

Abstract
Skeletal muscle is a major regulatory tissue of whole-body metabolism and is composed of a diverse mixture of cell (fiber) types. Aging and several diseases differentially affect the various fiber types, and therefore, investigating the changes in the proteome in a fiber-type specific manner is essential. Recent breakthroughs in isolated single muscle fiber proteomics have started to reveal heterogeneity among fibers. However, existing procedures are slow and laborious, requiring 2 h of mass spectrometry time per single muscle fiber; 50 fibers would take approximately 4 days to analyze. Thus, to capture the high variability in fibers both within and between individuals requires advancements in high throughput single muscle fiber proteomics. Here we use a single cell proteomics method to enable quantification of single muscle fiber proteomes in 15 min total instrument time. As proof of concept, we present data from 53 isolated skeletal muscle fibers obtained from two healthy individuals analyzed in 13.25 h. Adapting single cell data analysis techniques to integrate the data, we can reliably separate type 1 and 2A fibers. Ninety-four proteins were statistically different between clusters indicating alteration of proteins involved in fatty acid oxidation, oxidative phosphorylation, and muscle structure and contractile function. Our results indicate that this method is significantly faster than prior single fiber methods in both data collection and sample preparation while maintaining sufficient proteome depth. We anticipate this assay will enable future studies of single muscle fibers across hundreds of individuals, which has not been possible previously due to limitations in throughput.
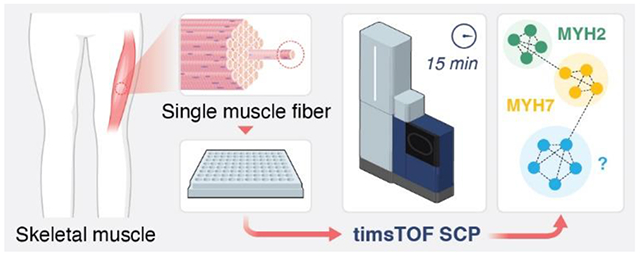
Graphical Abstract
INTRODUCTION
Skeletal muscle (skm), which comprises ~40% of human body mass, is characterized by remarkable heterogeneity of cell types that allows muscles to perform a wide variety of functional tasks. Three major skm cell types–commonly known as fibers–exist in human limb muscles that are differentiated primarily by their contractile kinetics from slow (type 1) to fast (type 2A and 2X) and metabolic profiles from more oxidative phosphorylation to more glycolytic.1,2 Slow fibers are involved primarily with low-intensity, continuous contractions, such as the maintenance of posture or walking, while fast fibers allow for rapid, forceful contraction. The differences in contractile kinetics between the fiber types are determined in large part by the myosin heavy chain (MYH) isoform that is expressed, namely MYH7 (type 1), MYH2 (type 2A), and MYH1 (type 2X).3 However, the fiber type diversity in human skm expands well beyond the contractile kinetics from the different MYH isoforms to include proteins involved in cell structure, excitation contraction coupling, regulatory proteins, and metabolism.1 Thus, to capture the cellular diversity in human skm and elucidate how the muscle adapts to acute and chronic perturbations (e.g., exercise training, bed rest, disease, or aging) in a fiber-type specific manner requires technological advancements in large-scale single cell proteomics analysis.
Recent breakthroughs in single fiber proteomics have allowed for isolation of proteins found solely within the skm fiber, without the contamination from adipocytes, connective tissue, blood vessels, and nerve cells that are all present in human skm biopsy lysates.3 These studies have overcome many of the major factors that complicate proteomic analysis of skm fibers, such as the predominance of the fiber protein mass taken up by the contractile proteins (myosin and actin) and the large variability in the size of fibers both within and across individuals,3-5 allowing for several key discoveries to be made in human skm biology. For example, it was found that the aging-related decrements in the size and contractile function of the fast fibers in older adults6 may be explained, in part, by a downregulation of proteins involved in sarcomere homeostasis and protein quality control of this fiber type.7 However, despite these recent successes, the current methods used in single fiber proteomics analysis are slow and laborious, limiting the number of fibers that can be studied per individual and making population scale single fiber proteomics studies impossible. Most previous studies also used MaxQuant’s match-between-runs (MBR) feature from larger muscle biopsy lysate analyses, which addresses missing peptides by allowing peptide peaks to be transferred between runs.8 However, without false identification transfer control, MBR may introduce false positives at a high rate, compromising protein quantification accuracy.8,9
Here we developed a method for high throughput single muscle fiber proteomics that we applied to 53 human single skm fibers obtained from the vastus lateralis of two female volunteers. Compared to prior single muscle fiber proteomics publications, our method uses streamlined proteomic sample preparation that does not require desalting along with MS data collection at a rate of 96 samples per day. Additionally, previous single skm proteomics papers have used Oribitrap mass spectrometers (ThermoScientific).3,5,7,10 In this study, we used a timsTOF SCP (Bruker) to collect diaPASEF11 data from single skm fibers, allowing nearly a 10-fold increase in throughput while still quantifying roughly the same number of proteins per single fiber compared to when MBR in MaxQuant was not used (see supplementary Figure 2 in Murgia et al., 2017).7 Additionally, we apply the python data analysis package scanpy, a tool designed for single-cell transcriptomics data, to perform unsupervised clustering of single skm fiber proteomics data.11,12 Our new workflow including sample preparation, data collection, and bioinformatics provides a rapid and complete platform for single muscle fiber proteomics that will enable large cohort studies of muscle proteome composition in the future.
METHODS
Participants.
Two female volunteers (22 and 69 years) participated in this study. Participants were self-reported to be healthy, community dwelling adults free of any known neurological, musculoskeletal, or cardiovascular diseases. Participants provided written informed consent, and procedures were approved by the Marquette University Institutional Review Board and conformed to the principles in the Declaration of Helsinki.
Skeletal Muscle Biopsy.
Percutaneous muscle biopsies were obtained from the vastus lateralis using the Bergstrom needle technique.13 Participants were instructed to abstain from strenuous exercise for 48 h prior to the biopsy procedure and to arrive at the laboratory fasted and without the consumption of caffeine. The biopsy site was cleaned with 70% ethanol, sterilized with 10% povidone–iodine, and anesthetized with 1% lidocaine HCl. A small ~1 cm incision was made overlying the distal one-third of the muscle belly, and the biopsy needle was inserted under local suction to obtain the tissue sample as described previously.14 Fiber bundles from the biopsy were separated, arranged longitudinally on a small notecard, frozen in liquid nitrogen-cooled isopentane (AC126470250; Acros Organics), and subsequently stored at −80 °C.
Single Muscle Fiber Isolation.
Skeletal muscle tissue samples were allowed to thaw in a standard Petri dish coated with Sylgard containing a cold (~4 °C) relaxing solution commonly used in single fiber contractile function experiments14,15 but with the addition of a protease and phosphatase inhibitor cocktail (78440; Thermo Scientific). The composition of the solution was 79.2 mM KCl, 20 mM imidazole, 7 mM EGTA, 5.44 mM MgCl2, 4.735 mM Na2ATP, and 14.5 mM Na2 phosphocreatine, and the pH of the solution adjusted to 7.0 with KOH. Single muscle fibers were manually isolated under a dissecting microscope with fine-tipped tweezers. Individual fibers were then carefully transferred into separate wells of a 384-well PCR plate (HSP3805; BioRad). Both the Petri dish and the 384-well PCR plate were maintained at ~4 °C during the muscle fiber isolation process with cold plates. The 384-well PCR plate was covered with aluminum sealing foil (MSF1001; BioRad) and stored at −80 °C until further analysis.
Sample Preparation.
Four hundred nanoliters of lysis buffer containing 100 mM triethylammonium bicarbonate (TEAB; 18597, Sigma; pH 8.5), 0.2% DDM, 10 ng/ μL trypsin (Promega) was dispensed into each well using the cellenONE system (cellenONE, Cellenion). After freezing and thawing for 5 min in −80 °C, 400 nL of trypsin (50 ng/ μL in 60 mM TEAB) was dispensed into each well again and the plate was incubated at 37 °C for 3.5 h. Digestion was quenched with 100 nL of 1% aqueous formic acid. The plate was stored at −80 °C until MS analysis.
Liquid Chromatography.
Dual trap single column (DTSC)16 was adapted for nanoflow to enable single cell analysis.17 The trapping columns were a 0.17 μL media bed (EXP2 from Optimize Technologies) packed with 10 μm diameter, 100 Å pore PLRP-S (Agilent) beads, and the analytical column was a PepSep 15 cm × 75 μm packed with 1.9 μm C18 stationary phase (Bruker). The configuration was installed on a Thermo Ultimate 3000 nanoRSLC equipped with one 10-port valve and one 6-port valve and a nano flow selector. Mobile phase A was 0.1% formic acid in water. Mobile phase B was 0.1% formic acid in acetonitrile. Peptides were separated using the following gradient all using linear transitions between conditions: starting conditions of 9% B at 500 nL/min; 22% B over 8 min; 37% B over 4.7 min; 1000 nL/min flow rate and 98% B over 0.2 min; hold at 98% B for 1 min; reduce to 9% B at 1,000 nL/min over 0.1 min; hold at 9% B at 1,000 nL/min for 0.9 min; return to 500 nL/min flow rate in 0.1 min (15 min total). Valves and trapping columns were heated to 55 °C, and the analytical column was heated to 60 °C. See Kreimer et al. for additional details about the dual trap chromatography setup.17
Mass Spectrometry.
The analytical column was directly connected to a 10 μm ZDV emitter (Bruker) inside of the Bruker captive source. The capillary voltage was set to 1700 V with the dry gas set at 3.0 L/min and 200 °C. Data was acquired by diaPASEF on a Bruker timsTOF SCP with the ion accumulation and trapped ion mobility ramp set to 166 ms. DIA scans were optimized and acquired with 90 m/z windows spanning 300 to 1200m/z and 0.6 to 1.43 1/K0. A 0.86 s cycle time included one full MS1 scan followed by 4 trapped ion mobility ramps to fragment all ions within the defined region.
Mass Spectrometry Data Analysis.
Peptide and Protein Quantification.
A spectral library was generated by a library-free search in DIA-NN 1.818,19 using a FASTA of the human proteome with one entry per gene downloaded on January 5, 2023, with 20,594 entries. The library from that first library-free search was used to analyze MS data from 53 single skm fibers each containing more than 500 proteins per fiber in the first search. Protein identifications by DIA-NN are based on unique peptides. The option for reannotation of identified peptides with the FASTA was used. The targeted library from the second search contained 1796 protein groups and 11,610 precursors in 9790 elution groups. Protein groups are formed when identified peptides cannot differentiate between multiple proteins with shared subsequences. Each search was conducted with C carbamidomethylation off, match-between-runs (MBR) enabled, double pass mode enabled, cross-run normalization disabled, speed and RAM usage set to optimal, and any LC (high-accuracy) quantification strategy. MBR in DIA-NN is not equivalent to MBR in MaxQuant; in DIA-NN, MBR means the spectral library for the first search is used for the second search. Precursor charge range was set between 2 and 4, mass accuracy was fixed to 15 ppm, and one missed cleavage was allowed in the database search. All other parameters were the default. Intensity-based absolute quantification (iBAQ)20 was manually implemented in python and used to calculate protein group quantities from peptide quantities. Peak intensities for all peptides corresponding to a protein group were summed and then divided by the number of possible unique peptides, which was calculated to allow for peptides with one missed cleavage and between 7 to 30 amino acids long to mirror DIA-NN settings. Resulting protein quantities in each well were normalized to alpha skeletal muscle actin (ACTA1, P68133) quantity in that well to control for fiber length and cross-sectional area, as previously described.3,5 Lastly, fibers were ranked by percent of their dominant MYH isoform. Pure MYH7, MYH2, or MYH1 myofibers were assigned if any of these three isoforms reached 80% of the total MYH signal for all isoforms.3 Fibers that did not have a single MYH isoform at more than 80% of the total MYH signal were labeled as one of two hybrids based on the top two most prevalent MYHs, either coexpression of MYH1 and MYH2 or MYH2 and MYH7. Protein intensities from MaxLFQ21 were also normalized to the quantity of ACTA1 in each well. An example extracted ion chromatogram for a random peptide was obtained from Skyline-daily22 version 22.2.1.351 to show the quality of chromatography. All data analysis was performed in python version 3.9.12.
Data Visualization.
Using scanpy version 1.9.1,23 we implemented a pipeline using Uniform Manifold Approximation & Projection (UMAP)24 and the Leiden method25 to reduce the data to a two-dimensional space and bin the data into discrete populations, respectively. Proteins appearing in less than 5% of fibers (or a minimum of 3 fibers) were filtered out, as these are rare. To obtain parameters leading to the best match to manually annotated cluster labels, we used a random search with 300 iterations optimizing number of principal components (PCs) that explain variance (range 1–50), neighbors that compute a cell’s local connectivity (range 1–15 or 30), Leiden resolution which controls coarseness of clustering (range 0–1 by a factor of 0.1), and whether to scale data to unit variance and zero mean. Using a variety of metrics, we measured similarity between the Leiden inferred cluster labels and author annotated myosin labels based on MYH percentage found in fibers associated with each cluster. The set of parameters with the highest normalized mutual information score (NMI) was used for the final UMAP and Leiden clustering. NMI, which ranges from 0 (similarity expected by chance) to 1 (perfect similarity), provides a measure of quality of clustering, and allows for comparison between data sets with different numbers of clusters. Multiple sets of parameters led to the same optimized NMI score of 0.7, and one set was chosen at random (no scaling, 33 PCs, 11 neighbors, 0.1 resolution). We also reported conditional entropy, which refers to the amount of information needed to describe a reference label given the Leiden clustering generated labels (low value means Leiden labels are very informative of reference labels) corresponding to the parameters with the highest NMI. A Chi squared test was used to test for a difference between young and old fibers between clusters.
Pathway Enrichment Analysis.
We first determined proteins that were most uniquely identifying clusters. Pairwise protein distribution comparisons were made between Leiden groups using a Wilcoxon Rank-Sum test and Benjamini–Hochberg (B–H) multiple testing correction with alpha = 0.01. Gene ontology (GO) Biological Process term enrichment analysis was applied to proteins with an adjusted p-value less than 0.01 (negative log10 adjusted p-value > 2) between Leiden groups. Term enrichment analysis was performed using ClueGO (version 2.5.9)26 application within Cytoscape (version 3.9.1; release date 5/25/2022).27 WikiPathways term enrichment was used to understand the most common pathways among all 744 proteins, and GO Biological Process using GO term fusion was used for network analysis of differentially expressed proteins between Leiden clusters. Only pathways with a p-value < 0.0001 were enriched for either pathway. For either analysis, we manually removed redundant terms to keep only those that connected all the proteins to the simplest network.
Code and Data Availability.
All mass spectrometer raw data, libraries, and outputs have been added to MassIVE database: ftp://MSV000091282@massive.ucsd.edu doi:10.25345/C54T6FC9Z (password: hello). JupyterLab notebooks can be found at xomicsdatascience/HT-Skm-Proteomics (github.com).
RESULTS
Our high throughput single skm fiber proteomics workflow enabled collection of high quality and high content protein data from 53 human fibers (31 from young and 22 from old), obtained from the vastus lateralis of two female volunteers (Figure 1A). Single fibers were manually isolated and carefully transferred to the bottom of an individual well of the 384-well plates. One pot sample preparation was then performed using a cellenONE for nanoliter liquid dispensing before directly loading 1/100th of the total sample from the same plate onto a timsTOF SCP MS for a 15 min total run time with diaPASEF data collection.
Figure 1.

Data and workflow overview. (A) Muscle biopsies were collected from two healthy female donors. Fibers were processed using one pot in-solution digestion in a 384-well plate, and data were acquired by DIA-PASEF on a Bruker timsTOF SCP. Proteins were quantified using library-free search with DIA-NN against the whole human proteome. Clustering allowed for visualization of similarly grouped fibers. Top ranked proteins distinguishing clusters were then used to understand biological pathways. (B) Number of proteins quantified per fiber. (C) Ten proteins with highest percent of total counts in each fiber. (D) WikiPathways term enrichment analysis summary of all 744 proteins identified in any fiber. (E) Number of peptides quantified per minute across binned mean retention times (RT). (F) Extracted ion chromatogram showing a typical full width at half-maximum (fwhm) of 5.2 s for example peptide KDFELNALNAR. (G) RT standard deviation in minutes across binned mean RTs.
At a false discovery rate (FDR) of 1%, we identified 868 unique protein groups present in at least one fiber. There were 124 protein groups with multiple members, which were filtered out to simplify subsequent analyses, resulting in 744 unique proteins (Table S1) corresponding to 8764 unique precursors. Proteins were included if they were identified in at least 3 fibers (>5%). An average of 629 ± 62 proteins were quantified per single muscle fiber (Figure 1B). Forty-one percent (305/744) of proteins were quantified in all 53 fibers. The most abundant protein across all fibers was ACTA1 (Figure 1C). WikiPathways enriched in the set of all proteins is depicted in Figure 1D; proteins involved with striated muscle contraction comprised the largest group of proteins (55%) detected from a pathway compared to all the proteins associated with that pathway, followed by mitochondrial fatty acid (FA) beta-oxidation (47%). Our chromatography method showed consistent instrument utilization across the elution gradient with more than 800 peptides quantified in each minute of data collection between 4 and 13 min (Figure 1E). Our chromatographic peaks were sharp with an example extracted ion chromatogram for a random peptide sequence (KDFELNALNAR) showing a full width at half-maximum (fwhm) of 5.2 s. Retention time (RT) standard deviations were small across binned mean RTs (Figure 1G). Despite the high sensitivity of the timsTOF SCP, our hundred-fold dilution of the samples resulted in almost no peptide signal in blank wells (Figure S1).
The 10 most abundant iBAQ-calculated proteins in pure type 2A or 1 fibers are plotted separately (Figure 2A,B), demonstrating the high abundance of ACTA1 and congruence of regulatory protein isoforms with their appropriate primary MYH isoform. Although MYL1 (a fast myosin regulatory light chain) was present among the top 10 most abundant proteins in both fiber types, its quantity was 2-fold higher in type 2A fibers (Figure 2C). Among the proteins with top 10 highest percentages of total counts per fiber from MaxLFQ quantification, ACTA1 was not as clearly more abundant (Figure S2).
Figure 2.

Ten most abundant proteins when quantities are computed by iBAQ. (A) Top ten proteins with highest percent of total counts in pure type 2A fibers quantified using iBAQ. (B) Top ten proteins with highest percent of total counts in pure type 1 fibers quantified using iBAQ. (C) MYL1 iBAQ-calculated quantity in type 1 versus 2A fibers. MYL11 is interchangeable with MYLPF and MLC2-fast.
We explored differences in ratios of MYH proteins per fiber between iBAQ and MaxLFQ, which are different protein quantification algorithms. A fiber was assigned as a pure type 1 or 2A if the fraction of MYH7 or MYH2, respectively, of all MYH isoforms combined was greater than 80%. Fibers not meeting this threshold were categorized as mixed fibers based on the top two most prevalent MYHs. Using MaxLFQ and a 80% cutoff for identifying pure fibers, there were 17 MYH7, 3 MYH1, and only 1 MYH2 fiber. The mixed fibers consisted of 5 MYH2/7, 6 MYH1/2, and 1 MYH1/7, and the remaining 20 fibers had a combination of MYH2 and MYH4 (Figure 3A, Table S2). The number of fibers expressing MYH4 is shown in Figure 3B. Initially there was one peptide (DEELDQLKR2+) mapped to MYH4 that differed from MYH2 by only the assignment of L versus I, which cannot be distinguished by mass alone, so this peptide was removed before subsequent iBAQ calculations (Figure S3). Use of unique peptides to quantify protein isoforms is often acceptable, but in this case, it appears DIA-NN has mistakenly assigned a I/L isobaric peptide as a unique peptide for quantification. Using DIA-NN with iBAQ calculated protein intensities, we found that most fibers had a single predominant MYH isoform (26 MYH2 and 18 MYH7) and the rest were mixed type (5 MYH2/7 and 4 MYH1/2) (Figure 3C, Table S3). Expression of MYH4 (Figure 3D), which is thought to be found in rodent skm and only in a specialized muscle type in humans,28 was 0.3% of total MYH content when protein quantities were computed using iBAQ, but 10% of total MYH when using MaxLFQderived protein quantities. Other single cell human skm studies also observe negligible or no MYH4 present in fibers.29 MYH6, which is found in cardiac muscle cells,30 was low in both quantification strategies: 0.3% of total MYH content when protein quantities were calculated using iBAQ and 0.8% of total MYH content when using MaxLFQ. The drastic reduction in MYH2 fibers, the unlikely MYH1/7 hybrid, and a large number of fibers expressing MYH4 (Figure 3B) indicate that protein quantities computed with iBAQ are preferred over those from MaxLFQ when computing ratios of proteins.
Figure 3.

Detected proportions of MYH isoforms across 53 skeletal muscle fibers. (A) Fraction of MYH proteins per fiber (sorted by lowest to highest MYH2 quantity from left to right across the 53 fibers) derived from MaxLFQ A horizontal line drawn at MYH fraction of 0.8 indicates the cutoff for assigning pure fibers. (B) Histogram of fibers per MYH fraction for each subtype using MaxLFQ. (C) Fraction of MYH proteins per fiber using protein quantities calculated using iBAQ (D) Histogram of fibers per MYH fraction for each subtype using protein quantities calculated using iBAQ. MYH4 y-axis limits are different from rest of panels due to much greater proportion of zeros.
We performed unsupervised Leiden clustering25 and visualization with UMAP using all proteins as input to better understand how proteome profiles would group the fibers. Given the manual groupings defined by MYH proportions, we optimized the data reduction and clustering to best match those groups. Three hundred random combinations of workflow parameters were tested with a random search (see Methods). We used NMI to measure the similarity between the data-inferred labels and the author-assigned labels. The parameters resulting in the highest NMI of 0.7 (no data scaling, 33 PCs, 11 neighbors, 0.1 resolution) were used for the final clustering and UMAP plot. The corresponding conditional entropy was 0.72. We did not expect perfect similarity scores because all proteins were used to generate the UMAP plots and the manual annotations are only based on four MYH subtypes.
The optimal Leiden clustering separated all protein level data into two clusters (Figure 4A). When colored by the manual MYH annotations, we see clear separation between pure MYH2 and MYH7 fibers, as well as mixed MYH2/7 and MYH1/2 fibers (Figure 4B). When comparing the two UMAP plots, Leiden cluster 1 aligns closely with MYH7 and MYH2/7 annotated fibers, and Leiden cluster 0 with both MYH2 and MYH1/2 annotated fibers. Figure 4C shows protein identifications per fiber are random within clusters (i.e., a cluster is not driven by a high or low count). The UMAP plot of fibers colored by iBAQ-calculated abundance of MYH1, −2, and −7 (Figure 4D) aligns with the manual MYH annotated fiber groups in Figure 4B.
Figure 4.

Clustering and dimension reduction of single fiber proteome data. (A) UMAP colored by Leiden clusters. (B) UMAP colored by author-annotated fiber labels. (C) UMAP colored by the number of proteins identified per fiber. (D) UMAP colored by iBAQ-calculated quantities of MYH1, MYH2, and MYH7 proteins.
Next, we performed a B–H corrected Wilcoxon Rank-Sum test with p-value < 0.01 to identify differentially expressed proteins between Leiden clusters 0 and 1. Ninety-four proteins were differentially expressed, of which 74 were upregulated in Leiden cluster 1 (mostly MYH7 fibers) and 20 were higher in Leiden cluster 0 (mostly MYH2 fibers). A volcano plot of the 94 proteins with an adjusted p-value < 0.01 is shown in Figure S4. There was no difference between young and old fiber distribution between Leiden clusters (65% young in Leiden 0 and 50% young in Leiden 1; p-value = 0.4), and although we are underpowered to compare fiber ratios between people, we note that four out of the five MYH2/7 hybrid fibers came from the older volunteer (Figure S5).
GO biological process term enrichment analysis performed using the 94 proteins with an adjusted p-value < 0.01 (Table S4) between Leiden 0 and 1 clusters revealed proteins associated with four main pathways, muscle contraction, muscle development (most of which are structural proteins or involved in excitation-contraction coupling or Ca2+ handling), oxidative phosphorylation (OXPHOS), and organic acid catabolic processes (most of which are involved in FA beta-oxidation) (Figure 5A). Many of these were canonical differences between fiber types, including skeletal troponin subunit isoforms (TNNI1, TNNT1, TNNC1 higher in slow fibers; TNNC2, TNNI2, TNNT3 higher in fast fibers), tropomyosins (TPM3 higher in slow fibers; TPM1 higher in fast fibers), myosin light chain proteins (MYL3, MYL5, MYL6B higher in slow fibers; MYL1 and MYL11, also known as MYLPF or MLC2-fast, higher in fast fibers), Z-line linking proteins (MYOZ2 higher in slow fibers; ACTN3 higher in fast fibers), sarcoplasmic reticulum proteins (CASQ2 and ATP2A2 higher in slow fibers; TRDN and ATP2A1 higher in fast fibers), and myofibril assembly/stability proteins (MYOM1, MYOM3, MYOT higher in slow fibers). This analysis also indicated that MYH14 (a nonmuscle MYH) was different between groups; however, MYH14 comprised 0.008% of total MYH content, and the highest MYH14 quantity in any fiber was 0.02% of total MYH in that fiber.
Figure 5.

Term enrichment analysis. (A) Network of Gene Ontology (GO) Biological Process enrichment of 94 differentially expressed proteins between Leiden clusters 0 and 1 showing four main pathways, muscle contraction, muscle development, oxidative phosphorylation, and organic acid catabolic process. The large circles indicate enriched GO terms, and the small circles show the proteins that are found in those pathways (green: proteins upregulated in Leiden cluster 0; salmon: proteins upregulated in Leiden cluster 1). (B) Raincloud plots of glycolytic proteins that were statistically higher in Leiden cluster 1 compared to 0 using an adjusted p-value cutoff of 0.05.
All proteins associated with OXPHOS, except ACTN3, had higher expression in the slow fibers as expected. OXPHOSenriched proteins were components of mitochondrial electronic transport chain, including complex IV (COX5A, COX7A2, and COX4I1), complex I (NDUFS1 and NDUFB5), and complex V (ATP5F1A). Although glycolysis was not an enriched pathway, there was one glycolytic protein, HK1, that was significantly higher in the fast fibers with an adjusted p-value < 0.01. An additional glycolytic protein, LDHA, was higher in the fast fibers but did not reach statistical significance (adjusted p-value 0.013). Distributions of log2 transformed iBAQ-calculated quantities for LDHA and HK1 are visualized using raincloud plots (Figure 5B,C). Lastly, enzymes involved in organic acid catabolic processes, including ACADS, HADHA, HADHB, DECR1, ACAA2, ACAT1, and HSD17B4, were upregulated in slow fibers as expected.
Tropomyosins TPM1 and TPM3, which regulate muscle contraction, were both present in every fiber despite their expected specificity for fast and slow fibers, respectively. Analysis of their relative proportions showed two groups where one isoform was dominant over the other (Figure S6A). Correlation between the fast and slow myosin and tropomyosin isoforms was assessed. The fast form of myosin (MYH2) was well correlated with the fast form of tropomyosin (TPM1) while the slow myosin (MYH7) was well correlated with the slow form of tropomyosin (TPM3). There was minimal correlation when comparing the quantities of mismatched fast and slow myosins and tropomyosins (Figure S6B), overall supporting the quality of protein quantification with our method.
DISCUSSION
Our high throughput single muscle fiber data acquisition and analysis is able to quantify and distinguish human type 1 and 2A fibers more quickly than previously described methods for skm single fiber proteomics. In ~1/10th the time, we are able to identify differences in protein quantities that are congruent with established skm physiology. We were also able to adapt a single cell data clustering tool originally designed for single cell transcriptomics data to streamline data interpretation. Furthermore, UMAP and Leiden clustering was able to separate slow and fast fibers, as well as “hybrid” fibers, into distinct groups. When this high throughput mass spectrometry method is paired with a clustering analysis tool, understanding of single skm fiber proteomes on the scale of a several hundred fibers analyzed per individual will be possible, which is a more accurate representation of the whole muscle than the current approach of <20 fibers per person.
Similar to other skm studies,7 we found most fibers (83%) were composed of a single dominant MYH isoform, namely pure MYH2 (type 2A) and MYH7 (type 1), and only a few fibers had mixed quantities of two MYH subtypes (Figure 3C). The mixed subtypes, or “hybrid” fibers, observed were MYH2/7 (1/2A) and MYH2/1 (2A/2X), which are the most observed hybrid fibers in humans when fiber types are determined with immunohistochemistry of muscle cross sections or SDS-PAGE of isolated single muscle fibers.2 We found that MYH protein quantities calculated using iBAQ from DIA-NN precursor data with subsequent actin normalization were more reliable in assigning MYH isoforms based on established knowledge of MYH subtype distributions within human skm compared to protein quantities from MaxLFQ. We characterized the differences in type 1 and 2A fiber types based on the quantities of the regulatory and structural proteins (troponins, tropomyosins, MYL proteins, Z-line linking proteins, sarcoplasmic reticulum proteins, and myofibril assembly/stability proteins) and found a majority of the proteins (Figure 2 and Figure 5) to also be congruent with previous findings.3,29 For example, MYL3, which we found to be significantly higher in Leiden 1 (mostly type 1 fibers), has been previously reported as 10 times more abundant in type 1 versus type 2A fibers.3 Other MYLs that we found to be differentially expressed in slow fibers, namely MYL6B and MYL5, were also reported to be significantly higher in human type 1 fibers.3 Additionally, the quantity of ACTN3, an actin binding protein, was previously found to be three times higher in type 2X compared to type 1 and 2A fibers.3 Although we did not find 2X fibers in our data set, we did observe that ACTN3 protein was differentially higher (log2 fold change of 2) in Leiden 0 (mostly type 2A) compared to Leiden 1 (mostly type 1). Together, these findings indicate that the depth and sensitivity of our approach was uncompromised by the nearly 10-fold decrease in data acquisition time.
In addition to the expected differences we observed in the regulatory and structural proteins between fiber types, we found several proteins differentially higher in the Leiden 1 cluster that play key roles in OXPHOS and organic acid catabolic processes, both of which occur in the mitochondria to generate ATP. These findings are consistent with established fiber type differences, as Leiden 1 is composed of primarily type 1 fibers which are well-known to have higher mitochondrial content, as they rely more on oxidative phosphorylation rather than glycolysis to generate ATP.1 An unexpected observation, however, was that we only found one glycolytic protein (HK1) that reached statistical significance between the two Leiden groups (Figure 5), as type 2A fibers are also known to rely more on glycolysis than type 1 fibers. One potential explanation for this observation is that we included fibers from both a young and older woman, and it was recently observed that proteins involved in glycolysis and glycogen metabolism were higher in slow fibers and lower in fast fibers from older compared with younger men.7 Thus, the inclusion of the older and younger women in our data set may have masked the expected fiber type differences in glycolytic protein content. Further studies are warranted to investigate if the metabolic protein profile differs between type 1 and 2A fibers from young and older women.
Our data indicate that MYH6 (alpha isoform in cardiac muscle), MYH4 (primarily in rodent limb muscles), and MYH14 (a nonmuscle myosin) were significantly different between Leiden groups; however, the abundance of each of these isoforms was <1% of the total MYH content. This is in line with the current literature, which shows MYH6 and MYH4 to be completely absent or in very low abundance in human skeletal muscle fibers.1,7,29 Nevertheless, the mean iBAQ-calculated quantity of MYH6 in our data was 5e-4 in Leiden group 1 and 8e-6 in Leiden group 0, which led to it being significantly different between groups despite the low amount. It is possible that the presence of these myosins is due to the allowed 1% false discovery rate in our data set. Misassignment of peptide sequences is especially problematic with MYH isoforms, where sequence homology is high; in this case the peptide contributing most of the MYH6 quantity is one amino acid different from MYH7 (a Y in MYH6 versus a F in MYH7). We had variable methionine oxidation off in our search due to the additional time cost in library-free DIA-NN searching; for this peptide, the tyrosine detected could actually be the F amino acid residues with neighboring oxidized M on the peptide from MYH7. This highlights a benefit of using the iBAQ approach for protein quantitation, which is that the reported quantities of proteins that are identified by a single peptide are penalized by the denominator that divides by the number of possible detectable peptides. In this way if these protein IDs are among our 1% false positives, their quantity will be very small because it is unlikely to get multiple false hits to the same protein. Further studies with Western blot or immunohistochemistry will reveal if there is in fact a small but significant increase in MYH6 in type 1 relative to type 2A fibers.
An unexpected observation in our study was that actin appeared at a much higher quantity than all other proteins in our data set (Figures 1 and 2). In contrast, a skm proteomics study on mice found myosin to be the most abundant protein, not actin.31 Upon further investigation, the high abundance of actin in our study appeared to be a consequence of the denominator in the iBAQ protein calculation, which in our implementation divides each protein by the number of potential unique observable peptides as a normalization of the summed peptide signal numerator. Specifically, actin is substantially shorter than myosins (377 amino acids in ACTA1 versus 1935 amino acids in MYH7), and thus the denominator used in the iBAQ calculation for ACTA1 was 7, while the denominator used for MYH7 was 120. This resulted in a difference in the ratio of unique observable peptides to amino acids to be 7/377 = 0.02 for ACTA1 compared with 120/1935 = 0.06 for MYH7. Therefore, the normalization ratio for ACTA1 was skewed relative to the protein’s length due to many nonunique peptides in ACTA1. This does not adversely affect the results of this study, because we only compared ratios between proteins with similar properties (e.g., myosins and tropomyosins). It is also generally well accepted that comparing ratios between different proteins from label free quantification proteomics data provides a ballpark estimate at best due to the differences in peptide ionization efficiency.
CONCLUSION
We report a complete workflow for high throughput single muscle fiber proteomics including sample preparation, data collection, and bioinformatic summary analysis. We also demonstrate that the iBAQ quantification algorithm for protein quantification better reflects the myosin heavy chain percentages based on known muscle physiology than those derived from MaxLFQ. The proteome data cleanly clusters itself into two groups that are mainly driven by MYH types. Comparing the two groups from clustering showed protein differences between the groups that are consistent with known muscle physiology. In the future our workflow will enable large scale studies of human muscle fiber heterogeneity.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grants R21AG074234 and R35GM142502 to J.G.M. and R01AG048262 to C.W.S. We thank H. Adam Steinberg and Dasom Hwang for graphic design assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.3c00072.
Table S1. iBAQ-calculated protein quantities normalized by quantity of ACTA1 per fiber for all 744 proteins and 53 fibers. Table S2. Fraction of each MYH isoform to total MYH per fiber calculated using protein quantities from iBAQ. Table S3. Fraction of each MYH isoform to total MYH per fiber calculated using MaxLFQ. Table S4. Means, log2(fold change) and −log10(B–H adjusted p-value) for all 744 proteins between Leiden clusters 0 and 1. Positive log2(fold change) means protein quantity was higher in Leiden cluster 0 compared to 1, and negative log2(fold change) means protein quantity was higher in Leiden cluster 1 compared to 0. Figure S1. Quality control metrics demonstrating little-to-no peptide counts in blank wells and robust peptide counts in sample wells. Figure S2. Ten most abundant proteins in slow and fast fibers when quantities are derived from MaxLFQ. Figure S3. iBAQ-calculated MYH fractions prior to removal of MYH4 peptide with overlap with MYH2 and description of peptide that was removed. Figure S4. Volcano plot highlighting 94 differentially expressed proteins between Leiden clusters 0 and 1. Figure S5. UMAP colored by old and young fiber types. Figure S6. Visualization of TPM1 and TPM3 per fiber and correlations with fast and slow fibers (PDF)
Supplementary table 1 (XLSX)
Supplementary table 2 (XLSX)
Supplementary table 3 (XLSX)
Supplementary table 4 (XLSX)
The authors declare no competing financial interest.
Contributor Information
Amanda Momenzadeh, Department of Computational Biomedicine, Cedars Sinai Medical Center, Los Angeles, California 90069, United States; Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
Yuming Jiang, Department of Computational Biomedicine, Cedars Sinai Medical Center, Los Angeles, California 90069, United States; Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
Simion Kreimer, Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
Laura E. Teigen, Department of Physical Therapy, Marquette University, Milwaukee, Wisconsin 53233, United States
Carlos S. Zepeda, Department of Physical Therapy, Marquette University, Milwaukee, Wisconsin 53233, United States
Ali Haghani, Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Department of Physical Therapy, Marquette University, Milwaukee, Wisconsin 53233, United States.
Mitra Mastali, Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
Yang Song, Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
Alexandre Hutton, Department of Computational Biomedicine, Cedars Sinai Medical Center, Los Angeles, California 90069, United States; Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States.
Sarah J. Parker, Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Department of Biomedical Sciences, Cedars Sinai Medical Center, Los Angeles, California 90048, United States
Jennifer E. Van Eyk, Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Department of Biomedical Sciences, Cedars Sinai Medical Center, Los Angeles, California 90048, United States
Christopher W. Sundberg, Department of Physical Therapy, Marquette University, Milwaukee, Wisconsin 53233, United States; Athletic and Human Performance Research Center, Marquette University, Milwaukee, Wisconsin 53233, United States
Jesse G. Meyer, Department of Computational Biomedicine, Cedars Sinai Medical Center, Los Angeles, California 90069, United States; Advanced Clinical Biosystems Research Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States; Smidt Heart Institute, Cedars Sinai Medical Center, Los Angeles, California 90048, United States
REFERENCES
- (1).Schiaffino S; Reggiani C Fiber Types in Mammalian Skeletal Muscles. Physiol. Rev 2011, 91 (4), 1447–1531. [DOI] [PubMed] [Google Scholar]
- (2).Murach KA; Dungan CM; Kosmac K; Voigt TB; Tourville TW; Miller MS; Bamman MM; Peterson CA; Toth MJ Fiber Typing Human Skeletal Muscle with Fluorescent Immunohistochemistry. J. Appl. Physiol 2019, 127 (6), 1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Murgia M; Nogara L; Baraldo M; Reggiani C; Mann M; Schiaffino S Protein Profile of Fiber Types in Human Skeletal Muscle: A Single-Fiber Proteomics Study. Skelet. Muscle 2021, 11 (1), 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gonzalez-Freire M; Semba RD; Ubaida-Mohien C; Fabbri E; Scalzo P; Højlund K; Dufresne C; Lyashkov A; Ferrucci L The Human Skeletal Muscle Proteome Project: A Reappraisal of the Current Literature. J. Cachexia Sarcopenia Muscle 2017, 8 (1), 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Murgia M; Nagaraj N; Deshmukh AS; Zeiler M; Cancellara P; Moretti I; Reggiani C; Schiaffino S; Mann M Single Muscle Fiber Proteomics Reveals Unexpected Mitochondrial Specialization. EMBO Rep. 2015, 16 (3), 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Grosicki GJ; Zepeda CS; Sundberg CW Single Muscle Fibre Contractile Function with Ageing. J. Physiol 2022, 600 (23), 5005–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Murgia M; Toniolo L; Nagaraj N; Ciciliot S; Vindigni V; Schiaffino S; Reggiani C; Mann M Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017, 19 (11), 2396–2409. [DOI] [PubMed] [Google Scholar]
- (8).Yu F; Haynes SE; Nesvizhskii AI IonQuant Enables Accurate and Sensitive Label-Free Quantification With FDRControlled Match-Between-Runs. Mol. Cell. Proteomics MCP 2021, 20, 100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lim MY; Paulo JA; Gygi SP Evaluating False Transfer Rates from the Match-between-Runs Algorithm with a Two-Proteome Model. J. Proteome Res 2019, 18 (11), 4020–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kallabis S; Abraham L; Müller S; Dzialas V; Türk C; Wiederstein JL; Bock T; Nolte H; Nogara L; Blaauw B; Braun T; Krüger M High-Throughput Proteomics Fiber Typing (ProFiT) for Comprehensive Characterization of Single Skeletal Muscle Fibers. Skelet. Muscle 2020, 10 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Meier F; Brunner A-D; Frank M; Ha A; Bludau I; Voytik E; Kaspar-Schoenefeld S; Lubeck M; Raether O; Bache N; Aebersold R; Collins BC; Röst HL; Mann M DiaPASEF: Parallel Accumulation-Serial Fragmentation Combined with Data-Independent Acquisition. Nat. Methods 2020, 17 (12), 1229–1236. [DOI] [PubMed] [Google Scholar]
- (12).Brunner A; Thielert M; Vasilopoulou C; Ammar C; Coscia F; Mund A; Hoerning OB; Bache N; Apalategui A; Lubeck M; Richter S; Fischer DS; Raether O; Park MA; Meier F; Theis FJ; Mann M Ultra-high Sensitivity Mass Spectrometry Quantifies Single-cell Proteome Changes upon Perturbation. Mol. Syst. Biol 2022, 18 (3). DOI: 10.15252/msb.202110798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bergstrom J. Percutaneous Needle Biopsy of Skeletal Muscle in Physiological and Clinical Research. Scand. J. Clin. Lab. Invest 1975, 35 (7), 609–616. [PubMed] [Google Scholar]
- (14).Sundberg CW; Hunter SK; Trappe SW; Smith CS; Fitts RH Effects of Elevated H+ and Pi on the Contractile Mechanics of Skeletal Muscle Fibres from Young and Old Men: Implications for Muscle Fatigue in Humans. J. Physiol 2018, 596 (17), 3993–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Teigen LE; Sundberg CW; Kelly LJ; Hunter SK; Fitts RH Ca2+ Dependency of Limb Muscle Fiber Contractile Mechanics in Young and Older Adults. Am. J. Physiol.-Cell Physiol 2020, 318 (6), C1238–C1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kreimer S; Haghani A; Binek A; Hauspurg A; Seyedmohammad S; Rivas A; Momenzadeh A; Meyer JG; Raedschelders K; Van Eyk JE Parallelization with Dual-Trap Single-Column Configuration Maximizes Throughput of Proteomic Analysis. Anal. Chem 2022, 94 (36), 12452–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kreimer S; Binek A; Chazarin B; Cho JH; Haghani A; Hutton A; Marbán E; Mastali M; Meyer JG; Mesquita T; Song Y; Van Eyk J; Parker S High-Throughput Single-Cell Proteomic Analysis of Organ-Derived Heterogeneous Cell Populations by Nanoflow Dual-Trap Single-Column Liquid Chromatography. Anal. Chem 2023, 95 (24), 9145–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Demichev V; Szyrwiel L; Yu F; Teo GC; Rosenberger G; Niewienda A; Ludwig D; Decker J; Kaspar-Schoenefeld S; Lilley KS; Mülleder M; Nesvizhskii AI; Ralser M Dia-PASEF Data Analysis Using FragPipe and DIA-NN for Deep Proteomics of Low Sample Amounts. Nat. Commun 2022, 13 (1), 3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Demichev V; Messner CB; Vernardis SI; Lilley KS; Ralser M DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17 (1), 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schwanhäusser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473 (7347), 337–342. [DOI] [PubMed] [Google Scholar]
- (21).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 2014, 13 (9), 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26 (7), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wolf FA; Angerer P; Theis FJ SCANPY: Large-Scale Single-Cell Gene Expression Data Analysis. Genome Biol. 2018, 19 (1), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).McInnes L; Healy J; Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2020, September 17 [Google Scholar]
- (25).Traag VA; Waltman L; van Eck NJ From Louvain to Leiden: Guaranteeing Well-Connected Communities. Sci. Rep 2019, 9 (1), 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bindea G; Mlecnik B; Hackl H; Charoentong P; Tosolini M; Kirilovsky A; Fridman W-H; Pagès F; Trajanoski Z; Galon J ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25 (8), 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13 (11), 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Harrison BC; Allen DL; Leinwand LA IIb or Not IIb? Regulation of Myosin Heavy Chain Gene Expression in Mice and Men. Skelet. Muscle 2011, 1 (1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Perez K; Ciotlos S; McGirr J; Limbad C; Doi R; Nederveen JP; Nilsson MI; Winer DA; Evans W; Tarnopolsky M; Campisi J; Melov S Single Nuclei Profiling Identifies Cell Specific Markers of Skeletal Muscle Aging, Frailty, and Senescence. Aging 2022, 14 (23), 9393–9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Schiaffino S. Muscle Fiber Type Diversity Revealed by Antimyosin Heavy Chain Antibodies. FEBS J. 2018, 285 (20), 3688–3694. [DOI] [PubMed] [Google Scholar]
- (31).Deshmukh AS; Murgia M; Nagaraj N; Treebak JT; Cox J; Mann M Deep Proteomics of Mouse Skeletal Muscle Enables Quantitation of Protein Isoforms, Metabolic Pathways, and Transcription Factors. Mol. Cell. Proteomics 2015, 14 (4), 841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.