

Abstract
To study the mechanisms by which Escherichia coli modulates the genotoxic effects of DNA damage, a novel system has been developed which permits quantitative measurements of various E. coli pathways involved in mutagenesis and DNA repair. Events measured include fidelity and efficiency of translesion DNA synthesis, excision repair, and recombination repair. Our strategy involves heteroduplex plasmid DNA bearing a single site-specific DNA adduct and several mismatched regions. The plasmid replicates in a mismatch repair-deficient host with the mismatches serving as strand-specific markers. Analysis of progeny plasmid DNA for linkage of the strand-specific markers identifies the pathway from which the plasmid is derived. Using this approach, a single 1,N6-ethenodeoxyadenosine adduct was shown to be repaired inefficiently by excision repair, to inhibit DNA synthesis by approximately 80 to 90%, and to direct the incorporation of correct dTMP opposite this adduct. This approach is especially useful in analyzing the damage avoidance-tolerance mechanisms. Our results also show that (i) progeny derived from the damage avoidance-tolerance pathway(s) accounts for more than 15% of all progeny; (ii) this pathway(s) requires functional recA, recF, recO, and recR genes, suggesting the mechanism to be daughter strand gap repair; (iii) the ruvABC genes or the recG gene is also required; and (iv) the RecG pathway appears to be more active than the RuvABC pathway. Based on these results, the mechanism of the damage avoidance-tolerance pathway is discussed.
Escherichia coli has evolved several strategies to cope with DNA damage, including a primitive form of cell cycle checkpoint control, nucleotide and base excision repair, UmuD′2C/RecA-assisted translesion synthesis, and damage avoidance-tolerance mechanisms such as recombination repair (6, 24). DNA damage induces SOS functions and halts progression through the cell cycle, providing time for DNA repair. However, some damage may escape repair and new damage may be introduced during DNA replication. These unrepaired lesions are believed to be the major sources of induced mutations (6).
Many DNA lesions block DNA synthesis, and translesion DNA synthesis (TLS) is often associated with replication errors. In E. coli, TLS, catalyzed by inducible SOS DNA polymerases (7, 10, 42), and DNA damage tolerance mechanisms (6) operate to overcome this block. RecA is activated upon DNA damage and facilitates autodigestion of LexA, a repressor of more than 20 SOS genes (6). RecA, UmuC (DNA polymerase V [pol V]), UmuD, DinB (DNA pol IV), and DNA pol II are encoded by prominent SOS genes. UmuD is converted to UmuD′ by proteolytic cleavage facilitated by RecA. An SOS DNA polymerase, DNA pol V, associates with two molecules of UmuD′ and catalyzes DNA synthesis across lesions (37). This synthesis is distributive, and pol V is thought to be replaced by DNA pol III after synthesis of a short stretch of DNA, during which pol V may incorporate an incorrect nucleotide opposite a lesion. Therefore, this pathway is error prone, inducing targeted point mutations.
E. coli has other mechanisms by which to overcome a DNA synthesis block. Restart of DNA synthesis downstream from the site of the block generates a gap in newly synthesized DNA. This gap subsequently may be filled with the parental strand of the sister molecule by recombination. The recA, recF, recO, recR, ruvA, ruvB, ruvC, and recG genes are believed to be involved in this process, termed daughter strand gap repair, in which the first four genes are involved in the presynapsis and synapsis stages and the remainder are involved in the postsynapsis stage (6, 12, 13). A hypothetical mechanism named template strand switching may also operate, in which the nascent strand switches its template to its sister molecule upon encountering a blocking DNA lesion and then switches back to the original template after bypassing the lesion (6). Both mechanisms are mechanistically error free.
The aim of this study was to determine the contributions of the various pathways described above to the overall response to DNA damage. Our strategy was to incorporate a single DNA adduct into double-stranded (ds) heteroduplex (HD) DNA bearing several mismatched regions. These mismatched regions serve as strand-specific markers. Following replication in a mismatch repair-deficient host cell, progeny plasmids are analyzed for each marker. Each pathway generates a unique linkage of markers, and the number of progeny plasmid derived from a given pathway represents its relative contribution to damage response in the host cell. The results are used to analyze mechanisms of responses to a DNA adduct. In this study, we conducted experiments using a single 1,N6-ethenodeoxyadenosine (ɛdA). ɛdA is one of four etheno DNA adducts, including 3,N4-etheno dC, N2,3-etheno dG, and 1,N2-etheno dG, that are produced by both endogenous and exogenous agents (23, 36). These adducts are mutagenic in vivo (2, 4, 14, 22, 25, 26). Although ɛdA is barely mutagenic in E. coli, it is strongly mutagenic in mammalian cells (26).
MATERIALS AND METHODS
E. coli strains and oligodeoxyribonucleotides.
The E. coli strains used in this study are shown in Table 1. All MO strains were constructed by P1 transduction (20). The alkA1 tag-1 genotype was confirmed by testing sensitivity to methyl methanesulfonate; the uvrA recF recO recR ruvABC and recA genotype was tested by determining sensitivity to UV irradiation; and mutS was tested by an increased spontaneous mutation frequency as determined by resistance to rifampin. The synthesis, purification, and characterization of the oligodeoxyribonucleotide containing ɛdA have been described previously (26).
TABLE 1.
E. coli strains used in this study
| Strain | Relevant genotype | Source and/or reference |
|---|---|---|
| AB1157 | EGSCa | |
| AB1886 | Same as AB1157 but uvrA | EGSC |
| AM208 | Same as AB1157 but recR256::kan | R. G. Lloyd; 17 |
| AM547 | Same as AB1157 but Δ(ruvA-ruvC)65 | R. G. Lloyd; 19 |
| AM888 | Same as AB1157 but ΔrusA::kan ΔruvAC65 | R. G. Lloyd; 18 |
| CS81 | Same as AB1157 but ruvB52 | R. G. Lloyd; 34 |
| DM2571 | Δ(recA-srl)306::Tn10 | EGSC |
| ES1481 | mutS215::Tn10 | EGSC; 35 |
| JC9239 | Same as AB1157 but recF143 | EGSC |
| MO199 | Same as AB1157 but mutS215::Tn10 | AB1157 × P1(ES1481) |
| MO200 | Same as JC9239 but mutS215::Tn10 | JC9239 × P1(ES1481) |
| MO201 | Same as RDK1541 but mutS215::Tn10 | RDK1541 × P1(ES1481) |
| MO202 | Same as AM208 but mutS215::Tn10 | AM208 × P1(ES1481) |
| MO204 | Same as CS81 but mutS201::Tn5 | CS81 × P1(RK1517) |
| MO206 | Same as AM547 but mutS215::Tn10 | AM547 × P1(ES1481) |
| MO207 | Same as TNM1072 but mutS215::Tn10 | TNM1072 × P1(ES1481) |
| MO208 | Same as MO206 but ΔrecG::kan | MO206 × P1(TNM1072) |
| MO210 | Same as MO206 but ΔrusA::kan | MO206 × P1(AM888) |
| MO219 | Same as SP254 but mutS215::Tn10 | SP254 × P1(ES1481) |
| MO220 | Same as NR9232 but mutS201::Tn5 | NR9232 × P1(RK1517) |
| MO933 | Same as MV1932 but mutS201::Tn5 | MV1932 × P1(RK1517) |
| MO934 | Same as MO933 but uvrA malE3::Tn10 | MO933 × P1(MV1185) |
| MO936 | Same as MO934 but mal+ | MO934 × P1(AB1886) |
| MO937 | Same as MO936 but Δ(recA-srl) 306::Tn10 | MO936 × P1(DM2571) |
| MV1185 | uvrA malE3::Tn10 | M. Volkert |
| MV1932 | alkA1 tag-1 | M. Volkert; 29 |
| NR9232 | mutD5 zaf-13::Tn10 | R. Schaaper; 31 |
| RDK1541 | Same as AB1157 but recO1504::Tn5 | R. Kolodner; 11 |
| RK1517 | Same as AB1157 but mutS201::Tn5 | R. Kolodner; 1 |
| SP254 | Same as AB1157 but recN262 | EGSC; 27 |
| TNM1072 | Same as AB1157 but ΔrecG263::kan | R. G. Lloyd; 19 |
EGSC, E. coli Genetic Stock Center, Yale University, New Heaven, Conn.
Plasmid vectors.
The construction of plasmid pSBK (8.4 kbp) has already been described (15). It is a shuttle vector containing the simian virus 40, BK, ColE1, and f1 origins of replication, the BK T-antigen gene, and the neomycin and ampicillin resistance genes. Strand-specific marker sequences were introduced by site-directed mutagenesis. Unique SpeI and NheI sites were constructed in one pSBK molecule, and unique AatII and BamHI sites were constructed in another. The SpeI and AatII sites were constructed at identical positions. BamHI and NheI sites are located 220 nucleotides upstream and 150 nucleotides downstream, respectively, from the site of the DNA adduct (see Fig. 3). The plasmid bearing SpeI and NheI sites and that bearing AatII and BamHI sites are named pS and pA, respectively (Fig. 1). Hybrid duplex DNA constructed from pS and pA forms mismatches at three regions (SpeI/AatII, NheI, and BamHI sites), which serve as strand-specific tags (see Fig. 3).
FIG. 3.

DNA sequence of regions containing strand-specific marker sequences and probes used for oligonucleotide hybridization. Marker sequences in the sequence-specific probes, A to D (overscored) and a to d (underlined), are highlighted. L and R probes (underlined) were used to identify progeny containing the 13-mer insert. Probes b (SA), ST, SG, SC, and SD were used to determine which base replaced ɛdA. –, direct connection of bases (e.g., G–G is GG); ∼, sequence interruption. Approximate distances between markers are shown in parentheses.
FIG. 1.

Structure of pA and pS. pA contains A, C, and D strand-specific markers, and pS contains a, c, and d marker sequences. B and b markers are generated during HD DNA construction; otherwise, pA and pS are identical. For the marker sequences, see Fig. 3. SV40, simian virus 40.
Construction of HD DNA containing a single ɛdA adduct.
The scheme for the procedure used to construct HD DNA containing a single ɛdA adduct is shown in Fig. 2. Detailed procedures have been described elsewhere (15); in brief, ds pA was digested with EcoRV (step I) and the linearized plasmid DNA was ligated to a blunt-ended duplex 13-mer, 5′ d(AGGTACGTAGGAG)/3′ d(TCCATGCATCCTC) containing a SnaBI site (5′TACGTA) (step II). Two constructs, each containing a single insert, with opposite orientations were isolated; one of these, pA106, was used in this study. Single-stranded (ss) pA106 was mixed with EcoRV-digested ds pS (steps III and IV). This mixture was treated with NaOH to denature ds pS and then neutralized to form ds DNA. Circular ss pA106 and its complementary strand from ds pS anneal to form HD DNA with a 13-nucleotide gap. Unmodified and modified 13-mers [3′ d(TCCATAXCTCCTC), where X is dA or ɛdA] were phosphorylated at the 5′ termini using T4 polynucleotide kinase and ATP, annealed to the gap, and ligated by T4 DNA ligase. The 13-mers are not perfectly complementary to the gap sequence, forming three base mismatches at and adjacent (5′ and 3′) to the site of ɛdA (Fig. 3). These mismatches are located opposite the SnaBI site and also serve as strand-specific markers. The ligation mixture was treated with SpeI and EcoRV to remove residual ds pS. Closed circular ds DNA was purified by ultracentrifugation in a CsCl-ethidium bromide solution. DNA was concentrated by Centricon 30 (Amicon, Beverly, Mass.), and the concentration was determined spectrophotometrically.
FIG. 2.

Construction of HD DNA containing a single ɛdA (X) residue. (I) EcoRV digestion of pA and pS. (II) Ligation of a duplex 13-mer containing a SnaBI site (5′ TACGTA). (III) Preparation of ss DNA. (IV) Preparation of gapped HD DNA. (V) Ligation of a modified 13-mer. Note that there are three contiguous base mismatches (highlighted) in the SnaBI site. There are also mismatches at the SpeI (d)/AatII (D), BamHI (a/A), and NheI (c/C) sites (not shown here for simplicity; see Fig. 3 for details). These mismatched regions serve as strand-specific markers.
In this HD construct, six and three base mismatches are formed at the SpeI/AatII and SnaBI sites, respectively. At each of the BamHI and NheI sites, one strand has six extra bases. These four mismatched regions serve as strand-specific markers: A/a (BamHI site), B/b (SnaBI site), C/c (NheI site), and D/d (AatII/SpeI sites). The unmodified complementary strand, derived from ss pA106, contains the A-B-C-D linkage. ɛdA was incorporated into the strand bearing the a-b-c-d linkage, which is the template for leading-strand synthesis.
Transformation of E. coli and analysis of progeny plasmid DNA.
Control or modified DNA (6 ng) was introduced into mismatch repair-deficient (mutS) MO strains by electroporation. The mutS mutation prevents mismatch repair. 2× YT medium (950 μl) was added to the electroporation mixture (50 μl) and then incubated for 20 min at 37°C (2 × YT medium contains [per liter] 16 g of tryptone, 10 g of yeast extract, and 5 g of NaCl [pH7]). A portion (10 to 50 μl of a 100× dilution) was plated onto a plate of 1× YT medium containing ampicillin (100 μg/ml) to determine the number of transformants in the mixture. The remaining mixture was incubated for 40 min and then added to 10 ml of 2× YT medium containing ampicillin. The mixture was cultured overnight, and progeny plasmid DNA was prepared by alkaline lysis. Purified plasmid DNA was used to transform E. coli DH5α. This second transformation segregates progeny plasmid DNA derived from each strand of HD DNA. Transformants were inoculated into 96-well plates and cultured for several hours. Bacteria were spotted onto filter paper placed on a 1× YT medium-ampicillin plate and cultured overnight. Filters were treated with 0.5 M NaOH, neutralized in 1 M Tris-HCl buffer (pH 7.4), washed in 1× SSC (0.15 M NaCl plus 0.015 M sodium citrate) and then in ethanol, and baked at 80°C for 2 h. To detect each strand-specific marker sequence, differential oligonucleotide hybridization (26) was conducted using the 32P-labeled probes shown in Fig. 3. An example of hybridization is shown in Fig. 4.
FIG. 4.

Oligonucleotide hybridization of E. coli transformants. MO199 was transformed with ɛdA-containing HD DNA. Progeny plasmids were used to transform E. coli DH5α. DH5α transformants were hybridized with all of the probes shown in Fig. 3.
Interpretation of results: progeny plasmid DNA derived from possible pathways.
When the HD construct bearing a single ɛdA residue is introduced into a mismatch repair-deficient host, several events may occur (Fig. 5). If the adduct is removed by excision repair before encountering the DNA replication apparatus, the 5′ and 3′ flanking mismatches are also removed and gap-filling DNA synthesis converts 5′ CXA (b) to 5′ ACG (B). As a result, progeny DNAs derived from the repaired strand contain the a-B-c-d linkage (progeny III) (step 1 of Fig. 5). If the construct is replicated without repair, progeny DNAs produced from the unmodified strand contain the A-B-C-D linkage (progeny I, step 2) while those from the modified strand show the a-b-c-d linkage (progeny II) when TLS occurs (step 3). When DNA synthesis is blocked by the adduct, it may be overcome by one of three mechanisms. One pathway involves UmuD′2C/RecA-assisted TLS. Other possibilities are template strand switching and daughter strand gap repair (6). In the former mechanism, upon encountering a blocking lesion, the 3′ end of the nascent strand switches its template to the newly synthesized strand of its sister molecule (step 9). When this mechanism operates, our construct will generate progeny with the a-B-C-d linkage (progeny IV, steps 9→8). In the daughter strand gap repair model, a ss gap, resulting from a synthesis block, is filled by strand transfer of the unmodified parental strand (step 4). The 3′ end of the blocked nascent strand is used to replicate the transferred region (step 5). These processes generate a Holliday junction. When the RuvA-RuvB complex migrates the Holliday junction and the Holliday junction-specific endonuclease, RuvC, resolves the Holliday junction (steps 6 and 7), four types of progeny arise, i.e., progeny with the linkages A-B-C-D (progeny I), a-B-C-d (progeny IV), a-B-C-D (progeny V), and A-B-C-d (progeny VI). Progeny I and progeny IV may also be produced when the Holliday junction is resolved by reverse branch migration catalyzed by RecG (step 8).
FIG. 5.

E. coli pathways involved in the processing of DNA damage. Modified input DNA (X represents ɛdA) is boxed. The ColE1 origin of replication (short vertical bars) and the direction of replication (short arrow) are shown. A to D and a to d correspond to those shown in Fig. 3. Progeny I to VI correspond to those listed in Tables 2 to 4. Excision repair of ɛdA (step 1) converts the sequence of the adducted region to B; replication of a repaired molecule produces progeny I and III. When modified DNA replicates (step 2), the complementary strand produces progeny I and the modified strand yields progeny II following TLS (step 3). When the adduct inhibits DNA synthesis, DNA damage avoidance mechanisms, daughter strand gap repair (steps 4 to 8) and/or template strand switching (steps 9 and 8), operate to overcome the inhibition. The daughter strand gap repair mechanism involves strand transfer (step 5), formation of a Holliday junction, gap-filling synthesis (step 5), and branch migration (step 6). Resolution of a Holliday junction by RuvC resolvase produces progeny I, IV, V, and VI (step 7). When a Holliday junction is resolved by reverse branch migration (step 8), progeny I and IV are produced. In the template strand switch mechanism, upon encountering a blocking lesion, the nascent strand switches its template to the sister molecule and synthesis continues (step 9). After bypassing the adducted region, the strand reassociates with the original template strand and continues synthesis (step 8). This mechanism produces progeny I and IV.
RESULTS
Inhibition of DNA synthesis and translesional DNA synthesis.
The HD construct was introduced into excision repair-deficient strain MO934 (alkA1 tag1 uvrA6) and strain MO937, a ΔrecA derivative of MO934 (Table 2). When the unmodified construct was used, more than 97% of the progeny consisted of progeny I and II, derived from the replication of each strand. In both strains, the number of progeny II was approximately 1.8-fold more than that of progeny I. A similar phenomenon has been reported by others (9). When the modified construct was used, the number of progeny II decreased markedly, indicating that ɛdA strongly blocks DNA synthesis. In excision repair- and recombination-deficient strain MO937, the approximate efficiency of TLS was 11% (7 of 63). The fraction of progeny II ranged from 7 to 14% in the strains used (Tables 2 to 4). To determine the base inserted opposite ɛdA, differential oligonucleotide hybridization was performed. This method allows the detection of a single base mismatch using oligonucleotide probes (38) and is frequently used in site-specific mutagenesis experiments (9, 21). Purified progeny plasmids were digested with SnaBI and used for transformation. This digestion results in enrichment in progeny II derived from the TLS pathway (Fig. 5). Hybridization using the b (SA), ST, SG, SC, and SD probes revealed that all of the 312 progeny II analyzed had dA at the position of ɛdA, indicating that the correct nucleotide, dTMP, had been inserted opposite this adduct. Thus, TLS is highly accurate and probe b will detect TLS events.
TABLE 2.
Linkage analysis for four marker sequences
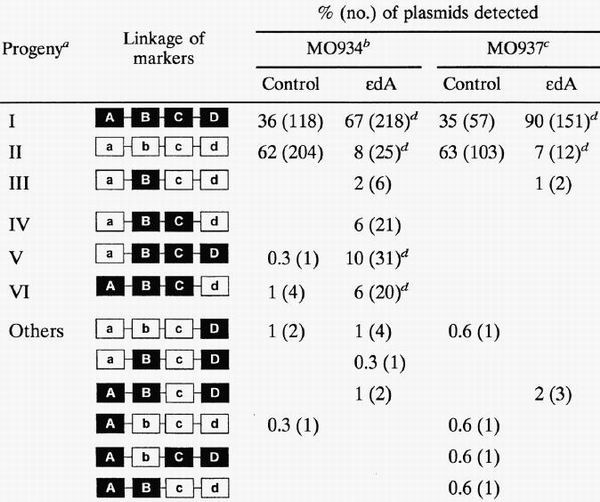 |
Progeny are derived from unmodified complementary-strand DNA (progeny I), translesion DNA synthesis (progeny II), excision repair (progeny III), and recombination repair (progeny I, IV, V, and VI). The origins of other progeny are not clear, but they may be generated by recombination between progeny I to VI. Refer to Fig. 5 for details.
alkA1 tag1 uvrA mutS.
alkA1 tag1 uvrA mutS ΔrecA.
TABLE 4.
Linkage analysis of E. coli strains having mutations in the genes involved in Holliday junction resolution
| Progeny | % (no.) of plasmids detected
|
|||||
|---|---|---|---|---|---|---|
| MO199 | MO207 ΔrecG | MO204 ruvB | MO206 ΔruvA-C | MO210 ΔruvA-C ΔrusA | MO208 ΔruvA-C ΔrecG | |
| I | 68 (178) | 72 (139) | 48 (90)a | 53 (199)a | 42 (192)a | 75 (192) |
| II | 10 (27) | 11 (21) | 12 (22) | 11 (40) | 13 (58) | 9 (23) |
| III | 4 (11) | 4 (7) | 6 (11) | 3 (11) | 8 (37)b | 8 (20)b |
| IV | 5 (14) | 3 (5) | 12 (23)a | 11 (41)a | 17 (76)a | 1 (3)a |
| V | 5 (13) | 6 (12) | 15 (29)a | 15 (56)a | 15 (67)a | 5 (12)a |
| VI | 6 (15) | 4 (7) | 4 (7) | 5 (20) | 4 (18) | 1 (2)a |
| Others | 2 (4)e | 0.5 (1)d | 4 (7)e | 2 (10)f | 2 (9)g | 2 (4)h |
P < 0.001.
P < 0.005.
a-B-c-D, 2; A-B-c-D, 1; A-b-C-D, 1.
a-b-C-D, 1.
a-b-c-D, 1; a-B-c-D, 1; A-B-c-D, 1; A-B-c-d, 3; a-b-C-D, 1.
a-b-c-D, 3; a-B-c-D, 2; A-B-c-D, 3; A-B-c-d, 2.
a-B-c-D, 3; A-B-c-d, 4; a-b-C-D, 1; a-b-C-d, 1.
a-b-c-D, 1; A-B-c-D, 2; A-B-c-d, 1.
Excision repair of ɛdA.
The fractions of progeny III having the linkage for excision repair events were >2% in excision repair-deficient strains (Table 2). The fractions increased marginally in repair-proficient strains, ranging from 3 to 8% (Tables 3 to 5), indicating slow repair of this adduct located in the mismatched region. This result is in contrast to that obtained with 3H-8-hydroxy-3-(β-d-2′-deoxyribofuranosyl)-5,6,7,8-tetrahydropyrido[3,2-a]purine-9-one [γ-(OH)-PdG], an endogenous DNA adduct that is repaired by nucleotide excision repair, for which progeny III accounts for >30% when a modified construct is introduced into excision repair-proficient cells (unpublished result). Our results suggest that ɛdA is not a good substrate for either base or nucleotide excision repair in E. coli. An in vitro study also has shown that ɛdA is repaired very inefficiently by base excision repair catalyzed by the E. coli alkA gene product, 3-methyladenine DNA glycosylase (30, 43).
TABLE 3.
Linkage analysis in E. coli mutants
| Progeny | % (no.) of plasmids detected
|
||||
|---|---|---|---|---|---|
| MO199 | MO200 recF | MO201 recO | MO202 recR | MO219 recN | |
| I | 68 (178) | 82 (216)a | 82 (157)a | 80 (145)a | 56 (102)a |
| II | 10 (27) | 11 (30) | 14 (27) | 14 (26) | 13 (23) |
| III | 4 (11) | 3.8 (10) | 3 (5) | 4 (7) | 5 (10) |
| IV | 5 (14) | 0.4 (1)a | 0a | 0a | 8 (15) |
| V | 5 (13) | 1.5 (4)a | 0.5 (1)a | 1 (2)a | 10 (18)a |
| VI | 6 (15) | 0a | 1 (2)a | 1 (2)a | 5 (10) |
| Others | 2 (4)b | 0.4 (1)c | 0 | 0 | 3 (5)d |
P < 0.001.
a-B-c-D, 2; A-B-c-D, 1; A-b-C-D, 1.
a-B-c-D, 1.
a-B-c-D, 2; a-b-c-D, 1; A-B-c-D, 1; A-B-c-d, 1.
TABLE 5.
Linkage analysis in SOS-induced E. coli and a mutD strain
| Progeny | % (no.) of plasmids detected
|
||
|---|---|---|---|
| MO199 | MO199 MCa | MO220 mutD5 | |
| I | 68 (178) | 47 (88)b | 57 (105)b |
| II | 10 (27) | 25 (47)b | 21 (38)b |
| III | 4 (11) | 6 (12) | 4 (7) |
| IV | 5 (14) | 7 (13) | 2 (5) |
| V | 5 (13) | 7 (13) | 5 (9) |
| VI | 6 (15) | 4 (8) | 8 (14) |
| Others | 2 (4)c | 4 (6)d | 3 (6)e |
MC, pretreatment of host with mitomycin C (5 μg/ml) for 30 min before preparation of an electrocompetent host.
P < 0.001.
a-B-c-D, 2; A-B-c-D, 1; A-b-C-D, 1.
a-b-c-D, 2; a-B-c-D, 2; A-b-C-D, 1.
a-b-c-D, 1; a-B-c-D, 1; A-B-c-D, 1; A-b-c-d, 1; A-b-c-d, 2.
Recombination repair.
Recombinants (progeny IV, V, VI, and others) accounted for <3% of the progeny when the unmodified control construct was introduced into MO934 (recA+) and MO937 (ΔrecA) (Table 2). The relative number of recombinants increased 10-fold (24%) when the ɛdA-modified construct was introduced into MO934. Progeny IV, V, and VI account for the majority (91%) of recombinants. When the ΔrecA mutant (MO937) was used, there was no increase in the number of recombinants. These results clearly indicate that progeny IV, V, and VI are generated by a RecA-dependent pathway(s) in response to ɛdA.
Genes involved in the damage tolerance-avoidance mechanism(s).
Recombinants may be derived from the daughter strand gap repair and/or strand switching pathways depicted in Fig. 5. First, we examined the requirement for genes hypothesized to be involved in the presynapsis and synapsis stages of daughter strand gap repair (11, 12). When the recF, recO, or recR gene was inactivated, the number of recombinants decreased markedly, revealing a requirement for these genes (Table 3). Inactivation of the recN gene, which is thought to be involved in ds break repair (12), showed no effect.
Next, effects of the inactivation of genes involved in the postsynapsis stage of daughter strand gap repair were examined (11, 12). RecG and RuvA-RuvB proteins are believed to independently catalyze branch migration of a Holliday junction (16, 40). RuvC protein, in concert with RuvAB, cleaves a Holliday junction. Inactivation of the recG gene did not affect the frequency of recombinant IV, V, or VI (Table 4). In contrast, inactivation of the ruvB gene (MO204) or all of the ruv genes (MO206) caused an approximately twofold increase in the total frequency of recombinants. This increase was ascribed mainly to the increase in the number of progeny IV and V. Additional inactivation of the rusA gene in MO206 (creating MO210), which codes for a cryptic Holliday junction resolvase (3), did not result in a further change in the number of recombinants. On the other hand, inactivation of the recG gene in MO206 (creating MO208) resulted in an 80% reduction in the number of recombinants, proving that the majority of recombinants observed in MO206 were generated by the RecG activity. Figure 6 summarizes the total frequency of recombinants (progeny IV, V, and VI) in various strains.
FIG. 6.

Frequency of recombinant progeny in various E. coli strains transformed with ɛdA-containing plasmid DNA. The y axis represents the percentage of recombinants (progeny IV, V, and VI) among all of the progeny. recA+, recA, and “wild” denote MO934, MO937, and MO199, respectively. Refer to Tables 3 to 5 for the other strains. mc indicates treatment with mitomycin C; refer to the legend to Table 5. An asterisk indicates P < 0.001, as determined by the χ2 test, compared with both controls (recA+ and “wild”).
Effects of SOS induction and inactivation of proofreading activity of pol III.
The induction of SOS functions by mitomycin C treatment increased the fraction of progeny II twofold, suggesting that inducible SOS DNA polymerases such as pol II, IV, and V may catalyze TLS accurately. Enhancement of the accurate TLS is also observed by the inactivation, due to mutD5, of the proofreading function of DNA pol III. The SOS induction or the mutD5 mutation did not influence the fraction of recombinants (Table 4).
DISCUSSION
Several pathways operate in E. coli in response to DNA damage (Fig. 5). To examine the individual contributions of these pathways, we have devised a novel plasmid-based approach using a single DNA adduct. Our strategy is to sort progeny plasmids according to the linkage of marker sequences, since each pathway produces progeny with a unique linkage. Results of this study indicate that ɛdA strongly inhibits DNA synthesis in E. coli; the efficiency of TLS was approximately 11%, and the induction of SOS functions mitigates the blocking effects. However, a significant fraction of DNA synthesis was still inhibited and a portion of the blocked DNA synthesis was rescued by daughter strand gap repair. Inactivation of the proofreading activity of DNA pol III also increased TLS, possibly by forcing pol III to perform TLS. TLS was highly accurate in the presence and in the absence of induced SOS functions and also in the presence of the mutD5 mutation. These findings suggest strongly that the correct nucleotide is almost exclusively incorporated opposite ɛdA in E. coli.
Our results clearly show, at the DNA sequence level, that the E. coli damage avoidance-tolerance mechanism operates to rescue nascent strands when progression of DNA synthesis is blocked by ɛdA (Table 2). Our results are consistent with daughter strand gap repair. We observed an increase in the number of progeny IV, V, and VI, which are predicted to emerge from steps 5 to 10 in Fig. 5. Production of these recombinants depends on the functions of the recA, recF, recO, and recR genes. Biochemical studies have revealed that the RecO-RecR complex facilitates binding of RecA protein to an ss DNA-binding protein-coated region of ss DNA and prevents the end-dependent disassembly of the RecA filament (8, 32). The RecF-RecR complex binds to ds DNA and is thought to inhibit excessive extension of a RecA filament into ds DNA (39). RecA is known to search for homology and to catalyze strand transfer. These biochemical activities explain the lack of recombinants in these mutants in our assay. RecF is also thought to be required for the resumption of DNA synthesis (5).
The results obtained in this study argue against the idea that the template strand switching mechanism plays a major role in damage avoidance of ɛdA. Our experimental system predicts the appearance of progeny IV if this mechanism were to operate (Fig. 5). Inactivation of the recA, recF, recO, and recR genes reduced markedly the numbers of all types of recombinants (Table 3). Therefore, it must be assumed that these genes are also required for any postulated template strand switching mechanism, if it exists.
Compared with the inactivation of genes involved in the presynapsis and synapsis stages, the effects of inactivation of the genes involved in the postsynapsis stage of recombination repair are complex. While inactivation of the recG gene does not affect the number of recombinants, inactivation of the ruvABC genes increased the incidence of recombinants (Table 4). RecG has ATPase and 3′→5′ helicase activity, binds a Holliday junction, and promotes branch migration (12). RuvA also binds a Holliday junction and targets RuvB (5′→3′ helicase) to a Holliday junction (12), which then promotes branch migration. RecG and RuvAB are thought to migrate a Holliday junction in opposite directions (41); RuvC resolves a Holliday junction by endonucleolytic cleavages (12), while RecG may resolve it by reverse branch migration (41). In considering these activities, it is understandable that the ΔrecG mutation had no effect on the incidence of progeny IV, V, and VI. On the other hand, the enhancing effects of mutations in the ruv genes (MO204, MO206, and MO210) are unexpected, suggesting that the RecG-catalyzed Holliday junction resolution pathway is more active than the RuvABC-catalyzed pathway and that the RuvABC pathway may suppress this RecG pathway. Enhanced daughter strand gap repair in the ruv mutants is an apparent paradox to the UV sensitivity of the ruv mutants. It is likely that a complicated combination of multiple Holliday junctions generated by daughter strand gap repair and double-strand break repair in the E. coli chromosome requires the endonucleolytic resolution mediated by RuvABC.
The second puzzle is how progeny V and VI are produced in the absence of RuvC resolvase. This is not due to the activity of cryptic RusA, since the introduction of ΔrusA into the ΔruvA-C mutant showed no effect. If the Holliday junction is resolved by reverse branch migration, progeny IV and I are expected. If the Holliday junction is not resolved, a plasmid dimer containing the linkage A-B-C-d–a-B-C-D is expected. Our analysis showed that recombinant progeny hybridized only one of the two probes at the A/a and D/d sites. Additionally, the 11 recombinants of progeny IV, V, and VI were confirmed to be monomers (8.4 kbp) by agarose gel electrophoresis.
The E. coli chromosome and ColE1 plasmid contain dif and cer sites, respectively, at which a dimer is monomerized by XerCD site-specific recombinase (33). Our pUC-based plasmid does not carry the cer or dif site. Therefore, a plasmid dimer can not be monomerized. This information suggests that progeny plasmids are monomerized at the completion of recombination events. Figure 7 shows our model for the generation of progeny V and VI by the RecG-catalyzed pathway. When a daughter strand gap is filled by strand transfer, the parental strand invades the adjacent ds regions to displace a marker: d is displaced by D in the left column, and a is displaced by A in the right column. A subsequent filling reaction and reverse branch migration will generate progeny V (left column) and VI (right column). In the latter case, the marker a must be removed by a nuclease before initiation of the gap-filling reaction. Since the number of progeny VI did not increase in the ruv mutants (Table 4), the latter pathway in Fig. 6 may be a rare event. If the high incidence of progeny V in the ruv mutants is ascribed to this pathway (left column), it is predicted that DNA synthesis is not resumed between marker c and the origin of replication, which is 3,100 nucleotides away. Hence, inactivation of the polB gene, which has been recently suggested to function in the resumption of DNA synthesis (28), will have no effect on the incidence of progeny V in the ruv strains. The introduction of another marker between c/C and d/D may help to answer this question.
FIG. 7.

Possible mechanisms generating progeny V and VI in ruv mutants. Refer to the legend to Fig. 5 and the text for an explanation.
When a ΔrecG mutation was introduced into ΔruvA-C strain MO208, the incidence of recombinants decreased markedly, clearly showing that RecG plays an essential role in resolving the Holliday junction in the absence of RuvABC. However, a significant number of recombinants derived from the daughter strand gap repair mechanism were observed in MO208 (ΔruvA-C ΔrecG). This suggests that RuvAB- and RecG-independent branch migration still occurs, although not efficiently, in E. coli.
ACKNOWLEDGMENTS
We thank A. Kuzminov for critical reading of the manuscript. We also thank M. Berlyn, R. Kolodner, R. G. Lloyd, R. M. Schaaper, and M. Volkert for E. coli strains.
This research was supported by U.S. Public Health Service grant CA76163 (M.M.), grant PO1CA47995 (A.P.G.), and in part by a Cancer Center grant (CA17613) while G.A.P. and M.M. were associated with the American Health Foundation.
G.A.P. and I.-Y.Y. contributed equally to this work.
REFERENCES
- 1.Au K G, Cabrera M, Miller J H, Modrich P. Escherichia coli mutY gene product is required for specific A-G→C:G mismatch correction. Proc Natl Acad Sci USA. 1988;85:9163–9166. doi: 10.1073/pnas.85.23.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basu A K, Wood M L, Niederhofer L J, Ramos L A, Essigmann J M. Mutagenic and genotoxic effects of three vinyl chloride-induced DNA lesions: 1,N6-ethenoadenine, 3,N4-ethenocytosine, and 4-amino-5-(imidazol-2-yl)imidazole. Biochemistry. 1993;32:12793–12801. doi: 10.1021/bi00210a031. [DOI] [PubMed] [Google Scholar]
- 3.Chan S N, Vincent S D, Lloyd R G. Recognition and manipulation of branched DNA by the RusA Holliday junction resolvase of Escherichia coli. Nucleic Acids Res. 1998;26:1560–1566. doi: 10.1093/nar/26.7.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng K C, Preston B D, Cahill D S, Dosanjh M K, Loeb L A. The vinyl chloride DNA derivative N2,3-ethenoguanine produces G→A transitions in Escherichia coli. Proc Natl Acad Sci USA. 1991;88:9974–9978. doi: 10.1073/pnas.88.22.9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Courcelle J, Carswell-Crumpton C, Hanawalt P C. recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc Natl Acad Sci USA. 1996;94:3714–3719. doi: 10.1073/pnas.94.8.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedberg E C, Walker G C, Siede W. DNA repair and mutagenesis. Washington, D.C.: American Society for Microbiology; 1995. [Google Scholar]
- 7.Friedberg E C, Gerlach V L. Novel DNA polymerases offer clues to the molecular basis of mutagenesis. Cell. 1999;98:413–416. doi: 10.1016/s0092-8674(00)81970-4. [DOI] [PubMed] [Google Scholar]
- 8.Hegde S P, Qin M-h, Li X-H, Atkinson M A L, Clark A J, Rajagopalan M, Madiraju M V V S. Interactions of RecF protein with RecO, RecR, and single-stranded DNA binding proteins reveal roles for the RecF-RecO-RecR complex in DNA repair and recombination. Proc Natl Acad Sci USA. 1996;93:14468–14473. doi: 10.1073/pnas.93.25.14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson K A, Fink S P, Marnett L J. Repair of propanodeoxyguanosine by nucleotide excision repair in vivo and in vitro. J Biol Chem. 1997;272:11434–11438. doi: 10.1074/jbc.272.17.11434. [DOI] [PubMed] [Google Scholar]
- 10.Johnson R E, Washington M T, Prakash S, Prakash L. Bridging the gap: a family of novel DNA polymerases that replicate faulty DNA. Proc Natl Acad Sci USA. 1999;96:12224–12226. doi: 10.1073/pnas.96.22.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolodner R, Fishel R A, Howard M. Genetic recombination of bacterial plasmid DNA: effect of RecF pathway mutations on plasmid recombination in Escherichia coli. J Bacteriol. 1985;163:1060–1066. doi: 10.1128/jb.163.3.1060-1066.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowalczykowski S C, Dixon D A, Eggleston A K, Lauder S D, Rehrauer W M. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langouët S, Mican A N, Müller M, Fink S P, Marnett L J, Muhle S A, Guengerich F P. Misincorporation of nucleotides opposite five-membered exocyclic ring guanine derivatives by Escherichia coli polymerases in vitro and in vivo: 1,N2-ethenoguanine, 5,6,7,9-tetrahydro-9-oxoimidazo[1,2-a]purine, and 5,6,7,9-tetrahydro-7-hydroxy-9-oxoimidazo[1,2-a]purine. Biochemistry. 1998;37:5184–5193. doi: 10.1021/bi972327r. [DOI] [PubMed] [Google Scholar]
- 15.Levine R L, Yang I-Y, Hossain M, Pandya G A, Grollman A P, Moriya M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 2000;60:4098–4104. [PubMed] [Google Scholar]
- 16.Lloyd R G, Sharples G J. Processing of recombination intermediates by the RecG and RuvAB proteins of Escherichia coli. Nucleic Acids Res. 1993;21:1719–1725. doi: 10.1093/nar/21.8.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahdi A A, Lloyd R G. Identification of the recR locus of Escherichia coli K-12 and analysis of its role in recombination and DNA repair. Mol Gen Genet. 1989;216:503–510. doi: 10.1007/BF00334397. [DOI] [PubMed] [Google Scholar]
- 18.Mahdi A A, Sharples G J, Mandal T N, Lloyd R G. Holliday junction resolvases encoded by homologous rusA genes in Escherichia coli K-12 and phage 82. J Mol Biol. 1996;257:561–573. doi: 10.1006/jmbi.1996.0185. [DOI] [PubMed] [Google Scholar]
- 19.Mandal T N, Mahdi A A, Sharples G J, Lloyd R G. Resolution of Holliday intermediates in recombination and DNA repair: indirect suppression of ruvA, ruvB, and ruvC mutations. J Bacteriol. 1993;175:4325–4334. doi: 10.1128/jb.175.14.4325-4334.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller J H. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1992. pp. 263–278. [Google Scholar]
- 21.Moriya M, Grollman A P. Mutations in the mutY gene of Escherichia coli enhance the frequency of targeted G:C→T:A transversions induced by a single 8-oxoguanine residue in single-stranded DNA. Mol Gen Genet. 1993;239:72–76. doi: 10.1007/BF00281603. [DOI] [PubMed] [Google Scholar]
- 22.Moriya M, Zhang W, Johnson F, Grollman A P. Mutagenic potency of exocyclic DNA adducts: marked differences between Escherichia coli and simian kidney cells. Proc Natl Acad Sci USA. 1994;91:11899–11903. doi: 10.1073/pnas.91.25.11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nair J, Barbin A, Velic I, Bartsch H. Etheno DNA-base adducts from endogenous reactive species. Mutat Res. 1999;424:59–69. doi: 10.1016/s0027-5107(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 24.Opperman T, Murli S, Smith B T, Walker G C. A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc Natl Acad Sci USA. 1999;96:9218–9223. doi: 10.1073/pnas.96.16.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palejwala V A, Simha D, Hymayun M Z. Mechanisms of mutagenesis by exocyclic DNA adducts. Transfection of M13 viral DNA bearing a site-specific adduct shows that ethenocytosine is a highly efficient RecA-independent mutagenic noninstructional lesion. Biochemistry. 1991;30:8736–8743. doi: 10.1021/bi00100a004. [DOI] [PubMed] [Google Scholar]
- 26.Pandya G, Moriya M. 1,N6-Ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry. 1996;35:11487–11492. doi: 10.1021/bi960170h. [DOI] [PubMed] [Google Scholar]
- 27.Picksley S M, Attfield P V, Lloyd R G. Repair of DNA double-strand breaks in Escherichia coli K12 requires a functional recN product. Mol Gen Genet. 1984;195:267–274. doi: 10.1007/BF00332758. [DOI] [PubMed] [Google Scholar]
- 28.Rangarajan S, Woodgate R, Goodman M F. A phenotype for enigmatic DNA polymerase II: a pivotal role for pol II in replication restart in UV-irradiated Escherichia coli. Proc Natl Acad Sci USA. 1999;96:9224–9229. doi: 10.1073/pnas.96.16.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santerre A, Britt A B. Cloning of a 3-methyladenine-DNA glycosylase from Arabidopsis thaliana. Proc Natl Acad Sci USA. 1994;91:2240–2244. doi: 10.1073/pnas.91.6.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saparbaev M, Kleibl K, Laval J. Escherichia coli, Saccharomyces cerevisiae, rat and human 3-methyladenine DNA glycosylases repair 1,N6-ethenoadenine when present in DNA. Nucleic Acids Res. 1995;23:3750–3755. doi: 10.1093/nar/23.18.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schaaper R M. Mechanisms of mutagenesis in the Escherichia coli mutator mutD5: role of DNA mismatch repair. Proc Natl Acad Sci USA. 1988;85:8126–8130. doi: 10.1073/pnas.85.21.8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shan Q, Bork J M, Webb B L, Inman R B, Cox M M. RecA protein filaments: end-dependent dissociation from ssDNA and stabilization by RecO and RecR proteins. J Mol Biol. 1997;265:519–540. doi: 10.1006/jmbi.1996.0748. [DOI] [PubMed] [Google Scholar]
- 33.Sherratt D J, Arciszewska L K, Blakely G, Colloms S, Grant K, Leslie N, McCulloch R. Site-specific recombination and circular chromosome segregation. Philos Trans R Soc Lond Ser B Biol Sci. 1995;347:37–42. doi: 10.1098/rstb.1995.0006. [DOI] [PubMed] [Google Scholar]
- 34.Shurvinton C E, Lloyd R G, Benson F E, Attfield P V. Genetic analysis and molecular cloning of the Escherichia coli ruv gene. Mol Gen Genet. 1984;194:322–329. doi: 10.1007/BF00383535. [DOI] [PubMed] [Google Scholar]
- 35.Siegel E C, Wain S L, Meltzer S F, Binion M L, Steinberg J L. Mutator mutations in Escherichia coli induced by the insertion of phage mu and the transposable resistance elements Tn5 and Tn10. Mutat Res. 1982;93:25–33. doi: 10.1016/0027-5107(82)90122-1. [DOI] [PubMed] [Google Scholar]
- 36.Swenberg J A, Bogdanffy M S, Ham A, Holt S, Kim A, Morinello E J, Ranasinghe A, Scheller N, Upton P B. Formation and repair of DNA adducts in vinyl chloride- and vinyl fluoride-induced carcinogenesis. In: Singer B, Bartsch H, editors. Exocyclic DNA adducts in mutagenesis and carcinogenesis. Lyon, France: IARC Scientific Publications; 1999. pp. 29–43. [PubMed] [Google Scholar]
- 37.Tang M, Shen X, Frank E G, O'Donnell M, Woodgate R, Goodman M F. UmuD′2C is an error-prone DNA polymerase, Escherichia coli pol V. Proc Natl Acad Sci USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wallace R B, Shaffer J, Murphy R F, Bonner J, Hirose T, Itakura K. Hybridization of synthetic oligodeoxyribonucleotides to ΦX174 DNA: the effect of single base pair mismatch. Nucleic Acids Res. 1979;6:3543–3557. doi: 10.1093/nar/6.11.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Webb B L, Cox M M, Inman R B. Recombinational DNA repair: the RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell. 1997;91:347–356. doi: 10.1016/s0092-8674(00)80418-3. [DOI] [PubMed] [Google Scholar]
- 40.West S C. The RuvABC proteins and Holliday junction processing in Escherichia coli. J Bacteriol. 1996;178:1237–1241. doi: 10.1128/jb.178.5.1237-1241.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitby M C, Ryder L, Lloyd R G. Reverse branch migration of Holliday junctions by RecG protein: a new mechanism for resolution of intermediates in recombination and DNA repair. Cell. 1993;75:341–350. doi: 10.1016/0092-8674(93)80075-p. [DOI] [PubMed] [Google Scholar]
- 42.Woodgate R. A plethora of lesion-replicating DNA polymerases. Genes Dev. 1999;13:2191–2195. doi: 10.1101/gad.13.17.2191. [DOI] [PubMed] [Google Scholar]
- 43.Wyatt M D, Allan J M, Lau A Y, Ellenberger T E, Samson L D. 3-Methyladenine DNA glycosylases: structure, function, and biological importance. Bioessays. 1999;21:668–676. doi: 10.1002/(SICI)1521-1878(199908)21:8<668::AID-BIES6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]