

Abstract
The isotype of epidermal growth factor receptor variant III (EGFRvIII) is often identified in glioblastomas. Previously, we created a mouse monoclonal antibody, 3C10 (IgG2b), that specifically recognized EGFRvIII, and a recombinant single‐chain variable fragment of 3C10. The aim of the current study was to develop genetically engineered T cells, termed T‐bodies, that express a chimeric receptor consisting of the 3C10 single‐chain variable fragment coupled to signaling modules such as the CD3zeta (ζ) chain, for the treatment of tumors expressing mutant EGFR. After successful construction of the chimeric 3C10/CD3ζ T‐cell receptor, its expression on the T‐body was observed using western blotting and flow cytometry. The specificity of the T‐body for EGFRvIII was evaluated using an interferon‐gamma Elispot assay and a standard 51Cr‐release cytotoxicity assay. Furthermore, we demonstrated that the systemically delivered T‐body infiltrated the intrabrain tumor and significantly delayed tumor growth. These results indicate that the T‐body expressing the chimeric 3C10/CD3ζ T‐cell receptor specifically recognized glioma cells expressing EGFRvIII. In conclusion, T‐body‐based immunotherapy appears to be a promising approach for the treatment of glioma. (Cancer Sci 2010; 101: 2518–2524)
The expression of epidermal growth factor receptor (EGFR) is amplified in approximately 50% of glioblastomas (GBM).( 1 ) The binding of a ligand to EGFR leads to receptor dimerization, autophosphorylation and activation of several downstream signaling pathways such as the Ras/Raf/MEK/ERK pathway, the PI3K/Akt pathway and the PLC‐gamma (γ)/PKC pathway, resulting in cell proliferation, motility and survival.( 2 ) Approximately 40–70% of brain tumors with EGFR amplification express mutant EGFR variant III (EGFRvIII); EGFRvIII has a deletion of exons 2–7 that causes a defect in the extracellular ligand‐binding domain and induces constitutive activation in a ligand‐independent manner.( 3 , 4 , 5 , 6 ) Notably, EGFRvIII is characterized by an 801‐base pair (bp) in‐frame deletion, which results in a unique sequence with a glycine residue at the fusion junction between amino acid residues 5 and 274. Epidermal growth factor receptor variant III is an attractive target antigen for cancer immunotherapy because it is not expressed in normal tissue and is associated with survival, invasion and angiogenesis in cancers.( 4 , 7 ) Previously, we generated the monoclonal antibody (mAb) 3C10 and a recombinant single‐chain variable fragment (scFv) antibody (Ab) that specifically recognizes EGFRvIII.( 8 , 9 , 10 )
Glioblastomas cannot be treated and result in death despite the extensive application of surgical excision and adjuvant chemo/radiotherapy. Consequently, various promising immunotherapy approaches for the treatment of glioma are being investigated.( 11 , 12 , 13 , 14 )
Cytotoxic T lymphocytes (CTL) are capable of effective recognition and destruction of tumor cells, and therefore cellular immunotherapy has been suggested for treating tumors in humans.( 15 ) However, it is difficult to obtain adequate quantities of tumor‐specific T cells. In addition, the isolation and ex vivo clonal expansion of tumor‐specific CTL from patients is a long and cumbersome process. As a result, general application of this approach has been limited.
Many of the limitations associated with cellular immunotherapy can be circumvented by arming polyclonal CTL with tumor‐specific chimeric T‐cell receptors (TCR), the so‐called ‘‘T‐body’’ approach.( 16 ) Chimeric TCR typically consist of a tumor antigen‐specific recognition scFv element derived from a mAb and components of TCR that mediate signal transduction in the CTL.( 17 ) The T‐body has the potential to recognize specific antigens in a major histocompatibility complex (MHC)‐independent manner; the applicability of this approach has been demonstrated both in vitro and in vivo.( 18 )
In the present study, we generated human T cells that expressed the scFv‐CD3zeta (ζ) chimeric antigen receptor (CAR) targeting the EGFRvIII antigen by using retroviral‐mediated transduction. The generated T‐body was able to secrete IFN‐γ and lyse GBM cells in an EGFRvIII‐dependent manner. Furthermore, systemic injection of the T‐body significantly inhibited intrabrain tumor growth in mice.
Materials and Methods
Cell lines. Human GBM cell lines U87MG, expressing wild‐type EGFR (EGFRwt), and U87‐EGFRvIII, stably expressing EGFRvIII, were kindly provided by Dr W. K. Cavenee (Ludwig Institute for Cancer Research, San Diego, CA, USA). The Jurkat T‐cell leukemia cell line was provided by Dr Y. Miyata (Department of Hematology, Nagoya University, Nagoya, Japan). All cell lines were maintained in RPMI‐1640 medium containing 10% fetal bovine serum (FBS) and penicillin/streptomycin. The U87‐EGFRvIII cell line was maintained in RPMI‐1640 medium containing 10% FBS and 400 μg/mL geneticin.
Sample collection and RNA extraction. Tumor specimens for molecular genetic analysis were obtained from 55 patients with malignant gliomas who underwent surgical procedures at Nagoya University Hospital or affiliated hospitals. The molecular genetic analysis performed in the study was approved by the Institutional Ethics Committee of Nagoya University, and all patients who registered for this study provided written informed consent. All tumors were histologically verified according to World Health Organization 2007 guidelines: 31 patients had GBM (grade IV), 10 had grade III gliomas, and 14 had grade II gliomas. RNA purification was performed using the standard TRIzol (Invitrogen, Carlsbad, CA, USA) method.
EGFR expression analysis by RT‐PCR. Epidermal growth factor receptor variant III expression was examined using RT‐PCR assays. First‐strand complementary DNA (cDNA) was synthesized from the total RNA (1 μg) extracted from 55 tumors and 20 normal tissues (Human Total RNA Master Panel II; Takara Bio, Otsu, Japan) by using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany). The cDNA was amplified by PCR using primers designed to flank the 801‐bp deleted region (exons 2–7) to detect both EGFRwt and EGFRvIII (Table 1). A 1044‐bp PCR product was obtained for EGFRwt, compared with a 243‐bp product for EGFRvIII.( 19 )
Table 1.
Primers used in the present study
| NheI‐leader‐VH | ACTGCTAGCACCGGTCCTTACAATGAAATGCA |
| AccIII‐linker‐VH | GTCCATGGCGCAAAGCTTATTAATTCCGGAACCACCACCACCGGAACCACCACCTCCTGAGGAGACTGTGAGAGTGGT |
| AccIII‐linker‐VL | GCATGGCTTCCGGAGGTGGTGGTTCACATATGGATGTTGTGATGACCCAGTCTCCACTCACTCTA |
| VspI‐VL | TTCCATGGCGCAAAGCTTATTAATGGATCCGCCGCCACCTGATCCGCCGCCTCCTGACCGTTTTATCTCCAGCTTGGTCCCTCCACC |
| VspI‐CD8 α | GACATTAATAGCAACTCCAT |
| BamHI‐CD8 α | TGCATGGATCCAGGAAGTCCAG |
| BamHI‐CD3ζ | TGCTGGATCCCAAACTCTGCT |
| EcoRI‐CD3ζ | GCTGGAATTCTGTTAGCGAGG |
| EGFR‐F | CTTCGGGGAGCAGCGATGCGAC |
| EGFR‐R | ACCAATACCTATTCCGTTACAC |
Underline indicates restriction enzyme sites. EGFR, epidermal growth factor receptor.
Construction of the anti‐EGFRvIII CAR. We constructed 3C10‐CAR, a CAR specific to EGFRvIII. 3C10‐CAR consists of the 3C10 scFv‐Ab against EGFRvIII, which is linked to the hinge portion of human CD8alpha (α) that is fused to the transmembrane and intracellular signaling domains of the CD3ζ chain (Fig. 1). The VH and VL cDNA fragments of 3C10 were subcloned into the plasmid vector pSRIG‐neo, and designated as pSRIG‐3C10VH_VL ( 10 , 20 ) The leader sequence VH and the first half of the linker domains were subcloned by high‐fidelity PCR amplification using pSRIG‐3C10VH_VL as a template, a sense primer including the NheI enzyme site (NheI‐leader‐VH primer; Table 1) and an antisense primer including the AccIII site (AccIII‐linker‐VH primer; Table 1). Similarly, the latter half of the linker domain and VL domain were amplified with an AccIII‐linker‐VL sense primer and a VspI‐VL antisense primer. The two fragments were ligated at the AccIII site and inserted into the TA cloning site of the pCR2.1TOPO vector (Invitrogen). The cDNA coding for the hinged portion of CD8α (aa 95–158) was amplified by PCR using CD8α cDNA (kindly provided by Dr E. Nakauchi, Department of Immunology, University of Tsukuba, Tsukuba, Japan) as a template and VspI‐CD8α sense and BamHI‐CD8α antisense primers (Table 1). The cDNA coding for the transmembrane and intracellular portions of CD3ζ (aa 8–142) was amplified by PCR using CD3 cDNA (kindly provided by Dr Weissman, National Cancer Institute, Bethesda, MD, USA) as a template and BamHI‐CD3ζ sense and EcoRI‐CD3ζ antisense primers (Table 1). These two fragments were ligated at the BamHI site and inserted into the pCR2.1TOPO vector. The leader sequence VH‐linker‐VL and the CD8α‐CD3ζ cDNA thus obtained were assembled into the pcDNA3.1 vector at three enzyme sites, NheI, VspI and EcoRI. The sequence of the final construct was confirmed bidirectionally by using the AccIII‐linker‐VL forward and EcoRI‐CD3ζ reverse primers.
Figure 1.
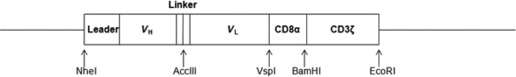
Construction of the anti‐epidermal growth factor receptor variant III (EGFRvIII) chimeric antigen receptor. The construct was composed of the VH and VL regions of the anti‐EGFRvIII mAb joined by a flexible linker, a membrane‐proximal hinge region of human CD8α, and the transmembrane and cytoplasmic regions of the human CD3ζ chain.
Construction of the retroviral vector expressing anti‐EGFRvIII CAR. The retroviral vector backbone pMEI‐5 neo vector (Takara Bio) was assembled with the 3C10‐CAR construct in pcDNA3.1‐3C10‐CAR. G3T‐hi cells were transfected with pMEI‐5 neo‐3C10‐CAR or pMEI‐5 neo‐green fluorescent protein (GFP) plasmids along with pGP and pE‐ampho packaging plasmids by using the Retrovirus Packaging Kit Ampho (Takara Bio). The cell‐free viral supernatants obtained were then frozen and stocked at −80°C.
Culture and retroviral transduction of primary human T cells and Jurkat cells. Freshly harvested peripheral blood mononuclear cells (PBMC) from healthy donors were separated over a monolayer of Ficoll (1000g for 20 min at 24°C). The PBMC were cultured in AIM‐V medium (Invitrogen) with 10% (v/v) human serum in the presence of interleukin 2 (IL‐2; 50 U/mL) (Shionogi, Osaka, Japan) and anti‐CD3 mAb (muromonab‐CD3, 100 ng/mL; Janssen Pharmaceutica, Titusville, NJ, USA) for 48 h. The PBMC were harvested, washed once, and resuspended at a density of 0.5 × 106 cells/mL in AIM‐V supplemented with 10% (v/v) human serum and IL‐2. On day 3, the PBMC (0.5 × 106 cells/mL) were harvested.
For transduction, we precoated a non‐tissue culture‐treated six‐well plate with the recombinant fibronectin fragment FN‐CH296 (RetroNectin; Takara Bio) at 100 μg per well. The cells were transduced with the retroviral vectors using the viral preloading method. Briefly, the FN‐CH296‐coated plates were loaded with the retroviral vector supernatant and incubated for 4 h at 37°C. The plate was then washed with phosphate‐buffered saline (PBS) and stimulated cells were added (2 mL per well). The cells were then incubated overnight at 37°C. After adding 4 mL of AIM‐V, the cells were transferred to a new six‐well plate and incubated with IL‐2. After an additional 24 h, 100 nM of Pep3 (LEEKKGNYVVTDHC), a 3C10‐specific peptide, was added to the cell culture. The cells were then expanded in the presence of 50 U/mL IL‐2 every other day for 1 month.
Jurkat cells (5 × 104 per well) were seeded in a 24‐well plate that was precoated with FN‐CH296 (25 μg per well) as described above. After incubation overnight at 37°C, the medium was replaced with RPMI‐1640 medium containing 10% FBS. Following an incubation period of 72 h, the transduced Jurkat cells were maintained in RPMI‐1640 medium containing 10% FBS and 400 μg/mL geneticin.
Western blot analysis. To confirm the transduction of 3C10‐CAR in Jurkat cells, western blot analysis was performed under both nonreducing and reducing conditions. After selection with geneticin, retrovirally transduced Jurkat cells were lysed with cell lysis buffer (Cell Signaling, Danvers, MA, USA), and the lysate was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis on 15% resolving gels using standard methods and subsequently blotted onto a polyvinylidene fluoride membrane. The membrane was probed with anti‐CD3ζ mAb (1:2000; BD Bioscience, Franklin Lakes, NJ, USA) and a horseradish peroxidase‐conjugated goat anti‐mouse Ab (1:3000), followed by visualization with enhanced chemiluminescence (GE Healthcare Japan, Osaka, Japan).
Flow cytometry. The expression of 3C10‐CAR on the cell surface of transduced Jurkat cells was examined using a FACS Calibur equipped with the CellQuest research software (Becton Dickinson, Mountain View, CA, USA). Jurkat cells were stained with biotinylated 3C10‐specific Pep3 followed by PE‐conjugated streptavidin (R&D Systems, Minneapolis, MN, USA).( 8 ) The PBMC were stained with fluorescein isothiocyanate (FITC)‐conjugated anti‐CD4 and PE‐conjugated anti‐CD8 antibodies (Beckman Coulter Japan, Tokyo, Japan).
IFN‐γ Elispot assays. Cells producing interferon‐gamma (IFN‐γ) were quantified by ELISpot (Mabtech, Nacka Strand, Sweden) according to the manufacturer’s instructions. Briefly, the PBMC (5.0 × 103, in triplicate wells) were cultured with anti‐CD3 Ab as a positive control or with U87MG or U87‐EGFRvIII glioma cells (1.0 × 104) and were incubated at 37°C for 24 h. The number of spots in the plate was counted by two observers.
Target cell lysis. The susceptibility of U87MG and U87‐EGFRvIII cells to PBMC retrovirally transduced with 3C10‐CAR was evaluated using a standard 4‐h 51Cr‐release assay at various effector:target (E:T) ratios. The percentage of specific lysis was calculated as follows:
A target inhibition assay was performed to confirm specific lysis. The PBMC transduced with 3C10‐CAR were pre‐incubated with various concentrations (0–25 μM) of Pep3 for 1 h. Cytotoxicity was assessed at an E:T ratio of 50:1, as described above.
Intracranial glioma xenograft. U87‐EGFRvIII cells (2.5 × 105) suspended in 5 μL PBS were injected stereotactically into 5‐ to 6‐week‐old NOD/SCID female mice (SLC, Shizuoka, Japan), as described previously.( 21 ) Mice bearing established tumors were randomly assigned to two different experimental groups. Four days after tumor inoculation, human PBMC transduced with 3C10‐CAR or non‐transduced PBMC (4 × 106 cells) were injected into the tail vein. Survival time was assessed after adoptive transfer of the PBMC. To evaluate tumor size, mice were killed at day 12, and brains were fixed in 10% formalin for 24 h and embedded in paraffin. Serial tissue sections (5 μm) were stained with hematoxylin and eosin. In order to determine whether the transferred PBMC infiltrated the tumor, paraffin‐embedded coronal sections were immunostained with PE‐conjugated anti‐human CD8 antibody (Dako, Glostrup, Denmark), and nuclei were counterstained with Hoechst 33342. Two perpendicular diameter measurements were obtained for each tumor with calipers. The tumor volume (V) was determined using the equation V = 0.5 × a × b 2, where a is the major axis and b is the minor axis. Survival and tumor size of treated and control tumors were analyzed using a one‐tailed Mann–Whitney test.
Results
Determination of EGFRvIII expression. Epidermal growth factor receptor variant III expression was analyzed in 55 gliomas using RT‐PCR. Figure 2A is a representative gel showing the EGFRwt and EGFRvIII PCR products from five gliomas and the U87MG and U87‐EGFRvIII cell lines. Epidermal growth factor receptor variant III expression was observed in nine of the 55 tumors. The expression frequency of EGFRvIII in gliomas of grades II, III and IV was 7.1%, 0% and 25.8%, respectively (Fig. 2B). This finding of a higher expression frequency in GBM (grade IV) is consistent with previous studies in which EGFRvIII was identified in approximately 17–57% of GBM.( 5 , 6 , 8 , 22 , 23 , 24 ) In addition, all 20 of the normal tissues tested were found to express EGFRwt but not EGFRvIII (Fig. 2C).
Figure 2.

Epidermal growth factor receptor variant III (EGFRvIII) expression in glioblastoma and normal tissues. (A) The detection of EGFRvIII expression in fresh‐frozen glioblastoma specimens by RT‐PCR. Primers flanking the deleted portion (exons 2–7) amplified cDNA fragments from both full‐length EGFR (1044 bp) and the truncated EGFRvIII (243 bp). U87MG (lane 1) and U87‐EGFRvIII (lane 2) were included as controls. A representative gel of the PCR products of tumor samples from five patients (Pt.) is shown. Patients (Pt) 1, 3, 4 and 5 had grade IV tumors, while patient 2 had a grade III tumor. (B) mRNA from 55 glioma samples was isolated and analyzed. The expression frequency of EGFRvIII in grade II, grade III and grade IV tumors was 7.1%, 0% and 25.8%, respectively. (C) EGFR expression in 20 normal tissues. All normal tissues expressed wild‐type EGFR but not EGFRvIII.
Expression of the anti‐EGFRvIII CD3ζ CAR on the surface of Jurkat cells. The expression of 3C10‐CAR was examined by western blot analysis with a mAb specific for the human CD3ζ chain (Fig. 3A). Nonreducing western blot analysis displayed the endogenous monomeric and homodimeric CD3ζ chains at the predicted molecular masses of 16 and 32 kDa, respectively. Reducing western blot analysis showed protein expression corresponding to the predicted mass of the monomeric 3C10‐CAR (50 kDa) in transduced cells only; nonreducing western blot analysis detected 3C10‐CAR homodimers at a molecular mass of approximately 100 kDa.
Figure 3.

The anti‐epidermal growth factor receptor variant III (EGFRvIII)‐CD3ζ chimeric antigen receptor (CAR) expression, cell surface trafficking and 3C10 binding on the surface of Jurkat cells. (A) Detection of anti‐EGFRvIII‐CD3ζ CAR expression in whole‐cell lysate derived from Jurkat cell transfectants by reducing and nonreducing western blotting analysis with a mAb specific for the human CD3ζ chain. The lower band (16 kDa) is the endogenous CD3ζ chain (white arrow), and the upper band (32 kDa) is its homodimer (black arrow). The anti‐EGFRvIII‐CD3ζ CAR was detected at the expected molecular weight of 50 kDa (white arrowhead). The anti‐EGFRvIII‐CD3ζ CAR homodimer was detected at 100 kDa (black arrowhead). C, control cell; T, transduced cells. (B) Jurkat cells were stained with biotin‐conjugated Pep3 and streptavidin‐FITC. Expression was not detected in the non‐transduced cells (black line). Data are representative of three independent experiments.
To investigate the cell‐surface expression of 3C10‐CAR, biotin‐conjugated Pep3 was used for fluorescence‐activating cell sorting analysis. Figure 3B clearly shows the surface expression of 3C10‐CAR on transduced Jurkat cells.
Superior IFN‐γ release and cytotoxicity mediated by 3C10‐CAR‐transduced T cells. The IFN‐γ release and cytotoxicity mediated by 3C10‐CAR‐transduced PBMC against U87‐EGFRvIII and U87MG target cell lines were assessed by Elispot and 51Cr‐release assays. A 2.5‐fold increase in IFN‐γ‐positive spots was observed for transduced PBMC co‐cultured with U87‐EGFRvIII compared with PBMC co‐cultured with U87MG cells (Fig. 4A).
Figure 4.

Epidermal growth factor receptor variant III (EGFRvIII)‐specific activation of receptor‐grafted T cells. (A) Interferon‐gamma (IFN‐γ) Elispot assay. A 2.5‐fold increase in IFN‐γ‐positive spots was observed for 3C10‐CAR‐transduced PBMC co‐cultured with U87‐EGFRvIII compared with those co‐cultured with U87MG cells. (B) 51Cr‐release assay. 3C10‐CAR‐transduced PBMC lysed EGFRvIII target cells (•) more significantly than other co‐cultures. (C) The addition of Pep3 inhibited cytotoxicity. Data are expressed as the mean ± standard deviation of three independent experiments.
To determine whether 3C10‐CAR is capable of conferring CTL‐mediated lysis against EGFRvIII‐positive cells, effector cells were mixed with 51Cr‐labeled target cells. The 3C10‐CAR‐expressing PBMC exerted significant lysis against U87‐EGFRvIII, although some alloreactivity was observed in other effector/target co‐cultures (Fig. 4B). The addition of Pep3, which competitively binds to EGFRvIII, inhibited the cytotoxic ability of the 3C10‐CAR‐expressing PBMC against U87‐EGFRvIII in a concentration‐dependent manner (Fig. 4C). These observations show that 3C10‐CAR‐expressing effector cells specifically recognize the EGFRvIII antigen.
The effect of 3C10‐CAR‐transduced T cells on EGFRvIII‐expressing brain tumor in vivo. Four days after intracranial injections of U87‐EGFRvIII cells (2.5 × 105), transduced or control PBMC (4 × 106) were adoptively transferred via the tail vein. The proportions of CD4‐ and CD8‐positive PBMC were 46% and 40%, respectively (Fig. 5A). Although complete tumor eradication was not observed, tumor growth was significantly retarded in mice injected with 3C10‐CAR‐transduced PBMC compared with the control (P = 0.0472; Fig. 5B). The number of CD8‐positive cells that had infiltrated the tumor was greater in mice injected with 3C10‐CAR‐transduced PBMC than in control mice (Fig. 5C). In addition, the survival time was remarkably prolonged in mice injected with 3C10‐CAR‐transduced cells (log rank test, P = 0.014; Fig. 5D).
Figure 5.

Systemic injection of 3C10‐CAR‐transduced PBMC retarded the growth of epidermal growth factor receptor variant III (EGFRvIII)‐positive tumors in the mouse brain. (A) Among the 3C10‐CAR‐transduced PBMC, the proportions of CD4+ and CD8+ cells were 45.99% and 39.4%, respectively. (B) At day 12 after tumor inoculation, the tumor volumes were determined. 3C10‐CAR‐transduced PBMC significantly inhibited tumor growth (P = 0.0472). (C) The number of CD8‐positive cells that had infiltrated the tumor was greater in mice injected with 3C10‐CAR‐transduced PBMC than in mice injected with control PBMC. (D) Survival of mice injected with 3C10‐CAR‐transduced and control PBMC. The survival time of mice injected with 3C10‐CAR‐transduced PBMC was higher than that of mice injected with control PBMC (log rank test, P = 0.014). The number of animals in each group is five.
Discussion
Generation of tumor‐specific T cells expressing CAR. Cellular immunotherapy involving the use of autologous tumor‐reactive or host‐compatible antigen‐specific T cells has significant potential in the treatment of malignant disease.( 25 ) However, the degree and persistence of the cellular immunity may be partly limited because of the poor immunogenicity of tumor cells, as evidenced by processes such as MHC silencing. As an alternative strategy, T‐bodies have been generated by the transfer of genes encoding CAR. Chimeric antigen receptors consist of a tumor antigen‐binding domain of an antibody that has been fused to intracellular signaling domains capable of activating T cells. Therefore, antigen recognition by the T‐body is not MHC restricted, and is directed to native cell surface structures. Eshhar et al. ( 16 ) were the first to create a CAR containing a hapten‐specific scFv and the CD3ζ chain or FcεRIγ chain as the intracellular domain. Several CAR directed against a variety of tumor antigens have been developed. Most of these CAR were transduced in primary mouse and human T cells.( 18 ) In addition, several clinical trials of T‐bodies as therapy for ovarian cancer, renal cancer, lymphoma and neuroblastoma are being carried out.( 26 , 27 , 28 , 29 , 30 , 31 )
EGFRvIII as a potential target in gliomas. There are a few fundamental reports relating to the use of T‐bodies for tumors of the central nervous system. T cells expressing a CAR consisting of a HER2‐specific scFv and domains of the CD3ζ chain or CTL expressing IL13‐zetakine, which is composed of an extracellular domain that contains the high‐affinity IL‐13 mutein and a cytoplasmic tail that contains a domain of the CD3ζ, have been shown to exert an antitumor effect against experimental medulloblastoma and glioma.( 32 , 33 , 34 ) It is important that target antigens chosen for clinical studies are limited to those expressed only by malignant cells and not by normal cells. A clinical study using T‐bodies in patients with renal carcinoma was terminated for the reason of cholestasis as an on‐target effect caused by high expression of the targeted antigen carbonic anhydrase in the biliary epithelium. Evidence shows that T‐bodies also injured normal cells expressing target molecules, resulting in unfavorable autoimmunity.( 27 )
The tumor‐specific antigens derived from tumor‐associated mutations in somatic genes are less likely to be associated with autoimmunity because they are absent in normal tissues. Epidermal growth factor receptor variant III is a rare example of a frequent and consistent tumor‐specific mutation that is central to the neoplastic process.( 2 , 3 ) In this study, the observed frequency of EGFRvIII expression in GBM was 26% (8/31), and this value is relatively lower than the frequency of 17–57% reported in previous studies;( 5 , 6 , 8 , 22 , 23 , 24 ) this finding can be partly attributed to the fact that the number of secondary GBM, in which EGFRvIII expression is less frequent, was large in this study. In addition, we found no EGFRvIII expression in the 10 grade III tumors, but the reason for this could be the small sample size. Nevertheless, the advantages of EGFRvIII include frequent expression in GBM, lack of expression in normal tissues and its importance in the oncogenic phenotype of tumors; these characteristics make EGFRvIII a potential target for antitumor immunotherapy.( 8 , 22 , 35 , 36 ) Therefore, an anti‐EGFRvIII CAR has potential as a therapeutic tool.
Future perspectives. Recent studies have focused on generating a more effective T‐body by improving CAR design. The in vivo activation of T‐bodies is an important part of this process. CD28, a T‐cell costimulatory factor, has been shown to induce T‐cell activation and proliferation in vivo.( 37 ) Chimeric antigen receptors with dual CD28‐CD3ζ signaling receptors may enable T cells to proliferate after repeated antigenic stimulation.( 38 , 39 , 40 ) Other costimulatory signaling domains, including 4‐1BB, OX40, DAP10 and ICOS, have been investigated.( 41 , 42 , 43 ) In addition, transduction efficiency of the vector should be improved. Intracellular IFN‐γ assay demonstrated that a proportion of CD8+/IFN‐γ+ cells in CAR‐transduced PBMC co‐cultured with U87EGFRvIII was 3–7% (data not shown). In order to improve transduction efficiency, lentiviral‐mediated transduction might be an alternate approach.
Another potent strategy involves the combination of the EGFRvIII T‐body with a kinase inhibitor of the receptor. Epidermal growth factor receptor variant III leads to constitutive activation of downstream signaling pathways, including second messenger pathways.( 2 , 3 ) Several clinical trials of erlotinib have been conducted in patients with glioma,( 44 , 45 , 46 ) and erlotinib has been found to be effective in a subset of patients whose tumors showed expression of EGFRvIII and phosphatase and tensin homolog deleted from chromosome 10 (PTEN), or high expression of EGFR and low phosphorylation of Akt.( 19 , 46 , 47 ) Combined treatment with an anti‐EGFRvIII T‐body and a tyrosine kinase inhibitor, which targets extracellular and intracellular domains of the receptor, may augment the potency of EGFR signaling inhibition.
In conclusion, we constructed an EGFRvIII‐targeted CAR gene and confirmed its successful retrovirus‐mediated expression. We demonstrated the activation and cytotoxicity of genetically engineered T cells both in vitro and in vivo. Further clinical trials should be conducted to determine the efficacy of T‐bodies in the treatment of EGFRvIII‐expressing gliomas.
Disclosure Statement
We declare there are no competing financial interests in relation to this work.
Acknowledgments
This work was supported by a Grant‐in‐Aid (B) (A.N.) and a Grant‐in‐Aid for Young Scientists (B) (M.I.) for Scientific Research from the Ministry of Health, Labor and Welfare, Japan.
References
- 1. Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo . Cancer Res 1991; 51: 2164–72. [PubMed] [Google Scholar]
- 2. Zandi R, Larsen AB, Andersen P, Stockhausen MT, Poulsen HS. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal 2007; 19: 2013–23. [DOI] [PubMed] [Google Scholar]
- 3. Nicholas MK, Lukas RV, Jafri NF, Faoro L, Salgia R. Epidermal growth factor receptor‐mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res 2006; 12: 7261–70. [DOI] [PubMed] [Google Scholar]
- 4. Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci USA 1990; 87: 8602–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000; 60: 1383–7. [PubMed] [Google Scholar]
- 6. Aldape KD, Ballman K, Furth A et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol 2004; 63: 700–7. [DOI] [PubMed] [Google Scholar]
- 7. Yamazaki H, Ohba Y, Tamaoki N, Shibuya M. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Jpn J Cancer Res 1990; 81: 773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Humphrey PA, Wong AJ, Vogelstein B et al. Anti‐synthetic peptide antibody reacting at the fusion junction of deletion‐mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci USA 1990; 87: 4207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okamoto S, Yoshikawa K, Obata Y et al. Monoclonal antibody against the fusion junction of a deletion‐mutant epidermal growth factor receptor. Br J Cancer 1996; 73: 1366–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakayashiki N, Yoshikawa K, Nakamura K et al. Production of a single‐chain variable fragment antibody recognizing type III mutant epidermal growth factor receptor. Jpn J Cancer Res 2000; 91: 1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Okada H, Kohanbash G, Zhu X et al. Immunotherapeutic approaches for glioma. Crit Rev Immunol 2009; 29: 1–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Izumoto S, Tsuboi A, Oka Y et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg 2008; 108: 963–71. [DOI] [PubMed] [Google Scholar]
- 13. De Vleeschouwer S, Fieuws S, Rutkowski S et al. Postoperative adjuvant dendritic cell‐based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res 2008; 14: 3098–104. [DOI] [PubMed] [Google Scholar]
- 14. Wheeler CJ, Black KL, Liu G et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res 2008; 68: 5955–64. [DOI] [PubMed] [Google Scholar]
- 15. Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today 1997; 18: 175–82. [DOI] [PubMed] [Google Scholar]
- 16. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody‐binding domains and the gamma or zeta subunits of the immunoglobulin and T‐cell receptors. Proc Natl Acad Sci USA 1993; 90: 720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer 2003; 3: 35–45. [DOI] [PubMed] [Google Scholar]
- 18. Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol 2009; 21: 215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mellinghoff IK, Wang MY, Vivanco I et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005; 2353: 2012–24. [DOI] [PubMed] [Google Scholar]
- 20. Akamizu T, Matsuda F, Okuda J et al. Molecular analysis of stimulatory anti‐thyrotropin receptor antibodies (TSAbs) involved in Graves’ disease. Isolation and reconstruction of antibody genes, and production of monoclonal TSAbs. J Immunol 1996; 157: 3148–52. [PubMed] [Google Scholar]
- 21. Natsume A, Wakabayashi T, Tsujimura K et al. The DNA demethylating agent 5‐aza‐2’‐deoxycytidine activates NY‐ESO‐1 antigenicity in orthotopic human glioma. Int J Cancer 2008; 122: 2542–53. [DOI] [PubMed] [Google Scholar]
- 22. Moscatello DK, Holgado‐Madruga M, Godwin AK et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 1995; 55: 5536–9. [PubMed] [Google Scholar]
- 23. Pelloski CE, Ballman KV, Furth AF et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol 2007; 25: 2288–94. [DOI] [PubMed] [Google Scholar]
- 24. Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor‐associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 1997; 57: 4130–40. [PubMed] [Google Scholar]
- 25. Yee C, Thompson JA, Byrd D et al. Adoptive T cell therapy using antigen‐specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA 2002; 99: 16168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kershaw MH, Westwood JA, Parker LL et al. A phase I study on adoptive immunotherapy using gene‐modified T cells for ovarian cancer. Clin Cancer Res 2006; 12: 6106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamers CH, Sleijfer S, Vulto AG et al. Treatment of metastatic renal cell carcinoma with autologous T‐lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 2006; 24: e20–2. [DOI] [PubMed] [Google Scholar]
- 28. Lamers CH, Langeveld SC, Groot‐van Ruijven CM, Debets R, Sleijfer S, Gratama JW. Gene‐modified T cells for adoptive immunotherapy of renal cell cancer maintain transgene‐specific immune functions in vivo. Cancer Immunol Immunother 2007; 56: 1875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Till BG, Jensen MC, Wang J et al. Adoptive immunotherapy for indolent non‐Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20‐specific T cells. Blood 2008; 112: 2261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pule MA, Savoldo B, Myers GD et al. Virus‐specific T cells engineered to coexpress tumor‐specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 2008; 14: 1264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park JR, Digiusto DL, Slovak M et al. Adoptive transfer of chimeric antigen receptor re‐directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther 2007; 15: 825–33. [DOI] [PubMed] [Google Scholar]
- 32. Ahmed N, Ratnayake M, Savoldo B et al. Regression of experimental medulloblastoma following transfer of HER2‐specific T cells. Cancer Res 2007; 67: 5957–64. [DOI] [PubMed] [Google Scholar]
- 33. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13‐zetakine redirected cytolytic T cells. Cancer Res 2004; 64: 9160–6. [DOI] [PubMed] [Google Scholar]
- 34. Stastny MJ, Brown CE, Ruel C, Jensen MC. Medulloblastomas expressing IL13Ralpha2 are targets for IL13‐zetakine+ cytolytic T cells. J Pediatr Hematol Oncol 2007; 29: 669–77. [DOI] [PubMed] [Google Scholar]
- 35. Garcia de Palazzo IE, Adams GP, Sundareshan P et al. Expression of mutated epidermal growth factor receptor by non‐small cell lung carcinomas. Cancer Res 1993; 53: 3217–20. [PubMed] [Google Scholar]
- 36. Wikstrand CJ, Hale LP, Batra SK et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res 1995; 55: 3140–8. [PubMed] [Google Scholar]
- 37. Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate‐specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate‐specific membrane antigen. Neoplasia 1999; 1: 123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Haynes NM, Trapani JA, Teng MW et al. Single‐chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 2002; 100: 3155–63. [DOI] [PubMed] [Google Scholar]
- 39. Hombach A, Wieczarkowiecz A, Marquardt T et al. Tumor‐specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL‐2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J Immunol 2001; 167: 6123–31. [DOI] [PubMed] [Google Scholar]
- 40. Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T‐lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 2002; 20: 70–5. [DOI] [PubMed] [Google Scholar]
- 41. Imai C, Mihara K, Andreansky M et al. Chimeric receptors with 4‐1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004; 18: 676–84. [DOI] [PubMed] [Google Scholar]
- 42. Marin V, Kakuda H, Dander E et al. Enhancement of the anti‐leukemic activity of cytokine induced killer cells with an anti‐CD19 chimeric receptor delivering a 4‐1BB‐zeta activating signal. Exp Hematol 2007; 35: 1388–97. [DOI] [PubMed] [Google Scholar]
- 43. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 2004; 172: 104–13. [DOI] [PubMed] [Google Scholar]
- 44. De Groot JF, Gilbert MR, Aldape K et al. Phase II study of carboplatin and erlotinib (Tarceva, OSI‐774) in patients with recurrent glioblastoma. J Neurooncol 2008; 90: 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brown PD, Krishnan S, Sarkaria JN et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol 2008; 26: 5603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Den Bent MJ, Brandes AA, Rampling R et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol 2009; 27: 1268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang MY, Lu KV, Zhu S et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN‐deficient and PTEN‐intact glioblastoma cells. Cancer Res 2006; 66: 7864–9. [DOI] [PubMed] [Google Scholar]