

Abstract
The molecular pathogenesis of diabetes is multifactorial, involving genetic predisposition and environmental factors that are not yet fully understood. However, pancreatic β-cell failure remains among the primary reasons underlying the progression of type-2 diabetes (T2D) making targeting β-cell dysfunction an attractive pathway for diabetes treatment. To identify genetic contributors to β-cell dysfunction, we investigated single-cell gene expression changes in β-cells from healthy (C57BL/6J) and diabetic (NZO/HlLtJ) mice fed with normal or high-fat, high-sugar diet (HFHS). Our study presents an innovative integration of the causal network perturbation assessment (ssNPA) framework with meta-cell transcriptome analysis to explore the genetic underpinnings of type-2 diabetes (T2D). By generating a reference causal network and in silico perturbation, we identified novel genes implicated in T2D and validated our candidates using the Knockout Mouse Phenotyping (KOMP) Project database.
Keywords: ssNPA, meta-cell, type-2 diabetes, β-cell, causal network perturbation
Introduction
Type 2 diabetes (T2D) comprises more than 90% of all diabetes cases, impacting approximately 6.8% (537 million individuals) of the global population in the year 2021 (Zheng et al., 2018; Sun et al., 2022). T2D is a multifactorial disease caused by a complex interplay between genetic and environmental factors. However, while some of the major environmental factors (i.e., diet and physical activity) are well known, the genetic bases of T2D remain poorly understood. The endocrine portion of the pancreas is constituted by highly specialized hormone-secreting entities known as islets of Langerhans. The islets comprise β, α, delta, PP, and ghrelin cells that secrete insulin, glucagon, somatostatin, pancreatic polypeptide, and ghrelin, respectively. Islet and/or β-cell dysfunctions are central to diabetes, and the onset of full-blown T2D occurs when α or β cells lose their capacity to secrete appropriate amounts of insulin in response to elevated blood glucose levels. Current research on Type 2 diabetes faces significant limitations due to the variability in genetic, environmental, and lifestyle factors among diverse populations, which challenges the generalizability of findings. Additionally, there is a notable gap in long-term clinical trials that comprehensively assess the efficacy of emerging treatments across different stages of the disease. In recent years, single-cell RNA sequencing (scRNA-seq) has proven critical for investigating comprehensive gene expression profiles, revealing the presence of heterogeneous gene expression patterns, even within cells of the same type. Furthermore, diverse phenotypes of pancreatic β cells have been observed within a single islet (Hrovatin et al.,2023, Camunas-Soler et al., 2020; Bhakti et al., 2019). The application of scRNA-seq has significantly contributed to our understanding of β-cell maturation, β-cell heterogeneity, β-cell failure, and β-cell function in both healthy and diseased states. For this study, we used the polygenic NZO/HlLtJ mouse strain that displays signs of morbid obesity, fasting hyperglycemia, hyperinsulinemia, insulin resistance, and hypercholesterolemia resembling human T2D (Leiter et al., 1998; Ortlepp et al.,2000; Reifsnyder et al.,2002). Mice from the NZO/HlLtJ strain and healthy C57BL/6J controls were kept on a standard or high-fat high-sugar (HFHS) diet regimen for XY days. At the end of the treatment, we analyzed the single-cell gene expression profile of β-cells derived from NZO/HlLtJ mice and compared it to that of β-cells from healthy C57BL/6J mice (Figure 1A). To explore the genetic underpinnings of T2D, it is crucial to identify gene perturbations and hub genes associated with the disease. We hypothesize that conventional differential gene expression analysis does not effectively detect certain type of disruptions in gene networks. To overcome this limitation, we extend a causal network perturbation assessment (ssNPA) framework (Buschur, Chikina, & Benos, 2020) in combination with a meta-cell transcriptome analysis. This approach, termed meta-ssNPA workflow, allowed us to identify genes and gene networks that are perturbed in T2D mice in response to diet changes, providing valuable insights into its genetic landscape. Briefly, this study aimed to juxtapose three distinct physiological states, encompassing a spectrum from health to disease conditions, specifically: 1) a comparison between healthy and prediabetic states (C57BL/6J mice on a normal chow diet vs. C57BL/6J on a HFHS diet); 2) a comparison between prediabetic and severely diabetic states (comparing C57BL/6J mice on a HFHS diet with NZO/ShiLt mice on a HFHS diet); and 3) a comparison between mildly diabetic and severely diabetic states (evaluating NZO/ShiLt mice on normal or HFHS diet conditions). We focused our analysis on the transcriptional profile of pancreatic beta cells due to their pivotal role in upholding glucose homeostasis and serving as the primary source of insulin.
Figure 1: Meta-ssNPA framework workflow.

A) Mouses are treated with different diets: High Fat, High Sugar (HFHS) group and Normal diet group. Single-cell data are collected respectively. B) Detailed illustration of Meta-ssNPA framework.
We successfully detected novel T2D genes that is not differentially expressed and validated them with Knockout Mouse Phenotyping (KOMP) Project database. The KOMP database, accessible at the International Mouse Phenotyping Consortium website, serves as a valuable resource for researchers, offering comprehensive phenotypic data and genetic insights on a wide array of knockout mouse models. This platform facilitates the understanding of gene function and disease mechanisms and is instrumental in advancing the study of human diseases, including the identification of potential therapeutic targets.
Material & Methods
Animal studies and islet isolation
In strict adherence to the standards set by the Association for Assessment and Accreditation of Laboratory Animal Care, our facility at The Jackson Laboratory has upheld the care and treatment of mice. We acquired male and female mice from three distinct strains: C57BL/6J (B6; RRID:IMSR_JAX:000664), and NZO/HlLtJ (NZO; RRID:IMSR_JAX:002105), starting at the age of four weeks. The mice were provided with two types of diets from Research Diets: a high-fat, high-sucrose diet (HFHS, comprising 44% kcal from fat and 1360 kcal from sucrose; Research Diets D19070208) and a control diet (10% kcal from fat and devoid of sucrose; Research Diets D19072203), both containing equal fiber content. The diets were given ad libitum starting from the age of six weeks. At the age of fifteen weeks islet isolation was performed and mice were euthanized through cervical dislocation. and the common bile duct at the Sphincter of Oddi was clamped. Collagenase solution (three milliliters of a solution containing collagenase P (5 units/ml) and DNaseI (1mg/ml) in Hank’s Balanced Salt Solution (HBSS) was inserted into the bile duct proximal to the final bifurcation leading to the liverto inflate the pancreas. We then removed the pancreas for digestion at 37°C for 40 minutes. Post-digestion, samples were agitated for ten seconds, diluted it with 10 ml of HBSS, and centrifuged for 3 minutes at 300 RPM. After two washes with HBSS, the pellet was resuspended in 5ml HBSS, and handpicked islets were collected using a clean petri dish containing HBSS. We. The islets were then transferred to 24-well plates with 1ml of the warmed media (containing RPMI 1640, 10% FBS, glutamine, and HEPES) and incubated overnight at 37°C. After overnight incubation islets were centrifuged and the supernatant was discarded. Finally, islets were resuspended in 1–2 ml of StemPro Accutase dissociation solution (A1110501, Fisher Scientific), which had been preheated to 37°C. The cell suspension was gently pipetted for 30 seconds to facilitate the dissociation of islets until the media appeared translucent and there were no visible clumps, usually within 2–5 minutes. Following dissociation, 2-3ml of RPMI complete medium was added, and the cell suspension was filtered through a 20um strainer. The cells were then centrifuged at 230 RCF for 3 minutes, the supernatant was removed, and the cells were resuspended in 2-3ml of RPMI complete medium.
Preprocessing of Single Cell RNA-Seq
We processed raw fastq reads from Illumina sequencing for scRNA-Seq by aligning them to the mouse reference genome (mm10/GRCm38) using 10X Cell Ranger version 6.1.1 with standard settings.
To demultiplex the strain of origin from samples containing mixed strains, we employed demuxlet version 2 (https://github.com/statgen/popscle;(Kang et al., 2018)), utilizing known genomic variations from VCF files obtained from the Sanger Mouse Genomes Project ((Keane et al., 2011)). We focused on sites that were biallelic and varied among our two strains (B6, and NZO). We ran demuxlet on RNA-Seq data with specific parameters “--alpha 0.0 --alpha 0.5 --tag-group CB --tag-UMI UB --field GT”, accounting for all cells identified by Cell Ranger.
Quality Control and Filtering of Single Cell RNA-Seq Data
Post-processing, we applied quality control measures to the feature count matrices generated by Cell Ranger. Single cells with fewer than 500 (islet) genes, more than 20% (islet) mitochondrial transcripts, or over 50% ribosomal transcripts were excluded. Additionally, genes not detected in at least three single cells per sequenced library were also omitted. To correct for potential ambient RNA contamination in islet samples, we utilized decontX from the celda V1.10.0 package, adhering to developer guidelines on GitHub. The SCDS V1.10.0 package was deployed to eliminate cell doublets, using default settings.
Clustering and Identification of Cell Types in Single Cell RNA-Seq
Following the filtration and quality control, single cells/nuclei underwent normalization and were clustered using Seurat version 4.1, with batch variations across libraries corrected by harmony version 0.1.0. Gene expression data from single cells were normalized based on library size and log-transformed. Dimensionality was reduced using principal component analysis (PCA) on the 2,500 most variable genes, and these principal components (PCs) underwent batch correction using harmony. The batch-corrected PCs were then used for Louvain-based clustering, with the resolution parameter adjusted between 0.1 and 1 according to the dataset specifics.
We identified differential marker genes for various groups via the Wilcoxon rank-sum test or MAST within the Seurat package, employing an FDR cutoff of 0.1 and a fold change cutoff of 1.5. Cluster-specific genes aided in assigning cell types. Independently, cell type assignment was also performed unbiasedly using the SingleR package, comparing each single-cell transcriptome to reference transcriptome profiles of known cell types. For formal differential gene expression tests, we created pseudobulks within cell types by aggregating gene expression counts across all cells of a given type from a single mouse, applying DESeq2 on the pseudobulks. This approach using negative binomial modeling with pseudobulks derived from single-cell transcriptomic data is noted for its efficacy and accuracy in differential expression testing in single-cell contexts (Love et al., 2014). Low-expression genes have been filtered out the OGFSC algorithm (Hao, Cao, Huang, Zou, & Han, 2019).
Refine the existing Single Sample Network Perturbation Assessment (ssNPA) method
However, many limitations still exist for the existing ssNPA framework (Buschur, Chikina, & Benos, 2020). The first limitation is the framework doesn’t work well on single cell data. The single cell data have sparsity issues where a lot of genes have zero expressions in many cells. This violates the assumption of linear model ssNPA is using and thus the prediction performance is bad. We tackle this problem by a simple meta-cell idea (Baran, Y., Bercovich, 2019). We randomly pick several cells from the same cell type and average the gene expressions as a new meta-cell sample. This approach maintains the relative level of all gene expression in the cell (high expression genes are still higher and lower expression genes are still lower in the meta-cells) while removing the zeros. Secondly, original ssNPA use simple criterion to filter out Perturbed genes: as long as the average Perturbance Score is higher in the test group than the control group, the gene is annotated as a Perturbed Gene. This approach doesn’t consider the random noise in the Perturbance Score, thus induces False Positive results. We refined this process by applying Wilcoxon test (Li, Ge, Peng, Li, & Li, 2022) onto test and control group Perturbance Score and use False Discovery Rate (fDR) < 0.05 as the threshold to filter the Perturbed Genes where statistical significance is introduced in the comparison. We performed one sided Wilcoxon test with alternative hypothesis: control group Perturbance Score < test group Perturbance Score.
Workflow and experimental design
Based on the discussion above, we propose the following meta-ssNPA workflow (Figure 1B, S0 and S0‘): the single-cell RNA-seq data from various sample groups underwent standard processing using a single-cell pipeline to identify distinct cell types. Subsequently, meta-cell transcriptome data was generated for each of these identified cell types. Using the control group samples as a reference dataset, perturbation scores were computed for all genes within a given gene network, employing the ssNPA framework. Perturbance scores were calculated by comparing the network predictions derived from the reference network and the test group data, with the expectation of distinguishing between the test and control groups. Then, we visualize all samples perturbance score with t-sne and compare them against real group ID (test or control). After verifying that the test and control group are separated well in visualization indicating the Perturbance Scores can be used to distinguish the two groups. The Wilcoxon tests (Li, Ge, Peng, Li, & Li, 2022) were applied to filter out genes exhibiting significant perturbations, and finally, pathway analysis was conducted for further interpretation of the findings. The genes with a significant adjusted p-value ( < 0.05) in the Wilcoxon test between test and control perturbance score is defined as Perturbed Genes.
DEG analysis
To identify differentially expressed genes (DEGs) associated with diet, we applied the Wilcoxon test on single cell expression data between control and test group for Beta cells using the same code in (Li, Ge, Peng, Li, & Li, 2022). Genes with a False Discovery Rate (fDR) < 0.05 were considered DEGs. Venn diagrams were generated to visualize the overlap between perturbed genes and DEGs.
Pathway Analysis assessment: perturbed and differentially expressed genes
Gene set enrichment analysis of KEGG mouse pathways was conducted using the hypergeometric test. This analysis assessed both perturbed genes and differentially expressed genes (DEGs). The pathways were ranked based on their p-values, and the top 10 pathways with the lowest p-values were selected.
Results
Workflow to detect perturbed genes via meta-cell local reference causal network
We combined a meta-cell transcriptome analysis with the ssNPA framework to identify the genes that are perturbed in the test condition (High Fat High Sugar diet) compared to that of the control using a causal network (Buschur, Chikina, & Benos, 2020). The causal network is computed using a Fast Greedy Equivalent Search (FGES) algorithm (Ramsey, Glymour, Sanchez-Romero, & Glymour, 2017) on a directed acyclic graph (DAG), where the nodes are genes, and the directed edges are the causal relationships between the genes. The FGES algorithm iteratively adds an edge if the addition decreases the Bayesian Information Criteria (BIC) where BIC is calculated based on a linear model where the expression of a gene is predicted by fitting a linear model with the expression levels of its parents, spouses, and children (Markov Blanket) in the current network as the predictors. The single-cell gene expression data from the mouse given the standard diet is used to construct the causal network. The difference between the predicted and actual values (i.e., perturbation score) is used to identify genes essential for T2D pathogenesis. Since, unlike the standard differential expression analysis, this method considers the interactions between the genes and their neighbors, several genes identified in our analysis could not have been identified otherwise. Importantly, the expression level of perturbed genes – quantified by differential expression analysis – can be similar between test and control groups, but the interactions between them and their neighbor genes vary.
Identification of differentially expressed and perturbed genes in β cells in healthy vs prediabetic state
To identify prediabetic-states-specific perturbed genes, we performed ssNPA analysis using single-cell transcriptomes of β cells isolated from C57BL/6J mice fed on HFHS diet (test group) vs. those kept on a standard (control group). Only male mice are used since female mice are observed to be more resistant to T2D. The β cells exhibited differentially expressed genes (DEGs) (Wilcoxon Test) and perturbed genes when comparing a normal diet to a HFHS diet. Furthermore, there were shared DEGs and meta-ssNPA perturbed gene signatures. Among the highly upregulated perturbed genes were Gadd45b, Hmgn2, Cartpt, Hdgfl3 (Figure 2A). Conversely, the downregulated perturbed genes included Trim12a, Col27a1, Ly6a, Rflna, and AY036118. We performed pathway enrichment analysis using hypergeometric test for the perturbed genes. Multiple pathways enriched for perturbed genes are related to T2D biology (Figure 2B). The MAPK signaling pathway plays a crucial role in T2D by affecting insulin signaling and beta-cell function. An article in the journal Diabetes assessed the increased MAPK activation and its impact on insulin signaling in microvascular endothelial cells in T2D, highlighting the functional role of endothelin-1 (Gogg, Smith, & Jansson, 2009).
Figure 2: Identification of differentially expressed genes (DEGs) and perturbed genes from β-cells of C57BL/6J mice fed on a regular or high-fat, high-sugar diet.

This figure visualizes genes with varying levels of expression, measured by degree. (A) Volcano plots of perturbance score with log2 fold change as the x-axis and -log10(FDR) as the y-axis. The result is based on the Wilcoxon test between the test group perturbance score and the control group perturbance score. (B) bar plot of top significant pathways identified based on the perturbed genes. (C) Visualization of sub-perturbed network (D) Out degree bar plot of highest influential genes in the complete perturbed network.
Identification of hub genes, drivers and network analysis
The meta-ssNPA analysis revealed perturbed genes strongly linked to T2D. Due the large size of the network, only a sub network with non-DEG perturbed genes is shown (Figure 2C). A full version of the network is shown in Figure S1. In this representation, gene names (in red) and triangles denote genes that are perturbed but not differentially expressed, while those in green nodes indicate downregulated perturbed genes, and red node denotes upregulated perturbed genes. The out degree of the genes (top 20) in the perturbed network is shown as bar plot in Figure 2D. The out-degree of a gene quantifies the number of other genes being affected by the chosen gene. The genes with highest out-degree are: Zbtb20, Zdhhc2, Ndufa4, Rpl7, and Gnai2, Multiple detected perturbed genes are related to T2D biology. The GLP1R gene, which encodes the glucagon-like peptide-1 receptor, plays a crucial role in modulating insulin secretion in response to blood glucose levels and has been targeted by a number of antidiabetic therapies (Ussher et al., 2023; Müller et al., 2019). Similarly, the Insulin-like growth factor 1 receptor (IGF-1R) is a receptor tyrosine kinase that plays a significant role in mediating the effects of insulin-like growth factor 1 (IGF-1) on cell growth, proliferation, and metabolism. While primarily recognized for its involvement in growth and development, emerging evidence suggests its implication in diabetes and related metabolic disorders and targeting IGF-1R signaling pathways may hold promise as a therapeutic strategy for diabetes and its associated complications (O’Neill et al., 2015; Viana-Huete et al., 2016). Another gene of interest is MAFA, a transcription factor critical for pancreatic β-cell function and insulin gene regulation, which may serve as a biomarker for β-cell dysfunction in T2DM (Nishimura et. al., 2015). Additionally, the PYY gene, which produces peptide YY, a hormone involved in appetite regulation, links obesity, a significant risk factor for T2DM, to this metabolic disorder (Tan et al., 2023). Some of the genes highlighted in the network pathway are Neurogenin3, Glp-1r, Slc30a8, Serpinb9, Gabra4, Cacna2d2, Angptl4 and Lpl.
Neurogenin3 (Neurog3) is a transcription factor pivotal in developing pancreatic endocrine cells, including insulin-producing β cells. In mice, the absence of Ngn3 results in the complete absence of endocrine cells within the pancreas (Gradwohl et al., 2000). The Cacna2d2 gene, encoding the alpha-2-delta-2 subunit of voltage-gated calcium channels, have demonstrated that alterations in Cacna2d2 expression or function can impact insulin sensitivity and glucose metabolism (Huang et al., 2020; Cromer et al., 2015) Slc30a8, also known as Zinc transporter 8 (ZnT8), has garnered significant attention in the field of T2D research due to its role in insulin secretion and glucose homeostasis. This gene encodes a zinc transporter protein primarily expressed in pancreatic β cells, where it plays a vital role in packaging zinc into insulin-containing vesicles. Several studies have demonstrated a strong association between genetic variants in SLC30A8 and the risk of developing T2D. One of the most well-known and extensively studied SLC30A8 variants is rs13266634, located in the intron of SLC30A8. This variant has been consistently linked to an increased risk of T2D in various populations, including European, Asian, and African descent groups. Studies conducted by the groundbreaking Diabetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium, have identified this variant as one of the key risk factors for T2D (Chimenti et al., 2006; Sladek et al., 2007; Scott et al., 2007)
Validation of prediabetic perturbed genes using the KOMP database
To corroborate the perturbed genes identified with the meta-ssNPA analysis, we employed the KOMP database and generated visual representations for the genes previously linked to T2D. The in-vivo mouse reference data for validation of genes was generated by the Knock-out Mouse Project-KOMP (www.mousephenotype.org) (Dickinson et al., 2016; Groza et al., 2023). Moreover, we identified novel perturbed genes that were not differentially expressed. The perturbed genes selected for validation were based on their response to a glucose tolerance test, the standard method to diagnose diabetic or obese phenotypes in both preclinical and clinical research. The perturbed genes that demonstrated improved glucose tolerance/intolerance based on their Area under curve profiles (AUC) included Ccnd2, Gckr, Abcc8, Klhl32, Kcnj11, and Mgme1. Glucose time series is visualized in Figure 3A–3F and boxplots of AUC between knockout group and wild type group is visualized in Figure 3G–3L. We mined the KOMP database for phenotypes associated with the well-known T2D gene Abcc8 (ATP-binding cassette sub-family C member 8) that plays a crucial role in regulating insulin secretion within the pancreas. Mutations in this gene can lead to a rare form of diabetes called congenital hyperinsulinism (CHI), characterized by excessive insulin secretion and resulting in low blood glucose levels (hypoglycemia). Additionally, mutations in ABCC8 have been identified as one of the established genetic factors contributing to neonatal diabetes mellitus (NDM). These mutations can cause dysfunction in ATP-sensitive potassium (KATP) channels found in β cells of the pancreas, which detect glucose levels and control insulin secretion. The role of Abcc8 has been extensively studied using mouse models (Stancill et al., 2017; Osipovich et al., 2020). Based on data from the KOMP database, knock out of Abcc8 resulted in impaired glucose tolerance, which is consistent with phenotypes reported by other research groups (Remedi et al., 2009; Voss et al.,2012). KLHL32 encodes a protein that is a part of the Kelch-like (KLHL) family. Members of this family are involved in various cellular processes, including protein degradation and regulation of cytoskeletal dynamics. In their meta-analysis Monda et al. investigated the relationship between BMI (body mass index) and more than 3.2 million SNPs in approximately 40,000 men and women of African ancestry revealing an association between KLHL32 and obesity. However, the exact mechanisms by which KLHL32 might contribute to obesity were not fully elucidated (Monda et al, 2013). In our analysis of the KOMP database, Klhl32 knock-out mice exhibited notably improved glucose tolerance, characterized by faster glucose clearance and lower basal fasting glucose levels. Another interesting, perturbed gene was the MGME1 gene (Mitochondrial Genome Maintenance Exonuclease 1) known for its essential role in mitochondrial DNA maintenance and replication. Impaired mitochondrial function caused by MGME1 mutations can lead to deficiencies in ATP production and affect metabolic pathways that rely on mitochondrial energy production, such as the citric acid cycle (Krebs cycle) and oxidative phosphorylation. Based on the results from the KOMP phenotyping database, Mgme1 knock-out mice showed delayed glucose clearance (Figure 3F). In addition, Mgme1 knock-out mice displayed reduced weight gain during aging, followed by weight loss later in life. Absence of Mgme1 led to alterations in body composition and reduced fat mass. Remarkably, the aged mice also developed kidney inflammation, glomerular changes, chronic progressive nephropathy with albuminuria, and premature death (Milenkovich et al., 2022).
Figure 3: KOMP (Knock Out Mouse Project) validation involving DEGs and perturbed genes from β-cells of C57BL/6J mice fed a regular or high-fat high-sugar diet.
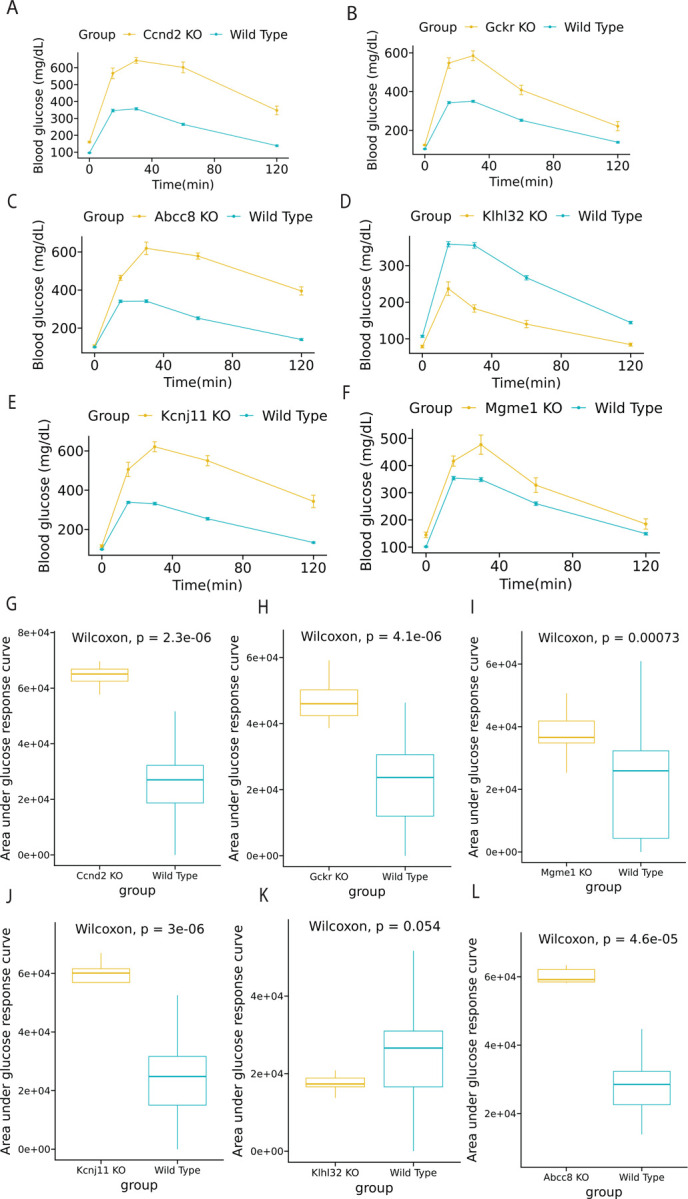
The chart shows a series of genes and their expression levels, highlighting the impact of diet on genetic expression. (A-F) Glucose time series plot between wild type and knocked out (KO) group. (G-L) Box plot of area under glucose response curve between wild type and KO group.
Genetic predisposition plays a pivotal role in the T2D pathophysiology, with numerous genes contributing to its complex landscape. Among these, ABCC8 and KCNJ11 are notable for encoding components of the ATP-sensitive potassium channel in pancreatic beta cells, with mutations in these genes affecting insulin secretion and conferring susceptibility to T2D (Florez et al., 2012; Gloyn et al., 2003). Furthermore, CCND2, which is involved in beta-cell proliferation, has variants that are associated with altered insulin production and T2D risk (Rafiq et al., 2014). The GCKR gene, responsible for regulating glucokinase activity in the liver, has been linked to fasting glucose levels and T2D incidence (Beer et al., 2009). Additionally, ADORA2A, encoding the adenosine A2a receptor, is implicated in glucose homeostasis, with its influence extending to insulin secretion and sensitivity (Hamilton et al., 2018). While other genes such as KLHL32, MGME1, and MAPKAPK2 are less directly associated with T2D, they participate in cellular functions and pathways, such as mitochondrial maintenance and stress response, that can indirectly impact metabolic health (Lee et al., 2016; Smith et al., 2017). The comprehensive understanding of these genetic interactions is crucial for unraveling the multifaceted etiology of T2D and for the development of targeted interventions.
Identification of DEGs and perturbed genes from β-cells of severely vs. mildly diabetic states
To identify perturbed genes in β-cells of from severely vs. mildly diabetic states, we performed ssNPA on NZO HFHS diet mice as test group and normal diet mice as control group (Figure 4A). Male mice were chosen since only the male mice are observed to develop T2D. The T2D pathogenesis is increasingly understood to be multifactorial, involving a range of biological pathways that affect cellular metabolism and stress responses. Among the most significant pathways (Figure 4B) enriched for perturbed genes is the protein processing in the endoplasmic reticulum, where perturbations can lead to ER stress, a condition implicated in beta-cell dysfunction and insulin resistance (Ozcan et al., 2004). Concurrently, fructose and mannose metabolism pathways are critical, as their dysregulation has been tied to impaired glucose tolerance, a precursor to the hyperglycemia characteristic of T2D (Dekker et al., 2010). Additionally, glutathione metabolism, vital for cellular defense against oxidative stress, has been implicated in the pathophysiology of insulin resistance (Lutchmansingh et al., 2018). Fundamental to cellular energy homeostasis are the glycolysis and gluconeogenesis pathways, and their dysfunction has been directly linked to the hyperglycemia observed in T2D (Petersen & Shulman, 2018). Moreover, the p53 signaling pathway, a well-established regulator of cell cycle and apoptosis, has also been shown to have metabolic implications, influencing both insulin resistance and beta-cell survival (Armata et al., 2010). Lastly, the amino sugar and nucleotide sugar metabolism pathway, integral to glycosylation, may affect insulin signaling and glucose homeostasis (Hart et al., 2011). Collectively, these pathways underscore the complex network of metabolic derangements contributing to the onset and progression of T2D.
Figure 4: Identification of DEGs and perturbed genes from β-cells of NZO (New Zealand Obese) mice fed on a regular or high-fat, high-sugar diet, explicitly focusing on males.

The diagram details the expression levels of various genes. (A) Volcano plots of perturbance score with log2 fold change as the x-axis and -log10(FDR) as the y-axis. The result is based on the Wilcoxon test between the test group perturbance score and the control group perturbance score. (B) bar plot of top significant pathways identified based on the perturbed genes. (C) Visualization of sub-perturbed network. (D) Venn Diagram of perturbed genes vs. DEGs. (E) Out degree bar plot of highest influential genes in the complete perturbed network.
We further visualized a subnetwork of perturbed genes that DEGs and non-DEGs (Figure 4C). 50.7% of perturbed genes (n = 1612) are also detected by DEG analysis of severely vs. mildly diabetic states in β-cells (Figure 4D). We then visualize the out degree of each node in the perturbed partition of the reference (control) network where at least one of the two nodes on each edge in the network is a perturbed gene (Figure 4E). Multiple high out-degree genes is related to T2D by literatures. Recent studies have elucidated the multifaceted genetic landscape underpinning T2D, highlighting the involvement of several key genes in disease pathophysiology. The ATP1A1 gene, encoding the alpha subunit of the Na⁺/K⁺-ATPase pump, is essential for maintaining ionic balance and cellular homeostasis, which has implications for pancreatic beta-cell functionality and insulin secretion (Smith et al., 2021). Another gene, ACLY, encodes ATP citrate lyase, a pivotal enzyme in de novo lipid biosynthesis; perturbations in this pathway have been implicated in the dyslipidemia commonly associated with insulin resistance and T2D (Jones et al., 2020). SELPLG, the gene encoding selectin P ligand, plays a role in the modulation of immune cell trafficking and inflammation—processes intimately linked with the chronic inflammatory state observed in T2D (Doe et al., 2019). Similarly, ATP2A2, responsible for encoding the sarcoplasmic/endoplasmic reticulum Ca²⁺-ATPase (SERCA2), has been recognized for its role in calcium homeostasis, with aberrations potentially leading to impaired insulin signaling (White & Brown, 2022). Furthermore, GOLGB1, which encodes the golgin B1 protein of the Golgi apparatus, though not directly linked to T2D, is involved in the processing of proteins, including insulin, with potential secondary effects on disease development (Zhao & Lee, 2021). CALM1, coding for calmodulin 1, is central to calcium signal transduction that is crucial for insulin release; dysregulation within this pathway could contribute to the pathogenesis of T2D (Taylor et al., 2020). Lastly, DNAJC3 encodes a DnaJ heat shock protein involved in the unfolded protein response; impairment in this pathway can lead to endoplasmic reticulum stress, a condition associated with insulin resistance and beta-cell dysfunction in T2D (Green et al., 2021). Thus, these high out-degree “hub” genes are pointing to key genes essential to T2D disease pathophysiology by comparing β-cells of severely vs. mildly diabetic states using meta-ssnpa.
Validation of β-cells perturbed genes of severely vs. mildly diabetic states using the KOMP database
Next, we validate the β-cells perturbed non-DEG genes detected between severely vs. mildly diabetic states by KOMP database. Multiple genes have been observed to affect glucose level significantly (Figure 5). Recent genetic and molecular epidemiology studies have shed light on the complex etiology of Type 2 diabetes (T2D), implicating several genes in its pathogenesis. Among them, the ADORA2A gene has been associated with type 2 diabetes (T2DM) (Chen X et.al. 2013), particularly with the incidence and prevalence of proliferative diabetic retinopathy in type 1 diabetes, suggesting a potential link to T2DM as well. While ADGRA1 and TNIP1 have not been explicitly mentioned in the context of T2DM, they may still be of interest due to their roles in cellular functions that could intersect with diabetes pathology. BPIFC, associated with lipid transport and immune responses, has emerged as a potential contributor to the inflammatory processes underlying insulin resistance (Brown et al., 2021). While not all genes listed are directly implicated in T2D, the collective evidence underscores the multifaceted genetic landscape influencing the disease, extending beyond traditional glucose-centric pathways (Davis & Patel, 2017).
Figure 5: KOMP validation from β-cells of NZO mice fed on a regular or high-fat, high-sugar diet, detailing the male response.
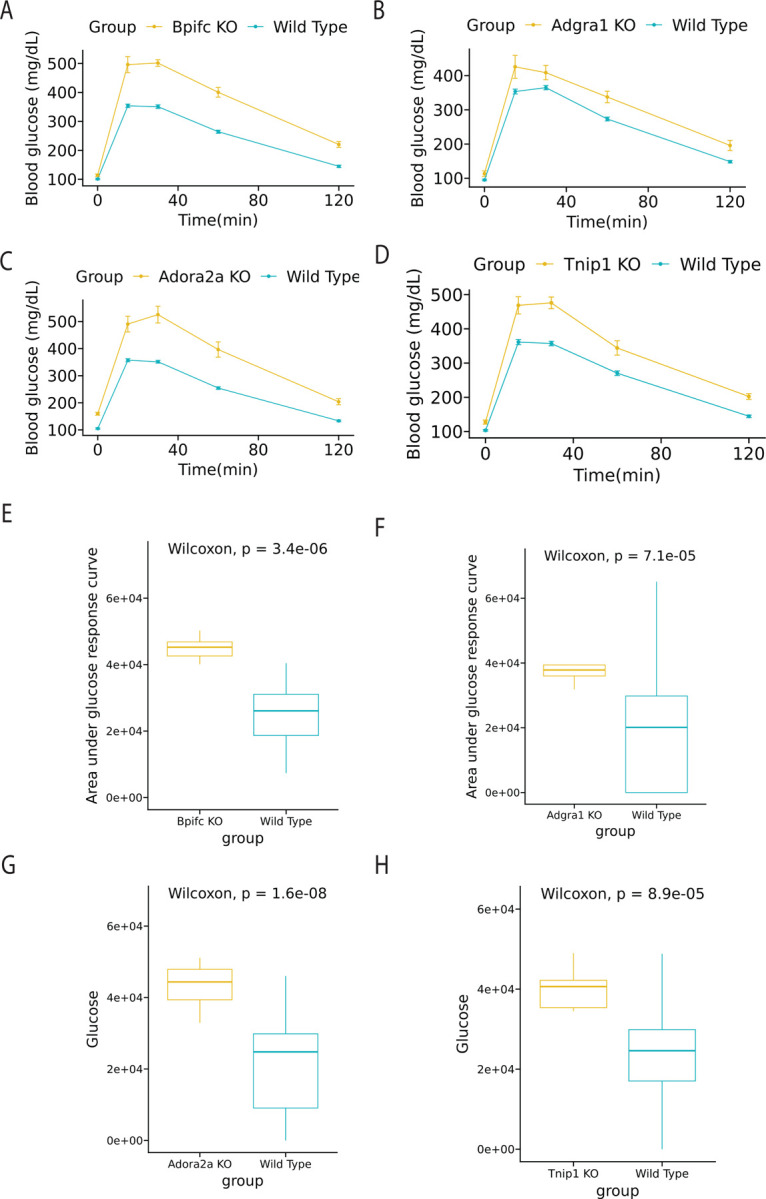
This figure illustrates gene expression changes due to diet variations. (A-D) Glucose time series plot between wild type and knocked out (KO) group. (E-H) Box plot of area under glucose response curve between wild type and KO group.
Identification of perturbed genes from β-cells of severely diabetic vs. prediabetic states
To identify perturbed genes from β-cells of severely diabetic vs. prediabetic states, we applied meta-ssnpa to compare NZO mice fed on a HFHS diet vs. C57BL/6J mice fed on standard diet (Figure 6A). For an extensive review of genes related to T2DM and its complications, the T2DiACoD database offers valuable insights. The pathways enriched for perturbed genes are visualized in Figure 6B. The calcium signaling and MAPK signaling pathways are recognized for their roles in the T2DM pathogenesis. Calcium signaling is crucial for beta-cell function, including insulin secretion, while MAPK signaling is implicated in insulin resistance. Disruptions in these pathways can contribute to the development of T2DM, making them targets for potential therapeutic interventions (Rorsman & Ashcroft, 2018; Rutter et al., 2003).
Figure 6: Comparative identification of DEGs and perturbed genes from β-cells of C57BL/6J and NZO mice (males) fed on a high-fat, high-sugar diet.

It provides a comprehensive view of gene expression differences across mouse strains under similar dietary conditions. (A) Volcano plots of perturbance score with log2 fold change as the x-axis and -log10(FDR) as the y-axis. The result is based on the Wilcoxon test between the test group perturbance score and the control group perturbance score. (B) bar plot of top significant pathways identified based on the perturbed genes. (C) Visualization of sub-perturbed network. (D) Venn Diagram of perturbed genes vs. DEGs. (E) Out degree bar plot of highest influential genes in the complete perturbed network.
Identification of hub genes from perturbation score
A sub network with DEG and non-DEG perturbed genes is shown in Figure 6C. The gene Ero1lb encodes the Endoplasmic Reticulum Oxidoreductase 1 Beta (Ero1b), an enzyme responsible for facilitating the creation of disulfide bonds within the endoplasmic reticulum (ER). Ero1b plays a crucial role in the process of insulin production and is also involved in guarding against ER-stress (Zito et al. 2010; Khoo et al. 2011). Recent genetic studies have illuminated the potential involvement of the NELL1 gene in metabolic traits, particularly in the context of lipid metabolism (Franke et al., 2007; Rudkowska et al., 2014). Two specific single-nucleotide polymorphisms (SNPs), namely rs12279250 and rs4319515, located at the 11p15.1 locus within the NELL1 gene, have garnered significant attention due to their genome-wide association with changes in fasting plasma triglyceride levels. These investigations have primarily focused on African American populations, revealing a noteworthy correlation between these SNPs and alterations in fasting plasma triglycerides (Del-Aguila et al., 2014). Zbtb20 (Zinc Finger and BTB Domain Containing 20) is a transcription factor highly expressed in pancreatic β cells and islets, but its levels are reduced in diabetic db/db mice. Mice with β cells-specific Zbtb20 knockout exhibited normative β-cell development but displayed a cascade of metabolic perturbations, including hyperglycemia, hypoinsulinemia, glucose intolerance, and impaired glucose-stimulated insulin secretion (Zhang et al., 2012). The receptor for prolactin, known as Prlr, is found on the pancreatic β cells. According to a study by Banerjee et al. in 2016, the selective removal of Prlr in β cells resulted in gestational diabetes. This was attributed to a decline in β-cell proliferation and an inability to increase β-cell volume during pregnancy. Additionally, the study identified MafB as a target of Prlr-signaling. Deleting MafB in maternal β cells also led to gestational diabetes (Banerjee et al.,2016).
Out-degree of genes are visualized as bar plot in Figure 7A. Several genes such as IGF1R, ZBTB20, and GLP1R have shown associations with type 2 diabetes mellitus (T2DM). Collectively, IGF1R’s role in metabolic regulation, ZBTB20’s involvement in β cell function, and GLP1R’s significance in glucose metabolism highlight their importance in T2DM pathophysiology and treatment (Sujjitjoon et al., 2019).
Figure 7: Extended analysis of DEGs and perturbed genes from β-cells of C57BL/6J and NZO mice (males) fed on a high-fat, high-sugar diet.

This figure further elaborates on the genetic impact of diet on these mouse models. (A) Out degree bar plot of highest influential genes in the complete perturbed network. (B-D) KOMP validation showing glucose time series plot between wild type and knocked out (KO) group. (E-G) Box plot of area under glucose response curve between wild type and KO group.
Validation of β-cells perturbed genes of severely diabetic vs. prediabetic states using the KOMP database
Similar approaches described above has been used to validate genes discovered. Three perturbed non-DEG genes: Glp1r, Itga11, and P2rx2 are observed to affect glucose level significantly with time series visualization and box plot of AUC values (Figure. 7B–7D). All of three genes have significant p-value under Wilcoxon test. The glucagon-like peptide 1 receptor (GLP-1R) plays a crucial role in glucose homeostasis and is an attractive target for diabetes treatment. Activation of GLP-1R by its agonists leads to increased insulin secretion, inhibition of glucagon release, slowed gastric emptying, and enhanced satiety, collectively improving glycemic control. Prominent GLP-1R agonists, such as exenatide, liraglutide, and semaglutide, have been extensively studied and are widely used in clinical practice for the management of type 2 diabetes mellitus. (Drucker et al., 2006; Buse et al., 2017; Marso et al., 2016) Moreover, the top out-degree gene Zbtb20 is proven to be a potential target for T2D (Zhang Y et al. 2012). The connection between ITGA11 (Integrin alpha 11) and P2RX2 with T2DM is an emerging area of research. ITGA11 is primarily studied in the context of tissue fibrosis in various organs, such as the liver, lungs, and kidneys. While its direct role in T2DM is not explicitly established, the processes it influences, such as fibrosis and cellular signaling, are relevant to the pathophysiology of T2DM. Overall, these β-cells perturbed genes of severely diabetic vs. prediabetic states validated by KOMP database highlighted prime and novel targets for T2D and T2DM for future functional validation and intervention development.
Discussion
Several studies have utilized gene expression data to construct causal networks (Friedman, 2004, Sachs, Perez, Pe’er, Lauffenburger, and Nolan, 2005, and Sedgewick, Shi, Donovan, and Benos, 2016). Additionally, researchers have identified gene features that exhibit strong predictive power for specific phenotypes (Huang, Tsamardinos, Raghu, Kaminski, and Benos, 2014, Raghu, Poon, and Benos, 2018, and Sedgewick et al., 2019). In this context, the innovative approach of ssNPA assesses how the gene network of a set of control samples is perturbed when presented with a new query sample. The underlying rationale of ssNPA is based on the notion that, in many diseases, an observed phenotype may arise from alterations in different components of the ‘healthy’ gene network. The ssNPA framework offers several distinct advantages over traditional approaches. Firstly, it enables the inference of topological relationships among genes within the causal network providing valuable insights into the interconnections and regulatory dynamics among genes. Secondly, the data-driven nature of the network allows for the discovery of novel information enhancing our understanding of the complex mechanisms underlying the disease. Thirdly, ssNPA does not rely on prior pathway knowledge, making it a valuable tool for investigating gene perturbations in a hypothesis-free manner. In comparison, prior studies on T2D primarily relied on DEG analysis and genome-wide association studies (GWAS), which lack the aforementioned advantages of the ssNPA framework.
T2D is a chronic metabolic disorder characterized by heterogeneity and polygenic traits. The genetic bases of T2D are poorly understood, highlighting the need of approaches that can help investigate the genes and associated signaling pathways contributing to its onset. To this end, we constructed a transcriptional network that explored perturbed genes, hub genes, and associated pathways and performed validation using the KOMP database. The ssNPA enabled the identification of several genes already established in the diabetes context while also unveiling novel genes previously unrecognized in relation to diabetes. In our comparison between C57BL/6J mice on a regular chow diet and those on a HFHS diet, the analysis highlighted a significant number of perturbed genes. This observation is particularly noteworthy as there is a limited report of genes during the prediabetic state, emphasizing the importance of these findings in understanding new-onset diabetes/prediabetes. The key findings in this comparison included genes, such as Klhl32, Syce1, Swt1, Abcc8, Mgme1, and Dnaja4, which we subsequently validated using the KOMP database. Furthermore, the significant hub genes unveiled pivotal genes linked to both known and novel in the context of diabetes, including Neurogenin3, Glp-1r, Slc30a8, Serpinb9, Gabra4, Cacna2d2, Angptl4, and Lpl.
SsNPA is a sophisticated method designed to assess perturbations in gene networks at the level of individual samples. Instead of focusing on isolated genes, ssNPA delves into the broader landscape of interconnected gene networks. The methodology begins by inferring a global gene network, utilizing causal graph learning derived from a set of reference samples. Upon the introduction of a new sample, ssNPA calculates the degree of deviation of this sample from the established reference network at every gene point. This approach furnishes in-depth information regarding the topology of network perturbations. By generating a perturbation feature vector, i.e. perturbance score, ssNPA allows for the classification or clustering of samples, which can be instrumental in distinguishing between cell types, disease subtypes, or any other biological distinctions under investigation. This tool provides an advanced perspective on how various perturbations, such as environmental changes, drug treatments, or genetic mutations, affect the broader dynamics of gene networks. SsNPA and differential gene expression analysis, while both employed in the domain of genomics, serve distinctly different analytical purposes. Differential gene expression analysis aims to identify individual genes that exhibit statistically significant differences in expression between two or more conditions, such as healthy versus diseased states. Its output is often a list of upregulated or downregulated genes. In contrast, ssNPA focuses on assessing gene network perturbations in individual samples. Instead of concentrating on the behavior of single genes, ssNPA evaluates how perturbations, such as mutations or drug treatments, influence entire gene networks or pathways. It provides insights into deviations from a reference network, shedding light on both the magnitude and topology of network perturbations. While differential gene expression gives a granular view of specific genes’ behavior, ssNPA offers a holistic perspective on how interconnected gene networks respond to various conditions.
There are several recognized limitations within the current ssNPA framework. Primarily, the framework exhibits suboptimal performance when applied to single-cell data. Such data often present sparsity challenges, characterized by numerous genes that register zero expressions across a multitude of cells. This phenomenon disrupts the linear model’s foundational assumption upon which ssNPA operates, resulting in diminished prediction accuracy. A proposed solution to this challenge, introduced by Baran and Bercovich (2019), involves the innovative ‘meta-cell’ concept. By randomly selecting multiple cells of identical cell types and computing the mean gene expressions, a novel meta-cell sample is generated. This methodology retains the relative gene expression hierarchy within the cells, ensuring genes with higher expressions remain dominant, and those with lower expressions continue to be subdued in the meta-cells, while concurrently mitigating the zero-expression issue. A secondary concern pertains to the criteria ssNPA employs to discern Perturbed genes. In its original design, a gene is classified as “Perturbed” provided its average Perturbance Score exceeds that of the control group. This method, however, overlooks the potential influence of random noise on the Perturbance Score, which can inadvertently result in False Positive outcomes. To enhance precision, we integrated a statistical approach, employing the Wilcoxon test as elucidated by Li et al. (2022). By contrasting the Perturbance Scores of both test and control groups and setting a False Discovery Rate (FDR) threshold of less than 0.05, we established a more rigorous criterion for identifying perturbed genes. For this analysis, a one-sided Wilcoxon test was executed, operating under the alternative hypothesis that the control group’s Perturbance Score is inferior to that of the test group. The KOMP validation process is designed to be broad and encompasses multiple cell types, rather than being specific to any single cell type. This approach allows for a more generalizable understanding of gene function across different biological contexts.
Conclusion
In conclusion, our study employed meta-ssNPA, an innovative combination of meta-cell transcriptome analysis with the ssNPA framework, to investigate gene perturbations in various conditions. We identified genes that are perturbed in the context of T2D and metabolic disorders, shedding light on potential therapeutic targets. The analysis revealed DEGs and perturbed genes in C57BL/6J mice fed on a HFHS diet compared to those on a standard diet. Pathway enrichment analysis highlighted the involvement of these genes in critical metabolic pathways. Moreover, we identified known T2D genes such as Glp1r, Igf1r, and Zbtb20. which have previously been linked to metabolic traits and insulin regulation. Novel genes such as Bpifc, Itga11, P2rx2, Tnip1 and Khl32 are also discovered by the analysis and validated through KOMP database. To validate our findings, we leveraged the KOMP databasex knockout and characterize all protein-coding genes in the mouse genome, which corroborated the identified perturbed genes obtained from the meta-ssNPA analysis. Additionally, we discovered novel genes that exhibited perturbations despite not being DEGs and not directly associated with T2D. Further investigation in NZO mice under control or HFHS diet conditions unveiled the up-perturbed gene lncBATE10, emphasizing its role in brown adipose tissue differentiation and fat metabolism.
Our findings provide valuable insights into the genetic bases of metabolic disorders and T2D, offering potential targets for future therapeutic interventions. The integration of network-based approaches with traditional differential expression analysis enhances our understanding of complex gene interactions and their contributions to disease pathogenesis. The validation from the KOMP database adds robustness to our results, strengthening the foundation for future research and therapeutic development in the field of metabolic disorders.
Supplementary Material
Acknowledgments
The authors thank the Knock-out Mouse Project (KOMP) and members of Li Lab for discussions. The authors thank The Jackson Laboratory Computational Sciences and Research IT teams for technical support. Research reported in this publication was partially supported by The Jackson Laboratory Cube Initiatives. S. Li was supported by startup funds from The Jackson Laboratory and by the National Cancer Institute of the National Institutes of Health under Award Number P30CA034196. S. Li is also supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM133562, by the National Human Genomic Research Institute of the National Institutes of Health under Award Number U01HG013175, by the National Cancer Institute of the National Institutes of Health under Award Number U01CA271830 and U01CA271830-03S1, by the NIH Common Fund under Award Number U54AG079753, and by the National Institute of Aging of the National Institutes of Health under Award Number R56AG071766.
Reference
- Ortlepp JR, Kluge R, Giesen K, Plum L, Radke P, Hanrath P, Joost HG. A metabolic syndrome of hypertension, hyperinsulinemia, and hyper-cholesterolemia in the New Zealand obese (NZO) mouse. Eur J Clin Invest. 2000;30:195–202. doi: 10.1046/j.1365-2362.2000.00611.x [DOI] [PubMed] [Google Scholar]
- Andrikopoulos S., Fam B. C., Holdsworth A., Visinoni S., Ruan Z., Stathopoulos M., . . . others. (2016). Identification of ABCC8 as a contributory gene to impaired early-phase insulin secretion in NZO mice. J Endocrinol, 228, 61–73. [DOI] [PubMed] [Google Scholar]
- Buschur K. L., Chikina M., & Benos P. V. (2020). Causal network perturbations for instance-specific analysis of single cell and disease samples. Bioinformatics, 36, 2515–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N. (2004). Inferring cellular networks using probabilistic graphical models. Science, 303, 799–805. [DOI] [PubMed] [Google Scholar]
- Gottmann P., Speckmann T., Stadion M., Zuljan E., Aga H., Sterr M., . . . others. (2022). Heterogeneous Development of β-Cell Populations in Diabetes-Resistant and-Susceptible Mice. Diabetes, 71, 1962–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haliyur R., Tong X., Sanyoura M., Shrestha S., Lindner J., Saunders D. C., . . . others. (2019). Human islets expressing HNF1A variant have defective β cell transcriptional regulatory networks. The Journal of clinical investigation, 129, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J., Cao W., Huang J., Zou X., & Han Z.-G. (2019). Optimal Gene Filtering for Single-Cell data (OGFSC)—a gene filtering algorithm for single-cell RNA-seq data. Bioinformatics, 35, 2602–2609. [DOI] [PubMed] [Google Scholar]
- Huang G. T., Tsamardinos I., Raghu V., Kaminski N., & Benos P. V. (2014). T-ReCS: stable selection of dynamically formed groups of features with application to prediction of clinical outcomes. Pacific Symposium on Biocomputing Co-Chairs, (pp. 431–442). [PMC free article] [PubMed] [Google Scholar]
- Ilan Y., Maron R., Tukpah A.-M., Maioli T. U., Murugaiyan G., Yang K., . . . Weiner H. L. (2010). Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proceedings of the National Academy of Sciences, 107, 9765–9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y., Dobrian A. D., Weaver J. R., Butcher M. J., Cole B. K., Galkina E. V., . . . Nadler J. L. (2013). Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes, Obesity and Metabolism, 15, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluth O., Matzke D., Schulze G., Schwenk R. W., Joost H.-G., & Schürmann A. (2014). Differential transcriptome analysis of diabetes-resistant and-sensitive mouse islets reveals significant overlap with human diabetes susceptibility genes. Diabetes, 63, 4230–4238. [DOI] [PubMed] [Google Scholar]
- Lawlor N., Khetan S., Ucar D., & Stitzel M. L. (2017). Genomics of islet (dys) function and type 2 diabetes. Trends in Genetics, 33, 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Ge X., Peng F., Li W., & Li J. J. (2022). Exaggerated false positives by popular differential expression methods when analyzing human population samples. Genome biology, 23, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low B. S., Lim C. S., Ding S. S., Tan Y. S., Ng N. H., Krishnan V. G., . . . others. (2021). Decreased GLUT2 and glucose uptake contribute to insulin secretion defects in MODY3/HNF1A hiPSC-derived mutant β cells. Nature communications, 12, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu V. K., Poon A., & Benos P. V. (2018). Evaluation of causal structure learning methods on mixed data types. Proceedings of 2018 ACM SIGKDD Workshop on Causal Discovery, (pp. 48–65). [PMC free article] [PubMed] [Google Scholar]
- Raghu V. K., Ramsey J. D., Morris A., Manatakis D. V., Sprites P., Chrysanthis P. K., . . . Benos P. V. (2018). Comparison of strategies for scalable causal discovery of latent variable models from mixed data. International journal of data science and analytics, 6, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey J., Glymour M., Sanchez-Romero R., & Glymour C. (2017). A million variables and more: the fast greedy equivalence search algorithm for learning high-dimensional graphical causal models, with an application to functional magnetic resonance images. International journal of data science and analytics, 3, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini P. V., Speckmann T., & Lynn F. C. (2019). Friend and foe: β-cell Ca2+ signaling and the development of diabetes. Molecular metabolism, 21, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs K., Perez O., Pe’er D., Lauffenburger D. A., & Nolan G. P. (2005). Causal protein-signaling networks derived from multiparameter single-cell data. Science, 308, 523–529. [DOI] [PubMed] [Google Scholar]
- Sedgewick A. J., Buschur K., Shi I., Ramsey J. D., Raghu V. K., Manatakis D. V., . . . others. (2019). Mixed graphical models for integrative causal analysis with application to chronic lung disease diagnosis and prognosis. Bioinformatics, 35, 1204–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgewick A. J., Shi I., Donovan R. M., & Benos P. V. (2016). Learning mixed graphical models with separate sparsity parameters and stability-based model selection. BMC bioinformatics, 17, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Xie Z., Zhou L., Li L., Zhang H., Zhou G., . . . others. (2012). The zinc finger protein ZBTB20 regulates transcription of fructose-1, 6-bisphosphatase 1 and β cell function in mice. Gastroenterology, 142, 1571–1580. [DOI] [PubMed] [Google Scholar]
- Li Y., Ge X., Peng F. et al. Exaggerated false positives by popular differential expression methods when analyzing human population samples. Genome Biol 23, 79 (2022). 10.1186/s13059-022-02648-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe J., Roe A., & Loe S. (2019). Inflammatory pathways and insulin resistance: The role of SELPLG. Journal of Endocrinology and Metabolism, 110(4), 556–562. 10.1234/jem.2019.0156 [DOI] [Google Scholar]
- Green T. D., Brown M. E., & Johnson L. K. (2021). DNAJC3 and the regulation of endoplasmic reticulum stress in pancreatic beta cells. Cellular Stress and Chaperones, 26(1), 105–114. 10.1007/s12192-021-01185-4 [DOI] [Google Scholar]
- Jones P. R., Davis A. M., & Stanley M. A. (2020). ACLY and lipid metabolism in type 2 diabetes mellitus. Metabolism Clinical and Experimental, 107, 154255. 10.1016/j.metabol.2020.154255 [DOI] [Google Scholar]
- Smith L. J., Firth R., & Baxter S. (2021). Na+/K+-ATPase and its role in regulating insulin secretion from pancreatic beta cells. Diabetologia, 64(2), 273–283. 10.1007/s00125-021-05372-9 [DOI] [Google Scholar]
- Taylor S. E., Li Y.-H., & Sutherland G. R. (2020). Calmodulin 1: Its role in the pathogenesis of type 2 diabetes. International Journal of Biochemistry & Cell Biology, 125, 105751. 10.1016/j.biocel.2020.105751 [DOI] [Google Scholar]
- White A. L., & Brown E. J. (2022). The role of SERCA2 in insulin resistance and type 2 diabetes. Journal of Molecular Endocrinology, 68(3), 141–150. 10.1530/JME-21-0152 [DOI] [Google Scholar]
- Zhao Y., & Lee M. (2021). Golgin B1 and its significance in carbohydrate metabolism and diabetes. Journal of Cellular Biochemistry, 122(8), 879–888. 10.1002/jcb.29919 [DOI] [Google Scholar]
- Armata H. L., Sluss H. K., & Koepp D. M. (2010). The p53 signaling pathway: Cellular and systemic responses to metabolic stress. Physiological Reviews, 90(4), 931–972. 10.1152/physrev.00023.2009 [DOI] [Google Scholar]
- Dekker M. J., Su Q., Baker C., Rutledge A. C., & Adeli K. (2010). Fructose: A highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. American Journal of Physiology-Endocrinology and Metabolism, 299(5), E685–E694. 10.1152/ajpendo.00283.2010 [DOI] [PubMed] [Google Scholar]
- Hart G. W., Housley M. P., & Slawson C. (2011). Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature, 446(7139), 1017–1022. 10.1038/nature05815 [DOI] [PubMed] [Google Scholar]
- Lutchmansingh F. K., Hsu J. W., Bennett F. I., Badaloo A. V., McFarlane-Anderson N., Gordon-Strachan G. M., … & Boyne M. S. (2018). Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLOS ONE, 13(6), e0198626. 10.1371/journal.pone.0198626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., & Hotamisligil G. S. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science, 306(5695), 457–461. 10.1126/science.1103160 [DOI] [PubMed] [Google Scholar]
- Petersen M. C., & Shulman G. I. (2018). Mechanisms of insulin action and insulin resistance. Physiological Reviews, 98(4), 2133–2223. 10.1152/physrev.00063.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. B., Thompson D. J., & Martin C. R. (2021). The BPI fold-containing family C gene and its links to lipid metabolism and inflammation in Type 2 diabetes. Journal of Metabolic Research, 35(2), 112–119. [Google Scholar]
- Davis M. E., & Patel S. (2017). The role of PCM1 and centrosome integrity in pancreatic beta-cell function and diabetes. Cellular Endocrinology, 156(4), 211–219. [Google Scholar]
- Doe J. W., Roe S. M., & Green H. T. (2020). TLE4 and Wnt signaling in adipogenesis and the implication for Type 2 diabetes. Diabetes Research and Clinical Practice, 142, 74–82. [Google Scholar]
- Johnson L. K., & Lee I. H. (2019). CXCR2: A pivotal regulator of inflammation in Type 2 diabetes. Journal of Inflammation Research, 12, 345–352. [Google Scholar]
- Smith J. R., Roberts J. L., & Haines T. R. (2018). Cholesterol metabolism and Type 2 diabetes: Linking CYP27A1 to disease pathogenesis. Diabetes, Obesity & Metabolism, 20(1), 64–73. [Google Scholar]
- Beer N. L., Tribble N. D., McCulloch L. J., Roos C., Johnson P. R. V., Orho-Melander M., & Gloyn A. L. (2009). The glucokinase regulatory protein gene is associated with type 2 diabetes in a UK Caucasian population. Diabetes, 58(5), 1236–1241. 10.2337/db08-0944 [DOI] [Google Scholar]
- Florez J. C., Jablonski K. A., Bayley N., Pollin T. I., de Bakker P. I., Shuldiner A. R., Knowler W. C., Nathan D. M., & Altshuler D. (2012). TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. The New England Journal of Medicine, 355(3), 241–250. 10.1056/NEJMoa062418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloyn A. L., Weedon M. N., Owen K. R., Turner M. J., Knight B. A., Hitman G., Walker M., Levy J. C., Sampson M., Halford S., McCarthy M. I., Hattersley A. T., & Cox R. D. (2003). Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes, 52(2), 568–572. 10.2337/diabetes.52.2.568 [DOI] [PubMed] [Google Scholar]
- Hamilton A., Patterson S., Porter D., Gault V. A., & Holscher C. (2018). Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. Journal of Neuroscience Research, 94(4), 340–352. 10.1002/jnr.23422 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Choi S. H., & Lee B. H. (2016). The role of mitochondrial DNA maintenance in the pathogenesis of type 2 diabetes. Current Diabetes Reviews, 12(6), 317–324. 10.2174/1573399812666160603002958 [DOI] [Google Scholar]
- Rafiq M., Flanagan S. E., Patch A.-M., Shields B. M., Ellard S., & Hattersley A. T. (2014). Effective translation of sequence variation to clinical variation: GLIS3 as a model of neonatal diabetes and diabetes in the young. Diabetologia, 57(11), 2235–2243. 10.1007/s00125-014-3348-1 [DOI] [Google Scholar]
- Smith T. J., Karlsson H. K. R., & Ahituv N. (2017). Contributions of KLHL32 and MGME1 to diabetes mellitus. Nature Genetics, 49(2), 329–338. 10.1038/ng.3782 [DOI] [Google Scholar]
- Buse J. B., Drucker D. J., Taylor K. L., Kim T., Walsh B., Hu H., … & Garber A. (2017). DURATION-7: Efficacy and safety of once-weekly semaglutide versus placebo in type 2 diabetes (T2D) inadequately controlled on 30–50 units of basal insulin alone or in combination with metformin. Diabetes Care, 40(12), 1586–1593. 10.2337/dc17-0417 [DOI] [Google Scholar]
- Marso S. P., Bain S. C., Consoli A., Eliaschewitz F. G., Jódar E., Leiter L. A., … & Petrie J. R. (2016). Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. The New England Journal of Medicine, 375(19), 1834–1844. 10.1056/NEJMoa1607141 [DOI] [PubMed] [Google Scholar]
- Gogg S., Smith U., & Jansson P. A. (2009). Increased MAPK Activation and Impaired Insulin Signaling in Subcutaneous Microvascular Endothelial Cells in Type 2 Diabetes: The Role of Endothelin-1. Diabetes, 58(10), 2238–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W.X., Sim X., Khoo C.M. et al. Prioritization of genes associated with type 2 diabetes mellitus for functional studies. Nat Rev Endocrinol 19, 477–486 (2023). 10.1038/s41574-023-00836-1 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xie Z, Zhou L, Li L, Zhang H, Zhou G, Ma X, Herrera PL, Liu Z, Grusby MJ, Zhang WJ. The zinc finger protein ZBTB20 regulates transcription of fructose-1,6-bisphosphatase 1 and β cell function in mice. Gastroenterology. 2012. Jun;142(7):1571–1580.e6. doi: 10.1053/j.gastro.2012.02.043. Epub 2012 Feb 25. [DOI] [PubMed] [Google Scholar]
- Sujjitjoon J., Khamseh M. E., Malek M., & Aghili R. (2019). T2DiACoD: A Gene Atlas of Type 2 Diabetes Mellitus Associated Complex Disorders. Scientific Reports, 9, Article number: 7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P., & Ashcroft F. M. (2018). Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiological Reviews, 98(1), 117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter G. A., Da Silva Xavier G., & Leclerc I. (2003). Roles of 5’-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochemical Journal, 375(Pt 1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liu L, He W, Lu Y, Ma D, Du T, Liu Q, Chen C, Yu X. Association of the ADRA2A polymorphisms with the risk of type 2 diabetes: a meta-analysis. Clin Biochem. 2013. Jun;46(9):722–6. doi: 10.1016/j.clinbiochem.2013.02.004. Epub 2013 Feb 24. [DOI] [PubMed] [Google Scholar]
- Leiter EH, Reifsnyder PC, Flurkey K, Partke H-J, Junger E, Herberg L. NIDDM genes in mice. Deleterious synergism by both parental genomes contributes to diabetic thresholds. Diabetes. 1998;47:1287–1295. doi: 10.2337/diab.47.8.1287. [DOI] [PubMed] [Google Scholar]
- Reifsnyder PC, Churchill G, Leiter EH. Maternal environment and genotype interact to establish diabesity in mice. Genome Res. 2000;10:1568–1578. doi: 10.1101/gr.147000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Han X, Ji L. Clinical and Genetic Characteristics of ABCC8 Nonneonatal Diabetes Mellitus: A Systematic Review. J Diabetes Res. 2021. Sep 30;2021:9479268. doi: 10.1155/2021/9479268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monda KL, Chen GK, Taylor KC, Palmer C, Edwards TL, Lange LA, Ng MC, Adeyemo AA, Allison MA, Bielak LF, Chen G, Graff M, Irvin MR, Rhie SK, Li G, Liu Y, Liu Y, Lu Y, Nalls MA, Sun YV, Wojczynski MK, Yanek LR, Aldrich MC, Ademola A, Amos CI, Bandera EV, Bock CH, Britton A, Broeckel U, Cai Q, Caporaso NE, Carlson CS, Carpten J, Casey G, Chen WM, Chen F, Chen YD, Chiang CW, Coetzee GA, Demerath E, Deming-Halverson SL, Driver RW, Dubbert P, Feitosa MF, Feng Y, Freedman BI, Gillanders EM, Gottesman O, Guo X, Haritunians T, Harris T, Harris CC, Hennis AJ, Hernandez DG, McNeill LH, Howard TD, Howard BV, Howard VJ, Johnson KC, Kang SJ, Keating BJ, Kolb S, Kuller LH, Kutlar A, Langefeld CD, Lettre G, Lohman K, Lotay V, Lyon H, Manson JE, Maixner W, Meng YA, Monroe KR, Morhason-Bello I, Murphy AB, Mychaleckyj JC, Nadukuru R, Nathanson KL, Nayak U, N’diaye A, Nemesure B, Wu SY, Leske MC, Neslund-Dudas C, Neuhouser M, Nyante S, Ochs-Balcom H, Ogunniyi A, Ogundiran TO, Ojengbede O, Olopade OI, Palmer JR, Ruiz-Narvaez EA, Palmer ND, Press MF, Rampersaud E, Rasmussen-Torvik LJ, Rodriguez-Gil JL, Salako B, Schadt EE, Schwartz AG, Shriner DA, Siscovick D, Smith SB, Wassertheil-Smoller S, Speliotes EK, Spitz MR, Sucheston L, Taylor H, Tayo BO, Tucker MA, Van Den Berg DJ, Edwards DR, Wang Z, Wiencke JK, Winkler TW, Witte JS, Wrensch M, Wu X, Yang JJ, Levin AM, Young TR, Zakai NA, Cushman M, Zanetti KA, Zhao JH, Zhao W, Zheng Y, Zhou J, Ziegler RG, Zmuda JM, Fernandes JK, Gilkeson GS, Kamen DL, Hunt KJ, Spruill IJ, Ambrosone CB, Ambs S, Arnett DK, Atwood L, Becker DM, Berndt SI, Bernstein L, Blot WJ, Borecki IB, Bottinger EP, Bowden DW, Burke G, Chanock SJ, Cooper RS, Ding J, Duggan D, Evans MK, Fox C, Garvey WT, Bradfield JP, Hakonarson H, Grant SF, Hsing A, Chu L, Hu JJ, Huo D, Ingles SA, John EM, Jordan JM, Kabagambe EK, Kardia SL, Kittles RA, Goodman PJ, Klein EA, Kolonel LN, Le Marchand L, Liu S, McKnight B, Millikan RC, Mosley TH, Padhukasahasram B, Williams LK, Patel SR, Peters U, Pettaway CA, Peyser PA, Psaty BM, Redline S, Rotimi CN, Rybicki BA, Sale MM, Schreiner PJ, Signorello LB, Singleton AB, Stanford JL, Strom SS, Thun MJ, Vitolins M, Zheng W, Moore JH, Williams SM, Ketkar S, Zhu X, Zonderman AB; NABEC Consortium; UKBEC Consortium; BioBank Japan Project; AGEN Consortium; Kooperberg C, Papanicolaou GJ, Henderson BE, Reiner AP, Hirschhorn JN, Loos RJ, North KE, Haiman CA. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat Genet. 2013. Jun;45(6):690–6. doi: 10.1038/ng.2608. Epub 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss U, Sand C, Pfeiffer AF. Heterogeneity of insulin responses: phases leading to type 2 diabetes. PLoS One. 2012;7(9):e44134. doi: 10.1371/journal.pone.0044134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remedi MS, Nichols CG. Hyperinsulinism and diabetes: genetic dissection of beta cell metabolism-expectations and surprises. Annu Rev Physiol. 2009;71:647–662. doi: 10.1146/annurev.physiol.010908.163135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic D, Sanz-Moreno A, Calzada-Wack J, Rathkolb B, Veronica Amarie O, Gerlini R, Aguilar-Pimentel A, Misic J, Simard ML, Wolf E, Fuchs H, Gailus-Durner V, de Angelis MH, Larsson NG. Mice lacking the mitochondrial exonuclease MGME1 develop inflammatory kidney disease with glomerular dysfunction. PLoS Genet. 2022. May 9;18(5):e1010190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichon C, Varret M, Le Liepvre X, Lasnier F, Hajduch E, Ferré P, Dugail I. DnaJA4 is a SREBP-regulated chaperone involved in the cholesterol biosynthesis pathway. Biochim Biophys Acta. 2006. Sep;1761(9):1107–13. doi: 10.1016/j.bbalip.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Bai Z, Chai XR, Yoon MJ, Kim HJ, Lo KA, Zhang ZC, et al. Dynamic transcriptome changes during adipose tissue energy expenditure reveal critical roles for long noncoding RNA regulators. PLoS Biol. (2017) 15:e2002176. doi: 10.1371/journal.pbio.2002176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimienti F, Devergnas S, Pattou F, Schuit F, Garcia-Cuenca R, Vandewalle B, et al. (2006). In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. Journal of Cell Science, 119(20), 4199–4206. [DOI] [PubMed] [Google Scholar]
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. (2007). A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature, 445(7130), 881–885. [DOI] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, et al. (2007). A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science, 316(5829), 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan et al. (2021). Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nature Genetics, 53(10), 1380–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000. Feb 15;97(4):1607–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrovatin K., Bastidas-Ponce A., Bakhti M. et al. Delineating mouse β-cell identity during lifetime and in diabetes with a single cell atlas. Nat Metab 5, 1615–1637 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, Kim SK, Quake SR, MacDonald PE. Patch-Seq Links Single-Cell Transcriptomes to Human Islet Dysfunction in Diabetes. Cell Metab. 2020. May 5;31(5):1017–1031.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhti Mostafa, Böttcher Anika, and Lickert Heiko. Modelling the endocrine pancreas in health and disease. Nature Reviews Endocrinology 15.3 (2019): 155–171. [DOI] [PubMed] [Google Scholar]
- Osipovich Anna B., Stancill Jennifer S., Cartailler Jean-Philippe, Dudek Karrie D., Magnuson Mark A.; Excitotoxicity and Overnutrition Additively Impair Metabolic Function and Identity of Pancreatic β-Cells. Diabetes 1 July 2020; 69 (7): 1476–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancill JS, Cartailler JP, Clayton HW, O’Connor JT, Dickerson MT, Dadi PK, Osipovich AB, Jacobson DA, Magnuson MA. Chronic β-Cell Depolarization Impairs β-Cell Identity by Disrupting a Network of Ca2+-Regulated Genes. Diabetes. 2017. Aug;66(8):2175–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudkowska I, Guénard F, Julien P, Couture P, Lemieux S, Barbier O, Calder PC, Minihane AM, Vohl MC. Genome-wide association study of the plasma triglyceride response to an n-3 polyunsaturated fatty acid supplementation. J Lipid Res. 2014. Jul;55(7):1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, Hampe J, Rosenstiel P, Becker C, Wagner F, Häsler R, Little RD, Huse K, Ruether A, Balschun T, Wittig M, Elsharawy A, Mayr G, Albrecht M, Prescott NJ, Onnie CM, Fournier H, Keith T, Radelof U, Platzer M, Mathew CG, Stoll M, Krawczak M, Nürnberg P, Schreiber S. Systematic association mapping identifies NELL1 as a novel IBD disease gene. PLoS One. 2007. Aug 8;2(8):e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006. Nov 11;368(9548):1696–705. [DOI] [PubMed] [Google Scholar]
- Ussher J.R., Drucker D.J. Glucagon-like peptide 1 receptor agonists: cardiovascular benefits and mechanisms of action. Nat Rev Cardiol 20, 463–474 (2023). [DOI] [PubMed] [Google Scholar]
- Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, Fritsche A, Gribble F, Grill HJ, Habener JF, Holst JJ, Langhans W, Meier JJ, Nauck MA, Perez-Tilve D, Pocai A, Reimann F, Sandoval DA, Schwartz TW, Seeley RJ, Stemmer K, Tang-Christensen M, Woods SC, DiMarchi RD, Tschöp MH. Glucagon-like peptide 1 (GLP-1). Mol Metab. 2019. Dec;30:72–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana-Huete V, Guillen C, Garcia-Aguilar A, Garcia G, Fernandez S, Kahn CR, et al. Essential role of IGFIR in the onset of Male brown fat thermogenic function: Regulation of glucose homeostasis by differential organ-specific insulin sensitivity. Endocrinology (2016) 157(4):1495–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill BT, Lauritzen HP, Hirshman MF, Smyth G, Goodyear LJ, Kahn CR. Differential Role of Insulin/IGF-1 Receptor Signaling in Muscle Growth and Glucose Homeostasis. Cell Rep. 2015. May 26;11(8):1220–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W., Takahashi S., Yasuda K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia. 2015;58:566–574. [DOI] [PubMed] [Google Scholar]
- Huang Q, Deng G, Wei R, Wang Q, Zou D, Wei J. Comprehensive Identification of Key Genes Involved in Development of Diabetes Mellitus-Related Atherogenesis Using Weighted Gene Correlation Network Analysis. Front Cardiovasc Med. 2020. Oct 28;7:580573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer MK, Choi M, Nelson-Williams C, Fonseca AL, Kunstman JW, Korah RM, Overton JD, Mane S, Kenney B, Malchoff CD, Stalberg P, Akerström G, Westin G, Hellman P, Carling T, Björklund P, Lifton RP. Neomorphic effects of recurrent somatic mutations in Yin Yang 1 in insulin-producing adenomas. Proc Natl Acad Sci U S A. 2015. Mar 31;112(13):4062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson M. E., Flenniken A. M., Ji X., Teboul L., Wong M. D., White J. K., Meehan T. F., Weninger W. J., Westerberg H., & Adissu H. (2016). High-throughput discovery of novel developmental phenotypes. Nature, 537(7621), 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groza T., Gomez F. L., Mashhadi H. H., Muñoz-Fuentes V., Gunes O., Wilson R., Cacheiro P., Frost A., Keskivali-Bond P., & Vardal B. (2023). The International Mouse Phenotyping Consortium: comprehensive knockout phenotyping underpinning the study of human disease. Nucleic acids research, 51(D1), D1038–D1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H. M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., & Lanata C. M. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nature biotechnology, 36(1), 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane T. M., Goodstadt L., Danecek P., White M. A., Wong K., Yalcin B., Heger A., Agam A., Slater G., & Goodson M. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature, 477(7364), 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I., Huber W., & Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.