

Summary
Although human cortical interneurons (cINs) are a minority population in the cerebral cortex, disruption of interneuron development is a frequent contributor to neurodevelopmental disorders. Here, we utilized a model for deriving cINs from human embryonic stem cells to profile chromatin state changes and generate an atlas of cis-regulatory elements (CREs) controlling human cIN development. We used these data to define candidate transcription factors (TFs) that may bind these CREs to regulate interneuron progenitor specification. Among these were RFX3 and RFX4, risk genes for autism spectrum disorder (ASD) with uncharacterized roles in human neuronal development. Using RFX3 and RFX4 knockdown models, we demonstrated new requirements for both genes in interneuron progenitor specification, with RFX3 deficiency causing precocious neuronal differentiation while RFX4 deficiency instead resulted in cessation of progenitor cell proliferation. Together, this work both defined central features of cis-regulatory control and identified new TF requirements for human interneuron development.
Subject areas: Biological sciences, Epigenetics, Neuroscience, Developmental neuroscience
Graphical abstract
Highlights
-
•
Built a cis-regulatory element (CRE) atlas for human cortical interneuron development
-
•
Progenitor CREs were enriched for RFX transcription factor binding sites
-
•
RFX3 deficiency caused precocious differentiation of interneuron progenitors
-
•
RFX4 deficiency caused cessation of interneuron progenitor proliferation
Biological sciences; Epigenetics; Neuroscience; Developmental neuroscience
Introduction
Although cortical interneurons (cINs) constitute only 20–30% of neurons in the human cerebral cortex, they play critical roles in balancing neuronal excitation to regulate cortical function.1 Accordingly, even minimal changes in their development can alter brain function, contributing to neurodevelopmental disorders (NDDs) including autism spectrum disorder (ASD) and intellectual disability (ID) syndromes.2,3 While critical for brain function, regulation of many aspects of cIN development remains uncharacterized, due to the inaccessibility of these cells during brain development. In recent years, we and others have developed in vitro models of cIN development using human pluripotent stem cells, providing an accessible model that recapitulates many aspects of this process.4,5,6,7
Regulation of gene expression during embryonic and fetal development is mediated by cis-regulatory elements (CREs) and the transcription factors (TFs) that bind them.8 CREs contain TF binding sites (TFBSs) that are bound by specific TFs to regulate target gene expression. While the locations of active CREs are frequently inferred by the presence of accessible chromatin,7 advancements in approaches for profiling post-translational histone modifications have refined methods for identifying CREs and studying their regulation.9 Modification of histone tails, including acetylation of histone H3 lysine 2710 (H3K27ac) and tri-methylation of histone H3 lysine 411 (H3K4me3), are commonly present at CREs and promoters associated with highly expressed genes. By contrast, histone H3 lysine 27 tri-methylation (H3K27me3) and histone H3 lysine 9 tri-methylation (H3K9me3) are associated with more repressive chromatin states and repression of gene expression. Furthermore, genomic locations enriched for both H3K27me3 and H3K4me3 (defined as “bivalent” chromatin) can poise genes for either rapid activation or repression.12
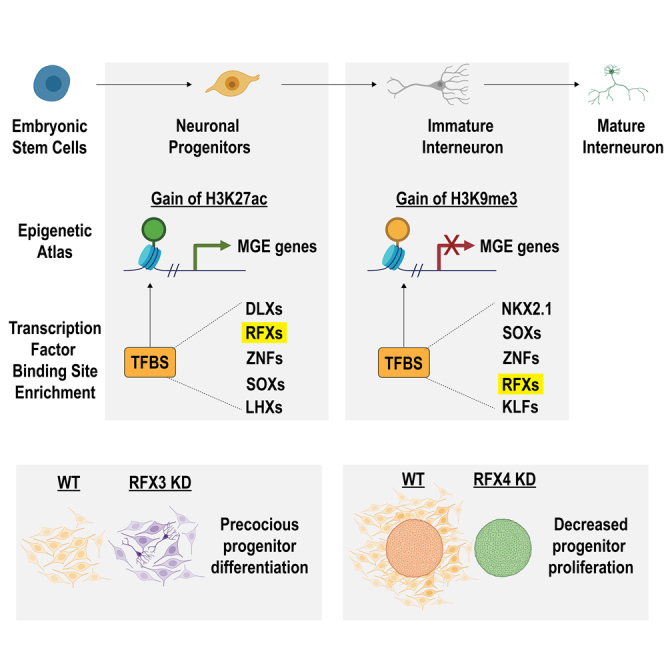
Defining how these histone modification states change across the genome during development provides essential information regarding the location and activity of CREs that regulate developmental gene expression. Defining changes in the epigenetic landscape during cIN development can also reveal how genetic variation in non-protein coding regions of the genome relates to disease etiology.13 Furthermore, mutations in genes encoding TFs14 and chromatin modifiers15 are frequent contributors to NDD etiology. While some NDD-associated genes in these categories have been characterized,5 sequencing continues to identify new contributors.16,17,18 However, a lack of understanding of gene regulatory control underlying the development of disease relevant neuronal cell types such as cINs has made it challenging to define their contribution to brain development. For example, while newly identified NDD risk genes such as the winged helix TFs RFX3, RFX4, and RFX7 are dynamically expressed during human brain development in vivo, their roles in these processes remain unknown.17
Here, we used an established model of cIN development from human embryonic stem cells (hESCs) to define changes in the epigenetic landscape accompanying cIN development. We used these data to derive an atlas of putative CREs that may regulate cIN specification and differentiation. We then examined TFBS enrichment within these CRE sequences to identify TFs that may control these processes, including RFX3 and RFX4. Accordingly, knockdown of these TFs revealed both synergistic and antagonistic requirements to regulate cIN differentiation timing, and suggested other TFs that may function downstream to regulate these processes. Together, this work provides insights into the transcriptional and epigenetic regulation of CRE activity during cIN development and demonstrates how these data can be used to identify TFs that control this process and are disrupted to cause NDDs.
Results
Gene expression changes associated with interneuron specification and differentiation
To understand the process of interneuron specification and differentiation, we used our previously developed protocol,4,5,19 focusing on three developmental transitions during hESC differentiation into mature cINs (Figure 1A). Substantial transcriptomic differences accompanied both hESC (D0) specification as MGE-like neuronal progenitors (D15) and differentiation into interneurons (D35), while fewer transcriptomic differences accompanied cIN maturation (D60) (Figure 1B).
Figure 1.

Gene expression changes during hESC differentiation into mature human cortical interneurons
(A) Schematic representation of the protocol used to produce matured human cortical interneurons (D60, green) from hESCs (D0, blue). Intermediate time points used to profile interneuron specification and differentiation were MGE-like neuronal progenitors (D15, orange) and immature cortical interneurons (D35, gray).
(B) Principal component analysis (PCA) highlighting variation in expression between D0 (blue, n = 4), D15 (orange, n = 4), D35 (gray, n = 4), and D60 (green, n = 3).
(C–F) Gene ontology (GO) analysis of genes with stage-specific enriched expression at: (C) D0, (D) D15, (E) D35, or (F) D60.
(G and H) Disease enrichment GO term analysis of genes with stage-specific enriched expression at (G) D35 or (H) D60.
(I) Transcription factor (TF) expression in the human fetal (13–18 weeks post conception20) MGE versus CGE and LGE, with the upper right quadrant identifying TFs with MGE-enriched expression. Some TFs of potential interest are highlighted.
(J) Expression of the subset of TFs with MGE-enriched expression at D0, D15, D35, and D60.
To better characterize these transitions, we examined differentially expressed genes (DEGs) with significantly higher gene expression at a single time point (Data S1A–S1G). Enrichment analysis of these genes revealed that “D0 unique” genes were associated with gene ontology (GO) terms related to basic cellular processes (Figure 1C and Data S2A), while “D15 unique” genes were instead associated with terms related to neural cell specification (Figure 1D and Data S2B). Finally, both genes with D35- and D60-enriched expression were associated with terms related to neuronal differentiation. Specifically, “D35 unique” genes were associated with more general aspects of neuron development (Figure 1E and Data S2C), while “D60 unique” genes were associated with neuronal properties, including “synaptic signaling” (Figure 1F and Data S2D). Finally, “D35 unique” genes were broadly enriched for genes associated with “neurodevelopmental disorders” (Figure 1G and Data S2C), while “D60 unique” genes were associated with diseases involving altered neuronal activity, including “seizures” and “epilepsy” (Figure 1H and Data S2D).
As many of these disease-related genes encode TFs, we next assessed which TFs might be candidate regulators of interneuron development. We identified TFs with significantly higher expression at D15, D35, or D60, versus D0, and then assessed their regional expression in the caudal (CGE) and lateral (LGE) versus the medial ganglionic eminences (MGEs) during human fetal brain development (post-conception weeks 13–1820) (Figure 1I and Data S2E). This defined 104 TFs with higher expression in MGE versus CGE or LGE; this encompassed established MGE markers (e.g., NKX2.1 and ASCL1) and other TFs, including the zinc finger (ZF) TFs ZNF25 and ZNF699 and homeodomain (HD) TFs NKX6.2 and DLX5. Finally, we examined how expression of these MGE-enriched TFs changed during our developmental time course, identifying four TF clusters with different temporal expression profiles (Figure 1J and Data S2F). While regional expression data remains limited for the developing human fetal brain at these stages, these results enabled us to define TFs that were dynamically and highly expressed both in our model and in the MGE during fetal brain development in vivo.
Chromatin landscape changes associated with interneuron specification and differentiation
To link these gene expression changes to CREs that may control them, we used CUT&Tag9 to profile four histone modifications (H3K27ac, H3K4me3, H3K27me3, and H3K9me3) that can broadly partition the chromatin landscape (Figure 2A). We first segregated the genome into seven clusters based on the presence each of these marks (Figure 2B). Cluster 1 constitutes chromatin lacking enrichment for any modification surveyed (“unmarked”), clusters 2–4 comprise active or open chromatin, clusters 6 and 7 constitute repressed or condensed chromatin, and cluster 5 captures bivalent chromatin (Figure 2B). We evaluated the relative proximity of each chromatin state to the nearest transcription start site (TSS), demonstrating that sites in clusters 3–5 are frequently found near the TSS, while sites in clusters 1, 2, 6, and 7 are often more distally located from the nearest TSS (Figure 2C).
Figure 2.

Chromatin-state transitions associated with interneuron specification and differentiation
(A) Graphical representation of the relationship between the histone marks profiled: H3K27ac (light green), H3K4me3 (dark green), H3K27me3 (red), and H3K9me3 (orange) and transcriptional state.
(B and C) (B) Emissions from ChromHMM model describing 7 clusters and (C) average occupancy of each cluster across all samples and distance to the nearest TSS.
(D) Percentage of occupancy of each cluster across the genome at D0 (blue, H3K4me3 n = 6, H3K27ac and H3K27me3 n = 5, H3K9me3 n = 3), D15 (orange, H3K4me3 n = 4, H3K27ac, H3K27me3, H3K9me3 n = 3), D35 (gray, H3K4me3, H3K27ac, and H3K9me3, n = 4, H3K27me3 n = 3), and D60 (green, H3K4me3 n = 6, all other histone marks N = 3).
(E) Heatmap representing loss of cluster 2 chromatin from D0, D15, and D35 to the subsequent time point, represented as a percentage of base pairs lost from cluster 2 at each time point.
(F) Width of chromatin segments assigned to cluster 2 at each time point, with median value marked by the dotted red line.
(G–J) Heatmaps representing the gain and loss of (G) cluster 6 or (I) cluster 7 chromatin from D0, D15, and D35 to the subsequent time point. Transitions from D0 and D15 are represented as a percentage of all base pairs gained or lost by either cluster 6 or 7 by the subsequent time point, whereas the D35 transition is represented as a percentage of all base pairs gained or lost from cluster 6 or 7 from D35 to D60. Width of chromatin segments assigned to (H) cluster 6 or (J) cluster 7 at D0, D15, D35, and D60, with the median value marked by the dotted black line.
We next considered the proportion of the genome assigned to each cluster across our time course. As expected, a majority was assigned to cluster 1, with the next largest clusters being 2, 6, and 7 (Figure 2D). The proportion of the genome assigned to clusters 2 and 5 decreased during differentiation (Figure 2D); by contrast, the proportion in repressive clusters 6 and 7 increased from D0 to D35, while declining from D35 to D60 (Figure 2D). Finally, the proportion of the genome in cluster 3 (H3K27ac/H3K4me3) increased from D0 to D35 and then decreased from D35 to D60, while cluster 4 (H3K4me3 only) had the converse trend (Figure 2D). These results suggest that a loss of H3K27ac and gain of repressive marks (H3K27me3/H3K9me3) are prominent trends accompanying interneuron differentiation (D0–D35), with subsequent loss of these repressive modifications and gain of H3K4me3 at some promoters upon interneuron maturation.
Further examining the loss of H3K27ac marked (cluster 2) chromatin during differentiation, we found that, at all transitions, the majority of H3K27ac chromatin transitioned to cluster 1 (“unmarked”; Figure 2E), while a minority (∼10%) transitioned into repressive chromatin (cluster 6/7; Figure 2E). The H3K27ac-marked chromatin peak width also declined during differentiation (Figure 2F). In a similar evaluation of clusters 6 and 7, there was a similar transition from unmarked chromatin to repressive (cluster 6/7) chromatin with a minimal gain of repressive marks from active (cluster 2) or bivalent (cluster 5) chromatin (Figures 2G and 2I). Finally, the loss of repressive marks from D35 to D60 again largely corresponded with a gain in unmarked chromatin, with minimal transitions from H3K27me3-marked chromatin to active (cluster 2) or repressive (cluster 7) chromatin (Figures 2G and 2I). H3K9me3 peak width generally decreased during differentiation, with an increase and subsequent decrease of H3K27me3 peak width from D15 to D60 (Figures 2H and 2J). Together, this analysis illustrates that the chromatin state changes profiled here most frequently involved transition to or from a chromatin state unmarked by these 4 modifications, with rarer transitions between the repressive, bivalent, and active chromatin states.
Developmental dynamics of H3K27ac enrichment identifies putative CREs controlling cIN specification and differentiation
To assess how stage specific gene expression was related to chromatin state transitions, we first examined changes in H3K27ac peaks and found that these were often associated with stage-enriched expression of the corresponding genes (Figures S1A–S1C). By contrast, H3K4me3 peak enrichment was more static and was not clearly associated with stage-enriched gene expression (Figures S2A–S2C). By focusing on changes in H3K27ac, we next identified significantly differentially acetylated regions (DARs) between D0 and D15, demonstrating that the majority of these (67%) were sites that gained H3K27ac signal from D0 to D15 (Figures 3A and S3A and Data S3A). GO analysis of the genes associated with these DARs (Data S3A) demonstrated strong enrichment for cell proliferation-related genes (Data S3B). To identify candidate CREs regulating differential gene expression from D0 to D15, we further related these DARs to genes with significant differential expression at D15 versus D0 and with expression of >1 RPKM at D15. We then examined expression of these genes across our full time course, defining three clusters with characteristic changes in gene expression (Figure S3B). We focused in particular on cluster 2 genes, which exhibited their highest expression at D15 (Figure S3B and Data S3C) and were related to neuronal development GO terms (Figure 3B, Data S3D). Performing TFBS enrichment analysis on cluster 2 gene-associated DARs revealed enriched TFBSs, including multiple homeobox TFs with roles in MGE development and TFs regulating more general aspects of neuronal differentiation (e.g., SOX21; Figure 3C and Data S3E).
Figure 3.

H3K27ac and H3K9me3 histone modification state transitions predict putative CREs during differentiation of hESCs into immature cINs
(A) H3K27ac peaks with significant changes in enrichment between D0 (blue, n = 5) and D15 (orange, n = 3).
(B and C) (B) GO term enrichment of associated genes and (C) TFBS enrichment under H3K27ac peaks that gained significant enrichment at D15 versus D0.
(D) H3K27ac peaks with significant differential enrichment between ventral telencephalic NPCs (orange, n = 3) versus dorsal NPCs (purple, n = 4).
(E and F) (E) GO term enrichment of associated genes and (F) TFBS enrichment under H3K27ac peaks with significant enrichment in ventral NPCs versus dorsal NPCs.
(G) H3K27ac peaks with significantly increased H3K27ac enrichment at D35 (gray, n = 4) versus D15 (orange, n = 3).
(H and I) (H) GO term enrichment of associated genes and (I) TFBS enrichment under H3K27ac peaks that gained significant enrichment at D35 versus D15.
(J) H3K9me3 peaks with significantly increased enrichment at D35 (gray, n = 4) versus D15 (orange, n = 3).
(K and L) (K) GO term enrichment of associated genes and (L) TFBS enrichment under peaks with significantly increased H3K9me3 at D35 versus D15. Color intensities and y axis scales for heat maps of histone mark signals are representative of read depth normalized per 10 million reads.
To assess the relevance of these putative CREs to MGE development, we next compared our ventral telencephalic MGE-like neural progenitors (ventral NPCs) to dorsal telencephalic-patterned NPCs (dorsal NPCs), using the previously described chromatin state clustering (Figure 2B); this revealed relatively minimal global differences, except for cluster 7 (H3K9me3), which occupied a larger proportion of the genome in ventrally versus dorsally patterned NPCs (Figure S3C). We then compared our significantly increased DARs at D15 versus D0 to significantly increased DARs in ventral NPCs versus dorsal NPCs (Figure S3D and Data S4A), identifying ventral NPC-specific DARs (Figure 3D). Examining genes associated with these DARs, we focused on genes with high D15 expression, finding those genes to be enriched for neurodevelopmental GO terms (Figure 3E and Data S4B). We also found that these genes were enriched for GO terms related to ASD and ID (Data S4B). Performing TFBS motif analysis on peaks associated with these genes revealed enrichment for ZF and high-mobility group (HMG) TFBSs, including ZNF711 and SOX21 (Figure 3F and Data S4C).
Finally, we identified sites that instead had increased H3K27ac signal at D35 versus D15 (Figures 3G and S3E and Data S5A), identifying genes associated with these DARs with high expression in cINs (D35/D60) (Figure S3F and Data S5B). These genes were enriched for GO terms related to neuronal differentiation and function (Figure 3H and Data S5C). Prominent TFBS enriched at these peaks included those recognized by bHLH, HD, and ZF TFs (Figure 3I and Data S5D). Together, these results show that changes in H3K27ac enrichment are associated with corresponding changes in the expression of genes regulating interneuron specification and differentiation. These candidate CREs also contained TFBS recognized by an overlapping set of TFs, which may function at these elements to regulate cIN development.
Developmental changes in H3K9me3 identify CREs involved in the maintenance of neuron progenitor fate and proliferation
While examining H3K27ac DARs provides insight into developmental CRE usage associated with activate gene expression, examining epigenetic changes that accompany CRE silencing is also important. Therefore, we focused on H3K9me3 as a marker of CRE repression, identifying sites with significantly greater H3K9me3 signal in ventral NPCs versus dorsal NPCs (Figures S4A and S4B and Data S6A). These sites were associated with genes enriched for GO terms related to “transcription factors” (particularly ZF TFs) and “glutamatergic synapses” (Data S6B). We related this regulation to general neuronal patterning by curating sites that significantly gained H3K9me3 in ventral and dorsal NPCs (Data S6A) and that also gained H3K9me3 at D15 versus D0 (Data S6A and S6B). Genes associated with these peaks were enriched for GO terms related to ectoderm development and growth factor (EGF/FGF) signaling (Data S6C).
We also assessed sites that significantly gained H3K9me3 at D35 versus D15, to identify CREs that may be active during MGE specification but are subsequently repressed during cIN differentiation. To parallel our prior analyses, we identified sites that significantly gained H3K9me3 at D35 versus D15 (Figure S4C and Data S7A) and examined the expression of genes associated with these sites. Hierarchical clustering identified several gene expression profiles of interest (Figure S4C and Data S7B), including cluster 3 genes that were most highly expressed as D15 and were enriched for GO terms related to ventral NPC specification (Data S7C). We also examined cluster 5 genes that were highly expressed at both D0 and D15, finding that these were enriched for GO terms related to “mitosis” and “DNA repair” (Figure 3K and Data S7D). Interestingly, TFBS enrichment analysis of these cluster 5 associated peaks revealed an enrichment for a key regulator of ventral NPC specification (NKX2.1) among other TFs (Figure 3L and Data S7E). Together, these data highlight roles for H3K9me3 in suppressing the expression of genes involved in alternate cell fates, including glutamatergic neuronal identities and earlier developmental roles (e.g., ectoderm) during NPC specification (D0–D15), and in suppressing mitosis and ventral NPC related genes during neuronal differentiation (D15–D35).
Identifying TFs associated with developmentally relevant CREs regulating cIN specification
To identify TFs that may bind the CREs identified in our in silico analysis, we next focused on three sets of putative CREs likely to regulate MGE specification and pooled TFBS enrichment results from each. These included (1) D15- and ventral NPC-enriched H3K27ac peaks associated with genes with high D15 expression (Figure 4A and Data S4A), (2) D35 specific-H3K27ac peaks associated with genes with high D15 and/or D35 expression (Figure 4B and Data S5A), and (3) sites that gained H3K9me3 at D35 versus D15 and were associated with genes with high D15 and low D35 expression (Figure 4C and Data S7A).
Figure 4.

Analysis of putative CREs and transcription factors with binding site enrichment under these peaks
(A–C) Examples from each set of putative CREs: (A) D15 enriched H3K27ac associated with high D15 gene expression, (B) D35 enriched H3K27ac associated with high D35 gene expression, and (C) D35 enriched H3K9me3 associated with high expression at early (D0 and D15) time points.
(D) Expression, during hESC-mature cIN differentiation, of TFs that recognize TFBS enriched in putative CREs. TFs with enriched expression at D15 and D35 are indicated in green text.
(E) Luciferase assay measures luminescence in D15 NPCs transfected with putative CRE-luciferase reporter plasmids, normalized to the unmodified (no CRE) luciferase vector. Dotted line represents basal expression from the no CRE control. CREs indicated in red are associated with an RFX binding motif. Data were analyzed with Student’s t test n = 4, ∗p value < 0.05, ∗∗p value < 0.01, ∗∗∗p value < 0.001 and ∗∗∗∗p value < 0.0001.
(F) Co-enrichment for target and co-occuring TFBSs within peak sets of interest, averaged across all peaks as a percentage of peak numbers with corresponding TFBS.
(G and H) Biological process and disease GO term enrichment for genes associated with a putative RFX3/4 TFBS-containing CRE within our peak set of interest.
By combining all of the TFs that were associated with a significantly enriched TFBS in these three CRE datasets, we identified candidate TFs that may function at these sites. Filtering this list to define the most likely candidate TFs; we restricted our TF list to those that were well-expressed at either D15 or D35 (RPKM > 1) and examined the gene expression changes of these TFs across cIN development. From this analysis, we identified 25 TFs as likely candidates to be functioning at our CREs of interest (Figure 4D, green).
We further refined our subset of putative CREs for further analysis by eliminating peaks which did not have a TFBS corresponding to our 25 candidate TFs (Data S8A). To test if these peaks had regulatory activity, we selected 29 peaks corresponding to 29 genes, and cloned a 0.4–1.5 kb fragment of genomic DNA containing the peak upstream of a thymidine kinase (TK) minimal promoter-luciferase reporter vector. At D15, 23 of these putative CREs had significant activity, with 21 enhancing and 3 repressing reporter expression (Figure 4E). By contrast, in hESCs only 12 of these 29 CREs exhibited significant enhancer activity, with 7 showing significantly lower enhancer activity in stem cells than in D15 NPCs, confirming that a majority of these CREs function as temporally specific enhancers (Figure S5A).
To further investigate potential interplay in gene regulation by our 25 candidate TFs, we also examined the frequency with which one TFBS occurred with a second in the same CRE (Figure 4F and Data S8B). This analysis identified clusters of TFBSs that frequently co-occurred, with some TFs recognizing TFBSs that were frequently proximal to other types of TFBSs, such as SMAD3 and NKX2.1 (Figure 4F and Data S8B). We also identified TFs that recognize TFBSs that do not frequently co-occur with other types of TFBSs, including the RFX factors (Figure 4F and Data S8B). These factors are of particular interest, as RFX3 and RFX4 have recently been identified as ID and ASD risk genes17 and both genes are expressed during specification of our hPSC-derived MGE-like NPCs (D15) and in the fetal MGE (Figure 4D and Data S2E). Returning to our luciferase assay results, 6 of the CREs that we tested had an RFX TFBS, and all exhibited significant enhancer activity (Figure 4E, red). Finally, we examined the genes associated with CREs with an RFX binding motif and found that these genes were enriched for ASD and ID risk genes (Figures 4G and 4H and Data S8C). These results suggest that RFX3 and RFX4 can function at developmentally relevant CREs and may regulate a suite of genes associated with NDDs.
Investigating the role of RFX3 and RFX4 in early interneuron specification
To investigate the roles of RFX3 and RFX4 during cIN development, we developed CRISPR inhibition models (KDs) for both genes (RFX3-G1 and RFX4-G1; Figure 5A). By comparison with control cells expressing dCas9-KRAB with no guide RNA (KRAB), both models exhibited significant reduction of the target gene mRNA and protein at D15 (Figures 5B–5E, S6A, and S6B). Therefore, we next used RNA sequencing (RNA-seq) analysis to examine gene expression changes between our RFX3 and RFX4 KDs compared to the KRAB control, identifying clear gene expression changes in both models (Figure 5F). We observed that, by comparison with the KRAB control, DEGs in the RFX3 and RFX4 KDs were largely non-overlapping, with a majority of DEGs exhibiting increased expression relative to the KRAB control in either model (Figure 5G, Data S9A and S9D). DEGs with increased expression in the RFX3 KD were enriched for GO terms related to neuronal function and “extracellular matrix” (Figure 5H and Data S9B). By contrast, genes with decreased expression in this model were enriched for earlier biological processes and TFs (Figure 5I and Data S9C). DEGs that were upregulated in the RFX4 KD were enriched for genes related to neuronal function (Figures 5H and 5J, Data S9B, and S9E), while downregulated DEGs in the RFX4 KD were instead involved in proliferation (Figure 5K and Data S9F). These data suggested that both RFX3 and RFX4 can prevent precocious differentiation, while RFX4 additionally may be required to promote NPC proliferation (Figures 5J and 5K, Data S9E and S9F).
Figure 5.

RFX3 and RFX4 CRISPRi models reveal convergent and divergent roles in cIN development
(A) Schematic representation of CRISPRi models using a two lentiviral expression construct system, with constitutive expression of both the dCas9-KRAB fusion and guide RNAs specific to the RFX3 or RFX4 promoter.
(B–E) Changes in RFX3 or RFX4 (B and C) mRNA and (D and E) protein expression in the RFX3 and RFX4 KD models. Data were analyzed with Student’s t test n = 4, ∗p value < 0.05, ∗∗p value < 0.01 and ∗∗∗p value < 0.001.
(F) Principal component analysis, highlighting variation between the KRAB only control (orange, n = 4), RFX3 (purple, n = 4), and RFX4 (green, n = 4) KD.
(G) Heatmap highlighting all significant DEGs in either the RFX3 or RFX4 KD models.
(H–K) GO enrichment analysis of DEGs significantly (H) up- or (I) downregulated in RFX3 KD samples, and (J) up- or (K) downregulated in RFX4 KD versus the KRAB control.
(L) GO enrichment analysis of common DEGs with opposing gene expression changes in the RFX3 versus RFX4 KD models, by comparison with the KRAB control.
(M) Heatmap of common DEGs in the “DNA-binding transcription activator activity” GO category with opposing gene expression changes in the RFX3 and RFX4 KD models.
(N) Heatmap of gene expression changes from D0–D60, for TFs that are significantly upregulated DEGs in the RFX4 KD and significantly downregulated DEGs in the RFX3 KD, versus the KRAB control.
(O) Changes in H3K27ac from D0–D60 at putative CREs that have significantly increased H3K27ac at D15 versus D0 and are associated with DEGs in the RFX3 and/or RFX4 KD.
(P) Heatmap of differentially expressed TFs in the RFX3 KD (purple), RFX4 KD (green), or both KDs (black), that are associated with a CRE both with an RFX TFBS and with an H3K27ac peak with significantly increased H3K27ac at D15 compared to D0.
Interestingly, and despite an apparently overlapping role for RFX3 and RFX4 in preventing precocious differentiation, many shared target genes were oppositely regulated in the RFX3 and RFX4 KD models (Figures 5F and 5G). To further examine this, we identified DEGs that were significantly up- or downregulated in the RFX3 KD and were oppositely regulated in the RFX4 KD model. This gene set was enriched for GO terms related to “neuron development” and for TFs (Figure 5L and Data S9G). Given our focus on the transcriptional regulation of interneuron development, we examined this set of oppositely regulated TFs in greater depth. Plotting expression of these oppositely regulated TFs, we observed that the majority were downregulated in the RFX3 KD but upregulated in the RFX4 KD; these included members of the FOS-JUN signaling pathway (e.g., FOSL1 and FOSL2) and other TFs involved in interneuron differentiation (e.g., ATF3, NKX6-1, and LHX9; Figure 5M and Data S9I). Consistent with this, most of these TFs were most highly expressed in D35 and/or D60 interneurons during our differentiation time course (Figure 5N and Data S9J). Although fewer TFs were upregulated in the RFX3 KD and downregulated in RFX4 KD, these also included genes that play important roles in interneuron neurogenesis (e.g., ASCL1 and HES5; Figure 5M and Data S8A). These results suggest that some of the divergent functionality of RFX3 and RFX4 may be the result of opposing regulation of shared TFs involved in interneuron neurogenesis and differentiation.
Since RFX3 and RFX4 appeared to differentially regulate the expression of some shared TFs, we used our histone modification state atlas and CRE data to identify TFs that could be direct targets of RFX3 and/or RFX4. We began by identifying putative direct TF targets of RFX3 and/or RFX4, focusing on H3K27ac peaks associated with DEGs in the RFX3 KD or RFX4 KD that also had increased H3K27ac signal at D15 versus D0 and that contained an RFX binding motif (Figure 5O and Data S9H). After identifying the TFs associated with these peaks, we eliminated those with low expression at D15 (<1 RPKM) to identify 20 TFs of interest; 12 of these were significant DEGs in both the RFX3 and RFX4 KDs (black, Figure 5P). In this work, we noted that RFX4 was associated with RFX TFBS containing H3K27ac enriched peaks at D15 and was significantly downregulated under both RFX3 KD and RFX4 KD conditions, suggesting that RFX4 may be directly regulated by RFX3. These results suggest that RFX3 and RFX4 likely play a direct role in regulating a large number of overlapping TFs. Furthermore, the identification of RFX4 as a likely RFX3 target suggests that some overlapping gene expression effects in the RFX3 and RFX4 KD models (e.g., negative regulation of genes with “gated channel activity”) could be due to reduced RFX4 expression.
Phenotypic consequences of RFX3 or RFX4 knockdown
To verify the gene expression changes observed in the aforementioned RFX3 and RFX4 KD models, we generated a second set of RFX3 KD and RFX4 KD models that utilized a second independent gRNA (RFX3-G2 and RFX4-G2). To alleviate concerns that additional selection for the gRNA plasmid, which was necessary in the RFX3-G1 and RFX4-G1 but not in the KRAB model, may have influenced the effects seen, we also derived a second set of models using a single vector system that expressed both the dCas9-KRAB fusion and each gRNA. We confirmed reduction of target gene expression in these RFX3-G2 and RFX4-G2 models at the mRNA and protein levels by comparison with KRAB controls (Figures S6C–S6H). To test our previous transcriptomic observation that RFX4 was also decreased in RFX3 KD models, we also performed western blotting for RFX4, finding a significant decrease in RFX4 protein levels in both RFX3 KD models (Figures S6I and S6J).
Using our RNA-seq data, we selected 16 genes that were upregulated in the RFX3-G1 model and belonged to the “adhesion” and “calcium ion binding” GO terms (Figure 5H and Data S9B). Differentiating both RFX3 KD models into ventral telencephalic NPCs, we then performed RT-qPCR for these genes, finding that all 16 genes were significantly upregulated in the RFX3-G1 KD, while the majority were also upregulated in the RFX3-G2 model (Figure 6A). Similarly, for the RFX4 KD model, we identified 8 upregulated genes associated with the “synapse” GO term and 8 downregulated genes associated with the “mitotic cell cycle” GO term (Figures 5J and 5K, Data S9E and S9F). Again, by performing RT-qPCR for these genes in ventral NPCs, we found that the “mitotic cell cycle genes” were all downregulated in both RFX4 KD models, while the majority of “synapse” genes assessed were upregulated in both RFX4 KDs (Figure 6B). Finally, to confirm the reciprocal effect of RFX3 and RFX4 KD on key TFs, we identified 8 TFs that were differentially regulated in the RFX3 and RFX4 KDs and performed RT-qPCR in all four models at D15 (Figure 5M). We validated findings for 4 TFs that were reciprocally regulated in the RFX3 and RFX4 KD models, as indicated by our RNA-seq analysis (Figure 6C); the other 4 TFs produced similar but non-significant changes in gene expression in at least one KD. This work validated key findings from our differential gene expression analysis in the RFX3-G1 and RFX4-G1 KD models by using independent KD cellular models with alternate gRNAs.
Figure 6.

Both RFX3 and RFX4 KD models exhibit deficits in human cortical interneuron differentiation
(A and B) RT-qPCR was used to confirm gene expression changes identified in the (A) RFX3 KD and (B) RFX4 KD models, using both the G1 and G2 KD models to test each gene at D15 of cortical interneuron differentiation.
(C) RT-qPCR was also used to investigate the reciprocal regulation of a selection of TFs previously identified as DEGs in the RFX3 KD and RFX4 KD models. Data are represented as mean fold change ± SEM.
(D–H) (D) Representative images indicating sphere size (marked by solid black lines) and outgrowth (marked by dotted black lines) and quantification of neurosphere size and relative outgrowth (outgrowth minus neurosphere size, relative to neurosphere size) from neurospheres at D12 of cortical interneuron differentiation in the RFX3 (E and F) and RFX4 (G and H) KD models. Data are represented as distributions, using all measures taken with quartiles (dotted lines) and means (black line) marked (scale bar: 100 μm).
(I–N) Representative images and quantification of the fraction of cells immunopositive for (I) Ki-67, (J) ASCL1, (K) NKX2.1, (L) DCX, (M) ATF3, and (N) FOSL1 in the KRAB and RFX3 KD models. Data are represented as mean ± SEM; data were analyzed by Student’s t-test, versus the paired KRAB controls. n = 4 biological replicate experiments for all conditions, ∗p value < 0.05, ∗∗p value < 0.01, ∗∗∗p value < 0.001 and ∗∗∗p value < 0.0001. Scale bars: 50 μm.
Given the decreased expression of proliferation genes in the RFX4 KD models and increased expression of cell adhesion genes in the RFX3 KD models, we also assessed the size and outgrowth of neurospheres at D12; abnormalities in neurosphere size or outgrowth can result from changes in either adhesion and/or proliferation. We observed increased sphere size and evidence for increased outgrowth in the RFX3 KD models (Figures 6D–6F), while the RFX4 KD NPCs had a contrasting cellular phenotype (Figures 6D, 6G, and 6H). For example, RFX4-G1 spheres had greatly reduced size and exhibited essentially no outgrowth (Figures 6D, 6G, and 6H). RFX4-G2 spheres exhibited a related but slightly less severe phenotype involving decreased sphere size and minimal outgrowth, with cells that did emerge from the NPC spheres exhibiting a characteristic neuronal rather than NPC morphology (Figures 6D, 6G, 6H, and S6K). These results indicate that there are distinct effects of RFX3 or RFX4 expression on neurosphere size and outgrowth, resulting in increased size and outgrowth from RFX3 KD spheres but a contrasting strong reduction of sphere size and outgrowth from RFX4 KD spheres.
The failure of RFX4 KD cells to proliferate precluded us from obtaining sufficient materials for more extensive analyses by immunofluorescence. However, to confirm that the lack of sphere outgrowth was due to a decrease in proliferation, as suggested by the transcriptomic data, and did not instead or additionally involve increased cell death, we also performed western blotting at D12 for both the mitotic marker Ki-67 and the apoptotic marker cleaved caspase-3. This analysis confirmed our hypothesis, as we observed a significant decrease in Ki-67 expression in RFX3 KDs, with no significant change in cleaved caspase-3 levels (Figures S6L–S6O).
To further examine the consequences of RFX3 deficiency on early cIN specification, we performed immunostaining in ventral NPCs at D17 of differentiation. First, we assessed whether increased neurosphere size and outgrowth was associated with increased proliferation by staining for Ki-67 and phospho-histone H3 (PH3), finding no significant differences between the KRAB control and RFX3 KD models (Figures 6I and S6P). As we had observed increased neurosphere size in the RFX3 KD models at day 12 (Figures 6D–6F), we hypothesized that this could instead involve increased growth during earlier stages of development. To test this hypothesis, we tracked KRAB and RFX3 KD neurosphere size from D0 to D10 of differentiation, finding a significantly greater increase in sphere size in both RFX3 KDs from D0-D10, relative to the KRAB control (Figures S6Q and S6R).
Next, we assessed several key markers of MGE specification, finding an increased fraction of cells that stained positive for each marker, with a ∼50% increase in the fraction of ASCL1 positive cells and a more moderate increase in the fraction of NKX2.1 and DLX2 positive cells (Figures 6J, 6K, and S6S). We also examined changes in SOX6 and LHX6 gene expression levels by using our transcriptomic data for RFX3 KD: while there was no significant change in LHX6 expression levels, expression of SOX6 was increased in the RFX3-G1 KD (Data S9A). To assess if these findings related to the fraction of cells that had transitioned into immature neurons, we assessed DCX-expressing cells and found an increase in the DCX-expressing cell fraction similar in magnitude to the finding for ASCL1 (Figure 6L).
Finally, we assessed three TFs that exhibited decreased expression upon RFX3 KD in our RNA-seq analysis (ATF3, FOSL1, and JUN), to assess whether the MGE, neurogenic, and immature neuron marker expression observed previously was accompanied by a decreased fraction of cells expressing these TFs. For ATF3 and FOSL1, the fraction of immunopositive cells was significantly reduced in both RFX3 KDs (Figures 6M and 6N), while the JUN-expressing fraction likewise decreased in both KD models, but was only significant for RFX3-G1 (Figure S6T). Together, these results suggest that RFX3 deficiency results in premature differentiation, which is coupled with the reduced expression of both TFs associated with earlier (stem cells, FOSL1) and later (neurons, ATF3 and JUN) developmental processes, suggesting a general dysregulation of stage-specific TF expression, which may impact later cIN development.
Discussion
In this study, we built an atlas to define the chromatin landscape changes that occur during human cIN development. We used this atlas to predict putative CREs that are activated or silenced during interneuron specification and differentiation. We subsequently used TFBS enrichment at these CREs, in combination with expression data for our hPSC-derived MGE-like NPCs and analogous fetal brain data, to identify candidate TFs that may bind these elements to control cIN development. From these candidate TFs, we focused on RFX3 and RFX4, as these TFs are highly expressed in both our MGE-like NPCs and in fetal MGE, while mutations in these genes are a newly described contributor to NDDs.17 We built models of RFX3 and RFX4 deficiency and used these to demonstrate that both genes play essential but distinct roles in early cIN development. This work illustrates the utility of the hPSC-derived cIN model to relate disease-associated TFs to sensitive cell populations and critical developmental time periods, and provides a relevant model of human cIN development that can be used to define gene regulatory networks underlying brain development and disease.
By examining changes in the chromatin landscape we determined that, during cIN differentiation, histone modifications associated with active and accessible chromatin decreased, while there was a concurrent increase in histone marks associated with repressive chromatin. These trends are reminiscent of chromatin state changes observed during early neural differentiation of mouse embryonic stem cell models, suggesting that they may broadly reflect chromatin state changes that accompany neuronal differentiation, agnostic of neuronal cell type or species.21,22,23 During interneuron maturation, we also observed reduction of both H3K27me3- and H3K9me3-associated chromatin relative to their levels in immature cINs, suggesting that accumulation of these marks is not required for interneuron maturation. We hypothesize that reduction in chromatin carrying these repressive marks may involve a transition to other mechanisms of repression, such as DNA methylation, which increases during neuronal maturation.24 Together, these findings elucidate regulatory events that underlie human cIN development and control key aspects of the development of this disease-relevant human neuronal cell type.
We next used this chromatin state atlas to first define and test a set of putative CREs likely to be involved in cIN development, and then to identify TFBS enrichment in selected CRE subsets and expression of TFs that recognize these TFBSs in our in vitro-derived MGE-like NPCs and in fetal MGE. This process identified CRE-enriched TFBSs and corresponding TFs with known functions during cIN development, including DLX2,25 SOX6,26 and NKX2.1,27 validating this workflow. Interestingly, in this work, NKX2.1 TFBSs were enriched primarily at putative CREs that gain repressive (H3K9me3) modifications during cIN differentiation. These results suggest that NKX2.1 may play a more central role in regulating MGE progenitor specification and proliferation, despite its continued expression during neuronal differentiation. This is consistent with mouse studies examining the role of NKX2.1,28,29 which have defined a central role in establishing MGE gene expression patterns which are then propagated and maintained by other factors such as LHX6. This work is also congruent with a role for NKX2.1 in regulating proliferation in other contexts, including early lung development and tumorigenesis.30 These results also suggest that deposition of H3K9me3 at key CREs may interfere with NKX2.1 binding to modulate target gene regulation during the transition from NPCs to immature neurons. In future work, it would be interesting to directly examine whether NKX2.1 binds CREs that regulate the proliferation-differentiation transition, and the dependence of this regulation on these changes in the chromatin landscape.
The identification of TFs with known roles in cIN development that are likely to function at our putative CREs illustrates the utility of this approach for predicting other TFs that may play critical roles in cIN development. Several additional MGE-expressed TFs with enriched TFBS in our selected CRE subset would be interesting candidates for further studies, to determine their requirements during human cIN development. This approach also has high utility for identifying TFs that are implicated in the etiology of NDDs, but for which the sensitive cell population is unknown. Mutations in genes encoding six of our 25 candidate TFs are associated with NDDs, including CUX1, DLX2, SOX6, POU3F3, and RFX3/4.17,31,32,33,34 Here, we focused further on the newly identified ASD and IDD risk genes RFX3 and RFX4, because their roles in human neurodevelopment, interneuron development, and disease biology have not yet been characterized. Studies in mouse models have defined roles for CUX135 and SOX636 in the development of specific interneuron subtypes (Reelin positive and Parvalbumin/Somatostatin positive interneurons, respectively) and important roles for DLX2 in the functional maturation of GABAergic interneurons.25 However, despite a greater understanding of the roles these TFs play in interneuron development, little is known about their relevance or contribution to disease etiology, or whether these relate to specific roles for these TFs in human interneuron development. Therefore, probing roles of these TFs specifically during human cIN development and relating these requirements to the consequences of pathogenic mutations in these genes that cause NDDs would be an interesting future area of study.
Here, we used multiple models of RFX3 or RFX4 deficiency to identify distinct roles for these TFs in regulating cIN development and to define a regulatory relationship between these TFs. While knockdown of either TF caused upregulation of some synaptic and neuronal differentiation genes, this work revealed that RFX4 is a critical regulator of ventral NPC proliferation, as even partial knockdown caused a nearly complete cessation of progenitor proliferation. By contrast, RFX3 knockdown had little effect on proliferation, but instead resulted in precocious upregulation of genes involved in neurogenesis and neuronal differentiation.
In keeping with their distinct phenotypic effects, few genes were similarly regulated by RFX3 versus RFX4, indicating that these related TFs have largely distinct roles. Our evidence of reciprocal regulation of key genes by RFX3 and RFX4 suggests a complex relationship between these two genes in regulating downstream networks of TFs that coordinate cell cycle exit and neuronal differentiation. They also suggest that either RFX3 or RFX4 deficiency results in an aberrant cell state in which expression of genes that regulate neuronal maturation is disrupted. Alterations in these aspects of neurodevelopment are likely contributors to the ASD and ID phenotypes seen in patients carrying these mutations. Interestingly, we also found that RFX4 is a likely target of RFX3, with diminished expression of RFX4 in the RFX3 KD potentially contributing to the effects seen. It will be interesting in future studies to identify the mechanisms by which pathogenic RFX3 and RFX4 mutations disrupt downstream TF networks to alter neuronal development and function in the etiology of these NDDs.
RFX3 and RFX4 were identified as ASD and attention-deficit/hyperactivity disorder risk genes in 2021 by Harris et al.,17 who examined a cohort of 38 individuals carrying variants in the RFX3, RFX4, or RFX7 genes. By examining fetal brain single-cell RNA-seq data, Harris et al.17 concluded that RFX3 was broadly expressed throughout the developing cortex, while RFX4 was predominantly restricted to proliferating radial glial progenitor cells. These expression profiles are congruent with our findings that RFX4 plays a critical role in maintaining interneuron progenitor proliferation, while RFX3 plays a more subtle role in preventing premature interneuron differentiation and maturation. These patterns of expression also suggest that the roles for RFX3 and RFX4 that we defined here may not be restricted to cIN development, with these genes potentially playing similar roles in cortical excitatory neuron development.
In addition to this recent link to NDDs, RFX3 loss of function in the mouse results in deficits in corpus callosum development, due to abnormal midline formation.37 Similar studies examining RFX4 loss of function in mouse models have shown that homozygous loss of either transcript variant 1 or 3 results in severe phenotypes38,39 and, in the case of transcript variant 3, a failure of dorsal midline brain structure formation and perinatal death.39 These more severe phenotypes are congruent with our finding that more deleterious consequences resulted from RFX4 KD, relative to RFX3 KD, during human interneuron development. However, these findings also provide evidence for more widespread roles for both RFX3 and RFX4 in other aspects of brain development. Together, our work and these prior findings in mouse models suggest that RFX3 and RFX4 have complex and multifaceted roles in multiple cell types during neuronal development. While this study provides insight into the roles of these genes in early cIN development, future work will be needed to assess mechanisms by which these TFs function and to define how dysregulation of these mechanisms causes NDDs.
Limitations of the study
Due to the inaccessibility of cortical GABAergic interneurons during human brain development, we instead performed these analyses in our human stem cell derived model of hcIN development, which is limited to recapitulating the relatively early developmental events of early to mid-fetal development. Furthermore, associating our CREs with TFs that may bind them is limited by existing data regarding TF binding site usage. Therefore, while we have identified key TFs with roles in hcIN development, it is reasonable to assume that this list is not exhaustive. While mutations in RFX3 and RFX4 are associated with NDDs, we examined the role of these genes using CRISPRi-mediated deficiency. This is a proxy for the haploinsufficiency seen in patients but is unlikely to fully mimic the consequences of the many different pathogenic RFX3 and RFX4 mutations that cause NDDs. Finally, while differential gene expression in the RFX3 and RFX4 KD models provides insight into the genes that these TFs may regulate, it is important to acknowledge that some of these transcriptomic differences may result from changes in the composition of the cell population or gene expression changes in a subpopulation of the cells profiled.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-ASCL1 | Abcam | Abcam Cat# ab211327; RRID: AB_2924270 |
| Rabbit monoclonal anti-ATF3 | Abcam | Abcam Cat# ab207434; RRID: AB_2734728 |
| Rabbit monoclonal anti-DCX | Abcam | Abcam Cat# ab207175; RRID: AB_2894710 |
| Rabbit monoclonal anti-FRA1 (FOSL1) | Abcam | Abcam Cat# ab252421; RRID: AB_2904513 |
| Rabbit monoclonal anti-KI67 | Abcam | Abcam Cat# ab16667; RRID: AB_302459 |
| Rabbit polyclonal anti-TTF1 (NKX2.1) | Abcam | Abcam Cat# ab204411 |
| Rabbit polyclonal anti-JUN | Abcam | Abcam Cat# ab10766 |
| Rabbit monoclonal anti-Cleaved Caspase 3 | Abcam | Abcam Cat# ab32042; RRID: AB_725947 |
| Rabbit polyclonal anti-DLX2 | Abcam | Abcam Cat# ab272902 |
| Rabbit polyclonal anti-Phospho Histone H3 | Abcam | Abcam Cat# ab80612; RRID: AB_2164915 |
| Mouse monoclonal anti-GAPDH | Abcam | Abcam Cat# ab8245; RRID: AB_2107448 |
| H3K27ac | Millipore-Sigma | Millipore Cat# 07-370; RRID: AB_310560 |
| H3K27me3 | Cell Signaling Technology | Cell Signaling Technology Cat# 9733; RRID: AB_2616029 |
| H3K4me3 | Millipore-Sigma | Millipore Cat# 07-473; RRID: AB_1977252 |
| H3K9me3 | Cell Signaling Technology | Cell Signaling Technology Cat# 13969; RRID: AB_2798355 |
| Rabbit IgG | Cell Signaling Technology | Cell Signaling Technology Cat# 2729; RRID: AB_1031062 |
| Guinea Pig Anti Rabbit Secondary | Antibodies-Online | Antibodies-Online Cat# ABIN101961; RRID: AB_10775589 |
| Chemicals, peptides, and recombinant proteins | ||
| SB431542 | Selleckchem | S1067 |
| SAG | Selleckchem | S6384 |
| LDN193189 | Tocris Biosciences | 6053/10 |
| XAV939 | Selleckchem | S1180 |
| Y27632 | Selleckchem | S6390 |
| DAPT | Selleckchem | S2215 |
| Palbociclib (PD0332991) | Selleckchem | S4482 |
| Ascorbic Acid | Sigma Aldrich | A5960 |
| Dibutyryl cAMP sodium salt | Sigma Aldrich | D0260 |
| BDNF | Peprotech | 450-02 |
| Cytarabine | Tocris Biosciences | 4520 |
| Critical commercial assays | ||
| Pierce™ Cypridina Luciferase Flash Assay Kit | Thermo Scientific | 16168 |
| Pierce™ Gaussia Luciferase Flash Assay Kit | Thermo Scientific | 16158 |
| Deposited data | ||
| Raw and analyzed CUT and Tag and RNA-seq data | This paper | GEO: GSE239481 |
| Experimental models: Cell lines | ||
| Human: H9 hESC line | McDonnell Genome Institute Genome Engineering & Stem Cell Center (GESC) | WA09 (RRID:CVCL_9773) |
| Oligonucleotides | ||
| Guide RNA: RFX3 G1-Forward | Sanson et al.40 | CACCGAGAGGGAGACACTCGCACG |
| Guide RNA: RFX3 G2-Forward | Sanson et al.40 | CACCGGGAGACACTCGCACGGGGA |
| Guide RNA: RFX3 G1-Reverse | Sanson et al.40 | CACCGGACACTCAGTAATCCAGCC |
| Guide RNA: RFX3 G2-Reverse | Sanson et al.40 | CACCGTTGACGGGTTTGGCTTTGCC |
| Guide RNA: RFX4 G1-Forward | Sanson et al.40 | AAACCGTGCGAGTGTCTCCCTCTC |
| Guide RNA: RFX4 G2-Forward | Sanson et al.40 | AAACTCCCCGTGCGAGTGTCTCCC |
| Guide RNA: RFX4 G1-Reverse | Sanson et al.40 | AAACGGCTGGATTACTGAGTGTCC |
| Guide RNA: RFX4 G2-Reverse | Sanson et al.40 | AAACGGCAAAGCCAAACCCGTCAAC |
| Luciferase Assay: CACNG8-Forward | This paper | TAGTCTCGAGCAGCTGCTGAACGATGGACTA |
| Luciferase Assay: CAMK2A-Forward | This paper | TAGTCTCGAGAACCTAGGGACCACTTGCCT |
| Luciferase Assay: CAMKV-Forward | This paper | TAGTCTCGAGCCTAAGGAGGCCTGTACCCT |
| Luciferase Assay: MST1R-Forward | This paper | TAGTCTCGAGACCGATAGCTCTGGACACCA |
| Luciferase Assay: KCNIP4-Forward | This paper | TAGTCTCGAGGAGAAGTGCTCCTCTGCCTG |
| Luciferase Assay: KCNN3-Forward | This paper | TAGTCTCGAGCCAAATGCAGAACAGGCACC |
| Luciferase Assay: EPHB2-Forward | This paper | TAGTCTCGAGGGAGGTTTAGGCCTGGTTCG |
| Luciferase Assay: BAG5-Forward | This paper | TAGTCTCGAGGAATTGGATGCGGTCTTGGC |
| Luciferase Assay: LCA5-Forward | This paper | TAGTCTCGAGCCACTTCCTGCTTAGTGGGG |
| Luciferase Assay: HDAC6-Forward | This paper | TAGTCTCGAGCTTTTACGTAGGCGCAGCAC |
| Luciferase Assay: FGF8-Forward | This paper | TAGTCTCGAGGCTTCCCACTAAGCCTCGAA |
| Luciferase Assay: LHX2-Forward | This paper | TAGTCTCGAGAGCGTGGATATGCTGGCTG |
| Luciferase Assay: BTG2-Forward | This paper | TAGTCTCGAGGCCAGTTAGGAGAGCGTCAA |
| Luciferase Assay: MEIS3-Forward | This paper | TAGTCTCGAGGTCCTTGAGCACAGACCCAG |
| Luciferase Assay: EFNB1-Forward | This paper | TAGTCTCGAGAACGCAGTAGCTCAGTCACC |
| Luciferase Assay: SERPINF1-Forward | This paper | TAGTCTCGAGCCAAATTGAGTGCAGGTCGC |
| Luciferase Assay: TUG1-Forward | This paper | TAGTCTCGAGCCACCGCTTTGTCTCCGATA |
| Luciferase Assay: SOX1-Forward | This paper | TAGTCTCGAGGGCCTTTCCTTTGCATGACG |
| Luciferase Assay: CRABP2-Forward | This paper | TAGTCTCGAGGACCTCCGACGGTCACTAGA |
| Luciferase Assay: EFNA5-Forward | This paper | TAGTCTCGAGTCTACGTGATTCACAGGCCG |
| Luciferase Assay: ZSWIM6-Forward | This paper | TAGTCTCGAGGGGAACAGGTGGCAACAGTA |
| Luciferase Assay: RTN4R-Forward | This paper | TAGTCTCGAGCCTCACCGATGAGCTCCTAA |
| Luciferase Assay: FOXO3-Forward | This paper | TAGTCTCGAGTTCTCGCCCGCTAGTGTTTT |
| Luciferase Assay: PIK3R1-Forward | This paper | TAGTCTCGAGCAGCGTACGCCCTCAGAAAA |
| Luciferase Assay: EHBP1-Forward | This paper | TAGTCTCGAGATAGCTTGCTGATGCGCCTA |
| Luciferase Assay: CACNG8-Reverse | This paper | CATTAAGCTTCCCCAGTACTCCCCTCTACG |
| Luciferase Assay: CAMK2A-Reverse | This paper | CATTAAGCTTCCATCAAGCTGCGAAATGGG |
| Luciferase Assay: CAMKV-Reverse | This paper | CATTAAGCTTTGAAAGAGGCCTGCTGCTAC |
| Luciferase Assay: MST1R-Reverse | This paper | CATTAAGCTTGCTCCTAATAGGGTGTGGCG |
| Luciferase Assay: KCNIP4-Reverse | This paper | CATTAAGCTTACTCCCTTAAGCCTCGTTGC |
| Luciferase Assay: KCNN3-Reverse | This paper | CATTAAGCTTGCTCTGGAGGTGAGACCAAC |
| Luciferase Assay: EPHB2-Reverse | This paper | CATTAAGCTTGGGAGGTTTGGCCAGTATCA |
| Luciferase Assay: BAG5-Reverse | This paper | CATTAAGCTTGACTGTTCGGGATAGGACGC |
| Luciferase Assay: LCA5-Reverse | This paper | CATTAAGCTTCGGACTAATGTTGGGGAGCA |
| Luciferase Assay: HDAC6-Reverse | This paper | CATTAAGCTTCTTCTTCCATTGCTTGGCCG |
| Luciferase Assay: FGF8-Reverse | This paper | CATTAAGCTTGACTGCGTCTTCACGGAGAT |
| Luciferase Assay: LHX2-Reverse | This paper | CATTAAGCTTTCCAGTTCGGAAGAAACGCT |
| Luciferase Assay: BTG2-Reverse | This paper | CATTAAGCTTCCCACCGCCTCTATTCAGTT |
| Luciferase Assay: MEIS3-Reverse | This paper | CATTAAGCTTAACAACCCCGTGATAGAGGC |
| Luciferase Assay: EFNB1-Reverse | This paper | CATTAAGCTTGCCAGTCCCATTAGTTTGCG |
| Luciferase Assay: SERPINF1-Reverse | This paper | CATTAAGCTTCGGCAAGTCTGAATCCTCCA |
| Luciferase Assay: TUG1-Reverse | This paper | CATTAAGCTTTGCTTTACACTGGGTGCCAT |
| Luciferase Assay: SOX1-Reverse | This paper | CATTAAGCTTATCATGCTGTACATCGGGGC |
| Luciferase Assay: CRABP2-Reverse | This paper | CATTAAGCTTCTGGAGGCTGAAGCTCTGTC |
| Luciferase Assay: EFNA5-Reverse | This paper | CATTAAGCTTCCCTAGATCGCCCCTAGGTT |
| Luciferase Assay: ZSWIM6-Reverse | This paper | CATTAAGCTTGTCCGAGAAGTGAGGATGCG |
| Luciferase Assay: RTN4R-Reverse | This paper | CATTAAGCTTCATGCCCCAGAATGGTCACT |
| Luciferase Assay: FOXO3-Reverse | This paper | CATTAAGCTTACTCCGACGAATCCGAGACG |
| Luciferase Assay: PIK3R1-Reverse | This paper | CATTAAGCTTATTAATAGCGCCCCGGGAAA |
| Luciferase Assay: EHBP1-Reverse | This paper | CATTAAGCTTCCTCCCCCTGTTAGCATCAC |
| Quantitative PCR: PAMR1-Forward | Wang et al.41 | GGGAAGTCGTGGGTTATACCA |
| Quantitative PCR: FAM20C-Forward | Wang et al.41 | CCTTCCAGAATTACGGGCAAG |
| Quantitative PCR: TESC-Forward | Wang et al.41 | GTCGGGAAACCCTCACATCG |
| Quantitative PCR: EFEMP1-Forward | Wang et al.41 | TGCAGACTGGCCGAAATAACT |
| Quantitative PCR: NOTCH1-Forward | Wang et al.41 | TGGACCAGATTGGGGAGTTC |
| Quantitative PCR: FBLN1-Forward | Wang et al.41 | AGAGCTGCGAGTACAGCCT |
| Quantitative PCR: CRELD1-Forward | Wang et al.41 | CCGGGACAACTTTGGAGGTG |
| Quantitative PCR: GSN-Forward | Wang et al.41 | AGATGGACTACCCCAAGCAGA |
| Quantitative PCR: SMOC1-Forward | Wang et al.41 | TCTATTCGTGTGACCAGGAGAG |
| Quantitative PCR: RASGRP1-Forward | Wang et al.41 | ACATCACCCAGTTCCGAATGA |
| Quantitative PCR: PCDHB5-Forward | Wang et al.41 | TTCCAAGGCGATGAAGTTACTC |
| Quantitative PCR: CLDN3-Forward | Wang et al.41 | AACACCATTATCCGGGACTTCT |
| Quantitative PCR: DSP-Forward | Wang et al.41 | GCAGGATGTACTATTCTCGGC |
| Quantitative PCR: ASS1-Forward | Wang et al.41 | CTTGGGGCCAAAAAGGTGTTC |
| Quantitative PCR: SPINT2-Forward | Wang et al.41 | AAGACCACTCCAGCGATATGT |
| Quantitative PCR: PCDHGB4-Forward | Wang et al.41 | CCCTGAGATGGTATTGAAGACAC |
| Quantitative PCR: ITGA2B-Forward | Wang et al.41 | GATGAGACCCGAAATGTAGGC |
| Quantitative PCR: GSC-Forward | Wang et al.41 | AACGCGGAGAAGTGGAACAAG |
| Quantitative PCR: PCDHB15-Forward | Wang et al.41 | CCCAGTAGGCTCCCTAGTTGT |
| Quantitative PCR: SOX9-Forward | Wang et al.41 | AGCGAACGCACATCAAGAC |
| Quantitative PCR: NR1D1-Forward | Wang et al.41 | ATCGTCCGCATCAATCGCAA |
| Quantitative PCR: STX1A-Forward | Wang et al.41 | TAAAGAGCATCGAGCAGTCCA |
| Quantitative PCR: KIF5A-Forward | Wang et al.41 | GACCAACAACGAATGTAGCATCA |
| Quantitative PCR: RAB3A-Forward | Wang et al.41 | GAGTCCTCGGATCAGAACTTCG |
| Quantitative PCR: LIN7A-Forward | Wang et al.41 | CTGTTAATGGCTGTCCCGAAT |
| Quantitative PCR: ITGA3-Forward | Wang et al.41 | CTACCACAACGAGATGTGCAA |
| Quantitative PCR: PLXND1-Forward | Wang et al.41 | TGAGTCTGTTGTACGCTGTGA |
| Quantitative PCR: CNTNAP2-Forward | Wang et al.41 | TCCCTCCACGTCCCAAAAATG |
| Quantitative PCR: AP1S2-Forward | Wang et al.41 | TTCAGACCGTTTTAGCACGGA |
| Quantitative PCR: NRG1-Forward | Wang et al.41 | AGAGCCTGTTAAGAAACTCGC |
| Quantitative PCR: ANLN-Forward | Wang et al.41 | TGCCAGGCGAGAGAATCTTC |
| Quantitative PCR: WDR62-Forward | Wang et al.41 | GCCTTCTCACCCAATATGAAGC |
| Quantitative PCR: CDCA2-Forward | Wang et al.41 | TCTGATTCGTTTCATTGCTCGG |
| Quantitative PCR: BIRC5-Forward | Wang et al.41 | AGGACCACCGCATCTCTACAT |
| Quantitative PCR: PLK1-Forward | Wang et al.41 | CCTGCACCGAAACCGAGTTAT |
| Quantitative PCR: PBK-Forward | Wang et al.41 | CCAAACATTGTTGGTTATCGTGC |
| Quantitative PCR: KI67-Forward | Wang et al.41 | GGGCCAATCCTGTCGCTTAAT |
| Quantitative PCR: KIF18A-Forward | Wang et al.41 | TGGACTTACTTTACACCAGCCC |
| Quantitative PCR: KIF11-Forward | Wang et al.41 | AGCAAGCTGCTTAACACAGTT |
| Quantitative PCR: SKA1-Forward | Wang et al.41 | CCTGAACCCGTAAAGAAGCCT |
| Quantitative PCR: ASCL1-Forward | Wang et al.41 | CCCAAGCAAGTCAAGCGACA |
| Quantitative PCR: PRDM16-Forward | Wang et al.41 | GTTCTGCGTGGATGCAAATCA |
| Quantitative PCR: HES5-Forward | Wang et al.41 | AAGAGAAAAACCGACTGCGGAA |
| Quantitative PCR: IRX5-Forward | Wang et al.41 | GGGCTACAACTCGCACCTC |
| Quantitative PCR: PROX1-Forward | Wang et al.41 | AAAGTCAAATGTACTCCGCAAGC |
| Quantitative PCR: KLF4-Forward | Wang et al.41 | CCCACATGAAGCGACTTCCC |
| Quantitative PCR: FOSL2-Forward | Wang et al.41 | CAGAAATTCCGGGTAGATATGCC |
| Quantitative PCR: ATF3-Forward | Wang et al.41 | CCTCTGCGCTGGAATCAGTC |
| Quantitative PCR: DDIT3-Forward | Wang et al.41 | GGAAACAGAGTGGTCATTCCC |
| Quantitative PCR: SNAI1-Forward | Wang et al.41 | TCGGAAGCCTAACTACAGCGA |
| Quantitative PCR: RFX3-Forward | Wang et al.41 | CACAGGCTCGACAGTGACC |
| Quantitative PCR: RFX4-Forward | Wang et al.41 | CACGGTGGTGAACATTGTCG |
| Quantitative PCR: GAPDH-Forward | Wang et al.41 | CTGGTAAAGTGGATATTGTTGCCAT |
| Quantitative PCR: PAMR1-Reverse | Wang et al.41 | GGCAGCTCTTGCAGTTTTCA |
| Quantitative PCR: FAM20C-Reverse | Wang et al.41 | TGCCTCTCGTAGTCAGAGAAAT |
| Quantitative PCR: TESC-Reverse | Wang et al.41 | CGAAGGTGATCCCCTCGTACA |
| Quantitative PCR: EFEMP1-Reverse | Wang et al.41 | CACACTGGATACGGTGGGAA |
| Quantitative PCR: NOTCH1-Reverse | Wang et al.41 | GCACACTCGTCTGTGTTGAC |
| Quantitative PCR: FBLN1-Reverse | Wang et al.41 | CGACATCCAAATCTCCGGTCT |
| Quantitative PCR: CRELD1-Reverse | Wang et al.41 | AGGCGGGTCTCACTGTCTT |
| Quantitative PCR: GSN-Reverse | Wang et al.41 | GGTCCCGCCAGTTCTTGAA |
| Quantitative PCR: SMOC1-Reverse | Wang et al.41 | GGATGACAATACCCTCACGGG |
| Quantitative PCR: RASGRP1-Reverse | Wang et al.41 | GCTGTCAATGAGATCGTCCAG |
| Quantitative PCR: PCDHB5-Reverse | Wang et al.41 | CAATGCCCTTTTCAGGCGAA |
| Quantitative PCR: CLDN3-Reverse | Wang et al.41 | GCGGAGTAGACGACCTTGG |
| Quantitative PCR: DSP-Reverse | Wang et al.41 | CCTGGATGGTGTTCTGGTTCT |
| Quantitative PCR: ASS1-Reverse | Wang et al.41 | GAGGTAGCGGTCCTCATACAG |
| Quantitative PCR: SPINT2-Reverse | Wang et al.41 | TCCTCAGAGCGGTAGCTGTT |
| Quantitative PCR: PCDHGB4-Reverse | Wang et al.41 | ATTGGCATCAGTCACTAGAACG |
| Quantitative PCR: ITGA2B-Reverse | Wang et al.41 | GTCTTTTCTAGGACGTTCCAGTG |
| Quantitative PCR: GSC-Reverse | Wang et al.41 | CTGTCCGAGTCCAAATCGC |
| Quantitative PCR: PCDHB15-Reverse | Wang et al.41 | GCTGCTTAGCTCAAAAGGTTTGT |
| Quantitative PCR: SOX9-Reverse | Wang et al.41 | CTGTAGGCGATCTGTTGGGG |
| Quantitative PCR: NR1D1-Reverse | Wang et al.41 | ATCGTCCGCATCAATCGCAA |
| Quantitative PCR: STX1A-Reverse | Wang et al.41 | GACATGACCTCCACAAACTTTCT |
| Quantitative PCR: KIF5A-Reverse | Wang et al.41 | AAATGGGGATGAACTTGTCTCC |
| Quantitative PCR: RAB3A-Reverse | Wang et al.41 | TGTCGTTGCGATAGATGGTCT |
| Quantitative PCR: LIN7A-Reverse | Wang et al.41 | GGCCTTCACTAGCTGCAAAAG |
| Quantitative PCR: ITGA3-Reverse | Wang et al.41 | CCGAAGTACACAGTGTTCTGG |
| Quantitative PCR: PLXND1-Reverse | Wang et al.41 | GCCCCTTTAGTTGGAGGCT |
| Quantitative PCR: CNTNAP2-Reverse | Wang et al.41 | TCTTGGCATAGCCGGGAGAA |
| Quantitative PCR: AP1S2-Reverse | Wang et al.41 | TGTCCTGATCCTCAATAGCACA |
| Quantitative PCR: NRG1-Reverse | Wang et al.41 | GTCCACTTCCAATCTGTTAGCA |
| Quantitative PCR: ANLN-Reverse | Wang et al.41 | CGCTTAGCATGAGTCATAGACCT |
| Quantitative PCR: WDR62-Reverse | Wang et al.41 | GCCACTACGATGTCTTTCTTCC |
| Quantitative PCR: CDCA2-Reverse | Wang et al.41 | ACATTTCGATACAGTGCAGGG |
| Quantitative PCR: BIRC5-Reverse | Wang et al.41 | AAGTCTGGCTCGTTCTCAGTG |
| Quantitative PCR: PLK1-Reverse | Wang et al.41 | CCGTCATATTCGACTTTGGTTGC |
| Quantitative PCR: PBK-Reverse | Wang et al.41 | GGCTGGCTTTATATCGTTCTTCT |
| Quantitative PCR: KI67-Reverse | Wang et al.41 | GTTATGCGCTTGCGAACCT |
| Quantitative PCR: KIF18A-Reverse | Wang et al.41 | GCTGTTTTGTCTTGTTGTCGC |
| Quantitative PCR: KIF11-Reverse | Wang et al.41 | CCTTCTTACGATCCAGTTTGGAA |
| Quantitative PCR: SKA1-Reverse | Wang et al.41 | TCATGTACGAAGGAACACCATTG |
| Quantitative PCR: ASCL1-Reverse | Wang et al.41 | AAGCCGCTGAAGTTGAGCC |
| Quantitative PCR: PRDM16-Reverse | Wang et al.41 | GGTGAGGTTCTGGTCATCGC |
| Quantitative PCR: HES5-Reverse | Wang et al.41 | TGCTCGATGCTGCTGTTGAT |
| Quantitative PCR: IRX5-Reverse | Wang et al.41 | CCCGTAAGGGTACGATCCCA |
| Quantitative PCR: PROX1-Reverse | Wang et al.41 | CTGGGAAATTATGGTTGCTCCT |
| Quantitative PCR: KLF4-Reverse | Wang et al.41 | CAGGTCCAGGAGATCGTTGAA |
| Quantitative PCR: FOSL2-Reverse | Wang et al.41 | GGTATGGGTTGGACATGGAGG |
| Quantitative PCR: ATF3-Reverse | Wang et al.41 | TTCTTTCTCGTCGCCTCTTTTT |
| Quantitative PCR: DDIT3-Reverse | Wang et al.41 | CTGCTTGAGCCGTTCATTCTC |
| Quantitative PCR: SNAI1-Reverse | Wang et al.41 | AGATGAGCATTGGCAGCGAG |
| Quantitative PCR: RFX3-Reverse | Wang et al.41 | GCACAGTCTGTACCTGCTGTA |
| Quantitative PCR: RFX4-Reverse | Wang et al.41 | TGAGTTAAGCTGTCAGGTAATGC |
| Quantitative PCR: GAPDH-Reverse | Wang et al.41 | TGGAATCATATTGGAACATGTAAACC |
| Recombinant DNA | ||
| pTK-Gaussia Luc Vector | Thermo Scientific | Thermo Scientific Cat# 16148 |
| pTK-Cypridina Luc Vector | Thermo Scientific | Thermo Scientific Cat# 16151 |
| Lenti-dCas9-KRAB-blast | Xie et al.42 | RRID:Addgene_89567 |
| CROP-seq-opti | Hill et al.43 | RRID:Addgene_106280 |
| Lenti-(BB)-EF1a-KRAB-dCas9-P2A-BlastR | Ferrer, J. | RRID:Addgene_118154 |
| Software and algorithms | ||
| Cutadapt Version 2.4 | Martin. | https://cutadapt.readthedocs.io/en/stable/ |
| STAR Version 2.5.4b | Dobin et al.44 | https://github.com/alexdobin/STAR |
| FeatureCounts Version 1.6.4 | Liao et al.45 | https://subread.sourceforge.net/featureCounts.html |
| DESeq2 Version 3.18 | Love et al.46 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| AIAP Version 1.1 | Liu et al.47 | https://github.com/Zhang-lab/ATAC-seq_QC_analysis |
| ChromHMM | Ernst et al.48 | http://compbio.mit.edu/ChromHMM/#:∼:text=ChromHMM%20is%20based%20on%20a,one%20or%20more%20cell%20types. |
| DiffBind Version 3.18 | Ross-Innes et al.49 | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| ChIPSeeker Version 1.32.1 | Yu et al.50 | https://github.com/YuLab-SMU/ChIPseeker |
| DeepTools Version 3.5.4 | Ramírez et al.51 | https://github.com/deeptools/deepTools |
| HOMER Version 4.11 | Heinz et al.52 | http://homer.ucsd.edu/homer/index.html |
| ToppFun Tool | ToppGene Suite | https://toppgene.cchmc.org/ |
| GraphPad Prism Version 9 | GraphPad Software | https://www.graphpad.com |
| Rstudio Version 3.5.1 with R Version 4.2.1 | Rstudio Software | https://www.rstudio.org |
| ImageJ | Schneider et al.53 | https://imagej.nih.gov/ij/ |
| μMANAGER version 2 | Edelstein et al.54 | https://micro-manager.org/ |
| MetaMorph | MetaMorph Software | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy |
| Chemidoc Image Lab Touch Software | Bio-Rad | https://www.bio-rad.com/en-us/product/chemidoc-mp-imaging-system?ID=NINJ8ZE8Z |
| StepOnePlus™ Real-Time PCR System with StepOne Software Version 2.3 | Applied Biosystems | https://www.thermofisher.com/order/catalog/product/4376600 |
| Biotek Synergy 2 | Biotek | 18531 |
| Other | ||
| BrainSpan RNA-seq Data for PCW 13-18 | Kang et al.20 | dbGaP Study Accession: phs000731.v2.p1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kristen Kroll (kkroll@wustl.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
-
•
RNA-seq and CUT&Tag datasets have been deposited in the Gene Expression Omnibus (GEO) repository and are publicly available as of the date of publication (GSE239481). Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines
Work with hESCs was performed in approval and accordance with the Washington University Embryonic Stem Cell Research Oversight Committee (ESCRO) under protocol #12-002. All hESCs were maintained under feeder-free conditions on vitronectin (Gibco™) in mTeSR™ Plus Medium (STEMCELL Technologies), and all experiments were carried out in the H9 hESC line and genetically modified derivatives. All cells were incubated in an incubator with 5% CO2 at 37°C.
Method details
Neuronal differentiation
For specification of hESCs as medial ganglionic eminence (MGE)-like progenitors and differentiation into cortical interneurons, embryoid bodies (EBs) were produced using AggreWell™800 Microwell Culture Plates (STEMCELL Technologies). hESCs were dissociated with Accutase (Life Technologies) and 1.5x106 cells were added per well to an AggreWell™800 Microwell Culture Plate. EBs were formed in AggreWell™ Embryoid Body Formation Medium (STEMCELL Technologies) for 3 days, before differentiation was started at (day (D) 0) by switching to ventral telencephalic neuroectoderm specification media (VSM). VSM contained Neurobasal-A (Life Technologies), B-27 supplement (without Vitamin A; Life Technologies), Glutamax (Life Technologies), β-Mercaptoethanol (Life Technologies), Non-Essential Amino Acids (Life Technologies), Penicillin-Streptomycin Solution (Life Technologies), 10 μM SB-431542 (Selleckchem), 100 nM LDN-193189 (Tocris Biosciences), 100 nM Smoothened Agonist (SAG; Selleckchem), and 2 μM XAV-939 (Selleckchem). For the first 4 days of differentiation media also contained 10 μM Y-27632 (Selleckchem). EBs were maintained on an orbital shaker at 80 rpm from D0 of differentiation.
On D10 of differentiation, EBs were plated onto Matrigel and laminin (5 μg/mL; Sigma) coated plates. At D15 of differentiation, cells were considered MGE-like progenitors, as assessed by marker expression in our prior work4,5 and were replated using accutase and cultured for a further 2 days in VSM before collection for immunofluorescence. For terminal differentiation, neurospheres were formed from MGE-like progenitors using AggreWell™800 Microwell Culture Plates as previously described, in ventral telencephalic neuroectoderm differentiation media (VDM) supplemented with 10 μM Y-27632. VDM contained Neurobasal-A (Life Technologies), B-27 supplement (without Vitamin A; Life Technologies), Glutamax (Life Technologies), β-Mercaptoethanol (Life Technologies), Non-Essential Amino Acids (Life Technologies) and Penicillin-Streptomycin Solution (Life Technologies). Neurospheres were maintained for 2 days in AggreWell™800 Microwell Culture Plates and then were maintained on an orbital shaker for 2 days at 80 rpm. After 19 days of differentiation from hESCs, neurospheres were plated onto Matrigel and laminin (5 μg/mL) coated plates and maintained in VDM supplemented with BDNF (20 ng/mL; PeproTech), with additional medium added 24 hours after plating.
From day 20-40 of differentiation, cultures were fed every 2 days by replacing half of the spent media. On days 22 and 24, media was supplemented with BDNF (20 ng/mL) and CultureOne™ Supplement (Life Technologies) after which media was supplemented with 200 μM ascorbic acid (Sigma-Aldrich), 20 ng/mL BDNF, 10 μM DAPT (Selleckchem) and 2 μM PD0332991 (Selleckchem) until day 30 of differentiation. From day 30-34 VDM was supplemented with 20 ng/mL BDNF, 200 μM ascorbic acid, 200 μM cAMP (Sigma-Aldrich) and 0.5 μM Cytarabine (Tocris Biosciences). Finally, from day 36, cells were fed with maturation media (MM), which was supplemented with 2 μg/mL Laminin on days 40 and 50. MM contained BrainPhys™ Neuronal Medium (Stem Cell Technologies), supplemented with B-27 plus supplement (Life Technologies) and penicillin-streptomycin solution (Life Technologies). Feedings after day 40 were done every 4 days. Cells were harvested at days 0, 15, 35 and 60 for assays, including RNA-sequencing (RNA-seq) and CUT&Tag. All experiments constitute a minimum of 3 biological replicates with each replicate constituting a separate and independent differentiation, with further details on precise numbers of biological replicates performed for each analysis detailed in the figure legends.
For specification of hESCs as dorsally patterned neuronal progenitors EBs were formed as previously described and were then transplanted into dorsal telencephalic neuroectoderm specification media (DSM) supplemented with 10 μM SB-431542, 100 nM LDN-193189, 10 μM Y-27632 and 2 μM XAV-939, constituting D0 of differentiation. DSM contained two-thirds DMEM/F12 (Gibco), one third Neurobasal™ Medium (Gibco), N2 supplement (Life Technologies), 0.5X B-27 supplement (without Vitamin A), Glutamax, β-Mercaptoethanol, Non-Essential Amino Acids and Penicillin-Streptomycin Solution. EBs were fed on D2 and D4 with DSM supplemented with 10 μM SB-431542, 100 nM LDN-193189 and 2 μM XAV-939, and on D6 and D8 with DSM supplemented with 10 μM SB-431542 and 100 nM LDN-193189. EBs were plated onto Matrigel, and laminin (5 μg/mL) coated plates on D10 and were fed every 2 days with un-supplemented DSM. Cells were collected on D20 as dorsally patterned neuronal progenitors for CUT&Tag processing.
CUT&Tag
CUT&Tag was performed on all samples using the protocols.io V.2. protocol,9 using 2-5x105 cells per sample. Nuclei were extracted and then adsorbed onto BioMag Plus Concanavalin beads (Bangs Laboratories), non-specific binding was blocked with 0.1% BSA, and primary antibodies (key resources table) were incubated overnight at 4°C. Secondary antibodies (key resources table) were applied for 30 minutes at room temperature before CUTANA™ pAG-Tn5 for CUT&Tag (Epicypher) was applied for 1 hour at room temperature and then activated and incubated for a further 1 hour at 37°C. Tagmentation was terminated using 0.5 M EDTA (Sigma-Aldrich), Proteinase K (New England Biosciences), and 10% SDS (Sigma-Aldrich) with a 1 hour incubation at 55°C. DNA extraction was performed using 25:24:1 Phenol/Chloroform/Isoamyl Alcohol (Sigma-Aldrich) and purified using the NucleoSpin® Gel and PCR Clean-up (MACHERY-NAGEL) using the manufacturer’s instructions for PCR clean-up. PCR amplification was performed with NEBNext® High-Fidelity 2X PCR Master Mix (New England Biosciences) and primers using i5 and i7 adapter sequences. Post PCR purification was done using AMPure XP Reagent (Beckman Coulter) in two stages, to remove small PCR artifacts and large genomic DNA. First, a 1:2 dilution of AMPure XP Reagent was added to the PCR reaction and then the AMPure XP Reagent was separated and discarded. A second round of purification was performed with a 1.25:1 mixture of AMPure XP Reagent: PCR reaction mix from the previous step and libraries were eluted in Molecular Biology Grade Water (Sigma-Aldrich). The quality of the final libraries was assessed using a 4150 TapeStation System (Agilent) with High Sensitivity D1000 reagents (Agilent). Peaks from 250-750 bp were combined to generate the concentration of each library.
Samples with unique dual-end indexes were pooled in equimolar concentrations and were sequenced by GTAC@MGI on the NovaSeq-6000 sequencer as 150 bp paired-end reads at a depth of ≥10 million (M) reads per sample. Raw reads from CUT&Tag samples were processed by AIAP (v1.1) using human genome hg38 as a reference to perform read quality control, alignment, quantification, and peak calling.47
Trimmed and aligned binary alignment and map files were used as input for ChromHMM clustering48 using IgG CUT&Tag samples as control input files. The LearnModel function was run to generate the model using a 100 bp bin size and 7 desired chromatin states. Changes in chromatin state coverage and occupancy were assessed using the bedtools55 intersectbed function.
Analysis of differential binding from CUT&Tag data was performed using DiffBind,49 using trimmed and aligned binary alignment and map files along with peaks identified by MACS2 within the AIAP (v1.1) workflow. Blacklisted sequences were removed and a minimum overlap of 2 samples was used to generate the consensus peak set for each biological condition. Read depth data was normalized using the reads in peaks library normalization method and significantly differentially bound peaks were identified using DESeq2, with a Benjamini and Hochberg FDR of <0.05. Complete result tables for significant peak changes in signal for each comparison are presented as supplementary tables. Peak sets, including differentially bound peaks, were annotated using ChIPseeker50 and defining the promotor-associated region as an area within 3 kb of the TSS. For downstream analysis only sites within 20 kb of the nearest TSS were used unless otherwise stated. Curated peak sets were visualized using deepTools,51 using the computeMatrix reference-point and plotHeatmap functions. Unless otherwise stated we filtered peaks to only those within 20 kb of the nearest TSS for visualization and downstream analysis.
Finally, TFBS enrichment analysis was performed using HOMER52 (v4.11, 10-24-2019) and the findMotifsGenome function. Peaks were condensed to 200 bp around peak centers and, unless otherwise stated, background data was GC content matched randomly generated sequences generated by HOMER. Raw results from the TFBS enrichment analysis are reported as supplementary tables. The annotatePeaks function of HOMER was also used to determine possible binding sites for TFs of interest within defined peak sets using available position weight matrices for the identified TFs.
RNA-seq
RNA was collected using a NuceloSpin® RNA kit (Macherey-Nagel) and RNA-seq library preparation was performed using the SMARTer Ultra Low RNA kit (Takara-Clontech). Reads were obtained using either an Illumina Hi-Seq 2500 or Illumina NovaSeq-6000 sequencer, to a depth of ∼30 million (M) reads per sample. Library preparation and sequencing was performed by the Washington University Genome Technology Access Center (GTAC) @McDonnell Genome Institute (MGI).
Raw reads from RNA-seq samples were quality trimmed using cutadapt (v2.4) with the options of quality-cutoff=15,10 minimum-length=36.56 Reads were aligned with STAR (v2.5.4b) using human genome hg38 as a reference with GENCODE V27 annotations.44 Reads were quantified at the gene level using featureCounts (v1.6.4) from the subread package.45 For differential expression analysis, only genes with >5 counts in >2 samples were included. Differential expression analysis was performed with DESeq2 in negative binomial mode, using counts for genes that passed the cutoff.46 A 1.5-fold linear expression change and a Benjamini and Hochberg false discovery rate of < 0.05 (FDR; adjusted p-value) were set as cutoff values for significance. Raw results from each DESeq2 analysis are presented as supplementary datasets. Expression data is presented and visualized using reads per kilobase of transcript per million mapped reads (RPKM) and comparisons to human fetal brain data were performed using study data downloaded from the dbGaP web site, under dbGaP accession phs000731.v2.p1 and processed as detailed above.
Gene ontology and pathway analysis
Gene ontology enrichment and pathway analysis was done using the ToppFun tool from the ToppGene suite57 and FDR adjusted p-values were generated using the probability density function methodology. Complete results for each gene set are presented as supplementary tables with manually selected results shown in figures representing FDR adjusted p-values.
Luciferase assays
Genomic DNA was isolated from D0 hESCs using the PureLink™ Genomic DNA Mini Kit (Invitrogen™) and 0.4-1.5kb fragments were cloned from the isolated DNA using primers surrounding putative CREs (key resources table) using Phusion™ High-Fidelity DNA Polymerase (Thermo Scientific™). Desired fragments were identified by gel electrophoresis and isolated using the Monarch® DNA Gel Extraction Kit (NEB). Purified fragments were cloned into pTK-Gaussia Luc Vector (Thermo Scientific™) using XhoI and HindIII restriction enzyme sites with successful cloning confirmed by colony PCR. Plasmids were heat shocked into NEB® Stable Competent E. coli and isolated using the PureLink™ HiPure Plasmid Midiprep Kit (Invitrogen™). pTK-Gaussia plasmids were transfected along with pTK-Cypridina Luc Vector at equal concentrations into either ventrally patterned NPCs or embryonic stem cells using Lipofectamine™ 3000 Transfection Reagent (Invitrogen™). Supernatant was collected 48-, 72- and 96-hours post transfection and luciferase signal from Cypridina luciferase was measured using Pierce™ Cypridina Luciferase Flash Assay Kit (Thermo Scientific™) and Gaussia luciferase was measured using Pierce™ Gaussia Luciferase Flash Assay Kit (Thermo Scientific™) on a Synergy2 instrument. All signals from Gaussia luciferase were normalized to signal from Cypridina luciferase from the cell sample and samples taken 48-, 72- and 96-hours post transfection from the same sample were averaged as technical replicates. Results are presented as signal relative to unmodified pTK-Gaussia (control).
Reverse Transcription and Quantitative PCR (RT-qPCR)
RNA was isolated using the NucleoSpin RNA kit (Macherey–Nagel) and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific) with ≥1 μg used to make cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Equal quantities of cDNA were used as template for the quantitative PCR (qPCR) using the Applied Biosystem StepOne Plus quantitative PCR system. GAPDH was used as an endogenous quantitative control and all primers used can be found in the key resources table. Data were generated from a minimum of 4 biological replicate samples, each of which consisted of 3 technical replicates. All statistics were performed using abundance relative to endogenous control, and data is presented as fold change by comparison with appropriate control.
Western blotting
Protein was isolated at D12 or D15 of differentiation in RIPA buffer (Abcam) with Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific) and western blotted using Bolt™ 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gels (Invitrogen) then was transferred to 0.2 μm nitrocellulose membrane (Bio-Rad). Nonspecific antibody binding was blocked by 5% milk solution, and primary antibodies (key resources table) were incubated with free floating membranes overnight at 4°C. Secondary antibodies were HRP conjugated Goat anti-Rabbit IgG and Goat anti-Mouse IgG (Invitrogen), each used at a 1:10,000 dilution. Visualization was performed by chemiluminescence using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Scientific), all band intensities were normalized to GAPDH loading controls, and a minimum of 3 biological replicates were averaged before statistical analysis. Statistical analysis was performed on normalized signal intensity, data is presented as fold change compared to appropriate controls, and complete blots are presented in supplementary figures.
Sphere size and outgrowth
Images were taken at from D0 to D12 of differentiation, using a 4x objective, of 20 neurospheres per biological replicate across 4 biological replicates per condition. Sphere size and outgrowth was measured using ImageJ and outgrowth is defined as the area of cells outgrown from the plated neurosphere normalized to corresponding sphere size.
Immunofluorescence
Cells were fixed in 4% Paraformaldehyde (Sigma-Aldrich) at D17 of differentiation before blocking by using 5% donkey serum (Sigma-Aldrich) and 0.1% Tween20 (Sigma-Aldrich). Primary antibodies (key resources table) were added overnight at 4°C. Cells were washed and incubated with secondary antibodies (donkey anti-mouse 555 and donkey anti-rabbit 488, Thermo Scientific) for 1 hour at room temperature. Cells were counterstained and mounted using ProLong™ Glass Antifade Mountant with NucBlue™ Stain (Invitrogen). Images were obtained using a spinning-disk confocal microscope, using MetaMorph software and image processing with ImageJ. Quantification was performed using a minimum of 3 random images taken from 3 independent biological replicates. Quantification was performed using CellProfiler,58 requiring a minimum of 60% overlap between immunofluorescence signal and DAPI staining for co-localization to be acknowledged.
Modeling RFX3 and RFX4 knockdown
To model RFX3 and RFX4 deficiency, CRISPRi lines were created by using H9 hESCs transduced with either dCas-KRAB (via Lenti-dCas9-KRAB-blast) and gRNAs (via CROP-seq-opti) for RFX3 or RFX4 or dCas9-KRAB and gRNA in a single vector (Lenti-(BB)-EF1a-KRAB-dCas9-P2A-BlastR). In both cases gRNAs for RFX3 and RFX4 were selected from the Dolcetto library of gRNAs40 at random. CROP-seq-opti was a gift from Jay Shendure43 (Addgene plasmid #106280), Lenti-dCas9-KRAB-blast was a gift from Gary Hon42 (Addgene plasmid #89567) and Lenti-(BB)-EF1a-KRAB-dCas9-P2A-BlastR was a gift from Jorge Ferrer (Addgene plasmid # 118154). Selected gRNAs (key resources table) were cloned into CROP-seq-opti or Lenti-(BB)-EF1a-KRAB-dCas9-P2A-BlastR using BsmBI (NEB). For transductions of all plasmids, lentivirus was produced in Lenti-X-293T cells (Takara Bio), by transfecting the cells with envelope (pMD2.G), packaging (psPAX2) and transfer plasmid at a ratio of 2:3:5 using Trans-IT Lenti transfection reagent (Mirus). pMD2.G and psPAX2 were gifts from Didier Trono (Addgene plasmids #12259 and #12260, respectively). Lenti-X-293T cells were grown in HEK media containing DMEM, high glucose, no glutamine (Gibco), supplemented with 10% FBS (Gibco), Glutamax, β-Mercaptoethanol, Non-Essential Amino Acids and Penicillin-Streptomycin Solution, Sodium Pyruvate (Life Technologies) and HEPES buffer (Life Technologies). Lentivirus was concentrated using Lenti-X-Concentrator (Takara Bio), as per the manufacturer's instructions, and was resuspended in Opti-MEM media (Gibco) and stored at -80°C. Transduction of hESCs was aided by the addition of 5 μg/mL polybrene (Sigma-Aldrich) immediately before the addition of lentivirus. Cells were allowed to recover for 48 hours before desired selection was performed by using 1 μg/mL blasticidin (Sigma-Aldrich) and 500 ng/mL puromycin (Life Technologies). Once cells were selected, they were maintained under selection for the duration of the required experiments.
Quantification and statistical analysis
Where appropriate, statistical analysis was carried out using a combination of GraphPad Prism version 9 (GraphPad Software; La Jolla, CA, USA, available from www.graphpad.com) and RStudio version 3.5.1 (RStudio: Integrated development environment for R; Boston, MA, USA. Available from www.rstudio.org). All technical replicates were averaged before statistical analysis and statistical tests used for each data analysis are detailed in the figure legends or in the method details section for specific analysis paradigms including differential gene expression and differential binding analysis. A minimum of 3 differentiations were used for each time point or biological condition with the number of differentiations used for each sample listed in figure legends as N. The results in Figures were presented as group mean and standard error (SE). Statistical significance is indicated with asterisks as follows: ns, no significance; ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
Acknowledgments
This work was supported by NIH grants R01NS114551, R01MH124808, and R01HD110556 to K.L.K., and by NIH P50HD103525 to Joseph Dougherty and Christina Gurnett (K.L.K. is project PI for the Human Cellular Models Unit of the Washington University Intellectual and Developmental Disabilities Research Center, with project effort funded by this grant). Foundations supporting this work included the Children’s Discovery Institute, Engelhart Family Foundation, Simons Foundation, M-CM Network, and WU Institute for Clinical and Translational Sciences to K.L.K.
Author contributions
G.C. and K.L.K. designed the study. G.C., K.K., and R.P. performed differentiation experiments for CUT&Tag and RNA-seq analysis, G.C. and J.D. performed the RFX3 and RFX4 knockdown experiments, H.J. generated immunofluorescence image data, and G.C performed the bioinformatic analyses. G.C. and J.D. analyzed the data and generated figures and G.C. and K.L.K. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 11, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109967.
Supplemental information
(A) Average expression (RPKM) of all human genes at D0, D15, D35, and D60 of cortical interneuron differentiation. Differential gene expression analysis comparing (B) D0–D15, (C) D0–D35, (D) D0–D60, (E) D15–D35, (F) D15–D60, and (G) D35–D60.
Results of gene ontology analysis on (A) D0, (B) D15, (C) D35, and (D) D60 specific genes. Identification of (E) lineage and (F) MGE specific transcription factors.
Results of differential H3K27ac binding analysis from (A) D0–D15 of cortical interneuron differentiation (B) and subsequent gene ontology analysis of genes associated with sites of increased H3K27ac signal at D15. (C) Clustering of genes associated with sites of increased H3K27ac signal at D15 based on gene expression changes during cortical interneuron differentiation and (D) the associated gene ontology analysis of cluster 2 genes and (E) TFBS enrichment analysis of peaks associated with cluster 2 genes.
(A) Results of differential H3K27ac binding analysis comparing dorsally and ventrally patterned neuronal progenitors and (B) subsequent gene ontology analysis of genes associated with sites of increased H3K27ac signal in ventrally patterned neuronal progenitors compared to dorsally patterned neuronal progenitors and (C) TFBS enrichment analysis of peaks associated with those genes.
Results of differential H3K27ac binding analysis from (A) D15–D35 of cortical interneuron differentiation and (B) clustering of genes associated with sites of increased H3K27ac signal at D35 based on gene expression changes during cortical interneuron differentiation with (C) subsequent gene ontology analysis of cluster 2 genes and (D) TFBS enrichment analysis of peaks associated with cluster 2 genes.
(A) Results of differential H3K9me3 binding analysis comparing dorsally and ventrally patterned neuronal progenitors and (B) differential H3K9me3 binding analysis from D0–D15 of cortical interneuron differentiation. (C) Subsequent gene ontology analysis of genes associated with sites of increased H3K9me3 signal at D15 and in ventrally patterned neuronal progenitors.
Results of (A) differential H3K9me3 binding analysis from D15-D35 of cortical interneuron differentiation and (B) clustering of genes associated with sites of increased H3K9me3 signal at D35, based on gene expression changes during cortical interneuron differentiation. Subsequent gene ontology analysis of (C) cluster 3 genes and (D) cluster 5 genes and (E) TFBS enrichment analysis of peaks associated with cluster 5 genes.
(A) Annotation of the instances of each TFBS in putative CREs for 25 candidate TFBSs (nd = no data) and (B) correlogram of the co-occurrence of TFBSs within CREs, indicated as a percentage of CREs with each TFBS. (C) Gene ontology analysis of genes associated with a putative CRE that contains either an RFX3 or RFX4 TFBS.
(A) Differential gene expression analysis of KRAB control versus RFX3-G1 KD in D15 ventrally patterned neuronal progenitors, with gene ontology analysis of genes significantly (B) up- or (C) downregulated in this comparison. (D) Differential gene expression analysis of KRAB control versus RFX4-G1 KD in D15 ventrally patterned neuronal progenitors, with gene ontology analysis of genes significantly (E) up- or (F) downregulated in this comparison. (G) Gene ontology analysis of genes with significant expression changes with the opposite directionality in the RFX3-G1 versus RFX4-G1 KD, with DEGs for each KD defined relative to the KRAB controls. (H) Peaks associated with genes differentially regulated in the RFX3-G1 KD versus RFX4-G1 KD models (with DEGs for each KD defined relative to the KRAB controls), and which showed significantly increased H3K27ac signal at D15 compared to D0 of cortical interneuron differentiation. (I) Gene expression data for differentially expressed TFs with opposite gene expression changes in RFX3 and RFX4 KD models and (J) gene expression changes during our differentiation time course for the subset of those TFs with increased expression in RFX4 and decreased expression in RFX3 KD models.
References
- 1.Wonders C.P., Anderson S.A. The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- 2.Filice F., Janickova L., Henzi T., Bilella A., Schwaller B. The Parvalbumin Hypothesis of Autism Spectrum Disorder. Front. Cell. Neurosci. 2020;14 doi: 10.3389/fncel.2020.577525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang J., Yang X., Tang K. Interneuron development and dysfunction. FEBS J. 2022;289:2318–2336. doi: 10.1111/febs.15872. [DOI] [PubMed] [Google Scholar]
- 4.Meganathan K., Lewis E.M.A., Gontarz P., Liu S., Stanley E.G., Elefanty A.G., Huettner J.E., Zhang B., Kroll K.L. Regulatory networks specifying cortical interneurons from human embryonic stem cells reveal roles for CHD2 in interneuron development. Proc. Natl. Acad. Sci. USA. 2017;114:E11180–E11189. doi: 10.1073/pnas.1712365115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis E.M.A., Chapman G., Kaushik K., Determan J., Antony I., Meganathan K., Narasimhan M., Gontarz P., Zhang B., Kroll K.L. Regulation of human cortical interneuron development by the chromatin remodeling protein CHD2. Sci. Rep. 2022;12 doi: 10.1038/s41598-022-19654-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maroof A.M., Keros S., Tyson J.A., Ying S.W., Ganat Y.M., Merkle F.T., Liu B., Goulburn A., Stanley E.G., Elefanty A.G., et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inglis G.A.S., Zhou Y., Patterson D.G., Scharer C.D., Han Y., Boss J.M., Wen Z., Escayg A. Transcriptomic and epigenomic dynamics associated with development of human iPSC-derived GABAergic interneurons. Hum. Mol. Genet. 2020;29:2579–2595. doi: 10.1093/hmg/ddaa150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wittkopp P.J., Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011;13:59–69. doi: 10.1038/nrg3095. [DOI] [PubMed] [Google Scholar]
- 9.Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A., et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H., Fan Z., Shliaha P.V., Miele M., Hendrickson R.C., Jiang X., Helin K. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature. 2023;615:339–348. doi: 10.1038/s41586-023-05780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Priego C., Hu R., Boshans L.L., Lalli M., Janas J.A., Williams S.E., Dong Z., Yang N. Mapping cis-regulatory elements in human neurons links psychiatric disease heritability and activity-regulated transcriptional programs. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.110877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayhan F., Konopka G. Regulatory genes and pathways disrupted in autism spectrum disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2019;89:57–64. doi: 10.1016/j.pnpbp.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eshraghi A.A., Liu G., Kay S.I.S., Eshraghi R.S., Mittal J., Moshiree B., Mittal R. Epigenetics and Autism Spectrum Disorder: Is There a Correlation? Front. Cell. Neurosci. 2018;12:78. doi: 10.3389/fncel.2018.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirzaa G.M., Chong J.X., Piton A., Popp B., Foss K., Guo H., Harripaul R., Xia K., Scheck J., Aldinger K.A., et al. De novo and inherited variants in ZNF292 underlie a neurodevelopmental disorder with features of autism spectrum disorder. Genet. Med. 2020;22:538–546. doi: 10.1038/s41436-019-0693-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris H.K., Nakayama T., Lai J., Zhao B., Argyrou N., Gubbels C.S., Soucy A., Genetti C.A., Suslovitch V., Rodan L.H., et al. Disruption of RFX family transcription factors causes autism, attention-deficit/hyperactivity disorder, intellectual disability, and dysregulated behavior. Genet. Med. 2021;23:1028–1040. doi: 10.1038/s41436-021-01114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos-Terra J., Deckmann I., Fontes-Dutra M., Schwingel G.B., Bambini-Junior V., Gottfried C. Transcription factors in neurodevelopmental and associated psychiatric disorders: A potential convergence for genetic and environmental risk factors. Int. J. Dev. Neurosci. 2021;81:545–578. doi: 10.1002/jdn.10141. [DOI] [PubMed] [Google Scholar]
- 19.Chapman G., Alsaqati M., Lunn S., Singh T., Linden S.C., Linden D.E.J., van den Bree M.B.M., Ziller M., Owen M.J., Hall J., et al. Using induced pluripotent stem cells to investigate human neuronal phenotypes in 1q21.1 deletion and duplication syndrome. Mol. Psychiatry. 2022;27:819–830. doi: 10.1038/s41380-021-01182-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M.M., Pletikos M., Meyer K.A., Sedmak G., et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrari F., Arrigoni L., Franz H., Izzo A., Butenko L., Trompouki E., Vogel T., Manke T. DOT1L-mediated murine neuronal differentiation associates with H3K79me2 accumulation and preserves SOX2-enhancer accessibility. Nat. Commun. 2020;11:5200. doi: 10.1038/s41467-020-19001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meshorer E., Yellajoshula D., George E., Scambler P.J., Brown D.T., Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Efroni S., Duttagupta R., Cheng J., Dehghani H., Hoeppner D.J., Dash C., Bazett-Jones D.P., Le Grice S., McKay R.D.G., Buetow K.H., et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437–447. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma A., Klein S.S., Barboza L., Lohdi N., Toth M. Principles Governing DNA Methylation during Neuronal Lineage and Subtype Specification. J. Neurosci. 2016;36:1711–1722. doi: 10.1523/JNEUROSCI.4037-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pla R., Stanco A., Howard M.A., Rubin A.N., Vogt D., Mortimer N., Cobos I., Potter G.B., Lindtner S., Price J.D., et al. Dlx1 and Dlx2 Promote Interneuron GABA Synthesis, Synaptogenesis, and Dendritogenesis. Cereb. Cortex. 2018;28:3797–3815. doi: 10.1093/cercor/bhx241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Azim E., Jabaudon D., Fame R.M., Macklis J.D. SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat. Neurosci. 2009;12:1238–1247. doi: 10.1038/nn.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nobrega-Pereira S., Kessaris N., Du T., Kimura S., Anderson S.A., Marin O. Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron. 2008;59:733–745. doi: 10.1016/j.neuron.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sandberg M., Flandin P., Silberberg S., Su-Feher L., Price J.D., Hu J.S., Kim C., Visel A., Nord A.S., Rubenstein J.L.R. Transcriptional Networks Controlled by NKX2-1 in the Development of Forebrain GABAergic Neurons. Neuron. 2016;91:1260–1275. doi: 10.1016/j.neuron.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandberg M., Taher L., Hu J., Black B.L., Nord A.S., Rubenstein J.L.R. Genomic analysis of transcriptional networks directing progression of cell states during MGE development. Neural Dev. 2018;13:21. doi: 10.1186/s13064-018-0119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tagne J.B., Gupta S., Gower A.C., Shen S.S., Varma S., Lakshminarayanan M., Cao Y., Spira A., Volkert T.L., Ramirez M.I. Genome-wide analyses of Nkx2-1 binding to transcriptional target genes uncover novel regulatory patterns conserved in lung development and tumors. PLoS One. 2012;7 doi: 10.1371/journal.pone.0029907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Platzer K., Cogné B., Hague J., Marcelis C.L., Mitter D., Oberndorff K., Park S.M., Ploos van Amstel H.K., Simonic I., van der Smagt J.J., et al. Haploinsufficiency of CUX1 Causes Nonsyndromic Global Developmental Delay With Possible Catch-up Development. Ann. Neurol. 2018;84:200–207. doi: 10.1002/ana.25278. [DOI] [PubMed] [Google Scholar]
- 32.Liu X., Novosedlik N., Wang A., Hudson M.L., Cohen I.L., Chudley A.E., Forster-Gibson C.J., Lewis S.M.E., Holden J.J.A. The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur. J. Hum. Genet. 2009;17:228–235. doi: 10.1038/ejhg.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tolchin D., Yeager J.P., Prasad P., Dorrani N., Russi A.S., Martinez-Agosto J.A., Haseeb A., Angelozzi M., Santen G.W.E., Ruivenkamp C., et al. De Novo SOX6 Variants Cause a Neurodevelopmental Syndrome Associated with ADHD, Craniosynostosis, and Osteochondromas. Am. J. Hum. Genet. 2020;106:830–845. doi: 10.1016/j.ajhg.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snijders Blok L., Kleefstra T., Venselaar H., Maas S., Kroes H.Y., Lachmeijer A.M.A., van Gassen K.L.I., Firth H.V., Tomkins S., Bodek S., et al. De Novo Variants Disturbing the Transactivation Capacity of POU3F3 Cause a Characteristic Neurodevelopmental Disorder. Am. J. Hum. Genet. 2019;105:403–412. doi: 10.1016/j.ajhg.2019.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cubelos B., Sebastián-Serrano A., Kim S., Redondo J.M., Walsh C., Nieto M. Cux-1 and Cux-2 control the development of Reelin expressing cortical interneurons. Dev. Neurobiol. 2008;68:917–925. doi: 10.1002/dneu.20626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Batista-Brito R., Rossignol E., Hjerling-Leffler J., Denaxa M., Wegner M., Lefebvre V., Pachnis V., Fishell G. The cell-intrinsic requirement of Sox6 for cortical interneuron development. Neuron. 2009;63:466–481. doi: 10.1016/j.neuron.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benadiba C., Magnani D., Niquille M., Morlé L., Valloton D., Nawabi H., Ait-Lounis A., Otsmane B., Reith W., Theil T., et al. The ciliogenic transcription factor RFX3 regulates early midline distribution of guidepost neurons required for corpus callosum development. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu P., Morrison J.P., Foley J.F., Stumpo D.J., Ward T., Zeldin D.C., Blackshear P.J. Conditional ablation of the RFX4 isoform 1 transcription factor: Allele dosage effects on brain phenotype. PLoS One. 2018;13 doi: 10.1371/journal.pone.0190561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang D., Zeldin D.C., Blackshear P.J. Regulatory factor X4 variant 3: a transcription factor involved in brain development and disease. J. Neurosci. Res. 2007;85:3515–3522. doi: 10.1002/jnr.21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanson K.R., Hanna R.E., Hegde M., Donovan K.F., Strand C., Sullender M.E., Vaimberg E.W., Goodale A., Root D.E., Piccioni F., Doench J.G. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018;9:5416. doi: 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X., Spandidos A., Wang H., Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012;40:D1144–D1149. doi: 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie S., Duan J., Li B., Zhou P., Hon G.C. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell. 2017;66:285–299.e5. doi: 10.1016/j.molcel.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 43.Hill A.J., McFaline-Figueroa J.L., Starita L.M., Gasperini M.J., Matreyek K.A., Packer J., Jackson D., Shendure J., Trapnell C. On the design of CRISPR-based single-cell molecular screens. Nat. Methods. 2018;15:271–274. doi: 10.1038/nmeth.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Y., Smyth G.K., Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S., Li D., Lyu C., Gontarz P.M., Miao B., Madden P.A.F., Wang T., Zhang B. AIAP: A Quality Control and Integrative Analysis Package to Improve ATAC-seq Data Analysis. Dev. Reprod. Biol. 2021;19:641–651. doi: 10.1016/j.gpb.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R., et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu G., Wang L.G., He Q.Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- 51.Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Edelstein A.D., Tsuchida M.A., Amodaj N., Pinkard H., Vale R.D., Stuurman N. Advanced methods of microscope control using muManager software. J. Biol. Methods. 2014;1 doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 57.Chen J., Bardes E.E., Aronow B.J., Jegga A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stirling D.R., Swain-Bowden M.J., Lucas A.M., Carpenter A.E., Cimini B.A., Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinf. 2021;22:433. doi: 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Average expression (RPKM) of all human genes at D0, D15, D35, and D60 of cortical interneuron differentiation. Differential gene expression analysis comparing (B) D0–D15, (C) D0–D35, (D) D0–D60, (E) D15–D35, (F) D15–D60, and (G) D35–D60.
Results of gene ontology analysis on (A) D0, (B) D15, (C) D35, and (D) D60 specific genes. Identification of (E) lineage and (F) MGE specific transcription factors.
Results of differential H3K27ac binding analysis from (A) D0–D15 of cortical interneuron differentiation (B) and subsequent gene ontology analysis of genes associated with sites of increased H3K27ac signal at D15. (C) Clustering of genes associated with sites of increased H3K27ac signal at D15 based on gene expression changes during cortical interneuron differentiation and (D) the associated gene ontology analysis of cluster 2 genes and (E) TFBS enrichment analysis of peaks associated with cluster 2 genes.
(A) Results of differential H3K27ac binding analysis comparing dorsally and ventrally patterned neuronal progenitors and (B) subsequent gene ontology analysis of genes associated with sites of increased H3K27ac signal in ventrally patterned neuronal progenitors compared to dorsally patterned neuronal progenitors and (C) TFBS enrichment analysis of peaks associated with those genes.
Results of differential H3K27ac binding analysis from (A) D15–D35 of cortical interneuron differentiation and (B) clustering of genes associated with sites of increased H3K27ac signal at D35 based on gene expression changes during cortical interneuron differentiation with (C) subsequent gene ontology analysis of cluster 2 genes and (D) TFBS enrichment analysis of peaks associated with cluster 2 genes.
(A) Results of differential H3K9me3 binding analysis comparing dorsally and ventrally patterned neuronal progenitors and (B) differential H3K9me3 binding analysis from D0–D15 of cortical interneuron differentiation. (C) Subsequent gene ontology analysis of genes associated with sites of increased H3K9me3 signal at D15 and in ventrally patterned neuronal progenitors.
Results of (A) differential H3K9me3 binding analysis from D15-D35 of cortical interneuron differentiation and (B) clustering of genes associated with sites of increased H3K9me3 signal at D35, based on gene expression changes during cortical interneuron differentiation. Subsequent gene ontology analysis of (C) cluster 3 genes and (D) cluster 5 genes and (E) TFBS enrichment analysis of peaks associated with cluster 5 genes.
(A) Annotation of the instances of each TFBS in putative CREs for 25 candidate TFBSs (nd = no data) and (B) correlogram of the co-occurrence of TFBSs within CREs, indicated as a percentage of CREs with each TFBS. (C) Gene ontology analysis of genes associated with a putative CRE that contains either an RFX3 or RFX4 TFBS.
(A) Differential gene expression analysis of KRAB control versus RFX3-G1 KD in D15 ventrally patterned neuronal progenitors, with gene ontology analysis of genes significantly (B) up- or (C) downregulated in this comparison. (D) Differential gene expression analysis of KRAB control versus RFX4-G1 KD in D15 ventrally patterned neuronal progenitors, with gene ontology analysis of genes significantly (E) up- or (F) downregulated in this comparison. (G) Gene ontology analysis of genes with significant expression changes with the opposite directionality in the RFX3-G1 versus RFX4-G1 KD, with DEGs for each KD defined relative to the KRAB controls. (H) Peaks associated with genes differentially regulated in the RFX3-G1 KD versus RFX4-G1 KD models (with DEGs for each KD defined relative to the KRAB controls), and which showed significantly increased H3K27ac signal at D15 compared to D0 of cortical interneuron differentiation. (I) Gene expression data for differentially expressed TFs with opposite gene expression changes in RFX3 and RFX4 KD models and (J) gene expression changes during our differentiation time course for the subset of those TFs with increased expression in RFX4 and decreased expression in RFX3 KD models.
Data Availability Statement
-
•
RNA-seq and CUT&Tag datasets have been deposited in the Gene Expression Omnibus (GEO) repository and are publicly available as of the date of publication (GSE239481). Accession numbers are listed in the key resources table. All other data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.