

Abstract
Lymphocyte activation gene 3 (LAG-3) is an inhibitory receptor that is highly expressed by exhausted T cells. LAG-3 is a promising immunotherapeutic target, with more than 20 LAG-3-targeting therapeutics in clinical trials and a fixed-dose combination of anti-LAG-3 and anti-PD-1 now approved to treat unresectable or metastatic melanoma. Although LAG-3 is widely recognized as a potent inhibitory receptor, important questions regarding its biology and mechanism of action remain. In this Perspective, we focus on gaps in the understanding of LAG-3 biology and discuss the five biggest topics of current debate and focus regarding LAG-3, including its ligands, signaling and mechanism of action, its cell-specific functions, its importance in different disease settings, and the development of novel therapeutics.
LAG-3 (CD223), identified in 1990 as a CD4 structural homolog1, is expressed by a diversity of lymphocytic and nonlymphocytic lineage cells2–4. LAG-3 was subsequently shown to be an inhibitory receptor in Lag3-knockout mice5,6. The majority of LAG-3 research to date has highlighted its role in T cell dysfunction, negatively regulating the immune response in tumors and chronic viral infections. However, LAG-3 is unconventional in a number of ways, leaving outstanding questions with respect to its ligands and function in disease settings.
Although knowledge gaps exist in our understanding of LAG-3 biology, anti-LAG-3 immunotherapeutics have been used to restore T cell function7. A phase 2/3 randomized trial reported that the anti-LAG-3 therapeutic relatlimab, in combination with nivolumab (anti-PD-1), achieved 47.7% 12-month progression-free survival (PFS) in patients with melanoma, compared to 36% with nivolumab monotherapy8. Following these results, a major landmark was achieved in 2022 when the US Food and Drug Administration (FDA) approved the relatlimab/nivolumab combination drug Opdualag for the treatment of unresectable or metastatic melanoma9. With this approval, LAG-3 became the third checkpoint inhibitor to show efficacy when targeted in the clinic, The response profile of Opdualag is comparable to the ipilimumab/nivolumab (anti-CTLA4/anti-PD-1) combination (47.7% versus 49% 12-month PFS), although 59% of patients had severe adverse events with ipilimumab/nivolumab compared with only 18.9% with Opdualag10,11. Combinatorial anti-LAG-3/anti-PD-1 has also been of benefit for patients who are not responsive to primary anti-PD-1/PD-L1 therapy12 in the neoadjuvant setting of resectable stage III/IV melanoma and visceral non-pulmonary metastases. Thus, given the superior toxicity profile, risk:benefit ratio and survival profile, relatlimab/nivolumab might become the preferred first-line therapy in advanced melanoma and neoadjuvant settings irrespective of mutational status.
Given the clinical relevance and efficacy of targeting LAG-3, further understanding of LAG-3–ligand structural biology, interactions and signaling is imperative. This Perspective summarizes important developments leading to the discovery, translation and regulatory approval of LAG-3. We also discuss gaps in our understanding of LAG-3 biology, providing insight into cell-specific functions of LAG-3 and how LAG-3 is regulated, along with mechanistic insights into LAG-3 signaling and function. We also discuss the future of LAG-3 therapeutics for the treatment of autoimmune and neurological diseases.
LAG-3 ligands
LAG-3 was initially shown to mediate T cell inhibition by interacting with its canonical ligand, major histocompatibility complex (MHC) class II. LAG-3–MHC class II interactions are mediated via loop 2 (103–112) of the LAG-3 D1 domain13,14. LAG-3 also seems to selectively recognize conformationally stable peptide–MHC class II complexes (pMHC class II)15. Additional ligands for LAG-3 have been suggested, including galectin-3 (Gal-3), liver and lymph node sinusoidal endothelial cell C-type lectin (LSECtin), fibrinogen-like protein 1 (FGL1), α-synuclein preformed fibrils (α-syn PFF) and, most recently, the T cell antigen receptor (TCR)–CD3 complex (Fig. 1). However, the relative importance of these alternative ligands is controversial.
Fig. 1 |. LAG-3 ligands.
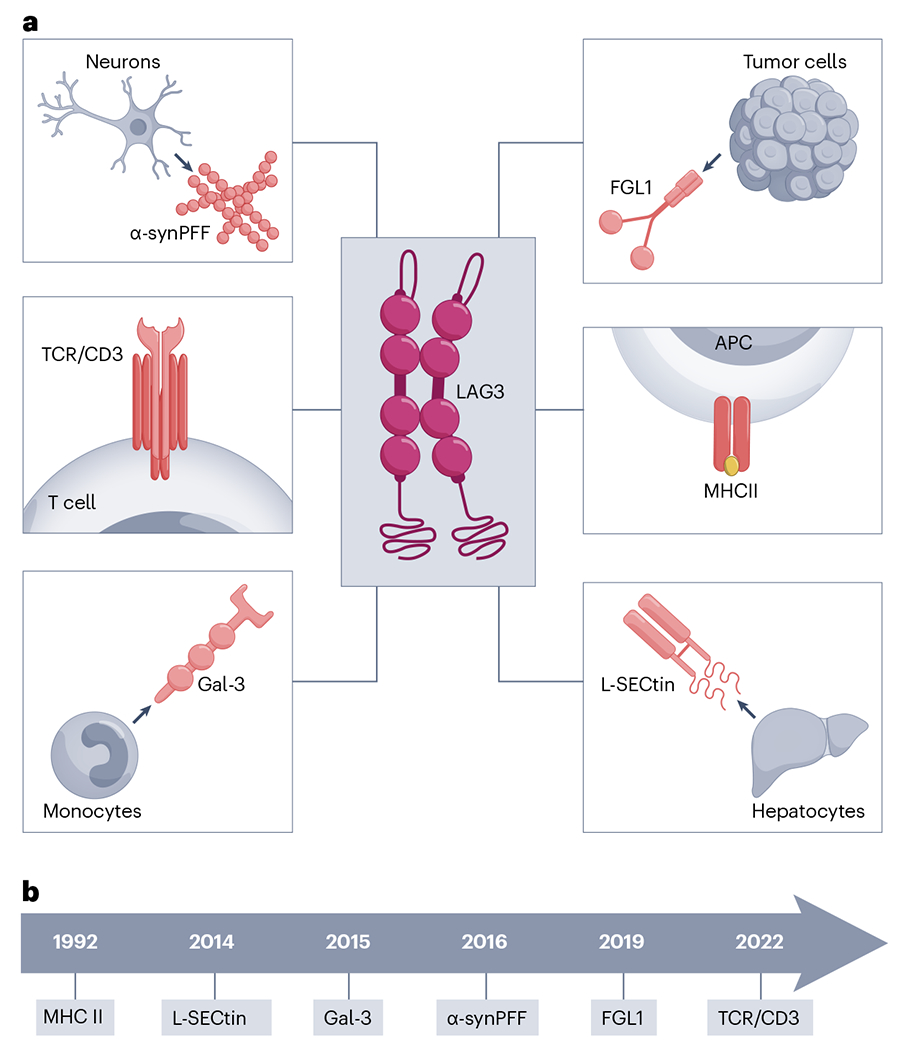
a, LAG-3 ligands and their source. APC, antigen-presenting cell; MHCII, MHC class II. b, A timeline of LAG-3 ligand discovery.
Nearly 10 years ago, two galactose-binding lectins, Gal-3 and LSECtin, were proposed as potential LAG-3 ligands, although data supporting the biological relevance of these ligands are limited. Gal-3 was reported to mediate suppression of effector CD8+ T cell function16,17 in endometrial carcinoma18, vulvar squamous neoplasia19 and multiple myeloma20. LSECtin suppresses effector T cell responses by downregulating expression of cell cycle kinases (CDK2, CDK4 and CDK6)21 in melanoma cells. Despite these reports, lectins remain undefined and are understudied in the context of LAG-3, so it is unknown if they are true LAG-3 ligands. If they are LAG-3 ligands, the physiological expression profile for lectins needs to be evaluated along with how they mediate regulatory effects.
FGL1 is a member of the fibrinogen family of proteins secreted by hepatocytes and tumor cells, with differential tumor-specific and site-specific expression22. FGL1 has been shown to bind the D1 domain of LAG-3, independently of MHC class II23, through a flexible loop in the C-terminal fibrinogen-like domain of FGL1 (ref. 24). FGL1 also binds to human LAG-3 (hLAG-3) and mouse LAG-3 (mLAG-3) via distinct molecular surfaces, which might be indicative of the distinct dimeric states of hLAG-3 versus mLAG-3 (ref. 24). LAG-3 was initially suggested to form dimers on the cell surface through biochemical analysis25, which has now been confirmed by structural analyses24.
Further studies are needed to characterize the functional relevance of FGL1 interactions with hLAG-3 and mLAG-3. Also, the importance of LAG-3–FGL1 interactions in different cell types, such as plasma cells, plasmacytoid dendritic cells (pDCs) and natural killer (NK) cells, is unclear. Another question is whether there is tumor-specific or tissue-specific expression of FGL1 and how that might affect interactions with, and the function of, LAG-3. Interestingly, one study investigated the relative contribution of LAG-3–pMHC class II versus LAG-3–FGL1 interactions using mice expressing LAG-3 mutants that lack the ability to bind to each of these ligands and found that the interaction with stable pMHC class II, but not FGL1, was required for maximal T cell inhibitory activity in vitro, as well as for limiting autoimmunity and preventing anticancer immunity13. However, the study did not completely rule out LAG-3–FGL1 interactions as being involved as it did not evaluate anti-LAG-3 in vivo, and it remains possible that the mutations used do not fully abrogate LAG-3–FGL1 in a physiological setting in vivo. In addition, the mutations used that abrogate LAG-3 interaction with stable pMHC class II might affect other, as-yet-unappreciated, ligand interactions or structural features that are important for LAG-3 function, such as TCR-CD3 engagement or dimerization, respectively.
LAG-3 has been shown to interact with the TCR–CD3 complex in the absence of MHC class II, suggesting that the TCR or CD3 function as important ligands for LAG-3 in cis rather than in trans26. LAG-3 was also shown to mediate its inhibitory activity by disrupting the association between the co-receptors CD4 and CD8 and the tyrosine kinase Lck, thereby limiting proximal TCR signaling and downstream T cell activation26. However, the LAG-3 domains and residues that mediate interaction with the TCR–CD3 complex and where LAG-3 binds to the TCR–CD3 complex are unknown.
LAG-3 is also expressed by neurons, where it might have non-immune functions. LAG-3 mediates cell–cell transmission in α-synucleinopathies through α-syn PFF27. Although papers have reported contradictory results with negligible expression of LAG-3 in mouse and human neurons28, despite expression in the brain5, α-syn PFF–LAG-3 interaction in disease settings cannot be ruled out. A major limitation of this initial work is that it did not rule out other α-syn PFF binding partners such as neurexin 1β and amyloid beta precursor-like protein 1 (APLP1), which might affect α-syn PFF transmission. In the absence of LAG-3, α-syn PFF binds neurexin 1β and APLP1, which raises the question of whether LAG-3 is a valid ligand in α-synucleinopathies or if LAG-3 functions in a synergistic manner with neurexin 1β and APLP1. LAG-3 might also bind to α-syn PFF–LAG-3 via another molecule. As neurexin 1β facilitates cell–cell transmission by forming intracellular junctions at neuron synapses, it would be worthwhile investigating the relative contributions of α-syn PFF binding partners (LAG-3, APLP1 and neurexin 1β) in neurological disorders. It will also be important to identify domains and residues that mediate LAG-3 binding to these ligands. Given these contradictory reports, additional validation studies are needed and could have implications for the potential of LAG-3 modalities in the treatment of neurodegenerative disorders.
Given the diversity of LAG-3 ligands, their hierarchy in terms of importance and function also needs to be defined. In addition, whether certain LAG-3–ligand interactions are disease specific and therapeutically targetable is important to delineate. Furthermore, we do not know if LAG-3 ligands function independently or have an additive/synergistic effect in modulating inhibitory function and if some ligands are more important than others for LAG-3 functionality. Given the potential importance of the TCR–CD3 complex as a ligand in cis that can function in the absence of MHC class II, we need to understand when interaction with the LAG-3 canonical ligand is important. For example, MHC class II might be involved when TCR ligand (pMHC) density is low and thus LAG-3 needs ‘help’ to maximize entry into the immune synapse. Future mechanistic dissection would benefit from understanding the relative contributions of canonical versus noncanonical ligand interactions. Lastly, characterizing a variety of anti-LAG-3 monoclonal antibody epitopes, their ability to block the various LAG-3 ligands and their subsequent effect in a variety of disease settings will help understand the relative importance of different LAG-3–ligand interactions and their effect on T cell function.
LAG-3 signaling and mechanism of action
Although LAG-3 has been studied extensively as a potential immunomodulatory target, its inhibitory signaling mechanism is not entirely clear. LAG-3 mediates inhibitory function via a cytoplasmic domain of four phylogenetically conserved motifs with undefined function: a membrane-proximal region FXXL motif, a putative serine phosphorylation site, a KIEELE motif and the repetitive C-terminal EP motif6,7,29,30. Early studies reported that LAP (LAG-3-associated protein), identified via a yeast two-hybrid screen, binds to the LAG-3 EP motif30. However, this finding has not been validated. Around the same time, a screen of several LAG-3 cytoplasmic domain mutants using a T cell hybridoma system identified the KIEELE motif as necessary for interleukin (IL)-2 production7.
Subsequent work indicated that LAG-3–pMHC class II interactions transduce two independent inhibitory signals via distinct intracellular motifs: the C-terminal EP repeat, and the FXXL motif in the membrane-proximal region29. The importance of the EP motif was further highlighted in a detailed dissection of the mechanism of LAG-3 function and how it affects TCR signaling. LAG-3 was shown to associate with the TCR–CD3 complex and track to the immunological synapse, where it subsequently disrupts the association of the CD4 and CD8 co-receptors with the tyrosine kinase p56lck (Lck). This effect is mediated by three unique features of the EP motif: (1) The highly acidic EP motif-containing LAG-3 cytoplasmic domain (pI: 4.75) associates via electrostatic interaction with the basic CD4 or CD8 cytoplasmic domains (pI: 11.25 and 11.62, respectively); (2) The EP motif lowers the local pH around the TCR–CD3–CD4/CD8 complex disrupting co-receptor–Lck interactions; and (3) The EP motif also binds and sequesters Zn2+ cations that are required for the co-receptor–Lck interaction. Collectively, these features of the EP motif disrupt co-receptor–Lck function, limiting CD3ζ and ZAP70 phosphorylation and downstream TCR signaling (Fig. 2)26.
Fig. 2 |. LAG-3 signaling.
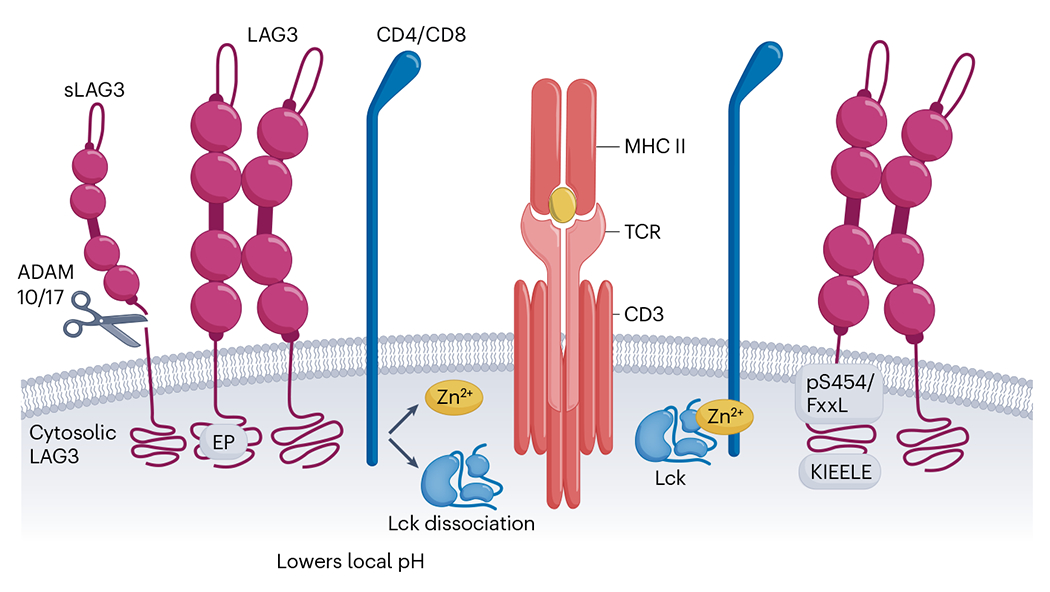
The ‘EP’, KIEELE’ and ‘pS454/FxxL’ motifs are highly conserved motifs within the cytoplasmic tail of LAG-3. The ‘EP’ motif causes Lck to dissociate from CD4/CD8 by lowering the local pH. The mechanisms of action for ‘KIEELE’ and ‘pS454/FxxL’ are unknown. sLAG3, soluble LAG-3; MHC II, MHC class II.
In neurons, LAG-3 might mediate cell–cell transmission through α-syn PFF in α-synucleinopathies in which the D1 loop of LAG-3 binds to the acidic α-syn C terminus. This seems to mediate internalization of exogenous misfolded α-syn PFF in a clathrin-dependent endocytic process31. LAG-3–α-syn colocalizes with Rab5, Rab7 and endosomal GTPases during neuronal tranmission27, facilitating pathological transmission of α-syn in neurons and transsynaptic synuclein transfer via LAG-3 and Rab5A32,33. As already discussed, there is controversy surrounding LAG-3 as the sole ligand responsible for α-syn PFF transmission. This issue is highlighted by the observation that APLP1, an α-syn PFF binding partner and member of the APP family, colocalizes with synaptic microvesicle protein (synaptophysin) and early endosome protein (Rab5) during endocytosis, indicating a role of APLP1 in neuron transmission.
In future studies, it will be important to look at the relative importance of the various LAG-3 cytoplasmic tail motifs in physiological and disease settings, and if there are other undiscovered motifs. For example, it is unknown if some known motifs, such as the serine phosphorylation site and KIEELE motif, mediate LAG-3 signaling and function in primary cells. We also do not know if the various motifs function independently or in conjunction with the EP or other motifs. Additionally, signaling pathways downstream of the EP motif need to be further explored, for example, to see if signaling molecules are associated with the cytoplasmic domain, as has been reported for LAP30.
One other important question is whether LAG-3–ligand interactions trigger conformational changes in LAG-3 extracellular domains and, if so, how these might be transmitted to LAG-3 intracellular motifs. Furthermore, whether different LAG-3 ligands mediate different signaling mechanisms via different LAG-3 motifs in presently unclear, as is an understanding of how LAG-3 mediates its inhibitory signal in cells that do not express CD4, CD8 and/or Lck, as well as cells that express Lck but not CD4/CD8. Given the apparent diversity of ligands and signaling motifs, multiple mechanisms might account for LAG-3 function depending on the cell, tissue and disease context.
Finally, a critical component for any signaling pathway is its termination. LAG-3 has a unique and rapid mechanism of signal termination via cell surface shedding by two disintegrin and metalloprotease family members: ADAM10 and ADAM17 (refs. 25,34). This mechanism of signal termination is far more rapid than transcriptional or translational downregulation. The physiological importance of this shedding has been highlighted by the demonstration that LAG-3 mutants that are resistant to ADAM-mediated shedding have greater inhibitory activity in vitro and in vivo25,35. These observations raise two important points that require further analysis. First, it was shown that there is a tight inverse relationship between LAG-3 surface expression and ADAM10 expression, and this relationship can directly affect PD-1 responsiveness to therapeutics and patient prognosis35. Given the small size of this limited dataset, it will be important to further assess if and how LAG-3 shedding might affect patient selection and responsiveness to anti-PD-1/PD-L1 and other combinatorial immunotherapies. Second, questions remain regarding the functional effect of soluble LAG-3 that is released following ADAM-mediated shedding. Although current data suggest that soluble LAG-3, which is a monomer, has low affinity for MHC class II and does not affect T cell function25, further analysis is needed to establish this point.
Cell-specific functions of LAG-3
LAG-3 negatively regulates T cell activation and function, but its importance for other cell types is unclear. LAG-3 is widely expressed by many cell types of both lymphocytic and nonlymphocytic lineage and its expression is a hallmark of exhausted CD4+ and CD8+ T cells in the context of persistent antigenic stimulation by tumors or chronic viral infections36–39. As most studies have focused on CD8+ exhausted T cells, we have a more limited understanding of LAG-3 function in CD4+ effector T cells. LAG-3 is expressed by several regulatory populations, including thymus-derived regulatory T (Treg) cells, in which it modulates their suppressive activity40–42, and CD4+ type 1 regulatory T (Tr1) cells43. LAG-3 is also expressed by subpopulations of NK cells, NKT cells, B cells and pDCs, although our understanding of its function in these cells is limited44. LAG-3 is also expressed in the brain, especially during early development, suggesting it might be important for normal brain development and function5. Given its role in neurodegenerative disorders27, more in-depth analysis of LAG-3 expression and function in the brain, and by neurons specifically, is warranted.
Ectopic expression of LAG-3 has been shown to make Treg cells immunosuppressive45. Intra-islet LAG-3-deficient Treg cells signal through the IKAROS family zinc finger 4 (Eos) pathway with enhanced IL-2–Stat5 signaling46. The IL-27–LAG-3 axis has also been reported to enhance Treg cell functionality in T cell-induced colitis41. Tr1 cells (CD49b+LAG-3+) secrete IL-10 and are regulated by early growth response gene 2 (Egr2)47, an important molecule involved in promoting anergy via TGF-β3 in an Egr2-dependent and Fas-dependent manner48. However, CD49b and LAG-3 are also coexpressed by other cells of the T cell lineage (for example, Treg cells and CD8+ T cells), indicating that the immunosuppressive function of Tr1 cells might be dependent on CD4+ differentiation stage49. As CD49b is expressed by memory T cells and LAG-3 by activated T cells, the CD49b+LAG-3+ phenotype captures a subset of activated memory cells instead of precursor and nonactivated Tr1 cells. This phenotypic and functional heterogeneity along with precise markers for identification of peripheral Tr1 cells is understudied. Further studies will help address if Tr1 cell subsets utilize distinct suppressive mechanisms. In addition, although there are studies of LAG-3+CD8+ exhausted T cells, a deeper understanding of how LAG-3 affects T cell function is required.
Given the diversity of cell types expressing LAG-3, it is important to understand if the role and impact of LAG-3 differs across different cell types. This is relevant as an in-depth analysis of LAG-3 signaling has revealed that it targets co-receptor–LCK signaling, thereby restricting this mode of action to CD4+ or CD8+ cells29. In the spleen and thymus of naive mice, ~10% of NK cells express LAG-3 (ref. 5). However, the importance of LAG-3 expression by NK cells and their role in tumor settings is unclear. Notably, LAG-3+ regulatory B cells promote an immunosuppressive environment via IL-10 and undergo development via Bruton’s tyrosine kinase and B cell receptor signaling, independent of T cells50.
LAG-3 is also expressed by other cell types, most notably pDCs4. LAG-3 seems to maintain homeostasis in a cell-intrinsic and cell-extrinsic manner and promotes immunosuppression in melanoma51 and head and neck squamous cell carcinoma52. In autoimmune disorders, LAG-3+ pDCs cause aberrant immune activation that drives inflammation51. Yet despite a decade of research on LAG-3+ pDCs, little is understood about how these cells function in different disease settings.
Finally, studies are needed to understand if there is a differential effect of LAG-3 immunotherapy on different cell subsets and how that might correlate with therapeutic response. If there is clinical value in targeting LAG-3 only on some cell types, then cell-specific therapeutics will also need to be developed.
Importance of LAG-3 in different disease settings
LAG-3 has a major function in tumors36,38,39, chronic viral infections37,53, autoimmune disorders54,55 and α-synucleinopathies27,56. Sustained LAG-3 expression has been directly correlated with an exhaustion phenotype in tumors and viral infections38,39,53, exemplified by impaired T cell proliferation and cytokine secretion thereby blocking antitumor and antiviral immunity. By contrast, LAG-3 also limits autoimmunity, as its deletion or blockade exacerbates autoimmune diabetes55,57,58. In tumors and viral infections, transcriptional analysis of exhausted T cells shows elevated expression of LAG-3 and other inhibitory receptors such as PD-1, TIGIT and TIM3. Antigen-specific tumor-infiltrating CD8+ T cells are negatively regulated by LAG-3 and PD-1 in multiple human tumors, including ovarian cancer59, head and neck squamous cell carcinoma60 and hepatocellular carcinoma61.
Similar results have been reported for intrahepatic hepatitis C virus infection62 and human T cell lymphotropic virus type I infection63. In chronic viral infection, CD4+ T cell exhaustion correlates with the loss of helper T cell function during infection with hepatitis B virus64 and with hepatitis C virus65. In non–small-cell lung cancer, a higher frequency of LAG-3+CD4+CD25− T cells infiltrates metastases than primary tumors66. Aside from the T cell compartment, high LAG-3 expression has also been reported for NKT cells in non–small-cell lung cancer67 and in chronic infection with human immunodeficiency virus68, although a major limitation is the challenge to define its role in these cells in a cancer setting. High LAG-3 expression also has been reported in mature B cells in chronic lymphocytic leukemia69.
Inhibitory receptors such as LAG-3, PD-1, TIM3, TIGIT and CTLA4 are important in mediating peripheral tolerance in autoimmunity45,70. Although many studies have addressed the role of LAG-3 in driving T cell exhaustion in tumor and chronic viral infections, LAG-3 also affects autoimmune disorders where it might be even more important. For example, two independent studies have reported a direct correlation between absence of LAG-3 and potentiation of autoimmune diabetes5,58. These findings highlight the potential importance of LAG-3 in autoimmunity where enhancing LAG-3 function could limit T cell-mediated pathology46,54. In the absence of LAG-3, high T cell infiltration is detected in the pancreatic islets of nonobese diabetic mice58. Additionally, these autoreactive intra-islet CD8+ T cells are transcriptionally, phenotypically and epigenetically restrained via an exhaustion-like program that is modulated by LAG-3 in nonobese diabetic mice55. As CD8+ T cell restraint is important in delaying disease onset, induction of this restrained phenotype in patients with type 1 diabetes might be a therapeutic option. Examination of this restrained CD8+ T cell subset in other autoimmune disorders would also be interesting. For example, kidney-infiltrating T cells in mouse lupus nephritis were also found to exhibit a restrained phenotype71. Identifying whether this restrained CD8+ T cell phenotype exists in autoimmune patients would enhance the translational value and relevance of these findings. Further insight into the mechanisms that promote and maintain a restrained phenotype may uncover new therapeutic approaches.
LAG-3 also modulates the function of Treg cells in autoimmunity. For example, LAG-3 limits Treg cell proliferation and function in mouse autoimmune diabetes46, whereas LAG-3+ Treg cells from patients with rheumatoid arthritis produce large amounts of IL-10 (ref. 72). In the mouse model collagen-induced arthritis, LAG-3+FOXP3− Treg cells induced by B cells are reported to ameliorate joint inflammation by driving IL-10 production73. A STAT6–LAG-3 signaling axis has also been suggested to be important for regulatory B cell function in Peyer’s patches74. Lastly, LAG-3+ T cells in systemic lupus erythematosus exhibiting regulatory function control B cell responses via Egr2-dependent and Egr3-dependent TGF-β production75. Given initial reports, it would be interesting to further explore LAG-3 expression on B cells and what function LAG-3 might have in different disease contexts.
As already noted, high LAG-3 expression in neurological disorders, such as Parkinson’s disease27 and prion diseases56, seems to affect disease but has been challenging to study. Given the potential role for LAG-3 in α-synucleinopathies, it will be important to clearly define the location of expression and the cell types (for example, neurons and supporting cells) that express LAG-3.
Development of LAG-3-targeting therapeutics
Two independent and noteworthy contributions have provided substantive insight into LAG-3 structure and mode of action, prompting a new perspective for the development of LAG-3-targeting therapeutics (Fig. 3). First, the structure of LAG-3 was solved providing fundamental insight into the molecular architecture of the LAG-3 ectodomain (D1–D4)24. Interestingly, LAG-3 forms a homodimer, confirming earlier biochemical data25, with a central axis via a conserved hydrophobic D2 domain with the remaining domains forming a V-shaped architecture24. Second, a primary mechanism of LAG-3 inhibitory signaling was identified26,76. Constitutive LAG-3 association with TCR–CD3 at the immunological synapse was shown to limit proximal TCR signaling via the co-receptor–Lck signaling pathway. Taken together, these insights indicate new possibilities to therapeutically target LAG-3 (ref. 76).
Fig. 3 |. LAG-3-specific therapeutics.
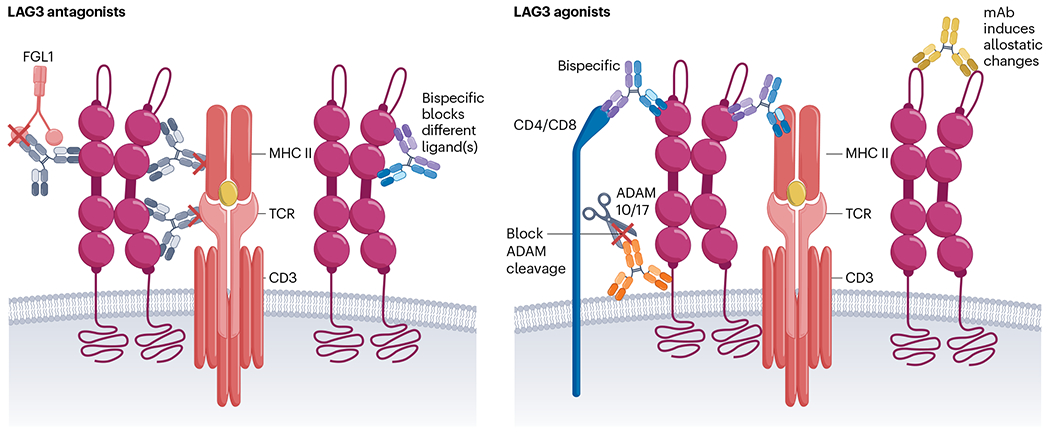
Mode of action for antagonists that mainly function by blocking LAG-3 ligand interaction with bispecifcs, providing an opportunity to simultaneously block two ligands (left). Mode of action for agonists by enhancing LAG-3 clustering to TCR–CD3, blocking ADAM10/ADAM17 (ADAM10/17)-mediated cleavage of LAG-3 and bispecifics to induce allosteric changes (right). MHC II, MHC class II; mAb, monoclonal antibody.
Although this initial LAG-3 structural insight is an important step, additional structural analysis of LAG-3 alone and in complex with its ligands and various therapeutic antibodies is needed. Further structural insight would facilitate precise mapping of LAG–3-MHC class II, LAG-3–FGL1 and LAG-3–TCR binding interfaces, providing valuable functional insight and how these interactions are disrupted by current therapeutic antibodies or could be improved with new therapeutics.
High-resolution structural insight will probably be important to generate new LAG-3 therapeutics, given that it is unclear if the current LAG-3 antagonists are optimal54,77 (Fig. 3), particularly in light of observations regarding the requirement for LAG-3 interaction with the TCR–CD3 complex26. In addition to LAG-3 therapeutics, several bispecific antagonists co-targeting LAG-3 and PD-1 or PD-L1 are also in the clinic. It will be interesting to see if these therapeutics have comparable efficacy to the current relatlimab/nivolumab combination or if there is any benefit or detriment to targeting two inhibitory receptor pathways simultaneously, and if they have different safety profiles. Another consideration is whether we need antagonists to optimally block all ligand interactions, including TCR–CD3.
Another important question relates to how to make optimal agonists for the treatment of autoimmune and inflammatory disorders. Inhibitory receptor agonists should be further explored. The precise mechanism that optimal LAG-3 agonists would need to induce needs to be deciphered for us to design optimal therapeutics. Selective depletion of LAG-3+ autoreactive T cells, while sparing LAG-3+ Treg cells, might be one therapeutic option. Whether autoimmune disorders can be managed effectively with LAG-3 agonists alone or synergistically with other inhibitory receptor agonists is unknown. Another major consideration for agonist development is the high frequency of LAG-3 shedding by ADAM metalloproteases25,34 (Fig. 3). Although this shedding could be beneficial for antagonists as a mode of action, extensive shedding could substantially limit the efficacy of agonists, suggesting that strategies to avoid it would be desirable54.
Finally, given the potential role of LAG-3 in neurological disorders, such as Parkinson’s disease27 and prion diseases56, LAG-3-targeting therapeutics might be useful, although these would need to be developed using technologies that enable therapeutic transmission across the blood–brain barrier.
Future directions
LAG-3 has emerged as a promising immunotherapeutic target. With the approval of relatlimab, a LAG-3-targeting therapeutic, opportunities exist for further clinical development. It also became the third checkpoint inhibitor to show efficacy when targeted in the clinic. Nevertheless, many questions remain, with further insight likely to affect optimal use of the current LAG-3-targeting therapeutics and the development of additional LAG-3-targeting therapeutics, especially for diseases other than cancer.
While this Perspective has focused on the five biggest topics of debate and contention surrounding LAG-3, there are many additional important questions related to how LAG-3 affects or is affected by other immune and nonimmune mechanisms that could affect optimal selection of new combinatorial treatments. Perhaps most important is that we gain a complete understanding of the mechanisms that drive the synergistic combination of therapeutics against LAG-3 and PD-1, given that this combination has had one approval from the US Food and Drug Administration and is advancing in multiple clinical trials targeting multiple tumor types. Further, what other therapeutic modalities should be combined with LAG-3 therapeutics for the treatment of different diseases? For example, given its mechanistic focus on limiting TCR signaling, LAG-3 therapies might combine well with those that target other inhibitory receptors such as TIM3 and TIGIT, co-stimulatory agonists, cytokines or chemotherapy.
One curious question is why LAG-3 is less potent than PD-1, given that LAG-3 targets TCR signaling, whereas PD-1 primarily targets the more subservient co-stimulatory signaling via CD28. This effect might partly be a result of broader expression of PD-1 versus LAG-3, but also might be related to much tighter control of LAG-3 expression via ADAM-mediated shedding34,35.
In summary, clinical approval of a LAG-3-targeting therapeutic has laid the foundation for further improvements in clinical care for cancer, but additional work is needed to maximize the potential of this therapeutic option such that the next generation of LAG-3-targeting therapeutics have better efficacy across multiple disease types, especially for autoimmune, inflammatory and neurological disorders.
Acknowledgements
The authors thank the members of the D.A.A.V. laboratory (https://www.vignali-lab.com/) for discussions and critically reading the manuscript. Figures were created using BioRender.com licensed to University of Pittsburgh. This study was supported by the US National Institutes of Health (P01 AI108545 and R01 AI144422 to D.A.A.V).
Competing interests
D.A.A.V. and C.J.W. declare patents covering LAG-3, with others pending, and are entitled to a share in net income generated from licensing of these patent rights for commercial development. D.A.A.V. is a cofounder and stock holder of Novasenta, Potenza, Tizona and Trishula and a stock holder of Oncorus and Werewolf; has patents licensed and royalties in BMS and Novasenta; is a scientific advisory board member of Tizona, Werewolf, F-Star, Bicara, Apeximmune and T7/Imreg Bio; is a consultant for BMS, Incyte, Regeneron, Ono Pharma and Avidity Partners; and receives research funding from BMS and Novasenta. The other authors declare no competing interests.
References
- 1. Triebel F. et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405 (1990). This study describes LAG-3.
- 2.Huard B, Gaulard P, Faure F, Hercend T & Triebel F Cellular expression and tissue distribution of the human LAG-3-encoded protein, an MHC class II ligand. Immunogenetics 39, 213–217 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Kisielow M, Kisielow J, Capoferri-Sollami G & Karjalainen K Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol 35, 2081–2088 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Workman CJ et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J. Immunol 182, 1885–1891 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Workman CJ, Rice DS, Dugger KJ, Kurschner C & Vignali DA Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur. J. Immunol 32, 2255–2263 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Workman CJ & Vignali DA The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol 33, 970–979 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Workman CJ, Dugger KJ & Vignali DA Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol 169, 5392–5395 (2002). [DOI] [PubMed] [Google Scholar]
- 8. Tawbi HA et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med 386, 24–34 (2022). The study reports the efficacy and safety of a phase 2/3 clinical trial of a relatlimab and nivolumab in patients with metastatic or unresectable melanoma. This combination resulted in 47.7% PFS in comparison to 36% PFS with nivolumab single therapy. No new safety signals with this combination therapy were reported.
- 9.US Food and Drug Administration. FDA approves Opdualag for unresectable or metastatic melanoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-opdualag-unresectable-or-metastatic-melanoma (2022).
- 10.Hodi FS et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 19, 1480–1492 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Long GV et al. Overall survival and response with nivolumab and relatlimab in advanced melanoma. N. Engl. J. Med 2, 4 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Ascierto PA et al. Nivolumab and relatlimab in patients with advanced melanoma that had progressed on anti-programmed death-1/programmed death ligand 1 therapy: results from the phase I/IIa RELATIVITY-020 trial. J. Clin. Oncol 41, 2724–2735 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maruhashi T. et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity 55, 912–924 (2022). This study highlights MHC class II as a critical functional ligand of LAG-3 in mouse models of autoimmunity and cancer.
- 14.Huard B. et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl Acad. Sci. USA 94, 5744–5749 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maruhashi T. et al. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol 19, 1415–1426 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Baixeras E. et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med 176, 327–337 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kouo T. et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res 3, 412–423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman LA, Ring KL & Mills AM LAG-3 and GAL-3 in endometrial carcinoma: emerging candidates for immunotherapy. Int. J. Gynecol. Pathol 39, 203–212 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Cocks MM & Mills AM The immune checkpoint inhibitor LAG-3 and its ligand GAL-3 in vulvar squamous neoplasia. Int. J. Gynecol. Pathol 41, 113–121 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Bae J. et al. Targeting LAG-3/GAL-3 to overcome immunosuppression and enhance antitumor immune responses in multiple myeloma. Leukemia 36, 138–154 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu F. et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T cell responses. Cancer Res. 74, 3418–3428 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Shi AP et al. Immune checkpoint LAG-3 and its ligand FGL1 in cancer. Front. Immunol 12, 785091 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347 (2019). FGL1 was demonstrated to be the major functional ligand for LAG-3 independently of its canonical ligand MHC class II. FGL1 was shown to inhibit antigen-specific T cell activation, and deletion of FGL1 promoted T cell immunity.
- 24. Ming Q. et al. LAG-3 ectodomain structure reveals functional interfaces for ligand and antibody recognition. Nat. Immunol 23, 1031–1041 (2022). This study reports the structure of LAG-3. The human and mouse LAG-3 ectodomains were identified as dimers via the D2 domain. The binding sites for FGL1 and MHC class II were infered to occur via the flexible loop 2 region in the LAG-3 D1 domain.
- 25.Li N, Workman CJ, Martin SM & Vignali DA Biochemical analysis of the regulatory T cell protein lymphocyte activation gene-3 (LAG-3; CD223). J. Immunol 173, 6806–6812 (2004). [DOI] [PubMed] [Google Scholar]
- 26. Guy C. et al. LAG-3 associates with TCR–CD3 complexes and suppresses signaling by driving co-receptor–Lck dissociation. Nat. Immunol 23, 757–767 (2022). This study provides clear mechanistic insight into the inhibitory function of LAG-3. It shows that LAG-3 could function independently of MHC class II and could instead use the TCR–CD3 complex as a ligand in cis within the immunological synapse. This led to dissociation of Lck from CD4 and CD8 co-receptors, resulting in reduced TCR signaling. This mechanism provides insight for the design of LAG-3-targeting immunotherapies.
- 27. Mao X. et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353, aah3374 (2016). This study demonstrates that neuron-to-neuron transmission of pathological alpha-synuclein is mediated by binding to LAG-3 via endocytosis of α-synPFF in Parkinson’s disease. Identification of α-synPFF as a ligand for LAG-3 provides new opportunities for targeting α-synucleinopathies.
- 28.Emmenegger M. et al. LAG-3 is not expressed in human and murine neurons and does not modulate alpha-synucleinopathies. EMBO Mol. Med 13, e14745 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeda TK, Sugiura D, Okazaki IM, Maruhashi T & Okazaki T Atypical motifs in the cytoplasmic region of the inhibitory immune co-receptor LAG-3 inhibit T cell activation. J. Biol. Chem 294, 6017–6026 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iouzalen N, Andreae S, Hannier S & Triebel F LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the downregulation of the CD3/TCR activation pathway. Eur. J. Immunol 31, 2885–2891 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Zhang S. et al. Mechanistic basis for receptor-mediated pathological alpha-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc. Natl Acad. Sci. USA 118, e2011196118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freeze B, Acosta D, Pandya S, Zhao Y & Raj A Regional expression of genes mediating trans-synaptic alpha-synuclein transfer predicts regional atrophy in Parkinson disease. Neuroimage Clin. 18, 456–466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu H. et al. Lymphocyte activation gene 3 (LAG-3) contributes to alpha-synucleinopathy in alpha-synuclein transgenic mice. Front. Cell Neurosci 15, 656426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li N. et al. Metalloproteases regulate T cell proliferation and effector function via LAG-3. EMBO J. 26, 494–504 (2007). This study shows that LAG-3 surface expression is controlled by two metalloproteases: ADAM10 and ADAM17. This shedding affected the inhibitory effect of LAG-3.
- 35. Andrews LP et al. Resistance to PD-1 blockade in the absence of metalloprotease-mediated LAG-3 shedding. Sci. Immunol 5, eabc2728 (2020). This study showed that expression of non-sheddable LAG-3 on CD4+ T cells limits the therapeutic efficacy of anti-PD-1 in a mouse model of cancer. Furthermore, analysis of patient samples highlighted the inverse correlation between surface LAG-3 expression and ADAM10 expression, and that high LAG-3 and low ADAM10 correlated with poor patient prognosis.
- 36.Andrews LP et al. A Cre-driven allele-conditioning line to interrogate CD4+ conventional T cells. Immunity 54, 2209–2217 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Blackburn SD et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol 10, 29–37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grosso JF et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J. Immunol 182, 6659–6669 (2009). This study shows that the combinatorial targeting of LAG-3 and PD-1 promoted sterilizing immunity against a chronic viral infection.
- 39. Woo SR et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012). This study shows that the combinatorial targeting of LAG-3 and PD-1 had synergistic antitumor activity, leading to the clinical development of LAG-3 therapeutics for the treatment of cancer.
- 40.Dadey RE, Workman CJ & Vignali DAA Regulatory T cells in the tumor microenvironment. Adv. Exp. Med. Biol 1273, 105–134 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Do JS et al. An IL-27/LAG-3 axis enhances Foxp3+ regulatory T cell-suppressive function and therapeutic efficacy. Mucosal Immunol. 9, 137–145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Overacre-Delgoffe AE & Vignali DAA Treg fragility: a prerequisite for effective antitumor immunity? Cancer Immunol. Res 6, 882–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gagliani N. et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med 19, 739–746 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Andrews LP, Marciscano AE, Drake CG & Vignali DA LAG-3 (CD223) as a cancer immunotherapy target. Immunol. Rev 276, 80–96 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang CT et al. Role of LAG-3 in regulatory T cells. Immunity 21, 503–513 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Zhang Q. et al. LAG-3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol 2, eaah4569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Okamura T. et al. CD4+CD25−LAG-3+ regulatory T cells controlled by the transcription factor Egr-2. Proc. Natl Acad. Sci. USA 106, 13974–13979 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okamura T. et al. TGF-β3-expressing CD4+CD25−LAG-3+ regulatory T cells control humoral immune responses. Nat. Commun 6, 6329 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang W, Solouki S, Carter C, Zheng SG & August A Beyond type 1 regulatory T cells: coexpression of LAG-3 and CD49b in IL-10-producing T cell lineages. Front. Immunol 9, 2625 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lino AC et al. LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity 49, 120–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Castelli C, Triebel F, Rivoltini L & Camisaschi C Lymphocyte activation gene-3 (LAG-3, CD223) in plasmacytoid dendritic cells (pDCs): a molecular target for the restoration of active antitumor immunity. Oncoimmunology 3, e967146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang LL et al. pDC depletion induced by CD317 blockade drives the antitumor immune response in head and neck squamous cell carcinoma. Oral Oncol. 96, 131–139 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Wherry EJ et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Grebinoski S & Vignali DA Inhibitory receptor agonists: the future of autoimmune disease therapeutics? Curr. Opin. Immunol 67, 1–9 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Grebinoski S. et al. Autoreactive CD8+ T cells are restrained by an exhaustion-like program that is maintained by LAG-3. Nat. Immunol 23, 868–877 (2022). Intra-islet CD8+ T cells in autoimmune diabetes have phenotypic, transcriptional, metabolic and epigenetic characteristics of exhausted T cells identified as the ‘restrained’ phenotype. CD8+ T cell–restricted deletion of LAG-3 perturbs the ‘restrained’ phenotype and accelerates disease phenotype, implicating LAG-3 as a target for autoimmune therapy.
- 56.Liu Y, Sorce S, Nuvolone M, Domange J & Aguzzi A Lymphocyte activation gene 3 (LAG-3) expression is increased in prion infections but does not modify disease progression. Sci. Rep 8, 14600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okazaki T. et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med 208, 395–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bettini M. et al. Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. J. Immunol 187, 3493–3498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsuzaki J. et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl Acad. Sci. USA 107, 7875–7880 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng WW et al. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology 5, e1239005 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo M. et al. Expression and clinical significance of LAG-3, FGL1, PD-L1 and CD8+ T cells in hepatocellular carcinoma using multiplex quantitative analysis. J. Transl. Med 18, 306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clements DM et al. Phenotypic and functional analyses guiding combination immune checkpoint immunotherapeutic strategies in HTLV-1 infection. Front. Immunol 12, 608890 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen N. et al. Lymphocyte activation gene 3 negatively regulates the function of intrahepatic hepatitis C virus-specific CD8+ T cells. J. Gastroenterol. Hepatol 30, 1788–1795 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Dong Y. et al. CD4+ T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 20, 27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J. et al. Elevated LAG-3 on CD4+ T cells negatively correlates with neutralizing antibody response during HCV infection. Immunol. Lett 212, 46–52 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Ma QY, Huang DY, Zhang HJ, Wang S & Chen XF Function and regulation of LAG-3 on CD4+CD25− T cells in non-small cell lung cancer. Exp. Cell. Res 360, 358–364 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Datar I. et al. Expression analysis and significance of PD-1, LAG-3 and TIM-3 in human non-small cell lung cancer using spatially resolved and multiparametric single-cell analysis. Clin. Cancer Res 25, 4663–4673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Juno JA et al. Elevated expression of LAG-3, but not PD-1, is associated with impaired iNKT cytokine production during chronic HIV-1 infection and treatment. Retrovirology 12, 17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kotaskova J. et al. High expression of lymphocyte-activation gene 3 (LAG-3) in chronic lymphocytic leukemia cells is associated with unmutated immunoglobulin variable heavy chain region (IGHV) gene and reduced treatment-free survival. J. Mol. Diagn 12, 328–334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang RY et al. LAG-3 and PD-1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 6, 27359–27377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tilstra JS et al. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Invest 128, 4884–4897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakachi S. et al. Interleukin-10-producing LAG-3+ regulatory T cells are associated with disease activity and abatacept treatment in rheumatoid arthritis. Arthritis Res. Ther 19, 97 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen SY, Hsu WT, Chen YL, Chien CH & Chiang BL Lymphocyte-activation gene 3+ (LAG-3+) forkhead box protein 3− (FOXP3−) regulatory T cells induced by B cells alleviates joint inflammation in collagen-induced arthritis. J. Autoimmun 68, 75–85 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Chu KH, Lin SY & Chiang BL STAT6 pathway is critical for the induction and function of regulatory T cells induced by mucosal B cells. Front. Immunol 11, 615868 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morita K. et al. Egr2 and Egr3 in regulatory T cells cooperatively control systemic autoimmunity through Ltbp3-mediated TGF-β3 production. Proc. Natl Acad. Sci. USA 113, E8131–E8140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hivroz C. LAG-3 disrupts the TCR signal by local acidification. Nat. Immunol 23, 649–651 (2022). [DOI] [PubMed] [Google Scholar]
- 77.Mullard A. LAG-3 pushes immuno-oncology’s leading edge. Nat. Rev. Drug Discov 21, 167–169 (2022). [DOI] [PubMed] [Google Scholar]