

Abstract
The epidermal growth factor (EGF) family and the EGF receptor (EGFR, ErbB) tyrosine kinase family have been spearheading the studies of signal transduction events that determine cell fate and behavior in vitro and in vivo. The EGFR family and their signaling pathways are giving us tremendous advantages in developing fascinating molecular target strategies for cancer therapy. Currently, two important types of EGFR inhibitors are in clinical use: neutralizing antibodies of EGFR or ErbB2, and synthetic small compounds of tyrosine kinase inhibitors designed for receptors. On the other hand, basic research of the EGF family ligands presents new challenges as membrane‐anchored growth factors. All members of the EGF family have important roles in development and diseases and are shed from the plasma membrane by metalloproteases. The ectodomain shedding of the ligands has emerged as a critical component in the functional transactivation of EGFRs in interreceptor cross‐talk in response to various shedding stimulants such as G‐protein coupled receptor agonists, growth factors, cytokines, and various physicochemical stresses. Among the EGFR‐ligands, heparin‐binding EGF‐like growth factor (HB‐EGF) is a prominent ligand in our understanding of the pathophysiological roles of ectodomain shedding in cancer, wound healing, cardiac diseases, etc. Here we focus on ectodomain shedding of the EGF family ligands, especially HB‐EGF by disintegrin and metalloproteases, which are not only key events of receptor cross talk, but also novel intercellular signaling by their carboxy‐terminal fragments to regulate gene expression directly. (Cancer Sci 2008; 99: 214–220)
Growth factors and their specific cell surface receptors harboring a tyrosine kinase activity are providing us unfathomable knowledge in understanding development, homeostasis and diseases. Epidermal growth factor (EGF) and its specific receptor (EGFR) and their relative members have been at the forefront of signaling and therapeutics. The EGFR family comprise four members: EGFR (ErbB1), ErbB2, ErbB3 and ErbB4; and the EGF family comprises 13 members: EGF, transforming growth factor‐α (TGF‐α), amphiregulin, heparin‐binding EGF‐like growth factor (HB‐EGF), betacellulin, epiregulin, epigen, neuregulin‐1 (NRG‐1), NRG‐2, NRG‐3, NRG‐4, NRG‐5 and NRG‐6.( 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 ) Under physiological conditions, activation of EGFRs is controlled by the spatiotemporal expression and post‐translational processing of the ligands. Recently, interreceptor cross‐talk has received significant attention as an essential element in understanding the increasingly complex signaling networks identified within cells. Among them, transactivation of EGFRs has been shown to play a crucial role in signaling by G‐protein coupled receptors (GPCRs), cytokine receptors, receptor tyrosine kinases, and integrins to a variety of cellular responses.( 21 , 22 ) Transactivation of EGFRs is mediated, at least in some cases, by the ligands, which are cleaved from their membrane‐anchored forms (proforms) in a process termed ‘ectodomain shedding’. Research on the transactivation mechanisms of EGFRs is not only driving forward functional analyses of ‘the ectodomain shedding’ of the ligands by a disintegrin and metalloproteases (ADAMs), but also uncovering intriguing functions of carboxy‐terminal fragments (CTFs) of the ligands, which are products of ectodomain shedding. Currently, we have advanced knowledge about the physiological significance of ectodomain shedding of proHB‐EGF, which couples two independent signaling pathways, EGFR signaling by the shed extracellular domain and CTF signaling by the remnant transmembrane peptide.( 23 , 24 )
In this review, we briefly summarize the current knowledge of the EGF and EGFR families in cancer research and in clinical settings, and would like to focus on a novel function of the EGFR ligands themselves beyond the concept of ‘growth factor’ in general.
The EGF and the EGFR families and their current advance in cancer
Epidermal growth factor receptor is a170‐kDa membrane protein first identified as a binding partner of 125I‐labeled EGF on cell surface of fibroblasts,( 25 ) and was found to be bearing tyrosin‐kinase activity,( 26 ) and phosphorylated itself (autophosphorylation) in response to EGF treatment in A‐431 epidermoid carcinoma cells.( 27 ) In the 1980s, the overexpression of EGFR in various epithelial tumors was widely reported and it has been substantiated that deregulation of EGFR itself and its signaling pathway plays an important role in human cancers. For example, EGFR mutations in human cancers have been analyzed intensively and found that the mutations resulted in increased tyrosine kinase activity of EGFR( 28 ) and the increased stimulation of EGFR through autocrine growth factor loops, in particular, through TGF‐α.( 29 ) Excessive signaling of EGFR is a hallmark of a wide variety of solid tumors. The continuous massive studies have revealed four members of the EGFR family and 13 members of their ligand family (the EGF family) described above. Amplification of EGFRs, for instance, was found in the majority of carcinomas, and amplification of the ErbB2d gene can be found in 20–30% of metastatic breast lesions. Thus, EGFRs are attractive candidates for targeted therapy, and, to date, anti‐EGFR and anti‐ErbB2 therapeutics have been developed, and some of them are in clinical use by using humanized neutralizing antibodies and synthetic small compounds of tyrosine kinase inhibitors (TKIs) (Table 1).( 30 , 31 , 32 ) The recent clinical application of EGFR‐TKIs was prospective well for the patients with the high levels of EGFR expression in non‐small cell lung cancers (NSCLCs). Unexpectedly, the response for EGFR‐TKIs in patients was quite low. These results have shown that the response for EGFR‐TKIs is not correlated to the expression level of EGFR. However, correlation analyses between mutations in the EGFR gene and response for an EGFR‐TKI gefitinib in clinical samples are providing intriguing results showing that the patient response was correlated with somatic mutations (either small, in‐frame deletions or amino acid substitutions) in the EGFR kinase domain to enhance kinase activity,( 33 , 34 ) and that in contrast, a single somatic mutation, T790M, of the EGFR kinase domain confers resistance to gefitinib treatment in NSCLC.( 35 ) Subsequently, it was found that this mutation is associated with inherited susceptibility to lung cancer.( 36 ) These reports strongly indicate that EGFR mutation analysis is helpful to predict sensitivity to gefitinib.
Table 1.
Approved epidermal growth factor receptors (EGFRs) targeted therapeutics
| Drug | Target | Type | Application |
|---|---|---|---|
| Cetuximab (Erbitux) | EGFR | Chimeric mAb | CRC, and head and neck cancer |
| Panitumumab (Vecitibix, ABX‐EGF) | EGFR | Fully human mAb | CRC |
| Gefinitib (Iressa, ZD1839) | EGFR | TKI | NSCLC |
| Erlotinib (Tarceva) | EGFR | TKI | NSCLC and panvreas cancer |
| Trastuzumab (Herceptin) | ErbB2 | Humanized mAb | Breast cancer |
CRC, colorectal cancer; mAb, monoclonal antibody; NSCLC, non‐small cell lung carcinoma; TKI, tyrosine kinase inhibitor.
On the other hand, accurate prediction of the side‐effects of TKIs is a big issue for developing more potent and safe drugs. For this purpose, it is becoming a focal point to profile the specificity of newly developed TKIs using kinase protein panels that are commercially available (http://www.carnabio.com/japanese/).
Ectodomian shedding of the ligands and EGFR transactivation in cancer
All members of the EGF family are type I transmembrane proteins and are expressed on cell surfaces that can be cleaved by cell surface proteases.( 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 37 , 38 ) However, in some instances (NRG‐1beta1 cleavage in Golgi apparatus),( 39 ) there is a step that leads to the release of soluble ligands. This cleavage, termed ectodomain shedding, is a crucial step in the control of ligand availability and receptor activation. EGFRs are often constitutively stimulated in cancer owing to the presence of EGF ligands in the tumors.( 40 ) The production of soluble EGF family ligands through ectodomain shedding occurs in response to various physiological and pharmacological agonists, including 12‐O‐tetradecanoylphorbol‐13‐acetate,( 41 , 42 ) calcium ionophores,( 43 ) GPCR ligands such as bombesin,( 44 ) angiotensin II,( 45 ) and bacterial lipoteicchoic acid,( 46 ) and cytokines and growth factors.( 47 , 48 ) These shedding stimuli evoke cleavage and release of soluble ligands, mostly HB‐EGF, leading to the rapid phosphorylation of EGFR and the following activation of intracellular signaling pathways. This process, termed EGFR transactivation, has important biological implications in normal and cancer cells.( 30 , 31 ) Therefore, it is essential to understand the mechanisms and functional potency of ectodomain shedding in the following three aspects: (i) what kinds of proteases catalyze the ectodomain shedding; (ii) how the responsible proteases are regulated by extracellular and intracellular signals; and (iii) what are the priority proteases and regulatory molecules for molecular‐based therapeutic drug development? Most proteases involved in the ectodomain shedding belong to the metalloproteases ADAMs family, and have also been examined in tumor cells. ADAMs, including ADAM8, ADAM9, ADAM10, ADAM12, ADAM15, ADAM17, ADAM 19, ADAM 28, ADAMTS1, ADAMTS4 and ADAMTS5 have been associated with various types of cancer.( 49 , 50 ) Most of them are involved in the shedding of distinct EGFR ligands. Activation mechanisms of ADAMs are still largely unknown, but some clues to elucidate the mechanisms have been raised from the aspects regarding phosphorylation and interactive proteins of ADAM tails,( 51 ) and G‐protein regulation.( 52 ) Protease activities of ADAMs are multiple‐regulated, not only by regulatory domains such as the pro‐domain and cyotoplasmic domains, but also by intrinsic metalloprotease inhibitors such as tissue inhibitor of metalloprotease,( 53 ) and reversion‐inducing‐cysteine‐rich protein with kazal motifs.( 54 ) Small compound inhibitors of ADAMs have been developed in several industries, and proved to be potent drugs for some diseases including cancer in mouse models. However, it is still far away from clinical application for cancer treatment. As an alternative way, a siRNA strategy for targeting ADAMs might be favorable. Much more precise studies of intracellular mechanisms underling ADAM activation by extracellular stimuli would provide us with more fascinating ways to develop ADAM inhibitors as a drug in the future. The molecular mechanism underlying the ligand shedding‐dependent EGFR transactivation mediated by ADAM activation is summarized in Fig. 1.
Figure 1.
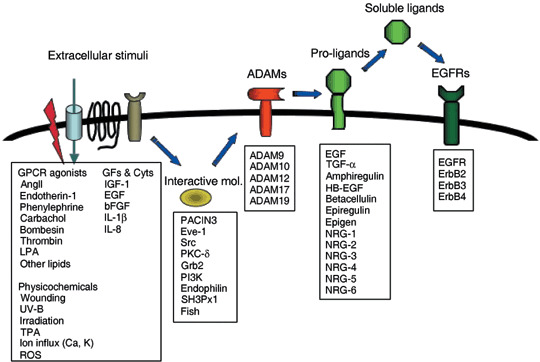
Ligand‐shedding dependent epidermal growth factor receptor (EGFR) transactivation. Disintegrin and metalloprotease (ADAM) proteins are activated by various stimuli including wounding, ion influx, G‐protein coupled receptor (GPCR) signaling, growth factor and cytokine signaling, protein kinase C (PKC) activation, and binding of cytoplasmic interactive proteins. EGFR ligand molecules are proteolytically cleaved by specific metalloprotease‐activity of ADAMs, resulting in the production of soluble ligands and stimulation of EGFR in autocrine and paracrine manners. AngII, angiotensin II; Cyts, cytokines; GFs, growth factors; HB‐EGF, Heparin‐binding epidermal growth factor carboxy‐terminal fragment; LPA, lysophosphatidic acid; MAPK, mitogen‐activated protein kinase; NRG, neuregulin; PI3K, phosphatidyl inositol 3‐kinase; PKC, protein kinase C; ROS, reactive oxygen species; TGF‐α, transforming growth factor‐α; TPA, 12‐O‐tetradecanoylphorbol‐13‐acetate.
Pathophysiology in disruption of proHB‐EGF shedding
Heparin‐binding EGF‐like growth factor is a leading molecule that has been linked to ligand shedding and EGFR transactivation, and is most intensively analyzed in its biological relevance in shedding in vitro and in vivo. Ligand shedding‐dependent EGFR transactivation was first described following activation of GPCR,( 55 ) and proHB‐EGF itself has been proved to be a crucial ligand and plays a central role in EGFR transactivation under various extracellular stimuli so far.( 56 , 57 )
Keratinocytes are the source of numerous growth factors (among which HB‐EGF is prominent) and play a central role in re‐epithelialization in skin wound healing in an autocrine manner as a migration factor, as well as a mitogen.( 58 , 59 ) Wounding stimuli evoked proHB‐EGF shedding enough for healing induction, which was abrogated by metalloprotease inhibitors to block the shedding.( 59 ) Aberration of the shedding of proHB‐EGF in soluble HB‐EGF and uncleavable proHB‐EGF knock‐in mice resulted in the severely abnormal developments of skin and heart.( 60 ) We also observed that skin wound healing is obviously retarded by the absence of keratinocyte migration in conditional Hb‐egf–/– mice.( 61 ) Over half of HB‐EGF‐null mice die during the first postnatal week, and survivors have dysfunctional hearts with grossly enlarged ventricular chambers and reduced life spans.( 62 , 63 ) Newborns lacking HB‐EGF have enlarged and malformed semilunar and atrioventricular heart valves. Furthermore, these phenotypes are similar to those observed in mice expressing an uncleavable form of proHB‐EGF (HBuc mice) generated by targeted gene replacement.( 60 ) The HBuc mice showed dilated cardiomyopathy in adult. Interestingly, dysregulated secretion of HB‐EGF in mice carrying transmembrane domain‐truncated mutant of proHB‐EGF (HBDtm mice) caused ventricular hypertrophy in heart.( 60 ) These findings indicate that the balanced shedding of proHB‐EGF is essential for the heart development and maintenance of the cardiac function,( 57 , 64 ) and that proHB‐EGF shedding is strictly controlled during development.
Ectodomain shedding of proHB‐EGF is induced by various stimuli including phorbol ester TPA,( 42 , 65 ) calcium ionophore,( 43 ) and various growth factors and cytokines.( 47 , 48 ) Protein kinase C (PKC) and mitogen‐activated protein kinase (MAPK) are involved in the intracellular signaling pathway for proHB‐EGF processing.( 66 , 67 ) GPCR agonists also stimulate proHB‐EGF processing, which mediates EGFR transactivation by GPCR signaling.( 44 ) The transactivation of EGFR induced, for example, by endothelin I, thrombin, lysophosphatidic acid (LPA), carbacol,( 44 ) insulin‐like growth factor‐1 (IGF‐1),( 68 ) basic fibroblast growth factor (bFGF), EGF,( 48 ) interleukin‐8 (IL‐8),( 47 ) estrogen,( 69 , 70 ) angiiotensin II, phenylephrine,( 45 , 71 , 72 ) Helicobacter pylori,( 73 )α2‐adrenergic receptor agonists,( 74 ) bacterial lipoteichoic acid,( 46 ) epoxyeicosatrienoic acid,( 75 ) and ultraviolet‐B,( 76 ) also apparently depends on proHB‐EGF shedding.
Metalloproteases are responsible for the proteolytic cleavage of proHB‐EGF as well as the other EGF family members because the ectodomain shedding of proHB‐EGF is efficiently inhibited by various metalloprotease inhibitors. Multiple members of the ADAM family are mainly implicated as shedding enzymes of proHB‐EGF. The ADAMs are characterized by a conserved domain structure, consisting of an N‐terminal signal sequence followed by pro‐domain, metalloprotease and disintegrin domains, a cycteine‐rich region, usually containing an EGF repeat, and finally a transmembrane domain and cytoplasmic tail.( 77 , 78 ) The overexpression of ADAM9 resulted in the increased shedding of proHB‐EGF without TPA and ADAM9 mutants of the metalloprotease domain inhibited TPA‐induced shedding in VeroH cells.( 67 ) TPA‐dependent proHB‐EGF shedding, however, remains unaffected in embryonic fibroblasts derived from mice lacking ADAM9.( 79 ) ADAM12 expression promoted proHB‐EGF shedding and the overexpression of dominant‐negative (metalloprotease domain deleted) ADAM12 but not ADAM9 abrogated the shedding by TPA treatment in HT1080 cells. Exogenous expression of dominant‐negative ADAM12 inhibited phenylephrine‐induced proHB‐EGF shedding in cardiomyocytes.( 45 ) TPA‐dependent proHB‐EGF shedding was largely impaired in embryonic fibroblasts derived from mice lacking ADAM12,( 80 ) and ADAM17.( 81 , 82 ) Transfection of wild‐type ADAM10, but not metalloprotease domain deleted ADAM10 stimulated the release of soluble HB‐EGF in COS7 cells,( 83 ) and ADAM10 was implicated in proHB‐EGF shedding and EGFR transactivation mediated by IL‐8,( 46 ) and bacterial lipoteichoic acid induced GPCR signaling.( 47 ) Reintroduction of ADAM17 into the immortalized fibroblasts derived from the mice eliminated zinc‐binding domain of ADAM17 resulted in the increased shedding of proHB‐EGF.( 81 , 82 )
The mechanism of ADAM activation has not yet been elucidated. However, we have identified two ADAM12‐docking proteins; Eve‐1,( 84 ) and PACSIN3,( 85 ) that upregulate TPA‐induced ADAM12 activation. Both Eve‐1 and PACSIN3 contain Src homology 3 (SH3) domains that can interact with the proline‐rich motifs of the ADAM12 cytoplasmic domain. Knockdown of each docking protein significantly reduces TPA‐ and angiotensin II‐induced proHB‐EGF shedding. Further, it has been reported that nardilysin (N‐arginine dibasic convertase (NRDc)), a metalloendopeptidase of the M16 family, specifically binds proHB‐EGF and enhances its shedding in cooperation with ADAM17.( 86 , 87 )
HB‐EGF‐CTF signaling beyond RTK activation
The ectodomain shedding of proHB‐EGF actually produces two fragments: an extracellular fragment (HB‐EGF) and a remnant fragment (HB‐EGF‐CTF). Recently we have identified promyelocytic leukemia zinc finger (PLZF) and B‐cell leukemia 6 (Bcl6) proteins as a binding protein of the cytoplasmic tail of proHB‐EGF.( 23 , 88 ) PLZF and Bcl6 are transcriptional repressors with structure domain homology of BR‐C, Itk, and kab and C2H2‐type Zn‐finger suppress cyclin A, c‐myc and HoxD, and macrophage inflammatory protein 1‐α, CD69 and CyclinD2 expression, respectively.( 89 , 90 , 91 , 92 , 93 , 94 ) These repressors negatively regulate the cell cycle. PLZF and Bcl6 produce transcriptional repression through recruitment of a repressor complex that contains N‐CoR, SMRT, Sin3a, and histone deacetylases.( 95 , 96 , 97 , 98 , 99 , 100 , 101 , 102 ) HB‐EGF‐CTF generated by proHB‐EGF shedding still contains a transmembrane region and is translocated from the plasma membrane into the reticular network of the endoplasmic reticulum (ER) and the nuclear envelope by retrograde membrane trafficking (Hieda and Higashiyama, 2007, unpublished observation). Since this process is impaired by the inhibition of metalloprotease activity, translocation of HB‐EGF‐CTF into the nuclear envelope is a shedding‐dependent event. HB‐EGF‐CTF has been visualized in the inner nuclear envelope under an electron microscope. Internalized HB‐EGF‐CTF associates with nuclear PLZF and Bcl6, which might occur at the nuclear periphery. One intriguing paper has just been published showing that proHB‐EGF itself upregulated E‐cadherin expression through suppression of ZEB1, a Zn‐finger typed transcriptional repressor, in pancreatic cells( 103 ) although the mechanism underlying such regulation was not discussed at all. We showed a possibility that proHB‐EGF itself translocated into the inner nuclear membrane and regulated transcriptional repressor activities (Hieda and Higashiyama, 2007, unpublished observation). We speculate that suppression of ZEB1 might be directly regulated by the cytoplasmic domain of the nuclear‐translocated proHB‐EGF.
Various recent studies have shown transcriptionally silent genes are located at or translocated to the nuclear periphery upon silencing.( 104 , 105 ) Indeed, interaction of HB‐EGF‐CTF with PLZF or Bcl6 results in the reversal of decreased expression of their target genes.( 23 , 88 ) Whether HB‐EGF‐CTF containing a transmembrane domain would directly regulate genes silenced by PLZF and Bcl6 at the nuclear periphery is still under investigation.
Coupling of derepression signaling with growth signaling
In parallel with HB‐EGF‐CTF production, proHB‐EGF shedding also generates HB‐EGF, a soluble ligand of EGFR. EGFR signaling promotes G1‐phase progression in the cell cycle by regulating the expression of cyclin D and c‐Myc via the Ras‐MAPK signaling cascade. Therefore, proteolytic cleavage of proHB‐EGF by ADAMs generates two types of mitogenic signaling molecules, and the coordination of the dual intracellular signals mediated by HB‐EGF and HB‐EGF‐CTF may be important for cell cycle progression.( 23 , 57 ) In fact, EGFR and FGFR‐mediated c‐Myc induction and cell cycle progression in primary cultured mouse embryonic fibroblasts are abrogated by knockout of the Hb‐egf, or by a metalloprotease inhibitor, although molecules downstream of the receptors are activated.( 48 ) Induction of c‐Myc expression by EGF or bFGF is recovered in Hb‐egf‐depleted mouse embryonic fibroblasts by overexpression of wild‐type proHB‐EGF, but no recovery was observed with an uncleavable mutant of proHB‐EGF. The uncleavable mutant also inhibited EGF‐induced acetylation of histone H3 at the mouse c‐Myc first intron region, which could negatively affect transcriptional activation. Thus, the signal transduction initiated by generation of HB‐EGF‐CTF in the shedding event plays an essential intermediary role in growth factor induced cell cycle progression.
Promyelocytic leukemia zinc finger and Bcl‐6 as well as HB‐EGF are expressed in a large number of tissues including the heart.( 106 , 107 , 108 ) This suggests that deregulation of PLZF and Bcl6 repressional activities by proHB‐EGF shedding is involved in regulation of cell proliferation and differentiation in various signaling cascades during the development and maintenance of adult tissues. Heart failure observed in HB‐EGF‐null, HBuc and HBDtm mice( 60 , 62 ) might in part be due to the loss of HB‐EGF‐CTF‐PLZF/Bcl6 signaling in the heart, where PLZF and Bcl6 are highly expressed and are suggested to be involved in maintaining cardiac function.( 108 ) HB‐EGF‐CTF has the ability to interact with these transcriptional repressors, which suggests that the integration of EGFR members and CTF signaling leads to various cellular responses by controlling gene transcription (Fig. 2).
Figure 2.
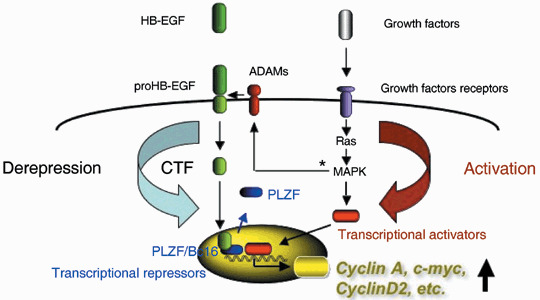
Heparin‐binding epidermal growth factor carboxy‐terminal fragments (HB‐EGF‐CTF) signaling couples with growth factor receptor signaling. ProHB‐EGF is shed by specific disintegrin and metalloproteases (ADAMs) and yield amino‐ and carboxy‐terminal fragments (HB‐EGF and HB‐EGF‐CTF). Produced HB‐EGF binds and activates epidermal growth factor receptor (EGFR), resulting in the activation of mitogen‐activated protein kinase (MAPK) cascade and various gene transcription. HB‐EGF‐CTF translocates into the inner nuclear membrane. HB‐EGF‐CTF and HB‐EGF generated by proHB‐EGF shedding mediate signaling into the nucleus directly (CTF signaling) and indirectly via EGFR signaling, respectively. Derepression signaling by HB‐EGF‐CTF and activation signaling through EGFR are coupled, regulate gene transcription and result in cellular responses to various stimuli. PLZF, promyelocytic leukemia zinc finger. *Activation of ADAMs by MAPK pathways has been reported in Gechtman et al. ( 66 ) and Umata et al. ( 116 )
BAG‐1 has been reported as the binding protein of the cytoplasmic domain of proHB‐EGF, and its interaction with proHB‐EGF leads to decreased cell adhesion, increased resistance to apoptosis, and rapid secretion of soluble HB‐EGF.( 109 ) BAG‐1 is a multifunctional protein that interacts with a diverse array of molecular targets including the Bcl‐2 apoptosis regulator, the 70 kDa heat shock proteins, Hsc70 and Hsp70, nuclear hormone receptors, the RAF kinase, components of the ubiquityinylation/proteasome machinery, and DNA.( 110 ) While BAG‐1 was first identified as a proHB‐EGF cytoplasmic binding protein in the cytoplasm and plasma membrane,( 109 ) BAG‐1 L, an isofom of BAG‐1, is localized in the nuclei of some types of cells.( 111 , 112 ) These findings suggest that BAG‐1 is a binding protein of nuclear HB‐EGF‐CTF and mediates the CTF signaling by proteolytic processing of proHB‐EGF.
Potential roles of cytoplasmic domains of EGF family members
Membrane‐anchored growth factors and their receptors provide the potential for bidirectional signaling, with a forward signal (receptor activation) mediated by the ectodomain and a reverse signal mediated by the intracellular domain of the ligand precursor. In the EGF family molecules, proTGF‐α activates an unidentified kinase in cells expressing it, when the precursor engages EGFR on neighboring cells.( 113 ) Recently, it has been reported that NRG‐1 is cleaved at the transmembrane domain and the released intracellular domain (NRG‐1‐ICD) enters the nucleus to repress the expression of several regulators of apoptosis.( 114 ) Currently, the same group has shown that in the mouse cochlea, synaptic activity increases the level of nuclear NRG‐ICD and upregulates postsynaptic density protein‐95 (PSD‐95), a scaffolding protein that is enriched in postsynaptic structures. NRG‐ICD enhances the transcriptional activity of the PSD‐95 promoter by binding to a zinc‐finger transcription factor Eos. The NRG‐ICD‐Eos complex induces endogenous PSD‐95 expression in vivo through a signaling pathway that is mostly independent of gamma‐secretase regulation. This upregulation of PSD‐95 expression by the NRG‐ICD‐Eos complex provides a molecular basis for activity‐dependent synaptic plasticity.( 115 )
While the machinery of CTF signaling of proHB‐EGF and NRG‐1 seems to be different, these findings suggest that the CTFs of the EGF family precursors have the ability to interact with transcriptional regulators in the nucleus and control gene expression in cells expressing the EGF family ligands. Indeed, we currently observed that other members of the EGF family are able to interact in many ways with transcriptional repressors (Morimoto and Higashiyama, 2007 unpublished observation).
Conclusion
Since EGF was discovered, EGF and its related ligands have been a driving force of the research of EGFRs and their signaling pathways for more than 40 years, which provides us tremendous information to understand growth mechanisms in normal and cancer cells. Therefore, we have paid little attention to the fact that all members of the EGF family are membrane‐anchored proteins, because their properties as ligands to activate receptor tyrosine kinases are fascinating enough. However, the interest of researchers in CTFs produced by ectodomain shedding are revealing ingenious molecular mechanisms of the EGF family ligands as growth factors, and soon we will able to fully understand why the EGF family ligands are membrane‐anchored factors. We believe that the knowledge obtained from the EGF family also provides new insights into the function of other membrane‐anchored growth factors and cytokines, and is quite helpful in unveiling unique properties of cancer.
Acknowledgments
We gratefully acknowledge Drs Daisuke Nanba, Yumi Kinugasa, Motonari Tanaka, and all members of Higashiyama's lab for helpful discussion.
References
- 1. Cohen S. Isolation and biological effects of an epidermal growth stimulating protein. Natl Cancer Inst Monogr 1964; 13: 13–27. [PubMed] [Google Scholar]
- 2. Derynck R, Roberts AB, Winkler ME, Chen EY, Goeddel DV. Human transforming growth factor‐α: precursor structure and expression in E. coli . Cell 1984; 38: 287–97. [DOI] [PubMed] [Google Scholar]
- 3. Shoyab M, Plowman GD, McDonald VL, Bradley JG, Todaro GJ. Structure and function of human amphiregulin: a member of the epidermal growth factor family. Science 1989; 243: 1074–6. [DOI] [PubMed] [Google Scholar]
- 4. Higashiyama S, Abraham JA, Miller J, Fiddes JC, Klagsbrun M. A heparin‐binding growth factor secreted by macrophage‐like cells that is related to EGF. Science 1991; 251: 936–9. [DOI] [PubMed] [Google Scholar]
- 5. Shing Y, Christfori G, Hanahan D et al . β‐cellulin: A mitogen from pancreatic B cell tumors. Science 1993; 259: 1604–14. [DOI] [PubMed] [Google Scholar]
- 6. Toyoda H, Komurasaki T, Uchida D et al . Epiregulin, a novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J Biol Chem 1995; 270: 7495–500. [DOI] [PubMed] [Google Scholar]
- 7. Strachan L, Murison JG, Prestidge RL, Sleeman MA, Watson JD, Kumble KD. Cloning and biological activity of epigen, a novel member of the epidermal growth factor superfamily. J Biol Chem 2001; 276: 18 265–71. [DOI] [PubMed] [Google Scholar]
- 8. Wen D, Peles E, Cupples R et al . Neu differentiation factor: a transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell 1992; 69: 559–72. [DOI] [PubMed] [Google Scholar]
- 9. Holmes WE, Sliwkowski MX, Akita RW et al . Identification of heregulin, a specific activator of p185erbB2. Science 1992; 256: 1205–10. [DOI] [PubMed] [Google Scholar]
- 10. Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell 1993; 72: 801–15. [DOI] [PubMed] [Google Scholar]
- 11. Marchionni MA, Goodearl AD, Chen MS et al . Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature 1993; 362: 312–18. [DOI] [PubMed] [Google Scholar]
- 12. Chang HD, Riese IIJ, Gillbert WD, Stern F, McMahan UJ. Ligands for ErbB‐family receptors encoded by a neuregulin related gene. Nature 1997; 387: 509–12. [DOI] [PubMed] [Google Scholar]
- 13. Carraway KL, Weber III JL, Unger MJ et al . Neuregulin‐2, a new ligand of ErbB3/ErbB4‐receptor tyrosine kinases. Nature 1997; 387: 512–16. [DOI] [PubMed] [Google Scholar]
- 14. Busfield SJ, Michnick DA, Chickering TW et al . Characterization of a neuregulin‐related gene, Don‐1, that is highly expressed in restricted region of the cerebellum and hippocampus. Mol Cell Biol 1997; 17: 4007–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higashiyama S, Horikawa M, Yamada K et al . A novel brain‐derived member of the epidermal growth factor family that interacts with ErbB3 and ErbB4. J Biochem 1997; 122: 675–80. [DOI] [PubMed] [Google Scholar]
- 16. Zhang D, Sliwkowski MX, Mark M et al . Neuregulin‐3 (NRG3): a novel neural tissue‐enriched protein that binds and activates ErbB4. Proc Natl Acad Sci USA 1999; 94: 9562–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harari D, Tzahar E, Romano J et al . Neuregulin‐4: a novel growth factor that acts through the ErbB4 receptor tyrosine kinase. Oncogene 1999; 18: 2681–9. [DOI] [PubMed] [Google Scholar]
- 18. Uchida T, Wada K, Akamatsu T et al . A novel epidermal growth factor‐like molecule containing two follistatin modules stimulates tyrosine phosphorylation of ErbB4 in MKN28 gastric cancer cells. Biochem Biophys Res Commun 1999; 266: 593–602. [DOI] [PubMed] [Google Scholar]
- 19. Kinugasa Y, Ohomoto H, Ishiguro H, Tokita Y, Oohira A, Higashiyama S. Neuroglycan C, a novel member of the neuregulin family. Biochem Biophys Res Commun 2004; 321: 1045–9. [DOI] [PubMed] [Google Scholar]
- 20. Citri A, Yarden Y. EGF–ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol 2006; 7: 505–16. [DOI] [PubMed] [Google Scholar]
- 21. Hackel PO, Zwick E, Prenzel N, Ullrich A. Epidermal growth factor receptors: critical mediators of multiple receptor pathways. Curr Opin Cell Biol 1999; 11: 184–9. [DOI] [PubMed] [Google Scholar]
- 22. Moghal NP, Sternberg W. Multiple positive and negative regulators of signaling by the EGF‐receptor. Curr Opin Cell Biol 1999; 11: 190–8. [DOI] [PubMed] [Google Scholar]
- 23. Nanba D, Mammoto A, Hashimoto K, Higashiyama S. Proteolytic release of the carboxy‐terminal fragment of proHB‐EGF causes nuclear export of PLZF. J Cell Biol 2003; 163: 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nanba D, Higashiyama S. Dual intracellular signaling by proteolytic cleavage of membrane‐anchored heparin‐binding EGF‐like growth factor. Cytokine Growth Factor Rev 2003; 15: 13–19. [DOI] [PubMed] [Google Scholar]
- 25. Carpenter G, Lembach KJ, Morrison MM, Cohen S. Characterization of the binding of 125I‐labeled epidermal growth factor to human fibroblasts. J Biol Chem 1975; 250: 4297–304. [PubMed] [Google Scholar]
- 26. Ushiro H, Cohen S. Identification of phosphotyrosine as a product of epidermal growth factor‐activated protein kinase in A‐431 cell membranes. J Biol Chem 1980; 255: 8363–5. [PubMed] [Google Scholar]
- 27. Carpenter G, King L Jr, Cohen S. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro . Nature 1978; 276: 409–10. [DOI] [PubMed] [Google Scholar]
- 28. Humphrey PA, Wong AJ, Vogelstein B et al . Anti‐synthetic peptide antibody reacting at the fusion junction of deletion‐mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci USA 1990; 87: 4207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sizeland AM, Burgess AW. Anti‐sense transforming growth factor α oligonucleotides inhibit autocrine stimulated proliferation of a colon carcinoma cell line. Mol Biol Cell 1992; 3: 1235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 2004; 4: 361–70. [DOI] [PubMed] [Google Scholar]
- 31. Hynes NE, Lane HA. ErbB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 2005; 5: 341–54. [DOI] [PubMed] [Google Scholar]
- 32. Bubil EM, Yarden Y. The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr Opin Cell Biol 2007; 19: 124–34. [DOI] [PubMed] [Google Scholar]
- 33. Lynch TJ, Bell DW, Sordella R et al . Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 34. Paez JG, Janne PA, Lee JC et al . EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 35. Kobayashi S, Boggon TJ, Dayaram T et al . EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 36. Bell DW, Gore I, Okimoto RA et al . Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 2005; 37: 1315–16. [DOI] [PubMed] [Google Scholar]
- 37. Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res 2003; 284: 2–13. [DOI] [PubMed] [Google Scholar]
- 38. Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res 2003; 284: 14–30. [DOI] [PubMed] [Google Scholar]
- 39. Yokozeki T, Wakatsuki S, Hatsuzawa K, Black RA, Wada I, Sehara‐Fujisawa A. Meltrin β/ADAM19 mediates ectodomain shedding of neuregulin β1 in the Golgi apparatus: fluorescence correlation spectroscopic observation of the dynamics of ectodomain shedding in living cells. Genes Cells 2007; 12: 329–43. [DOI] [PubMed] [Google Scholar]
- 40. Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor‐related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 1995; 19: 183–232. [DOI] [PubMed] [Google Scholar]
- 41. Pandiella A, Massague J. Cleavage of the membrane precursor for transforming growth factor alpha is a regulated process. Proc Natl Acad Sci USA 1991; 88: 1726–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goishi K, Higashiyama S, Klagsbrun M, Ishikawa M, Mekada E, Taniguchi N. Phorbol ester induces the rapid processing of cell surface heparin‐binding EGF‐like growth factor: conversion from juxtacrine to paracrine growth factor activity. Mol Biol Cell 1995; 6: 967–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dethlefsen SM, Raab G, Moses MA, Adam RM, Klagsbrun M, Freeman MR. Extracellular calcium influx stimulates metalloproteinase cleavage and secretion of heparin‐binding EGF‐like growth factor independently of protein kinase C. J Cell Biochem 1998; 69: 143–53. [DOI] [PubMed] [Google Scholar]
- 44. Prenzel N, Zwick E, Daub H et al . EGF receptor transactivation by G‐protein‐coupled receptors requires metalloproteinase cleavage of proHB‐EGF. Nature 1999; 402: 884–8. [DOI] [PubMed] [Google Scholar]
- 45. Asakura M, Kitakaze M, Takashima S et al . Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB‐EGF. metalloproteinase inhibitors as a new therapy. Nat Med 2002; 8: 35–40. [DOI] [PubMed] [Google Scholar]
- 46. Lemjabbar H, Basbaum C. Platelet‐activating factor receptor and ADAM10 mediate responses to Staphylococcus aureus in epithelial cells. Nat Med 2002; 8: 41–6. [DOI] [PubMed] [Google Scholar]
- 47. Tanida S, Joh T, Itoh K et al . The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology 2004; 127: 559–69. [DOI] [PubMed] [Google Scholar]
- 48. Nanba D, Inoue H, Shigemi Y, Shirakata Y, Hashimoto K, Higashiyama S. An intermediary role of proHB‐EGF shedding in growth factor‐induced c‐Myc gene expression. J Cell Physiol 2007; 214: 465–75. [DOI] [PubMed] [Google Scholar]
- 49. Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci 2007; 98: 621–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kenny PA, Bissell MJ. Targeting TACE‐dependent EGFR ligand shedding in breast cancer. J Clin Invest 2007; 117: 337–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Seals DF, Courtneidge SA. The ADAMs family of mettalloproteases: multidomain proteins with multiple functions. Genes Dev 2003; 17: 7–30. [DOI] [PubMed] [Google Scholar]
- 52. Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G‐protein‐coupled receptor. Am J Physiol Cell Physiol 2006; 291: C1–C10. [DOI] [PubMed] [Google Scholar]
- 53. Murphy G, Knauper V, Lee MH et al . Role of TIMPs (tissue inhibitors of metalloproteinases) in pericellular proteolysis: the specificity is in the detail. Biochem Soc Symp 2003; 70: 65–80. [DOI] [PubMed] [Google Scholar]
- 54. Muraguchi T, Takegami Y, Ohtsuka T et al . RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat Neurosci 2007; 10: 838–45. [DOI] [PubMed] [Google Scholar]
- 55. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G‐protein‐coupled receptors. Nature 1996; 379: 557–60. [DOI] [PubMed] [Google Scholar]
- 56. Higashiyama S. Metalloproteinase‐mediated shedding of Heparin‐binding EGF‐like growth factor and its pathophysiological roles. Protein Peptide Lett 2004; 11: 443–50. [DOI] [PubMed] [Google Scholar]
- 57. Higashiyama S, Nanba D. ADAM mediated ectodomain shedding of the EGFR ligands in receptor cross talk. Biochemica Biophysica Acta 2005; 751: 110–7. [DOI] [PubMed] [Google Scholar]
- 58. Hashimoto K, Higashiyama S, Asada H et al . Heparin‐binding EGF‐like growth factor is an autocrine growth factor for human keratinocytes. J Biol Chem 1994; 269: 20 060–6. [PubMed] [Google Scholar]
- 59. Tokumaru S, Higashiyama S, Endo T et al . Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing. J Cell Biol 2000; 151: 209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yamazaki S, Iwamoto R, Saeki K et al . Mutant mice with defects in the ectodomain shedding of HB‐EGF show severe abnormal development. J Cell Biol 2003; 163: 469–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shirakata Y, Kimura R, Nanba D et al . Heparin‐binding EGF‐like growth factor is essential for keratinocyte migration in skin wound healing. J Cell Sci 2005; 118: 2363–70. [DOI] [PubMed] [Google Scholar]
- 62. Iwamoto R, Yamazaki S, Asakura M et al . HB‐EGF and ErbB signaling is essential for heart function. Pro Natl Acad Sci USA 2003; 100: 3221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jackson LF, Qiu TH, Sunnarborg SW et al . Defective valvulogenesis in HB‐EGF and TACE‐null mice is associated with aberrant BMP signaling. EMBO J 2003; 22: 2704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iwamoto R, Mekada E. ErbB and HB‐EGF signaling in heart development and function. Cell Struct Funct 2006; 31: 1–14. [DOI] [PubMed] [Google Scholar]
- 65. Raab G, Higashiyama S, Hetelekidis S et al . Biosynthesis and processing by phorbol ester of the cells surface‐associated precursor form of heparin‐binding EGF‐like growth factor. Biochem Biophys Res Commun 1994; 204: 592–7. [DOI] [PubMed] [Google Scholar]
- 66. Gechtman Z, Alonson JL, Raab G, Ingber DE, Klagsbrun M. The shedding of membrane‐anchored heparin‐binding epidermal‐like growth factor is regulated by the raf/mitogen‐activated protein kinase cascade and by cell adhesion and spreading. J Biol Chem 1999; 274: 28 828–35. [DOI] [PubMed] [Google Scholar]
- 67. Izumi Y, Hirata M, Hasuwa H et al . A metalloprotease‐disintegrin, MDC9/meltrin‐γ/ADAM9 and PKCδ are involved in TPA‐induced ectodomain shedding of membrane‐anchored heparin‐binding EGF‐like growth factor. EMBO J 1998; 17: 7260–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Roudabush FL, Pierce KL, Maudsley S, Khan KD, Luttrell LM. Transactivation of the EGF receptor mediates IGF‐1‐stimulated shc phosphorylation and ERK1/2 activation in COS‐7 cells. J Biol Chem 2000; 275: 22 583–9. [DOI] [PubMed] [Google Scholar]
- 69. Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen‐induced activation of Erk‐1 and Erk‐2 requires the G protein‐coupled receptor homolog, GPR30, and occurs via trans‐activation of the epidermal growth factor receptor through release of HB‐EGF. Mol Endocrinol 2000; 14: 1649–60. [DOI] [PubMed] [Google Scholar]
- 70. Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem 2003; 278: 2701–12. [DOI] [PubMed] [Google Scholar]
- 71. Fujiyama S, Matsubara H, Nozawa Y et al . Angiotensin AT(1) and AT(2) receptors differentially regulate angiopoietin‐2 and vascular endothelial growth factor expression and angiogenesis by modulating heparin binding‐epidermal growth factor (EGF) ‐mediated EGF receptor transactivation. Circ Res 2001; 88: 22–9. [DOI] [PubMed] [Google Scholar]
- 72. Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease‐dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem 2001; 276: 7957–62. [DOI] [PubMed] [Google Scholar]
- 73. Wallasch C, Crabtree JE, Bevec D, Robinson PA, Wagner H, Ullrich A. Helicobacter pylori‐stimulated EGF receptor transactivation requires metalloprotease cleavage of HB‐EGF. Biochem Biophys Res Commun 2002; 295: 695–701. [DOI] [PubMed] [Google Scholar]
- 74. Cussac D, Schaak S, Denis C, Paris H. Alpha 2B‐adrenergic receptor activates MAPK via a pathway involving arachidonic acid metabolism, matrix metalloproteinases, and epidermal growth factor receptor transactivation. J Biol Chem 2002; 277: 19 882–8. [DOI] [PubMed] [Google Scholar]
- 75. Chen JK, Capdevila J, Harris RC. Heparin‐binding EGF‐like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc Natl Acad Sci USA 2002; 99: 6029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Seo M, Lee M‐J, Heo JH et al . G protein beta gamma subunits augment UVB‐induced apoptosis by stimulating the release of soluble heparin binding EGF‐like growth factor from human keratinocytes. J Biol Chem 2007; 282: 24 720–30. [DOI] [PubMed] [Google Scholar]
- 77. Black RA, White JM. ADAMs: focus on the protease domain. Curr Opin Cell Biol 1998; 10: 654–9. [DOI] [PubMed] [Google Scholar]
- 78. Schlöndorff J, Blobel CP. Metalloprotease‐disintegrins: modular proteins capable of promoting cell–cell interactions and triggering signals by protein‐ectodomain shedding. J Cell Sci 1999; 112: 3603–17. [DOI] [PubMed] [Google Scholar]
- 79. Weskamp G, Cai H, Brodie TA et al . Mice lacking the metalloprotease‐disintegrin MDC9 (ADAM9) have no evident major abnormalities during development or adult life. Mol Cell Biol 2002; 22: 1537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kurisaki T, Masuda A, Sudo K et al . Phenotypic analysis of Meltrin alpha (ADAM12) ‐deficient mice: involvement of Meltrin alpha in adipogenesis and myogenesis. Mol Cell Biol 2003; 23: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sahin U, Weskamp G, Kelly K et al . Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 2004; 164: 769–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Horiuchi K, Le Gall S, Schulte M et al . Substrate selectivity of epidermal growth factor‐receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell 2007; 18: 176–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yan Y, Shirakabe K, Werb Z. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein‐coupled receptors. J Cell Biol 2002; 158: 221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mori S, Tanaka M, Nanba D et al . PACSIN3 binds ADAM12/meltrin alpha and upregulates ectodomain shedding of heparin‐binding EGF‐like growth factor. J Biol Chem 2003; 278: 46 029–34. [DOI] [PubMed] [Google Scholar]
- 85. Tanaka M, Mori S, Nanba D et al . ADAM12‐binding protein Eve‐1 is required for ectodomain shedding of heparin‐binding epidermal growth factor‐like growth factor. J Biol Chem 2004; 279: 41 950–9. [DOI] [PubMed] [Google Scholar]
- 86. Nishi E, Prat A, Hospital V, Elenius K, Klagsbrun M. N‐arginine dibasic convertase is a specific receptor for heparin‐binding EGF‐like growth factor that mediates cell migration. EMBO J 2001; 20: 3342–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nishi E, Hiraoka Y, Yoshida K, Okawa K, Kita T. Nardilysin enhances ectodomain shedding of heparin‐binding epidermal growth factor‐like growth factor through activation of tumor necrosis factor‐alpha‐converting enzyme. J Biol Chem 2006; 281: 31 164–72. [DOI] [PubMed] [Google Scholar]
- 88. Kinugasa Y, Hieda M, Hori M, Higashiyama S. The carboxyl‐terminal fragment of pro‐HB‐EGF reverses Bcl6‐mediated gene repression. J Biol Chem 2007; 282: 14 797–806. [DOI] [PubMed] [Google Scholar]
- 89. Yeyati PL, Shaknovich R, Boterashvili S et al . Leukemia translocation protein PLZF inhibits cell growth and expression of cyclin A. Oncogene 1999; 18: 925–34. [DOI] [PubMed] [Google Scholar]
- 90. Barna M, Merghoub T, Costoya JA et al . Plzf mediates transcriptional repression of HoxD gene expression through chromatin remodeling. Dev Cell 2002; 3: 499–510. [DOI] [PubMed] [Google Scholar]
- 91. McConnell MJ, Chevallier N, Berkofsky‐Fessler W et al . Growth suppression by acute promyelocytic leukemia‐associated protein PLZF is mediated by repression of c‐myc expression. Mol Cell Biol 2003; 23: 9375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fernández de Mattos S, Essafi A, Soeiro I et al . FoxO3a and BCR‐ABL regulate cyclin D2 transcription through a STAT5/BCL6‐dependent mechanism. Mol Cell Biol 2004; 24: 10 058–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shaffer ALYuX, He Y, Boldrick J, Chan EP, Staudt LM. BCL‐6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity 2000; 13: 199–212. [DOI] [PubMed] [Google Scholar]
- 94. Chattopadhyay A, Tate SA, Beswick RW, Wagner SD, Ko Ferrigno P. A peptide aptamer to antagonize BCL‐6 function. Oncogene 2006; 25: 2223–33. [DOI] [PubMed] [Google Scholar]
- 95. Hong SH, David G, Wong CW, Dejean A, Privalsky ML, Privalsky . SMRT corepressor interacts with PLZF and with the PML‐retinoic acid receptor alpha (RARalpha) and PLZF‐RARalpha oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci USA 1997; 94: 9028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. David G, Alland L, Hong SH, Wong CW, DePinho RA. Dejean Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia‐associated PLZF protein. Oncogene 1998; 16: 2549–56. [DOI] [PubMed] [Google Scholar]
- 97. Grignani F, De Matteis S, Nervi C et al . Fusion proteins of the retinoic acid receptor‐alpha recruit histone deacetylase in promyelocytic leukaemia. Nature 1998; 391: 815–8. [DOI] [PubMed] [Google Scholar]
- 98. Guidez F, Ivins S, Zhu J, Söderström M, Waxman S, Zelent A. Reduced retinoic acid‐sensitivities of nuclear receptor corepressor binding to PML‐ and PLZF‐RARalpha underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood 1998; 91: 2634–42. [PubMed] [Google Scholar]
- 99. He LZ, Guidez F, Tribioli C et al . Distinct interactions of PML‐RARalpha and PLZF‐RARalpha with co‐repressors determine differential responses to RA in APL. Nat Genet 1998; 18: 126–35. [DOI] [PubMed] [Google Scholar]
- 100. Lin RJ, Nagy L, Inoue S, Shao W, Miller WH Jr, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature 1998; 391: 811–4. [DOI] [PubMed] [Google Scholar]
- 101. Huynh KD, Bardwell VJ. The BCL‐6 POZ domain and other POZ domains interact with the co‐repressors N‐CoR and SMRT. Oncogene 1998; 17: 2473–84. [DOI] [PubMed] [Google Scholar]
- 102. Huynh KD, Fischle W, Verdin E, Bardwell VJ. BCoR, a novel corepressor involved in BCL‐6 repression. Genes Dev 2000; 14: 1810–23. [PMC free article] [PubMed] [Google Scholar]
- 103. Wang F, Sloss C, Zhang X, Lee SW, Cusack JC. Membrane‐bound heparin‐binding epidermal growth factor like growth factor regulates E‐cadherin expression in pancreatic carcinoma cells. Cancer Res 2007; 67: 8486–93. [DOI] [PubMed] [Google Scholar]
- 104. Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 1997; 91: 845–54. [DOI] [PubMed] [Google Scholar]
- 105. Kosak ST, Skok JA, Medina KL et al . Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 2002; 296: 158–62. [DOI] [PubMed] [Google Scholar]
- 106. Senbonmatsu T, Saito T, Landon EJ et al . A novel angiotensin II type 2 receptor signaling pathway: possible role in cardiac hypertrophy. EMBO J 2003; 22: 6471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Cook M, Gould A, Brand N et al . Expression of the zinc‐finger gene PLZF at rhombomere boundaries in the vertebrate hindbrain. Proc Natl Acad Sci USA 1995; 92: 2249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yoshida T, Fukuda T, Hatano M et al . The role of Bcl6 in mature cardiac myocytes. Cardiovasc Res 1999; 42: 670–9. [DOI] [PubMed] [Google Scholar]
- 109. Lin J, Hutchinson L, Gaston SM, Raab G, Freeman MR. BAG‐1 is a novel cytoplasmic binding partner of the membrane form of heparin‐binding EGF‐like growth factor: a unique role for proHB‐EGF in cell survival regulation. J Biol Chem 2001; 267: 30 127–32. [DOI] [PubMed] [Google Scholar]
- 110. Townsend PA, Cutress RI, Sharp A, Brimmell M, Packham G. BAG‐1. a multifunctional regulator of cell growth and survival. Biochim Biophys Acta 2003; 1603: 83–98. [DOI] [PubMed] [Google Scholar]
- 111. Knee DA, Froesch BA, Nuber U, Takayama S, Reed JC. Structure‐function analysis of Bag1 proteins. Effects on androgen receptor transcriptional activity. J Biol Chem 2001; 276: 12 718–24. [DOI] [PubMed] [Google Scholar]
- 112. Hague A, Packham G, Huntley S, Shefford K, Eveson JW. Deregulated Bag‐1 protein expression in human oral squamous cell carcinomas and lymph node metastases. J Pathol 2002; 197: 60–71. [DOI] [PubMed] [Google Scholar]
- 113. Shum L, Reeves SA, Kuo AC, Fromer ES, Derynck R. Association of the transmembrane TGF‐alpha precursor with a protein kinase complex. J Cell Biol 1994; 125: 903–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bao J, Wolpowitz D, Role LW, Talmage DA. Back signaling by the Nrg‐1 intracellular domain. J Cell Biol 2003; 161: 1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bao J, Lin H, Ouyang Y et al . Activity‐dependent transcription regulation of PSD‐95 by neuregulin‐1 and Eos. Nat Neurosci 2004; 7: 1250–8. [DOI] [PubMed] [Google Scholar]
- 116. Umata T, Hirata M, Takahashi T et al . A dual signaling cascade that regulates the ectodomain shedding of heparin‐binding epidermal growth factor‐like growth factor. J Biol Chem 2001; 276: 30 475–82. [DOI] [PubMed] [Google Scholar]