI] [PubMed] [Google Scholar]
- Bang C. G.; Jensen J. F.; Cohrt E. O.; Olsen L. B.; Siyum S. G.; Mortensen K. T.; Skovgaard T.; Berthelsen J.; Yang L.; Givskov M.; Qvortrup K.; Nielsen T. E. A Linker for the Solid-Phase Synthesis of Hydroxamic Acids and Identification of HDAC6 Inhibitors. ACS Comb. Sci. 2017, 19, 657–669. 10.1021/acscombsci.7b00068. [DOI] [PubMed] [Google Scholar]
- Sinatra L.; Bandolik J. J.; Roatsch M.; Sonnichsen M.; Schoeder C. T.; Hamacher A.; Scholer A.; Borkhardt A.; Meiler J.; Bhatia S.; Kassack M. U.; Hansen F. K. Hydroxamic Acids Immobilized on Resins (HAIRs): Synthesis of Dual-Targeting HDAC Inhibitors and HDAC Degraders (PROTACs). Angew. Chem., Int. Ed. 2020, 59, 22494–22499. 10.1002/anie.202006725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley C. J.; Homer J. A.; Gialelis T. L.; Barrow A. S.; Koelln R. A.; Moses J. E. Accelerated SuFEx Click Chemistry For Modular Synthesis. Angew. Chem., Int. Ed. 2022, 61, e202112375 10.1002/anie.202112375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobera M.; Madauss K. P.; Pohlhaus D. T.; Wright Q. G.; Trocha M.; Schmidt D. R.; Baloglu E.; Trump R. P.; Head M. S.; Hofmann G. A.; Murray-Thompson M.; Schwartz B.; Chakravorty S.; Wu Z. N.; Mander P. K.; Kruidenier L.; Reid R. A.; Burkhart W.; Turunen B. J.; Rong J. X.; Wagner C.; Moyer M. B.; Wells C.; Hong X.; Moore J. T.; Williams J. D.; Soler D.; Ghosh S.; Nolan M. A. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- Bürli R. W.; Luckhurst C. A.; Aziz O.; Matthews K. L.; Yates D.; Lyons K. A.; Beconi M.; McAllister G.; Breccia P.; Stott A. J.; Penrose S. D.; Wall M.; Lamers M.; Leonard P.; Müller I.; Richardson C. M.; Jarvis R.; Stones L.; Hughes S.; Wishart G.; Haughan A. F.; O’Connell C.; Mead T.; McNeil H.; Vann J.; Mangette J.; Maillard M.; Beaumont V.; Munoz-Sanjuan I.; Dominguez C. Design, Synthesis, and Biological Evaluation of Potent and Selective Class IIa Histone Deacetylase (HDAC) Inhibitors as a Potential Therapy for Huntington’s Disease. J. Med. Chem. 2013, 56, 9934–9954. 10.1021/jm4011884. [DOI] [PubMed] [Google Scholar]
- Mak J. Y. W.; Wu K. C.; Gupta P. K.; Barbero S.; McLaughlin M. G.; Lucke A. J.; Tng J.; Lim J.; Loh Z.; Sweet M. J.; Reid R. C.; Liu L. G.; Fairlie D. P. HDAC7 Inhibition by Phenacetyl and Phenylbenzoyl Hydroxamates. J. Med. Chem. 2021, 64, 2186–2204. 10.1021/acs.jmedchem.0c01967. [DOI] [PubMed] [Google Scholar]
- Maolanon A. R.; Madsen A. S.; Olsen C. A. Innovative Strategies for Selective Inhibition of Histone Deacetylases. Cell Chem. Biol. 2016, 23, 759–768. 10.1016/j.chembiol.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Chakrabarti A.; Oehme I.; Witt O.; Oliveira G.; Sippl W.; Romier C.; Pierce R. J.; Jung M. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. 10.1016/j.tips.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Yang H.; Chen L.; Sun Q.; Yao F.; Muhammad S.; Sun C. The role of HDAC11 in obesity-related metabolic disorders: A critical review. J. Cell. Physiol. 2021, 236, 5582–5591. 10.1002/jcp.30286. [DOI] [PubMed] [Google Scholar]
- Bai P.; Liu Y.; Yang L.; Ding W.; Mondal P.; Sang N.; Liu G.; Lu X.; Ho T. T.; Zhou Y.; Wu R.; Birar V. C.; Wilks M. Q.; Tanzi R. E.; Lin H.; Zhang C.; Li W.; Shen S.; Wang C. Development and Pharmacochemical Characterization Discover a Novel Brain-Permeable HDAC11-Selective Inhibitor with Therapeutic Potential by Regulating Neuroinflammation in Mice. J. Med. Chem. 2023, 66, 16075–16090. 10.1021/acs.jmedchem.3c01491. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Andrade R.; Hanna A. A.; Pflum M. K. H. Evidence that HDAC7 acts as an epigenetic ″reader″ of AR acetylation through NCoR-HDAC3 dissociation. Cell Chem. Biol. 2022, 29, 1162–1173. 10.1016/j.chembiol.2022.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younker J. M.; Hengge A. C. A mechanistic study of the alkaline hydrolysis of diaryl sulfate diesters. J. Org. Chem. 2004, 69, 9043–9048. 10.1021/jo0488309. [DOI] [PubMed] [Google Scholar]
- Szeler K.; Williams N. H.; Hengge A. C.; Kamerlin S. C. L. Modeling the Alkaline Hydrolysis of Diaryl Sulfate Diesters: A Mechanistic Study. J. Org. Chem. 2020, 85, 6489–6497. 10.1021/acs.joc.0c00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Yruela C.; Fass D. M.; Cheng C. L.; Herz J.; Olsen C. A.; Haggarty S. J. Kinetic Tuning of HDAC Inhibitors Affords Potent Inducers of Progranulin Expression. ACS Chem. Neurosci. 2019, 10, 3769–3777. 10.1021/acschemneuro.9b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina D. M.; Jafari R.; Ignatushchenko M.; Seki T.; Larsson E. A.; Dan C.; Sreekumar L.; Cao Y. H.; Nordlund P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341, 84–87. 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Jafari R.; Almqvist H.; Axelsson H.; Ignatushchenko M.; Lundbäck T.; Nordlund P.; Molina D. M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Nielsen A. L.; Rajabi N.; Kudo N.; Lundo K.; Moreno-Yruela C.; Bæk M.; Fontenas M.; Lucidi A.; Madsen A. S.; Yoshida M.; Olsen C. A. Mechanism-based inhibitors of SIRT2: structure-activity relationship, X-ray structures, target engagement, regulation of α-tubulin acetylation and inhibition of breast cancer cell migration. RSC Chem. Biol. 2021, 2, 612–626. 10.1039/D0CB00036A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troelsen K. S.; Bæk M.; Nielsen A. L.; Madsen A. S.; Rajabi N.; Olsen C. A. Mitochondria-targeted inhibitors of the human SIRT3 lysine deacetylase. RSC Chem. Biol. 2021, 2, 627–635. 10.1039/D0CB00216J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabi N.; Hansen T. N.; Nielsen A. L.; Nguyen H. T.; Baek M.; Bolding J. E.; Bahlke O. O.; Petersen S. E. G.; Bartling C. R. O.; Strømgaard K.; Olsen C. A. Investigation of Carboxylic Acid Isosteres and Prodrugs for Inhibition of the Human SIRT5 Lysine Deacylase Enzyme. Angew. Chem., Int. Ed. 2022, 61, e202115805 10.1002/anie.202115805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolding J. E.; Nielsen A. L.; Jensen I.; Hansen T. N.; Ryberg L. A.; Jameson S. T.; Harris P.; Peters G. H. J.; Denu J. M.; Rogers J. M.; Olsen C. A. Substrates and Cyclic Peptide Inhibitors of the Oligonucleotide-Activated Sirtuin 7. Angew. Chem., Int. Ed. 2023, 62, e202314597 10.1002/anie.202314597. [DOI] [PubMed] [Google Scholar]
- You Y.; Kim H. S.; Park J. W.; Keum G.; Jang S. K.; Kim B. M. Sulfur(vi) fluoride exchange as a key reaction for synthesizing biaryl sulfate core derivatives as potent hepatitis C virus NS5A inhibitors and their structure-activity relationship studies. RSC Adv. 2018, 8, 31803–31821. 10.1039/C8RA05471A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gude M.; Ryf J.; White P. D. An accurate method for the quantitation of Fmoc-derivatized solid phase supports. Lett. Pept. Sci. 2002, 9, 203–206. 10.1007/bf02538384. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.