

Summary
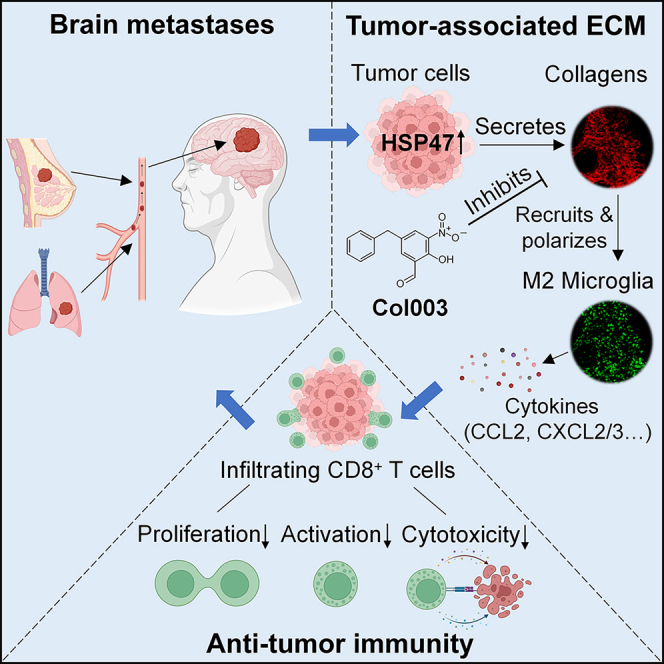
Brain metastases (BrMs) are the leading cause of death in patients with solid cancers. BrMs exhibit a highly immunosuppressive milieu and poor response to immunotherapies; however, the underlying mechanism remains largely unclear. Here, we show that upregulation of HSP47 in tumor cells drives metastatic colonization and outgrowth in the brain by creating an immunosuppressive microenvironment. HSP47-mediated collagen deposition in the metastatic niche promotes microglial polarization to the M2 phenotype via the α2β1 integrin/nuclear factor κB pathway, which upregulates the anti-inflammatory cytokines and represses CD8+ T cell anti-tumor responses. Depletion of microglia reverses HSP47-induced inactivation of CD8+ T cells and abolishes BrM. Col003, an inhibitor disrupting HSP47-collagen association restores an anti-tumor immunity and enhances the efficacy of anti-PD-L1 immunotherapy in BrM-bearing mice. Our study supports that HSP47 is a critical determinant of M2 microglial polarization and immunosuppression and that blocking the HSP47-collagen axis represents a promising therapeutic strategy against brain metastatic tumors.
Keywords: HSP47, collagen, microglia, brain metastases, anti-tumor immunity
Graphical abstract
Highlights
-
•
HSP47 upregulation in lung cancer and breast cancer drives brain metastases (BrMs)
-
•
Collagen deposition in BrMs promotes M2 microglial polarization and immunosuppression
-
•
Col003, an inhibitor of HSP47, enhances anti-tumor CD8+ T cell responses
-
•
Col003 enhances the efficacy of PD-L1 blockade therapy in BrMs
Wang et al. demonstrate that HSP47 upregulation in tumor cells promotes collagen deposition and brain metastasis by promoting M2 microglial polarization and immunosuppression. Blocking the HSP47-collagen axis by Col003 restores the anti-tumor immunity and enhances the efficacy of anti-PD-L1 immunotherapy in brain metastases.
Introduction
Brain metastases (BrMs) are an increasingly common malignancy of the central nervous system (CNS) that occurs in over 20% of all cancer patients.1 The most common primary tumors associated with BrMs are lung cancer (20%–50%), breast cancer (5%–20%), and melanoma (7%-16), although any type of cancer can metastasize to the brain.1,2 Despite advances in systemic therapies, including a combination of surgery, chemotherapy, radiotherapy, targeted therapies, and immunotherapies, prognosis after the development of BrMs remains very poor, with a 2-year overall survival rate of less than 10%.2,3 Understanding the molecular characteristics of BrMs relative to the primary tumors and the mechanisms that drive BrMs will help to develop preventive therapeutic strategies against this deadly disease.
After invading the CNS, tumor cells are confronted with the highly complex and specialized brain microenvironment that is fundamentally different from the primary site, including cellular components, matrix composition, and immune landscape.4,5 The ability of tumor cells to rapidly co-opt niche cells in the brain to block or evade anti-tumor immune response is a critical determinant for successful metastatic colonization.6,7 In established BrMs, microglia are the most abundant non-cancerous cell type, constituting up to 30% of the total tumor mass.8 Microglia have the ability to polarize to either a “classical” proinflammatory phenotype (M1), characterized by increased levels of proinflammatory cytokines and the ability to elicit an anti-tumor T cell immune response, or an “alternative” anti-inflammatory phenotype (M2), which is immunosuppressive and promotes tumor growth by secreting growth factors or facilitating angiogenesis.7,8 A large proportion of microglial cells in intracranial tumors exhibit an M2-like phenotype,7,9 and targeting M2 microglia represents a promising strategy for intracranial tumors.10,11 However, how the metastatic tumor cells modulate microglial polarization and create the immunosuppressive local milieu in the brain remains largely unclear.
The extracellular matrix (ECM) is a critical determinant of the tumor microenvironment (TME), which modifies the external microenvironment and is the main mediator of cancer cell adaptation, controlling the development and progression of cancer.12,13,14 To reveal the ECM proteins that may contribute to BrM, we analyzed their expression levels in BrMs in comparison to the primary tumors, which identified HSP47 as a critical regulator of BrM. We found that HSP47-mediated collagen deposition in the brain metastatic lesions induces M2 microglial polarization, leading to a repressed CD8+ T cell anti-tumor immune response. Blocking the HSP47-collagen axis restores an anti-tumor brain immune microenvironment and augments the efficacy of anti-PD-L1 immunotherapy, indicating a premising therapeutic strategy for brain metastatic tumors.
Results
HSP47 is overexpressed in BrMs and high-level HSP47 predicts poor survival in patients with cancer
ECM remodeling is crucial for cancer development and progression.13,14 To identify a potential ECM protein that may promote BrM, we first compared the expression levels of the ECM genes (89 genes) in primary breast cancer and lung cancer, the two most common primary cancers that metastasize to the brain, with the corresponding brain metastatic tumors, respectively. The result showed that a total of 15 genes were significantly co-upregulated in lung cancer and breast cancer BrMs compared to the primary tumors (Figures 1A and 1B). To further explore the role of these genes in BrM, we constructed brain metastatic cell lines through five rounds of intracardiac inoculation and recovery of BrMs in mice using LLC1 and 4T1 cells and obtained LLC1-BMT5 and 4T1-BMT5 cells, respectively (Figure 1C). Compared to the parental cells, the brain metastatic cells were more prone to metastasize to the brain, with the probability of BrM elevated from 12.5% to 100% for both 4T1-BMT5 cells and LLC1-BMT5 cells (Figures 1D and 1E). Notably, the LLC1-BMT5 cells showed significant morphological changes compared to the parental cells, exhibiting the properties of cancer stem cells in fetal bovine serum-containing medium (Figure S1A).
Figure 1.

HSP47 is overexpressed in brain metastasis and high-level HSP47 predicts poor survival in cancer patients
(A) Heatmaps showing the differential expression of ECM genes in breast cancer BrMs (n = 97) compared to primary tumors (n = 137) (GEO: GSE184869, GSE12276, GSE29431, GSE46928, and GSE100534; GitHub: JNCI_2018) and in lung cancer BrMs (n = 144) compared to primary tumors (n = 166) (GEO: GSE14108 and GSE13213; OMIX: OMIX575; BioProject: PRJNA681304; ArrayExpress: E_MTAB_8659).
(B) Venn diagram showing the overlapping genes that are co-upregulated in BrMs in (A).
(C) Diagram showing the in vivo selection procedure of brain metastatic cell populations (BMT5) from 4T1 breast cancer cells and LLC1 lung cancer cells, respectively.
(D) 4T1-BMT5 and the parental 4T1 cells (5 × 104 cells/mouse) expressing the firefly luciferase were intracardially implanted into immunocompetent BALB/c mice, while LLC1-BMT5 and the parental LLC1 cells (5 × 104 cells/mouse) were implanted into C57/BL6 mice, respectively. BrMs were monitored by bioluminescence imaging (BLI) at 28 (4T1 and 4T1-BMT5 cells) or 35 days (LLC1 and LLC1-BMT5 cells) after cell implantation. Representative BLI images are shown (left). BLI photon fluxes in the brain for each mouse were quantified (right, mean ± SD of n = 8 mice, two-sided Mann-Whitney test). ∗∗∗p < 0.001.
(E) Quantification of the percentages of mice that developed bone metastases or BrMs after intracardial implantation of the tumor cells.
(F) RT-qPCR assays detected the expression of the 15 co-upregulated genes in 4T1-BMT5 and LLC1-BMT5 cells compared to the parental cells (mean ± SEM of n = 3 independent experiments, two-tailed Student’s t test). GAPDH was used as an internal control. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(G) Immunostaining of HSP47 in BrMs and the paired primary breast (top) or lung cancer tissues (bottom). Representative images of two patients for each cancer type are shown. Scale bars, 100 μm. The expression scores of HSP47 in BrMs were compared with those of the paired primary tumors (n = 12 for BRCA and n = 26 for LUAD, paired Student’s t test).
(H) Kaplan-Meier analysis showing the correlation between HSP47 mRNA levels and overall survival of patients with breast cancer (top) and lung cancer (bottom).
We next detected the expression of these 15 ECM genes in LLC1-BMT5 and 4T1-BMT5 cells, and the result showed that HSP47 (SERPINH1) was the only gene upregulated in both of the brain metastatic cell lines compared to the corresponding parental cells (Figure 1F). This result was validated by immunoblotting using brain metastatic cells generated from different mice (Figure S1B). Moreover, public datasets also showed a great upregulation of HSP47 in human BrMs from breast cancer and lung cancer (Figure S1C). To determine the expression of HSP47 in different cell types in BrMs and the primary tumors, we analyzed the single-cell RNA sequencing data. The results showed that HSP47 is low in tumor cells of the primary tumors, although some non-cancer cells show high levels of HSP47, especially in cancer-associated fibroblasts (CAFs) (Figures S1D and S1F), which is consistent with previous reports.15,16 However, the level of HSP47 is significantly upregulated in tumor cells in BrMs compared to the primary tumors (Figures S1E and S1G). Importantly, tumor cell is the most abundant cell type expressing HSP47 in BrMs. In breast invasive carcinoma (BRCA) BrMs, tumor cells account for 48.6% and CAFs account for 4.5%. Similarly, in lung adenocarcinoma (LUAD) BrMs, tumor cells and CAFs account for 50.3% and 2.2%, respectively (Figures S1E and S1G). Further, immunostaining of brain metastatic tissues and the paired primary tumor tissues verified that HSP47 levels were significantly higher in BrMs than in primary tumors (Figure 1G). Accordingly, high-level HSP47 predicted worse overall survival rates in patients with lung cancer or breast cancer (Figure 1H). Together, these results demonstrated that HSP47 is upregulated in brain metastatic tumors and that high-level HSP47 is associated with poor survival of patients with cancer.
HSP47 is required for BrM
To explore the role of HSP47 in breast cancer and lung cancer BrMs, parental 4T1 and LLC1 cells were engineered to stably express HSP47 (Figure 2A). We found that overexpression of HSP47 significantly promoted the formation of BrMs after intracardiac inoculation of the cancer cells into BALB/C and C57BL/6 mice, as detected by in vivo bioluminescence imaging (Figures 2B and 2C), and thus markedly shortened the BrM-free survival (BMFS) of the BrMs-bearing mice (Figure 2D). Hematoxylin and eosin (H&E) staining of the mouse brain tissues confirmed that HSP47 overexpression greatly increased the number of BrMs derived from both 4T1 and LLC1 cells (Figures 2E and 2F). In view of the high expression level of HSP47 in brain metastatic tumor cells, we next depleted HSP47 in 4T1-BMT5 and LLC1-BMT5 cells using short hairpin RNAs (shRNAs) (Figure 2G). We found that depletion of HSP47 significantly repressed BrMs of both 4T1-BMT5 and LLC1-BMT5 cells (Figure 2H) and thus notably prolonged the survival time of the BrM-bearing mice (Figure 2I). H&E staining of brain tissues confirm that HSP47 depletion decreased the number of BrMs (Figure 2J). Together, these observations strongly supported that HSP47 is required for breast cancer and lung cancer BrMs.
Figure 2.

HSP47 is required for BrM
(A) 4T1 and LLC1 cells expressing HSP47 were analyzed by immunoblotting (top) and RT-qPCR (bottom), respectively. The RT-qPCR data were normalized to GAPDH (mean ± SEM of n = 3 independent experiments, two-tailed Student’s t test). ∗∗∗p < 0.001.
(B) 4T1 and LLC1 cells (5 × 104 cells/mouse) expressing HSP47 were intracardially injected into mice, respectively. Representative BLI images of two mice for each group are shown. BLI photon fluxes in the brain for each mouse were quantified (mean ± SD, n = 8 mice for each group, two-sided Mann-Whitney test). ∗∗p < 0.01.
(C) The percentages of mice forming brain and bone metastases after intracardial cell implantation.
(D) The BMFS of mice was evaluated (n = 8 mice for each group, Kaplan-Meier model with two-sided log-rank test). ∗∗p < 0.01.
(E and F) H&E staining showing the BrMs in (B). Representative images are shown (E). Scale bars, 200 μm. Insets: high-magnification images corresponding to the areas marked by dotted red lines. The number of BrMs were quantified (F). Large metastases are >300 μm on the longest axis (mean ± SD, n = 8 mice, two-sided Mann-Whitney test). ∗∗∗p < 0.001.
(G) 4T1-BMT5 and LLC1-BMT5 cells expressing HSP47 shRNAs were analyzed by immunoblotting (top) and RT-qPCR (bottom). For RT-qPCR, GAPDH was used as an internal control. Data are expressed as mean ± SEM, n = 3 independent experiments, two-tailed Student’s t test. ∗∗∗p < 0.001.
(H) 4T1-BMT5 and LLC1-BMT5 cells expressing HSP47 shRNAs were intracardially injected into mice, respectively. Representative BLI images of mice are shown. BLI photon fluxes in the brain for each mouse were quantified (mean ± SD, n = 8 mice for each group, two-sided Mann-Whitney test). ∗∗∗p < 0.001.
(I) The BMFS of mice was evaluated (n = 8 mice for each group, Kaplan-Meier model with two-sided log rank test). ∗∗∗p < 0.001.
(J) H&E staining of the BrMs in (H). Representative images are shown. Scale bars, 200 μm. Insets: high-magnification images corresponding to the areas marked by dotted red lines. The number of BrMs were quantified (mean ± SD, n = 8 mice, two-sided Mann-Whitney test). ∗∗∗p < 0.001.
HSP47 promotes microglial polarization to M2 phenotype and immunosuppression in BrMs
Because microglia/macrophages are the most abundant immune cells in the brain7,17 and have been previously reported to regulate the development and progression of brain tumors,8 we subsequently investigated whether HSP47 controls BrM by regulating microglia/macrophages. In mouse brain metastatic tissues, we found that the population of microglial cells (CD11b+CD45low) was significantly higher than that of macrophages (CD11b+CD45high) (Figure S2A), which is consistent with the literature.18 Depletion of HSP47 significantly decreased the population of CD206+CD11b+CD45low microglial cells (M2) but increased the population of CD11c+CD11b+CD45low microglial cells (M1), as determined by fluorescence-activated cell sorting assays (Figures 3A and S2A). This result was confirmed by immunostaining (Figures 3B and S2B). Accordingly, HSP47 depletion dramatically increased the infiltration of CD8+ T cells in brain metastatic lesions (Figures 3C, S2C, and S2D). Moreover, depleting CD8+ T cells using an anti-CD8α monoclonal antibody significantly reversed the effect of HSP47 depletion on BrM (Figure 3D), indicating that HSP47 promotes BrM by repressing anti-tumor CD8+ T cell immune responses.
Figure 3.

HSP47 promotes M2 microglial polarization and immunosuppression in brain metastatic niche
(A) Representative fluorescence-activated cell sorting (FACS) plots and quantification of CD206+CD11b+CD45low and CD11c+CD11b+CD45low cells in the mouse BrMs (mean ± SEM, n = 5 independent experiments, Student’s t tests). ∗∗∗p < 0.001.
(B) Mouse BrMs derived from 4T1-BMT5 cells were double stained with anti-HSP47 and anti-CD206 antibodies. Representative images are shown. Scale bars, 100 μm. The density of CD206+ cells in each microscope filed was counted (mean ± SEM, n = 6 randomly selected fields, Student’s t tests). ∗∗∗p < 0.001.
(C) Representative FACS plots and quantification of CD8+CD3+CD45+ cells in the mouse BrMs derived from 4T1-BMT5 cells. Results are presented as mean ± SEM of n = 5 independent experiments, Student’s t tests. ∗∗∗p < 0.001.
(D) LLC1-BMT5 cells expressing HSP47 shRNAs were intracardially injected into mice. Mice were then injected intraperitoneally with a CD8α monoclonal antibody (mAb; 100 μg/mouse) or immunoglobulin G2b (IgG2b) (100 μg/mouse). Representative BLI images and quantification of BLI photon fluxes in the brain are shown (mean ± SD, n = 8 mice for each group, two-sided Mann-Whitney test). ∗∗∗p < 0.001.
(E) Mice intracardially injected with 4T1 or LLC1 cells expressing HSP47 were kept on a standard diet or fed with PLX3397 (290 mg/kg) for 3 weeks. Representative BLI images are shown. BLI photon fluxes in the mouse brain were quantified (mean ± SD of n = 8 mice for each group, two-sided Mann-Whitney test). ∗p < 0.05 and ∗∗p < 0.01.
(F) H&E staining of BrMs in mice. Scale bars, 200 μm. Large metastases are >300 mm on the longest axis (mean ± SD of n = 8 mice, Student’s t test). ∗p < 0.05 and ∗∗p < 0.01.
(G) The percentages of mice forming brain or bone metastases were quantified.
(H) Representative FACS plots and quantification of CD11b+CD45lowcells in mouse BrMs (mean ± SEM, n = 5 independent experiments, Student’s t tests). ∗∗∗p < 0.001.
(I) Representative FACS plots and quantification of CD8+CD3+CD45+ cells in mouse BrMs (mean ± SEM, n = 5 independent experiments, Student’s t tests). ∗∗∗p < 0.001.
(J–M) The populations of Ki67+, CD69+, granzyme B (GZMB)+, interferon (IFN)-γ+, and TNF-α+ CD8+ T cells and CD25+FOXP3+CD4+ Tregs in (I) were quantified (mean ± SEM, n = 5 independent experiments, Student’s t tests), respectively. ∗∗∗p < 0.001.
(N) Immunofluorescence (IF) co-staining of HSP47 and CD206 in brain metastatic lesions of patients with breast cancer. Representative images of two tumors are shown. Scale bars, 100 μm. The expression correlation between HSP47 and CD206 was analyzed (n = 10 randomly selected fields, Pearson correlation coefficient).
To determine whether HSP47 promotes BrM by regulating microglial polarization, we depleted microglia in mice and then detected their effect on BrM. PLX3397, a specific inhibitor of CSF1R,19 was shown to efficiently eliminate microglial cells in brain tissues (Figure S2E). We found that PLX3397 reversed the effect of HSP47 overexpression on BrM derived from both 4T1 and LLC1 cells (Figures 3E–3G). Nevertheless, PLX3397 had a limited effect on bone metastasis (Figures 3E and 3G), which further indicated that HSP47 promoted tumor BrM in a microglia-dependent manner. PLX3397 significantly inhibited the M1-to-M2 microglial polarization induced by HSP47 overexpression (Figure 3H). Accordingly, PLX3397 markedly increased the infiltration of CD8+ T cells that were repressed by HSP47 overexpression (Figure 3I). Further, HSP47 overexpression significantly decreased the populations of proliferating (Ki67+), activated (CD69+), and cytolytic (granzyme B+, interferon-γ+, tumor necrosis factor α [TNF-α]+) CD8+ T cells while increasing the population of CD4+CD25+FOXP3+ regulatory T cells (Tregs) in the brain metastatic lesions, and these effects were reversed by administration of PLX3397 (Figures 3J–3M and S2F–S2L). In human BrM tissues of breast cancer, high-level HSP47 was positively correlated with the infiltration of CD206+ microglial cells (Figure 3N). Together, these results demonstrate that HSP47 promotes BrM by promoting M2 microglial polarization and inhibiting the anti-tumor CD8+ T cell immunity.
COL1 mediates HSP47-induced M2 microglial polarization and immunosuppression in BrMs
Because HSP47 is a major chaperone that is essential for the correct folding of procollagen and secretion of mature collagens,20 we next explored whether the effect of HSP47 on microglia was mediated by collagens. Type I collagen (COL1) is the most abundant collagen in the tumor matrix and has recently been reported to modulate an immunosuppressive tumor-associated macrophages (TAM) in vitro.21 We found that 4T1-BMT5 and LLC1-BMT5 cells secreted a higher level of COL1 compared to the parental cells (Figure 4A) and that depletion of HSP47 decreased the levels of COL1 in the culture medium (Figure 4B). Consistently, immunostaining results demonstrated that COL1 was the predominant type of collagen induced by HSP47 overexpression in the metastatic lesions (Figure S3A). Moreover, the cell conditioned medium (CM) of 4T1 or LLC1 cells overexpressing HSP47 significantly promoted the Transwell migration of BV2 microglial cells (Figures S3B and S3C). Accordingly, the CM of 4T1-BMT5 or LLC1-BMT5 cells with HSP47 depletion sharply inhibited the migration of BV2 cells, which was reversed by adding COL1 (Figure 4C), suggesting that HSP47 promotes microglia attraction through COL1.
Figure 4.

COL1 mediates HSP47-induced microglia recruitment and polarization
(A) ELISA quantification of the COL1 levels in the CM of 4T1-BMT5 and LLC1-BMT5 cells compared to the corresponding parental cells.
(B) ELISA quantification of the COL1 levels in the CM of 4T1-BMT5 and LLC1-BMT5 cells expressing HSP47 shRNAs.
(A and B) Data are expressed as mean ± SD of n = 3 independent experiments, Student’s t tests. ∗∗p < 0.01 and ∗∗∗p < 0.001.
(C) BV2 cells were cultured in the CM of 4T1-BMT5 or LLC1-BMT5 cells expressing HSP47 shRNA and then treated with 4 mg/mL COL1 for 24 h. Cells that migrated onto the lower surface of the Transwell chamber were stained by crystal violet. Representative images are shown. Scale bars, 100 μm. Data are presented as mean ± SEM, n = 6 randomly selected microscope fields of two independent experiments, Student’s t tests. ∗∗∗p < 0.001.
(D) BV2 cells cultured in the CM of 4T1-BMT5 or LLC1-BMT5 cells expressing HSP47 shRNA were treated with COL1 for 48 h. The cell lysates were analyzed by immunoblotting using anti-CD206 and anti-Arg1 antibodies.
(E) BV2 cells were treated as in (D) and the expression of CD206 was analyzed by FACS. Representative FACS plots are shown. FACS data were statistically analyzed (mean ± SD, n = 3 independent experiments, Student’s t tests). ∗∗p < 0.01 and ∗∗∗p < 0.001.
(F) 4T1-BMT5 cells expressing HSP47 shRNA were treated with COL1 for 24 h and then intracardially injected into mice. Representative BLI of two mice for each group are shown. BLI photon fluxes in the brain for each mouse were quantified (mean ± SD, n = 8 mice for each group, two-sided Mann-Whitney test). ∗p < 0.05.
(G) The BMFS of mice was evaluated (n = 8 mice for each group, Kaplan-Meier model with two-sided log-rank test). ∗∗p < 0.01.
(H) 4T1-BMT5 cells expressing control shRNA or HSP47 shRNA were engineered to express GFP and then intracardially injected into mice. Consecutive mouse BrM tissues were immunostained with the antibodies against GFP, HSP47, COL1-4, CD206, or CD8α. Scale bars, 200 μm. Insets: high-magnification images corresponding to the areas marked by dotted red lines.
(I) CD8+ T cells were cultured overnight in the CM of tumor cells overexpressing HSP47 or 4 mg/mL COL1. Cells that migrated onto the lower surface of the Transwell chamber were counted by FACS.
(J) BV2 cells were treated with the CM of tumor cells expressing HSP47 or 4 mg/mL COL1 overnight. The medium was replaced with fresh medium and then used to treat CD8+ T cells. The T cells that migrated onto the lower surface of the Transwell chamber were counted by FACS.
(I and J) Representative FACS plots are shown. FACS data were statistically analyzed (mean ± SD, n = 3 independent experiments, Student’s t tests). ∗p < 0.05 and ∗∗∗p < 0.001.
(K and L) IF double staining of COL1 and CD206 in human BrMs from patients with breast or lung cancer. Scale bars, 100 μm. The staining density of COL1 and CD206 cells in each tissue was analyzed (n = 10 tissues for breast cancer BrMs and n = 16 tissues for lung cancer BrMs, Pearson correlation coefficient).
We further examined the effect of the HSP47-COL1 axis on the expression of polarization markers in microglia. COL1 treatment upregulated the level of CD206 in BV2 cells (Figure S3D). As we expected, the CMs derived from 4T1 and LLC1 cells overexpressing HSP47 significantly upregulated the levels of M2 polarization makers including CD204, CD206, and ARG1, but decreased the levels of M1 polarization makers such as CD11c, CD32, and CD86, in BV2 cells (Figures S3E–S3G). Moreover, the CMs of 4T1-BMT5 and LLC1-BMT5 cells with HSP47 depletion sharply inhibited the expression of M2 polarization markers in BV2 cells, and this effect was reversed by treatment with COL1 (Figures 4D and 4E).
To further validate that HSP47-induced M2 microglial polarization and BrM are dependent on COL1, we performed in vivo rescue experiments. 4T1-BMT5 cells expressing HSP47 shRNA were coated with COL1 and then intracardially injected into mice. We found that treatment with COL1 at least partially rescued the effect of HSP47 depletion on BrM (Figures 4F and S3H) and, accordingly, shortened the BMFS of mice (Figure 4G). Immunostaining of mouse brain metastatic tissues revealed that HSP47 depletion inhibited COL1 deposition and M2 microglial polarization and promoted CD8+ T cell infiltration in the metastatic lesions, and these effects were reversed by COL1 pre-treatment (Figures 4H and S3I). Notably, we found that the CM of 4T1 or LLC1 cells overexpressing HSP47 did not significantly inhibit the Transwell migration of CD8+ T cells and that the addition of COL1 only moderately inhibited CD8+ T cell infiltration (Figures 4I and S3J). However, the CM of microglial cells after treatment by the CM of cancer cells expressing HSP47 or by COL1 greatly inhibited the Transwell migration of CD8+ T cells (Figures 4J and S3K). These results suggest that HSP47-mediated collagen deposition represses CD8+ T cell infiltration mainly through regulating microglial polarization. Moreover, in human BrMs from patients with breast cancer or lung cancer, high-level COL1 deposition is positively correlated with the infiltration of CD206+ microglial cells (Figures 4K and 4L). Collectively, these results indicate that HSP47 promotes M2 microglial polarization and immunosuppression through COL1 in BrMs.
COL1 promotes M2 microglial polarization through the integrin α2β1/NF-κB pathway
To elucidate the mechanism underlying COL1 promoting M2 microglial polarization, we cultured BV2 cells in a 3D COL1 gel and then profiled gene expression by transcriptome sequencing. The Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis indicated that the changed genes were significantly enriched in the TNF pathway, nuclear factor κB (NF-κB) signaling pathway, and cytokine-cytokine receptor interaction (Figure 5A). Gene enrichment analysis (GSEA) confirmed that the enriched genes are related to M2 macrophage polarization and the NF-κB signaling pathway (Figure 5B). A large number of cytokine genes involved in M2 macrophage polarization showed significant upregulation after COL1 treatment (Figure 5C) and were functionally connected to the NF-κB interaction network, as judged by the GeneMANIA database (Figure S4A).
Figure 5.

HSP47-mediated COL1 biosynthesis promotes M2 microglial polarization through β-integrin/NF-κB signaling
(A) BV2 cells were treated with COL1 and then subjected to RNA sequencing analysis (n = 3 biological replicates for each group). The Kyoto Encyclopedia of Genes and Genomes analysis shows the top 11 enriched pathways with significant difference.
(B) Gene set enrichment analysis of the changed genes after COL1 treatment. NES (normalized enrichment score) and p value are shown.
(C) Heatmap showing the most significantly upregulated cytokine genes in COL1-treated BV2 cells vs. control BV2 cells.
(D) BV2 cells transfected with p65 small interfering RNA (siRNA) were treated with COL1, and the cell lysates were analyzed by immunoblotting. The levels of selected cytokine genes were analyzed by RT-qPCR.
(E) BV2 cells under COL1 treatment were treated with TC-I-15. Cell lysates were analyzed by immunoblotting, and the levels of selected cytokine genes were detected by RT-qPCR.
(D and E) GAPDH was used as an internal control. Data are expressed as mean ± SEM of n = 3 independent experiments. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(F) CUT&Tag analysis of p65 binding at CCL2, CXCL2, and CXCL3 promoters in BV2 cells after treatment by COL1. Spike-in was used as an internal control. Data are expressed as mean ± SEM, n = 3 independent experiments, two-tailed Student’s t test. ∗∗∗p < 0.001.
(G) BV2 cells under COL1 treatment were transfected with p65 siRNA or treated with TC-I-15 and the expression of CD206 was analyzed by FACS. Representative FACS plots are shown. The FACS data were statistically analyzed (mean ± SEM, n = 3 independent experiments). ∗∗∗p < 0.001.
We next sought to explore whether COL1 promotes M2 microglial polarization through the NF-κB pathway. To this end, we transfected p65 small interfering RNA into BV2 cells and then treated cells with COL1. We found that COL1 upregulated the level of phosphorylated p65 (S536) and significantly induced the expression of the cytokine genes (Figure 5D). Importantly, depletion of p65 reversed the effect of COL1 on cytokine gene expression (Figure 5D), indicating that COL1 induces their expression through NF-κB p65. It has been reported that integrin α2β1 is a major COL1 receptor in multiple cell types.22 To determine whether integrin α2β1 mediates COL1-induced M2 microglial polarization, BV2 cells cultured in COL1 3D gel were treated with TC-I-15, an inhibitor of integrin α2β1. As with p65 depletion, TC-I-15 remarkably attenuated COL1-induced p65 phosphorylation and the expression of these cytokine genes in BV2 cells (Figure 5E). It has been reported that NF-κB can bind to the promoters of cytokine genes, including CCL2, CXCL2, and CXCL3 (Figure S4B), and induce their expression in lung cancer cells.23 We verified that p65 was indeed bound to the promoter regions of these genes in BV2 cells, which were enhanced by COL1 treatment (Figure 5F). Moreover, depletion of p65 or TC-I-15 treatment attenuated M2 microglial polarization induced by COL1 (Figure 5G). Together, these results suggest that COL1 promotes microglial polarization to the M2 phenotype through the integrin α2β1/NF-κB signaling.
Targeting HSP47-mediated collagen biosynthesis inhibits BrM
We next evaluated the effect of targeted inhibition of HSP47 on BrM. Col003, a recently developed selective inhibitor of HSP47 that competitively binds to the collagen binding sites of HSP47,24 was selected for investigation (Figure 6A). We verified in vitro that Col003 treatment of 4T1-BMT5 and LLC1-BMT5 cells significantly decreased the level of COL1 in the cell culture medium in a dose-dependent manner, with half-maximal inhibitory concentrations of 3.96 and 4.68 μM, respectively (Figure 6B). Col003 has a molecular weight of 247, but its permeability to the blood-brain barrier (BBB) has not been evaluated. To this end, Col003 (20 mg/kg) was intravenously injected into mice, and the concentration of Col003 in the cerebrospinal fluid (CSF) was quantified at different time points. We found that Col003 in the CSF sharply increased at 30 min after injection, peaked at about 5 μg/mL at 2 h, and remained at 1 μg/mL at least for even 12 h after administration (Figure 6C). This result indicates that Col003 has a relatively good BBB penetration and in vivo tolerability.
Figure 6.

Targeting HSP47-mediated COL1 biosynthesis inhibits BrM
(A) Molecular formula of Col003.
(B) 4T1-BMT5 or LLC1-BMT5 cells were treated with the indicated concentration of Col003 for 24 h and COL1 level in the CM was quantified by ELISA (mean ± SD, n = 3 independent experiments). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
(C) CSF was extracted from mice at different time points after intravenous injection of Col003 (20 mg/kg) and the concentration of Col003 was determined by high-performance liquid chromatography (mean ± SD, n = 3 mice for each time point).
(D) 4T1-BMT5 or LLC1-BMT5 cells were intracardially injected into mice. Seven days later, Col003 was injected (5 or 20 mg/kg) into mice via vein every third day for 1 month. Representative BLI images are shown. BLI photon fluxes in the brain for each mouse were quantified (mean ± SD, n = 8 mice for each group, two-sided Mann-Whitney test). ∗∗∗p < 0.001.
(E) H&E staining of BrM tissues in mice. Representative images are shown (left). Scale bars, 200 μm. Large metastases are >300 mm on the longest axis (mean ± SD, n = 8 mice, two-sided Mann-Whitney test). ∗∗p < 0.01 and ∗∗∗p < 0.001.
(F) The BMFS of mice was evaluated (n = 8 mice for each group, Kaplan-Meier model with two-sided log-rank test). ∗∗∗p < 0.001.
(G) Representative FACS plots and quantification of CD8+CD3+CD45+ cells in the mouse BrM tissues.
(H) Representative FACS plots and quantification of CD206+CD11b+CD45low and CD11c+CD11b+CD45low cells in the BrM tissues.
(I–L) The populations of Ki-67+, CD69+, GZMB+, IFN-γ+, and TNF-α+ CD8+ T cells and CD25+FOXP3+CD4+ Tregs were analyzed by FACS.
(G–L) Data are expressed as mean ± SEM, n = 5 independent experiments, Student’s t tests. ∗∗∗p < 0.001.
(M) Consecutive mouse BrM tissues were analyzed by immunohistochemistry using the antibodies against GFP, COL1, and CD8α. Representative images are shown. Scale bars, 200 μm.
To determine the effect of Col003 on BrM, Col003 was intravenously injected into mice every third day after intracardiac injection of brain metastatic cells. Compared with vehicle control, Col003 potently inhibited BrMs of both 4T1-BMT5 and LLC1-BMT5 cells in a concentration-dependent manner (Figures 6D and 6E) and thus prolonged the BMFS of the mice (Figure 6F). H&E staining of brain tissues confirmed that Col003 decreased the number and size of brain metastatic lesions (Figure 6E). Flow cytometry analysis showed that the population of M2 microglia was significantly decreased, while the population of M1 microglia was significantly increased, in the Col003-treated group (Figure 6H), which was validated by immunostaining (Figures S5A and S5B). Moreover, Col003 significantly increased the infiltration of CD8+ T cells as determined by flow cytometry (Figure 6G). Further studies demonstrated that Col003 significantly increased the populations of proliferating, activated, and cytolytic CD8+ T cells while suppressing the population of CD4+ Tregs in the brain metastatic lesions (Figures 6I–6L and S5C–S5H). In consecutive mouse brain tissues, we confirmed that Col003 inhibited the COL1 deposition and promoted the infiltration of CD8+ T cells (Figure 6M). Together, these results indicate that targeting the HSP47-COL1 axis by Col003 inhibits BrM by suppressing M2 microglial polarization and restoring anti-tumor immunity.
Col003 augments the responsiveness of BrMs to anti-PD-L1 treatment
Because Col003 increases the populations of proliferating, activated, and cytolytic CD8+ T cells in metastatic lesions, we further explored the potential synergistic effect of Col003 plus immune checkpoint blockade in the treatment of BrMs. To this end, BrM-bearing mice derived from 4T1-BMT5 and LLC1-BMT5 cells were treated with anti-PD-L1 antibody alone or anti-PD-L1 antibody in combination with Col003 (Figure 7A, top). We found that anti-PD-L1 antibody alone only moderately inhibited BrM (Figures 7A and S6A) and had a modest effect on the survival of the mice compared to the vehicle control (Figure 7B). However, the anti-PD-L1 antibody in combination with Col003 substantially inhibited BrM (Figures 7A and S6A) and dramatically extended mouse survival (median survival duration of 21 days in anti-PD-L1 alone vs. 63 days in anti-PD-L1+Col003 for 4T1-BMT5 cells and 25 vs. 74 days for LLC1-BMT5 cells) (Figure 7B). In the mouse BrMs, anti-PD-L1 alone also slightly decreased the population of M2 microglial cells (Figure 7C), which is consistent with previous reports that blocking PD-L1 inhibits M2 macrophage polarization.25 Moreover, the anti-PD-L1 antibody in combination with Col003 significantly decreased the population of M2 microglial cells and increased the population of M1 microglial cells (Figure 7C). Accordingly, Col003 in combination with PD-L1 blockade substantially promoted the infiltration and activation of CD8+ T cells (Figures 7D, 7E, and S6B). Collectively, these results demonstrate that blocking the HSP47-COL1 axis by Col003 enhances the efficacy of PD-L1 blockade therapy in BrMs.
Figure 7.

Col003 augments the therapeutic efficacy of anti-PD-L1 antibody in BrMs
(A) 4T1-BMT5 or LLC1-BMT5 cells were intracardially implanted into mice. Seven days after cell injection, mice were intraperitoneally injected with anti-PD-L1 antibody alone (200 μg/mouse) every third day four consecutive times or with anti-PD-L1 plus Col003 (20 mg/kg) (top). Representative BLI images of each group are shown. BLI photon fluxes in the brain for each mouse were quantified (mean ± SD, n = 8 mice for each group, two-sided Mann-Whitney test). ∗∗∗p < 0.001.
(B) The BMFS of mice was evaluated (n = 8 mice for each group, Kaplan-Meier model with two-sided log-rank test). ∗∗∗p < 0.001.
(C) Representative FACS plots and quantification of CD206+CD11b+CD45low and CD11c+CD11b+CD45low cells in the BrM tissues.
(D) Representative FACS plots and quantification of CD45+CD3+CD8+cells in the BrM tissues.
(C and D) Data are expressed as mean ± SEM of n = 5 independent experiments, Student’s t tests. ∗∗∗p < 0.001.
(E) Co-staining of GZMB and CD8α in mouse BrM tissues. Representative images are shown (left). Scale bars, 100 μm. The GZMB+ and CD8α+ positive cells in each microscope field were counted (mean ± SEM, n = 6 randomly selected fields for each group, Student’s t tests). ∗∗∗p < 0.001.
(F) Schematic diagram shows the findings revealed in this study. Upregulation of HSP47 in tumor cells promotes collagen deposition in the brain metastatic niches, which promotes M2 microglial polarization and expression of anti-inflammatory cytokines through the integrin α2β1/NF-κB pathway, resulting in repression of anti-tumor CD8+ T cell immunity.
Discussion
Due to the unique microenvironment in the brain, the development of BrMs is a highly complex and selective process. In this study, we have identified HSP47 as a critical determinant of BrM from breast and lung cancers. We revealed that HSP47-mediated collagen deposition in metastatic lesions promotes M2 microglial polarization, leading to repressed anti-tumor CD8+ T cell responses. Moreover, blocking the HSP47-collagen axis by Col003 restores anti-tumor immunity in brain and augments the efficacy of anti-PD-L1 immunotherapy for the treatment of BrMs (Figure 7F).
Interaction of cancer cells with the TME is critical for metastatic colonization and outgrowth.26 The anti-tumor T cell responses are inhibited by the highly immunosuppressive brain TME even in the context of immune checkpoint blockade.7,26 Microglia are the major brain resident immune cells involved in innate immune processes and maintaining CNS homeostasis.8 Microglia are extremely heterogeneous and can be polarized into the tumor-suppressive M1 phenotype or the tumor-promoting M2 phenotype in response to various microenvironmental stimuli, including those from the invading tumor cells.18,27 In this study, we verified that microglia are dominated by the M2 phenotype in BrMs in mouse models and human patients. We found that HSP47 upregulation in the metastatic tumor cells promotes COL1 biosynthesis and deposition in the metastatic lesion, which drives M2 microglial polarization and inhibits the anti-tumor CD8+ T cell responses. Thus, blocking HSP47-mediated collagen deposition will reverse the M1-to-M2 phenotypic shift of microglial cells and turn the “cold” immunosuppressive TME to the “hot” immunologically beneficial TME in BrMs.
Collagen is a major ECM component in cancer and critically implicated in metastatic tumor initiation and progression.28 Collagen deposition and remodeling in the ECM directly promote tumor cell proliferation, death resistance, replicative immortality, and invasion.29 A recent report demonstrated that the density of collagen promotes an immunosuppressive TAM.21 Moreover, elevated collagen in lung cancer correlates with increased subpopulations of exhausted CD8+ T cells through receptor LAIR1 on T cells.30 We identified in this study that COL1 is the predominant type of collagen induced by HSP47 overexpression. Moreover, although collagen deposition can act as a direct barrier for T cell infiltration,31,32,33 we proved that COL1-mediated M2 polarization and cytokine expression of microglial cells play a major role in repressing CD8+ T cell infiltration and activation. Thus, HSP47-induced collagen deposition and remodeling represent a critical mechanism that enables the metastatic cells to create an immunosuppressive microenvironment in the brain.
As an endoplasmic reticulum-resident specific molecular chaperone of procollagens, HSP47 is indispensable for the molecular maturation of collagens and is a promising target for the treatment of collagen-related disorders such as fibrosis of liver, lung, and other organs.20,34 HSP47 has been reported to be upregulated in various kinds of cancers and is required for tumor growth, epithelial-to-mesenchymal transition, and invasion.35,36 Further, HSP47-mediated collagen deposition also promotes cancer cell extravasation and metastatic colonization by recruiting platelets.37 Until now, the role of HSP47 in tumor immunity has been uncertain. Although we cannot exclude that HSP47 may regulate BrM through other steps of the metastatic process, we conclude from this study that HSP47-collagen-axis-mediated M2 microglial polarization and immunosuppression play a key role in BrM formation for the following reasons. Firstly, HSP47 depletion or blockade significantly decreases the population of M2 microglial cells and increases the infiltration and anti-tumor activity of CD8+ T cells, thereby inhibiting BrMs, while neutralization of CD8+ T cells can reverse this effect. Second, HSP47 expression is positively correlated with the infiltration of M2 microglia in BrMs. Finally, depleting microglia by PLX3397 reverses the effects of HSP47 overexpression on CD8+ T cell immune responses and brain metastatic niche formation. Thus, our study highlights the underlying mechanism for HSP47 in tumor progression and metastasis.
Col003 is a recently identified HSP47 inhibitor that competitively blocks the binding of HSP47 to collagens and efficiently decreases collagen secretion in cultured cells, representing a potential remedy for fibrosis.24,38 It has been reported that Col003 significantly inhibits thrombus formation in FeCl3-induced rat carotid arterial thrombus and thus can be used as an anti-platelet strategy to treat ischemic stroke.39 A more recent study also demonstrated that HSP47 ablation by Col003 contributes to thromboprotection in bears, patients with spinal cord injury, and mice.40 We demonstrated in this study that Col003 is able to cross the BBB and has good in vivo tolerability. Col003 administration in mice inhibits M2 microglial polarization and promotes the proliferation, activation, and cytotoxicity of CD8+ T cells. Thus, Col003 in combination with PD-L1 blockade can restore an anti-tumor CD8+ T cell immunity and eliminate BrM, thus representing a promising therapeutic strategy for brain metastatic tumors.
In conclusion, our study reveals that HSP47-mediated collagen deposition is required for BrM by modulating an immunosuppressive brain microenvironment. Targeting the HSP47-collagen axis by Col003 restores the anti-tumor immune response and enhances the efficacy of PD-L1 blockade, strengthening its therapeutic application for brain metastatic tumors.
Limitations of the study
In this study, we demonstrate that HSP47 upregulation in lung cancer and breast cancer, two of the most common primary tumors associated with BrMs, is a critical driver of BrM. However, it remains to be investigated whether HSP47 upregulation is a universal mechanism of BrM for other primary tumors with high brain metastatic potential, such as melanoma, colorectal cancer, etc. Moreover, we were limited by the number of paired patients with BrM, especially paired breast cancer BrMs. A larger sample size may have contributed to this study, including the influence of factors such as primary tumor subtype, gender, and age.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HSP47 | proteintech | Cat# 10875-1-AP; RRID: AB_2301500 |
| Anti-HSP47 | Santa Cruz | Cat# sc-5293; RRID: AB_627757 |
| Anti-N-cadherin | proteintech | Cat# 22018-1-AP; RRID: AB_2813891 |
| Anti-E-cadherin | proteintech | Cat# 20874-1-AP; RRID: AB_10697811 |
| Anti-Snail | proteintech | Cat# 13099-1-AP; RRID: AB_2191756 |
| Anti-Collagen I | proteintech | Cat# 14695-1-AP; RRID: AB_2082037 |
| Anti-Collagen I | proteintech | Cat# 67288-1-Ig; RRID: AB_2882554 |
| Anti-Collagen II | proteintech | Cat# 28459-1-AP; RRID: AB_2881147 |
| Anti-Collagen III | proteintech | Cat# 22734-1-AP; RRID: AB_2879158 |
| Anti-Collagen IV | proteintech | Cat# 19674-1-AP; RRID: AB_2878595 |
| Anti-GFP-Tag | Abclonal | Cat# AE012; RRID: AB_2770402 |
| Anti-Arg1 | proteintech | Cat# 16001-1-AP; RRID: AB_2289842 |
| Anti-CD163 | proteintech | Cat# 16646-1-AP; RRID: AB_2756528 |
| Anti-CD206 | proteintech | Cat# 18704-1-AP; RRID: AB_10597232 |
| Anti- CD11c | proteintech | Cat# 17342-1-AP; RRID: AB_2129787 |
| Anti-IBA1 | proteintech | Cat# 10904-1-AP; RRID: AB_2224377 |
| Anti-CD8 | Abcam | Cat# ab217344; RRID: AB_2890649 |
| Anti-CD8 | Santa Cruz | Cat# sc-7970; RRID: AB_627208 |
| Anti-P65 | CST | Cat# 8242; RRID: AB_10859369 |
| Anti-Phospho-P65 | CST | Cat# 3033; RRID: AB_331284 |
| Anti-Tubulin | Ray antibody | Cat# RM2007; RRID: AB_2934267 |
| CD11b-BV605 | Biolegend | Cat# 101257; RRID: AB_2565431 |
| CD11c-APC | Biolegend | Cat# 117310; RRID: AB_313779 |
| CD206-PE | Biolegend | Cat# 141706; RRID: AB_10895754 |
| CD45-PE/cy7 | Biolegend | Cat# 103114; RRID: AB_312979 |
| CD45-APC/cy7 | BD Pharmingen | Cat# 557659; RRID: AB_396774 |
| CD3-PerCP/cy5.5 | Biolegend | Cat# 100218; RRID: AB_1595492 |
| CD3-FITC | Biolegend | Cat# 100204; RRID: AB_312661 |
| CD4-PE | Biolegend | Cat# 100408; RRID: AB_312693 |
| CD8-APC/Fire™ 750 | Biolegend | Cat# 100766; RRID: AB_2572113 |
| CD8− PerCP/cy5.5 | BD Pharmingen | Cat# 551162; RRID: AB_394081 |
| GZMB-BV421 | Biolegend | Cat# 396414; RRID: AB_2810603 |
| TNF-α-BV650 | Biolegend | Cat# 506333; RRID: AB_2562450 |
| IFN-γ-BV510 | Biolegend | Cat# 505842; RRID: AB_2734494 |
| Ki67-BV786 | BD Pharmingen | Cat# 563756; RRID: AB_2732007 |
| CD69-AF700 | Biolegend | Cat# 104539; RRID: AB_2566304 |
| CD25-BV605 | BD Pharmingen | Cat# 563061; RRID: AB_2737982 |
| FOXP3-AF647 | BD Pharmingen | Cat# 560401; RRID: AB_1645201 |
| Bacterial and virus strains | ||
| EF1A-MCS-SV40-firefly_Luciferase-IRES-Puromycin | Genechem | https://www.genechem.com.cn/ |
| pCLenti-EF1-Neo-CMV-HSP47-WPRE | HanBio | https://hanbio.net/ |
| Tet-pLKO-neo | YouBio | http://www.youbio.cn/vector |
| CMV-MCS-3flag-EF1-GFP-T2A-Blasticidin | Genechem | https://www.genechem.com.cn/ |
| Biological samples | ||
| Human primary breast cancer tissues and paired brain metastatic tissues | Guangdong Provincial People’s Hospital of Southern Medical University (Guangzhou, China) | N/A |
| Human primary lung cancer tissues and paired brain metastatic tissues | Guangdong Provincial People’s Hospital of Southern Medical University (Guangzhou, China) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Puromycin | Thermo Fisher Scientific | Cat# A1113803 |
| Neomycin | Solarbio | Cat# IN0130 |
| Blasticidin | Selleck | Cat# S7419 |
| Col003 | MedChemExpress | Cat# HY-124817 |
| TC-I 15 | MedChemExpress | Cat# HY-107588 |
| InVivoMAb anti-mouse CD8a | Selleck | Cat# A2102 |
| IgG2b isotype control-InVivo | Selleck | Cat# A2116 |
| InVivoMAb anti-mouse PD-L1 (B7-H1) | Bio X Cell | Cat# BE0361; RRID: AB_2927503 |
| PLX3397 | MolDiets | Cat# M18072603 |
| Cultrex rCollagen I | R&D Systems | Cat# 3440-100-01 |
| Collagenase 1 | Worthington | Cat# LS004197 |
| ACK Lysis Buffer | LEAGENE | Cat# CS0001 |
| Dnase 1 | Solarbio | Cat# D8071 |
| DAPI | Abcam | Cat# ab104139 |
| Citrate Sodium Buffer, pH 6.0 | Solarbio | Cat# C1010 |
| Tris-EDTA buffer, pH 9.0 | Elabscience | Cat# E-IR-R104 |
| Fetal Bovine Serum | Gibco | Cat# 10099141 |
| Tween 20 | Beyotime | Cat# ST1727 |
| Triton X-100 | Beyotime | Cat# ST797 |
| RIPA buffer | Beyotime | Cat# P0013B |
| Trizol reagent | Invitrogen | Cat# 15596026 |
| D-Luciferin potassium salt | Beyotime | Cat# ST196 |
| Lipofectamine 2000 reagent | Invitrogen | Cat# 11668019 |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| PrimeScript™ RT reagent Kit | TAKARA | Cat# RR037A |
| CUT&Tag Kit | YEASEN | Cat# 12598ES12 |
| True-Nuclear Transcription Factor Buffer Set | Biolegend | Cat# 424401 |
| Mouse Collagen Type I ELISA Kit | CUSABIO | Cat# CSB-E08083m |
| Mouse PBMC Isolation Kit | IPHASE | Cat# 071E100.11 |
| Mouse CD8+ T cell Isolation Kit | IPHASE | Cat# 071E203.11 |
| Deposited data | ||
| RNA-seq of BV2 cells | This paper | GEO: GSE261941 |
| Experimental models: Cell lines | ||
| 4T1 | Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) | N/A |
| LLC1 | Type Culture Collection of Chinese Academy of Sciences (Shanghai, China) | N/A |
| BV2 | BeNa Culture Collection (Beijing, China) | N/A |
| 293FT | BeNa Culture Collection (Beijing, China) | N/A |
| Experimental models: Organisms/strains | ||
| BALB/C mice | Guangdong Medical Laboratory Animal Center (Foshan, China) | N/A |
| C57BL/6 mice | Guangdong Medical Laboratory Animal Center (Foshan, China) | N/A |
| Oligonucleotides | ||
| Primer sequences are provided in the Table S1 | This paper | N/A |
| Software and algorithms | ||
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
| Prism, v8 | GraphPad | https://www.graphpad.com/scientificsoftware/prism/ |
| ImageJ | NIH Image | https://imagej.nih.gov/ij/index.html |
| R (version 4.3.2) | The R Project for Statistical Computing | https://www.r-project.org/ |
| R Studio | PBC | https://www.rstudio.com/ |
| ComplexHeatmap (R package, version 2.16.0) | N/A | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| circlize (R package, version 0.4.15) | N/A | https://jokergoo.github.io/circlize_book/book/ |
| tidyverse (R package, version 2.0.0) | N/A | https://www.tidyverse.org/learn/ |
| ggplot2 (R package, version 3.4.4) | N/A | https://ggplot2.tidyverse.org |
| UMAP | N/A | https://arxiv.org/abs/1802.03426 |
| ggvenn (R package, version 0.1.10) | N/A | https://CRAN.R-project.org/package=ggvenn |
| AnnotationDbi (R package, version 1.62.2) | Bioconductor | https://bioconductor.org/packages/AnnotationDbi |
| clusterProfiler (R package, version 4.8.3) | Wu et al.39 | https://yulab-smu.top/biomedical-knowledge-mining-book/ |
| enrichplot (R package, version 1.20.3) | Bioconductor | https://bioconductor.org/packages/release/bioc/html/enrichplot.html |
| FastQC v0.11.9 | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trim galore v0.6.7 | N/A | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Hisat2 v2.2.1 | Kim et al.41 | http://daehwankimlab.github.io/hisat2/ |
| DEseq2 (R package, version 1.40.2) | Love et al.42 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| infercnvpy v0.4.3 | N/A | https://infercnvpy.readthedocs.io/en/latest/ |
| Seurat (R package, version 5.0.1) | N/A | http://satijalab.org/seurat |
| scanpy v1.9.3 | N/A | https://scanpy.readthedocs.io/en/stable/ |
| Other | ||
| PVDF membrane | Millipore | Cat# IPVH00010 |
| 24-well plate | Corning | Cat# 3524 |
| 0.45 μm filter | Sorfa | Cat# 622120 |
| 70 μm cell filter | ABCbio | Cat# ABC60070 |
| Transwell insert | Corning | Cat# 3422 |
Resource availability
Lead contact
Further information and request of resources can be directed to and will be fulfilled by the lead contact, Aidong Zhou (aidern0927@smu.edu.cn).
Materials availability
All unique models generated in this study are available upon request to the lead contact with a completed materials transfer agreement.
Data and code availability
-
•
RNA-seq data generated in this study have been deposited at GEO database and are publicly available as of the date of publication. Accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Cell culture and treatment
Mouse breast cancer cell line 4T1 and lung cancer cell line LLC1 were obtained from the cell bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Mouse microglia cell line BV2 and human embryonic kidney cell line 293FT were purchased from BeNa Culture Collection (Beijing, China). All cells were incubated with 95% humidified air and 5% CO2 incubator at 37°C. 4T1 cells were grown in RPMI-1640 (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco). LLC1, BV2 and 293FT cells were grown in DMEM (Gibco) supplemented with 10% FBS. Cell lines were authenticated by short tandem repeat profiling and were routinely tested for mycoplasma contamination. No cell lines used in this study were found in the database of commonly misidentified cell lines that is maintained by NCBI Biosample.
Animal studies
All animal experiments were processed in accordance with the Southern Medical University Animal Protection Guidelines, and the protocol was approved by the Southern Medical University Health Facility Animal Care and Use Committee. Female BALB/C mice and C57BL/6 mice (6–8 weeks old) were maintained in the specific-pathogen-free facility of the Laboratory Animal Center of Southern Medical University (Guangzhou, China). The mice in each experiment were randomly grouped. Mice were anesthetized with sodium pentobarbital (1.0%, 50 mg/kg), and 4T1 or LLC1 cells (5×104 cells/mouse) were intracardially injected into BALB/c mice or C57BL/6 mice, respectively. The progression of metastasis in mice was monitored by in vivo BLI.
To deplete CD8+ T cells, 7 days after cell injection, CD8α mAb (Selleck, 100 μg/mouse) or IgG2b (Selleck, 100 μg/mouse) was administered via intraperitoneal injection every other day for four consecutive times. To deplete microglia, mice were fed chow containing PLX3397 (290 mg/kg, Moldiets) for three weeks prior to the injection of cancer cells. To assess the effect of PD-L1 blockage on the incidence of BrMs, an anti-PD-L1 antibody (200 μg/mouse, BioXCell) was administered via intraperitoneal injection every third day for four consecutive times. To determine the effect of blocking the HSP47-collagens axis on brain metastasis, Col003 (20 mg/kg, MedChemExpress) was administered by vein injection every third day for one month. The combination group was treated with both anti-PD-L1 antibody and Col003, and the control group was injected with IgG control at the experimental endpoint, mice were humanely killed and their brains were fixed in 4% formaldehyde and used for subsequent histopathological evaluation.
Human samples
Human primary breast or lung cancer tissues and paired brain metastatic tissues were obtained from Guangdong Provincial People’s Hospital of Southern Medical University (Guangzhou, China). All tissue samples were collected in accordance with the hospital’s informed consent policy and with approval from the hospital’s institutional research ethics committee. The patient characteristics are listed in Table S1.
Method details
Derivation of brain metastatic tumor cell lines
For bioluminescence tracking, tumor cells were stably transfected with luciferase with a puromycin resistance gene (Genechem Co, Ltd. Shanghai). 4T1-or LLC1-luc cells (5×104 cells/mouse) in a volume of 100 μL was injected into the left cardiac ventricle of anesthetized BALB/C and C57BL/6 mice (6–8 weeks old), respectively. Tumor development was monitored by BLI. Brain metastatic lesions were minced with sterile surgical scissors and digested in RPMI1640 culture medium containing antibiotic–antimycotic agents medium supplemented with 1 mg/mL collagenase 1 (Worthington), and then placed on a rotator at 37°C for 30 min. The tissues were minced and placed in centrifuge tubes, cells were briefly centrifuged, resuspended in 0.025% trypsin–EDTA (Gibco), and incubated for an additional 10 min at 37 °C. Undigested tissue clumps were removed through a 70 μm cell filter (ABCbio) to collect the cells. Cells were placed in 25 cm2 culture flasks and incubated at 37°C in an incubator. After 24 h, brain metastatic cells were screened by puromycin (4 mg/mL) for one week. Brain metastatic cell lines (4T1-BMT1 and LLC1-BMT1) were reintroduced into mice, and 4T1-BMT5 and LLC1-BMT5 cells were obtained after four screens.
Stable cell line establishment
Lentiviral expression plasmid, packaging plasmid psPAX2 and envelope plasmid pMD2.G (4:3:1) were co-transfected into 293FT cells using Lipofectamine 2000 reagent (Invitrogen). The supernatants containing lentiviruses were collected and filtered (0.45 μm) at 48 h post-transfection. Tumor cells were infected by lentiviruses with 4 μg/mL polybrene for 24 h, and sable cells were selected by neomycin (200 μg/mL) for one week.
CD8+ T cell isolation
Mouse peripheral blood mononuclear cells (PBMC) were obtained using the PBMC Isolation Kit (IPHASE). Briefly, PBMC were isolated from mouse blood using Ficoll gradient centrifugation. Erythrocytes were lysed with ACK lysis buffer for 3 min at room temperature, centrifuged and the cells were resuspended by adding PBMC washing buffer. Mouse CD8+ T cells were then obtained from PBMC using the CD8+ T cell Isolation Kit (IPHASE). IPHASE Mouse CD8 Biotin-Antibody Cocktail was mixed with PBMC and incubated on ice for 15 min, IPHASE CD8 SA Nanobeads were added and mixed, and the incubation was continued on ice for 15 min. The centrifuge tubes were then placed in a magnetic rack and magnetically separated for 5 min to obtain mouse CD8+ T cells. After isolation, the purity of the cells was assessed by flow cytometry for PerCP/Cyanine5.5-conjugated CD3 (Biolegend), APC/Fire 750-conjugated CD8α (Biolegend). CD8+ T cells with >80% purity were used for subsequent experiments.
Transwell migration assay
To investigate the effect of tumor-derived CM on microglial polarization, we harvested CM from tumor cells, centrifuged at 2000 rpm for 15 min and filtered (0.45 μm), and then treated microglia with the CM. BV2 cells or CD8+ T cells (2×104) in 200 μL FBS-free media were loaded into the top chamber of transwell inserts (Corning). Cells were stimulated with tumor-derived CM or M2 microglia-derived CM for 24 h, and then incubated at 37°C overnight. BV2 cells migrated onto the lower chamber were stained with crystal violet blue and then counted under a bright-field microscope, while CD8+ T cells were counted by flow cytometry.
3D culture cells in COL1 gels
COL1 gels were prepared according to the manufacturer’s protocol (R&D Systems). Briefly, cells were suspended in cell culture medium and then placed on ice. COL1 was mixed with 10×DMEM with Phenol Red (Sigma Aldrich), 7.5% Sodium Bicarbonate and distilled water in a sterile tube. The cell suspension was mixed with COL1 gel, ensuring that the final concentration of COL1 gel is 4 mg/mL, and then immediately plated onto 24-well plates (Corning). Plates were placed in a 37°C incubator for 1 h to promote gel formation, and then fresh cell culture medium (2mL/well) was added.
Real-time/quantitative PCR
Total RNA was extracted using Trizol reagent (Invitrogen) and was reverse transcribed into complementary DNA (cDNA) using PrimeScript RT reagent Kit (TAKARA). qRT–PCR was performed using TB Green Premix Ex Taq (TAKARA) in a Roche LightCycler 480 System. GAPDH was used as an internal control. The expression levels of target RNAs relative to control was computed by 2-ΔΔCT. The primers used to amplify target genes are listed in Table S2.
RNA-seq analysis
After treatment with 4 mg/mL COL1 for 24 h, BV2 cells were collected and total RNA was extracted using the RNeasy Mini Kit (Qiagen). RNA samples were then sequenced on an Illumina Novaseq platform. FastQC (v 0.11.9) was utilized to assess the sequencing quality of the fastq files and ensure that all samples exhibited high quality. Illumina adapter sequences were trimmed using Trim galore (v 0.6.7). 150 bp paired-end reads were aligned to mouse reference genome (GRCm39) using hisat2 (v2.2.1) method.41 Comparative analysis of differentially expressed genes based on reads was performed using DEseq2 (v1.40.2).42 GSEA and KEGG pathway enrichment analysis were performed using the "clusterProfiler" R package (v 4.8.3) and the data were visualized by the ggplot2 package (v3.4.4). The differentially expressed genes are listed in Table S3.
Cleavage under target & tagmentation (CUT&Tag)
CUT & Tag is a method for studying protein-DNA interactions.43 BV2 cells were harvested after treatment with COL1 and treated according to the manufacturer’s protocol (YEASEN). Briefly, cell lysates were incubated with ConA beads and then sequentially incubated with a primary antibody against p65 (0.5 μg, CST) for 2 h, a secondary antibody for 30 min, and pA/G-Tn5 transposase for 1 h. After incubation with proteinase K at 55°C for 30 min, DNAs were extracted and then detected by quantitative PCR. DNA spike-in was used as an internal control. The sequences of the oligonucleotides used in the study are listed in Table S2.
Immunoblotting
Cells were lysed in RIPA buffer (Beyotime) containing 1×protease inhibitors (Roche) and phosphatase inhibitors (Roche). 20 μg protein lysate was loaded onto 8% or 10% SDS-PAGE gels for separation and was transferred to PVDF membranes (Millipore). The membranes were incubated with appropriate antibodies and then analyzed with an ECL system (BLT GelView 6000). Details of all antibodies are listed in key resources table.
Enzyme-linked immunosorbent assay (ELISA)
After 24 h of routine culture, tumor cells were starved overnight in serum-free medium, and then treated with different concentrations of Col003. After 48 h, the cell culture supernatant was harvested, centrifuged at 10000 × g for 5 min at 4°C, and filtered (0.45 μm) to clear all dead cells and debris. The levels of COL1 in cell culture medium of tumor cells were quantified by ELISA kits (CUSABIO) according to the manufacturer’s protocol.
Immunohistochemistry and immunofluorescence
Immunostaining was performed as we previously described.44,45 For immunohistochemistry staining, BrM tissues were deparaffined and hydrated through an alcohol series. Antigen retrieval was performed in an autoclave for 20 min using citrate buffer (pH 6.0, Solarbio) or EDTA buffer (pH 9.0, Elabscience). After blocking with goat serum for 1 h at room temperature, tissue slides were incubated with primary antibodies at 4°C overnight, followed by treatment with secondary antibodies. The slides were detected with DAB and counterstained with Hematoxylin. Images were taken using an ECLIPSENi-U microscope (NIKON). For immunofluorescence, the slides were incubated overnight at 4°C with primary antibodies. After washing with PBST, the slides were incubated with a fluorescent-conjugated second antibody (Alexa Fluor 594, Life Technologies, 1:500, Alexa Fluor 488, Life Technologies, 1:500) for 1 h at room temperature. Coverslips were mounted on slides using fade-resistant mounting medium containing DAPI (Abcam). Images were taken using a deconvolution microscope (Zeiss).
Flow cytometry
Fresh mouse brain metastatic tissues were cut up and digested with 0.5 mg/mL type 1 collagenase and 0.1 mg/mL DNase I (Solarbio) at 37°C for 30 min and filtered (70 μm) to obtain single cell suspensions. Erythrocytes were lysed with ACK Lysis Buffer (LEAGENE). Surface staining was then performed with directly conjugated antibodies against CD45, CD11b, CD11c, CD206, CD3, CD4, CD8, CD25, CD69 for 30 min on ice. For intracellular cytokine and transcription factor staining, after surface staining, cells were fixed and permeabilized with True-Nuclear Transcription Factor Buffer Set (Biolegend) and incubated with fluorescently conjugated antibodies against GZMB, TNF-α, IFN-γ, Ki67, FOXP3 for 50 min on ice. After washing with Perm Wash buffer and PBS, cells were resuspended in 0.2 mL PBS. The data were obtained by BD Accuri C6 (BD Pharmingen) and analyzed with FlowJo software (version 10.8). Details of all markers are listed in key resources table.
CSF collection
BALB/C mice were injected with Col003 (20 mg/kg) into the tail vein, and the CSF was harvested at various time points after injection. The mice were anesthetized with isoflurane, and then the heads of the mice were fixed with a brain stereotaxic device. A 1.5 cm incision was made between the ears with sterile surgical scissors, and the foramen magnum of the occipital bone was exposed to the field of view by bluntly peeling away the muscle interposition with surgical forceps. The muscles on either side of the incision are clamped and propped open with hemostatic forceps to allow complete exposure of the surgical field. A sharp glass needle was slowly inserted into the median line below the occipital bone, and CSF (5 μL/mouse) was extracted. CSF was placed in 100 μL PCR tubes and quickly transferred to storage at −80°C. The CSF was lyophilized to powder by a freeze dryer (Christ) and dissolved in acetonitrile. The concentrations of Col003 in CSF were analyzed by high-performance liquid chromatograph (Agilent).
Quantification and statistical analysis
All analysis was performed using GraphPad Prism 8 software. Data are presented as means ± standard deviation (SD) or standard errors of the mean (SEM). For all quantitative data, the statistical tests used are shown in the Figure Legends. Kaplan-Meier plots were used to analyze survival curve, and log rank tests were used to compare two survival curves. Statistical significance was determined by one way ANOVA and Student’s t test. Statistical significance was defined as a p-value of less than 0.05.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31871406 and 81901250), the Natural Science Foundation of Guangdong Province (2021A1515011067, 2023A1515010419, 2022A1515012540, and 2019A1515010104), the Guangzhou Science and Technology Plan Project (202201011639), the High-level Hospital Construction Project of Guangdong Provincial People's Hospital (DFJH201924) and Funds for Construction of High-Level Universities in Guangdong Province. The graphical abstract was created with BioRender.com, with permission.
Author contributions
L.W. and C.L. designed and performed most of the experiments. H.Z. assisted in the bioinformatic analysis. S.L., C.X., Y. Zou, Y.X., and Z.Z. assisted in some in vitro experiments. K.Z., S.Y., Y. Zeng, X.C., and G.L. assisted in the mouse experiments. Z.H., Dexiang Zhou, Dong Zhou, and Y.Y. provided tissue samples, cell lines, reagents, and conceptual advice. A.Z. and L.W. wrote and revised the manuscript. A.Z. conceived and supervised the study. All authors discussed the results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 13, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101533.
Contributor Information
Dong Zhou, Email: zhoudong5413@163.com.
Yong Yang, Email: yangyong@gdph.org.cn.
Aidong Zhou, Email: aidern0927@smu.edu.cn.
Supplemental information
References
- 1.Achrol A.S., Rennert R.C., Anders C., Soffietti R., Ahluwalia M.S., Nayak L., Peters S., Arvold N.D., Harsh G.R., Steeg P.S., Chang S.D. Brain metastases. Nat. Rev. Dis. Primers. 2019;5:5. doi: 10.1038/s41572-018-0055-y. [DOI] [PubMed] [Google Scholar]
- 2.Suh J.H., Kotecha R., Chao S.T., Ahluwalia M.S., Sahgal A., Chang E.L. Current approaches to the management of brain metastases. Nat. Rev. Clin. Oncol. 2020;17:279–299. doi: 10.1038/s41571-019-0320-3. [DOI] [PubMed] [Google Scholar]
- 3.Soffietti R., Ahluwalia M., Lin N., Rudà R. Management of brain metastases according to molecular subtypes. Nat. Rev. Neurol. 2020;16:557–574. doi: 10.1038/s41582-020-0391-x. [DOI] [PubMed] [Google Scholar]
- 4.Schulz M., Salamero-Boix A., Niesel K., Alekseeva T., Sevenich L. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front. Immunol. 2019;10:1713. doi: 10.3389/fimmu.2019.01713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambert A.W., Pattabiraman D.R., Weinberg R.A. Emerging Biological Principles of Metastasis. Cell. 2017;168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fares J., Ulasov I., Timashev P., Lesniak M.S. Emerging principles of brain immunology and immune checkpoint blockade in brain metastases. Brain. 2021;144:1046–1066. doi: 10.1093/brain/awab012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strickland M.R., Alvarez-Breckenridge C., Gainor J.F., Brastianos P.K. Tumor Immune Microenvironment of Brain Metastases: Toward Unlocking Antitumor Immunity. Cancer Discov. 2022;12:1199–1216. doi: 10.1158/2159-8290.CD-21-0976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soto M.S., Sibson N.R. The Multifarious Role of Microglia in Brain Metastasis. Front. Cell. Neurosci. 2018;12:414. doi: 10.3389/fncel.2018.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q., Abdo R., Iosef C., Kaneko T., Cecchini M., Han V.K., Li S.S.C. The spatial transcriptomic landscape of non-small cell lung cancer brain metastasis. Nat. Commun. 2022;13:5983. doi: 10.1038/s41467-022-33365-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin Y., Kang Y., Wang M., Wu B., Su B., Yin H., Tang Y., Li Q., Wei W., Mei Q., et al. Targeting polarized phenotype of microglia via IL6/JAK2/STAT3 signaling to reduce NSCLC brain metastasis. Signal Transduct. Target. Ther. 2022;7:52. doi: 10.1038/s41392-022-00872-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caffarel M.M., Braza M.S. Microglia and metastases to the central nervous system: victim, ravager, or something else? J. Exp. Clin. Cancer Res. 2022;41:327. doi: 10.1186/s13046-022-02535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kai F., Drain A.P., Weaver V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell. 2019;49:332–346. doi: 10.1016/j.devcel.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winkler J., Abisoye-Ogunniyan A., Metcalf K.J., Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020;11:5120. doi: 10.1038/s41467-020-18794-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J., Zhang L., Wan D., Zhou L., Zheng S., Lin S., Qiao Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021;6:153. doi: 10.1038/s41392-021-00544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyamura T., Sakamoto N., Ishida K., Kakugawa T., Taniguchi H., Akiyama Y., Okuno D., Hara A., Kido T., Ishimoto H., et al. Presence of heat shock protein 47-positive fibroblasts in cancer stroma is associated with increased risk of postoperative recurrence in patients with lung cancer. Respir. Res. 2020;21:234. doi: 10.1186/s12931-020-01490-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shirai T., Tsukiji N., Sasaki T., Oishi S., Yokomori R., Takano K., Suzuki-Inoue K. Cancer-associated fibroblasts promote venous thrombosis through podoplanin/CLEC-2 interaction in podoplanin-negative lung cancer mouse model. J. Thromb. Haemost. 2023;21:3153–3165. doi: 10.1016/j.jtha.2023.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Wu S.Y., Watabe K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci. 2017;22:1805–1829. doi: 10.2741/4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S.Y., Xing F., Sharma S., Wu K., Tyagi A., Liu Y., Zhao D., Deshpande R.P., Shiozawa Y., Ahmed T., et al. Nicotine promotes brain metastasis by polarizing microglia and suppressing innate immune function. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green K.N., Crapser J.D., Hohsfield L.A. To Kill a Microglia: A Case for CSF1R Inhibitors. Trends Immunol. 2020;41:771–784. doi: 10.1016/j.it.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito S., Nagata K. Biology of Hsp47 (Serpin H1), a collagen-specific molecular chaperone. Semin. Cell Dev. Biol. 2017;62:142–151. doi: 10.1016/j.semcdb.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Larsen A.M.H., Kuczek D.E., Kalvisa A., Siersbæk M.S., Thorseth M.L., Johansen A.Z., Carretta M., Grøntved L., Vang O., Madsen D.H. Collagen Density Modulates the Immunosuppressive Functions of Macrophages. J. Immunol. 2020;205:1461–1472. doi: 10.4049/jimmunol.1900789. [DOI] [PubMed] [Google Scholar]
- 22.Boraschi-Diaz I., Wang J., Mort J.S., Komarova S.V. Collagen Type I as a Ligand for Receptor-Mediated Signaling. Front. Phys. 2017;5 doi: 10.3389/fphy.2017.00012. ARTN 12. [DOI] [Google Scholar]
- 23.Shanmugam G., Sunny J.S., Rakshit S., George M., Leela K.V., Sarkar K. Involvement of inflammatory cytokines and epigenetic modification of the mtTFA complex in T-helper cells of patients? suffering from non-small cell lung cancer and chronic obstructive pulmonary disease. Mol. Immunol. 2022;151:70–83. doi: 10.1016/j.molimm.2022.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Ito S., Ogawa K., Takeuchi K., Takagi M., Yoshida M., Hirokawa T., Hirayama S., Shin-Ya K., Shimada I., Doi T., et al. A small-molecule compound inhibits a collagen-specific molecular chaperone and could represent a potential remedy for fibrosis. J. Biol. Chem. 2017;292:20076–20085. doi: 10.1074/jbc.M117.815936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei Y., Liang M., Xiong L., Su N., Gao X., Jiang Z. PD-L1 induces macrophage polarization toward the M2 phenotype via Erk/Akt/mTOR. Exp. Cell Res. 2021;402 doi: 10.1016/j.yexcr.2021.112575. [DOI] [PubMed] [Google Scholar]
- 26.de Visser K.E., Joyce J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. 2023;41:374–403. doi: 10.1016/j.ccell.2023.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Xing F., Liu Y., Wu S.Y., Wu K., Sharma S., Mo Y.Y., Feng J., Sanders S., Jin G., Singh R., et al. Loss of XIST in Breast Cancer Activates MSN-c-Met and Reprograms Microglia via Exosomal miRNA to Promote Brain Metastasis. Cancer Res. 2018;78:4316–4330. doi: 10.1158/0008-5472.CAN-18-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu S., Xu H., Wang W., Li S., Li H., Li T., Zhang W., Yu X., Liu L. The role of collagen in cancer: from bench to bedside. J. Transl. Med. 2019;17:309. doi: 10.1186/s12967-019-2058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nissen N.I., Karsdal M., Willumsen N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019;38:115. doi: 10.1186/s13046-019-1110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng D.H., Rodriguez B.L., Diao L., Chen L., Wang J., Byers L.A., Wei Y., Chapman H.A., Yamauchi M., Behrens C., et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat. Commun. 2020;11:4520. doi: 10.1038/s41467-020-18298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han X., Li Y., Xu Y., Zhao X., Zhang Y., Yang X., Wang Y., Zhao R., Anderson G.J., Zhao Y., Nie G. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat. Commun. 2018;9:3390. doi: 10.1038/s41467-018-05906-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salmon H., Franciszkiewicz K., Damotte D., Dieu-Nosjean M.C., Validire P., Trautmann A., Mami-Chouaib F., Donnadieu E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 2012;122:899–910. doi: 10.1172/jci45817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartmann N., Giese N.A., Giese T., Poschke I., Offringa R., Werner J., Ryschich E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res. 2014;20:3422–3433. doi: 10.1158/1078-0432.Ccr-13-2972. [DOI] [PubMed] [Google Scholar]
- 34.Bellaye P.S., Burgy O., Bonniaud P., Kolb M. HSP47: a potential target for fibrotic diseases and implications for therapy. Expert Opin. Ther. Targets. 2021;25:49–62. doi: 10.1080/14728222.2021.1861249. [DOI] [PubMed] [Google Scholar]
- 35.Zhu J., Xiong G., Fu H., Evers B.M., Zhou B.P., Xu R. Chaperone Hsp47 Drives Malignant Growth and Invasion by Modulating an ECM Gene Network. Cancer Res. 2015;75:1580–1591. doi: 10.1158/0008-5472.can-14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoneda A., Minomi K., Tamura Y. HSP47 promotes metastasis of breast cancer by interacting with myosin IIA via the unfolded protein response transducer IRE1α. Oncogene. 2020;39:4519–4537. doi: 10.1038/s41388-020-1311-7. [DOI] [PubMed] [Google Scholar]
- 37.Xiong G., Chen J., Zhang G., Wang S., Kawasaki K., Zhu J., Zhang Y., Nagata K., Li Z., Zhou B.P., Xu R. Hsp47 promotes cancer metastasis by enhancing collagen-dependent cancer cell-platelet interaction. Proc. Natl. Acad. Sci. USA. 2020;117:3748–3758. doi: 10.1073/pnas.1911951117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito S., Saito M., Yoshida M., Takeuchi K., Doi T., Nagata K. A BRET-based assay reveals collagen?Hsp47 interaction dynamics in the endoplasmic reticulum and small-molecule inhibition of this interaction. J. Biol. Chem. 2019;294:15962–15972. doi: 10.1074/jbc.RA119.010567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu S., Liang C., Xie X., Huang H., Fu J., Wang C., Su Z., Wang Y., Qu X., Li J., Liu J. Hsp47 Inhibitor Col003 Attenuates Collagen-Induced Platelet Activation and Cerebral Ischemic-Reperfusion Injury in Rats. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.792263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thienel M., Müller-Reif J.B., Zhang Z., Ehreiser V., Huth J., Shchurovska K., Kilani B., Schweizer L., Geyer P.E., Zwiebel M., et al. Immobility-associated thromboprotection is conserved across mammalian species from bear to human. Science. 2023;380:178–187. doi: 10.1126/science.abo5044. [DOI] [PubMed] [Google Scholar]
- 41.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li S., Chen Y., Xie Y., Zhan H., Zeng Y., Zeng K., Wang L., Zhan Z., Li C., Zhao L., et al. FBXO7 Confers Mesenchymal Properties and Chemoresistance in Glioblastoma by Controlling Rbfox2-Mediated Alternative Splicing. Adv. Sci. 2023;10 doi: 10.1002/advs.202303561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng K., Zeng Y., Zhan H., Zhan Z., Wang L., Xie Y., Tang Y., Li C., Chen Y., Li S., et al. SEC61G assists EGFR-amplified glioblastoma to evade immune elimination. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2303400120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
RNA-seq data generated in this study have been deposited at GEO database and are publicly available as of the date of publication. Accession number is listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.