

Abstract
Limb girdle muscular dystrophy (LGMD) 2F is caused by mutations in the δ-sarcoglycan (SG) gene. Previously, we have shown successful application of a recombinant adeno-associated virus (AAV) vector for genetic and biochemical rescue in the Bio14.6 hamster, a homologous animal model for LGMD 2F (J. Li et al., Gene Ther. 6:74–82, 1999). In this report, we show efficient and long-term δ-SG expression accompanied by nearly complete recovery of physiological function deficits after a single-dose AAV vector injection into the tibialis anterior muscle of the dystrophic hamsters. AAV vector treatment led to more than 97% recovery in muscle strength for both the specific twitch force and the specific tetanic force, when compared to the age-matched control. Vector treatment also prevented pathological muscle hypertrophy and resulted in normal muscle weight and size. Finally, vector-treated muscle showed substantial improvement of the histopathology. This is the first report of successful functional rescue of an entire muscle after AAV-mediated gene delivery. This report also demonstrates the feasibility of in vivo gene therapy for LGMD patients by using AAV vectors.
Limb girdle muscular dystrophies (LGMD) are a group of heterogeneous inherited neuromuscular diseases. The severe and early-age onset phenotypes are often caused by mutations in sarcoglycan (SG) genes α (LGMD 2D), β (LGMD 2E), γ (LGMD 2C), and δ (LGMD 2F) (for a review, see reference 10 and references therein). These small transmembrane proteins associate in equal stoichiometry on the muscle cell membrane to form a heterotetramer, termed the SG complex. Primary deficiency of any single SG protein generally results in partial or complete disappearance of the entire SG complex on the sarcolemma, leading to muscular dystrophy. The lack of effective treatment for LGMD necessitates the search for innovative therapeutic strategies, such as gene therapy (8, 12, 17). Although LGMD 2F itself is a rare and genetically recessive disease, the success of gene therapy in treating this disease will benefit other genetic disorders as well.
The first available animal model for LGMD is the naturally occurring cardiomyopathic Syrian hamster Bio14.6 (13), which has a deletion in the δ-SG gene (25, 27). This primary genetic impairment causes secondary biochemical deficiency of the entire SG complex on sarcolemma of myofibers. In addition, no SG proteins can be detected within the muscle cells due to rapid degradation in the absence of the SG complex, although the mRNA transcripts are normal for α, β, and γ components (25, 27). Besides severe cardiomyopathy, the Bio14.6 hamsters also suffer from skeletal muscle myopathy, with degeneration and regeneration, necrosis, and central nucleation of myofibers. Muscle physiology studies have revealed overt pathological hypertrophy accompanied by profound deficits in contractile forces, compared to normal F1B hamsters (J. F. Watchko, J. Li, M. J. Daood, E. P. Hoffman, and X. Xiao, submitted for publication). Since the disease in the hamster is genetically and biochemically similar to that in human LGMD 2F patients, the Bio14.6 hamster provides an excellent animal model to develop gene therapy for LGMD.
Vectors based on the nonpathogenic and defective adeno-associated virus (AAV) (2, 23, 34) have proven to be the most successful in vivo gene transfer system currently available for muscle-directed gene therapy (15, 35). AAV vectors are capable of efficiently and stably transducing both mature and immature muscle cells (26a) but incapable of eliciting host cytotoxic T-lymphocyte immune responses against vector-transduced cells (14, 35). The vectors can also integrate into the host chromosome DNA, therefore rendering long-term gene transfer. These features have been employed for gene transfer of both cellular and secretable proteins by using muscle as a platform (1, 7–9, 15, 17, 22, 30). Previously, we have shown efficient transduction of the human δ-SG gene by an AAV vector and restoration of the SG complex to the sarcolemma in the Bio14.6 dystrophic hamsters (17). In addition, others have shown a protective effect at the myofiber level after δ-SG gene transfer with either adenoviral (12) or AAV vectors (8). However, no previous report has shown functional rescue in an entire muscle tissue, e.g., muscle weakness and pathological hypertrophy. Here we report the first evidence of efficient and long-term rescue of muscle functional deficits in Bio14.6 hamsters after a single administration of an AAV vector containing the δ-SG gene. Specifically, direct intramuscular injection of the AAV–δ-SG vector into the tibialis anterior (TA) muscle resulted in extensive gene transfer and high levels of δ-SG expression, leading to sustained restoration of the SG complex throughout the muscle tissue. More importantly, biochemical recovery, as a result of AAV vector-mediated gene therapy, produced therapeutic efficacy at the physiological levels, including rescue of the contractile force deficits, correction of pathological hypertrophy, and major improvement in muscle histology. Our results demonstrate feasibility for clinical application of AAV vector-mediated gene therapy for SG deficiency in human patients.
MATERIALS AND METHODS
Production of AAV vectors.
Construction of an AAV vector containing human δ-SG cDNA under the control of the cytomegalovirus (CMV) promoter (AAV–CMV–δ-SG) has been reported previously (17). The recombinant viral vector stocks were produced by cotransfection methods as described by Xiao et al. (36). The AAV vector was purified twice through CsCl density gradient purification according to the previously published protocols (29, 35). The vector titers of viral particle number were determined by the DNA dot blot method and were in the range of 2 × 1012 to 5 × 1012 viral particles per ml.
Intramuscular injection of AAV vectors.
The dystrophic Bio14.6 hamsters were purchased from Bio Breeders (Fitchburg, Mass.) and handled in accordance with the institutional guidelines of the University of Pittsburgh. Before AAV vector injection, 40-day-old hamsters were anesthetized with 2.5% Avertin intraperitoneally. The TA muscle was exposed after a small skin incision. Two 30-μl doses of AAV–CMV–δ-SG (5 × 1010 viral particles each) were injected into the middle region of the TA muscle. The two injection sites were about 5 mm apart from each other. The skin was sutured after vector injection. At 3 weeks and 4 months postintramuscular vector injection, the hamsters were euthanized and the muscle tissues were harvested for further analyses.
Western analysis.
Western analysis was carried out according to a previously published method (17). Briefly, 50 mg of the TA muscle was homogenized and lysed in radioimmunoprecipitation assay buffer (10 mM Tris-Cl [pH 8.2], 1% Triton X-100, 1% sodium dodecyl sulfate, 150 mM NaCl). The samples were separated on sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. After blocking in 10% nonfat dry milk in Tris-buffered saline (TBS) buffer (50 mM Tris-Cl [pH 7.5], 200 mM NaCl) for 1 h, the membranes were incubated with primary antibodies in TBS containing 0.5% Tween 20 at room temperature for 1 h. The polyclonal antibody recognizing a conserved epitope of human, mouse, and hamster δ-SG was used in this experiment with a 1:5,000 dilution (27). Following primary antibody incubation and rinses, the membranes were incubated with the secondary antibody, goat anti-rabbit immunoglobulin conjugated with horseradish peroxidase (Sigma), with 1:5,000 dilution in 2% dry milk and TBS buffer. After a 1-h antibody incubation and three washes with TBS buffer containing 0.5% Tween 20 and one wash with TBS, the δ-SG protein band was visualized with a chemiluminescence reagent (DuPont) and exposed to X-ray film.
Immunofluorescence staining.
Cryostat sectioning of the TA muscle tissue was performed at 5 μm thickness with an IECMinotone (International Equipment Company). For immunofluorescence staining, the unfixed muscle cryosections were immediately blocked in 10% horse serum and phosphate-buffered saline (PBS) at room temperature for 1 h. Monoclonal antibodies against α-SG and δ-SG (Novocatra Laboratories) or a polyclonal antibody to β-SG and γ-SG (21) was diluted 1:100 in 10% horse serum–PBS and incubated with the cryosections for 2 h at room temperature. After three washes, the sections were incubated with Cy-3-labeled antimouse or antirabbit secondary antibodies at 1:500 dilution in 10% horse serum–PBS (Jackson Immuno Research Laboratories). After three washes, the samples were mounted in 90% glycerol–PBS or Gelmount (Fisher). Photographs were taken with a Nikon Microphot-FXA microscope, using an Optronics DEI750 color-integrating digital camera.
Muscle physiology.
The TA muscle was isolated for in vitro study by removing the overlying bicep femoris and gently opening the fascia of the anterior compartment. The distal portion of the TA tendon was secured with 5-0 silk suture, the tendon was cut from its insertion, and the entire TA was removed with its tibial origin intact. The TA was mounted in a vertical tissue chamber which was constantly perfused with mammalian Ringers solution aerated with 95% O2–5% CO2 and maintained at 25°C. The TA tibial origin was fixed by securing the head of the tibia with a vascular clamp mounted in series to a micropositioner near the base of the tissue chamber. Care was taken to position the tibia in a vertical orientation so that TA fibers were in vertical alignment. The tendinous insertion was secured with a microaneurism clip (Fine Science Tools, Inc.) which was connected to a force transducer and length servosystem (Model 305B, dual mode; Aurora Scientific) via fine wire, providing a noncompliant attachment to the force transducer.
The muscle was stimulated (Grass model S-88 stimulator and current amplifier) by use of monophasic rectangular pulses of cathodal current (1.0-ms duration) delivered through platinum plate electrodes placed ∼1 cm apart. The TA was positioned midway between the two electrodes. To ensure supramaximal stimulation, the current was increased by 50% over the current necessary to obtain peak twitch force (∼250 to 300 mA). Muscle fiber length was adjusted incrementally by using a micropositioner until maximal isometric twitch force (Pt) responses were obtained (i.e., optimal fiber length [Lo]). Lo was measured with a microcaliper accurate to 0.1 mm (Fisher Scientific). The dependence of force generation on the rate of stimulation and maximum tetanic force (Po) was assessed by use of a range of stimulation frequencies (20, 50, 75, and 100 pulses per second) delivered in 500-ms duration trains with 2 min intervening between each train. Following these measurements, the stimulated muscle was weighed on an analytic balance (model 2100; Fisher Scientific) after tendon and bone attachments were removed and the muscle blotted dry. All forces (Newtons) were normalized for a muscle cross-sectional area (CSA), the latter estimated on the basis of the following formula: muscle weight (in grams)/[Lo (in centimeters) × 1.056 (in grams per cubic centimeter)]. The estimated CSA was used to determine specific twitch (Pt/CSA) and specific tetanic (Po/CSA) forces of the muscles.
RESULTS
Efficient and sustained restoration of the SG complex.
In our previous studies, we have demonstrated highly efficient gene transfer in the dystrophic muscle of the Bio14.6 hamster by an AAV vector carrying the therapeutic δ-SG gene (17). Although efficient restoration of the missing SG complex was demonstrated in the gastrocnemius muscle, the studies were carried out for only a short term and in limited regions of the muscle group. In this study, we chose the TA muscle, an easily isolated muscle of appropriate size, for vector injection as well as for force measurement.
The TA muscles of Bio14.6 hamsters were injected in two sites with a total dose of 1011 AAV–CMV–δ-SG vector particles at the age of 40 days. At 4 months post vector injection, the experimental animals were sacrificed and the TA muscle was isolated. After in vitro muscle physiology measurement (see below), the TA muscles were subjected to biochemical examinations by Western blot and immunofluorescence analyses. As shown in Fig. 1, high levels of human δ-SG cDNA expression were observed by Western blot analysis in the vector-injected TA muscle. AAV vector-mediated δ-SG gene expression is significantly higher than that in the same muscle from the wild-type F1B control hamster (Fig. 1, lane 1). The results show efficient and stable gene transfer by AAV vectors in the dystrophic muscle.
FIG. 1.

Western analysis of δ-SG expression in the TA muscle in the wild-type F1B hamster (lane 1), Bio14.6 dystrophic hamster (lane 2), and Bio14.6 dystrophic hamsters treated with the AAV–CMV–δ-SG vector (lanes 3 to 8) 4 months earlier. Note various high levels of δ-SG gene expression in all TA muscles which were treated with the recombinant AAV vector containing human δ-SG cDNA.
To assess the distribution of δ-SG expression within the vector-injected muscle, immunofluorescence staining was performed. The TA muscles from wild-type, vector-treated, and untreated dystrophic hamsters were cryosectioned and stained with antibodies against the four individual SG proteins (Fig. 2a). The TA muscle from the wild-type F1B hamster revealed uniform sarcolemma staining (Fig. 2a, top row) with antibodies against α-, β-, and γ-SG. Absence of δ-SG staining in the wild-type hamster muscle is due to the nature of the monoclonal antibody, which recognizes only human (not hamster) δ-SG protein. As expected, the negative control Bio14.6 TA muscle that was not treated with the AAV vector revealed no immunofluorescence staining for all four SG proteins (Fig. 2a, bottom row). However, the AAV vector-treated TA muscle of the Bio14.6 hamster showed widespread positive staining by all four antibodies against the individual SG proteins (Fig. 2a, middle row). These results indicate that sustained and widespread expression of human δ-SG cDNA from the AAV vector compensated the genetic defect in the dystrophic muscle, resulted in restoration of the entire SG complex, and lead to positive staining for all four SG proteins on the sarcolemma.
FIG. 2.

Immunofluorescence analysis of the SG complex in the TA muscle. (a) Cryosections of the TA muscle from the wild-type (WT) F1B hamster (top row), Bio14.6 hamster treated with the recombinant AAV vector (middle row), and untreated Bio14.6 hamster (bottom row) were immunofluorescently stained with antibodies against the four components of the SG complex (α-SG, β-SG, γ-SG, and δ-SG). Note that the anti-δ-SG monoclonal antibody is specific to the human δ-SG protein. Therefore, it was unable to detect the δ-SG in the wild-type hamster, but it was able to detect the human δ-SG expressed in the recombinant AAV vector-treated Bio14.6 hamster muscle. (b) Cryosections of the TA muscle from another Bio14.6 hamster treated with the recombinant AAV vector. The consecutive sections were immunofluorescently stained with antibodies either against δ-SG (A) or against α-SG (B). Note that overexpression of δ-SG in different regions of the vector-treated muscle did not interfere with the distribution of α-SG on the myofiber sarcolemma.
It is noteworthy that in our previous short-term (3 weeks) study with the gastrocnemius muscle, we observed overexpression of the δ-SG gene and accumulation of δ-SG protein in cytosol in numerous muscle fibers (17). In this study, a subset of myofibers in the TA muscle also revealed overexpression of δ-SG with the inappropriate cytoplasmic localization. Examples of the persistent overexpression of δ-SG are shown in Fig. 2b, as well as in Fig. 2a. Despite large amounts of δ-SG protein in those fibers, no detectable muscle impairment or immune consequences, such as lymphocyte infiltration, were observed. Interestingly, during the course of our present study, a duration of 4 months, overexpression of δ-SG in those myofibers apparently persisted. These findings suggest that overexpression of human δ-SG in a dystrophic hamster muscle can fulfill its normal functions without causing detrimental effects.
Correction of muscle hypertrophy and morphology.
A comparative evaluation of muscle weight in a group of age-matched (4 to 5 months old) hamsters was performed by using TA muscles isolated from normal F1B hamsters, untreated Bio14.6 hamsters, and vector-treated Bio14.6 hamsters. Since the lengths of TA muscles from all the above hamsters are identical (Watchko et al., submitted), the muscle weight differences reflect the degree of hypertrophy. As shown in Table 1, the TA muscle from the normal F1B hamster weighed 117 ± 9 mg, while TA from Bio14.6 weighed 172 ± 13 mg. However, after vector treatment the Bio14.6 TA muscle weighed 111 ± 7 mg, essentially corrected to the levels of normal F1B hamsters. This result indicated that the hypertrophy was fully reversed. Morphologically, the vector-treated TA muscle exhibited indistinguishable gross appearance from that of the wild type, while the untreated TA muscle was apparently hypertrophied.
TABLE 1.
Measurements of TA muscle weighth and forcesa
| Hamster | Muscle weight in mg (n) | Specific twitch force in N/cm2 (n) | Specific tetanic force in N/cm2 (n) |
|---|---|---|---|
| F1B (wild type) | 117 ± 9 (8) | 9.3 ± 1.5 (8) | 22.8 ± 2.3 (8) |
| Bio14.6 (untreated) | 172 ± 13 (9) | 5.5 ± 1.2 (9) | 15.6 ± 2.8 (9) |
| Bio14.6 (AAV vector treated) | 111 ± 7 (4) | 9.2 ± 0.8 (4) | 22.6 ± 1.7 (4) |
Forty-day-old male Bio14.6 hamsters were injected with 1011 particles of an δ-SG–AAV vector into the TA muscle. At 4 months after vector injection, the animals were sacrificed and the TA muscle was isolated. Age-matched F1B hamsters (wild type) and Bio14.6 hamsters (mutant) not treated with the recombinant AAV vector were used as controls. The in vitro muscle force measurement was performed according to standard protocols (see Materials and Methods). The weight of the TA muscle was determined on an analytic balance after force measurement. All values in the table include standard deviations.
Histological examination of the normal, vector-treated, and untreated dystrophic TA muscle samples yielded results consistent with the gross morphology data. Hematoxylin and eosin staining of cross sections from the normal hamster TA muscle displayed uniform myofiber sizes and peripherally localized nuclei (Fig. 3A), while the untreated Bio14.6 hamster muscle suffered focal necrosis and displayed uniform central nucleation (Fig. 3C), an indication of muscle degeneration and regeneration. However, the age-matched AAV vector-treated TA muscle of the Bio14.6 hamster showed little evidence of degeneration and regeneration with more consistent fiber size. Necrosis was absent in the vector-transduced muscle, and a majority of the centrally localized nuclei were reversed to the normal peripheral location (Fig. 3B). It is noteworthy that at the age of vector injection (40 days), degeneration and regeneration and central nucleation were already extensive in most of the myofibers of the Bio14.6 hamster TA muscle (Fig. 3D). Similar partial reversal of the central nuclei to the peripheral location has been observed in a genetically normal mouse muscle after an acute degeneration-regeneration process was stopped (19).
FIG. 3.

Histological staining (hematoxylin and eosin) of the TA muscle samples from the 5-month-old wild-type F1B hamster (A), 5-month-old AAV-treated Bio14.6 hamster (B), 5-month-old untreated Bio14.6 hamster (C), and 40-day-old untreated Bio14.6 hamster (D). Note the extensive degeneration and regeneration and focal necrosis in the untreated TA muscles.
Recovery of muscle force deficits.
A major functional deficit in muscular dystrophy patients is the loss of muscle strength. In our previous physiological study with the Bio14.6 hamster TA muscle, we revealed profound muscle force deficits (Watchko et al., submitted), similar to those seen in the dystrophic patients. Although long-term and efficient restoration of the δ-SG complex and substantial improvement of muscle morphology and histology have been accomplished, the most important criterion is to see whether the muscle force can be subsequently recovered as a result of AAV vector gene therapy in the dystrophic TA muscle. Therefore, we performed in vitro contractile force measurement by using the TA muscles from age-matched (4 to 5 months old) normal F1B hamsters, untreated Bio14.6 hamsters, and vector-treated Bio14.6 hamsters. The latter had received an intramuscular administration of 1011 viral vector particles 4 months before. The TA muscles were carefully separated from the hind legs and subjected to in vitro electrophysiological stimulation and contractile measurement on a force transducer (see Materials and Methods).
Two different muscle contractile forces were tested, i.e., peak twitch force and maximal tetanic force. In those tests, the untreated Bio14.6 TA muscle displayed significant lower muscle force than the F1B muscle when the forces were normalized for the muscle CSA. The specific peak twitch force in Bio14.6 hamsters was 5.5 ± 1.2 N/cm2 versus 9.3 ± 1.5 N/cm2 in F1B normal hamsters, showing a 41% deficit. Similarly, the specific tetanic force in Bio14.6 hamsters was 15.6 ± 2.8 N/cm2 versus 22.8 ± 2.3 N/cm2 in F1B normal hamsters, showing a 32% deficit. However, AAV–CMV–δ-SG vector-treated Bio14.6 muscles regained muscle strength, generating a specific twitch force of 9.2 ± 0.8 N/cm2 and a specific tetanic force of 22.6 ± 1.7 N/cm2. Quantitation of the force recovery score demonstrated almost complete regain of muscle strength in the vector-treated TA muscle to wild-type F1B levels. The specific peak twitch force recovered to 97.4% of the wild-type level (versus 59% in the untreated muscle). The specific tetanic force recovered to 97.2% of the wild-type level (versus 68% in the untreated muscle). The results are summarized in Table 1 and depicted in Fig. 4. The calculations of the specific peak twitch and specific tetanic force recovery scores are adopted from Deconinck et al. (4) and are respectively illustrated as follows:
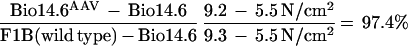 |
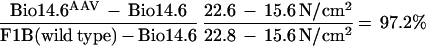 |
FIG. 4.

Comparison of TA muscle mass, twitch force, and tetanic force in wild-type F1B hamsters (dotted bar; n = 8), untreated Bio14.6 hamsters (white bar; n = 9), and AAV vector-treated Bio14.6 hamsters (hatched bar; n = 4) (see Table 1 for details).
DISCUSSION
In this report we describe the first evidence of muscle force recovery and reversal of pathological hypertrophy in the δ-SG-deficient dystrophic Bio14.6 hamsters after intramuscular injection of an AAV vector containing the human δ-SG cDNA. The therapeutic effects were achieved as a result of sustained and widespread restoration of the δ-SG complex on sarcolemma by one-time AAV vector treatment in the TA muscle of this LGMD animal model. The contractile force recovery rates of the vector-treated TA muscle were 97.4% for specific twitch force and 97.2% for specific tetanic force. Thus, the vector treatment essentially restored the contractile forces of the dystrophic muscle to the levels of the age-matched wild-type control hamsters. In addition, the pathologic hypertrophy was also fully reversed. The vector-treated muscle exhibited normal-size weight and morphology when compared to those of the wild-type age-matched control hamsters.
Recent studies with the Bio14.6 hamsters have demonstrated the feasibility of gene therapy for LGMD. Efficient restoration of the missing SG complex in the TA and gastrocnemius muscles was achieved by either adenoviral vector (12)- or AAV vector (8, 17)-mediated in vivo gene transfer of the human δ-SG gene in this hamster model. Furthermore, the protective effect of muscle cell membrane integrity has also been observed in myofibers positive for vector gene transfer, as judged by the fluorescent dye leakage tests and improvement of muscle histology (8, 12). However, because loss of muscle strength is the most direct and disabling deficiency in muscular dystrophy patients, the ultimate test to evaluate therapeutic effects in dystrophic muscle should include contractile force recovery. Partial muscle contractile force recovery has been shown in the mouse model (mdx) for Duchenne muscular dystrophy after adenoviral vector-mediated dystrophin gene transfer (3). Nonetheless, no force recovery has been previously reported in Bio14.6 hamsters after gene vector treatment, possibly due to relatively low vector transduction efficiency (8).
The Bio14.6 hamster is an excellent LGMD animal model for muscle force deficit studies and for gene therapy strategy development. The dystrophic TA muscle displays significant deficiency in muscle strength. Thus, the specific twitch force and tetanic force are 59 and 68% of those of the wild-type hamster, respectively. In addition, the TA muscle also displayed extensive central nucleation and pathological hypertrophy, which are common signs of dystrophy. Furthermore, this muscle is large enough to readily perform intramuscular vector injection but small enough to carry out in vitro contractile force measurement. Nonetheless, the TA muscle in the Bio14.6 hamster does not seem ideal for an in vivo myofiber membrane leakage test, because in vivo administration of Evans Blue dye did not reveal significant myofiber leakage under the unstressed condition (data not shown and references 8 and 31). On the contrary, the gastrocnemius muscle endured much more significant cell membrane leakage under the same unstressed condition (12). Due to the large size and large force generated beyond our instrument scale, we could not determine whether the gastrocnemius muscle suffers more force deficits than the TA muscle or how much contractile force the former recovers after AAV vector treatment (17).
AAV vectors are based on a nonpathogenic and replication-defective human parvovirus (2, 23). A large body of previous studies has demonstrated that not only wild-type AAV but also recombinant AAV vectors can integrate into the host chromosomes both in vitro and in vivo (7, 16, 18, 20, 24, 28, 33, 35). Recently, circular episomal forms of vector DNA have been discovered to also persist in vivo (5, 6). Stable vector DNA persistence and lack of host cytotoxic T-lymphocyte reactions enable efficient and long-term in vivo gene delivery by AAV vectors to treat genetic diseases. In particular, AAV can efficiently and stably transduce normal, regenerating, immature, and mature muscles (35; Pruchnic et al., submitted). This is especially suitable for muscle-orientated gene therapy for both muscular and nonmuscular diseases. Due to the small particle size and abundant receptors, the AAV vector can render widespread intramuscular gene delivery far beyond the injection needle track. In this study, immunofluorescence analysis revealed a high percentage of myofibers displaying the SG complex on the sarcolemma of vector-treated dystrophic TA muscles. Due to operation variability, gene transfer rates varied among individual samples, ranging from 60% to more than 90% of the myofibers transduced within the injected TA muscles (data not shown). Interestingly, despite the variation in transduction percentages, muscle forces as well as hypertrophy data showed nearly complete recovery in the TA muscles examined with little variation (Table 1). These results suggest that correction of over half of the diseased myofibers in a muscle will confer nearly full recovery of physiological functions in a dystrophic muscle tissue. This notion is consistent with the observations in Duchenne muscular dystrophy patients (11) as well as in Duchenne muscular dystrophy animal model mdx mice (26), where approximately 20% of the normal dystrophin expression can confer normal muscle functions.
It is noteworthy that certain areas adjacent to the injection site or certain muscle fibers had supranormal levels of transgene expression (Fig. 2b). A similar phenomenon was also observed in the gastrocnemius muscle in our previous studies (17). This overexpression might be due either to high doses of vector uptake in areas near the injection sites or to high receptor levels in a certain subtype of myofibers (32; Pruchnic et al., submitted). However, the overexpression of the human δ-SG gene has persisted in numerous myofibers through the duration of the 4-month period. No deleterious consequences to the muscle cells were detected due to the overexpression. No cellular immune responses, such as lymphocyte infiltration, were detected in the transduced muscle. These observations support the safety profile of intramuscular injection of the AAV vector that expresses the human δ-SG protein in the immunocompetent animals. Because of the short life span of Bio14.6 hamsters, which is reportedly 146 days on average (13), we did not carry out any long-term studies on hamsters beyond 5 months of age. However, recently the vender Bio Breeders informed us that under the improved animal care conditions, Bio14.6 hamsters can have prolonged life spans of up to 10 to 12 months, much longer than that originally reported about 30 years ago. This will allow us to carry out longer-term safety studies. Since muscle is one of the largest organs in humans, gene therapy through direct intramuscular injection requires the production and in vivo delivery of enormous amounts of viral vectors. This challenging task awaits novel technological advancement in vector production and delivery, as well as preclinical and clinical safety studies. Interestingly, a novel vector delivery methodology has been developed recently which administers the vectors through the blood vessel into the muscle tissue. Such a systemic gene delivery method in combination with high-titer AAV vectors (36) should render more widespread and uniform transduction than direct intramuscular injection in the target muscle tissues (8).
ACKNOWLEDGMENTS
We thank L. W. Sun and R. Pruchnic for assistance with the graphics.
This work is supported by grants from the National Institutes of Health (RO1AR45967-01 and AR45925-01 to X. Xiao).
REFERENCES
- 1.Barton-Davis E R, Shoturma D I, Musaro A, Rosenthal N, Sweeney H L. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci USA. 1998;95:15603–15607. doi: 10.1073/pnas.95.26.15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berns K I, Giraud C. Biology of adeno-associated virus. Curr Top Microbiol Immunol. 1996;218:1–23. doi: 10.1007/978-3-642-80207-2_1. [DOI] [PubMed] [Google Scholar]
- 3.Deconinck N, Ragot T, Marechal G, Perricaudet M, Gillis J M. Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene. Proc Natl Acad Sci USA. 1996;93:3570–3574. doi: 10.1073/pnas.93.8.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deconinck N, Tinsley J, De Backer F, Fisher R, Kahn D, Phelps S, Davies K, Gillis J M. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med. 1997;3:1216–1221. doi: 10.1038/nm1197-1216. [DOI] [PubMed] [Google Scholar]
- 5.Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y, Fisher K J, Engelhardt J F. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol. 1998;72:8568–8577. doi: 10.1128/jvi.72.11.8568-8577.1998. . (Erratum, 73:861, 1999.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duan D, Yue Y, Yan Z, McCray P B, Jr, Engelhardt J F. Polarity influences the efficiency of recombinant adenoassociated virus infection in differentiated airway epithelia. Hum Gene Ther. 1998;9:2761–2776. doi: 10.1089/hum.1998.9.18-2761. [DOI] [PubMed] [Google Scholar]
- 7.Fisher K J, Jooss K, Alston J, Yang Y, Haecker S E, High K, Pathak R, Raper S E, Wilson J M. Recombinant adeno-associated virus for muscle directed gene therapy. Nat Med. 1997;3:306–312. doi: 10.1038/nm0397-306. [DOI] [PubMed] [Google Scholar]
- 8.Greelish J P, Su L T, Lankford E B, Burkman J M, Chen H, Konig S K, Mercier I M, Desjardins P R, Mitchell M A, Zheng X G, Leferovich J, Gao G P, Balice-Gordon R J, Wilson J M, Stedman H H. Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat Med. 1999;5:439–443. doi: 10.1038/7439. [DOI] [PubMed] [Google Scholar]
- 9.Herzog R W, Hagstrom J N, Kung S H, Tai S J, Wilson J M, Fisher K J, High K A. Stable gene transfer and expression of human blood coagulation factor IX after intramuscular injection of recombinant adeno-associated virus. Proc Natl Acad Sci USA. 1997;94:5804–5809. doi: 10.1073/pnas.94.11.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman E P. Counting muscular dystrophies in the post-molecular census. J Neurol Sci. 1999;164:3–6. doi: 10.1016/s0022-510x(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman E P, Brown R H, Kunkel L M. Dystrophin: the protein product of the Duchene muscular dystrophy locus. Biotechnology. 1992;24:457–466. [PubMed] [Google Scholar]
- 12.Holt K H, Lim L E, Straub V, Venzke D P, Duclos F, Anderson R D, Davidson B L, Campbell K P. Functional rescue of the sarcoglycan complex in the BIO14.6 hamster using delta-sarcoglycan gene transfer. Mol Cell. 1998;1:841–848. doi: 10.1016/s1097-2765(00)80083-0. [DOI] [PubMed] [Google Scholar]
- 13.Homburger F, Baker J R, Nixon C W, Whitney R. Primary, generalized polymyopathy and cardiac necrosis in an inbred line of Syrian hamsters. Med Exp. 1962;6:339–345. doi: 10.1001/archinte.1962.03620230106015. [DOI] [PubMed] [Google Scholar]
- 14.Jooss K, Yang Y, Fisher K J, Wilson J M. Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J Virol. 1998;72:4212–4223. doi: 10.1128/jvi.72.5.4212-4223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kessler P D, Podsakoff G M, Chen X, McQuiston S A, Colosi P C, Matelis L A, Kurtzman G J, Byrne B J. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci USA. 1996;93:14082–14087. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotin R M, Siniscalco M, Samulski R J, Zhu X D, Hunter L, Laughlin C A, McLaughlin S, Muzyczka N, Rocchi M, Berns K I. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci USA. 1990;87:2211–2215. doi: 10.1073/pnas.87.6.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Dressman D, Tsao Y P, Sakamoto A, Hoffman E P, Xiao X. rAAV vector-mediated sarcoglycan gene transfer in a hamster model for limb girdle muscular dystrophy. Gene Ther. 1999;6:74–82. doi: 10.1038/sj.gt.3300830. [DOI] [PubMed] [Google Scholar]
- 18.Lubovy M, McCune S, Dong J Y, Prchal J F, Townes T M, Prchal J T. Stable transduction of recombinant adeno-associated virus into hematopoietic stem cells from normal and sickle cell patients. Biol Blood Marrow Transplant. 1996;2:24–30. [PubMed] [Google Scholar]
- 19.Martin H, Ontell M. Regeneration of dystrophic muscle following multiple injections of bupivaccine. Muscle Nerve. 1988;11:588–596. doi: 10.1002/mus.880110611. [DOI] [PubMed] [Google Scholar]
- 20.Miao C H, Snyder R O, Schowalter D B, Patijn G A, Donahue B, Winther B, Kay M A. The kinetics of rAAV integration in the liver. Nat Genet. 1998;19:13–15. doi: 10.1038/ng0598-13. [DOI] [PubMed] [Google Scholar]
- 21.Mizuno Y, Noguchi S, Yamamoto H, Yoshida M, Nonaka I, Hirai S, Ozawa E. Sarcoglycan complex is selectively lost in dystrophic hamster muscle. Am J Pathol. 1995;146:530–536. [PMC free article] [PubMed] [Google Scholar]
- 22.Monahan P E, Samulski R J, Tazelaar J, Xiao X, Nichols T C, Bellinger D A, Read M S, Walsh C E. Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophilia. Gene Ther. 1998;5:40–49. doi: 10.1038/sj.gt.3300548. [DOI] [PubMed] [Google Scholar]
- 23.Muzyczka N. Use of adeno-associated virus as a general transduction vector for mammalian cells. Curr Top Microbiol Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]
- 24.Nakai H, Iwaki Y, Kay M A, Couto L B. Isolation of recombinant adeno-associated virus vector-cellular DNA junctions from mouse liver. J Virol. 1999;73:5438–5447. doi: 10.1128/jvi.73.7.5438-5447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nigro V, Okazaki Y, Belsito A, Piluso G, Matsuda Y, Politano L, Nigro G, Ventura C, Abbondanza C, Molinari A M, Acampora D, Nishimura M, Hayashizaki Y, Puca G A. Identification of the Syrian hamster cardiomyopathy gene. Hum Mol Genet. 1997;6:601–607. doi: 10.1093/hmg/6.4.601. [DOI] [PubMed] [Google Scholar]
- 26.Phelps S F, Hauser M A, Cole N M, Rafael J A, Hinkle R T, Faulkner J A, Chamberlain J S. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum Mol Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- 26a.Pruchnic, R., B. H. Cao, Z. Qu, X. Xiao, J. Li, R. J. Samulski, M. Epperly, and J. Huard. The use of adeno-associated virus to circumvent the maturation dependent viral transduction of muscle fiber. Hum. Gene Ther., in press. [DOI] [PubMed]
- 27.Sakamoto A, Ono K, Abe M, Jasmin G, Eki T, Murakami Y, Masaki T, Toyo-oka T, Hanaoka F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: an animal model of disrupted dystrophin-associated glycoprotein complex. Proc Natl Acad Sci USA. 1997;94:13873–13878. doi: 10.1073/pnas.94.25.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samulski R J, Zhu X, Xiao X, Brook J D, Housman D E, Epstein N, Hunter L A. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J. 1991;10:3941–3950. doi: 10.1002/j.1460-2075.1991.tb04964.x. . (Erratum, 11:1228, 1992.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Snyder R, Xiao X, Samulski R J. Production of recombinant adeno-associated viral vectors. In: Dracopoli N, Haines J, Krof B, Moir D, Seidman C, Seidman J S, editors. Current protocols in human genetics. New York, N.Y: John Wiley & Sons Ltd.; 1996. pp. 12.1.1–12.2.23. [Google Scholar]
- 30.Song S, Morgan M, Ellis T, Poirier A, Chesnut K, Wang J, Brantly M, Muzyczka N, Byrne B J, Atkinson M, Flotte T R. Sustained secretion of human alpha-1-antitrypsin from murine muscle transduced with adeno-associated virus vectors. Proc Natl Acad Sci USA. 1998;95:14384–14388. doi: 10.1073/pnas.95.24.14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Straub V, Rafael J A, Chamberlain J S, Campbell K P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Summerford C, Samulski R J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu P, Phillips M I, Bui J, Terwilliger E F. Adeno-associated virus vector-mediated transgene integration into neurons and other nondividing cell targets. J Virol. 1998;72:5919–5926. doi: 10.1128/jvi.72.7.5919-5926.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao X, Li J, McCown T J, Samulski R J. Gene transfer by adeno-associated virus vectors into the central nervous system. Exp Neurol. 1997;144:113–124. doi: 10.1006/exnr.1996.6396. [DOI] [PubMed] [Google Scholar]
- 35.Xiao X, Li J, Samulski R J. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao X, Li J, Samulski R J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]