

Abstract
Spliceostatin A (SSA) is a methylated derivative of an antitumor natural product FR901464, which specifically binds and inhibits the SF3b spliceosome sub‐complex. To investigate the selective antitumor activity of SSA, we focused on the regulation of vascular endothelial growth factor (VEGF) mRNA, since VEGF is a key regulatory component in tumor angiogenesis and known for the intricate regulation of mRNA processing, such as alternative splicing. We found that in HeLa cells SSA reduced the amount of both mRNA and protein of VEGF. Spliceostatin A not only inhibited the splicing reaction of VEGF pre‐mRNA but also reduced the total amount of VEGF’s transcripts, while SSA affected GAPDH mRNA to a lesser extent. Given a significant reduction in VEGF gene expression, SSA was expected to possess anti‐angiogenic activity in vivo. Indeed, SSA inhibited cancer cell‐derived angiogenesis in vivo in a chicken chorioallantoic membrane (CAM) assay. The inhibition of angiogenesis with SSA was abolished by addition of exogenous VEGF. We also performed global gene expression analyses of HeLa cells and found that the expression levels of 38% of total genes including VEGF decreased to <50% of the basal levels following 16 h of SSA treatment. These results suggest that the global interference of gene expression including VEGF in tumor cells is at least one of the mechanisms by which SSA (or FR901464) exhibits its strong antitumor activity. (Cancer Sci 2010; 101: 2483–2489)
Angiogenesis is the process of forming new blood vessels, which sprout from pre‐existing microvasculatures.( 1 ) Because the malignant neoplasms constantly require sufficient nutrients and oxygen from the blood supply to keep growing and/or metastasizing, the inhibition of tumor‐associated neoangiogenesis represents a promising target for antineoplastic therapy.( 1 ) Since vascular endothelial growth factor (VEGF) is the most critical inducer of tumor angiogenesis,( 2 , 3 , 4 ) inhibiting VEGF action has been a successful strategy for therapeutic intervention.( 5 ) The VEGF binds to its cognate receptors including Flt‐1, KDR/Flk‐1 and Flt‐4 to activate the MAPK and Akt signaling pathways, which collectively induce endothelial cell growth, migration, homeostasis and tube formation. Therefore, many anti‐angiogenic agents, currently being used clinically and/or under phase II or III examination, are either an antibody against VEGF or target VEGF signaling by inhibiting VEGF receptor (VEGFR) tyrosine kinase activity.( 6 , 7 , 8 ) In addition, multiple anti‐angiogenic agents that target the VEGF signaling pathway have also obtained US FDA approval.( 9 )
Vascular endothelial growth factor gene expression is mainly regulated by hypoxia‐inducing factor‐1,( 10 ) but recent studies have also shown its control at the post‐transcriptional levels. First, there are two opposed families of VEGF isoforms, pro‐angiogenic and anti‐angiogenic VEGF (such as VEGF165 and VEGF165b, respectively), which are produced by alternatively spliced forms of mRNA depending on the splice site choice of exon 8.( 11 , 12 ) The second example is a riboswitch found in human VEGF mRNA 3′ UTR, which undergoes a binary conformational change in response to environmental signals such as interferon–γ and hypoxia, and regulates VEGF translation in myeloid cells.( 13 )
In eukaryotes, protein‐coding sequences in primary transcripts (pre‐mRNA) are interrupted by introns, which are eliminated by a multiprotein complex known as the spliceosome.( 14 , 15 ) Recently, two small molecules, spliceostatin A (SSA; Fig. 1)( 16 , 17 ) and pladienolide,( 18 ) were independently reported to target the SF3b complex, a sub‐component of the spliceosome, and inhibit the splicing reaction both in vitro and in vivo. Spliceostatin A was shown to prevent not only splicing but also nuclear retention of a subset of unspliced RNA, thereby producing some aberrant proteins. Indeed, the accumulation of unspliced pre‐mRNA of p27 cyclin‐dependent kinase (CDK) inhibitor resulted in the production of C‐terminally truncated protein (p27*), which could be responsible for the cell‐cycle arrest caused by SSA, since p27* is resistant to proteasomal degradation.( 16 ) Importantly, these compounds were originally isolated by means of their antitumor activities. However, the molecular mechanisms by which splicing inhibition causes selective killing of tumor cells remain to be elucidated. The cell‐cycle arrest by p27* expression is one of the possible reasons for growth inhibition of cancer cells.
Figure 1.
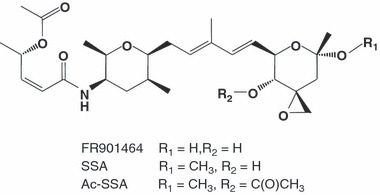
Chemical structure of FR901464, spliceostatin A (SSA) and its inactive derivative, acetyl‐spliceostatin A (Ac‐SSA).
In this study, we focused on VEGF as another possible target of SSA because of: (i) an important role of VEGF in tumorigenesis; (ii) regulation of VEGF activity by alternative splicing; and (iii) the reported inhibition of hypoxia‐induced transactivation of the human VEGF promoter by pladienolide B.( 19 , 20 ) Here, we report that SSA exerts a strong anti‐angiogenic activity in vivo due to the inhibition of transcription and splicing of VEGF mRNA.
Materials and Methods
Materials and cultured cell lines. Preparations and characterizations of FR901464, SSA, and Ac‐SSA (Fig. 1) were described previously.( 16 ) Acyclic retinoid (ACR) was a generous gift from Kowa (Tokyo, Japan).( 21 ) HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Wako Pure Chemical Industries, Osaka, Japan) supplemented with 10% fetal bovine serum (FBS) and grown in a humidified incubator with 5% CO2 at 37°C.
RT‐PCR. HeLa cells grown to confluence were treated with vehicle (0.1% methanol), SSA or acetylated‐SSA (Ac‐SSA) and total RNA was prepared with TRIzol (Invitrogen, Carlsbad, CA, USA). Total RNA was reverse‐transcribed by random 9‐mer primers using a RNA PCR Kit (AMV) Ver.3.0 (Takara Bio Inc., Shiga, Japan). cDNA fragments were analyzed by either a PCR kit with Phusion DNA High‐Fidelity DNA polymerase (AR BROWN, Tokyo, Japan) followed by 10% PAGE gel electrophoresis or qPCR using SYBR Premix Ex Taq II (Takara Bio Inc.) and LightCycler480 (Roche, Mannheim, Germany). In qPCR, all primer pairs were designed by Primer Express software (Applied Biosystems, Foster City, CA, USA) or Universal ProbeLibrary Assay Design Center (Roche Applied Science, Tokyo, Japan), and unique products from qPCR were confirmed by melt‐curve analysis. The sequences of the primers are available on request.
Western blotting. Cell lysates were fractionated using SDS‐PAGE and immunoblot analysis as described.( 22 ) Antibodies to VEGF (sc‐507) and GAPDH (#MAB374) were obtained from Santa Cruz (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and Millipore (Billerica, MA, USA), respectively.
Enzyme‐linked immunosorbent assay (ELISA). HeLa cells were grown in 2 mL DMEM+10% FBS in each 6‐well culture plates, and the medium was collected after treatment with vehicle (0.1% methanol), SSA or Ac‐SSA for 12 and 24 h. The VEGF levels in the medium (100 μL out of 2 mL) of each sample were quantitated using a Human VEGF ELISA Kit (RayBiotech, Norcross, GA, USA) and they were normalized by cell numbers.
Chicken chorioallantoic membrane (CAM) assay. A CAM assay was performed as previously described.( 21 , 23 , 24 ) To monitor tumor‐induced angiogenesis, gelatin sponges (Gelfoam, Upjohn Company, Kalamazoo, MI, USA) were cut and placed on top of a growing CAM at day 8.( 21 , 25 ) The sponges were then adsorbed with 20 μL of cell suspension of HeLa cells (3 × 105 cells per sponge) supplemented with chemicals with or without 100 ng/mL VEGF (Santa Cruz). After further incubation for 4 days, the CAM were photographed using a Leica model M420 microscope (Leica Microsystems, Wetzlar, Germany) at ×16 magnification. Quantitative evaluations of the angiogenic response were carried out by counting microvessels inside the border region around the sponge converging toward the implant under the microscope.
Chromatin immunoprecipitation (ChIP) analysis. HeLa cells were treated with 200 nM SSA for the indicated times, and protein‐DNA complexes were cross‐linked with formaldehyde and sonicated to yield 500‐bp fragments (Bioruptor, CosmoBio., Tokyo, Japan). Protein‐DNA complexes were precipitated by the specific antibodies, and precipitated DNA were purified with Nucleospin Extract II kit (Macherey‐Nagel, Duren, Germany) and subjected to qPCR. The antibodies against RNA polymerase II (Pol II) phosphorylated at Ser‐2 or Ser‐5 in the C‐terminal region of the large subunit (CTD) were purchased from Covance (Princeton, NJ, USA). The conditions for qPCR in ChIP are the same as those in RT‐qPCR. Specific primer sequences for ChIP are available on request.
Microarrays. Total RNA was extracted from HeLa cells using an RNAeasy kit (Qiagen, Hilden, Germany). cDNA synthesis, cRNA amplification, biotinylation and fragmentation were performed with the Poly‐A RNA Control Kit, One‐Cycle cDNA Synthesis Kit, Sample Cleanup Module and IVT Labeling Kit (Affymetrix, Santa Clara, CA, USA). Labeled target RNA was hybridized onto Human Genome U133 Plus 2.0 GeneChip expression arrays (Affymetrix). For statistical analysis, one‐sided Wilcoxon’s signed rank test was used to assess probe‐pair saturation, calculate a P‐value for changes of gene expression and then classify each gene as “increase” (change in P‐value <0.002667), “no change” or “decrease” (change in P‐value >0.997333). All analyses were performed using the Affymetrix GeneChip Operating System (GCOS) Ver.1.4 at the Support Unit for Bio‐material Analysis in RIKEN BSI Research Resources Center (RRC).
Statistical analyses. The data were expressed as the means ± standard deviations (SD) and analyzed using Student’s t test. General linear models (analysis of variance) were used to study differences among the groups. Post hoc pairwise comparisons were performed with Bonferroni’s test if the overall test was significant. Differences with P < 0.05 were considered statistically significant. Asterisks indicate significant differences as follows: ***P < 0.001; **P < 0.01; *P < 0.05.
Results
Inhibition of VEGF expression by SSA. To see whether the production of VEGF mRNA is affected by inhibiting cellular splicing activity, we first treated HeLa cells with SSA followed by RNA extraction and RT‐PCR. Primers to detect both the pro‐angiogenic isoform (VEGF165, ∼200‐bp amplicon) and the anti‐angiogenic isoform (VEGF165b, ∼150‐bp amplicon) were used in PCR. As a negative control experiment, we used an SSA derivative acetylated at the C4 hydroxyl group (Ac‐SSA; Fig. 1); this compound fails to induce p27*, a product of pre‐mRNA of p27, and is unable to compete with biotin‐SSA for interaction with SF3b.( 16 ) As a result, SSA, but not Ac‐SSA, decreased the amount of VEGF165 in a time‐dependent manner (Fig. 2a). We could not detect the VEGF165b isoform in HeLa cells regardless of the presence or absence of SSA. We next tried to confirm that production of VEGF protein is also impaired by treatment with SSA. Western blot analysis showed that the amount of intracellular VEGF protein decreased by treatment with SSA (Fig. 2b) or its mother compound FR901464. We measured the amounts of VEGF protein secreted into medium by HeLa cells treated with the compounds (Fig. 2c). When cells were challenged with SSA or FR901464, the amount of secreted VEGF was significantly reduced to 30–40% of the untreated level, whereas Ac‐SSA had no effect. These results suggest that the splicing inhibitor prominently suppresses pro‐angiogenic VEGF production through the inhibition of mature mRNA production, which might be due to its action of inhibiting splicing.
Figure 2.

Spliceostatin A (SSA) suppressed vascular endothelial growth factor (VEGF) production. (a) HeLa cells were treated with 200 nM SSA or inactive acetylated‐SSA (Ac‐SSA) for the indicated times, and RT‐PCR analysis was performed using primers that detect both pro‐angiogenic VEGF165 (∼200‐bp) and anti‐angiogenic VEGF165b, (∼150‐bp). (b) HeLa cells were treated with 200 nM SSA or FR901464 (FR) for 3 or 12 h, and then western blot analysis using anti‐VEGF antibody was performed. The GAPDH was a loading control. (c) HeLa cells were treated with vehicle (0.1% methanol; MeOH), 200 nM FR901464, 200 nM SSA or Ac‐SSA for the indicated times. The levels of secreted VEGF protein in the medium were determined by ELISA and were normalized by cell numbers in the same dishes. The results are expressed as percentages of the control cells treated with vehicle as the means ± SD of three independent experiments, each performed in duplicate. ***P < 0.001; *P < 0.05.
Spliceostatin A inhibits the splicing of VEGF mRNA. To confirm the inhibition of splicing in VEGF pre‐mRNA by SSA, we estimated the level of either spliced (mature) and unspliced (pre‐mRNA) VEGF mRNA production by RT‐qPCR. To detect mature mRNA, we determined the relative mRNA amounts by using a primer pair flanking a splicing border (exon 1/exon 2) of the VEGF gene (Fig. 3a). The levels of mature mRNA were decreased by approximately 80% in the presence of SSA at concentrations ≥20 nM. In contrast, the same PCR products were not affected by Ac‐SSA and only partial suppression was observed at 200 nM. Reciprocally, RT‐qPCR using a primer pair positioned within intron 1 (intron 1/intron 1) of the VEGF gene indicated that pre‐mRNA accumulated in the presence of SSA but not Ac‐SSA (Fig. 3b). The extent of accumulation of unspliced transcripts of VEGF was obviously lower than that of decrease in mature transcripts, suggesting that SSA not only compromised the splicing efficiency but also affected the transcriptional efficiency or mRNA turnover. Consistent with this, PCR using a primer pair positioned within exon 1 (exon 1/exon 1), by which we can logically detect the sum of spliced and unspliced mRNA, showed a significant reduction of the total level of transcripts by SSA (Fig. 3c). Splicing of GAPDH transcripts was scarcely inhibited by SSA (Fig. 3d), suggesting that the SSA’s inhibitory action on splicing preferentially targets a subset of genes including VEGF. We therefore hypothesized that SSA has anti‐tumor angiogenesis activity by suppressing VEGF production, and explored this idea by investigating the relationship between VEGF expression and tumor angiogenesis in in vivo models.
Figure 3.
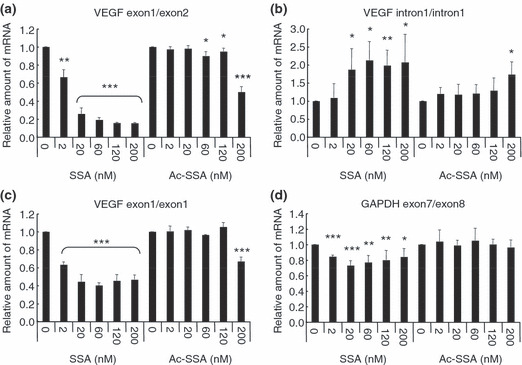
Spliceostatin A (SSA) inhibited production of vascular endothelial growth factor (VEGF) mRNA in a dose‐dependent manner. HeLa cells treated with various concentrations of SSA or inactive acetylated‐SSA (Ac‐SSA) for 6 h. RT‐qPCR analyses were performed for mature mRNA (a), pre‐mRNA (b) and the sum of both (c) of VEGF, as well as for mature mRNA of GAPDH (d). The results are the means ± SD of three independent experiments. ***P < 0.001; **P < 0.01; *P < 0.05.
Spliceostatin A inhibits in vivo tumor angiogenesis. We performed a CAM assay to see the effect of SSA on tumor‐induced angiogenesis in vivo (4, 5). In this assay, HeLa cells in a gelatin sponge implanted on top of the growing CAM stimulates angiogenesis of membrane vessels by secreting VEGF. Four days after implantation of the sponge, newly formed microvessels were detectable in control CAM (Fig. 4a). The numbers of microvessels were significantly reduced when a sponge was supplemented with 2 nM SSA or a reference compound ACR( 21 ) at 5 μM. In contrast, FR901464 failed to inhibit microvessel formation at the same concentration as SSA, and only a weak inhibition was detected at a concentration 10 times higher (Fig. 4a). As FR901464 is chemically unstable,( 16 ) the weaker activity than SSA is probably due to its in vivo instability. Quantitative evaluation showed that SSA reduced microvessel formation by approximately 50% of the control, which is almost the same level as the medium alone (Fig. 4b). Furthermore, the reduced number of microvessels in the presence of SSA was restored to the control level by adding 100 ng/mL of exogenous VEGF (Fig. 5). In contrast, Ac‐SSA did not show such inhibitory activity on angiogenesis. These CAM results suggest that the anti‐angiogenic activity of SSA observed here is due to the inhibition of VEGF production.
Figure 4.
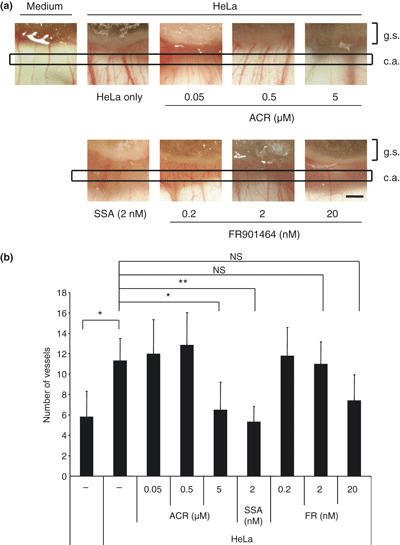
Comparison among spliceostatin A (SSA), FR901464 and ACR in their ability to suppress in vivo blood vessel formation induced by HeLa cells in chicken chorioallantoic membrane (CAM) tissues (gelatin sponge‐CAM assay). (a) HeLa cells were delivered at 3 × 105 cells per embryo onto the top of the CAM at day 8 using a gelatin sponge implant, supplemented with or without SSA (2 nM), FR901464 (0.2, 2 and 20 nM) and acyclic retinoid (ACR) (0.05, 0.5 and 5 μM) as a positive control. At day 12, newly formed microvessels sprouting towards the implant were observed under the microscope. Representative results are shown. g. s., represents the area of gelatin sponge; c. a., represents the count area meaning the area in which we counted microvessels. Black scale bars represent 500 μm. (b) The numbers of the microvessels inside the same area (surrounded by squares) were counted. Each datum represents the average ±SD (n = 6–7). **P < 0.01; *P < 0.05. NS, not significant.
Figure 5.

Suppression of in vivo blood vessel formation induced by HeLa cells in chicken chorioallantoic membrane (CAM) tissues by spliceostatin A (SSA). (a) HeLa cells were delivered at 3 × 105 cells per embryo onto the top of the CAM at day 8 using a gelatin sponge implant, supplemented with or without 2 nM SSA, acetylated‐SSA (Ac‐SSA) or 100 ng/mL vascular endothelial growth factor (VEGF). At day 12, newly formed microvessels sprouting towards the implant were observed under the microscope. Representative results are shown. g. s., represents the area of gelatin sponge; c. a., represents the count area meaning the area in which we counted microvessels. (b) The numbers of the microvessels inside the same area were counted. Each datum represents the average ±SD (n = 6–7). ***P < 0.001; *P < 0.01.
Effects of splicing inhibition on gene transcription. Given the significant decrease in the sum of spliced and unspliced mRNA of VEGF (Fig. 3c), we postulated that SSA reduced not only splicing but also the transcriptional efficiency of the VEGF gene. To see whether SSA treatment affects the transcription, we determined the levels of Pol II recruitment by using ChIP. The C‐terminal domain of a Pol II large subunit is specifically modified during transcriptional initiation and elongation: Ser‐5 is phosphorylated by transcription factor IIH (TFIIH) upon initiation, whereas Ser‐2 is phosphorylated by p‐TEFb during elongation.( 26 ) We found that the recruitment of Pol II phosphorylated at Ser‐5 especially on the promoter, and the exon 1 region was drastically reduced by SSA treatment (Fig. 6a). The recruitment of Pol II phosphorylated at Ser‐2 to the VEGF gene region was also impaired by SSA. These results imply that the decrease in the VEGF mRNA level by SSA is ascribed to not only splicing inhibition but also inhibition of the transcriptional processes including Pol II recruitment.
Figure 6.
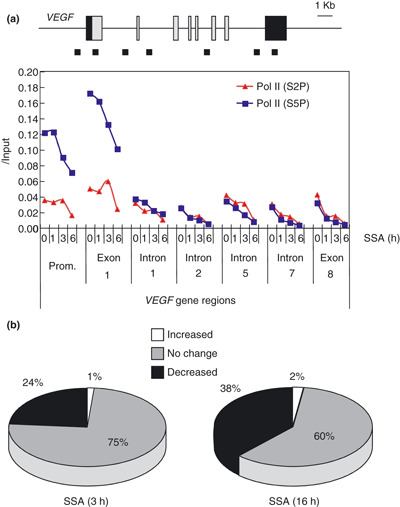
(a) HeLa cells were treated with 200 nM spliceostatin A (SSA) for the indicated times, and chromatin immunoprecipitation (ChIP) analysis with anti‐RNA polymerase II (Pol II) antibody phosphorylated at Ser‐2 or Ser‐5 in C‐terminal domain (CTD) (red or blue lines, respectively) was performed. Schematic representation of the designed primer pairs targeting seven different sites of the vascular endothelial growth factor (VEGF) gene region used in ChIP are shown above as small squares. The intensity of signals of each sample was calculated from the ratio to those of the input sample. The experiments were performed independently at least three times, and a representative result is shown. (b) Summary of the results of the microarray analyses in HeLa cells treated with 200 nM SSA for 3 or 16 h. Total numbers of genes found on Human Genome U133 Plus 2.0 GeneChip expression arrays (Affymetrix, Santa Clara, CA, USA) are presented as either no change (gray), decreased (black) or increased (white).
We further investigated whether the inhibition of splicing reaction affects global gene expression. We analyzed genome‐wide transcription using DNA microarrays to monitor the change in the gene expression pattern in HeLa cells following the SSA challenge, and found that 12,967 (24%) and 20,815 (38%) of transcripts were downregulated by treatment with 200 nM SSA for 3 h and 16 h, respectively (Fig. 6b). These results indicate that gene transcription is globally affected by SSA and suggest a link between splicing and transcription. Because SSA has potent antitumor activity, we extracted the downregulated genes whose molecular functions are involved in cell proliferation, anti‐apoptosis or angiogenesis as classified by Gene Ontology (Table 1). Among the many genes possibly involved in tumorigenesis, VEGF was ranked as fifth of the cell proliferation‐related genes and top of the angiogenesis‐related genes. Thus, the inhibition of not only splicing but also transcription of the VEGF gene appears to be responsible, at least in part, for the antitumor activity of SSA, although we cannot rule out the possible contribution of other genes.
Table 1.
List of genes downregulated by spliceostatin A (SSA) treatment in HeLa. Genes relevant to tumorigenesis (proliferation, anti‐apoptosis and angiogenesis) whose expression was down‐regulated following treatment with 200 nM SSA for 3 and 16 h in the microarray analyses are summarized. The extent of changes in the expression levels of each gene are presented on the left side as log ratios, and listed from the top according to the extent of changes on cells treated for 3 h. Genes whose expression levels were downregulated >2.8‐fold (signal log ratio ≤−1.5) are listed
| 3 h | 16 h | Gene title |
|---|---|---|
| Cell proliferation (GO : 0008283) | ||
| −3.8 | −3.8 | Cyclin‐dependent kinase 3 |
| −3.1 | −2.8 | Patched homolog 1 (Drosophila) |
| −3.1 | −2.5 | CDC‐like kinase 1 |
| −3.0 | −6.7 | Interleukin 6 receptor |
| −2.5 | −5.0 | Vascular endothelial growth factor A |
| −2.0 | −3.1 | Mature T‐cell proliferation 1 |
| −1.9 | −1.6 | MAX dimerization protein 1 |
| −1.8 | −5.1 | KIT ligand |
| −1.7 | −4.0 | TRAF interacting protein |
| −1.7 | −1.5 | Tumor necrosis factor (ligand) superfamily, member 9 |
| −1.7 | −2.4 | Taxilin alpha |
| −1.7 | −2.6 | Zinc finger E‐box binding homeobox 1 |
| −1.6 | −2.7 | CDNA clone IMAGE:5311370 |
| −1.5 | −6.3 | Interferon regulatory factor 2 |
| −1.5 | −2.6 | Transforming growth factor, alpha |
| −1.5 | −1.9 | Tripartite motif‐containing 27 |
| Anti‐apoptosis (GO : 0006916) | ||
| −3.9 | −3.1 | Neuregulin 2 |
| −3.2 | −5.2 | CASP8 and FADD‐like apoptosis regulator |
| −3.0 | −4.0 | T‐box 3 (ulnar mammary syndrome) |
| −2.7 | −7.3 | NLR family, apoptosis inhibitory protein |
| −2.3 | −1.6 | Chromobox homolog 4 (Pc class homolog, Drosophila) |
| −2.2 | −3.9 | Baculoviral IAP repeat‐containing 4 |
| −2.2 | −4.6 | NLR family, apoptosis inhibitory protein |
| −2.1 | −1.9 | BCL2/adenovirus E1B 19kDa interacting protein 1 |
| −1.9 | −2.2 | Retinoic acid receptor, alpha |
| −1.7 | −3.8 | Cytokine induced apoptosis inhibitor 1 |
| −1.6 | −3.1 | Insulin‐like growth factor 1 receptor |
| −1.6 | −4.0 | Phosphoinositide‐3‐kinase, catalytic, alpha polypeptide |
| −1.5 | −2.7 | Phosphoprotein enriched in astrocytes 15 |
| −1.5 | −4.8 | Sema domain, immunoglobulin domain (Ig), transmembrane domain (TM) and short cytoplasmic domain, (semaphorin) 4D |
| −1.5 | −2.4 | BCL2‐like 2 |
| −1.5 | −3.1 | Megakaryoblastic leukemia (translocation) 1 |
| Angiogenesis (GO : 0001525) | ||
| −2.5 | −5.0 | Vascular endothelial growth factor A |
| −2.1 | −3.8 | Secretogranin II (chromogranin C) |
| −2.0 | −4.1 | Angiogenic factor with G patch and FHA domains 1 |
| −1.6 | −3.5 | Jagged 1 (Alagille syndrome) |
| −1.5 | −2.6 | Transforming growth factor, alpha |
Discussion
We demonstrated that SSA inhibited the production of VEGF from cancer cells and suppressed cancer angiogenesis. HeLa cells, epithelial‐like cells derived from cervical cancers, secrete a significant amount of VEGF as a paracrine factor to promote formation of new blood vessels from the vicinity. On the other hand, vascular endothelial cells consistently express VEGF receptors, and thus have been considered as the primary recipients of cancer‐cell‐derived VEGF. It is extensively studied that interference of VEGF expression or its signaling prevents cancer angiogenesis.( 5 , 7 ) As VEGF is mandatory to cancer angiogenesis not only through the angiogenic cascade of paracrine VEGF signaling between cancer cells and endothelial cells, but also through supporting the maintenance or survival of vascular cells as an autocrine factor produced in endothelial cells,( 27 ) it is essential for proliferation, survival and differentiation of tumor blood vessels.( 5 ) We showed that SSA reduced the production of VEGF as a paracrine factor in cancer cells. Therefore, the results strongly suggest that SSA’s anticancer activity is, at least in part, mediated by its anti‐angiogenesis activity, although a large number of genes other than VEGF could be involved in the anti‐carcinogenic activity of SSA (Table 1).
There are a number of steps in regulating VEGF post‐transcriptionally: alternative splicing and riboswitch.( 13 ) The delicate regulatory mechanisms at the mRNA levels might be associated with the higher sensitivity of VEGF to splicing inhibition. Initially, we speculated that SSA changes the pattern of alternative splicing to give rise to a predominant population of anti‐angiogenic VEGF, as is the case for the inhibitor of splicing factor kinases, SRPK1/2.( 28 ) However, it was not the case for SSA, because HeLa cells express only pro‐angiogenic VEGF165 but not anti‐angiogenic VEGF165b (Fig. 2a). Instead, the present study suggests that SSA greatly reduced VEGF gene transcription probably at the initiation step, which might also affect elongation (3, 6). Indeed, pladienolide B, another splicing inhibitor, was originally discovered as an inhibitor of hypoxia‐induced transactivation of the human VEGF promoter.( 19 , 20 ) Furthermore, microarray analysis showed that SSA treatment affected global gene expression, suggesting a link between splicing and transcription, although its mechanism remains elusive. On the contrary, there are a few but significant number of tumor‐related genes whose expression is upregulated by SSA; five genes in cell proliferation (IL‐8, MAPKKK 11, basic leucine zipper nuclear factor 1 [JEM‐1], IL‐6, androgen‐induced proliferation inhibitor), one gene in anti‐apoptosis (tumor necrosis factor, alpha‐induced protein 3), and one gene in angiogenesis (IL‐8). These effects might be less significant than the remarkable activity of SSA to downregulate many genes, but it seems important to elucidate the molecular mechanism underlying the SSA‐induced gene activation. More importantly, the sensitivity to SSA was varied in individual genes, for example, splicing and transcription of GAPDH was fairly resistant to SSA activity, compared with VEGF. It is obviously necessary to elucidate the mechanisms underlying the selective inhibition of splicing and/or transcription by SSA for better understanding of the selective toxicity to tumor cells by splicing inhibition.
In summary, we found that SSA significantly suppressed production of VEGF in cancer cells at both the mRNA and protein levels, and exhibited a potent in vivo anti‐angiogenesis activity in CAM assays. Our finding suggests a promising strategy for anti‐angiogenic therapy by splicing inhibitors.
Disclosure Statement
The authors declare no conflict of interest.
Acknowledgments
The authors are indebted to the members of Research Resources Center, RIKEN Brain Science Institute for microarray analysis. This work was supported in part by a Grant‐in‐Aid for Scientific Research (S) from the Ministry of Education, Culture, Sports, Science and Technology, and by “Chemical Genomics Research Project” from RIKEN.
References
- 1. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti‐VEGF antibody for treating cancer. Nat Rev Drug Discov 2004; 3(5): 391–400. [DOI] [PubMed] [Google Scholar]
- 2. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000; 407(6801): 249–57. [DOI] [PubMed] [Google Scholar]
- 3. Folkman J. Anti‐angiogenesis: new concept for therapy of solid tumors. Ann Surg 1972; 175(3): 409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matsumoto T, Claesson‐Welsh L. VEGF receptor signal transduction. Sci STKE 2001; 2001(112): RE21. [DOI] [PubMed] [Google Scholar]
- 5. Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 2009; 29(6): 789–91. [DOI] [PubMed] [Google Scholar]
- 6. De Bazelaire C, Alsop DC, George D et al. Magnetic resonance imaging‐measured blood flow change after antiangiogenic therapy with PTK787/ZK 222584 correlates with clinical outcome in metastatic renal cell carcinoma. Clin Cancer Res 2008; 14(17): 5548–54. [DOI] [PubMed] [Google Scholar]
- 7. Seiwert TY, Cohen EE. Targeting angiogenesis in head and neck cancer. Semin Oncol 2008; 35(3): 274–85. [DOI] [PubMed] [Google Scholar]
- 8. William WN Jr, Kies MS, Fossella FV et al. Phase 2 study of carboplatin, docetaxel, and bevacizumab as frontline treatment for advanced nonsmall‐cell lung cancer. Cancer 2010; 116(10): 2401–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zangari M, Fink LM, Elice F, Zhan F, Adcock DM, Tricot GJ. Thrombotic events in cancer patients receiving antiangiogenesis agents. J Clin Oncol 2009; 27(29): 4865–73. [DOI] [PubMed] [Google Scholar]
- 10. Maxwell PH, Dachs GU, Gleadle JM et al. Hypoxia‐inducible factor‐1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA 1997; 94(15): 8104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harper SJ, Bates DO. VEGF‐A splicing: the key to anti‐angiogenic therapeutics? Nat Rev Cancer 2008; 8(11): 880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bates DO, Cui TG, Doughty JM et al. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down‐regulated in renal cell carcinoma. Cancer Res 2002; 62(14): 4123–31. [PubMed] [Google Scholar]
- 13. Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress‐responsive RNA switch regulates VEGFA expression. Nature 2009; 457(7231): 915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kramer A. The structure and function of proteins involved in mammalian pre‐mRNA splicing. Annu Rev Biochem 1996; 65: 367–409. [DOI] [PubMed] [Google Scholar]
- 15. Nagai K, Muto Y, Pomeranz Krummel DA et al. Structure and assembly of the spliceosomal snRNPs. Novartis Medal Lecture. Biochem Soc Trans 2001; 2: 15–26. [DOI] [PubMed] [Google Scholar]
- 16. Kaida D, Motoyoshi H, Tashiro E et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre‐mRNA. Nat Chem Biol 2007; 3(9): 576–83. [DOI] [PubMed] [Google Scholar]
- 17. Lo CW, Kaida D, Nishimura S et al. Inhibition of splicing and nuclear retention of pre‐mRNA by spliceostatin A in fission yeast. Biochem Biophys Res Commun 2007; 364(3): 573–7. [DOI] [PubMed] [Google Scholar]
- 18. Kotake Y, Sagane K, Owa T et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 2007; 3(9): 570–5. [DOI] [PubMed] [Google Scholar]
- 19. Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y. Pladienolides, new substances from culture of Streptomyces platensis Mer‐11107. I. Taxonomy, fermentation, isolation and screening. J Antibiot (Tokyo) 2004; 57(3): 173–9. [DOI] [PubMed] [Google Scholar]
- 20. Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y. Pladienolides, new substances from culture of Streptomyces platensis Mer‐11107. II. Physico‐chemical properties and structure elucidation. J Antibiot (Tokyo) 2004; 57(3): 180–7. [DOI] [PubMed] [Google Scholar]
- 21. Komi Y, Sogabe Y, Ishibashi N et al. Acyclic retinoid inhibits angiogenesis by suppressing the MAPK pathway. Lab Invest 2010; 90(1): 52–60. [DOI] [PubMed] [Google Scholar]
- 22. Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci USA 2001; 98(1): 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komi Y, Suzuki Y, Shimamura M et al. Mechanism of inhibition of tumor angiogenesis by beta‐hydroxyisovalerylshikonin. Cancer Sci 2009; 100(2): 269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Komi Y, Ohno O, Suzuki Y et al. Inhibition of tumor angiogenesis by targeting endothelial surface ATP synthase with sangivamycin. Jpn J Clin Oncol 2007; 37(11): 867–73. [DOI] [PubMed] [Google Scholar]
- 25. Ribatti D, Nico B, Vacca A, Presta M. The gelatin sponge‐chorioallantoic membrane assay. Nat Protoc 2006; 1(1): 85–91. [DOI] [PubMed] [Google Scholar]
- 26. Hampsey M, Reinberg D. Tails of intrigue: phosphorylation of RNA polymerase II mediates histone methylation. Cell 2003; 113(4): 429–32. [DOI] [PubMed] [Google Scholar]
- 27. Helotera H, Alitalo K. The VEGF family, the inside story. Cell 2007; 130(4): 591–2. [DOI] [PubMed] [Google Scholar]
- 28. Nowak DG, Amin EM, Rennel ES et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro‐angiogenic to anti‐angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem 2010; 285(8): 5532–40. [DOI] [PMC free article] [PubMed] [Google Scholar]