

Summary
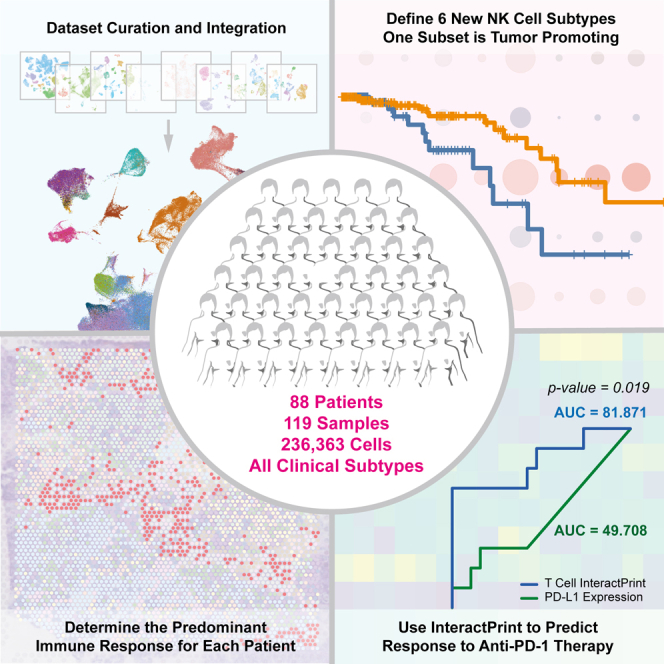
We present an integrated single-cell RNA sequencing atlas of the primary breast tumor microenvironment (TME) containing 236,363 cells from 119 biopsy samples across eight datasets. In this study, we leverage this resource for multiple analyses of immune and cancer epithelial cell heterogeneity. We define natural killer (NK) cell heterogeneity through six subsets in the breast TME. Because NK cell heterogeneity correlates with epithelial cell heterogeneity, we characterize epithelial cells at the level of single-gene expression, molecular subtype, and 10 categories reflecting intratumoral transcriptional heterogeneity. We develop InteractPrint, which considers how cancer epithelial cell heterogeneity influences cancer-immune interactions. We use T cell InteractPrint to predict response to immune checkpoint inhibition (ICI) in two breast cancer clinical trials testing neoadjuvant anti-PD-1 therapy. T cell InteractPrint was predictive of response in both trials versus PD-L1 (AUC = 0.82, 0.83 vs. 0.50, 0.72). This resource enables additional high-resolution investigations of the breast TME.
Graphical abstract
Highlights
-
•
Generated a large single-cell RNA sequencing primary breast tumor atlas
-
•
Identified six new NK cell subsets in the breast tumor microenvironment
-
•
Determined how cancer cell heterogeneity influences immune response in the breast TME
-
•
InteractPrint predicts patient response to immunotherapy across subtypes
Xu, Saunders, and Huang et al. generate a large, single-cell RNA sequencing dataset of the breast tumor microenvironment. They use this real-world data atlas to further characterize NK cells and create InteractPrint. InteractPrint is a method that accounts for tumor heterogeneity and accurately predicts response to immunotherapy across breast cancer subtypes.
Introduction
Breast cancer is the most common cancer among women.1 The development of breast cancer is driven by both cancer epithelial cell-intrinsic factors2,3,4 and the tumor microenvironment (TME).5,6 The medical treatment of breast cancer therefore targets these diverse cell populations and includes traditional chemotherapy, targeted agents inhibiting cancer cell hormone receptors, kinases, cell cycle entry, and immune cell modulators. To further improve these therapies, a deeper understanding of the cellular and molecular composition of breast tumors is required.
Single-cell RNA sequencing (scRNA-seq) technology has been applied to better characterize tumor microenvironments. For breast cancer, several scRNA-seq studies have been performed to identify key immune, cancer cell, and stromal populations of the breast TME.7,8,9,10,11,12,13,14 These studies provided insight into molecular phenotypes of cancer cells, multiple immune populations, and other stromal cells. However, each study was limited by the number of samples and cells analyzed. This poses challenges to performing comprehensive analysis of heterogeneous cell populations and their cellular interactions in the TME.
For example, natural killer (NK) cells are innate lymphoid immune cells critical to anti-tumor defense. In breast cancer, tumor-infiltrating NK cells are rare,15,16,17,18,19 representing 1%–6% of total tumor cells in published scRNA-seq datasets of primary breast tumors.7,8,9,11,12,13,14 Their cytotoxic activity is regulated by a series of functionally activating and inactivating receptors. After tumor exposure, the balance of NK cell-activating and -inactivating receptors can change, and they can lose their cytotoxic activity or proliferative capacity or even become tumor promoting.20,21,22 Because of the small numbers of NK cells processed in most human studies, scRNA-seq analyses of NK cells are often underpowered to capture their distinct functional phenotypes. Additionally, breast cancer is known to have substantial heterogeneity within the tumor of a single patient and between patients of a clinical subtype.23,24 Therefore, comprehensive analysis of cancer epithelial cell heterogeneity requires large and diverse datasets with adequate numbers of samples from all clinical breast cancer subtypes.
In this study, we created an integrated scRNA-seq atlas of the breast TME, consisting of 236,363 cells from 119 biopsy samples across 8 publicly available datasets.7,8,9,10,11,12,13,14 This resource enables separation of cell populations within primary breast tumors and robust characterization of cellular heterogeneity at the single-cell level. This integrated dataset is more statistically powerful than traditional meta-analyses of original source datasets and enables evaluation of correlations with clinical features. We used this resource to define immune and cancer epithelial cell heterogeneity along with their interactions. It is the first, to our knowledge, to define NK cell subsets in breast cancer and provides evidence that cancer epithelial cell heterogeneity influences immune interactions and response to anti-PD-1 therapy. This dataset provides a comprehensive resource to better understand the composition of the breast TME.
Results
An integrated scRNA-seq dataset of breast cancer samples reveals distinct NK cell subsets that exhibit diverse functional characteristics
To develop a high-resolution atlas of the breast TME, we analyzed scRNA-seq data from 119 samples collected from primary tumor biopsies of 88 patients across 8 publicly available breast cancer datasets (Figures 1A and S1A–S1C; Data S1).7,8,9,10,11,12,13,14 After processing each dataset separately to filter out low-quality cells and doublets, we integrated a total of 236,363 cells across all clinical subtypes and a wide spectrum of clinical features (Data S1). We assessed batch effect to ensure no cluster was driven by a single dataset or technology (STAR Methods; Figures S1D–S1L and S2A–S2I). Cell types were identified by taking the top call resulting from a three-step process that labeled clusters based on a signature score of canonical cell markers, marker count coupled with average expression, and greatest average expression of the marker genes alone (Data S2; STAR Methods). Uniform manifold approximation and projection (UMAP) visualization showed clustering of cells by lineage. Immune and stromal cell populations clustered together across clinical subtypes, while epithelial cells showed separation by subtype (Figures 1B and S1F), which is consistent with other studies.11,13 For all datasets, single-cell copy number variant (CNV) profiles were estimated to distinguish cancer from normal epithelial cells (Figures S3A–S3D).
Figure 1.

Integrated scRNA-seq dataset of primary breast cancer identifies six NK cell subsets in breast cancer
(A) Brief overview of the processing and integration pipeline for 8 primary breast cancer datasets.
(B) UMAP visualization of 236,363 cells across 119 samples from 88 patients analyzed by scRNA-seq.
(C) UMAP visualization showing major subsets of natural killer (NK) cells.
(D) Bubble heatmap showing expression of upregulated differentially expressed genes for each major NK cell subset (Bonferroni-adjusted p < 0.05).
(E) Boxplot showing expression of the rNK cell signature in each NK cell subset. NK-1 was significantly different from all other clusters (Kruskal-Wallis p < 0.0001, with post hoc Dunn test p values shown; ∗∗∗∗p < 0.0001).
(F) MA plot of differentially expressed genes between rNK and non-rNK cells (Bonferroni-adjusted p < 0.05).
(G) Boxplot showing the expression level of the rNK signature by clinical subtype. No significant difference was found between subtypes (Kruskal-Wallis p > 0.05).
(H) Circos plots showing representative predictive receptor-ligand pairs between rNK cells and all cancer epithelial cells separated by clinical subtype. Shared receptors across all subtypes are colored in red.
(I) Boxplot showing the Pearson correlations of rNK signature gene expression in reprogrammed NK (rNK) cells compared with non-rNK cells versus rNK cells compared with rNK cells (across all clinical subtypes of breast cancer). Pearson correlations between rNK cells and rNK cells are higher than those between rNK cells and non-rNK cells (two-sided Wilcoxon test, ∗∗∗∗p < 0.0001).
(J) Scatterplot showing the Pearson correlation of age and proportion of rNK cells by sample (p <0.01).
(K) Kaplan-Meier plot showing worse clinical outcome in breast cancer patients with high expression of the rNK cell gene signature (log rank test, p < 0.05).
(L) Bar plot showing relative proportions of NK subsets across tumor samples and clinical subtypes.
See also Figures S1–S5 and Data S1 and S4.
Because the number of cells in this dataset permits statistically powered analysis of rare immune cell populations in human breast cancers, we first leveraged the integrated dataset to better characterize the heterogeneity of NK cells. While NK cells are key mediators of anti-tumor control, our understanding of their varied phenotype and function in the breast TME is limited and incomplete. To our knowledge, there are no prior studies that dissect NK cell subsets in the human breast TME. To address this gap, we re-clustered NK cells from the integrated dataset (Figure S4A). Unsupervised graph-based clustering uncovered 6 clusters of NK cells, designated NK-0 through NK-5 (Figures 1C, S4B, and S4C).
Differential gene expression analysis between clusters revealed upregulated genes defining each NK subset (Figures 1D and S4D; Table 1; Data S3; STAR Methods). NK-0 and NK-2 express high levels of FCGR3A (CD16) and cytolytic molecules (granzymes and PRF1), which suggests that they are similar to CD56dim NK cells.25,26,27,28,29 NK-0 is enriched for KLRC2, ETS1, and effector genes (GZMH and CCL5), which closely resembles gene expression profiles described previously for “memory-like” NK cells.25 NK-2 is defined by increased expression of cytotoxicity-related genes (GZMA, GZMB, PRF1, and SPON2) and S1PR5, which has been described previously in CD56dim bone marrow NK cells.25 NK-4 is predominated by genes involved in interferon signaling (IFI6 and ISG15), suggesting that this subset may be influenced by interferon-high tumor microenvironments and consists of activated NK cells involved in the direct anti-tumor response.30 NK-3 cells appear to have features of tissue-resident NK cells, with upregulated expression of SELL, IL7R, and GZMK as well as reduced expression of cytolytic genes and FCGR3A (CD16).31 In contrast, genes of inactivity and reduced cytotoxicity were upregulated in clusters NK-1 and NK-5. Most notably, NK-1 was marked by genes related to the NR4A family,32,33 JUN, FOS, and DUSP1. NR4A is a family of orphan nuclear receptors that act as transcription factors; they are thought to negatively regulate T cell cytotoxicity32 and have been described as marking specific NK cells with reduced interferon gamma production.29,33 NK-5 had reduced expression of cytolytic genes and FCGR3A (CD16) and increased expression of KLRC1 and CD96, which are inactivators of NK cell activity.34,35 To further define the function of NK cell subsets, we performed gene set enrichment analysis of individual clusters, which confirmed their functional phenotypes (Figure S4E).
Table 1.
Marker genes for 6 NK cell subsets
| NK subset | Gene |
|---|---|
| NK-0 | FCGR3A |
| NK-0 | PRF1 |
| NK-0 | FGFBP2 |
| NK-0 | GZMH |
| NK-0 | ETS1 |
| NK-1 | NR4A1 |
| NK-1 | NR4A2 |
| NK-1 | DUSP1 |
| NK-1 | DUSP2 |
| NK-1 | FOS |
| NK-1 | JUN |
| NK-2 | FCGR3A |
| NK-2 | PRF1 |
| NK-2 | FGFBP2 |
| NK-2 | GZMA |
| NK-2 | GZMB |
| NK-2 | CXCF1 |
| NK-2 | SPON2 |
| NK-2 | CX3CR1 |
| NK-2 | S1PR5 |
| NK-3 | GZMK |
| NK-3 | SELL |
| NK-3 | IL7R |
| NK-3 | LTB |
| NK-4 | ISG15 |
| NK-4 | IFI6 |
| NK-4 | IFIT3 |
| NK-4 | IFI44L |
| NK-5 | CCL5 |
| NK-5 | HLA-DRB1 |
| NK-5 | KLRC1 |
| NK-5 | CD74 |
| NK-5 | MYADM |
| NK-5 | HSPE1 |
See also Data S3.
Reprogrammed NK cells are most similar to the NK-1 subset and are observed in patient samples independent of subtype
Previously in ex vivo and mouse models, we observed that NK cells can be “reprogrammed” after exposure to malignant mammary epithelial cells to promote tumor outgrowth.20,21 To determine the human significance of this finding, we first generated a signature of mouse reprogrammed NK (rNK) cells based on an experiment20 comparing the transcriptomes of healthy NK cells with tumor-exposed NK cells that we found to be tumor promoting and reprogrammed (Figure S5A). We next converted the original signature to the human analog (Figure S5B; Table 2) and applied it to the NK cell subsets. NK-1 scored significantly higher for the rNK signature than all other NK cell subsets (p < 0.0001) (Figure 1E). Differential gene expression analysis of rNK cells compared with non-rNK cells revealed that the NR4A family (NR4A1, NR4A2, and NR4A3), FOS, JUN, and DUSP1 were among the most differentially expressed genes (Figure 1F; Data S4; STAR Methods), similar to the transcriptional profile of the NK-1 subset.
Table 2.
rNK cell signature with upregulated and downregulated genes
| Upregulated rNK genes | Downregulated rNK genes |
|---|---|
| ABCA1 | AHRR |
| ALOX12 | ALDH1B2 |
| CALD1 | ASB2 |
| CAVIN2 | ASNS |
| CCL4 | ATF5 |
| CLU | AVIL |
| CMKLR1 | BCAT1 |
| CR2 | CARS1 |
| CX3CR1 | CDH1 |
| DTX1 | CDKN1A |
| DUSP1 | CEMIP2 |
| F5 | CHAC1 |
| FAM81A | CISH |
| FOS | CLBA1 |
| FOSB | COX6A2 |
| GAS2L1 | CXCR6 |
| GFRA2 | EXYL1 |
| GP6 | FMNL2 |
| HEATR9 | GPT2 |
| HES1 | HMOX1 |
| ITGAX | HPGDS |
| JUN | ISG20 |
| KLRG1 | ITGA1 |
| LTBP1 | LGALS3 |
| MID1 | LHFPL2 |
| MPIG6B | ME1 |
| NHSL2 | MTHFD2 |
| NR4A1 | NEK6 |
| NR4A2 | NQO1 |
| NR4A3 | OSBPL1A |
| NYLK | OSGIN1 |
| PARVB | PACSIN1 |
| PLXNA4 | PMEPA1 |
| RASGRP2 | PPP2R2C |
| RHPN1 | PYCR1 |
| SCD | RN7SL1 |
| SLC6A4 | SCN3B |
| THBS1 | SH3PXD2B |
| TMTC1 | SLC1A4 |
| TNFAIP3 | SLC6A9 |
| TUBB1 | SLC7A3 |
| VWF | SLC7A5 |
| XDH | SNORA23 |
| SSTR2 | |
| TBC1D16 | |
| TRIB3 | |
| ZNF503 |
See also Data S4.
To test whether rNK cells were associated with a specific breast cancer subtype, we examined the expression of rNK cells across clinical subtypes. We found no significant differences in rNK cell expression across all subtypes (p > 0.05, n = 3,720 NK cells total) (Figures 1G and S5C). Additionally, we found shared receptor-ligand pairs between NK cells and cancer epithelial cells across all subtypes (Figure 1H), including LGALS3_SPN, RPS19_ICAM1, and HSP90B1_TNFRSF1B. Further, the average Pearson correlation in gene expression levels between rNK cells was greater than between rNK and non-rNK cells (p < 0.0001) (Figures 1I and S5D). Together, these findings demonstrate that rNK cells are not defined by specific breast cancer subtype biology but suggest that a shared but still unknown mechanism contributes to NK cell reprogramming.
To further investigate the clinical significance of rNK cells, we observed that higher expression of rNK cells correlates with older age (R = 0.33, p < 0.01) (Figure 1J). Survival analysis was performed on patients in The Cancer Genome Atlas (TCGA) breast cancer cohort, and we first confirmed that age was not a confounder of this analysis (Figure S5E). Given the limitations of applying the rNK cell signature to bulk RNA-seq samples from TCGA, which include a substantial fraction of non-NK cells, only samples with a relatively high fraction of tumor-infiltrating NK cells were selected for analysis (STAR Methods). Increased expression of the rNK cell signature in tumors with a high fraction of NK cells correlates with worse overall survival (p < 0.05) (Figures 1K and S5F).
We then asked whether NK cell subsets were uniformly expressed across individuals and breast cancer subtypes. To answer this question, we characterized the degree of NK cell heterogeneity across patients in the integrated dataset. We observed remarkable heterogeneity in the proportions of NK cell subsets across patients (Figure 1L). Additionally, no NK cell subset was driven by a single patient, and all NK cell subsets were present across each breast cancer clinical subtype. However, NK cell subset heterogeneity as quantified using ROGUE analysis was observed to be significantly higher in certain clinical subtypes than others (Figure S5G). While there have been multiple reports of NK cell subsets in other cancers,28,29 none have yet explored the diversity of NK cell subsets within individual patient samples. Our findings provide further evidence of the diverse phenotypes of NK cells within individual primary breast tumors.
Individual breast tumors have varying degrees of cancer epithelial cell heterogeneity
Because we observed that NK cell heterogeneity is associated with certain clinical subtypes of breast cancer (Figure 1L), we reasoned that heterogeneity within breast cancer subtypes would be important when further characterizing the breast TME. We then used our dataset to explore the heterogeneity of cancer epithelial cells at different resolutions: at the level of single gene expression, molecular subtypes, and then 10 categories of cancer epithelial cells that reflect intratumoral transcriptional heterogeneity (ITTH).
Cancer epithelial cells are well known to demonstrate substantial intertumoral and intratumoral heterogeneity in primary breast tumors at the single-gene level.8,9,13,14,23,24 For example, heterogeneous expression of therapeutic targets could have clinical implications. Newer anti-HER2 and anti-TROP2 agents have shown benefit in patients across heterogeneous RNA and protein expression of their targets.36,37 This highlights an opportunity to better understand ERBB2 (HER2) and TACSTD2 (TROP2) expression heterogeneity in cancer epithelial cells using transcriptomics data. In contrast to bulk RNA-seq, which aggregates expression levels across all cell types and thus offers limited resolution for studying intratumoral heterogeneity,38 the integrated dataset can be used evaluate ERBB2 and TACSTD2 heterogeneity in cancer epithelial cells at the single-cell level across tumor samples. To do so, epithelial cells in the integrated dataset were re-clustered and re-integrated to account for technology-driven batch effects (Figures S6A–S6C). Cancer epithelial cells were distinguished from normal epithelial cells (Figures S3A and S3B). Consistent with prior studies,11,13 epithelial cells demonstrated stratification by patient (Figure S6A).
Previous bulk RNA-seq and immunohistochemistry (IHC) studies have reported expression of the ERBB2 gene or HER2 protein in up to 70% of HER2-negative breast tumors.39,40 We detect ERBB2 expression in 92% of samples independent of clinical subtype at the single-cell level (Figures 2A and S6D). For TACSTD2, we similarly observed notable heterogeneity (Figures 2B and S6E). In particular, TACSTD2 expression was observed across all subtypes in 94% of samples. This provides additional evidence at single-cell resolution of what has been previously described in bulk RNA-seq and IHC studies, which report TROP2 positivity in 50%–93% of breast cancer samples.41,42,43 Interestingly, the proportion of ERBB2Hi and ERBB2Med cells and TACSTD2Hi and TACSTD2Med cells also varied between samples, reflecting heterogeneous RNA expression at the cellular level. We next asked how other clinically relevant target genes were related to ERBB2 expression. We found that PIK3CA, ERBB3, and FGFR expression was highest in ERBB2Hi cells (Figure 2C). In contrast, TACSTD2 and CD274 expression levels were highest in ERBB2Med cells and notably lower in ERBB2Hi cells. Upon analysis of target genes related to TACSTD2, we found that EGFR, CDK, and NTRK expression was elevated in TACSTD2Hi cells (Figure 2D). ERBB2, ERBB3, PIK3CA, and AR expression was highest in TACSTD2Med cells. Additionally, we observed that TACSTD2Med cells highly express NECTIN2, a ligand related to TIGIT, which hints at potential synergy with anti-TROP2 therapeutics and immune checkpoint inhibition.
Figure 2.

Cancer epithelial cells demonstrate substantial ITTH
(A) Bar plot showing proportions of ERBB2Hi, ERBB2Med, and ERBB2Lo cells by sample.
(B) Bar plot showing proportions of TACSTD2Hi, TACSTD2Med, and TACSTD2Lo cells by sample.
(C) Heatmap of Z-scored average expression of clinically actionable targets in ERBB2Hi, ERBB2Med, and ERBB2Lo cells.
(D) Heatmap of Z-scored average expression of clinically actionable targets in TACSTD2Hi, TACSTD2Med, and TACSTD2Lo cells.
(E) MA plot showing differentially expressed genes between ERBB2Hi vs. ERBB2Med and ERBB2Lo cells (Bonferroni-adjusted p < 0.05).
(F) MA plot showing differentially expressed genes between TACSTD2Hi vs. TACSTD2Med and TACSTD2Lo cells (Bonferroni-adjusted p < 0.05).
(G) Boxplot showing the proportion of ERBB2-expressing cells per sample by nodal status (two-sided Wilcoxon test, p > 0.05).
(H) Boxplot showing the proportion of TACSTD2-expressing cells per sample by nodal status (two-sided Wilcoxon test, p < 0.05).
(I) Percentage of cancer epithelial cells by molecular subtype, sorted by sample score by the ROGUE metric.
(J) Plot showing discordance in predicted heterogeneity by molecular subtype and by ROGUE metric by sample. Samples with >50% difference between the normalized ROGUE metric and the maximum percentage of cells within the sample that belonged to a single molecular subtype are classified as discordant.
See also Figures S6 and S7 and Data S5 and S6.
Next, we characterized the heterogeneity of molecular features between ERBB2Hi, ERBB2Med, and ERBB2Lo populations. We performed gene set enrichment analysis for the ERBB2 and TACSTD2 groups to further characterize function (Figures S6F and S6G) and differential gene expression analyses between the groups (Figures 2E, S6F, and S6G). Of the upregulated genes for ERBB2Hi cells, 47 genes have been shown to be direct interactors with ERBB2 (Data S5). Differentially expressed genes in ERBB2Med cells compared with ERBB2Hi and ERBB2Lo cells may provide insight into molecular features associated with ERBB2 heterogeneity and HER2-low tumors (Figures 2E, S6H, and S6I). For instance, CEACAM6,44 DUSP6,45 and ITGB646 were found to be upregulated in in ERBB2Med cells, which is consistent with prior reports of their expression in HER2+ cancer cells (Figure 2E). For TACSTD2Hi, TACSTD2Med, and TACSTD2Lo cells, differential gene expression analyses (Figures 2F, S6J, and S6K; Data S6) identified KRT14 and KRT17 as significantly upregulated genes in TACSTD2Hi cells. These genes have been implicated as markers for highly metastatic breast cancer cells.47 When assessing for correlation with clinical features, the proportion of ERBB2-expressing cells (ERBB2Hi or ERBB2Med) within non-HER2+ tumors did not show significant association with nodal status (p = 0.25) (Figures 2G, S7A, and S7B). However, tumors with an increased proportion of TACSTD2-expressing cells were significantly associated with higher nodal status (p = 0.015) (Figure 2H). When performing this analysis separately in each cohort, the combined result by Fisher’s combined probability was not statistically significant, though it trended toward significance (X = 11.227, p = 0.08) (Figure S7C). This again highlights the value of our data integration approach, which creates a more statistically powered dataset and enables evaluation of correlations with clinical features over traditional meta-analysis methods.
Our study joins several reports noting the heterogeneous expression of single genes within single tumors.48,49,50,51 Recognizing that intratumoral heterogeneity occurs beyond single genes, we next characterized the ITTH of cancer epithelial cells in primary breast tumors. To do so, we applied a well-characterized SC50 molecular subtype classifier13 that scores the four molecular subtypes (luminal A, luminal B, Her2, and basal) to cancer epithelial cells in the integrated dataset. We found that each patient tumor expressed differing proportions of cells from each molecular subtype with a varied degree of concordance with the clinical subtype diagnosis (Figure 2I). This finding prompted us to explore how cancer epithelial cell ITTH may be influenced by features beyond molecular subtype. We quantified the degree of heterogeneity across all cancer epithelial cells in a patient tumor using ROGUE analysis (Figure 2I).52 The ROGUE score for each individual tumor sample also reflected molecular subtype heterogeneity to some degree; however, we noticed discordance in 33.3% of samples, which demonstrated homogeneity based on molecular subtype but high heterogeneity based on ROGUE score (Figure 2J; STAR Methods). This suggests that other factors beyond molecular subtype-associated genes drive the observed heterogeneity and underscores a need for different approaches to study cancer epithelial cell ITTH at higher resolution than that of existing subtype classifiers.
Cancer epithelial cell heterogeneity can be defined by 10 unifying groups of gene signatures
To develop a high-resolution classifier of heterogeneous cancer epithelial cells, we first performed unsupervised clustering on all cancer epithelial cells in the integrated dataset to generate signatures of upregulated genes that capture distinct molecular features of cancer epithelial cell clusters. Next, supervised classification was performed based on expression of 12 clinical therapeutic targets (ESR1, ERBB2, ERBB3, PIK3CA, NTRK1/NTRK2/NTRK3, CD274, EGFR, FGFR1/FGFR2/FGFR3/FGFR4, TACSTD2, CDK4/CDK6, AR, and NECTIN2) to ensure that clinically relevant associations were captured by upregulated gene signatures (STAR Methods). The motivation for including therapeutic targets was to create classifications grounded in relevant clinical approaches. We additionally supervised classification of all cancer epithelial cells based on molecular subtype to generate upregulated gene signatures that reflect subtype features. Consensus clustering of all generated gene signatures identified 10 unifying groups, which we defined as “gene elements” (GEs) (Figures S8A and S8B). We defined each GE by the top 100 genes that occurred most frequently across gene signatures assigned to the group (Table 3; STAR Methods). We scored each cancer epithelial cell by the individual 10 GEs and assigned GE-based cell labels (Figure 3A; STAR Methods).
Table 3.
Gene lists consisting of 100 genes for 10 GEs
| GE1 | GE2 | GE3 | GE4 | GE5 | GE6 | GE7 | GE8 | GE9 | GE10 |
|---|---|---|---|---|---|---|---|---|---|
| AC090498-1 | ALDH3B2 | A2M | ANLN | AIF1 | ADIRF | AC093001-1 | ADIRF | AC093001-1 | AGR2 |
| AC105999-2 | ALOX15B | ACTA2 | ANP32E | ALOX5AP | ANAPC11 | ADIRF | AFF3 | ADIRF | APOD |
| ADIRF | APOD | ACTG2 | ARL6IP1 | ANXA1 | ATP5ME | AGR2 | ALCAM | AGR2 | AREG |
| AGR2 | AZIN1 | ANGPTL4 | ASF1B | APOC1 | AZGP1 | AGR3 | ANKRD30A | AGR3 | AZGP1 |
| AGR3 | B2M | ANXA1 | ASPM | APOE | BLVRB | ANKRD37 | ANXA2 | APOD | B2M |
| ALDH2 | BNIP3 | APOD | ATAD2 | AREG | BST2 | APOD | AR | AQP1 | BST2 |
| ANKRD30A | C1orf21 | APOE | AURKA | C1ORF162 | CALM1 | AQP3 | ARFGEF3 | AQP5 | BTG2 |
| ARL6IP1 | CALD1 | BGN | BIRC5 | C1QA | CCND1 | ARC | ASAH1 | AREG | C15ORF48 |
| ARMT1 | CALU | C6ORF15 | BUB1B | C1QB | CD9 | AREG | ATP1B1 | ASCL1 | CCL20 |
| ATAD2 | CAPG | CALD1 | CCNB1 | C1QC | CETN2 | ATF3 | AZGP1 | AZGP1 | CD74 |
| AZGP1 | CD24 | CALML5 | CCNB2 | CARD16 | CISD3 | AZGP1 | BTG1 | BMPR1B | CEBPD |
| BATF | CD59 | CAV1 | CDC20 | CCL3 | CLDN7 | BAMBI | CD59 | C15ORF48 | CHI3L1 |
| BMPR1B | CD74 | CAVIN1 | CDC6 | CCL4 | COX6C | BTG1 | CDK12 | CALML5 | CHI3L2 |
| BST2 | CD99 | CAVIN3 | CDCA3 | CCL5 | CRABP2 | BTG2 | CEBPD | CCL28 | CP |
| BTG2 | CDKN2B | CCL28 | CDCA8 | CD2 | CRACR2B | C15ORF48 | CLDN3 | CD55 | CRISP3 |
| C15ORF48 | CFD | CCN2 | CDK1 | CD27 | CRIP1 | CALML5 | CLDN4 | CEACAM6 | CSTA |
| CCDC74A | CKB | CD24 | CDKN2A | CD37 | CRIP2 | CCDC74A | CLTC | CFD | CTSC |
| CEBPD | CLDN3 | CDKN2A | CDKN3 | CD3D | CSTB | CCN1 | CLU | CLIC3 | CTSD |
| CFD | CLDN4 | CHI3L1 | CENPA | CD3E | CYB5A | CD55 | CNN3 | CLU | CTSS |
| CLDN4 | CNN3 | COL1A2 | CENPE | CD48 | CYBA | CDKN1A | CTNNB1 | COX6C | CXCL1 |
| CLU | COL12A1 | COL6A1 | CENPF | CD52 | CYC1 | CEBPB | CTNND1 | CSTB | CXCL17 |
| COX6C | COX6C | COL6A2 | CENPK | CD53 | DBI | CEBPD | EFHD1 | CTSD | CYBA |
| CPB1 | CRIP1 | COTL1 | CENPM | CD69 | DCXR | CFD | EGR1 | CXCL14 | DEFB1 |
| CRIP1 | CSRP1 | CRYAB | CENPU | CD7 | DSTN | CLDN3 | ELF3 | CXCL17 | FDCSP |
| CST3 | CSRP2 | CSTA | CENPW | CD74 | EEF1B2 | CLDN4 | EPCAM | DHRS2 | GBP1 |
| CTHRC1 | CTNNB1 | CXCL2 | CIP2A | CD83 | ELOC | CST3 | ERBB2 | DSCAM-AS1 | GBP2 |
| CXCL14 | CTTN | DEFB1 | CKAP2 | CELF2 | EMP2 | CTD-3252C9-4 | ESR1 | DUSP1 | HLA-A |
| DHRS2 | CYSTM1 | DEPP1 | CKLF | COL1A2 | FXYD3 | CTSK | EVL | ERBB2 | HLA-B |
| DSCAM-AS1 | DDIT4 | EFEMP1 | CKS1B | CORO1A | GPX4 | DHRS2 | FOSB | FADS2 | HLA-C |
| ELF3 | DHRS2 | FABP5 | CKS2 | CREM | GSTM3 | DNAJB1 | GATA3 | FAM3D | HLA-DMA |
| ELP2 | DLX5 | FBXO32 | CTHRC1 | CST7 | H2AJ | DUSP1 | GRB7 | FHL2 | HLA-DPA1 |
| ERBB4 | DSC2 | FDCSP | DEK | CTSL | H2AZ1 | EDN1 | H4C3 | GDF15 | HLA-DPB1 |
| ESR1 | EFHD1 | FGFBP2 | DLGAP5 | CTSW | HINT1 | EGR1 | HES1 | GLYATL2 | HLA-DQA1 |
| EVL | EFNA1 | FN1 | DTYMK | CXCR4 | HMGB1 | ELF3 | HLA-B | GPX1 | HLA-DQA2 |
| FABP3 | ELF5 | GABRP | DUT | CYBB | HSPE1 | ELOVL2 | HNRNPH1 | GSN | HLA-DQB1 |
| FHL2 | ENO1 | GSTP1 | ECT2 | CYTIP | IDH2 | ESR1 | HSPA1A | GSTP1 | HLA-DRA |
| FKBP5 | FAM229B | HLA-A | FAM111A | DUSP2 | JPT1 | FHL2 | HSPA1B | HDC | HLA-DRB1 |
| FSIP1 | FASN | HLA-B | FAM111B | EMP3 | KDELR2 | FOS | IGFBP5 | HSPB1 | HLA-DRB5 |
| GJA1 | GJA1 | ID1 | GGH | FCER1G | KRT10 | FOSB | INTS6 | IGFBP5 | HLA-E |
| GSTM3 | GRIK1-AS1 | IFI27 | GTSE1 | FN1 | KRT18 | GATA3 | ITGB1 | ISG20 | ID3 |
| HES1 | GSTP1 | IGFBP3 | H1-2 | FYB1 | KRT19 | GDF15 | ITGB6 | ITM2A | IFI16 |
| HSPB1 | H2AJ | IGFBP5 | H1-3 | GIMAP7 | KRT7 | GRB7 | ITM2B | KRT23 | IFI27 |
| IFI27 | HILPDA | IGFBP7 | H2AZ1 | GMFG | KRT8 | GSTM3 | JUN | KRT7 | IFI44L |
| IFI6 | HNRNPH1 | IL32 | H2AZ2 | GPR183 | LGALS1 | H1-2 | KLF6 | LGALS1 | IFI6 |
| IFITM1 | HSPA5 | KLK5 | H2BC11 | GPSM3 | LGALS3 | HES1 | KRT7 | LGALS3 | IFIT1 |
| IFITM2 | IFI27 | KLK7 | H4C3 | GZMA | LSM3 | ICAM1 | LDLRAD4 | LY6E | IFIT2 |
| IFITM3 | IFITM3 | KRT14 | HELLS | GZMK | LSM4 | ID2 | LMNA | MARCKS | IFIT3 |
| IGFBP4 | IGKC | KRT15 | HMGB1 | HCST | LY6E | IER2 | LRATD2 | MFGE8 | IFITM1 |
| INPP4B | JPT1 | KRT16 | HMGB2 | HLA-DPA1 | MARCKSL1 | IER3 | MAGED2 | MGP | IFITM2 |
| ISG15 | KCNC2 | KRT17 | HMGB3 | HLA-DPB1 | MIEN1 | IFITM1 | MAL2 | MS4A7 | IFITM3 |
| JUNB | KRT15 | KRT5 | HMGN2 | HLA-DQA1 | MIF | IGFBP4 | MARCKS | MT-ATP8 | IGFBP7 |
| KCNE4 | KRT23 | KRT6A | HMMR | HLA-DRA | MPC2 | IGFBP5 | MT-ND4L | MTCO2P12 | IL32 |
| KCNJ3 | KRT7 | KRT6B | IQGAP3 | HLA-DRB1 | MRPL12 | IRF1 | MT2A | MUC5B | IRF1 |
| KRT18 | LAPTM4B | KRT81 | KIF20B | IGSF6 | MRPL51 | JUN | MUC1 | MUCL1 | ISG15 |
| KRT19 | LDHB | LAMB3 | KIF23 | IL2RG | MRPS34 | JUNB | MYH9 | NDRG2 | KRT15 |
| LDLRAD4 | LMO4 | LCN2 | KIF2C | IL32 | MTDH | KLF4 | NEAT1 | NFKBIZ | KRT19 |
| MAGED2 | LTF | LTF | KNL1 | IL7R | MUCL1 | KLF6 | NFIB | NPW | KRT5 |
| MDK | MAFB | LY6D | KPNA2 | ISG15 | NDUFB9 | KRT15 | PERP | NR4A1 | KRT7 |
| MESP1 | MAL2 | MFAP5 | LGALS1 | ITGB2 | NDUFC2 | KRT18 | PKM | NUDT8 | LCN2 |
| MGP | MAOB | MFGE8 | MAD2L1 | KLRB1 | NME1 | LGALS3 | PLAT | PALMD | LGALS1 |
| MGST1 | MFAP2 | MGP | MKI67 | LAPTM5 | PAFAH1B3 | MAFB | PMEPA1 | PDZK1IP1 | LGMN |
| MRPS30 | MGST1 | MIA | MT2A | LCK | PFDN2 | MAGED2 | PSAP | PERP | LTF |
| MRPS30-DT | MRPL15 | MMP7 | MYBL2 | LIMD2 | PFN1 | MGP | RAD21 | PHGR1 | LUM |
| MS4A7 | MT1X | MT1X | MZT1 | LSP1 | PIP | NAMPT | RBP1 | PIP | LY6D |
| MT-ATP8 | MUCL1 | MT2A | NEK2 | LST1 | POLR2K | NCOA7 | RHOB | PLAT | LYZ |
| NOVA1 | MYBPC1 | MYL9 | NUF2 | LTB | PPDPF | NFKBIA | RUNX1 | PRSS21 | MAFB |
| PEG10 | NME2 | MYLK | NUSAP1 | LY96 | PSMA7 | NFKBIZ | S100A10 | PSCA | MARCKS |
| PHGR1 | NUPR1 | NDRG1 | PBK | LYZ | PSMB3 | NR4A1 | SAT1 | PTHLH | MGP |
| PI15 | PCSK1N | NDUFA4L2 | PCLAF | MEF2C | PSME2 | NR4A2 | SCARB2 | PYDC1 | MIA |
| PIP | PFN2 | NFKBIA | PCNA | MNDA | RAN | PERP | SCD | RGS10 | MMP7 |
| PLAAT4 | PHGDH | NNMT | PLK1 | MS4A6A | RANBP1 | PLAT | SDC1 | RGS2 | MRPS30-DT |
| PLAT | PRSS23 | PDLIM4 | PRC1 | MSR1 | RBIS | PMAIP1 | SERHL2 | RHCG | MX1 |
| PRSS23 | PSMB3 | PLS3 | PRR11 | NKG7 | REEP5 | PRSS23 | SH3BGRL3 | RP11-53O19-2 | NNMT |
| PSD3 | PTHLH | POSTN | PTTG1 | PTPRC | ROMO1 | REL | SHISA2 | S100A1 | PI3 |
| PVALB | PTPN1 | PRNP | RACGAP1 | RAC2 | RPS26 | RHOV | SLC38A2 | S100A10 | PIGR |
| RAMP1 | RAMP1 | PTN | RAD21 | RGCC | S100A14 | RND1 | SLC39A6 | S100A6 | RAMP2 |
| RBP1 | RAMP3 | RARRES1 | RHEB | RGS1 | S100A16 | S100P | SLC40A1 | S100A7 | RARRES1 |
| RHOBTB3 | RBP1 | RCAN1 | RNASEH2A | RGS2 | SEC61G | SAT1 | SOX4 | S100A8 | RHCG |
| SCGB3A1 | RSU1 | RGS2 | RPL39L | RNASE1 | SELENOP | SLC39A6 | SYTL2 | S100A9 | RNASE1 |
| SCUBE2 | S100A10 | S100A2 | RRM2 | S100A4 | SH3BGRL | SLC40A1 | TACSTD2 | S100P | RSAD2 |
| SEMA3C | S100A6 | S100A4 | SMC4 | S100A6 | SLC9A3R1 | SOCS3 | TCAF1 | SAA2 | S100A8 |
| SERPINA1 | SCUBE2 | S100A6 | SPC25 | SEPTIN6 | SMIM22 | SOX4 | TCIM | SCGB1D2 | S100A9 |
| SH3BGRL | SFRP1 | S100A8 | STMN1 | SLC2A3 | SNRPB | SOX9 | TFAP2B | SCGB2A1 | S100P |
| SLC39A6 | SH3BGRL | S100A9 | TFDP1 | SMAP2 | SNRPG | STC2 | TIMP1 | SCGB2A2 | SAA2 |
| SLC40A1 | SLC39A4 | SAA1 | TK1 | SOCS1 | SPINT2 | TACSTD2 | TM4SF1 | SDC2 | SCGB1D2 |
| SNCG | SLC40A1 | SAA2 | TMEM106C | SPARC | SQLE | TCIM | TMC5 | SERHL2 | SCGB2A1 |
| STC2 | SOX4 | SBSN | TMPO | SPP1 | SRP9 | TFF1 | TMEM123 | SERPINA1 | SERPING1 |
| TCEAL4 | STC2 | SERPING1 | TOP2A | SRGN | STARD10 | TIMP3 | TPM1 | SLC12A2 | SLC39A6 |
| TCIM | STOM | SFRP1 | TPX2 | STK4 | TCEAL4 | TM4SF1 | TRPS1 | SLC18A2 | SOD2 |
| TFF1 | TCIM | SGK1 | TROAP | TMSB4X | TMCO1 | TNFRSF12A | TSC22D1 | SLPI | SPATS2L |
| TFF3 | TFF3 | SLC25A37 | TTK | TNFAIP3 | TMEM14B | TSC22D3 | TSPYL1 | SYNM | TCIM |
| TIMP1 | TMSB4X | SLPI | TUBA1B | TRAC | TPI1 | TUBA1A | TUBA1A | TACSTD2 | TFF1 |
| TMC5 | TTYH1 | SPARC | TUBA1C | TRBC1 | TPM1 | VASN | VEGFA | TFF1 | TFF3 |
| TPM1 | TUBA1A | SPARCL1 | TUBB | TRBC2 | TSPAN13 | VEGFA | WSB1 | TFF3 | TMEM45A |
| TPRG1 | UBE2V2 | TAGLN | TUBB4B | TREM2 | TUBA1B | VTCN1 | XIST | TM4SF1 | TNFAIP6 |
| VSTM2A | VIM | THBS1 | TYMS | TYROBP | TUBB | XBP1 | YBX1 | TMC5 | TNFSF10 |
| VTCN1 | YBX1 | TPM2 | UBE2C | VIM | UQCRQ | ZFAND2A | YBX3 | TSC22D3 | TXNIP |
| WFDC2 | YBX3 | TSHZ2 | UBE2S | WIPF1 | XBP1 | ZFP36 | ZFP36L1 | TSPAN1 | WFDC2 |
| XBP1 | YWHAH | VIM | UBE2T | ZEB2 | YBX1 | ZFP36L1 | ZFP36L2 | TXNIP | XBP1 |
| ZFP36L1 | YWHAZ | ZFP36L2 | ZWINT | ZNF331 | ZNF706 | ZFP36L2 | ZNF292 | XBP1 | ZFP36 |
Figure 3.

Cancer epithelial cell heterogeneity can be defined by 10 GEs that influence immune cell interactions
(A) Heatmap of Z-scored signature scores of the 10 identified gene elements (GEs) representing all cancer epithelial cells, ordered based on the maximum Z-scored GE signature score. Annotations represent dataset origin, clinical subtype, PAM50 subtype, and SC50 subtype. The “sample” annotation was included to demonstrate that no individual patient sample contributed heavily to a particular GE.
(B) Percentage of cancer epithelial cells assigned to each GE by molecular subtype.
(C) Gene set enrichment using ClusterProfiler of the differentially expressed genes by GE. Significantly enriched gene sets from the MSigDB Hallmark collection are shown (Benjamini-Hochberg-adjusted p < 0.05).
(D) Heatmap of the scaled number of curated predicted receptor-ligand pairs between cancer epithelial cells by GE and interacting immune and stromal cells.
(E) Scatterplots showing Spearman correlations of expression of NK-cell related GE1 and GE6 with sensitivity to NK cell killing (Benjamini-Hochberg-adjusted p < 0.05).
(F) Circos plots showing curated receptor-ligand pairs between cancer epithelial cells that highly express NK cell-related GE1 and GE6 with NK cells. NK cell activating receptor-ligand pairs are colored blue; NK cell inactivating receptor-ligand pairs are colored red.
See also Figure S8.
When assessing for molecular subtypes, GE3-labeled cells were predominantly assigned to the basal subtype, while the majority of GE9-labeled cells were assigned to the Her2 subtype (Figure 3B). Cells labeled by GE1 and GE7 were almost exclusively assigned as luminal A and luminal B. In contrast, GE5- and GE10-labeled cells were assigned to all molecular subtypes. Next, we used gene set enrichment analysis (Figure 3C) to identify functional annotations for each GE. This analysis identified shared and distinct functional features for all GEs. GE4 was uniquely enriched for cell cycle and proliferation hallmarks (MKI67, PCNA, and CDK1). GE2 and GE3 contained hallmark genes of EMT (VIM and ACTA2). GE1, GE6, GE7, and GE9 contained genes associated with estrogen response (ESR1, AREG, and TFF3). GE5 and GE10 were enriched for hallmarks of allograft rejection (HLA-DRA and HLA-DRB1) and complement (C1QA/B/C and C1R).
To assess how GE-based cell labels allow us to characterize cancer epithelial cell heterogeneity within a tumor sample, we applied our GEs to the integrated dataset to deconstruct each individual patient tumor into the 10 GEs (Figure S8C). Notably, GE-based heterogeneity was not constrained by clinical or molecular subtype. This again confirms that significant cancer epithelial cell ITTH exists even within cells from a tumor labeled by a single clinical or molecular subtype. Overall, we generated 10 GEs to characterize cancer epithelial cell ITTH and deconstruct a heterogeneous tumor into its diverse cellular phenotypes.
GEs predict individual patient predominant immune response
To examine how cancer epithelial cell ITTH influences immune interactions in the TME, we generated a decoder matrix of predicted GE-immune interaction strength. GE-immune interaction strength is determined based on the scaled number of predicted receptor-ligand pairings between GEs and immune cells (Figures 3D and S8D; STAR Methods).
To experimentally validate the decoder matrix, we tested these predictions with human breast cancer cell lines. In the decoder matrix, cancer epithelial cells labeled by GE1 and GE6 were predicted to highly interact with NK cells (GE1 and GE6 have the highest scaled number of curated receptor-ligand pairings). We applied the GEs to human breast cancer cell lines from the Cancer Cell Line Encyclopedia to quantify GE expression across cell lines (Figure S8E). Given that GE1 and GE6 have the greatest predicted interaction strength with NK cells (Figure 3D), we hypothesized that expression of these GEs will have a significant influence on NK cell function (i.e., sensitivity or resistance of cancer cell lines to NK cell killing). To test this, we selected breast cancer cell lines with differing expression of GE1 and GE6. BT-474 had increased expression of GE1 and GE6, while MDA-MB-436 had decreased expression of GE1 and GE6. Using these selected cell lines, we assessed the relationship between GE1 and GE6 expression and sensitivity to NK cell killing. We co-cultured BT-474 (GE1 and GE6 high) and MDA-MB-436 (GE1 and GE6 low) with NK-92, a human NK cell line. As hypothesized, GE1 and GE6 expression had a statistically significant impact on NK cell function. NK cell cytotoxicity against BT-474 at 24 h was significantly reduced (p < 0.0001) compared with NK cell cytotoxicity against MDA-MB-436 (Figure S8F). This finding suggests that GE1 and GE6 confer resistance to NK cell cytotoxicity. Next, to expand on these experimental findings, we used a study by Sheffer et al.,53 which reports experimental sensitivity or resistance of 26 breast cancer cell lines to NK cell cytotoxicity (STAR Methods).53,54Increased GE1 and GE6 expression was significantly correlated with increased resistance to NK cell killing (R = −0.54, p < 0.05 for GE1; R = −0.54, p < 0.05 for GE6) (Figure 3E), consistent with the decoder matrix and our experimental findings. Other GEs with fewer predicted NK cell interactions in the decoder matrix did not have statistically meaningful correlations with sensitivity to NK cells (Figure S8G). To investigate interactions that contribute to these phenotypes, we assessed predicted receptor-ligand pairs between cells that highly express GE and NK cells (Figure 3F). We observed that GE1- and GE6-labeled cells were predicted to have receptor-ligand pairs that have been characterized as inactivators of NK cell activity (e.g., NECTIN2_TIGIT, THBS1_CD47, and CD320_TGFRB2). These functional studies validate two of the predictions made by the decoder matrix by showing that GE1 and GE6 are predictive of significant resistance to NK cell killing for breast cancer cell lines.
Overall, this decoder matrix provides a blueprint for quantifying the degree of interactions between each GE and different immune cell types. Moreover, this decoder matrix curates key activating and inhibitory receptors that can be used to infer how GE-immune interactions affect immune cell behavior.
Spatial mapping of GEs reflects predicted immune interactions
To validate the predicted interactions curated by the decoder matrix, we used a spatial transcriptomics dataset containing published data from 10× Genomics and from Wu et al.13 We first deconvoluted the underlying composition of cell types through integration of the spatial transcriptome data with the integrated dataset (STAR Methods). Because T cell infiltration was relatively high across spatial transcriptomics samples (Figure S9A), we chose to explore T cell interactions using this dataset. To do so, we applied the 10 GEs to each sample in the dataset. Using the decoder matrix, we inferred which GE-labeled cells interact with T cells and which ones do not. Thus, we hypothesized that these GE-labeled cells and CD8+ T cells would be spatially organized in breast tumors. To test this, we examined the co-expression of the GEs and the presence of neighboring CD8+ T cells. Notably, GE5 expression demonstrated positive correlations with CD8+ T cells in all samples (mean R = 0.33, all p < 0.0001) (Figure 4A). In one representative image, we determined the co-localization of CD8+ T cells with GE5 expression (Figure 4B). For areas with high presence of CD8+ T cells, we observed increased colocalization of select curated receptor-ligand pairs (ITGB2_ITGAL, LTB_TNFRSF1A, and ALOX5AP_ALOX5) (Figure 4B). As expected, GEs with limited predicted interactions did not consistently co-localize with CD8+ T cells (Figure S9B).
Figure 4.

GE-immune interactions predict response to anti-PD-1 therapy
(A) Heatmap of Pearson correlations between expression of each of the 10 GEs and the presence of CD8+ T cells for 6 spatial transcriptomics samples across spots containing CD8+ T cells (n.s., Benjamini-Hochberg-adjusted p > 0.05).
(B) For a representative TNBC sample, pathological annotation of morphological regions into distinct categories. UCell signature scores of CD8+ T cells are overlaid onto spatial tumor sample spots (red). A UCell signature score of GE5 (a CD8+ T cell activating GE) is overlaid onto tumor sample spots (red). A colocalization score for ITGB2_ITGAL, LTB_TNFRSF1A, and ALOX5AP_ALOX5 (predicted receptor-ligand pairs for GE5 and CD8+ T cells) is overlaid onto tumor sample spots (red).
(C) Heatmap of average expression of each of the 10 GEs across cancer epithelial cells in each sample from Bassez et al.57 T cell InteractPrint is shown below.
(D) Boxplot showing T cell InteractPrint prediction of response to anti-PD-1 therapy across all clinical subtypes in Bassez et al.57 (R, responder; NR, non-responder; p < 0.05). Also shown is the AUC of ROC comparing the performance of T cell InteractPrint (AUC = 81.87) and of PD-L1 expression (AUC = 49.71) in Bassez et al.57 samples (bootstrap test with n = 10,000, p < 0.05).
(E) Heatmap of average expression of each of the 10 GEs across cancer epithelial cells in each sample from the I-SPY2 trial. T cell InteractPrint is shown below.
(F) Boxplot showing T cell InteractPrint prediction of response to anti-PD-1 therapy across all clinical subtypes in I-SPY2 trial samples (two-sided Wilcoxon test p <0.0001). Also shown is the AUC of ROC comparing the performance of T cell InteractPrint (AUC = 83.02) and of PD-L1 expression (AUC = 72.33) in the I-SPY2 trial (bootstrap test with n = 10,000, p <0.05).
(G) Schematic of T cell InteractPrint to predict patient response to anti-PD-1 therapy.
See also Figure S9.
InteractPrint: A weighted score to predict the predominant tumor-interacting immune cell for an individual patient tumor
We then hypothesized that the GE-immune interaction decoder matrix could be applied to individual tumor tissues. To account for how cancer epithelial cell ITTH within a tumor influences immune cell interactions, we developed InteractPrint. InteractPrint reflects interactions between the predominant tumor-responsive immune cells from the decoder matrix and cancer cells that highly express each GE, weighted by the GE composition of an individual patient tumor. This approach permits real-world application of InteractPrint since it accounts for heterogeneity of GEs within a tumor.
InteractPrint predicts anti-PD-1 therapeutic response
We then sought to use InteractPrint to characterize the predominant immune response within patients for therapeutically targeted immune cells. Because current immune checkpoint inhibitors (ICI) target CD8+ T cell-driven cancers, we developed T cell InteractPrint to predict who might respond to ICI. For the comparator, average PD-L1 expression on cancer epithelial cells was selected, as PD-L1 remains the main biomarker used clinically to determine who should receive ICI for many solid tumors, including patients with recurrent unresectable or metastatic triple-negative breast cancer (TNBC).55,56
We applied our approach to a separate scRNA-seq dataset published by Bassez et al.,57 which contains tumor biopsies from breast cancer patients pre and post anti-PD-1 therapy (Figures S8C and S8D). Deconstruction of each individual patient tumor into the 10 GEs revealed considerable cancer epithelial cell ITTH prior to anti-PD-1 treatment (Figure 4C), similar to what was observed in the integrated dataset (Figure S8C). To assess the capacity of the T cell InteractPrint to predict responders to anti-PD-1 therapy, we derived receiver operating characteristic (ROC) curves in this dataset (Figure 4D). Across clinical subtypes of breast cancer, the T cell InteractPrint demonstrated an area under the curve (AUC) of 81.87% (p = 0.0061) in predicting response to anti-PD-1 therapy, inferred from T cell clonotype expansion.53 This was a significant improvement (p = 0.019) over average PD-L1 expression on cancer epithelial cells, the current clinical biomarker to predict patients who will respond to anti-PD-1 therapy in breast cancer, which had an AUC of 49.71% (p > 0.05).
Next, we applied our predictor to a separate validation dataset containing results from the I-SPY2 trial. I-SPY2 is an ongoing, multicenter, open-label, adaptively randomized phase 2 trial of neoadjuvant chemotherapy for early-stage breast cancer at high risk of recurrence.57 In this trial, patients with breast cancer received anti-PD-1 therapy (the same as patients from Bassez et al.57) combined with paclitaxel. We applied the 10 GEs to microarray data from pre-treatment tumor samples from the I-SPY2 trial and observed levels of heterogeneity that were comparable with those described in the scRNA-seq datasets (Figure 4E). In the I-SPY2 trial dataset, T cell InteractPrint (AUC = 83.02%, p = 8.1 × 10−7) demonstrated significant improvement (p = 0.034) over average PD-L1 expression on cancer epithelial cells (AUC = 72.33%, p = 0.001) in predicting response to anti-PD-1 therapy (Figure 4F).
Across two trials, T cell InteractPrint demonstrated significant improvement over PD-L1 at predicting response to anti-PD-1 therapy. This highlights the ability of T cell InteractPrint to decode how cancer epithelial cell ITTH impacts CD8+ T cell response for each individual patient.
Discussion
In this study, we present an atlas resource that integrates scRNA-seq data of 236,363 cells that represent the breast TME. This resource enables high-resolution characterization of rare immune cell and cancer epithelial cell heterogeneity and demonstrates how heterogeneity influences immune cell interactions that have not been evaluated previously.
First, we leveraged the statistical power of this integrated dataset to demonstrate how NK cells, a population of rare immune cells that have not been classified in the breast TME, can be studied further. We identified six subsets of NK cells, which consist of activated and cytotoxic, exhausted, and rNK cells. Identification of rNK cells in most but not all samples (i.e., 72% of samples) provides a subtype-independent approach to identify patients who may benefit from rNK cell-directed therapies. We also performed receptor-ligand analysis on rNK cells and tumor cells to identify potential interactions that could lead to this phenotype (Figure 1H). Interestingly, KLRG1 is among the identified interactions between HER2+ cancer epithelial cells and rNK cells. KLRG1 was previously validated as a potential regulator of rNK cell function.20 Ongoing and future experimental work by is needed to determine mechanisms that drive this distinct and functional rNK cell phenotype. Our findings add to the growing body of literature on distinct NK cell subsets and phenotypes. In particular, the gene expression profile of the cytotoxic NK-2 subset aligns with CD56dim subsets identified previously in bone marrow by Crinier et al.25 and Yang et al.,27 in peripheral blood by Smith et al.,26 and in human melanoma metastases by de Andrade et al.28 The NK-0 subset closely resembles previously described “memory-like” NK cells derived from bone marrow by Crinier et al.25 and have been described after viral or tumor exposure. Our description of NK-4 aligns with prior observations of “inflamed” interferon (IFN)-responding NK cells in the bone marrow by Yang et al.27 and in peripheral blood by Smith et al.26 NK-3 demonstrated features consistent with prior studies of tissue-resident NK cells derived from bone marrow by Yang et al.27 and from melanoma metastases by de Andrade et al.28 The unique transcriptional profile of the NK-5 subset has been described previously as exhausted.34,35 Last, expression profiles (e.g., upregulated NR4A family, DUSP1, FOS, and JUN) similar to the rNK-1 subset have been described in peripheral blood by Smith et al.,26 in bone marrow by Yang et al.,27 and in human head and neck cancers by Moreno-Nieves et al.,29 as well as in our prior studies on metastasis-promoting NK cells derived from ex vivo and mouse models.20 Additionally, our present study is the first to identify six subsets of NK cells in human primary breast tumors, which can now be quantified and measured in response to prospective therapeutics.
Through this analysis, we observed that NK cell heterogeneity is associated with breast cancer clinical subtypes. These clinical subtypes are well known to harbor substantial heterogeneity.23,24,40 This led us to use this resource to further understand clinically relevant heterogeneity within the breast TME and cancer epithelium at resolutions higher than studied previously. At the single-cell resolution, we quantified the heterogeneity of single-gene expression (i.e., ERBB2 and TACSTD2) across tumors and found that the majority of samples across all breast cancer subtypes expressed ERBB2 and TACSTD2. These findings prompt further functional investigation of what degree of transcriptomic expression correlates with clinical efficacy of anti-HER2 and anti-TROP2 antibody-drug conjugates. The new class of antibody-drug conjugates targeting these proteins has recently demonstrated efficacy across breast cancer subtypes. For HER2/ERBB2, high concordance between proteomic HER2 status and ERBB2 mRNA expression has been reported in the literature,58,59,60,61,62,63 and we corroborate these findings in the integrated dataset (Figure S6D). Similarly, for TROP2/TACSTD2, concordance between proteomic TROP2 and TACSTD2 mRNA expression has been reported in various solid tumors, including breast.64,65,66,67,68,69 Further, examining genes that are positively correlated with ERBB2 and TACSTD2 uncovers other potential clinical targets that can synergize with current anti-HER2 and anti-TROP2 therapies and provides a rationale for novel combination approaches. Then, we characterized cancer epithelial cell heterogeneity by using unsupervised clustering and supervised classification based on breast cancer molecular subtypes and clinical therapeutic target gene expression. While discrepancies between clinical and molecular subtyping have been well documented, we provide an approach to defining cancer epithelial cell heterogeneity at the single-cell level by using 10 GEs. This approach enables high-resolution characterization of cancer epithelial cell ITTH and deconstruction of a heterogeneous tumor into its diverse epithelial phenotypes.
To further demonstrate how this resource facilitates analysis of the breast TME, we then use information from the 10 GEs to identify how cancer epithelial cell heterogeneity influences interactions with immune populations. Current ICI biomarker approaches mainly focus on the expression of single targets, resulting in an incomplete characterization of the TME complexity. Our approach for T cell InteractPrint score calculates cancer epithelial cell heterogeneity within a tumor sample and the number of predicted interactions between heterogeneous cancer epithelial cells and CD8+ T cells (Figure 4G). This captures how heterogeneous expression of GEs shifts the predicted strength of T cell interactions for an individual patient’s tumor. Across two trials and all subtypes of breast cancer, T cell InteractPrint predicted response to T cell immune checkpoint inhibition. This finding is significant because anti-PD-1 therapy is not effective in HR+ disease70 and has limited efficacy in TNBC disease71 compared with the response seen in other solid tumors.72,73,74,75 The development of InteractPrint from this resource serves as another example of how this resource can be used to uncover biology that, once validated, could inform response to ICI in breast cancer.
The breast TME is a complex ecosystem that encompasses diverse cell phenotypes, heterogeneous interactions among cells, and varied expression of clinically targetable features. The development of this resource and examples of its utility uncovered information about NK cells and how heterogeneous cancer epithelial cells and their predicted immune interactions can predict immune checkpoint therapy responses. Future use of this resource is likely to yield additional impactful findings.
Limitations of the study
A limitation of our study is that we compared InteractPrint with PD-L1 by transcriptomic expression in early-stage breast cancer trials. PD-L1 expression by IHC is approved in the setting of recurrent unresectable or metastatic TNBC disease for selection of patients to receive ICI.76 However, PD-L1 expression has been associated with increased response rates in neoadjuvant trials,77,78,79,80,81 and concordance between PD-L1 mRNA and proteomic expression has been shown.82,83,84,85 While this provides the rationale behind our selection of PD-L1 transcriptomic expression as the comparator for T cell InteractPrint, a discussion of the limitations of this comparator is necessary. First, assessment of PD-L1 expression based on mRNA levels rather than proteomic expression is not widely used in the clinic. Second, across the neoadjuvant trials, differences in study design, patient enrollment, and subgroup analyses make it difficult to reconcile mixed findings around the role of PD-L1 as a biomarker in early-stage breast cancer. Last, evaluation of other exploratory biomarkers is ongoing.86,87,88,89 There is still an outstanding need for improved patient selection to maximize efficacy and minimize exposure to adverse events associated with ICIs. These limitations define a need for future prospective studies to compare T cell InteractPrint and PD-L1 gene and protein expression, along with other exploratory biomarkers, to predict response to ICI.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-huCD56 BV605, Clone HCD56 (mouse IgG1k) | BioLegend | Cat#318334; RRID: AB_2561912 |
| Chemicals, peptides, and recombinant proteins | ||
| 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) | Invitrogen | Cat#D1306 |
| Dimethyl sulfoxide (DMSO) | Sigma Aldrich | Cat#D2650 |
| Dulbecco′s Phosphate Buffered Saline (DPBS) | Sigma Aldrich | Cat#D8537 |
| Fetal Bovine Serum (FBS) | Corning | Cat#35-011-CV |
| Human IL-2 IS, premium grade | Miltenyi Biotec | Cat#130-097-746 |
| RPMI-1640 | Corning | Cat#10-040-CV |
| Penicillin-Streptomycin (100X) | Cytiva HyClone | Cat#SV30010 |
| Sodium Pyruvate (100 mM) | Gibco | Cat#11360070 |
| Minimum Essential Media (MEM) non-essential amino acids (NEAA) (100X) | Gibco | Cat#11140050 |
| GlutaMAX Supplement | Gibco | Cat#35050061 |
| 2-mercaptoethanol (50 mM) | Gibco | Cat#21985023 |
| Critical commercial assays | ||
| UltraComp eBeads Plus Compensation Beads | Invitrogen | Cat#01-3333-42 |
| Deposited data | ||
| Primary breast tumor atlas | This paper | https://doi.org/10.5281/zenodo.10672250 |
| Experimental models: Cell lines | ||
| NK-92 | ATCC | Cat#CRL-2407, RRID: CVCL_2142 |
| BT-474 | ATCC | Cat#HTB-20, RRID: CVCL_0179 |
| MDA-MB-436 | ATCC | Cat#HTB-130, RRID: CVCL_0623 |
| K-562 | ATCC | Cat#CCL-243, RRID: CVCL_0004 |
| Software and algorithms | ||
| limma (v3.50.1) | Ritchie et al.90 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| org.Hs.e.g.,.db (v3.14.0) | Carlson et al.91 | https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html |
| DoubletFinder (v2.0.3) | McGinnis et al.92 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Seurat (v4.1.0) | Hao et al.93 | https://satijalab.org/seurat/ |
| MAST (v1.20.0) | Finak et al.94 | https://www.bioconductor.org/packages/release/bioc/html/MAST.html |
| SCTransform (v0.3.2.9008) | Hafemeister et al.95 | https://github.com/satijalab/sctransform |
| UCell (v1.99.1) | Andreatta et al.96 | https://github.com/carmonalab/UCell |
| clusterProfiler (v4.2.2) | Wu et al.97 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| msigdbr (v7.5.1) | Dolgalev et al.98 | https://cran.r-project.org/web/packages/msigdbr/vignettes/msigdbr-intro.html |
| TCGAbiolinks (v2.18.0) | Colaprico et al.99 | https://bioconductor.org/packages/release/bioc/html/TCGAbiolinks.html |
| DESeq2 (v1.34.0) | Love et al.100 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| inferCNV (v.0.99.7) | Tickle et al.101 | https://github.com/broadinstitute/infercnv |
| ROGUE | Liu et al.52 | https://github.com/PaulingLiu/ROGUE |
| cola (v2.0.0) | Gu et al.102 | https://www.bioconductor.org/packages/release/bioc/html/cola.html |
| NicheNet (v1.1.0) | Browaeys et al.103 | https://github.com/saeyslab/nichenetr |
| CellChat (v0.0.1) | Jin et al.104 | https://github.com/jinworks/CellChat |
| BisqueRNA (v1.0.5) | Jew et al.105 | https://github.com/cozygene/bisque |
| pROC (v1.18.0) | Robin et al.106 | https://cran.r-project.org/web/packages/pROC/index.html |
| Other | ||
| Resource website for the primary breast tumor atlas publication containing dataset and analyses | This paper | https://github.com/ChanLab-UTSW/BreastCancer_Integrated |
| Original source dataset of immune cells in primary breast tumors | Azizi et al.7 | GEO: GSE114727 |
| Original source dataset of primary TNBC tumors | Karaayvaz et al.8 | GEO: GSE118389 |
| Original source dataset of primary breast tumors | Pal et al.9 | GEO: GSE161529 |
| Original source dataset of T cells in primary TNBC tumors | Savas et al.107 | GEO: GSE110686 |
| Original source dataset of primary breast tumors | Wu et al.13 | GEO: GSE176078 |
| Original source dataset with primary breast tumors | Xu et al.14 | GEO: GSE180286 |
| Original source dataset with primary breast tumors | Qian et al.11 | https://lambrechtslab.sites.vib.be/en/pan-cancer-blueprint-tumour-microenvironment-0 |
| Original source dataset of primary TNBC tumors | Wu et al.12 | https://singlecell.broadinstitute.org/single_cell/study/SCP1106/stromal-cell-diversity-associated-with-immune-evasion-in-human-triple-negative-breast-cancer |
| Original source dataset of PD-1 treated primary breast tumors | Bassez et al.108 | https://lambrechtslab.sites.vib.be/en/single-cell |
| Breast cancer cell line data from DepMap 22Q2 public release | Ghandi et al.109 | https://depmap.org/portal/download/all/ |
| Spatially resolved data for 6 primary breast tumors | Wu et al.12 | https://doi.org/10.5281/zenodo.4739739 |
| Spatially resolved data from 5 primary breast tumors | 10x Genomics | https://www.10xgenomics.com/datasets/human-breast-cancer-ductal-carcinoma-in-situ-invasive-carcinoma-ffpe-1-standard-1-3-0; https://www.10xgenomics.com/datasets/human-breast-cancer-visium-fresh-frozen-whole-transcriptome-1-standard; https://www.10xgenomics.com/datasets/human-breast-cancer-block-a-section-1-1-standard-1-1-0; https://www.10xgenomics.com/datasets/human-breast-cancer-whole-transcriptome-analysis-1-standard-1-2-0; https://www.10xgenomics.com/products/xenium-in-situ/preview-dataset-human-breast |
| I-SPY2-990 mRNA and clinical data for I-SPY2 trial | Nanda et al.57 | GEO: GSE194040 |
Resources availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dr. Isaac S. Chan (isaac.chan@utsouthwestern.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code will be deposited at https://github.com/ChanLab-UTSW/BreastCancer_Integrated and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
NK-92
NK-92 cells are a human NK cell line derived from a 50-year-old male with malignant non-Hodgkin’s lymphoma. These cells were cultured in RPMI-1640 with 10% FBS, 1% penicillin-streptomycin, 1% sodium pyruvate, 1% MEM-NEAA, 1% GlutaMAX, 0.01% 2-mercaptoethanol, and 100 IU/mL of human IL-2 at 37°C and 5% CO2.
BT-474
BT-474 cells are a human mammary duct cell line derived from a 60-year-old female with invasive ductal carcinoma. These cells were cultured in RPMI-1640 with 10% FBS and 1% penicillin-streptomycin at 37°C and 5% CO2.
MDA-MB-436
MDA-MB-436 cells are a human mammary gland cell line derived from a 43-year-old female with adenocarcinoma. These cells were cultured in RPMI-1640 with 10% FBS and 1% penicillin-streptomycin at 37°C and 5% CO2.
K-562
K-562 cells are a human bone marrow-derived lymphoblastic cell line from a 53-year-old female with chronic myelogenous leukemia. These cells were cultured in RPMI-1640 with 10% FBS and 1% penicillin-streptomycin at 37°C and 5% CO2.
NK-92 cell line media was used in all coculture conditions.
Cell line authentication was performed at the UT Southwestern DNA Genotypic Core facility.
Method details
Processing of single-cell RNA-seq datasets
We obtained 119 primary breast tumor samples across 8 publicly available datasets from 88 untreated female patients 32 to 90 years of age. All gene names were converted to the official gene alias by the HUGO Gene Nomenclature Committee (HGNC) using limma (v3.50.1) and org.Hs.e.g.,.db (v3.14.0) packages.90,91 Cells were filtered by percent mitochondrial transcripts, percent hemoglobin genes, number of RNA molecules, and number of features. In brief, cells below the 5th percentile and above the 95th percentile of each metric were removed, as well as cells with over 15% mitochondrial content. We used the DoubletFinder (v2.0.3) package to identify and remove doublets from the dataset.92 Doublet rates were estimated using reported rates from the original technology used and cell loadings provided by original studies.
Integration of primary breast tumor datasets
119 untreated primary samples were integrated via reference-based integration using Seurat (v4.1.0) to remove batch effects. To prevent over-correction, SCTransform (v0.3.2.9008) was used.95 The 10x datasets were chosen as the reference and rann was chosen for FindNeighbors. Success of batch effect correction was determined by ensuring that no single technology, cohort, or subtype was driving any clusters (Figures S1D–S1L, S2A–S2I; Data S8).
Cell type annotation and clustering
Initial cell type annotations were identified using canonical and literature-derived cell markers as specified in Data S2.96,110,111,112,113 Three methods were used to refine the annotations. The first utilized cluster-level annotations via the UCell (v1.99.1) package96; the second labeled cells based on thresholds of number of markers, and then clustered and calculated the average expression of those markers to refine the cell identities8; the third took highest average expression of select markers. The annotation with highest agreement across the three methods was selected. If all methods disagreed, the cluster-level annotation was chosen.
For the cluster-level method, all cell markers were aggregated into a single score using the AddModuleScore_UCell function from the UCell (v1.99.1) package.93,96 Clusters with the highest score for a given cell type were labeled, isolated, and re-integrated to account for batch effects. Subtype-specific cell markers were applied (e.g., CD4 for CD4+ T cells).
For the second method, cell type annotations were identified based on the number of cell type markers with non-zero expression for a given cell. In brief, epithelial cells labeled if they had two epithelial markers or at least one of EPCAM, KRT8, KRT18, or KRT19. Immune cells were labeled if they had at least two markers of that immune cell type and no other type, PTPRC and at least one marker of that type and no other, or at least three markers for that type and at most one marker of a different immune type. Stromal cells were labeled if they had only cell-type-specific markers or at least three cell-type-specific markers and at most one endothelial marker. Finally, endothelial cells were labeled if they had only endothelial markers or at least three endothelial markers and at most one marker associated with a stromal cell type.
Lastly, we examined log-normalized expression values of the selected cell type markers for each cell. Each cell was annotated with the cell type that had the highest average expression for their markers across all features. T and myeloid subsets were identified in the same manner once the cells were identified as T cells or myeloid cells respectively.
The final cell call was determined based on the highest consensus or defaulted to the cluster-level annotation. Of the 116,346 cells which had original source annotations, 93% had concordant annotations between the original source and our analysis (Data S7).
Identification of natural killer cell subsets
The NK cell cluster was isolated and re-integrated (Figure S4A). Given the higher dimensionality of the dataset containing only NK cells (i.e., number of features ≫ number of NK cells), the Manhattan distance metric was used. FindMarkers in Seurat (v4.1.0) and MAST (v1.20.0) were used to identify differentially expressed genes for each cluster, with absolute log2 fold change (log2FC) cutoff of 0.56 (Bonferroni adjusted p value <0.05).93,94 Marker genes for each NK cell subset are included in Table 1.
To identify human tumor-promoting rNK cells, we previously developed a gene signature based on genes upregulated in tumor-exposed NK cells compared to healthy NK cells in MMTV-PyMT and WT FVB/n mice.20 In our prior study, primary healthy and tumor-exposed NK cells were isolated, and total RNA was extracted and sequenced using Illumina NextSeq 500. Bulk RNA-seq paired-end reads were aligned and mapped using hisat2114 and HTSeq115 respectively, and DESeq2 was used for differential gene expression analysis. In the current study, mouse genes were converted into their human aliases using BioMart (v2.50.0).116 Because the MMTV-PyMT mouse strain used in the previous study most closely resembles the luminal A/luminal B and basal subtypes, these subtypes were first analyzed for the presence of rNK cells. NK cells in the top 75th percentile for the 90-gene signature were labeled as rNK cells. We identified 841 total rNK cells in the integrated dataset.
Gene set enrichment analysis across the NK cell subsets was performed using clusterProfiler (v4.2.2) and the Hallmark gene set collection from msigdbr (v7.5.1).97,98 Only genes with log2FC > 0 were considered. Samples with fewer than 10 NK cells were omitted. For visualization of differentially expressed genes, the log2FC cutoff was increased to 1.5 and a false discovery rate (FDR) cutoff was set to 0.05. To examine expression of the rNK signature within NK cell subsets and across clinical subtypes, Kruskal-Wallis and pairwise post-hoc Dunn tests were performed. For similarity analysis of rNK cells, the expression matrix was reduced to genes in the rNK cell signature, and the Pearson correlation coefficient was calculated for all pairwise combinations of rNK cells with rNK cells and for rNK cells with non-rNK cells. These analyses were also stratified by age to ensure that age was not a confounder (Figures S5D–S5F).
Survival analysis
To assess survival outcomes, we obtained the primary solid tumor samples from the breast cancer cohort of The Cancer Genome Atlas (TCGA).117 Expression data was normalized using TCGAbiolinks (v2.18.0) package and transformed using DESeq2 (v1.34.0) with default parameters.99,100 For all breast cancer samples, we applied NK-specific genes (NCAM1, FGFBP2, KLRD1, FCGR3A, KLRK1) and the 44 upregulated genes of the rNK signature. Of the 1,098 total patients in the dataset, we labeled the top 300 patients with highest rNK signature expression as ‘rNK-high,’ and the remaining 798 patients were labeled as ‘rNK-low.’ Next, we selected samples with high fraction of tumor-infiltrating NK cells (activated or resting NK cells predicted to be greater than a relative fraction of 0.015 of tumor-infiltrating immune cells in the sample), as determined by Xu et al.14 This selected 349 patients for the survival analysis (excluded 749 patients with low NK cell infiltrate). Kaplan-Meier survival curves were generated using survival (v2.44-1.1)118 and assessed using log rank test statistics. Patients ≥45yo demonstrated worse outcomes with increased rNK cell signature expression (p < 0.05) (Figure S5F); survival analysis for patients <45yo did not show significance, though there was a similar trend. To ensure age was not a confounder, correlation between age at initial diagnosis and survival was also assessed (R = −0.11, p > 0.05) (Figure S5E).
Identification of epithelial cell clusters
Epithelial cells were re-clustered and re-integrated to account for batch effects (Figures S6A–S6C). Copy number variant (CNV) profile analysis was used for cancer (malignant) versus normal (non-malignant) assignments. The CNV signal for individual cells was estimated using inferCNV (v.0.99.7) with a 100-gene sliding window; genes with mean count less than 0.1 across all cells were filtered out, and the signal was denoised using a dynamic threshold of 1.3 s.d. from the mean.101 Non-T cell immune cells were used for the reference cell profiles. Epithelial cells were classified as normal (non-malignant), cancer (malignant), or unassigned using a previously described method.119 Briefly, inferred changes at each genomic locus were scaled (between −1 and +1) and the mean of the squares of these values was used to define a CNV signal for each cell. For each sample, an average CNV profile was created, and each cell in the sample was then correlated to this profile for the CNV correlation score. Epithelial cells were classified cancer vs. normal based on CNV signal and CNV correlation, with thresholds of 0.4 for CNV correlation and 0.02 for CNV signal (Figures S3A–S3B). This assigned 75,883 cancer, 3,524 normal, and 4,997 unassigned epithelial cells.
Within cancer epithelial cells, ERBB2-positive and TACSTD2-positive cells were chosen due to clinical relevance. ERBB2 and TACSTD2 expression levels are calculated using UCell (v1.99.1)96. ERBB2Hi cells were defined by ERRB2 expression above the 97.5th percentile of all cells, ERBB2Med cells by expression at or below the 97.5th percentile, and ERBB2Lo cells by zero expression. TACSTD2Hi cells were defined by positive TACSTD2 expression above the 95th percentile of all cells, TACSTD2Med cells by expression at or below the 95th percentile, and TACSTD2Lo cells by zero expression. FindMarkers in Seurat (v4.1.0) and MAST (v1.20.0) were used to identify differentially expressed genes (>5 cells per cluster, detected in >20% of cells in a cluster, log2FC cutoff of 1.5, FDR cutoff of 0.05).93,94 Gene set enrichment analysis was performed using clusterProfiler (v4.2.2) and the Hallmark gene set collection from msigdbr (v7.5.1),97,98 using a 0.1 cutoff for absolute difference in percent expression between the pairwise populations. Expression levels of clinically actionable targets for each subset of cells was estimated by AverageExpression by Seurat (v4.1.0).93 For visualization of differentially expressed genes (Figures S6H–S6K), log2FC cutoff of 1.5 and FDR cutoff of 0.05 were used. To explore associations with clinical features, linear regression and Pearson correlations were calculated between the proportion of ERBB2-positive or TACSTD2-positive cells per sample and age or nodal status, and these analyses were stratified by subtype in Figures S7A–S7C. We additionally explored associations between % TACSTD2+ cells and nodal status in each cohort and then combined the results using Fisher’s combined probability test, which was found to not be statistically significant (Figure S7C; Fisher’s combined probability X = 11.227, p = 0.08). In contrast, for the integrated dataset, there was a statistically significant association between % TACSTD2+ cells per sample and nodal across all samples with nodal status clinical data (p < 0.05, n = 38).
Molecular subtype of samples using SCSubtype
To identify molecular breast cancer subtypes, we used the SC50 subtype gene signature described in Wu et al.13 In brief, the mean read counts for each signature were determined and the highest mean was assigned as the subtype for that cell. To determine the molecular subtype for each tumor, we determined the number of cells classified under each SC50 subtype, and then selected the subtype with the highest number of cells to be the tumor molecular subtype, following the method of Wu et al.13
Cancer epithelial cell heterogeneity analysis
For each tumor sample with over 50 cancer epithelial cells, heterogeneity was assessed using ROGUE, an entropy-based statistic that enables accurate and sensitive assessment of cluster purity.52 To identify samples with discordance between heterogeneity as characterized by the ROGUE score versus by molecular subtype, we calculated the difference between the normalized ROGUE score and the highest percentage of cells of a single subtype. Samples with difference over 50% were determined to be discordant.
To identify gene expression patterns across cancer epithelial cells, unsupervised clustering and supervised classification of all cancer epithelial cells for tumor samples with more than 50 cancer epithelial cells were performed. We generated an exhaustive collection of gene signatures that reflect molecular features of different cancer epithelial cells.
For unsupervised clustering, cancer epithelial cells were clustered at 15 resolutions (0.01, 0.05, 0.08, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 1.0, 1.3, 1.6, 1.8, 2.0) utilizing Seurat (v4.1.0)93. FindMarkers in Seurat (v4.1.0) and MAST (v1.20.0) were used to identify differentially expressed genes (>5 cells per cluster, only test genes with >25% difference in the fraction of detection between the clusters, log2FC > 0.25).93,94 Dataset-wide unsupervised clustering returned 519 gene signatures. Unsupervised clustering was also performed on the sample level, which returned 5,546 gene signatures.
For supervised classification by SC50 molecular subtype, cancer epithelial cells were grouped by SC50 subtype.13 FindMarkers in Seurat (v4.1.0) and MAST (v1.20.0) were then used to identify differentially expressed genes in each group (>5 cells per group, only test genes detected in >20% of cells in a group, log2FC > 0.1).93,94 Supervised classification based on SC50 molecular subtype returned 4 gene signatures.
For supervised classification based on clinical therapeutic targets, expression of 12 clinical therapeutic targets was considered: ESR1, ERBB2, ERBB3, PIK3CA, NTRK1/NTRK2/NTRK3, CD274, EGFR, FGFR1/FGFR2/FGFR3/FGFR4, TACSTD2, CDK4/CDK6, AR, and NECTIN2. Cancer epithelial cells grouped based on high (expression level above the 90th percentile), medium (expression level below the 90th percentile but non-zero), and low (no or zero expression) expression of clinical targets. FindMarkers in Seurat (v4.1.0) and MAST (v1.20.0) were then used to identify differentially expressed genes in each group (>5 cells per group, only test genes detected in >20% of cells in a group, log2.93,94 Supervised classification based on clinical target expression returned 32 gene signatures.
The 12 clinical therapeutic targets were selected based on availability of corresponding therapeutic agents that are approved or under clinical development for the treatment of breast cancer. ESR1 encodes estrogen receptor, the target for hormone therapies such as selective estrogen receptor modulators (e.g., tamoxifen) and selective estrogen receptor degraders (e.g., fulvestrant). ERBB2 encodes HER2, the target for anti-HER2 therapies (e.g., trastuzumab, margetuximab, pertuzumab) and ADCs (e.g., trastuzumab-deruxtecan, T-DM1). ERBB3 encodes HER3, the target for anti-HER3 monoclonal antibodies (e.g., patritumab, seribantumab, lumretuzumab), bispecific antibodies (e.g., EGFR/HER3 duligotuzumab, HER2/HER3 zenocutuzumab, HER3/IGF-1R isitarumab), and ADCs (e.g., patritumab deruxtecan), all currently under clinical development. PIK3CA encodes PI3 kinase, the target for PI3K inhibitors (e.g., alpelisib). NTRK1, NTRK2, and NTRK3 are genes involved in chromosomal rearrangements (NTRK fusions) targeted by TRK inhibitors (e.g., larotrectinib, entrectinib). CD274 encodes PD-L1, the target for PD-L1 inhibitors (e.g., atezolizumab, durvalumab) and PD-1 inhibitors (e.g., pembrolizumab). EGFR encodes the EGFR protein, the target for small molecule inhibitors (e.g., lapatinib, gefitinib, erlotinib, osimertinib) and monoclonal antibodies (e.g., cetuximab, panitumumab). FGFR1, FGFR2, FGFR3, and FGFR4 encode fibroblast growth factor receptors, targeted by pan-FGFR tyrosine kinase inhibitors (e.g., AZD-4547, futibatinib, erdafitinib) currently under clinical development. TACSTD2 encodes TROP2, the target for anti-TROP2 ADCs (e.g., sacituzumab govitecan). CDK4 and CDK6 encode the cyclin-dependent kinases 4 and 6, the targets for CDK4/6 inhibitors (e.g., abemaciclib, palbociclib, and ribociclib). AR encodes the androgen receptor (AR), the target for AR inhibitors (e.g., enzalutamide), CYP17 inhibitors which inhibit production of androgens (e.g., abiraterone, seviteronel), and selective androgen receptor modulators or SARMs (e.g., enobosarm or GTx-024). NECTIN2 encodes NECTIN2 or CD112, which binds TIGIT on T and NK cells and is the target for anti-TIGIT monoclonal antibodies (e.g., tiragolumab, ociperlimab, pembrolizumab/vibostolimab) which are currently under investigation.
Only gene signatures containing over 20 genes were kept. Additionally, signature redundancy was reduced by comparing all unsupervised gene signatures and removing pairs with Jaccard similarity index >0.9. A total of 1,101 gene signatures were generated.
Consensus clustering of the Jaccard similarities between gene signatures (using spherical k-means clustering, metric ATC, implemented with cola (v2.0.0)) was used to identify 10 groupings (Figures S8A–S8B).102 For each grouping, we took the top 100 genes with highest frequency of occurrence across clusters. These were defined as a ‘gene element’ (GE) and were named GE1 to GE10. GE signature expression was calculated for each cancer epithelial cell using UCell (v1.99.1).96 GE signature expression was Z score normalized across all cancer epithelial cells, and cells were assigned to the GE with the highest z-scored expression.
Receptor-ligand pairing analysis
To identify interactions that may influence NK cell reprogramming, NicheNet analyses were run between rNK cells and cancer epithelial cells separated by clinical subtype. rNK cells were set as the ‘sender’ population, and non-rNK cells were set as the ‘reference’103. Receptor-ligand regulatory potential scores for the top 50 predicted ligands and top 200 predicted targets were calculated and for each predicted receptor-ligand pair, an R-L interaction score was calculated as a product of ligand expression (fold change in average expression of the ligand in cancer epithelial cells of that clinical subtype) and receptor expression (percent of the rNK population that has positive expression of the receptor). For the top 20 R-L pairs selected based on this interaction score, circos plots were generated.
To identify interactions between cancer epithelial cells and interacting cells (i.e., CD4+ T cells, CD8+ T cells, regulatory T cells, B cells, plasma cells, myeloid cells, mast cells, MDSCs, NK cells, rNK cells, fibroblasts, myoepithelial cells, endothelial cells, perivascular-like cells), receptor-ligand pairing analysis was performed using NicheNet (v1.1.0) and CellChat (v0.0.1).103,104 For each GE, separate NicheNet analyses were run between cancer epithelial cells assigned to that GE (‘sender’) and each interacting cell population (‘receiver’). The top 50 predicted ligands and top 200 predicted targets were used for the R-L interaction score, which was the product of ligand expression (fold change in average expression on cancer epithelial cells with high vs. low GE expression) and receptor expression (percent of the interacting cell subset with positive receptor expression). For the top 20 receptor-ligand pairs selected based on this R-L interaction score, circos plots were generated. In addition to NicheNet analysis, cancer epithelial cell and interacting cell communication analysis was conducted using CellChat (v0.0.1) using default parameters.104 For each GE, the cell-cell communication network between GE-labeled cancer epithelial cells and interacting cells was visualized using CellChat (v0.0.1) (104). Receptor-ligand pairings with significant (Bonferroni adjusted p value <0.05) probability of interaction were selected as a curated list.
The number of curated receptor-ligand interactions for each GE and interacting cell population was used to infer the degree of interaction between the GEs and interacting cell populations. First, the entire list of R-L interactions predicted by NicheNet was filtered. For each interacting cell population, the top 2,000 predicted R-Ls were selected based on Nichenet prediction for regulatory potential. Then, of those selected pairs, the top 400 predicted R-Ls for each GE were selected based on ligand expression (fold change in average expression of the ligand on cancer epithelial cells with high vs. low GE expression). Lastly, all overlapping R-L interactions that were predicted by both NicheNet and CellChat for a GE and interacting cell pair were selected. We combined the list of overlapping R-L interactions and the list of selected NicheNet R-L interactions to generate a list of curated R-L interactions for each GE and interacting cell population. For each GE and interacting cell pair, the number of curated R-L interactions was normalized across each interacting cell pair. This scaled number of R-L interactions was used to infer the degree of interaction between the GE and the interacting cell population. We visualized the scaled number of curated receptor-ligand interactions in our GE-immune interaction decoder matrix (Figure 3E). We also visualized the absolute number of curated receptor-ligands between each GE and interacting cell (Figure S8D).
Breast cancer cell line exploration
To explore cancer epithelial cell heterogeneity and NK cell sensitivity, we obtained bulk RNA-seq data from the Broad Cancer Cell Line Encyclopedia (CCLE) DepMap portal for human breast cancer cell lines.53,54 Bulk RNA-seq data from CCLE containing TPM values of protein-coding genes were inferred using the RSEM tool and loaded into Seurat (v.4.1.0) and log-normalized.13,93 For each cell line, GE expression was calculated by the UCell (v1.99.1) score of the 100-gene GE signature. For each GE, UCell (v1.99.1) scores were Z score normalized across all breast cancer cell lines.
We experimentally confirmed NK cell cytotoxicity against select human breast cancer cell lines. We selected the BT-474 cell line which had increased expression of NK-resistant GEs (GE1 and GE6) and the MDA-MB-436 cell line which had decreased expression of NK-resistant GEs. Additionally, the K562 cell line (derived from human myelogenous leukemia) is known to be sensitive to NK cell killing and therefore served as a positive control.53,120,121 The NK-92 cell line, a highly cytotoxic NK cell line, was cultured in media with IL-2. To determine killing function of NK cells against cancer cell lines, BT-474, MDA-MB-436, and K562 cells were cocultured with NK-92 cells at a ratio of 1:2 in 96-well round-bottom plates (50,000 cancer cells per well and 100,000 NK-92 cells per well) for 24 h at 37°C. Cells were stained for CD56 (BV605, Clone HCD56, Biolegend 318334) and DAPI in DPBS with 3% FBS. FACS analysis was performed on the Cytek Aurora. Higher NK cytotoxicity was inferred based on increased % DAPI+ in CD56-negative cancer cells.
From Sheffer et al.,53 breast cancer cell line sensitivity to NK cell killing was assessed using reported 24-h AUC values. Briefly, Sheffer et al. performed a PRISM-based phenotypic screen with pools of DNA-barcoded cell lines to quantify NK cell cytotoxicity against cancer cell lines using the AUC of tumor cell survival. Please refer to the original study53 for additional information. For breast cancer cell lines, NK cell sensitivity was based on the reported 24-h AUC values from the Sheffer et al. study. Spearman correlation was used to assess the relationship between GE expression and NK cell sensitivity for breast cancer cell lines.
Spatial transcriptomics analysis
Processed spatial transcriptomics count matrices for 6 samples from Wu et al. and 5 samples from 10x were loaded into Seurat (v.4.1.0).13,93 We deconvoluted the underlying composition of cell types using the anchor-based Seurat integration workflow (Figure S9A). The resulting annotations calculated the fraction of each cell type per given spot and mapped the spatial distribution of cell types, which we further corroborated by the spatial expression of marker genes (Data S2). Spots labeled as normal tissue or artifact by pathologist annotation were excluded.
To investigate interactions between cancer epithelial cells and immune or stromal cells, spots were first filtered based on presence of cancer epithelial cells (spots with less than 10% predicted cancer epithelial cells excluded).93 Each spot containing cancer epithelial cells was scored for expression of each of the 10 GEs using UCell (v1.99.1).96 For immune and stromal cell populations, spots were filtered based on presence of their respective cell types (spots with 0% predicted cells excluded).93 Each spot containing the respective cell type was scored for expression of that cell using canonical and literature-derived cell markers (Data S2). To assess colocalization, Pearson correlations were computed across spots containing between the expression of each GE and the expression of CD8+ T cell markers. For cell signaling predictions between select GE ligands and CD8+ T cell receptors, R-L co-localization scores were defined as the product of the ligand and receptor normalized expression levels.
Development of InteractPrint
For each sample, the average expression of each GE was calculated as the average of the scaled UCell (v1.99.1) (scaled across all cancer epithelial cells in the dataset) score.96 Next, the number of curated R-L pairs in the GE-immune decoder matrix between each GE and CD8+ T cells was used to infer the degree of interaction between cancer epithelial cells and CD8+ T cells. GE1, GE6, GE7, GE8, and GE9 were designated as “inactivating” based on the presence inactivating CD8+ T cell receptors (e.g., NECTIN2_TIGIT) in the list of curated receptor-ligand interactions for those GEs.
T cell InteractPrint was calculated as the average of the number of curated CD8+ T cell R-L interactions in the GE-immune interaction decoder matrix, weighted by average expression of each GE and a factor of −1 for inactivating GEs.
Weighted CD8+ T cell interaction score for a patient’s tumor
GE (ranges from 1 to 10)
Average GE expression (average of Z score normalized UCell scores for the GE
across all cancer epithelial cells in the sample)
Number of curated R-L pairs (from GE-immune interaction decoder matrix)
Multiplier for activating or inactivating GE ( for CD8+ T cell activating
GEs; for CD8+ T cell inactivating GEs)
Validation of InteractPrint
To assess the predictive value of the T cell InteractPrint, we applied our method to a publicly available scRNA-seq dataset containing 29 primary breast tumors from patients who received pembrolizumab (Bassez et al.).108 In Bassez et al., response was inferred based on T cell clonal expansion, as determined by sTCR-seq of pre- and on-treatment samples.108 To determine cancer epithelial cells in the Bassez et al. dataset, CNV analysis was performed (Figures S3C–S3D). GE signature expression and T cell InteractPrint were calculated for each pre-treatment sample.
We applied our method to the I-SPY2 microarray dataset containing 69 primary breast tumors from patients who received combination paclitaxel and pembrolizumab.57 The data was loaded using limma (v3.15), and the batch-corrected and normalized expression data provided by the authors was inserted into the object.90 Genes names were converted using the same method described in the scRNA-seq processing section. Microarray data was deconvoluted with BisqueRNA (v1.0.5) using marker-based devolution with the 10 GE signatures to estimate the relative abundance of the GEs within each sample.105 GE signature expression and T cell InteractPrint were compared for responders and non-responders.
On both datasets, we assessed the predictive value of the T cell InteractPrint compared to average expression levels of PD-L1 on cancer epithelial cells. ROC curves and AUC statistics were generated using the pROC (v1.18.0).106 Bootstrap method (n = 10,000) in pROC (v1.18.0) was used for significance testing between T cell InteractPrint ROC and PD-L1 ROC curves.
Quantification and statistical analysis
Statistical significance was determined using the Wilcoxon Rank-Sum test unless otherwise stated in the figure legend. Where appropriate, p values were adjusted using the Bonferroni correction where appropriate for multiple testing, unless otherwise stated in the figure legend. All boxplots depict the first and third quartiles as the lower and upper bounds, respectively. The whiskers represent 1.5x the interquartile range, and the center depicts the median. All statistical tests with statistical parameters used are defined in the figure legends; p values <0.05 were considered significant.
Acknowledgments
We thank Drs. Carlos Arteaga, Suzanne Conzen, and John Minna for reading our manuscript and providing helpful feedback. This study was supported by funding provided by METAvivor, Susan G. Komen (CCR231010879), the NIH (1K08CA270188-01A1), the Peter Bradley Carlson Trust, the Theresa's Research Foundation, and the NCI/UTSW Simmons Cancer Center (P30 CA142543). Special thanks to all members of the Chan Lab.
Author contributions
Conceptualization, L.X., K.S., S.-P.H., J.M., C.H., and I.S.C.; methodology, L.X., K.S., S.-P.H., H.K., K.C., L.X., and I.S.C.; investigation, L.X., K.S., S.-P.H., H.R., K.M.-A., I.T., K.C., and I.S.C.; visualization, L.X., K.S., S.-P.H., and I.S.C.; writing – original draft, L.X., K.S., S.M.R., E.T.R.T., J.M., C.H., L.X., and I.S.C.; writing – revision, L.X., K.S., H.L.M., S.M.R., E.T.R.T., J.M., C.H., L.X., and I.S.C.; funding acquisition, I.S.C.; supervision, I.S.C.
Declaration of interests
I.S.C., L.X., and K.S. are co-inventors on a pending patent application for a method to determine a predominant immune signal in a breast tumor microenvironment.
Published: April 12, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101511.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Hanker A.B., Sudhan D.R., Arteaga C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell. 2020;37:496–513. doi: 10.1016/j.ccell.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Hajj M., Wicha M.S., Benito-Hernandez A., Morrison S.J., Clarke M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng Y., Spezia M., Huang S., Yuan C., Zeng Z., Zhang L., Ji X., Liu W., Huang B., Luo W., et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018;5:77–106. doi: 10.1016/j.gendis.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Place A.E., Jin Huh S., Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 2011;13:227. doi: 10.1186/bcr2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polyak K. Breast cancer: origins and evolution. J. Clin. Invest. 2007;117:3155–3163. doi: 10.1172/JCI33295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M., et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308.e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karaayvaz M., Cristea S., Gillespie S.M., Patel A.P., Mylvaganam R., Luo C.C., Specht M.C., Bernstein B.E., Michor F., Ellisen L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-06052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pal B., Chen Y., Vaillant F., Capaldo B.D., Joyce R., Song X., Bryant V.L., Penington J.S., Di Stefano L., Tubau Ribera N., et al. A single-cell RNA expression atlas of normal, preneoplastic and tumorigenic states in the human breast. EMBO J. 2021;40 doi: 10.15252/embj.2020107333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savas P., Loi S. Expanding the Role for Immunotherapy in Triple-Negative Breast Cancer. Cancer Cell. 2020;37:623–624. doi: 10.1016/j.ccell.2020.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Qian J., Olbrecht S., Boeckx B., Vos H., Laoui D., Etlioglu E., Wauters E., Pomella V., Verbandt S., Busschaert P., et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020;30:745–762. doi: 10.1038/s41422-020-0355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu S.Z., Roden D.L., Wang C., Holliday H., Harvey K., Cazet A.S., Murphy K.J., Pereira B., Al-Eryani G., Bartonicek N., et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020;39 doi: 10.15252/embj.2019104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu S.Z., Al-Eryani G., Roden D.L., Junankar S., Harvey K., Andersson A., Thennavan A., Wang C., Torpy J.R., Bartonicek N., et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021;53:1334–1347. doi: 10.1038/s41588-021-00911-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu K., Wang R., Xie H., Hu L., Wang C., Xu J., Zhu C., Liu Y., Gao F., Li X., et al. Single-cell RNA sequencing reveals cell heterogeneity and transcriptome profile of breast cancer lymph node metastasis. Oncogenesis. 2021;10 doi: 10.1038/s41389-021-00355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma C., Kaewkangsadan V., Eremin J.M., Cowley G.P., Ilyas M., El-Sheemy M.A., Eremin O. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): differential restoration of blood profiles by NAC and surgery. J. Transl. Med. 2015;13:180. doi: 10.1186/s12967-015-0535-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouzidi L., Triki H., Charfi S., Kridis W.B., Derbel M., Ayadi L., Sellami-Boudawara T., Cherif B. Prognostic Value of Natural Killer Cells Besides Tumor-Infiltrating Lymphocytes in Breast Cancer Tissues. Clin. Breast Cancer. 2021;21:e738–e747. doi: 10.1016/j.clbc.2021.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Gu-Trantien C., Loi S., Garaud S., Equeter C., Libin M., de Wind A., Ravoet M., Le Buanec H., Sibille C., Manfouo-Foutsop G., et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Invest. 2013;123:2873–2892. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruffell B., Au A., Rugo H.S., Esserman L.J., Hwang E.S., Coussens L.M. Leukocyte composition of human breast cancer. Proc. Natl. Acad. Sci. USA. 2012;109:2796–2801. doi: 10.1073/pnas.1104303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rezaeifard S., Talei A., Shariat M., Erfani N. Tumor infiltrating NK cell (TINK) subsets and functional molecules in patients with breast cancer. Mol. Immunol. 2021;136:161–167. doi: 10.1016/j.molimm.2021.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Chan I.S., Knútsdóttir H., Ramakrishnan G., Padmanaban V., Warrier M., Ramirez J.C., Dunworth M., Zhang H., Jaffee E.M., Bader J.S., Ewald A.J. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J. Cell Biol. 2020;219 doi: 10.1083/jcb.202001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan I.S., Ewald A.J. The changing role of natural killer cells in cancer metastasis. J. Clin. Invest. 2022;132 doi: 10.1172/jci143762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melaiu O., Lucarini V., Cifaldi L., Fruci D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.03038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polyak K. Heterogeneity in breast cancer. J. Clin. Invest. 2011;121:3786–3788. doi: 10.1172/jci60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turashvili G., Brogi E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017;4:227. doi: 10.3389/fmed.2017.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crinier A., Dumas P.-Y., Escalière B., Piperoglou C., Gil L., Villacreces A., Vély F., Ivanovic Z., Milpied P., Narni-Mancinelli É., Vivier É. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia. Cell. Mol. Immunol. 2021;18:1290–1304. doi: 10.1038/s41423-020-00574-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith S.L., Kennedy P.R., Stacey K.B., Worboys J.D., Yarwood A., Seo S., Solloa E.H., Mistretta B., Chatterjee S.S., Gunaratne P., et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv. 2020;4:1388–1406. doi: 10.1182/bloodadvances.2019000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang C., Siebert J.R., Burns R., Gerbec Z.J., Bonacci B., Rymaszewski A., Rau M., Riese M.J., Rao S., Carlson K.-S., et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 2019;10:3931. doi: 10.1038/s41467-019-11947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Andrade L.F., Lu Y., Luoma A., Ito Y., Pan D., Pyrdol J.W., Yoon C.H., Yuan G.-C., Wucherpfennig K.W. Discovery of specialized NK cell populations infiltrating human melanoma metastases. JCI Insight. 2019;4 doi: 10.1172/jci.insight.133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreno-Nieves U.Y., Tay J.K., Saumyaa S., Horowitz N.B., Shin J.H., Mohammad I.A., Luca B., Mundy D.C., Gulati G.S., Bedi N., et al. Landscape of innate lymphoid cells in human head and neck cancer reveals divergent NK cell states in the tumor microenvironment. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2101169118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheon H., Borden E.C., Stark G.R. Interferons and their stimulated genes in the tumor microenvironment. Semin. Oncol. 2014;41:156–173. doi: 10.1053/j.seminoncol.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dogra P., Rancan C., Ma W., Toth M., Senda T., Carpenter D.J., Kubota M., Matsumoto R., Thapa P., Szabo P.A., et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell. 2020;180:749–763.e13. doi: 10.1016/j.cell.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J., López-Moyado I.F., Seo H., Lio C.-W.J., Hempleman L.J., Sekiya T., Yoshimura A., Scott-Browne J.P., Rao A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature. 2019;567:530–534. doi: 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou F., Drabsch Y., Dekker T.J.A., De Vinuesa A.G., Li Y., Hawinkels L.J.A.C., Sheppard K.-A., Goumans M.-J., Luwor R.B., De Vries C.J., et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014;5:3388. doi: 10.1038/ncomms4388. [DOI] [PubMed] [Google Scholar]
- 34.Chan C.J., Martinet L., Gilfillan S., Souza-Fonseca-Guimaraes F., Chow M.T., Town L., Ritchie D.S., Colonna M., Andrews D.M., Smyth M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014;15:431–438. doi: 10.1038/ni.2850. [DOI] [PubMed] [Google Scholar]
- 35.Braud V.M., Allan D.S., O'Callaghan C.A., Söderström K., D'Andrea A., Ogg G.S., Lazetic S., Young N.T., Bell J.I., Phillips J.H., et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 36.Modi S., Jacot W., Yamashita T., Sohn J., Vidal M., Tokunaga E., Tsurutani J., Ueno N.T., Prat A., Chae Y.S., et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022;387:9–20. doi: 10.1056/nejmoa2203690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rugo H.S., Bardia A., Marmé F., Cortes J., Schmid P., Loirat D., Tredan O., Ciruelos E., Dalenc F., Gómez Pardo P., et al. Primary results from TROPiCS-02: A randomized phase 3 study of sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (Pts) with hormone receptor–positive/HER2-negative (HR+/HER2-) advanced breast cancer. J. Clin. Oncol. 2022;40:LBA1001. doi: 10.1200/JCO.2022.40.17_suppl.LBA1001. [DOI] [Google Scholar]
- 38.Li X., Wang C.-Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021;13:36. doi: 10.1038/s41368-021-00146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan R.S.Y.C., Ong W.S., Lee K.-H., Lim A.H., Park S., Park Y.H., Lin C.-H., Lu Y.-S., Ono M., Ueno T., et al. HER2 expression, copy number variation and survival outcomes in HER2-low non-metastatic breast cancer: an international multicentre cohort study and TCGA-METABRIC analysis. BMC Med. 2022;20 doi: 10.1186/s12916-022-02284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schettini F., Chic N., Brasó-Maristany F., Paré L., Pascual T., Conte B., Martínez-Sáez O., Adamo B., Vidal M., Barnadas E., et al. Clinical, pathological, and PAM50 gene expression features of HER2-low breast cancer. npj Breast Cancer. 2021;7 doi: 10.1038/s41523-020-00208-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vidula N., Yau C., Rugo H. Trophoblast Cell Surface Antigen 2 gene (TACSTD2) expression in primary breast cancer. Breast Cancer Res. Treat. 2022;194:569–575. doi: 10.1007/s10549-022-06660-x. [DOI] [PubMed] [Google Scholar]
- 42.Ambrogi F., Fornili M., Boracchi P., Trerotola M., Relli V., Simeone P., La Sorda R., Lattanzio R., Querzoli P., Pedriali M., et al. Trop-2 Is a Determinant of Breast Cancer Survival. PLoS One. 2014;9 doi: 10.1371/journal.pone.0096993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aslan M., Hsu E.-C., Garcia-Marques F.J., Bermudez A., Liu S., Shen M., West M., Zhang C.A., Rice M.A., Brooks J.D., et al. Oncogene-mediated metabolic gene signature predicts breast cancer outcome. npj Breast Cancer. 2021;7:141. doi: 10.1038/s41523-021-00341-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rizeq B., Zakaria Z., Ouhtit A. Towards understanding the mechanisms of actions of carcinoembryonic antigen-related cell adhesion molecule 6 in cancer progression. Cancer Sci. 2018;109:33–42. doi: 10.1111/cas.13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanda Y., Mizuno A., Takasaki T., Satoh R., Hagihara K., Masuko T., Endo Y., Tanabe G., Sugiura R. Down-regulation of dual-specificity phosphatase 6, a negative regulator of oncogenic ERK signaling, by ACA-28 induces apoptosis in NIH/3T3 cells overexpressing HER2/ErbB2. Gene Cell. 2021;26:109–116. doi: 10.1111/gtc.12823. [DOI] [PubMed] [Google Scholar]
- 46.Desai K., Nair M.G., Prabhu J.S., Vinod A., Korlimarla A., Rajarajan S., Aiyappa R., Kaluve R.S., Alexander A., Hari P.S., et al. High expression of integrin β6 in association with the Rho-Rac pathway identifies a poor prognostic subgroup within HER2 amplified breast cancers. Cancer Med. 2016;5:2000–2011. doi: 10.1002/cam4.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheung K.J., Gabrielson E., Werb Z., Ewald A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seol H., Lee H.J., Choi Y., Lee H.E., Kim Y.J., Kim J.H., Kang E., Kim S.-W., Park S.Y. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: its clinicopathological significance. Mod. Pathol. 2012;25:938–948. doi: 10.1038/modpathol.2012.36. [DOI] [PubMed] [Google Scholar]
- 49.Janiszewska M., Liu L., Almendro V., Kuang Y., Paweletz C., Sakr R.A., Weigelt B., Hanker A.B., Chandarlapaty S., King T.A., et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat. Genet. 2015;47:1212–1219. doi: 10.1038/ng.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soucheray M., Capelletti M., Pulido I., Kuang Y., Paweletz C.P., Becker J.H., Kikuchi E., Xu C., Patel T.B., Al-Shahrour F., et al. Intratumoral Heterogeneity in EGFR-Mutant NSCLC Results in Divergent Resistance Mechanisms in Response to EGFR Tyrosine Kinase Inhibition. Cancer Res. 2015;75:4372–4383. doi: 10.1158/0008-5472.can-15-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muzumdar M.D., Dorans K.J., Chung K.M., Robbins R., Tammela T., Gocheva V., Li C.M.-C., Jacks T. Clonal dynamics following p53 loss of heterozygosity in Kras-driven cancers. Nat. Commun. 2016;7 doi: 10.1038/ncomms12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu B., Li C., Li Z., Wang D., Ren X., Zhang Z. An entropy-based metric for assessing the purity of single cell populations. Nat. Commun. 2020;11:3155. doi: 10.1038/s41467-020-16904-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheffer M., Lowry E., Beelen N., Borah M., Amara S.N.-A., Mader C.C., Roth J.A., Tsherniak A., Freeman S.S., Dashevsky O., et al. Genome-scale screens identify factors regulating tumor cell responses to natural killer cells. Nat. Genet. 2021;53:1196–1206. doi: 10.1038/s41588-021-00889-w. [DOI] [PubMed] [Google Scholar]
- 54.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Network N.C.C. Breast Cancer. 2022 https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (v4.2022) [Google Scholar]
- 56.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nanda R., Liu M.C., Yau C., Shatsky R., Pusztai L., Wallace A., Chien A.J., Forero-Torres A., Ellis E., Han H., et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020;6:676–684. doi: 10.1001/jamaoncol.2019.6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu N.C., Wong W., Ho K.E., Chu V.C., Rizo A., Davenport S., Kelly D., Makar R., Jassem J., Duchnowska R., et al. Comparison of central laboratory assessments of ER, PR, HER2, and Ki67 by IHC/FISH and the corresponding mRNAs (ESR1, PGR, ERBB2, and MKi67) by RT-qPCR on an automated, broadly deployed diagnostic platform. Breast Cancer Res. Treat. 2018;172:327–338. doi: 10.1007/s10549-018-4889-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denkert C., Huober J., Loibl S., Prinzler J., Kronenwett R., Darb-Esfahani S., Brase J.C., Solbach C., Mehta K., Fasching P.A., et al. HER2 and ESR1 mRNA expression levels and response to neoadjuvant trastuzumab plus chemotherapy in patients with primary breast cancer. Breast Cancer Res. 2013;15 doi: 10.1186/bcr3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z., Portier B.P., Gruver A.M., Bui S., Wang H., Su N., Vo H.-T., Ma X.-J., Luo Y., Budd G.T., Tubbs R.R. Automated Quantitative RNA in Situ Hybridization for Resolution of Equivocal and Heterogeneous ERBB2 (HER2) Status in Invasive Breast Carcinoma. J. Mol. Diagn. 2013;15:210–219. doi: 10.1016/j.jmoldx.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 61.Press M.F., Finn R.S., Cameron D., Di Leo A., Geyer C.E., Villalobos I.E., Santiago A., Guzman R., Gasparyan A., Ma Y., et al. HER-2 Gene Amplification, HER-2 and Epidermal Growth Factor Receptor mRNA and Protein Expression, and Lapatinib Efficacy in Women with Metastatic Breast Cancer. Clin. Cancer Res. 2008;14:7861–7870. doi: 10.1158/1078-0432.Ccr-08-1056. [DOI] [PubMed] [Google Scholar]
- 62.Kurozumi S., Yamaguchi Y., Matsumoto H., Inoue K., Kurosumi M., Oyama T., Horiguchi J., Fujii T., Shirabe K. Comparing protein and mRNA expressions of the human epidermal growth factor receptor family in estrogen receptor-positive breast cancer. Med. Mol. Morphol. 2019;52:90–98. doi: 10.1007/s00795-018-0206-y. [DOI] [PubMed] [Google Scholar]
- 63.Vassilakopoulou M., Togun T., Dafni U., Cheng H., Bordeaux J., Neumeister V.M., Bobos M., Pentheroudakis G., Skarlos D.V., Pectasides D., et al. In Situ Quantitative Measurement of HER2mRNA Predicts Benefit from Trastuzumab-Containing Chemotherapy in a Cohort of Metastatic Breast Cancer Patients. PLoS One. 2014;9 doi: 10.1371/journal.pone.0099131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coates J.T., Sun S., Leshchiner I., Thimmiah N., Martin E.E., McLoughlin D., Danysh B.P., Slowik K., Jacobs R.A., Rhrissorrakrai K., et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021;11:2436–2445. doi: 10.1158/2159-8290.Cd-21-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chou J., Trepka K., Sjöström M., Egusa E.A., Chu C.E., Zhu J., Chan E., Gibb E.A., Badura M.L., Contreras-Sanz A., et al. TROP2 Expression Across Molecular Subtypes of Urothelial Carcinoma and Enfortumab Vedotin-resistant Cells. Eur. Urol. Oncol. 2022;5:714–718. doi: 10.1016/j.euo.2021.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohmachi T., Tanaka F., Mimori K., Inoue H., Yanaga K., Mori M. Clinical significance of TROP2 expression in colorectal cancer. Clin. Cancer Res. 2006;12:3057–3063. doi: 10.1158/1078-0432.Ccr-05-1961. [DOI] [PubMed] [Google Scholar]
- 67.Bignotti E., Todeschini P., Calza S., Falchetti M., Ravanini M., Tassi R.A., Ravaggi A., Bandiera E., Romani C., Zanotti L., et al. Trop-2 overexpression as an independent marker for poor overall survival in ovarian carcinoma patients. Eur. J. Cancer. 2010;46:944–953. doi: 10.1016/j.ejca.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 68.Stepan L.P., Trueblood E.S., Hale K., Babcook J., Borges L., Sutherland C.L. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J. Histochem. Cytochem. 2011;59:701–710. doi: 10.1369/0022155411410430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bardia A., Rugo H.S., Cortés J., Tolaney S.M., Schmid P., Motwani M., Yoon O.K., Boice J., Zhuo L., Marmé F. Trop-2 mRNA expression and association with clinical outcomes with sacituzumab govitecan (SG) in patients with HR+/HER2– metastatic breast cancer (mBC): Biomarker results from the phase 3 TROPiCS-02 study. J. Clin. Oncol. 2023;41:1082. doi: 10.1200/JCO.2023.41.16_suppl.1082. [DOI] [Google Scholar]
- 70.Rugo H.S., Delord J.P., Im S.A., Ott P.A., Piha-Paul S.A., Bedard P.L., Sachdev J., Le Tourneau C., van Brummelen E.M.J., Varga A., et al. Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer. Clin. Cancer Res. 2018;24:2804–2811. doi: 10.1158/1078-0432.Ccr-17-3452. [DOI] [PubMed] [Google Scholar]
- 71.Kwa M.J., Adams S. Checkpoint inhibitors in triple-negative breast cancer (TNBC): Where to go from here. Cancer. 2018;124:2086–2103. doi: 10.1002/cncr.31272. [DOI] [PubMed] [Google Scholar]
- 72.Gandhi L., Rodríguez-Abreu D., Gadgeel S., Esteban E., Felip E., De Angelis F., Domine M., Clingan P., Hochmair M.J., Powell S.F., et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 73.Garon E.B., Rizvi N.A., Hui R., Leighl N., Balmanoukian A.S., Eder J.P., Patnaik A., Aggarwal C., Gubens M., Horn L., et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 74.Schachter J., Ribas A., Long G.V., Arance A., Grob J.J., Mortier L., Daud A., Carlino M.S., McNeil C., Lotem M., et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006) Lancet. 2017;390:1853–1862. doi: 10.1016/s0140-6736(17)31601-x. [DOI] [PubMed] [Google Scholar]
- 75.Bellmunt J., de Wit R., Vaughn D.J., Fradet Y., Lee J.L., Fong L., Vogelzang N.J., Climent M.A., Petrylak D.P., Choueiri T.K., et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017;376:1015–1026. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cortes J., Rugo H.S., Cescon D.W., Im S.-A., Yusof M.M., Gallardo C., Lipatov O., Barrios C.H., Perez-Garcia J., Iwata H., et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022;387:217–226. doi: 10.1056/NEJMoa2202809. [DOI] [PubMed] [Google Scholar]
- 77.Mittendorf E.A., Zhang H., Barrios C.H., Saji S., Jung K.H., Hegg R., Koehler A., Sohn J., Iwata H., Telli M.L., et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396:1090–1100. doi: 10.1016/S0140-6736(20)31953-X. [DOI] [PubMed] [Google Scholar]
- 78.Gianni L., Huang C.S., Egle D., Bermejo B., Zamagni C., Thill M., Anton A., Zambelli S., Bianchini G., Russo S., et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple-negative, early high-risk and locally advanced breast cancer: NeoTRIP Michelangelo randomized study. Ann. Oncol. 2022;33:534–543. doi: 10.1016/j.annonc.2022.02.004. [DOI] [PubMed] [Google Scholar]
- 79.Loibl S., Schneeweiss A., Huober J., Braun M., Rey J., Blohmer J.U., Furlanetto J., Zahm D.M., Hanusch C., Thomalla J., et al. Neoadjuvant durvalumab improves survival in early triple-negative breast cancer independent of pathological complete response. Ann. Oncol. 2022;33:1149–1158. doi: 10.1016/j.annonc.2022.07.1940. [DOI] [PubMed] [Google Scholar]
- 80.Loibl S., Untch M., Burchardi N., Huober J., Sinn B.V., Blohmer J.U., Grischke E.M., Furlanetto J., Tesch H., Hanusch C., et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019;30:1279–1288. doi: 10.1093/annonc/mdz158. [DOI] [PubMed] [Google Scholar]
- 81.Schmid P., Cortes J., Pusztai L., McArthur H., Kümmel S., Bergh J., Denkert C., Park Y.H., Hui R., Harbeck N., et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020;382:810–821. doi: 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 82.Conroy J.M., Pabla S., Nesline M.K., Glenn S.T., Papanicolau-Sengos A., Burgher B., Andreas J., Giamo V., Wang Y., Lenzo F.L., et al. Next generation sequencing of PD-L1 for predicting response to immune checkpoint inhibitors. J. Immunother. Cancer. 2019;7:18. doi: 10.1186/s40425-018-0489-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kang S.Y., Heo Y.J., Kwon G.Y., Kim K.M. Expression of CD274 mRNA Measured by qRT-PCR Correlates With PD-L1 Immunohistochemistry in Gastric and Urothelial Carcinoma. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.856444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tretiakova M., Fulton R., Kocherginsky M., Long T., Ussakli C., Antic T., Gown A. Concordance study of PD-L1 expression in primary and metastatic bladder carcinomas: comparison of four commonly used antibodies and RNA expression. Mod. Pathol. 2018;31:623–632. doi: 10.1038/modpathol.2017.188. [DOI] [PubMed] [Google Scholar]
- 85.Li Y., Vennapusa B., Chang C.W., Tran D., Nakamura R., Sumiyoshi T., Hegde P., Molinero L. Prevalence Study of PD-L1 SP142 Assay in Metastatic Triple-negative Breast Cancer. Appl. Immunohistochem. Mol. Morphol. 2021;29:258–264. doi: 10.1097/pai.0000000000000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.El Bairi K., Haynes H.R., Blackley E., Fineberg S., Shear J., Turner S., de Freitas J.R., Sur D., Amendola L.C., Gharib M., et al. The tale of TILs in breast cancer: A report from The International Immuno-Oncology Biomarker Working Group. npj Breast Cancer. 2021;7:150. doi: 10.1038/s41523-021-00346-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cristescu R., Aurora-Garg D., Albright A., Xu L., Liu X.Q., Loboda A., Lang L., Jin F., Rubin E.H., Snyder A., Lunceford J. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: a pan-tumor retrospective analysis of participants with advanced solid tumors. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iwase T., Blenman K.R.M., Li X., Reisenbichler E., Seitz R., Hout D., Nielsen T.J., Schweitzer B.L., Bailey D.B., Shen Y., et al. A Novel Immunomodulatory 27-Gene Signature to Predict Response to Neoadjuvant Immunochemotherapy for Primary Triple-Negative Breast Cancer. Cancers. 2021;13:4839. doi: 10.3390/cancers13194839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gonzalez-Ericsson P.I., Wulfkhule J.D., Gallagher R.I., Sun X., Axelrod M.L., Sheng Q., Luo N., Gomez H., Sanchez V., Sanders M., et al. Tumor-Specific Major Histocompatibility-II Expression Predicts Benefit to Anti-PD-1/L1 Therapy in Patients With HER2-Negative Primary Breast Cancer. Clin. Cancer Res. 2021;27:5299–5306. doi: 10.1158/1078-0432.Ccr-21-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carlson M. org.Hs.eg.db: Genome wide annotation for Human. version 3.8.2R package. 2019 [Google Scholar]
- 92.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Finak G., McDavid A., Yajima M., Deng J., Gersuk V., Shalek A.K., Slichter C.K., Miller H.W., McElrath M.J., Prlic M., et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Andreatta M., Carmona S.J. UCell: Robust and scalable single-cell gene signature scoring. Comput. Struct. Biotechnol. J. 2021;19:3796–3798. doi: 10.1016/j.csbj.2021.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dolgalev I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format. R package version. 2022;7 [Google Scholar]
- 99.Colaprico A., Silva T.C., Olsen C., Garofano L., Cava C., Garolini D., Sabedot T.S., Malta T.M., Pagnotta S.M., Castiglioni I., et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44:e71. doi: 10.1093/nar/gkv1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tickle T.,T.I., Georgescu C., Brown M., Haas B. 2019. inferCNV of the Trinity CTAT Project. [Google Scholar]
- 102.Gu Z., Schlesner M., Hübschmann D. cola: an R/Bioconductor package for consensus partitioning through a general framework. Nucleic Acids Res. 2021;49:e15. doi: 10.1093/nar/gkaa1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 104.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.-H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jew B., Alvarez M., Rahmani E., Miao Z., Ko A., Garske K.M., Sul J.H., Pietiläinen K.H., Pajukanta P., Halperin E. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 2020;11:1971. doi: 10.1038/s41467-020-15816-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Savas P., Virassamy B., Ye C., Salim A., Mintoff C.P., Caramia F., Salgado R., Byrne D.J., Teo Z.L., Dushyanthen S., et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018;24:986–993. doi: 10.1038/s41591-018-0078-7. [DOI] [PubMed] [Google Scholar]
- 108.Bassez A., Vos H., Van Dyck L., Floris G., Arijs I., Desmedt C., Boeckx B., Vanden Bempt M., Nevelsteen I., Lambein K., et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat. Med. 2021;27:820–832. doi: 10.1038/s41591-021-01323-8. [DOI] [PubMed] [Google Scholar]
- 109.Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., Barretina J., Gelfand E.T., Bielski C.M., Li H., et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pliner H.A., Shendure J., Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods. 2019;16:983–986. doi: 10.1038/s41592-019-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Quan F., Liang X., Cheng M., Yang H., Liu K., He S., Sun S., Deng M., He Y., Liu W., et al. Annotation of cell types (ACT): a convenient web server for cell type annotation. Genome Med. 2023;15:91. doi: 10.1186/s13073-023-01249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gky900. D721-d728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Crinier A., Milpied P., Escalière B., Piperoglou C., Galluso J., Balsamo A., Spinelli L., Cervera-Marzal I., Ebbo M., Girard-Madoux M., et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity. 2018;49:971–986.e5. doi: 10.1016/j.immuni.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Durinck S., Moreau Y., Kasprzyk A., Davis S., De Moor B., Brazma A., Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21:3439–3440. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 117.Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V., et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Therneau T. A Package for Survival Analysis in R. R package version. 2022;3.3.1 [Google Scholar]
- 119.Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M., et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835–849.e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zamai L., Mariani A.R., Zauli G., Rodella L., Rezzani R., Manzoli F.A., Vitale M. Kinetics of in vitro natural killer activity against K562 cells as detected by flow cytometry. Cytometry. 1998;32:280–285. doi: 10.1002/(sici)1097-0320(19980801)32:4<280::aid-cyto4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 121.Tremblay-McLean A., Coenraads S., Kiani Z., Dupuy F.P., Bernard N.F. Expression of ligands for activating natural killer cell receptors on cell lines commonly used to assess natural killer cell function. BMC Immunol. 2019;20:8. doi: 10.1186/s12865-018-0272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
-
•
All original code will be deposited at https://github.com/ChanLab-UTSW/BreastCancer_Integrated and is publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.