

Abstract
The interferon (IFN) protein is a cytokine with pleiotropic biological functions that include induction of apoptosis, inhibition of angiogenesis and immunomodulation. We previously examined the two antitumor mechanisms, taking advantage of the fact that IFN‐α did not show cross‐species activity in its in vivo effect. In a nude mouse subcutaneous xenograft model using human pancreatic cancer cells, the expression of human IFN‐α effectively induced cell death of human pancreatic cancer cells, whereas mouse IFN‐α augmented antitumor immunity by stimulation of natural killer cells. Here, we extended our investigation to a syngeneic pancreatic cancer model, so that the integrated antitumor activity of local IFN‐α gene therapy, including the antiproliferative, proapoptotic, antiangiogeneic and immunomodulatory effects, can be evaluated rigorously. When a recombinant hamster IFN‐α adenovirus was injected into syngeneic subcutaneous tumors of hamster pancreatic cancer (PGHAM‐1) cells in Syrian hamster, tumor growth was significantly suppressed due to cell death and T cell‐ and natural killer cell‐mediated antitumor immunity. Moreover, in this case, tumor regression was observed not only for the injected subcutaneous tumors but also for the untreated tumors both in the peritoneal cavity and at distant sites. No significant systemic toxicity was observed in the treated hamsters. Moreover, the subcutaneous rechallenge of PGHAM‐1 cells was rejected in three of four cured hamsters from the initial tumor challenge. This study further demonstrated that local IFN‐α gene therapy is a promising therapeutic strategy for pancreatic cancer, due to its multiple mechanisms of antitumor activity and its lack of significant toxicity. (Cancer Sci 2007; 98: 455–463))
Pancreatic cancer remains one of the most formidable challenges in oncology today despite the recent advances in therapeutic and diagnostic modalities.( 1 , 2 , 3 ) It is expected to continue to be one of the five leading causes of cancer‐related death in Japan, with less than 5% of patients alive 5 years after diagnosis.( 4 ) Its high mortality is due to the high incidence of metastatic disease at the time of diagnosis, a rapidly progressive clinical course, and the lack of adequate systemic therapies.( 1 , 2 , 3 ) Pancreatic cancer therefore must often be considered as a rapidly progressive disease, and new therapeutic approaches that can effectively target the spread of this cancer in vivo are urgently needed.( 4 , 5 , 6 )
The interferon (IFN)‐α protein is a cytokine with pleiotropic biological properties that include antiviral activity, regulation of cell proliferation, induction of apoptosis and immunomodulation.( 7 ) The cytokine has been used worldwide for treatment of a variety of cancers including certain hematological malignancies such as chronic myeloid leukemia and solid tumors such as melanoma and renal carcinoma.( 7 , 8 ) However, clinical experiences with IFN protein therapy for many other solid cancers have generally not been encouraging.( 7 ) In the conventional regimen of IFN clinical trials, the recombinant IFN‐α protein is administered systemically through subcutaneous or intramuscular routes. However, because the protein is degraded rapidly in the blood circulation and only 0.01% of subcutaneously injected IFN‐α can reach the target organs,( 9 ) delivery of the IFN‐α protein might be insufficient or might result in an unsustainable level in the tumor site, which may be the cause of the diminished antitumor effect in previous clinical trials based on the IFN‐α protein. In contrast, because gene transfer allows an increased and sustained local concentration of IFN‐α in the target sites with minimal leakage of the cytokine into the systemic blood circulation,( 10 , 11 ) the use of IFN‐α is expected to improve the therapeutic effect and safety in the context of gene therapy. In fact, reports show that direct IFN‐α gene transfer into tumors suppresses growth of various cancers such as breast cancer, prostate cancer, renal cancer, hepatocellular carcinoma, basal cell carcinoma and leukemia.( 12 , 13 , 14 , 15 )
We reported for the first time that pancreatic cancer cells are characteristically susceptible to expression of the IFN‐α transgene, which effectively induces cell death in cancer cells.( 11 ) We also demonstrated that in addition to direct cytotoxicity in the injected site, gene transfer of IFN‐α augmented antitumor immunity by stimulation of natural killer (NK) cells in subcutaneous xenografts of human pancreatic cancer cells in nude mice.( 16 ) However, because the activity of NK cells is much elevated in nude mice compared with immune‐competent mice, one might argue that the xenograft model in nude mice is quite different from the human clinical setting. However, IFN can also induce the upregulation of major histocompatibility complex (MHC) class I expression, cause an increase in cytotoxic T lymphocyte activity and enhance the generation of T helper cells,( 17 ) the effect of which could not be evaluated in the nude mice system. Previous reports demonstrated that IFN‐α gene therapy induces antitumor immunity in tumor models of immune‐competent mice such as breast cancer, renal cancer, melanoma and squamous cell carcinoma.( 15 , 18 , 19 ) Furthermore, whether IFN‐α gene transfer can induce a significant antitumor effect is highly dependent on the kind of cancer, so it is crucial to use the target cancer in proper animal models as a preclinical study, and a pancreatic cancer model in mouse is not generally available. Therefore, in the present study, to explore the integrated antitumor activity of IFN‐α gene therapy, including direct cytotoxicity and immune activation in immune‐competent animals, we examined hamster IFN‐α gene delivery into hamster syngeneic pancreatic cancer tumors in Syrian hamsters. The hamster IFN‐α gene transduction showed significant inhibition of tumor growth not only at the injection sites but also at distant sites, which was attributed to an IFN‐α‐induced stimulation of systemic cellular immunity. The present study suggests that IFN‐α gene therapy is a promising therapeutic strategy for pancreatic cancer due to its multiple mechanisms of antitumor activity.
Materials and Methods
Cell lines. The PGHAM‐1 cell line was derived from an N‐nitrosobis(2‐oxopropyl)amine (BOP)‐induced pancreatic tumor in a Syrian hamster. The tumor showed a remarkable similarity to human pancreatic ductal adenocarcinoma and had a moderately to well‐differentiated histology. PGHAM‐1 contains a K‐ras point mutation at codon 12 (GGT to GAT),( 20 ) which is one of the most frequent types of K‐ras mutation in human pancreatic cancers. The HPD1NR and HPD2NR cell lines were established from a transplantable hamster ductal adenocarcinoma induced by N‐nitrosobis(2‐hydroxypropyl)amine (BHP).( 21 ) The DDT1‐MF2 is a Syrian hamster‐derived leiomyosarcoma cell line and was purchased from American Type Culture Collection (Manassas, VA, USA). PGHAM‐1 cells were cultured in RPMI‐1640 medium supplemented with 10% fetal bovine serum (FBS), and HPD1NR, HPD2NR and DDT1‐MF2 cells were cultured in Dulbecco's modified Eagle's medium with 10% FBS.
Construction of recombinant adenovirus vectors. The recombinant adenovirus vectors expressing hamster IFN‐α (AdCV‐hamIFN), alkaline phosphatase (AP) (AdCV‐AP) and no transgene (Ad‐ΔE1) were prepared as described previously.( 22 , 23 ) To construct AdCV‐hamIFN, a hamster IFN‐α cDNA fragment was amplified from Syrian hamster liver RNA by polymerase chain reaction (PCR), and the cDNA was cloned into the adenoviral shuttle plasmid downstream of the cytomegalovirus immediate early promoter. The recombinant adenovirus was prepared by a Cre/loxP‐mediated recombination of a sub360 adenoviral cosmid and the hamster IFN‐α plasmid. The sub360 cosmid contains an Ad5 genome with an entire deletion of the E1 and a partial deletion of the E3 regions.( 23 ) A cesium chloride‐purified virus solution was desalted using a sterile Bio‐Gel P‐6 DG chromatography column (Econopac DG 10; Bio‐Rad, Hercules, CA, USA). Viral preparations were confirmed to be free of E1+ adenovirus by PCR assay.( 24 )
In vitro growth analysis. The PGHAM‐1 cells were seeded at 2 × 103 per well in 96‐well plates and infected with AdCV‐hamIFN or Ad‐ΔE1 at multiplicity of injections of 30, 100 and 300. The cell numbers were assessed by a colorimetric cell viability assay using a water‐soluble tetrazolium salt (Tetracolor One; Seikagaku Corp., Tokyo, Japan) 5 days after infection. The absorbance was determined by spectrophotometry using a wavelength of 450 nm with 595 nm as a reference. The assays (carried out in eight wells) were repeated a minimum of two times and the mean ± SD was plotted. The data were expressed as the relative cell growth (OD450 of AdCV‐hamIFN‐infected cells/OD450 of Ad‐ΔE1‐infected cells).
Annexin V assay and cell cycle analysis. Cultured cells were infected with AdCV‐hamIFN or AdCV‐AP in a 60‐mm dish and treated with 2 mM ethylenediaminetetracetic acid at day 3, followed by staining with Annexin‐V–fluorescein‐isothiocyanate (Medical and Biological Laboratories Co., Nagoya, Japan), which detects the phosphatidylserine of inverted plasma membranes. To analyze the DNA contents, cultured cells were infected with AdCV‐hamIFN or Ad‐ΔE1 in a 60‐mm dish, followed by staining with propidium iodide (PI; Sigma, St Louis, MO, USA). The cells were examined by flow cytometry using a FACScan system (BD Biosciences, San Jose, CA, USA).
Reverse transcription–polymerase chain reaction of MHC class I antigen. To examine the expression of MHC class I α‐chain antigen on PGHAM‐1 cells, reverse transcription (RT)‐PCR amplification was carried out using total RNA from the cells, which were infected with AdCV‐hamIFN or Ad‐ΔE1 for 2 days, in a 50‐µL PCR mixture containing 1.5 mM MgCl2, 0.2 mM dNTPs, 1 IU recombinant Taq DNA polymerase and the following primer set: MHC class I (Hm‐1‐a haplotype) upstream (5′‐CGCTGCGGTATTTCTACATC‐3′) and downstream (5′‐AAAGGCACATGTGACCCATC‐3′) primers; and β‐actin upstream (5′‐GACTACCTCATGAAGATCCT‐3′) and downstream (5′‐GCGGATGTCCACGTCACACT‐3′) primers. In total, 35 cycles (β‐actin: 25 cycles) of the PCR were carried out at 95°C for 30 s, 61°C for 30 s and 72°C for 1 min. The PCR products were fractionated on a 2.0% agarose gel and visualized by ethidium bromide staining under ultraviolet transillumination.
PCR and RT‐PCR of the hamster IFN‐α gene. To detect the IFN‐α DNA of AdCV‐hamIFN in the injected tumors, DNA was extracted from tumors at 3, 7, 14, 21, 28 and 35 days after the injection of AdCV‐hamIFN or Ad‐ΔE1 by the Agglutination Partition method (SepaGene; Sanko Junyaku Co., Tokyo, Japan). PCR amplification was carried out using DNA from the tumors in a 50‐µL PCR mixture with the following primer set: AdCV‐hamIFN upstream (5′‐AAATGGGCGGTAGGCGTGTA‐3′) and downstream (5′‐TTCCTCTCTCAGGTACACAGTG‐3′) primers, which detect only IFN‐α DNA of AdCV‐hamIFN and not the endogenous IFN‐α gene. To examine the expression of IFN‐α on the vector‐injected tumors, RT‐PCR amplification was carried out using total RNA from the tumors in a 50‐µL PCR mixture with the following primer set: IFN‐α upstream (5′‐GATGAGCTACTGGTCAACCTG‐3′) and downstream (5′‐TTCCTCTCTCAGGTACACAGTG‐3′) primers. In total, 30 cycles (β‐actin: 25 cycles) of the PCR were carried out at 95°C for 30 s, 60°C for 30 s and 72°C for 1 min. The PCR products were fractionated on a 2.0% agarose gel.
Animal models. Four‐week‐old female Syrian hamsters were obtained from Japan SLC (Hamamatsu, Japan) and kept in a specific pathogen‐free environment. A syngeneic tumor model was established by injecting the PGHAM‐1 or DDT1‐MF2 cell suspensions (100 µL of 1 × 108 cells) subcutaneously into the right flank. When the tumor mass was established (∼10 mm in diameter), 100 µL (1.0 × 109 plaque forming units [pfu]) of a viral solution was injected into the tumor with a 26‐gauge hypodermic needle, and the animals were observed for tumor growth. In our previous report using a subcutaneous tumor model in nude mouse, the injection of 1 × 108 pfu of virus into a tumor ∼5 mm in diameter significantly suppressed tumor growth.( 16 ) In this study, the diameter of the subcutaneous tumor in the hamster was ∼10 mm at the time of virus injection. Because the tumor volumes in the hamster model were approximately eight‐fold those in the mouse model, we injected 10‐fold virus particles into the subcutaneous tumors. The short (r) and long (l) diameters of the tumors were measured and the tumor volume of each was calculated as (r2 × l)/2. Tumor volumes were presented as the mean ± SD. For a distant metastasis treatment model, 1 × 108 PGHAM‐1 cells were injected subcutaneously into the right flank at day 0. When the tumor mass was established, 1 × 108 PGHAM‐1 cells were injected simultaneously into the left flank at day 4. One day later, 1.0 × 109 pfu of the viral solution was injected once into the subcutaneous tumor on the right flank, and the animals were observed for tumor growth on both flanks, and for survival. For a peritoneal dissemination treatment model, 1 × 108 cells of PGHAM‐1 cell suspensions were injected subcutaneously into the right flank at day 0. When the tumor mass on the right flank was established, 1 × 106 cells of PGHAM‐1 cell suspensions were injected into the peritoneal cavity at day 10. One day later, 1.0 × 109 pfu of the viral solution were injected once into the subcutaneous tumor. Fourteen days after the PGHAM‐1 injection, the hamsters were killed and examined for peritoneal dissemination. Histological sections (3 µm) were made from the brain, lung, liver, heart, pancreas, spleen, kidney, stomach, small intestine, colon, ovary and skeletal muscle and analyzed for the presence of tumors and for any pathological changes. The remaining animals were observed for survival. Animal studies were carried out according to the Guidelines for Animal Experiments, drawn up by the Committee for Animal Experiments of the National Cancer Center, which meet the ethical standards required by law and the guidelines about experimental animals in Japan.
Cytotoxicity assays. Using 24‐well plates (Nunc A/S, Roskilde, Denmark), responder splenocytes (5 × 106 cells/mL) were cultured with 30 Gy‐irradiated PGHAM‐1 stimulators (3–4 × 105/mL) in complete RPMI containing 50 µM 2‐mercaptoethanol for 5 days. The responder cells were then collected and used as effector cells in a 4‐h chromium release assay against the indicated target cells. Indicated target cells were labeled by combining 5 × 106 cells with 185 MBq51Cr (PerkinElmer Japan Co., Kanagawa, Japan) in a total volume of 0.2 mL of complete RPMI for 1 h at 37°C, followed by three washes with plain RPMI. For a chromium release assay, effector cells were mixed with 2 × 104 target cells in a total volume of 0.2 mL of complete RPMI in 96‐well round‐bottom plates (Becton Dickinson, Franklin Lakes, NJ, USA). Supernatants were harvested with the Skatron harvesting system (Skatron, Sterling, VA, USA) and counted in a gamma counter (Packard Bioscience Company, Meriden, CT, USA). Percentage cytotoxicity was calculated as:
| ([experimental cpm − spontaneous cpm]/[maximum cpm − spontaneous cpm]) × 100. |
Spontaneous cpm was obtained from targets cultured in medium alone, and maximum cpm was obtained from targets incubated in 1% Nonidet P‐40. Each assay was carried out in triplicate.
Histology. At 14–21 days after the injection of viral solutions, the hamsters were killed and subcutaneous tumors were collected. The tumors were fixed with formalin, and sections were stained with hematoxylin–eosin or processed for immunohistochemistry with antimouse CD3ɛ antibody (sc‐1127; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or antimouse NK antibody (PK136; BD Biosciences). We concluded that both antibodies react with the hamster immune cells based on the similarity of staining patterns and relative percentages in the hamster spleen to those observed in mice. Bilbo et al. also reported that the same antimouse NK antibody shows cross‐reactivity with hamster NK cells.( 25 ) Immunostaining was carried out using the streptoavidin–biotin–peroxidase complex (Nichirei, Tokyo, Japan). Parallel negative controls without primary antibodies were run in all cases. The sections were counterstained with methylgreen, and the cells staining positive were counted in 10 high‐power fields (×400) under the microscope.
Measurement of hemoglobin content. The tumors were excised 3 days after the intratumoral injection of AdCV‐hamIFN or Ad‐ΔE1, and homogenized in distilled water. After centrifugation at 2500g for 20 min at 4°C, we determined the hemoglobin concentration of the supernatant using a hemoglobin assay kit (Hemoglobin B‐test; Wako Pure Chemical Industries, Osaka, Japan).
Statistical analysis. Comparative analysis of in vivo tumor volume and in vitro cell proliferation was carried out using Student's t‐test; mouse survival was analyzed by Kaplan–Meier survival plot followed by a Logrank (Mantel–Cox) test. Differences were considered statistically significant when the P‐value was less than 0.05.
Results
Cytotoxic effect of interferon‐α gene transduction into hamster pancreatic cancer cells. To study whether expression of the hamster IFN‐α gene effectively inhibits cell growth, PGHAM‐1 cells were infected with the AdCV‐hamIFN adenoviral vector. The cell lysate of AdCV‐hamIFN‐infected PGHAM‐1 cells showed an elevated level of 2′,5′‐oligoadenylate synthetase, which is regulated by type I IFN and induces growth suppression and cell death (Fig. 1a). The growth of PGHAM‐1 cells infected with AdCV‐hamIFN was significantly suppressed compared with cells infected with Ad‐ΔE1 (Fig. 1b). The Annexin‐V and TdT‐mediated dUTP‐biotin nick end labeling assays showed that the infection of AdCV‐hamIFN induced apoptotic cell death in PGHAM‐1 cells (Fig. 1c,d). The flow cytometry of AdCV‐hamIFN‐infected PGHAM‐1 cells stained with PI showed that IFN‐α significantly increased the proportion of cells in G0/G1 phase and the subdiploid fraction of apoptosis compared with control virus infection (Fig. 1e).( 17 ) The fractions of cells in the S and G2/M phases in AdCV‐hamIFN‐infected cells tended to decrease compared with those in Ad‐ΔE1‐infected cells. The results suggested that IFN‐α gene transduction induces cell cycle arrest at G0/G1 and apoptotic cell death in PGHAM‐1 cells.
Figure 1.
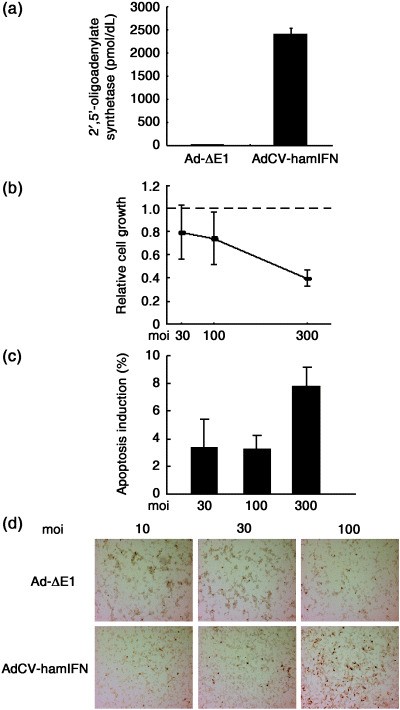
Cytotoxic effect of interferon (IFN)‐α expression in PGHAM‐1 cells. Cells were infected with AdCV‐hamIFN or control vector. (a) Induction of 2‐5AS in AdCV‐hamIFN‐infected PGHAM‐1 cells. 2’,5’‐oligoadenylate synthetase expression was determined by radioimmunoassay. (b) The growth suppression of hamster IFN‐α‐transduced PGHAM‐1 cells. The cells were infected with AdCV‐hamIFN or Ad‐ΔE1 at multiplicity of injections of 30, 100 and 300. Cell growth was determined by cell proliferation assay 5 days after infection. (c) Apoptotic cell death in AdCV‐hamIFN‐infected PGHAM‐1 cells. Cells were infected with AdCV‐hamIFN or AdCV‐AP at moi of 30, 100 and 300, and apoptotic cells were analyzed by Annexin‐V assay 3 days after infection. The data were expressed as specific cell death (%) (cell death fraction induced by AdCV‐hamIFN – that by AdCV‐AP). The assays were carried out in triplicate, and the mean ± SD was plotted. Each result is representative of at least three independent experiments. (d) TdT‐mediated dUTP‐biotin nick end labeling staining of PGHAM‐1 cells. PGHAM‐1 cells were infected with AdCV‐hamIFN or Ad‐ΔE1 at moi of 10, 30 and 100. Brown cells are TUNEL positive. (e) Cells in the cell cycle phases. PGHAM‐1 cells were infected with AdCV‐hamIFN or Ad‐ΔE1 at moi of 30, 100 and 300, and DNA contents were analyzed by staining with propidium iodide 3 days after infection. The data were expressed as the relative proportion of cells in the cell cycle phases (%) (fractions induced by AdCV‐hamIFN – those by Ad‐ΔE1). The assays were carried out in triplicate, and the mean ± SD was plotted. *P ≤ 0.05 for cells treated with AdCV‐hamIFN versus cells treated with Ad‐ΔE1. (f) The growth suppression of hamster IFN‐α‐transduced HPD1NR and HPD2NR cells. The cells were infected with AdCV‐hamIFN or Ad‐ΔE1 at moi of 1 and 3, as adenovirus infection was toxic to the cells. Cell growth was determined by cell proliferation assay for 5–6 days after infection.
Furthermore, we added an examination of the antiproliferative effect of IFN‐α gene transfer in two other hamster pancreatic cancer cell lines. As shown in Fig. 1f, infection of AdCV‐hamIFN resulted in 30–50% inhibition of cell growth in the HPD1NR and HPD2NR cell lines.
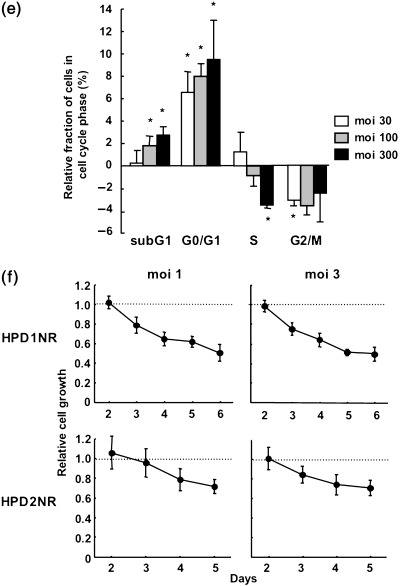
Direct antitumor effect of hamster IFN‐α gene transduction into PGHAM‐1 tumors. First, we examined whether hamster IFN‐α induces the expression of MHC class I α‐chain on PGHAM‐1 cells in vitro. RT‐PCR analysis showed that the injection of AdCV‐hamIFN upregulated MHC class I expression compared with the mock or Ad‐ΔE1 vectors (Fig. 2a). Then, the in vivo antitumor effect of hamster IFN‐α gene transduction was examined in PGHAM‐1 subcutaneous tumors. The intratumoral injection of AdCV‐hamIFN showed remarkable tumor‐suppressive effects, whereas the control adenovirus failed to produce any specific therapeutic response (Fig. 2b). An in vitro cytotoxicity assay was carried out to assess antitumor cytotoxic T‐lymphocyte responses induced by IFN‐α gene transfer in the hamsters. IFN‐α gene transfer significantly enhanced the cytotoxic activity of splenocytes for PGHAM‐1 cells but not for DDT1‐MF2 cells (Fig. 2c).
Figure 2.
Direct antitumor effect of hamster interferon (IFN)‐α gene transduction into PGHAM‐1 subcutaneous tumors. (a) Reverse transcription (RT) –polymerase chain reaction (PCR) analysis of major histocompatibility complex class I gene expression on PGHAM‐1 cells infected with AdCV‐hamIFN or Ad‐ΔE1. β‐actin was used as an internal control. Mock means that nothing was injected into the tumor. (b) Growth inhibition of subcutaneous tumors injected with AdCV‐hamIFN. When the PGHAM‐1 subcutaneous tumor was established on the right flank, the tumor was injected once with 1.0 × 109 plaque forming units of AdCV‐hamIFN or Ad‐ΔE1 (n = 6). *P < 0.05 for tumors treated with AdCV‐hamIFN versus tumors treated with Ad‐ΔE1. (c) In vitro cytotoxicity assay of AdCV‐hamIFN‐injected mice. Splenocytes were collected from each group of mice (n = 3–4 per group) 10 days after the vector injection and were evaluated for their cytotoxicity against PGHAM‐1 and DDT1‐MF2 cells after stimulation with irradiated PGHAM‐1 cells. (d) PCR detection of the IFN‐α gene in tumors. DNA was extracted from tumors at 3, 7, 14, 21, 28 and 35 days after injection of AdCV‐hamIFN or Ad‐ΔE1, and adenoviral IFN‐α DNA was amplified by PCR with vector‐specific primers (upper panel). The endogenous IFN‐α gene was also amplified with IFN‐α primers (lower panel). (e) RT‐PCR detection of IFN‐α expression in the tumors. RNA was extracted from tumors at 3, 14, 21, 28 and 35 days after injection of viruses, and the expression of IFN‐α mRNA was analyzed by RT‐PCR with IFN‐α primers (upper panel). The β‐actin gene was also amplified as an internal control (lower panel).
We examined the presence of adenoviral IFN‐α DNA and expression of the IFN‐α gene in the AdCV‐hamIFN‐injected tumors by PCR. The adenoviral IFN‐α DNA was detected by vector‐specific primers until 21 days after injection (Fig. 2d). The mRNA of IFN‐α was detected in the AdCV‐hamIFN‐injected tumors until 14 days after injection, whereas IFN‐α expression was detected at day 3 and disappeared at day 14 in Ad‐ΔE1‐injected tumors (Fig. 2e), which might be an endogenous response to a viral infection. IFN‐α expression was detected again at days 28 and 35 in the AdCV‐hamIFN‐injected tumors, in spite of the lack of vector‐derived IFN‐α DNA. The infiltrated immune cells in the tumors might express IFN‐α.
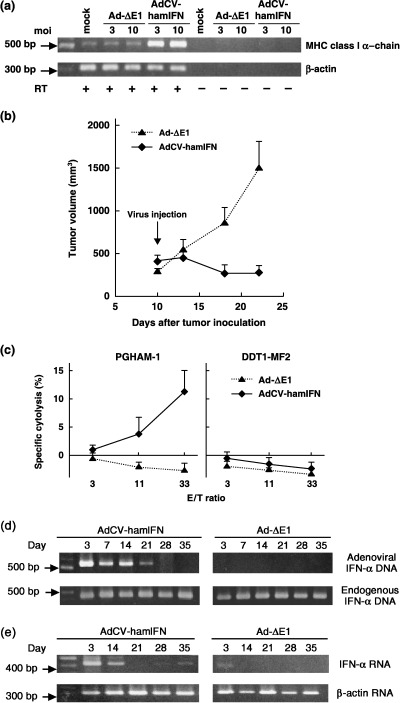
We then infected the leiomyosarcoma cell line (DDT1‐MF2) with AdCV‐hamIFN to examine the effect of IFN‐α on other cancer cells. The in vitro cell growth inhibition was less than 30% (Fig. 3a) and in the subcutaneous tumor model, IFN‐α gene transfer showed some tumor growth inhibition, but it was less than that in PGHAM‐1 tumors (Fig. 3b). To examine the gene transduction efficiency into PGHAM‐1 and DDT1‐MF2 cells, the cells were infected with adenovirus‐expressing green fluorescent protein (GFP) in vitro. Flow cytometry showed that the proportion of GFP‐positive cells was 48.3, 49.8 and 73.9% for PGHAM‐1 cells at moi 30, 100 and 300, respectively, whereas for DDT1‐MF2 the respective values were 33.7, 63.0 and 76.1, suggesting that the infectivity of type 5 adenovirus to DDT1‐MF2 is rather better than to PGHAM‐1. The results suggested that IFN‐α gene transfer could elicit a strong antitumor effect against pancreatic cancer compared with leiomyosarcoma.
Figure 3.
Hamster interferon (IFN)‐α gene transduction into DDT1‐MF2 cells. (a) Growth suppression of DDT1‐MF2 cells in vitro. The cells were infected with AdCV‐hamIFN or Ad‐ΔE1 at multiplicity of injections of 3, 10 and 30, as adenovirus infection was toxic to the cells. Cell growth was determined by cell proliferation assay for 5 days after infection. (b) Antitumor effect of hamster IFN‐α gene transduction into DDT1‐MF2 subcutaneous tumors. The cells were injected subcutaneously into the right flank of hamsters, and then 1.0 × 109 plaque forming units of viral solution were injected once into the tumor (n = 6). The animals were observed for tumor growth. *P < 0.05 for tumor volumes of AdCV‐hamIFN‐treated hamsters versus those of Ad‐ΔE1‐treated hamsters.
No significant systemic toxicity after intratumoral injection of AdCV‐hamIFN. To characterize the potential toxicity of local IFN‐α gene therapy, the animals were killed 8 days after the intratumoral injection of an adenovirus. All treated hamsters looked healthy during the course of the experiment. All of the selected serum biochemical markers were within normal limits. For AdCV‐hamIFN‐treated and Ad‐ΔE1‐injected hamsters these were: albumin, 2.43 ± 0.15 and 2.5 ± 0.2 g/dL; AP, 911 ± 211 and 897 ± 673 IU/L; amylase, 8733 ± 820 and 8083 ± 1303 IU/L; blood urea nitrogen, 28.3 ± 2.6 and 27.6 ± 2.2 mg/dL; creatinine, 0.19 ± 0.01 and 0.2 ± 0.01 mg/dL; AST, 46.6 ± 4.5 and 59.3 ± 22.9 IU/L; ALT, 42 ± 20 and 46 ± 24 IU/L; LDH, 284 ± 74 and 349 ± 168 IU/L; and total bilirubin, 0.05 ± 0.01 and 0.05 ± 0.01 mg/dL. Furthermore, there were no pathological changes in major organs in any of the treated animals, suggesting that systemic toxicity of local IFN‐α gene therapy is minimal.
Suppression of tumors at distant sites by intratumoral injection of AdCV‐hamIFN. We next examined whether hamster IFN‐α gene transduction has an effect on untreated tumors at distant sites. PGHAM‐1 cells were injected into both flanks of the hamsters, and when tumors were established, 1 × 109 pfu of AdCV‐hamIFN was injected once into the tumor on the right flank. A regression of both the treated tumor on the right flank and the untreated tumor on the opposite flank was observed, whereas injection of control Ad‐ΔE1 did not affect the growth of tumors on either flank (Fig. 4a). The treatment with AdCV‐hamIFN resulted in a statistically significant improvement (P < 0.05) in the survival of treated mice compared with the control Ad‐ΔE1‐treated animals (Fig. 4b).
Figure 4.
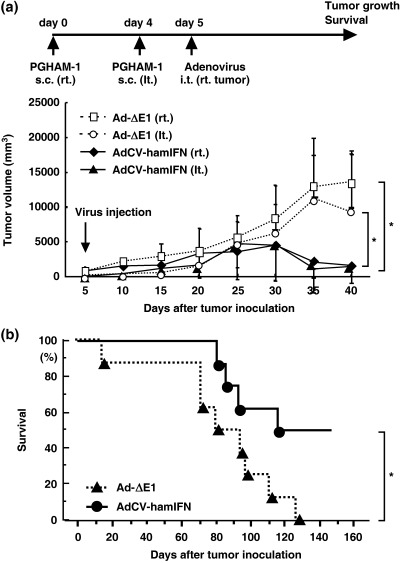
Suppression of contralateral subcutaneous tumors by intratumoral injection of AdCV‐hamIFN. The PGHAM‐1 cells were injected subcutaneously into both flanks, and then 1.0 × 109 plaque forming units of viral solution were injected once into the right tumor (n = 8). The animals were observed for tumor growth and survival. (a) Tumor volumes on both flanks. *P < 0.01 for tumor volumes of AdCV‐hamIFN‐treated hamsters versus those of Ad‐ΔE1‐treated hamsters. (b) Survival curves of hamsters. *P < 0.05 for survival rate of AdCV‐hamIFN‐treated hamsters versus that of Ad‐ΔE1‐treated hamsters.
Immunohistochemical staining showed that significant numbers of CD3‐positive cells and NK cells infiltrated into the vector‐injected and contralateral untreated tumors in AdCV‐hamIFN‐treated hamsters, whereas those cells were sparse in the subcutaneous tumors of Ad‐ΔE1‐treated hamsters (Fig. 5a). The average CD3‐positive cell counts in 10 representative high‐power view fields (×400) were: tumors injected with AdCV‐hamIFN, 23.4 ± 8.2; contralateral untreated tumors of AdCV‐hamIFN‐treated hamsters, 33.8 ± 13.4; tumors infected with Ad‐ΔE, 16.7 ± 3.9; and contralateral untreated tumors of Ad‐ΔE1‐injected hamsters, 4.1 ± 4.1. Similarly, the average of NK cell counts were: tumors injected with AdCV‐hamIFN, 69.1 ± 17.4; contralateral untreated tumors of AdCV‐hamIFN‐treated hamsters, 31.4 ± 13.6; tumors infected with Ad‐ΔE1, 5.3 ± 5.2; and contralateral untreated tumors of Ad‐ΔE1‐injected hamsters, 3.3 ± 2.4 (Fig. 5b). The results suggested that both T cells and NK cells are effectors that induce antitumor immunity. Although RT‐PCR analysis showed that PGHAM‐1 cells express MHC class I antigens in in vitro culture, MHC class I expression may be heterogeneous in an in vivo tumor mass.
Figure 5.
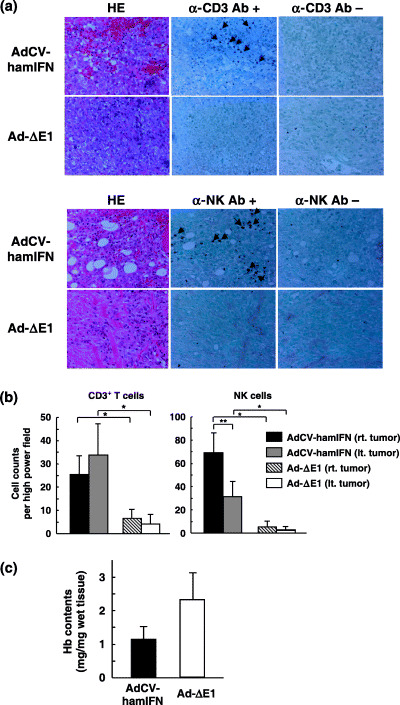
CD3+ T cells and natural killer (NK) cells in subcutaneous tumors. (a) The immunohistochemical staining of PGHAM‐1 tumors. At 14–21 days after intratumoral injection of AdCV‐hamIFN or Ad‐ΔE1, the subcutaneous tumors were fixed with formalin for hematoxylin–eosin (HE) staining, and immunohistochemistry with anti‐CD3ɛ (upper panel) or anti‐NK cells (lower panel) (×400). Ab, antibody. (b) Count of CD3+ T cells and NK cells. The CD3‐positive and NK cells were counted under a microscope in 10 high‐power fields (×400), and the mean ± SD are presented. *P < 0.01 for cell numbers of AdCV‐hamIFN‐treated hamster versus those of Ad‐ΔE1‐treated hamster. **P < 0.01 for cell numbers of right vector‐injected tumors versus left tumors in AdCV‐hamIFN‐treated hamsters. (c) Hemoglobin contents of the subcutaneous tumors. At 3 days after injection of viruses, the tumors were excised and the hemoglobin contents measured as a parameter for angiogenesis.
We next measured the hemoglobin content as a parameter of angiogenesis in the subcutaneous tumors 3 days after the virus injection. The hemoglobin content was reduced in the tumors injected with AdCV‐hamIFN compared with those injected with Ad‐ΔE1 (Fig. 5c). However, it was not determined whether the loss of vessels was caused indirectly by the apoptosis‐ and immune‐mediated marked cell death or directly by the antiangiogeneic activity of IFN‐α.
As another model of distant metastasis, we injected PGHAM‐1 cells into the peritoneal cavity following injection into the right flank. Again, when a tumor mass was established on the right flank, 1 × 109 pfu of adenovirus were injected once into the tumor. In the Ad‐ΔE1‐injected hamsters, tumor nodules were disseminated on the mesentery and pancreas, whereas AdCV‐hamIFN significantly suppressed peritoneal dissemination in the hamsters (Table 1) without significant changes in blood chemistry. Histological examination at day 28 revealed no pathological change in major organs of the vector‐infected hamsters (data not shown). Furthermore, treatment with AdCV‐hamIFN significantly improved survival of the treated mice compared with Ad‐ΔE1 (P < 0.01) (Fig. 6). All of the dead animals were confirmed to have disseminated tumors in the peritoneal cavity. These data suggested that the antitumor effect of local IFN‐α gene therapy is not limited to a locally injected tumor site but that it can induce systemic immunity against pancreatic cancer cells.
Table 1.
Peritoneal tumors after intratumoral injection of recombinant adenovirus vectors expressing hamster interferon‐α into subcutaneous tumors in hamsters
| Adenovirus (1 × 109 pfu) | Hamster no. | Tumor on the mesentery | Tumor on the pancreas |
|---|---|---|---|
| 1 | ++ | – | |
| 2 | ++ | – | |
| 3 | ++ | – | |
| 4 | – | + | |
| 5 | + | + | |
| Ad‐ΔE1 | 6 | ++ | + |
| 7 | + | – | |
| 8 | + | – | |
| 9 | + | – | |
| 10 | ++ | – | |
| 11 | – | – | |
| 12 | – | – | |
| 13 | – | – | |
| 14 | – | – | |
| AdCV‐hamIFN | 15 | – | – |
| 16 | – | – | |
| 17 | – | – | |
| 18 | ++ | – | |
| 19 | – | – | |
| 20 | – | – |
+, ≤10 in number; ++, >10 in number. pfu, plaque forming units. No. of hamsters with tumor/total: 10/10 in Ad‐ΔE1; 1/10 in AdCV‐hamIFN.
Figure 6.
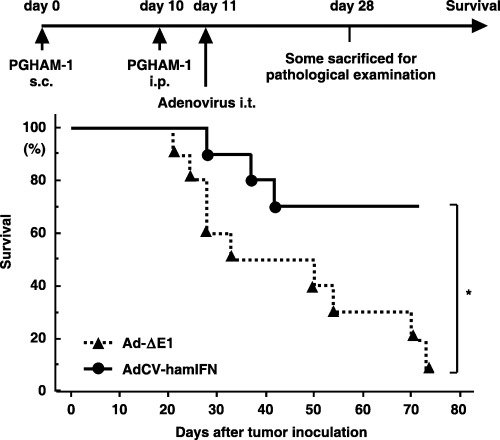
Suppression of intraperitoneal tumors by intratumoral injection of AdCV‐hamIFN. When the PGHAM‐1 subcutaneous tumor was established on the right flank, 2 × 106 cells of PGHAM‐1 cell suspensions were injected into the peritoneal cavity and, 1 day later, 1.0 × 109 plaque forming units of viral solution were injected once into the subcutaneous tumor. AdCV‐hamIFN, n = 10; Ad‐ΔE1, n = 10. *P < 0.01 for survival rate of AdCV‐hamIFN‐treated hamsters versus that of Ad‐ΔE1‐treated hamsters.
Tumor‐specific immunity induced by IFN‐α gene transfer is long lasting in vivo. The initial PGHAM‐1 subcutaneous tumors disappeared in several hamsters treated with AdCV‐hamIFN. To examine how long tumor‐specific immunity lasts in vivo, four hamsters that survived the initial PGHAM‐1 challenge with complete tumor remission were again inoculated with PGHAM‐1 cells on the right flank and DDT1‐MF2 cells on the left flank 3 months after the injection of AdCV‐hamIFN. The DDT1‐MF2 cells formed a tumor mass in all hamsters, whereas PGHAM‐1 cells were rejected in three of four hamsters. This finding demonstrated that the tumor‐specific immunity induced by IFN‐α gene therapy is potentially long lasting in the immune‐competent animals.
Discussion
It has been reported that the antitumor responses caused by IFN‐α gene therapy manifest three aspects: direct antiproliferative effect,( 12 , 13 , 26 , 27 , 28 ) stimulation of antitumor immunity,( 15 , 18 , 19 ) and antiangiogenic activity.( 29 , 30 ) Our previous reports using a human pancreatic cancer xenograft model in nude mice showed that the recombinant IFN‐α adenovirus caused direct apoptosis induction and NK cell‐mediated cytolysis of pancreatic cancer cells. In the present study, we examined the integrated antitumor effects including direct cytotoxicity, immune stimulation of NK cells and CTL and the antiangiogenic effects of local IFN‐α gene therapy, and confirmed that intratumoral IFN‐α gene transfer induces a marked regional antitumor effect and systemic immunity in syngeneic immune‐competent animals. The systemic immunity is not attributable to the elevated level of IFN‐α in the systemic circulation because we found some leakage of the cytokine following the intratumoral injection of IFN‐expressing adenoviral vector.( 16 )
Immunohistochemical analyses in the present study suggested that both T cells and NK cells have significant roles in inducing an antitumor effect. The recognition of targets by NK cells is regulated through a balance of activating or inhibitory signals: only tumor cells that have lost MHC class I expression or have a dominant activating ligand are predicted to be susceptible to NK‐cell cytolysis.( 31 , 32 ) However, CTL recognizes the antigen peptides bound to MHC class I molecules and induces cell death via cytotoxic effector molecules. Although IFN‐α is known to upregulate the cell‐surface expression of MHC class I molecules, favoring the antitumor activity of CTL, the cytokine also augments tumor necrosis factor‐related apoptosis inducing ligand or the Fas ligand and the release of perforin( 17 ) to support NK cells as well as T cells. Thus, IFN‐α expression may result in effective antitumor immunity in clinical cases of pancreatic cancer, in which heterogeneous MHC expression coexists in the tumor mass.( 33 )
In the present study we used a subcutaneous tumor as a model of locally advanced pancreatic cancer, which is the main target of local IFN‐α gene therapy. Although locally advanced pancreatic cancer is defined as surgically unresectable with no evidence of distant metastasis, it is highly prone to metastasis during or after standard chemoradiotherapy. To provide a significant impact on long‐term survival, locally advanced cancer also requires the improvement of both regional control and effective systemic treatment against the occurrence of distant metastasis. As shown in 4, 6, IFN‐α gene transfer into tumors significantly suppressed the growth of vector‐injected tumors and induced the antitumor immunity of untreated subcutaneous and peritoneal tumors, suggesting that local IFN‐α gene therapy is a highly promising strategy for locally advanced pancreatic cancer. In the future, systemic chemotherapies, such as gemcitabine and fluorouracil‐based agents,( 4 , 34 ) and new targeted agents, such as antibodies against vascular endothelial growth factor and epidermal growth factor receptor,( 3 ) might be combined with local IFN‐α gene therapy to further improve systemic antitumor activity in advanced cases of pancreatic cancer.
With respect to the safety of the therapy, it is noteworthy that IFN‐α gene transfer showed very limited antiproliferative effects on normal cells such as hepatocytes and vascular endothelial cells.( 11 ) In addition, there was a large difference in the IFN‐α protein concentration of vector‐injected tumor tissue compared with serum.( 11 , 16 ) In fact, no serum abnormalities were detected 8 days after the intratumoral administration of an adenovirus into the hamsters. Therefore, we expect the toxicity of local IFN‐α gene therapy to be tolerable in human clinical trials. In summary, our preclinical study suggests that a regional adenovirus‐mediated gene transfer of IFN‐α is a promising new approach to treating pancreatic cancer. The strategy should be evaluated in future clinical trials for this highly intractable cancer.
Acknowledgments
We thank Dr Yoichi Konishi for providing HPD1NR and HPD2NR cell lines, and Ms Miwa Kushida for her technical support. This work was supported in part by a grant‐in‐aid for the 3rd Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labour and Welfare of Japan, and by grants‐in‐aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan. H. Hara and M. Ohashi are awardees of a Research Resident Fellowship from the Foundation for Promotion of Cancer Research.
References
- 1. Lygidakis NJ, Jain S, Sacchi M, Vrachnos P. Adenocarcinoma of the pancreas − past, present and future. Hepatogastroenterology 2005; 52: 1281–92. [PubMed] [Google Scholar]
- 2. Lockhart AC, Rothenberg ML, Berlin JD. Treatment for pancreatic cancer: current therapy and continued progress. Gastroenterology 2005; 128: 1642–54. [DOI] [PubMed] [Google Scholar]
- 3. Willett CG, Czito BG, Bendell JC, Ryan DP. Locally advanced pancreatic cancer. J Clin Oncol 2005; 23: 4538–44. [DOI] [PubMed] [Google Scholar]
- 4. Okusaka T, Matsumura Y, Aoki K. New approaches for pancreatic cancer in Japan. Cancer Chemother Pharmacol 2004; 54 (Suppl 1): S78–82. [DOI] [PubMed] [Google Scholar]
- 5. Yoshida T, Ohnami S, Aoki K. Development of gene therapy to target pancreatic cancer. Cancer Sci 2004; 95: 283–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tamada K, Wang XP, Brunicardi FC. Molecular targeting of pancreatic disorders. World J Surg 2005; 29: 325–33. [DOI] [PubMed] [Google Scholar]
- 7. Pfeffer LM, Dinarello CA, Herberman RB et al. Biological properties of recombinant α‐interferons: 40th anniversary of the discovery of interferons. Cancer Res 1998; 58: 2489–99. [PubMed] [Google Scholar]
- 8. Gutterman JU. Cytokine therapeutics: lessons from interferon α. Proc Natl Acad Sci USA 1994; 91: 1198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salmon P, Le Cotonnec JY, Galazka A, Abdul‐Ahad A, Darragh A. Pharmacokinetics and pharmacodynamics of recombinant human interferon‐β in healthy male volunteers. J Interferon Cytokine Res 1996; 16: 759–64. [DOI] [PubMed] [Google Scholar]
- 10. Suzuki K, Aoki K, Ohnami S et al. Adenovirus‐mediated gene transfer of interferon α improves dimethylnitrosamine‐induced liver cirrhosis in rat model. Gene Ther 2003; 10: 765–73. [DOI] [PubMed] [Google Scholar]
- 11. Hatanaka K, Suzuki K, Miura Y et al. Interferon‐α and antisense K‐ras RNA combination gene therapy against pancreatic cancer. J Gene Med 2004; 6: 1139–48. [DOI] [PubMed] [Google Scholar]
- 12. Zhang JF, Hu C, Geng Y et al. Treatment of a human breast cancer xenograft with an adenovirus vector containing an interferon gene results in rapid regression due to viral oncolysis and gene therapy. Proc Natl Acad Sci USA 1996; 93: 4513–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Iqbal Ahmed CM, Johnson DE, Demers GW et al. Interferon α2b gene delivery using adenoviral vector causes inhibition of tumor growth in xenograft models from a variety of cancers. Cancer Gene Ther 2001; 8: 788–95. [DOI] [PubMed] [Google Scholar]
- 14. Hottiger MO, Dam TN, Nickoloff BJ, Johnson TM, Nabel GJ. Liposome‐mediated gene transfer into human basal cell carcinoma. Gene Ther 1999; 6: 1929–35. [DOI] [PubMed] [Google Scholar]
- 15. Coleman M, Muller S, Quezada A et al. Nonviral interferon α gene therapy inhibits growth of established tumors by eliciting a systemic immune response. Hum Gene Ther 1998; 9: 2223–30. [DOI] [PubMed] [Google Scholar]
- 16. Ohashi M, Yoshida K, Kushida M et al. Adenovirus‐mediated interferon α gene transfer induces regional direct cytotoxicity and possible systemic immunity against pancreatic cancer. Br J Cancer 2005; 93: 441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chawla‐Sarkar M, Lindner DJ, Liu YF et al. Apoptosis and interferons: role of interferon‐stimulated genes as mediators of apoptosis. Apoptosis 2003; 8: 237–49. [DOI] [PubMed] [Google Scholar]
- 18. Horton HM, Anderson D, Hernandez P, Barnhart KM, Norman JA, Parker SE. A gene therapy for cancer using intramuscular injection of plasmid DNA encoding interferon α. Proc Natl Acad Sci USA 1999; 96: 1553–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li S, Zhang X, Xia X et al. Intramuscular electroporation delivery of IFN‐α gene therapy for inhibition of tumor growth located at a distant site. Gene Ther 2001; 8: 400–7. [DOI] [PubMed] [Google Scholar]
- 20. Yanagi K, Onda M, Uchida E. Effect of angiostatin on liver metastasis of pancreatic cancer in hamsters. Jpn J Cancer Res 2000; 91: 723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mori T, Tsutsumi M, Noguchi O et al. Characterization of three cloned cell lines from a N‐nitrosobis(2‐hydroxypropyl) amine‐induced transplantable hamster pancreatic ductal adenocarcinoma. Int J Pancreatol 1994; 16: 171–7. [DOI] [PubMed] [Google Scholar]
- 22. Nakano M, Aoki K, Matsumoto N et al. Suppression of colorectal cancer growth using an adenovirus vector expressing an antisense K‐ras RNA. Mol Ther 2001; 3: 491–9. [DOI] [PubMed] [Google Scholar]
- 23. Aoki K, Barker C, Danthinne X, Imperiale MJ, Nabel GJ. Efficient generation of recombinant adenoviral vectors by Cre‐lox recombination in vitro . Mol Med 1999; 5: 224–31. [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang WW, Koch PE, Roth JA. Detection of wild‐type contamination in a recombinant adenoviral preparation by PCR. Biotechniques 1995; 18: 444–7. [PubMed] [Google Scholar]
- 25. Bilbo SD, Dhabhar FS, Viswanathan K, Saul A, Yellon SM, Nelson RJ. Short day lengths augment stress‐induced leukocyte trafficking and stress‐induced enhancement of skin immune function. Proc Natl Acad Sci USA 2002; 99: 4067–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ahmed CM, Sugarman BJ, Johnson DE et al. In vivo tumor suppression by adenovirus‐mediated interferon α2b gene delivery. Hum Gene Ther 1999; 10: 77–84. [DOI] [PubMed] [Google Scholar]
- 27. Ahmed CM, Wills KN, Sugarman BJ et al. Selective expression of nonsecreted interferon by an adenoviral vector confers antiproliferative and antiviral properties and causes reduction of tumor growth in nude mice. J Interferon Cytokine Res 2001; 21: 399–408. [DOI] [PubMed] [Google Scholar]
- 28. Demers GW, Johnson DE, Machemer T et al. Tumor growth inhibition by interferon‐α using PEGylated protein or adenovirus gene transfer with constitutive or regulated expression. Mol Ther 2002; 6: 50–6. [DOI] [PubMed] [Google Scholar]
- 29. Indraccolo S, Habeler W, Tisato V et al. Gene transfer in ovarian cancer cells: a comparison between retroviral and lentiviral vectors. Cancer Res 2002; 62: 6099–107. [PubMed] [Google Scholar]
- 30. Indraccolo S, Gola E, Rosato A et al. Differential effects of angiostatin, endostatin and interferon‐α(1) gene transfer on in vivo growth of human breast cancer cells. Gene Ther 2002; 9: 867–78. [DOI] [PubMed] [Google Scholar]
- 31. Long EO, Barber DF, Burshtyn DN et al. Inhibition of natural killer cell activation signals by killer cell immunoglobulin‐like receptors (CD158). Immunol Rev 2001; 181: 223–33. [DOI] [PubMed] [Google Scholar]
- 32. Liao NS, Bix M, Zijlstra M, Jaenisch R, Raulet D. MHC class I deficiency: susceptibility to natural killer (NK) cells and impaired NK activity. Science 1991; 253: 199–202. [DOI] [PubMed] [Google Scholar]
- 33. Scupoli MT, Sartoris S, Tosi G et al. Expression of MHC class I and class II antigens in pancreatic adenocarcinomas. Tissue Antigens 1996; 48: 301–11. [DOI] [PubMed] [Google Scholar]
- 34. Okusaka T, Kosuge T. Systemic chemotherapy for pancreatic cancer. Pancreas 2004; 28: 301–4. [DOI] [PubMed] [Google Scholar]