

Abstract
Dendritic cell (DC)‐based immunotherapy is rapidly emerging as a promising treatment in cancer therapy. We had previously shown that DC pulsed with either defined mRNA of tumor antigen (Ag) such as α‐fetoprotein (AFP), or total RNA of hepatocellular carcinoma (HCC) could elicit Ag‐specific cytotoxic T lymphocyte (CTL) response. Therefore, we suggested a novel DC‐based therapeutic method, in which DCs derived from CD34+ cells enriched peripheral blood mononuclear cells were pulsed with liposome‐coated AFP and mutant P53 (mtP53) fused gene pEGFP‐C3/AFP‐mtP53 to induce bi‐targeted specific CTL responses against HCC. Three different genotype HCC cell lines, HepG2 (human histocompatibility leukocyte antigens (HLA) A2 positive, AFP expressing positive, P53 expressing negative), SMMC7721 (HLA A2 positive, neither AFP nor P53 expressing positive), and HMCC97 (HLA A2 positive, both AFP and P53 expressing positive) were selected as targets for CTL responses. An important finding was that DCs pulsed with the liposome‐coated fused gene could evoke more intensive bi‐targeted Ag‐specific CTL responses against HMCC97 than DCs pulsed with either AFP or P53 single gene (P < 0.05). This experimental therapeutic model provides a new promising cytotherapeutic approach, in that DCs pulsed with the fused gene of different Ags might induce more extensive multitargeted antitumor immunity. (Cancer Sci 2008; 99: 1420–1426)
Abbreviations:
- AEC
3‐amino‐9‐ethyl carbazole
- AFP
α‐fetoprotein
- Ag
antigen
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- DMEM
Dulbecco's minimum essential medium
- EGFP
enhanced green fluorescent protein
- FITC
fluorescein‐isothiocyanate
- G‐CSF
granulocyte colony‐stimulating factor
- HCC
hepatocellular carcinoma
- HLA
histocompatibility leukocyte antigens
- IL
interleukin
- mtP53
mutant P53
- PBMC
peripheral blood mononuclear cells
- PCR
polymerase chain reaction
- RT‐PCR
reverse transcription–polymerase chain reaction
HCC is a malignant disease with an annual global incidence of 1.2 million, and the incidence is rising rapidly in both Asia and Western countries because of the global epidemic of hepatitis B and C infection.( 1 ) In most cases of multinodular HCC, surgical ablative intervention is generally inapplicable and expected survival is less than 6 months.( 2 ) Liver transplantation is regarded as an efficacious therapy but only in some selected patients and the long‐term value still under investigation.( 3 ) Trans‐artery catheterized embolization is a primary therapeutic intervention for newly diagnosed HCC patients who lose the chance of surgical ablation.( 4 ) Therefore, to sustain the clinical effectiveness or decrease the relapse rate is still an urgent requirement. Specific immunotherapy against HCC as well as gene therapy is focused on some specific Ag genes, intervention using suicide genes( 5 ) or tumor suppression genes,( 6 ) and cytokine‐based strategies.( 7 )
DCs are regarded as professional Ag‐presenting cells and could prime naive T cells to induce Ag‐specific immunity in an HLA‐restricted manner. It has been shown that DCs could induce potent antitumor immunity both in vitro and in vivo.( 8 ) On the basis of these early observations, DC‐based immunotherapy and DC vaccines have been studied in several stage I or II clinical trials as an active immunotherapy for malignant diseases such as melanoma, renal cancer, prostate cancer, and other tumors.( 9 , 10 , 11 , 12 , 13 ) The primary results were encouraging in that DC vaccines could induce tumor‐specific immune responses, and tumor lesions regressed in some patients. 9 , 10 , 13
However, DC vaccines are occasionally transitory and unsuccessful in vivo because of tumor heterogeneity and weak immunogenicity of most tumor Ags.( 14 ) The range of potential tumor Ags is wide and expanding because of the sequencing of the cancer genome and gene‐expression profiling of cancer subsets.( 15 ) Tumor‐specific Ags include the idiotypic immunoglobulin of B‐cell lymphomas, and these safe Ags have been investigated in several vaccination programs.( 16 , 17 , 18 ) However, most candidate Ags are expressed to some degree by normal cells and most clinical trials of solid tumors have still focused on either cancer–testis Ags or lineage‐specific Ags.( 19 )
Therefore, DC vaccine formulation and immunological principles were taken into account to induce effective immunity. Several strategies had been employed to load DCs with tumor Ags, including co‐incubating DCs with Ag peptides( 20 ), fusing DCs with tumor cells,( 21 ) incubating DCs with tumor‐derived proteins or RNA,( 22 ) and transfecting DCs with tumor Ag genes.( 23 ) DCs loaded with tumor cell lysate, total RNA, and fusion cells are convenient and have the advantages of potentially inducing a polyclonal immune response against multiple targets on the cancer cells, and reducing the possibility of tumor escape from immune recognition. The disadvantages are the risk of inducing autoimmunity, the requirement for available tumor tissue, as well as the fact that the efficiency of DC‐induced immune responses depends on the concentration of immunogenic tumor Ag. Also, the evaluation of the immune response becomes more difficult as the specific Ag epitopes most frequently are unidentified.( 24 ) Although the selection of vectors becomes problematic, DCs pulsed with DNA or RNA of certain Ag could lead to prolonged presentation of Ag and generate a high‐affinity tumor‐reactive CTL response.( 25 , 26 )
Overexpression of AFP could be detected in approximately 60–80% HCC,( 27 ) and mtP53 in approximately 40–70% HCC. It has been proved that both AFP‐ and P53‐loaded DCs could induce specific CTL responses.( 28 , 29 )
In our group, we had previously shown that AFP mRNA‐transfected DCs could induce CD4+ and CD8+ mediated T‐cell responses against HCC, and total RNA of the HCC cell line HepG2 could elicit an Ag‐specific CTL response. Fusing DCs with HepG2 cells was more efficient than RNA in priming the T helper type 1 response, whereas RNA‐loaded DCs were more effective in CTL priming.( 30 , 31 , 32 ) Furthermore, antitumor immunity against hepatitis B virus‐infected HCC was also induced by DCs pulsed with adenovirus‐delivered hepatitis B surface Ag gene.( 33 ) These issues primed the idea that DCs pulsed with fused genes of candidate Ag AFP and mtP53 could elicit more intensive and extensive antitumor immunity through a bi‐targeted CTL response.
Meterials and Methods
Patient characteristics and blood donation. In this study, we collected PBMCs donated by hepatic carcinoma patients (HLA A2 positive). The study comprised eight patients ≥18 years old who had a histologically confirmed diagnosis of unresectable or metastatic hepatic cancer, Karnofsky Performance Status ≥80%, and anticipated survival ≥3 months. Key exclusion criteria included: evidence of being immunocompromised; past or present diagnosis of autoimmune disease; steroid use within 1 month; evidence of active, uncontrolled infection; or receipt of immunotherapy or biotherapy. Written informed consent was obtained from all patients prior to donation.
Patients were receiving pacilitaxel (Sanofi‐Aventis, France) 80 mg/m2 and doxorubicin 25 mg/m2 treatment weekly. PBMCs were mobilized with human recombinant G‐CSF (Kirin Brewery, Tokyo, Japan) at a dose of 5 mg/kg for 3–5 days after chemotherapy when the amount of peripheral blood leukocytes decreased to approximately 1.0 × 109/L. PBMCs were separated by density centrifugation using the COBE Spectra cell separator (Gambro BCT, Lakewood, CO). And 5 mL PBMCs were used for generating DCs and T cells.
Cytokines and chemicals. Human recombinant granulocyte‐macrophage colony‐stimulating factor was obtained from Kirin Brewery. Human recombinant IL‐4 and tumor necrosis factor‐α were purchased from R&D Systems (Minneapolis, MN). Trizol, avian myeloblastosis virus reverse transcriptase, oligo(dT), Lipofectamine 2000, and DMEM were products of Invitrogen (Carlsbad, CA). AEC and 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide were purchased from Sigma (St Louis, MO). T4 DNA ligase, PCR reagents and polymerase were purchased from Promega (Madison, WI). Restriction digestion enzymes were from TaKaRa Bio (Shiga, Japan).
Cell lines. Human HCC cells HepG2 (HLA A2 positive, AFP secreting positive, P53 secreting negative), SMMC7721 (HLA A2 positive, neither AFP nor P53 secreting positive), HMCC97 (HLA A2 positive, both AFP and P53 secreting positive), and human embryo kidney HEK293 cells were all cultured in DMEM supplemented with 10% fetal calf serum (Gibco Invitrogen, Grand Island, NY) and antibiotics.
Plasmid vector and RT‐PCR reaction. Plasmid vector pEGFP‐C3 was purchased from BD Biosciences (San Jose, CA) and mtP53 cDNA was donated by Dr Zhe Wang (Fourth Military Medical University, Xian, China). The vector was sequenced to be correct and the mtP53 gene was cloned between restriction digestion enzyme sites HindIII and EcoRI. AFP cDNA was amplified by the RT‐PCR method from HepG2 cells. The RT reaction was carried out at 37 C for 60 min and the PCR reaction was according to the manufacturer's protocol. The sense primer was 5′‐TAAAgATCTAgCCACCATggCACTgCATAgAAATgAATATg‐3′ (with Kozak sequence and restriction enzyme digestion site BglII) and the antisense primer wass 5′‐gAAAAgCTTAACTCCCAAAgCAgCACgAg‐3′ (with restriction enzyme digestion site HindIII but no stop code). The PCR products, AFP cDNAs, were cloned into vector pEGFP‐C3/mtP53 and the reconstructed vector was named pEGFP‐C3/AFP‐mtP53.
Transecting HEK293 cells with pEGFP‐C3/AFP‐mtP53. Plasmid pEGFP‐C3/AFP‐mtP53 was extracted according to the instructions of the Mini Wizard DNA Extraction Kit (Promega). 4 micrograms of pEGFP‐C3/AFP‐mtP53 and 10 µL Lipofectamine 2000 were diluted in 250 µL DMEM, mixed together and placed for 10 min at room temperature. Then the mixture was added into HEK293 cells cultured in DMEM. HEK293 cells were incubated for another 24 h and were monitored by fluorescent microscopy. Untransfected HEK293 cells were cultured as the control.
Expression of target Ags in transfected HEK293 cells. Mouse anti‐human AFP monoclonal antibody and rabbit anti‐human P53 monoclonal antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). HEK293 cells transfected with pEGFP‐C3/AFP‐mtP53 were placed on slides. Because green fluorescent protein may cause false results, the immunocytochemical stain method was used to detect AFP expression first according to regular procedure of the SABC Kit (Boster, Wuhan, China). AEC was used as the chromogen for AFP detection. FITC‐conjugated goat anti‐mouse antibody (Boster) was used for AFP detection according to regular immunofluorescence protocol and Cy3 conjugated goat antirabbit antibody (Boster) was used for mtP53 detection. The untransfected HEK293 cells were cultured as the control.
Culture of DCs. CD34+ cell‐enriched PBMCs were washed and resuspended in X‐VIVO 15 (Cambrex) to yield 10 × 106 cells/mL. 2 mL of cell suspension was incubated in each well of a 6‐well flat‐bottom plate (Corning Costar, Cambridge, MA) at 37°C for 2 h. Non‐adherent cells were removed gently for generating T cells. Adherent cells were cultured in X‐VIVO 15 medium supplemented with 100 µg/L of granulocyte‐macrophage colony‐stimulating factor and 10 µg/L IL‐4 for 7–8 days. 2.5 µg/L of tumor necrosis factor‐α was supplied at day 5.
Flow cytometry and phenotype of cultured DCs. Phenotypic changes of DCs were monitored by flow cytometric analysis. Human monoclonal antibodies for flow cytometric analysis included anti‐CD1a, anti‐CD11c conjugated by phycoerythrin, anti‐CD80, anti‐CD86, and anti‐HLA‐DR (conjugated by FITC) purchased from BD Biosciences. DCs were labeled with FITC‐ or phycoerythrin‐conjugated antibodies directly and analyzed by an EPICS XL‐MCL flow cytometer (Beckman‐Coulter, Miami, FL, US). Unlabeled DCs were used as the control.
DCs pulsed with liposome‐coated pEGFP‐C3/AFP‐mtP53. Plasmid pEGFP‐C3/AFP‐mtP53 was extracted according to the instructions of the Mini Wizard DNA Extraction Kit (Promega). 4 µg of pEGFP‐C3/AFP‐mtP53 plasmid and 10 µL Lipofectamine were diluted in 250 µL X‐VIVO 15 medium, mixed together and placed for 30 min at room temperature. Then the mixture was added into DCs and incubated for another 24 h. If fluorescent microscopy showed that green fluorescent protein was successfully expressed, DCs were then irradiated (30 Gy) for subsequent experiments, given within 60 min. mRNA of the fused gene AFP‐mtP53 was amplified by RT‐PCR and the phenotypic changes of DCs pre‐ and post‐transfection were monitored by flow cytometric analysis.
Generation of activated T cells. Non‐adherent PBMCs cells (2 × 106 cells/mL) were cultured in X‐VIVO 15 medium supplemented with 20 IU/mL IL‐2 for 3 days in T‐75 culture flasks (Corning Costar). The activated T cells were then harvested and washed before use in the following experiments.
Incubation of DCs with activated autologous T cells. Activated autologous T cells of HCC patients were placed in each well of a 6‐well flat‐bottom plate at a concentration of 2 × 106 cells/mL. DCs (2 × 106 cells/mL) were then added to each well at a rate of 1:20 (DCs vs. T cells). The plates were incubated for 3 days at 37°C in 5% CO2. Potential changes in the morphology of DCs or T cells during the course of the experiments were monitored by phase contrast microscopy. The DC and T cell mixtures were harvested and washed by DMEM for downstream exam.
CTL response and assays. HepG2 cells, HMCC97 cells, and SMMC7721l cells cultured in DMEM were added to wells of a 96‐well plate (Corning Costar), respectively, at a concentration of 2 × 106 cells/mL. The DC and T cell mixture was diluted by DMEM to a concentration of 2 × 106 cells/mL. The mixture was added to each well of cancer cells at a rate of 1:20 (DC and T cell mixture vs. cancer cells). The cells were incubated for 24 h at 37°C with 5% CO2. The DC and T cell mixture and cancer cells were cultured in the same conditions as the controls.
3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide was added into each well at a concentration of 0.5 mg/mL, and then cells were incubated for 4 h and centrifuged for 4 min. The suspension was discarded and 150 µL DMSO added to each well to dissolve the purple deposition. Then the absorption of each well was assayed at 490 nm.
In order to obtain integrated data, DCs pulsed with plasmid pEGFP‐C3, pEGFP‐C3/mtP53, and pEGFP‐C3/AFP were set as the controls of the CTL response. pEGFP‐C3/AFP was obtained by replacing the mtP53 fragment with AFP cDNA, which was also amplified by RT‐PCR reaction but using different primers: sense primer, 5′‐TAAAgATCTAgCCACCATggCACTgCATag‐AAATgAATATg‐3′; antisense primer, 5′‐TTggTCgACTTAAACTCCCAAAgCAgCACgAg‐3′ (involving stop code TAA). The reconstructed plasmid pEGFP‐C3/AFP underwent sequencing to be validated.
Statistical analysis. The data of CTL responses were expressed as the mean ± standard error (SE). Differences between groups were analyzed by Student–Newman–Keuls’ test. Statistical significance was considered as P < 0.05.
Results
Reconstruction of fused gene with AFP and mtP53. A length of 1773 bp AFP cDNA without the stop code was amplified by the RT‐PCR method (Fig. 1a). The fused gene of AFP and mtP53 of approximately 3.0 kb was cloned into plasmid pEGFP‐C3 and the reconstructed plasmid was named pEGFP‐C3/AFP‐mtP53 (Fig. 1b). The inserted fused gene was sequenced to be valid.
Figure 1.
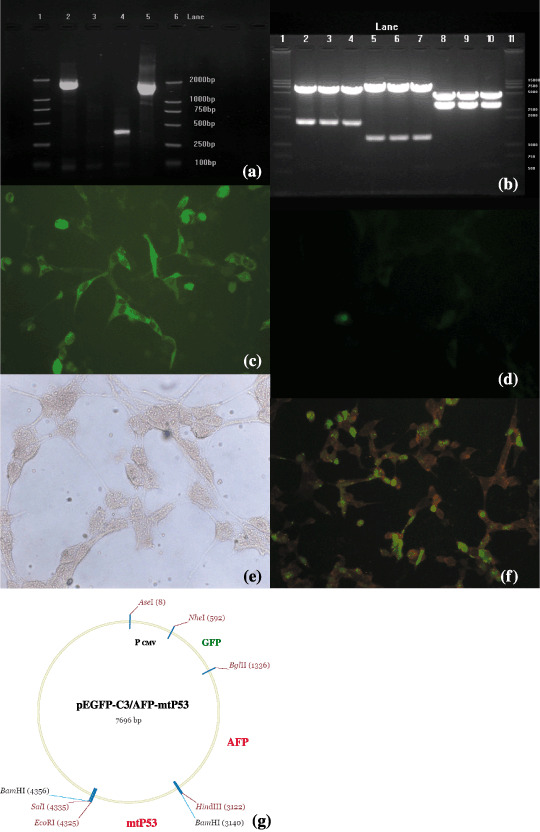
Reconstruction of enhanced green fluorescent protein/α‐feroprotein–mutant P53 plasmid (pEGFP‐C3/AFP‐mtP53) and the expression of the AFP‐mtP53 gene in eukaryotic cells. (a and b) Gel electrophoretic patterns of reconstructed plasmid pEGFP‐C3/AFP‐mtP53. (a) Reverse transcription–polymerase chain reaction amplification results. Lanes 1 and 6, molecular weight; lane 2, 1.8 kb AFP cDNA amplified; lanes 3 and 4, other samples; lane 5, positive control. (b) Double restriction enzyme reaction results of pEGFP‐C3/AFP‐mtP53 in three samples (no. 1–3). Lanes 1 and 11, molecular weight; lanes 2–4, 1.8 kb AFP cDNAs (sample 1–3); lanes 5–7, 1.2 kb mtP53 cDNA (samples 1–3); lanes 8–10, 3.0 kb AFP‐mtP53 fusion gene in samples 1–3. (c–f) Fluorescent microscopy analysis of human embryonic kidney HEK293 cells transfected with plasmid pEGFP‐C3/AFP‐mtP53 (×200). (c) HEK293 cells transfected with pEGFP‐C3/AFP‐mtP53 emitted green fluorescence 24 h after transfection. (d) The expression of AFP by the immunocytochemical staining method showed that there were red stains in transfected HEK293 cells. (e) Immunofluorescence assay showed that transfected HEK293 cells expressed orange fluorescence, a compound of green fluorescence (fluorescein‐isothiocyanate‐conjugated AFP antibody) and red fluorescence (Cy3‐conjugated mtP53 antibody). (f) Blank HEK293 cells. (g) Construction map of pEGFP‐C3/AFP‐mtP5. The AFP gene is located between 1336 and 3122 bp, and the p53 gene is located between 3140 and 4325 bp.
Expression of AFP‐mtP53 fused gene in HEK293 cells. The fused gene pEGFP‐C3/AFP‐mtP53 was transfected into HEK293 cells for inducing expression. The transfection rate reached 70% and green fluorescence was detected 24 h after transfection (Fig. 1c). Immunocytochemical staining was used to detect AFP expression by the streptavidin–biotin‐complex method and AEC was used as the colorant. There were red stains in transfected HEK293 cell, but not in control HEK293 cells (Fig. 1e). Immunofluorescence assay was used to detect expression of AFP and mtP53 and results showed that transfected HEK293 cell released orange fluorescence (Fig. 2f), whereas no fluorescence was observed in control HEK293 cells (Fig. 1d).
Figure 2.
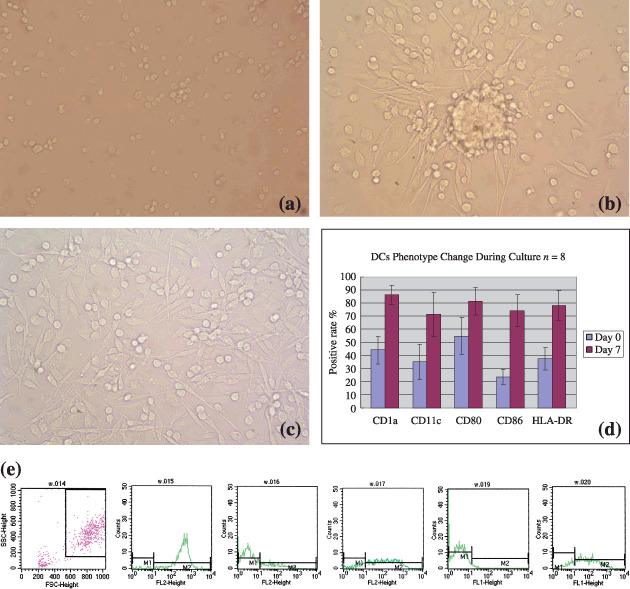
Dendritic cell (DC) generation and the morphology and phenotypical character of DCs during culture and results of flow cytometry. (a) DCs at day 0 were all small. (b) DCs at day 5, cells congregated and formed clusters. (c) DCs at day 7. (d and e) Flow cytometric analysis of DCs cultured from CD34+ cell‐enriched peripheral blood mononuclear cells harvested from hepatocellular carcinoma patients after mobilized with chemotherapy and granulocyte colony‐stimulating factor. (d) The status of five membrane molecules, CD1a, CD11c, CD80, CD86, and HLA‐DR, was 44.2 ± 10.7%, 35.1 ± 13.5%, 54.9 ± 14.2%; 23.7 ± 5.8%; and 37.5 ± 8.5%, respectively, at day 0; at day 7, the status was 86.2 ± 7.5%, 71.4 ± 16.8%, 81.3 ± 10.4%; 74.2 ± 12.4%; and 78.2 ± 11.3%, respectively (P < 0.05). (e) Flow cytometry curves of DCs cultured for 7 days. From left to right: selection gate, CD1a, CD11c, CD80, CD86, and HLA‐DR.
Phenotype of DCs derived from CD34+ cell‐enriched PBMCs. The amount of PBMCs of apheresis mobilized from HLA A2 +HCC patients by chemotherapy and G‐CSF was 45.37 ± 12.41 × 109 cells/L, in which the amount of CD34+ cells was about 0.2% (9.87 ± 3.93 × 104 cells/mL). The morphology of DCs changed daily following the culture (Fig. 2a–c). After 7 days of culture, the population of large dendritic‐like cells was 74.6 ± 17.3% and that of small lymphoid‐like cells was 25.4 ± 17.3%. The expression of surface molecules CD1a, CD11c, CD80, CD86, and HLA‐DR of dendritic cells assayed by flow cytometry during culture in eight samples were 44.2 ± 10.7%, 35.1 ± 13.5%, 54.9 ± 14.2%, 23.7 ± 5.8%, and 37.5 ± 8.5%, respectively, at first, and after culture with cytokines, the status was 86.2 ± 7.5%, 71.4 ± 16.8%, 81.3 ± 10.4%, 74.2 ± 12.4%, and 78.2 ± 11.3%, respectively, with statistical difference (P < 0.05) (Fig. 2d,e).
DCs pulsed with liposome‐coated pEGFP‐C3/AFP‐mtP53. A transfection rate of nearly 20% was achieved by prolonged incubation of PBMC‐derived DCs with lipofectamine‐coated pEGFP‐C3/AFP‐mtP53. Green fluorescence was detected in fused gene‐loaded DCs 24 h after transfection; no fluorescence was seen in the control group. (Fig. 3a,b). RT‐PCR assay showed that transfected DCs expressed mRNA of fused gene AFP‐mtP53 (Fig. 3c,d). The phenotype of DCs after transfection was slightly changed compared with that before transfection. However, this change had no statistical difference (Fig. 3e,f).
Figure 3.
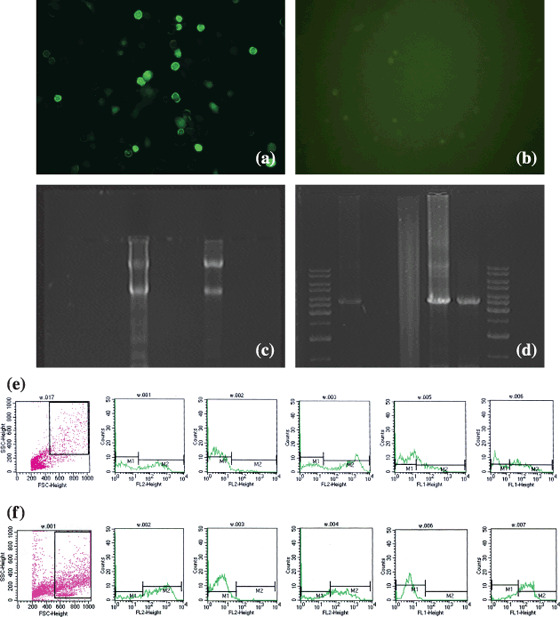
Photograph of dendritic cells (DCs) transfected with enhanced green fluorescent protein/α‐feroprotein–mutant P53 plasmid (pEGFP‐C3/AFP‐mtP53), reverse transcription–polymerase chain reaction (RT‐PCR) results of AFP‐P53 fused gene, and flow cytometry analyses pre‐ and post‐transfection. (a and b) Fluorescent microscopy photograph of DCs transfected with pEGFP‐C3/AFP‐mtP53 (×200). (a) DCs expressing green fluorescence, 24 h after transfection. (b) No fluorescence was observed in blank DCs. (c and d) RT‐PCR assay of mRNA of AFP‐mtP53 fused gene. (c) Total RNA extracted from transfected DCs. (d) Gel electrophoretic pattern of RT‐PCR results. Lanes 1–7: molecular weight (DNA ladder 1 kb); sample 1 (3.7 kb containing GFP, AFP, and p53); negative control; glyceraldehyde‐3‐phosphate dehydrogenase; positive control; sample 2; and molecular weight. (e and f) Flow cytometry analysis of DCs pre‐ and post‐transfection. (e) Phenotypes of DCs pre‐transfection. (f) Phenotypes of DCs post‐transfection. From left to right: selection gate, CD1a, CD11c, CD80, CD86, and HLA‐DR.
CTL response induced by DCs pulsed with fused gene. Eight samples of DCs, derived from PBMCs of HCC patients, were pulsed with different tumor Ags and DC‐induced Ag‐specific CTL responses were assayed in three hepatic cell lines, HepG2 (HLA A2 positive, AFP positive, P53 negative), SMMC7721 (HLA A2 positive, both AFP and P53 negative), and HMCC97 (HLA A2 positive, both AFP and mtP53 positive). The inhibitory rates of HepG2 cells in pEGFP‐C3/AFP‐mtP53, pEGFP‐C3/AFP, pEGFP‐C3/mtP53, pEGFP‐C3, and the control group were 51.6 ± 4.2, 48.3 ± 3.6, 32.5 ± 3.9, 28.1 ± 3.1, and 23.7 ± 2.7, respectively. The inhibitory rate of SMMC7721 cells were 23.1 ± 2.7, 21.7 ± 3.1, 18.5 ± 2.6, 20.2 ± 2.4, and 15.7 ± 3.3, respectively, and that of HMCC97 cells were 65.2 ± 4.8, 50.2 ± 4.2, 47.7 ± 3.8, 24.5 ± 2.6, and 26.5 ± 3.1, respectively (Table 1 and Fig. 4).
Table 1.
Cytotoxic T lymphocyte responses in different hepatic cancer cells
| Groups | Cells | |||
|---|---|---|---|---|
| n | Growth inhibition rate (%) | |||
| HepG2 | SMCC7721 | HMCC97 | ||
| pEGFP‐C3/AFP‐mtP53 | 8 | 51.6 ± 4.2 | 23.1 ± 2.7 | 65.2 ± 4.8 |
| pEGFP‐C3/AFP | 8 | 48.3 ± 3.6 | 21.7 ± 3.1 | 50.2 ± 4.2 |
| pEGFP‐C3/mtP53 | 8 | 32.5 ± 3.9 | 18.5 ± 2.6 | 47.7 ± 3.8 |
| pEGFP‐C3 | 8 | 28.1 ± 3.1 | 20.2 ± 2.4 | 24.5 ± 2.6 |
| Control | 8 | 23.7 ± 2.7 | 15.7 ± 3.3 | 26.5 ± 3.1 |
AFP, α‐fetoprotein; EGFP, enhanced green fluorescent protein; mtP53, mutant P53.
Figure 4.
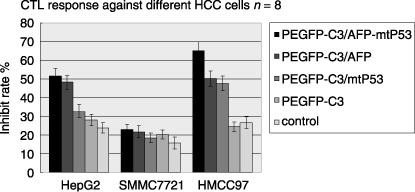
Dendritic cells (DCs) induced cytotoxic T lymphocyte responses against hepatocellular carcinoma cells. Left to right in each column, enhanced green fluorescent protein/α‐feroprotein–mutant P53 plasmid (pEGFP‐C3/AFP‐mtP53), pEGFP‐C3/AFP, pEGFP‐C3/mtP53, pEGFP‐C3, and control. The growth inhibitory rates of HepG2 cells were 51.6 ± 4.2, 48.3 ± 3.6, 32.5 ± 3.9, 28.1 ± 3.1, and 23.7 ± 2.7, respectively. The growth inhibitory rates of pEGFP‐C3/AFP‐mtP53 and pEGFP‐C3/AFP groups were higher than those of other groups with significant difference (P < 0.05). The growth inhibitory rates of SMMC7721 cells were 23.1 ± 2.7, 21.7 ± 3.1, 18.5 ± 2.6, 20.2 ± 2.4, and 15.7 ± 3.3, respectively. There was no statistical difference in any group (P > 0.05). The growth inhibitory rates of HMCC97 cells were 65.2 ± 4.8, 50.2 ± 4.2, 47.7 ± 3.8, 24.5 ± 2.6, and 26.5 ± 3.1, respectively. The rate of the pEGFP‐C3/AFP‐mtP53 group was the highest in all groups with significant difference compared with other groups (P < 0.05). The rates of the pEGFP‐C3/AFP and pEGFP‐C3/mtP53 groups were also higher than those of the pEGFP‐C3 and control groups (P < 0.05). Although the rate of the pEGFP‐C3/AFP group was higher than that of the pEGFP‐C3/mtP53 group, there was no statistical difference (P > 0.05).
AFP‐specific CTL responses were detected in HepG2 cells and HMCC97 cells expressing AFP Ag (P < 0.05). P53‐specific CTL responses were also detected in HMCC97 cells. SMMC7721 cells, expressing neither AFP nor P53, were refractory to CTL responses induced by DCs pulsed with either the fused gene or single genes. HepG2 cells also showed higher inhibition to DCs pulsed with the AFP gene and AFP‐mtP53 fused gene compared to DCs pulsed with the single mtP53 gene (P < 0.05). The inhibition of HMCC97 cells was detected in CTL responses induced by DCs pulsed with mtP53, AFP, and AFP‐mtP53 and a statistical difference was seen compared with the paralleled groups (P < 0.05). Furthermore, in HMCC97 cells, the highest inhibitory rate was seen in DCs pulsed with AFP‐mtP53 compared with DCs pulsed with a single gene (P < 0.05), which showed that both AFP and mtP53 Ag‐specific CTL responses were induced by DCs pulsed with the fused gene. Although the inhibitory rate for HMCC97 cells was higher in the AFP group than in the mtP53 group, there was no statistical difference (P > 0.05).
Discussion
HCC is a common malignant tumor worldwide, especially in Asia where it is generally associated with liver cirrhosis, frequently caused by chronic infection with hepatitis B and C. To maintain the clinical efficacy of surgical ablation is still a challenge. The lack of effective treatment modalities for inoperable multinodular HCC has led to the search for new therapeutic options, such as adoptive immunotherapy. DCs are professional presenting cells that could stimulate naive T lymphocytes to initiate innate and adoptive immunity. DC‐based immunotherapy and DC vaccines have been proved an active immunotherapy for malignant diseases both in vitro and in vivo.( 9 , 10 , 11 , 12 , 13 )
AFP is a transcriptionally regulated protein expressed by most HCC patients. It had been previously held that high plasma levels of this oncofetal protein could suppress body immune responses. However, further studies have shown that DCs could present AFP‐derived HLA‐A2 binding peptides to T cell repertories and induce complete immunity.( 34 , 35 ) We also previously proved that DCs were capable of inducing further differentiation into mature DCs after fusion with HepG2 cells or transfection with HepG2 cell total RNA to elicit specific T‐cell responses in vitro. DCs transfected with AFP mRNA could evoke CD4+ and CD8+ mediated T‐cell responses against HCC, and total RNA of HepG2 could elicit more effective Ag‐specific CTL responses than DCs fused with HepG2 cells, even if fusion loading was more efficient in priming the T helper type 1 response.( 30 , 31 , 32 )
Mutations of the P53 gene have been frequently observed in a variety of human malignancies including approximately 40–70% of HCC. It had been proven that P53‐derived HLA‐A2+ binding peptides were able to activate human T cells and the generated effector T cells are cytotoxic to human HLA‐A2+, P53+ tumor cells.( 36 , 37 , 38 ) In addition, DCs loaded with P53 peptides could induce P53‐specific cytotoxic T cells both in vivo and in vitro.( 39 , 40 )
Vaccination strategies using only one tumor Ag could impose selection pressure on the tumor, which could result in Ag‐loss variants. The tumor cells that lost expression of that certain tumor Ag might then escape from immune rejection.( 41 ) Therefore, DC vaccination with several different tumor Ags might be superior for priming more effective anti‐tumor immune responses.( 42 ) Based on these studies, we have postulated that loading DCs with the liposome‐coated AFP‐mtP53 fused gene could evoke the bi‐targeted CTL response of extensive activities against HCC. The fused gene was constructed by linking each single gene directly without an extra linker. Theoretically, it should have multiple epitopes, possibly even the structure could be reconstructed. The tridimensional structure of each protein could be influenced and some Ag epitopes may be lost. However, this would not apply to all epitopes because AFP and mtP53 are large protein molecules and, after digested by protease, only short Ag peptides are capable of binding with major histocompatibility complex molecules. This is quite different from other fused protein studies. In our study, immunocytochemical staining and immunofluorescence assays proved that HEK293 cells transfected with the AFP‐mtP53 fused gene could express AFP‐mtP53 simultaneously without different intracellular expression levels of each gene with maintained immunogenicity (Fig. 1). DCs pulsed with the fused gene could prime a bi‐targeted Ag‐specific CTL response against each Ag‐expressing hepatocellular cell line separately. There was a trend that CTL responses induced by AFP gene‐loaded DCs seemed more effective than that of mtP53 gene‐loaded DCs vaccine even without statistical significance. The most important finding of our study was that more intensive and extensive CTL responses were observed by DCs pulsed with the AFP‐mtP53 fused gene (Fig. 4). This finding is important and valuable for further immunotherapeutic treatment because of the lack of uniformity of tumor‐associated Ags in tumor cells. Compared to DC vaccines loaded with cancer cell lysate, total RNA, and tumor‐DC fused cells, DC vaccines pulsed with fused or combined tumor Ags might induce more extensive multitargeted antitumor immunity without the risk of autoimmune reaction.
In our study, we have employed chemotherapy and G‐CSF to mobilize peripheral blood hematopoietic stem cells for enrichment of CD34+ cells of HCC patients as a resource of DCs. The method provides the possibility of obtaining a large number of CD34+ cell‐enriched PBMCs for generating DCs. This strategy was as effective as generating DCs from several progenitor cells involving PBMCs,( 43 ) umbilical blood mononuclear cells,( 44 ) hematopoietic stem cells,( 45 ) and even leukemia cells.( 46 )
Successfully delivering DNA or tumor Ag gene into DCs remains problematic. Viral vehicles have a relatively higher transfection rate and might stimulate innate and adaptive immunity in vivo. But such stimulation frequently activates immature DCs regardless of reducing the immunostimulatory activity of the DC vaccine due to either immunodominance or immunoregulation of viral proteins.( 47 , 48 ) Liposome‐mediated nucleic acid delivery has been successfully used to transfer DNA or RNA into certain cells in vitro. In our study the transfection rate of HEK293 cells was approximately 70%. Considering the intake ability of DCs, it had been supposed that liposome‐mediated transfection should have a relatively higher transfection rate. However, the rate of transfecting plasmid DNA into DCs was much lower for limited differentiation and proliferation ability of mature DCs. Recently, cationic lipids, liposomes, and other adjuvants were reported to effectively deliver Ag epitopes and plasmid DNA into DCs and elicit specific antitumor immune responses in vivo.( 49 ) In addition, cationic liposomes alone could stimulate DCs, leading to the expression of co‐stimulatory molecules, CD80 and CD86, which are crucial to induce complete immunity.( 50 ) For these reasons and further studies, we have used liposome‐coated plasmids to deliver the AFP‐mtP53 fusion gene into DCs. In this study, when the incubation time of fused gene plasmid and liposome in DC culture media was prolonged, a transduction rate of approximately 20% was reached. The mechanisms of a relatively higher transfection rate might be the result of longer incubation time, but this still needs to be verified in further studies.
In conclusion, the idea of DCs pulsed with liposome‐coated Ag‐fused genes provides a new promising therapeutic model to induce more extensive multi‐targeted anti‐tumor immunity. Several aspects of vaccine optimization, including Ag selection, are still required for further studies. If the initially promising results presented here are confirmed, such DC‐based immunotherapy, adopting a multi‐Ag strategy, could serve as a novel therapeutic model for cancer.
Acknowledgments
This work was supported in part by the Teaching and Research Award for Outstanding Young Teachers in High Institutes (TRAOYT1999‐016), Ministry of Education, China. We thank Dr Liwen Li of Xijing Hospital and Dr Lei Shi of the First Hospital of Xian Jiaotong University for their technical support.
References
- 1. Schafer DF, Sorrell MF. Hepatocellular carcinoma. Lancet 1999; 353: 1253–7. [DOI] [PubMed] [Google Scholar]
- 2. Liu CL, Fan ST. Nonresectional therapies for hepatocellular carcinoma. Am J Surg 1997; 173: 358–65. [DOI] [PubMed] [Google Scholar]
- 3. Fuster J, Charco R, Llovet JM et al . Liver transplantation in hepatocellular carcinoma. Transpl Int 2005; 18: 278–82. [DOI] [PubMed] [Google Scholar]
- 4. Bruix J, Sala M, Llovet JM. Chemoembolization for hepatocellular carcinoma. Gastroenterology 2004; 127: S179–88. [DOI] [PubMed] [Google Scholar]
- 5. Su H, Chang JC, Xu SM et al . Selective killing of AFP‐positive hepatocellular carcinoma cells by adeno‐associated virus transfer of the herpes simplex virus thymidine kinase gene. Hum Gene Ther 1996; 7: 463–70. [DOI] [PubMed] [Google Scholar]
- 6. Habib NA, Ding SF, El‐Masry R et al . Preliminary report: the short‐term effects of direct P53 DNA injection in primary hepatocellular carcinomas. Cancer Detect Prev 1996; 20: 103–7. [PubMed] [Google Scholar]
- 7. Bui LA, Butterfield LH, Kim JY et al . In vivo therapy of hepatocellular carcinoma with a tumor‐specific adenoviral vector expressing interleukin‐2. Hum Gene Ther 1997; 8: 2173–82. [DOI] [PubMed] [Google Scholar]
- 8. William KD, Dongxia X, Elizabeth JS. Dendritic cell immunotherapy for the treatment of neoplastic disease. Biol Blood Marrow Transplant 2006; 12: 113–25. [DOI] [PubMed] [Google Scholar]
- 9. Schuler‐Thurner B, Schultz ES, Berger TG et al . Rapid induction of tumor‐specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide‐loaded monocyte‐derived dendritic cells. J Exp Med 2002; 195: 1279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holtl L, Zelle‐Rieser C, Gander H et al . Immunotherapy of metastatic renal cell carcinoma with tumor lysate‐pulsed autologous dendritic cells. Clin Cancer Res 2002; 8: 3369–76. [PubMed] [Google Scholar]
- 11. Fong L, Brockstedt D, Benike C et al . Dendritic cell‐based xenoantigen vaccination for prostate cancer immunotherapy. J Immunol 2001; 167: 7150–6. [DOI] [PubMed] [Google Scholar]
- 12. Ladhams A, Schmidt C, Sing G et al . Treatment of non‐resectable hepatocellular carcinoma with autologous tumor‐pulsed dendritic cells. J Gastroenterol Hepatol 2002; 17: 889–96. [DOI] [PubMed] [Google Scholar]
- 13. Maier T, Tun‐Kyi A, Tassis A et al . Vaccination of patients with cutaneous T‐cell lymphoma using intranodal injection of autologous tumor‐lysate–pulsed dendritic cells. Blood 2003; 102: 2338–44. [DOI] [PubMed] [Google Scholar]
- 14. Morisaki T, Matsumoto K, Onishi H et al . Dendritic cell‐based combined immunotherapy with autologous tumor‐pulsed dendritic cell vaccine and activated T cells for cancer patients: rationale, current progress, and perspectives. Hum Cell 2003; 16: 175–82. [DOI] [PubMed] [Google Scholar]
- 15. Kruger T, Schoor O, Lemmel C et al . Lessons to be learned from primary renal cell carcinomas: novel tumor antigens and HLA ligands for immunotherapy. Cancer Immunol Immunother 2005; 54: 826–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCarthy H, Ottensmeier CH, Hamblin TJ, Stevenson FK. Anti‐idiotype vaccines. Br J Haematol 2003; 123: 770–81. [DOI] [PubMed] [Google Scholar]
- 17. Bendandi M, Gocke CD, Kobrin CB et al . Complete molecular remissions induced by patient‐specific vaccination plus granulocyte‐monocyte colony‐stimulating factor against lymphoma. Nat Med 1999; 5: 1171–7. [DOI] [PubMed] [Google Scholar]
- 18. Timmerman JM, Czerwinski DK, Davis TA et al . Idiotype‐pulsed dendritic cell vaccination for B‐cell lymphoma: clinical and immune responses in 35 patients. Blood 2002; 99: 1517–26. [DOI] [PubMed] [Google Scholar]
- 19. Stevenson FK, Rice J, Zhu D. Tumor vaccines. Adv Immunol 2004; 82: 49–103. [DOI] [PubMed] [Google Scholar]
- 20. Murphy GP, Tjoa BA, Simmons SJ et al . Infusion of dendritic cells pulsed with HLA‐A2‐specific prostate‐specific membrane antigen peptides: a phase II prostate cancer vaccine trial involving patients with hormone‐refractory metastatic disease. Prostate 1999; 38: 73–8. [DOI] [PubMed] [Google Scholar]
- 21. Gong J, Avigan D, Chen D, Wu Z, Koido S, Kashiwaba M, Kufe D. Activation of antitumor cytotoxic T lymphocytes by fusions of human dendritic cells and breast carcinoma cells. Proc Natl Acad Sci USA 2000; 97: 2715–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Induction of tumor immunity and cytotoxic T lymphocyte responses using dendritic cells transfected with messenger RNA amplified from tumor cells. Cancer Res 2000; 60: 1028–34. [PubMed] [Google Scholar]
- 23. Ribas A, Butterfield LH, Hu B et al . Generation of T‐cell immunity to a murine melanoma using MART‐1‐engineered dendritic cells. J Immunother 2000; 23: 59–66. [DOI] [PubMed] [Google Scholar]
- 24. Reichardt VL, Brossart P, Kanz L et al . Dendritic cells in vaccination therapies of human malignant disease. Blood Rev 2004; 18: 235–43. [DOI] [PubMed] [Google Scholar]
- 25. Liao X, Li Y, Bonini C et al . Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high‐affinity tumor‐reactive cytotoxic T lymphocytes. Mol Ther 2004; 9: 757–64. [DOI] [PubMed] [Google Scholar]
- 26. Oh ST, Kim CH, Park MY et al . Dendritic cells transduced with recombinant adenoviruses induce more efficient anti‐tumor immunity than dendritic cells pulsed with peptide. Vaccine 2006; 24: 2860–8. [DOI] [PubMed] [Google Scholar]
- 27. Johnson PJ. Role of α‐fetoprotein in the diagnosis and management of hepatocellular carcinoma. J Gastroenterol Hepatol 1999; 14: S32–6. [DOI] [PubMed] [Google Scholar]
- 28. Butterfield LH, Ribas A, Meng WS et al . T‐cell responses to HLA‐A*0201 immunodominant peptides derived from α‐fetoprotein in patients with hepatocellular cancer. Clin Cancer Res 2003; 9: 5902–8. [PubMed] [Google Scholar]
- 29. Svane IM, Pedersen AE, Johnsen HE et al . Vaccination with P53‐peptide‐pulsed dendritic cells, of patients with advanced breast cancer: report from a phase I study. Cancer Immunol Immunother 2004; 53: 633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang L, Zhang H, Liu W et al . Specific antihepatocellular carcinoma T cells generated by dendritic cells pulsed with hepatocellular carcinoma cell line HepG2 total RNA. Cell Immunol 2005; 238: 61–6. [DOI] [PubMed] [Google Scholar]
- 31. Zhang HM, Zhang LW, Ren J, Fan L, Si XM, Liu WC. Induction of α‐fetoprotein‐specific CD4‐ and CD8‐mediated T‐cell response using RNA‐transfected dendritic cells. Cell Immunol 2006; 239: 144–50. [DOI] [PubMed] [Google Scholar]
- 32. Zhang HM, Zhang LW, Liu WC, Cheng J, Si XM, Ren J. Comparative analysis of DC fused with tumor cells or transfected with tumor total RNA as potential cancer vaccines against hepatocellular carcinoma. Cytotherapy 2006; 8: 580–8. [DOI] [PubMed] [Google Scholar]
- 33. Yang JY, Cao DY, Liu WC, Zhang HM, Teng ZH, Ren J. Dendritic cell generated from CD34+ hematopoietic progenitors can be transfected with adenovirus containing gene of HBsAg and induce antigen‐specific cytotoxic T cell responses. Cell Immunol 2006; 240: 14–21. [DOI] [PubMed] [Google Scholar]
- 34. Vollmer CM Jr, Eilber FC, Butterfield LH et al . α‐Fetoprotein‐specific genetic immunotherapy for hepatocellular carcinoma. Cancer Res 1999; 59: 3064–7. [PubMed] [Google Scholar]
- 35. Butterfield LH, Meng WS, Koh A et al . T cell responses to HLA‐A*0201‐restricted peptides derived from human α‐fetoprotein. J Immunol 2001; 166: 5300–8. [DOI] [PubMed] [Google Scholar]
- 36. Barfoed AM, Petersen TR, Kirkin AF, Thor SP, Claesson MH, Zeuthen J. Cytotoxic T‐lymphocyte clones, established by stimulation with the HLA‐A2 binding P5365‐73 wild type peptide loaded on dendritic cells in vitro, specially recognize and lyse HLA‐A2 tumour cells overexpressing the P53 protein. Scand J Immunol 2000; 51: 128. [DOI] [PubMed] [Google Scholar]
- 37. Ropke M, Hald J, Guldberg P et al . Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild‐type P53‐derived peptide. Proc Natl Acad Sci USA 1996; 93: 14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wurtzen PA, Claesson MH. A HLA‐A2 restricted human CTL line recognizes a novel tumor cell expressed P53 epitope. Int J Cancer 2002; 99: 568. [DOI] [PubMed] [Google Scholar]
- 39. Petersen TR, Buus S, Brunak S, Nissen MH, Sherman LA, Claesson MH. Identication and design of P53‐derived HLA‐A2‐binding peptides with increased CTL immunogenicity. Scand J Immunol 2001; 53: 357. [DOI] [PubMed] [Google Scholar]
- 40. Nikitina EY, Clark JI, Van Beynen J et al . Dendritic cells transduced with full‐length wild‐type P53 generate antitumor cytotoxic T lymphocytes from peripheral blood of cancer patients. Clin Cancer Res 2001; 7: 127–35. [PubMed] [Google Scholar]
- 41. Schreiber H. Wu TH, Nachman J, and Kast WM. Immunodominance and tumor escape. Semin Cancer Biol 2002; 12: 25. [DOI] [PubMed] [Google Scholar]
- 42. Palmowski MJ, Choi EM, Hermans IF et al . Competition between CTL narrows the immune response induced by prime‐boost vaccination protocols. J Immunol 2002; 168: 4391. [DOI] [PubMed] [Google Scholar]
- 43. Paquette RL, Hsu NC, Kiertscher SM et al . Interferon‐α and granulocyte‐macrophage colony‐stimulating factor differentiate peripheral blood monocytes into potent antigen‐presenting cells. J Leukoc Biol 1998; 64: 358–67. [DOI] [PubMed] [Google Scholar]
- 44. Bender A, Sapp M, Schuler G, Steinman RM, Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J Immunol Meth 1996; 196: 121–35. [DOI] [PubMed] [Google Scholar]
- 45. Cao H, Verge V, Baron C et al . In vitro generation of dendritic cells from human blood monocytes in experimental conditions compatible for in vivo cell therapy. J Hematother Stem Cell Res 2000; 9: 183–94. [DOI] [PubMed] [Google Scholar]
- 46. Vuillier F, Maloum K, Thomas EK, Jouanne C, Dighiero G, Scott‐Algara D. Functional monocyte‐derived dendritic cells can be generated in chronic lymphocytic leukaemia. Br J Haematol 2001; 115: 831–44. [DOI] [PubMed] [Google Scholar]
- 47. Miller G, Lahrs S, Pillarisetty VG, Shah BA, DeMatteo PR. Adenovirus infection enhances dendritic cell immunostimulatory properties and induces natural killer and T‐cell‐mediated tumor protection. Cancer Res 2002; 62: 5260–6. [PubMed] [Google Scholar]
- 48. Jonuleit H, Tuting T, Steitz J et al . Efficient transduction of mature CD83+ dendritic cells using recombinant adenovirus suppressed T cell stimulatory capacity. Gene Ther 2000; 7: 249–54. [DOI] [PubMed] [Google Scholar]
- 49. U’Ren L, Kedl R, Dow S. Vaccination with liposome – DNA complexes elicits enhanced antitumor immunity. Cancer Gene Ther 2006; 13 (11): 1033–44. [DOI] [PubMed] [Google Scholar]
- 50. Vangasseri DP, Cui Z, Chen W, Hokey DA, Falo LD, Huang L. Immunostimulation of dendritic cells by cationic liposomes. Mol Membr Biol 2006; 23: 385–95. [DOI] [PubMed] [Google Scholar]