

Abstract
Hereditary diffuse gastric cancer is a cancer syndrome caused by germline mutations in the gene for the cell adhesion protein E‐cadherin (CDH1). E‐cadherin plays a central role in the maintenance of cell polarity and its loss during tumorigenesis is associated with poorly differentiated cancers and a poor prognosis. Hereditary diffuse gastric cancer is dominated by diffuse‐type gastric adenocarcinoma, often with signet ring cell morphology. Large numbers of stage T1a signet ring cell carcinomas exist in the stomachs of CDH1 mutation carriers from a young age, and these foci sometimes show enrichment to the transition zone between the body and antrum. Generally these signet ring cell carcinomas are hypoproliferative, lack Wnt pathway activation, and are relatively indolent. However, a small proportion of the T1a foci contain cells that are poorly differentiated, display mesenchymal features, and express activated c‐Src and its downstream targets. These same features are observed in more advanced stages of hereditary diffuse gastric cancer progression, suggesting that an epithelial–mesenchymal transition is required for tumor invasion beyond the muscularis mucosae. Hereditary diffuse gastric cancer initiation requires somatic down‐regulation of the second CDH1 allele, which in most cases is caused by DNA promoter hypermethylation. Subsequent to CDH1 down‐regulation, lost polarity in gastric stem or progenitor cells would be predicted to interfere with mitotic spindle orientation and the segregation of cell fate determinants. We predict that this disruption of cell division results in daughter cells being deposited in the lamina propria where their population expands and partially differentiates, resulting in the formation of foci of signet ring cells. (Cancer Sci 2009; 100: 1151–1157)
E‐cadherin is a homophilic cell‐to‐cell adhesion protein that is expressed ubiquitously at the adherens junction of epithelial tissue and effectively acts as a bridge between the cytoskeletons of adjacent cells. Abrogation of its expression leads to a loss of apical–basal cell polarity and consequent disruption of the numerous fundamental processes that require spatial asymmetry such as specialized membrane function, cytoskeletal organization, intracellular vesicle trafficking, stem cell division, and cell migration.
Somatic mutation or down‐regulation of the E‐cadherin gene (CDH1) is common in sporadic tumors and is associated with a poorly differentiated phenotype and poor clinical outcome. Its loss is typified by weakly adherent cells that do not form glands and are often observed in isolation, small clusters, or ribbons.( 1 ) E‐cadherin loss is generally considered to be a late event in tumorigenesis; hence, the protein is frequently described as a suppressor of invasion and metastasis.( 2 ) However, the identification of germline mutations in CDH1 and the characterization of the associated cancer syndrome, hereditary diffuse gastric cancer (HDGC), have demonstrated an etiological role for E‐cadherin loss in tumor initiation. Here, we describe HDGC's molecular and clinical characteristics and review data pertinent to E‐cadherin's role in the initiation of gastric cancer.
Germline CDH1 Mutation Spectrum
To date, more than 100 different germline CDH1 mutations have been described in HDGC families. Of the identified mutations, about half are frameshift or nonsense.( 3 ) Functional assays have also confirmed the deleterious effect of several mis‐sense changes.( 4 , 5 ) In addition to the coding sequence mutations, specific polymorphisms in the CDH1 promoter (‐160A) and intron 2 regulatory region (163 + 37235A) are also associated with an increased incidence of diffuse gastric cancer.( 6 , 7 ) In the homozygous state, these low penetrance alleles increase relative risk by between two and 20‐fold.
Although inactivating germline CDH1 mutations have been observed in a diverse range of ethnic groups, it is notable that they appear to occur less frequently in countries with high sporadic gastric cancer rates such as Japan, Korea, Italy, and Portugal. This observation may relate to chance clusters of sporadic gastric cancer obscuring the identity of the families with an inherited susceptibility. Alternatively, the effect may be explained by the population frequencies of low penetrance alleles such as those discussed above; conceivably, the co‐existence of low penetrance CDH1 polymorphisms and inactivating germline CDH1 mutations may be associated with reduced embryonic viability in these populations.
HDGC Clinical Features and Pathology
The clinical history of HDGC families is dominated by diffuse‐type gastric adenocarcinoma (Lauren classification( 8 )), often with signet ring cell (SRC) morphology.( 3 ) The mean age at diagnosis of advanced gastric cancer, prior to the recent introduction of endoscopic surveillance programs and prophylactic surgery in HDGC families, has been 40 years (range 14–85 years).( 3 )
The anatomical mapping of stomachs from patients with HDGC who have undergone total gastrectomy has shown that almost all patients have multiple independent foci of stage T1a signet ring cell carcinoma (SRCC).( 9 , 10 ) These foci, here termed ‘early HDGC’ (eHDGC),( 11 ) have penetrated through the basement membrane into the lamina propria but have not invaded the underlying muscularis mucosae (Fig. 1). In one New Zealand series, the average number of eHDGC foci per stomach was in excess of 100, with the largest number (487) being observed in a 16‐year‐old female (Charlton et al.;( 9 ) A. Charlton and V. Blair, unpubl. data, 2008). The number of foci per stomach was not correlated with the age of the patient (V. Blair, pers. comm., 2008), suggesting that a proportion of these foci are transient. The mapped foci ranged in diameter from 0.1 to 10 mm, with most being less than 1 mm.
Figure 1.
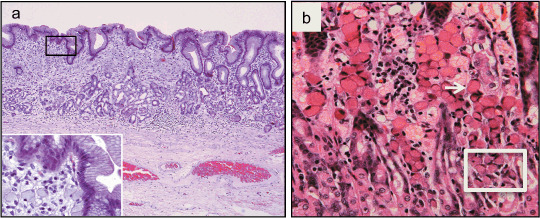
Stage T1a signet ring cell carcinomas (SRCC) in CDH1 germline mutation carrier. (a) A 9 mm focus (the left two thirds of the frame) occupies the full thickness of the mucosa under an intact epithelium (hematoxylin–eosin stain, original magnification 40 ×). Inset frame shows signet ring cells in the lamina propria (original magnification 400 ×). Adapted from Charlton et al.( 9 ) (b) A SRCC showing typical SRCs in the luminal part (one SRC is marked with an arrow) and smaller, less‐differentiated cells at the base of the foci (box). Adapted from Humar et al.( 11 )
In addition to the multifocal T1a SRCC, in situ SRCCs and two layered structures composed of an inner layer of benign mucous cells and an outer layer of SRCs (Pagetoid spread) are also occasionally observed in the gastric glands of germline CDH1 mutation carriers.( 12 ) The relationship between these in situ carcinomas and the T1a foci has not yet been established.
In general, eHDGC lack Wnt pathway activation( 13 ) and are hypo‐proliferative, as evidenced by the relative absence of mitotic cells and lower Ki67 expression than surrounding non‐malignant tissue.( 11 ) This apparent indolence is supported by the differentiated state of the SRCs( 11 ) and the observation that, despite the near 100% penetrance of multifocal disease at a young age, the lifetime risk of advanced diffuse gastric cancer is only around 70%. Thus, the vast majority of T1a carcinomas fail to progress to an advanced state.
In some,( 9 ) but not all( 10 , 12 , 14 , 15 ) mutation carriers’ stomachs, the foci of eHDGC are larger and occur at highest density in the transition zone between the antrum and body of the stomach (Fig. 2). Transition zone enrichment has been observed previously in gastric cancer models: dogs administered with the carcinogen N‐ethyl‐N′‐nitro‐N‐nitrosoguanidine (ENNG) for three months developed SRCCs exclusively in the antral mucosa immediately adjacent to the transition zone.( 16 ) In mice with conditional inactivation of the bone morphogenetic protein receptor Bmpr1a, tumors develop specifically at both the gastric squamocolumnar and gastrointestinal transition zones.( 17 ) Similarly, mice that have been exposed to the carcinogen N‐nitroso‐N‐butylurea( 18 ) or have a specific Smad4 mutation( 19 ) also develop tumors which are observed primarily in the gastrointestinal transition zone. It is not yet known why this region is particularly sensitive to tumorigenesis, although it may relate to its higher proliferation rate.( 17 ) Alternatively, the indecision in cell fate commitment in this zone may correspond to a state of epigenetic plasticity that renders the cells vulnerable to epigenetic dysregulation.
Figure 2.

Stomach map showing size and location of foci of SRCC and mucosal zones in a 28‐year‐old‐male CDH1 germline mutation carrier. Foci are to scale, except foci less than 1 mm are shown as 1 mm for visibility. This patient had 214 foci with a strong clustering in the transition zone. Adapted from Charlton et al.( 9 )
In addition to the stomach, germline CDH1 mutation carriers have developed cancers at other diverse sites including the breast, lung, salivary gland, and colorectum,( 3 , 20 ) but only lobular breast cancer occurs at a frequency significantly above the risk of the general population.( 21 , 22 ) Analogous to diffuse gastric cancer, lobular breast cancer does not form glands and is marked by isolated cells or small clusters and ribbons of poorly differentiated malignant cells.( 1 )
It is unclear why germline CDH1 mutation predisposes predominantly to gastric cancer. One possibility is increased genetic and epigenetic damage occurring in the gastric epithelium due to higher carcinogen exposure, relative hypoxia, chronic inflammation, or Helicobacter pylori infection. A second possibility relates to the gastric mucosa's inherently high cellular turnover and capacity for tissue remodeling or repair; in this active setting, fewer mutational or epi‐mutational events may be required to shift a cell to a poorly controlled invasive, proliferative state. Finally, as discussed later, proteolytic enzyme(s) produced specifically by gastric cells may be required for the penetration of the early, E‐cadherin‐deficient carcinomas through the epithelial basement membrane.
The Second CDH1 Hit
Both the multifocal eHDGC and late stage diffuse gastric tumors observed in HDGC have low E‐cadherin expression compared to the surrounding non‐malignant mucosa, demonstrating that the non‐mutated (second) CDH1 allele has also been down‐regulated or lost.( 11 ) Decreased E‐cadherin expression in eHDGC is associated with reduced membranous staining of other proteins that form part of the adherens junction, providing evidence that loss of the second CDH1 allele precipitates the disintegration of the adherens junction and the subsequent loss of normal cell polarity. Studies in both advanced HDGC tumors( 5 , 23 ) and eHDGC( 13 ) have shown that this ‘second hit’ is caused by promoter hypermethylation in at least 50% of cases. Mutation and LOH‐mediated second hits are reported to occur less frequently.( 24 ) Of interest, the methylation patterns observed in the eHDGCs are monoallelic and specific for each focus,( 13 ) indicating that the second hit has affected a single cell that has expanded clonally. This places epigenetic silencing of the CDH1 promoter at the beginning of eHDGC evolution. Notably, promoter methylation appears to also be a major mechanism underlying CDH1 down‐regulation in sporadic diffuse gastric cancers( 25 ) and lobular breast carcinomas.( 26 )
Promoter hypermethylation is likely to be just one mechanism in a hierarchy of inter‐related epigenetic silencing events implicated in HDGC. In particular, histone modifications are also likely to induce the second CDH1 hit, even if only transiently. The known transcriptional repressors of CDH1 (Snail, Slug, ZEB1, ZEB2,( 27 ) and probably also Twist and E47( 28 )) all act through histone deacetylase activity, and over‐expression of the Polycomb repression complex protein EZH2 causes transcriptional silencing of CDH1 by trimethylation of lysine 27 on histone 3 (H3K27).( 29 ) Repression of CDH1 transcription by these factors may decrease E‐cadherin levels below the critical threshold required for functionality. Since active transcription is crucial to maintaining CpG islands in an unmethylated state,( 30 , 31 , 32 ) short‐term, reversible CDH1 repression may eventually become ‘fixed’ through DNA methylation. This possibility is supported by recent studies with the vHMEC‐ras breast cancer cell line model.( 33 ) Culturing vHMEC‐ras cells in serum‐rich media induces an epithelial–mesenchymal transition (EMT) that is associated with the up‐regulation of Snail and the down‐regulation of CDH1. CDH1 down‐regulation appears to be mediated by histone deacetylation in the early passages after serum exposure and by promoter methylation in later passages. The promoter methylation is permanent and heritable, remaining in place following the withdrawal of serum.
Triggers for the Second CDH1 Hit
Factors which promote the second CDH1 hit in germline CDH1 mutation carriers are likely to trigger SRC formation and contribute to the variable penetrance of HDGC. Several physiological and pathological processes are capable of inducing sustained E‐cadherin down‐regulation in human tissue, including repair processes,( 34 , 35 ) H. pylori infection,( 36 ) the inflammatory response,( 27 , 37 ) and hypoxia.( 38 , 39 ) Perhaps surprisingly, H. pylori infection, which is equally associated with the diffuse and intestinal types of gastric cancer,( 40 ) and severe gastritis are not obvious features of HDGC patients at the time of gastrectomy. This suggests that either these mechanisms are not major triggers of the second CDH1 hit, or that transient episodes of H. pylori infection or gastritis are sufficient to provoke stable DNA methylation.
Proposed Mechanism of HDGC Initiation
The simultaneous occurrence of up to several hundred foci of diffuse gastric cancer in the stomachs of germline CDH1 mutation carriers argues that specific mutations in other genes are unlikely to be required for HDGC initiation, although more widespread epigenetic dysregulation is probable. Therefore, eHDGCs may be regarded as simple neoplasms whose initial existence owes more to the inherent characteristics of the tissue than to widespread mutation and disorganization of the genome.
Lineage labeling experiments with gastric differentiation markers and the proliferation marker Ki67 are consistent with an initial development of eHDGC from the upper isthmus of the neck region of the gastric gland.( 11 , 13 ) The neck region is thought to be the location of the gastric stem and progenitor cells.( 41 ) Initiation appears to be followed by movement towards the luminal surface and expression of mucin markers characteristic of differentiated surface pit cells.( 11 , 13 ) A comparable pattern of SRCC development has been described by Sunagawa et al. in the ENNG‐induced canine gastric cancer model( 16 ) and by Sugihara et al. in human sporadic SRCC.( 42 ) Both groups also noted the formation of an early double‐layered structure in the gastric glands as described in HDGC families by Carneiro et al.( 12 )
We propose that the initiation of eHDGC is caused by the abrogation of E‐cadherin's critical role in cell polarity and epithelial tissue architecture. E‐cadherin‐mediated cell‐to‐cell adhesion provides a spatial cue that promotes cellular asymmetry and polarity.( 43 , 44 ) Without this correct spatial organization, the fundamental processes that regulate cell division, such as the orientation of the mitotic spindle, are disrupted. Since the plane of cell division is parallel to the plane of the mitotic spindle, the orientation of the spindle directs the positioning of the daughter cells. Fleming et al.( 45 ) recently used microtubule fluorescence imaging of the intestinal tissue of adult mice to predict that an apically located spindle with an angle greater than 30° to the apical surface would be likely to produce one daughter cell that had lost contact with the basement membrane. In Drosophila, the mitotic spindle of the dividing pIIa cell in the developing sensory organ of the dorsal cortex rotates to align with the Drosophila E‐cadherin (DE‐cadherin) complex, ensuring the daughter cells retain the orientation of the mother cell.( 46 ) However, partial loss of DE‐cadherin disrupts orientation of the spindle, leading to irregularity in the orientation of cell division and displacement of daughter cells out of the epithelial plane. Similarly, in the developing mouse embryo, knockout of α‐catenin (an intracellular component of the adherens junction) results in randomization of spindle alignment and mis‐oriented cell division in epithelial cells of the skin.( 47 )
Degradation of the basement membrane would be required before misaligned daughter cells could be deposited in the lamina propria (Fig. 3a). Membrane degradation may be induced by the over‐expression of specific matrix metalloproteinases (MMPs). Down‐regulation of E‐cadherin is correlated with up‐regulation of the type IV collagenases MMP‐2 and MMP‐9.( 48 , 49 ) Although eHDGC does not strongly express these proteases (B. Humar, unpubl. data, 2007), limited secretion of these MMPs from the basolateral surface of unpolarized, CDH1‐deficient cells may be sufficient to breach the basement membrane. Alternatively, degradation may be the result of deregulated zymogen secretion. Mucous neck cells, the presumed origin of eHDGC,( 11 ) are precursors of pepsinogen‐producing chief cells (Fig. 3b) and already transcribe pepsinogen mRNA.( 50 ) Early neoplastic cells may secrete sufficient pepsinogen from the basolateral surface for focal perforation of the basement membrane. Accordingly, we have observed a belt of pepsinogen surrounding unpolarized SRCs in eHDGC (Fig. 3c). Focal degradation of type IV collagen α chains has been observed previously in other subtypes of minimally invasive gastric intramucosal neoplastic lesions.( 51 )
Figure 3.
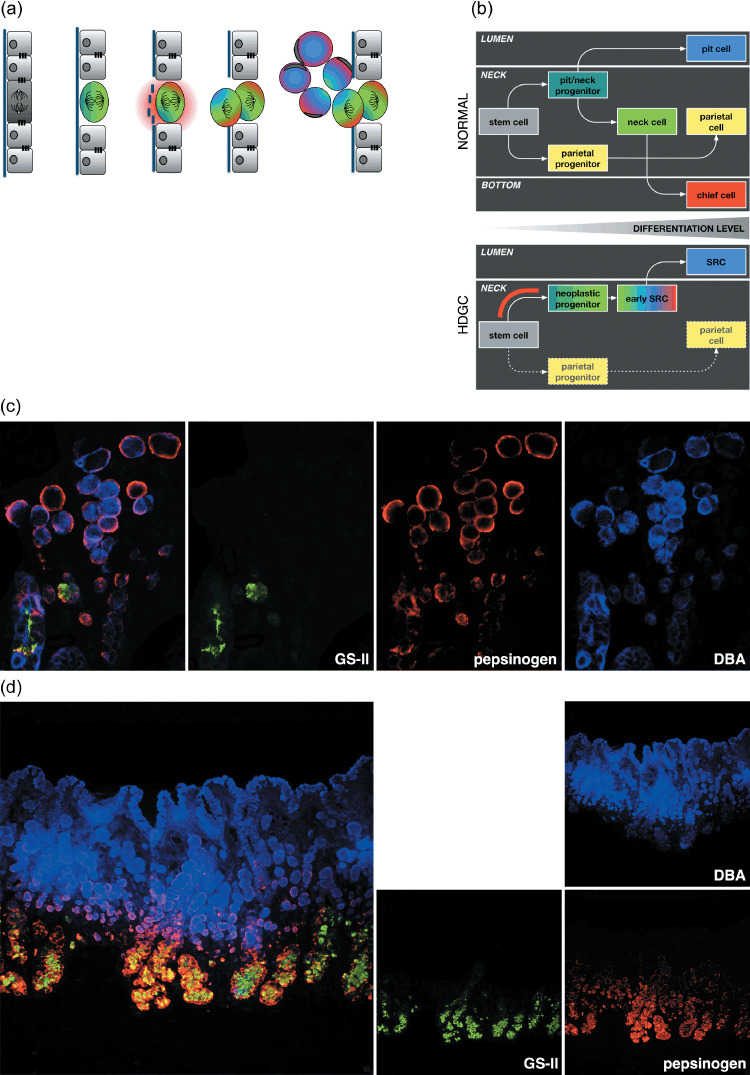
Model of the early development and differentiation of eHDGC. (a) Mis‐orientation of the mitotic spindle and enzymatic digestion of the basement membrane may result in daughter cells dividing out of the epithelial plane and accumulating in the lamina propria. Colors refer to the different gastric lineages as detailed in (b–d). (b) Developmental program of gastric epithelium. The gastric stem cell is thought to reside at the upper isthmus (the upper part of the gastric proliferative zone, the mucous neck region) and give rise to two precursor cells: the mucous neck cell/surface pit cell progenitor and the parietal progenitor. The parietal progenitor produces the lineage of terminally differentiated, acid‐secreting parietal cells found mostly around the neck region of gastric glands. The neck/pit progenitor gives rise to two separate lineages: the terminally differentiated surface pit cells located at the gastric lumen, and the proliferative mucous neck cells at the gastric neck. The neck cells further mature into pepsinogen‐secreting, terminally differentiated chief cells located at the bottom of gastric units (upper). In HDGC, E‐cadherin deficiency causes loss of polarity and, as a consequence, mis‐distribution of gastric cell fate factors (lower). Accordingly, gastric lineages often do not become separated, with neoplastic SRCs expressing markers of up to three distinct cell types including the co‐expression of differentiation markers normally observed at the lumen (pit) or the bottom (chief) of gastric units. Red bar indicates approximate developmental stage at which the second hit can occur. The fate of the parietal lineage during HDGC development is not known. Either the second hit does not affect the gastric stem cell, or E‐cadherin deficiency is not compatible with the survival of the parietal lineage. Notably, diffuse gastric cancer is characterized by a lack of parietal‐type cells. (c) Fluorescence image showing the co‐expression of up to three gastric lineage markers in individual SRCs (GSII/green, mucous neck cells; pepsinogen/red, chief cells; DBA/blue, pit cells).( 11 ) Note simultaneous expression of pit and chief cell markers in the large, luminal SRCs and the peripheral, unpolarized expression of pepsinogen in the SRCs. (d) Birth of intramucosal SRCs from the proliferating region (green) of the gastric antrum. The gastric antrum lacks the typical bottom part of gastric glands enriched with chief cells. Note expression of pepsinogen in the luminal, pit‐type SRCs.
In addition to the failing in spindle orientation, abrogated cell polarity also leads to the disruption of cell fate determination.( 47 , 52 ) Somatic stem cells have the defining capacities of self‐renewal and differentiation. Self‐renewal describes the stem cell's ability to preserve itself by either ‘symmetrical division’ in which both daughter cells are stem cells, or ‘asymmetrical division’ in which one daughter cell remains a stem cell and the other becomes a ‘progenitor cell’ that is locked into a differentiation pathway. The fate of the daughter cells from asymmetric divisions is governed by proteins that migrate and accumulate asymmetrically in response to cues established within polarized cells. Following cell division, the presence or absence of these cell fate proteins in the daughter cells dictates either self‐renewal capability or a pathway to differentiation. This process of fate determination has been well illustrated in Drosophila neuroblasts, where the tumor suppressor proteins Numb, Brat, and the homeobox transcription factor Prospero segregate asymmetrically in the polarized neuroblast and inhibit self‐renewal in one of the two daughter cells.( 53 ) Numb is a repressor of the Notch signaling pathway, Brat represses the transcription factor Myc, and Prospero acts to repress cell cycles mediators including cyclin A and E, as reviewed in Wodarz and Gonzalez( 54 ). Together, these proteins ensure this daughter cell exits the cell cycle and moves into a pathway that generates terminally differentiated neurons. In mutants that lack asymmetrical segregation of Numb, Brat, or Prospero, neither daughter cell is repressed, resulting in proliferation and the development of large metastasizing brain tumors, as reviewed in Knoblich( 55 ). In mammalian neuroepithelial cells, deletion of Numb causes dispersion of neuronal progenitors, along with a mislocalization of E‐cadherin and the disorganization of adherens junctions, resulting in the formation of structures similar to those found in primitive neuroepithelial human tumors. Importantly, silencing of Cdh1 in the neuroepithelium results in a phenotype similar to that of Numb mutants.( 56 )
Further evidence for a profound polarity defect in eHDGC comes from the observation that eHDGC cells can aberrantly express markers of normally separate gastric lineages. The gastric stem cell is thought to produce a precursor that gives rise to both the surface pit cell lineage and the mucous neck cell lineage, which further differentiates into zymogenic chief cells (Fig. 3b).( 57 ) Of note, early eHDGC cells can express markers for pit, neck, and chief cells simultaneously, consistent with a defect in the asymmetric distribution of cell fate determinants (Fig. 3d).( 11 ) Moreover, this observation places the timing, in some instances at least, of the initiating second CDH1 hit at the root of the gastric developmental program, either just before the progenitor cell separates into the pit and the neck lineages, or as early as in the gastric stem cell itself.
In HDGC, the presence of large numbers of indolent, hypo‐proliferative foci of signet ring cells is consistent with the displacement into the lamina propria of daughter cells that are committed to a differentiated fate.( 11 ) Consequently, these foci of SRCC may be relatively short‐lived or transient. However, the unpolarized localization of cell fate determinants and abnormal mitotic spindle orientation may also occasionally result in the displacement of cells with self‐renewal capability. The presence of self‐renewing cells in the lamina propria may lead to the formation of SRCCs which have the capacity for sustained cell division and the potential to progress. The stochastic nature of these initiating events, and perhaps the requirement for additional mutations, may explain the unpredictable progression of these intramucosal foci to advanced cancer.
Progression of eHDGC Beyond the Gastric Mucosa
Invasion of the indolent eHDGC into extramucosal tissue is associated with a phenotypic shift from signet ring morphology to a poorly differentiated state. A small percentage of eHDGC foci contain an underlying population of smaller, poorly differentiated cells that display features of an EMT. The mechanisms behind this EMT are not known; however, they may involve the kinase c‐Src, an established promoter of the mesenchymal state; c‐Src and its downstream targets focal adhesion kinase and Stat‐3 are active specifically in the intramucosal, poorly differentiated cells, but not in the overlying SRCs.( 11 ) Poorly differentiated cells with activated c‐Src increasingly dominate the histology of more advanced (>T1a) disease stages,( 11 ) while the signet ring cells remain concentrated in the region above the neck.
Recent studies have suggested that both normal and neoplastic ‘stem‐like’ cells express markers associated with an EMT.( 58 , 59 ) This observation would support the contention that the population of eHDGC that displays features of an EMT have self‐renewing stem cell characteristics that will enable their survival, expansion, and invasion.
Concluding Remarks
The characterization of HDGC provides several chemopreventative and therapeutic approaches for the clinical management of this cancer syndrome that may have broader implications for sporadic diffuse gastric cancer and lobular breast cancer.( 26 ) Firstly, administration of demethylating agents and/or histone deacetylase (HDAC) inhibitors in a chemopreventative setting would be predicted to maintain expression of the second CDH1 allele and prevent the loss of cell polarity. This might inhibit the development of SRCC, or reduce the number of existing foci. The DNA demethylating agent 5‐aza‐2′‐deoxycytidine, which has been FDA‐approved for the treatment of myelodysplastic syndrome, re‐expresses CDH1 in numerous cell types, and histone deacetylase inhibitors including sodium butyrate( 60 ) and the well‐characterized anticonvulsant valproic acid( 61 ) also promote up‐regulation of CDH1. Interestingly, aspirin has also recently been shown to repress CDH1 methylation in human gastric epithelia.( 62 )
Given the likely importance of stem cells, or stem‐cell characteristics, in HDGC development, agents that interfere with stem cell pathways might provide therapeutic options for both the hereditary and sporadic forms of diffuse gastric cancer. Of the pathways implicated in stem cell biology, Notch and Hedgehog present promising candidates, because they are thought to be required for both the maintenance of tumor‐initiating cells( 63 , 64 ) and the development of the embryonic stomach.( 65 ) Both the Notch and Hedgehog pathways are over‐expressed in diffuse gastric cancer and have been correlated with disease progression.( 66 , 67 ) Finally, inhibitors of the EMT‐associated proteins c‐Src, focal adhesion kinase, and Stat‐3 may also act to inhibit the progression of SRCC.
Acknowledgments
The preparation of this manuscript was supported by the Health Research Council of New Zealand. We acknowledge the many valuable contributions from our colleagues in the field of HDGC research that we were unable to include in this review because of space constraints. The authors are grateful to M. Aspiring for critical reading of the manuscript.
References
- 1. Chan JK, Wong CS. Loss of E‐cadherin is the fundamental defect in diffuse‐type gastric carcinoma and infiltrating lobular carcinoma of the breast. Adv Anat Pathol 2001; 8: 165–72. [DOI] [PubMed] [Google Scholar]
- 2. Berx G, Staes K, Hengel JV et al . Cloning and characterization of the human invasion suppressor gene E‐cadherin (CDH1). Genomics 1995; 26: 281–9. [DOI] [PubMed] [Google Scholar]
- 3. Blair V, Martin I, Shaw D et al . Hereditary diffuse gastric cancer: diagnosis and management. Clin Gastroenterol Hepatol 2006; 4: 262–75. [DOI] [PubMed] [Google Scholar]
- 4. Suriano G, Seixas S, Rocha J, Seruca R. A model to infer the pathogenic significance of CDH1 germline missense variants. J Mol Med 2006; 84: 1023–31. [DOI] [PubMed] [Google Scholar]
- 5. Corso G, Roviello F, Paredes J et al . Characterization of the P373L E‐cadherin germline missense mutation and implication for clinical management. Eur J Surg Oncol 2007. [DOI] [PubMed]
- 6. Humar B, Graziano F, Cascinu S et al . Association of CDH1 haplotypes with susceptibility to sporadic diffuse gastric cancer. Oncogene 2002; 21: 8192–5. [DOI] [PubMed] [Google Scholar]
- 7. Nasri S, More H, Graziano F et al . A novel diffuse gastric cancer susceptibility variant in E‐cadherin (CDH1) intron 2: a case control study in an Italian population. BMC Cancer 2008; 8: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lauren P. The two histological main types of gastric carcinoma: diffuse and so‐called intestinal‐type carcinoma. An attempt at a histo‐clinical classification. Acta Pathol Microbiol Scand 1965; 64: 31–49. [DOI] [PubMed] [Google Scholar]
- 9. Charlton A, Blair V, Shaw D, Parry S, Guilford P, Martin IG. Hereditary diffuse gastric cancer: predominance of multiple foci of signet ring cell carcinoma in distal stomach and transitional zone. Gut 2004; 53: 814–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huntsman DG, Carneiro F, Lewis FR et al . Early gastric cancer in young, asymptomatic carriers of germ‐line E‐cadherin mutations. N Engl J Med 2001; 344: 1904–9. [DOI] [PubMed] [Google Scholar]
- 11. Humar B, Fukuzawa R, Blair V et al . Destabilized adhesion in the gastric proliferative zone and c‐Src kinase activation mark the development of early diffuse gastric cancer. Cancer Res 2007; 67: 2480–9. [DOI] [PubMed] [Google Scholar]
- 12. Carneiro F, Huntsman DG, Smyrk TC et al . Model of the early development of diffuse gastric cancer in E‐cadherin mutation carriers and its implications for patient screening. J Pathol 2004; 203: 681–7. [DOI] [PubMed] [Google Scholar]
- 13. Humar B, Blair V, Charlton A, More H, Martin I, Guilford P. E‐cadherin deficiency initiates gastric signet‐ring cell neoplasia in mice and man. Cancer Res (in Press) 2009; 69: 2050–6. [DOI] [PubMed] [Google Scholar]
- 14. Rogers WM, Dobo E, Norton JA et al . Risk‐reducing total gastrectomy for germline mutations in E‐cadherin (CDH1): pathologic findings with clinical implications. Am J Surg Pathol 2008; 32: 799–809. [DOI] [PubMed] [Google Scholar]
- 15. Barber ME, Save V, Carneiro F et al . Histopathological and molecular analysis of gastrectomy specimens from hereditary diffuse gastric cancer patients has implications for endoscopic surveillance of individuals at risk. J Pathol 2008; 216: 286–94. [DOI] [PubMed] [Google Scholar]
- 16. Sunagawa M, Takeshita K, Nakajima A, Ochi K, Habu H, Endo M. Duration of ENNG administration and its effect on histological differentiation of experimental gastric cancer. Br J Cancer 1985; 52: 771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bleuming SA, He XC, Kodach LL et al . Bone morphogenetic protein signaling suppresses tumorigenesis at gastric epithelial transition zones in mice. Cancer Res 2007; 67: 8149–55. [DOI] [PubMed] [Google Scholar]
- 18. Ward JM, Weisburger EK. Intestinal tumors in mice treated with a single injection of N‐nitroso‐N‐butylurea. Cancer Res 1975; 35: 1938–43. [PubMed] [Google Scholar]
- 19. Hohenstein P, Molenaar L, Elsinga J et al . Serrated adenomas and mixed polyposis caused by a splice acceptor deletion in the mouse Smad4 gene. Genes Chromosomes Cancer 2003; 36: 273–82. [DOI] [PubMed] [Google Scholar]
- 20. More H, Humar B, Weber W et al . Identification of seven novel germline mutations in the human E‐cadherin (CDH1) gene. Hum Mutat 2007; 28: 203. [DOI] [PubMed] [Google Scholar]
- 21. Schrader KA, Masciari S, Boyd N et al . Hereditary diffuse gastric cancer: association with lobular breast cancer. Fam Cancer 2008; 7: 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kaurah P, MacMillan A, Boyd N et al . Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007; 297: 2360–72. [DOI] [PubMed] [Google Scholar]
- 23. Grady WM, Willis J, Guilford PJ et al . Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 2000; 26: 16–7. [DOI] [PubMed] [Google Scholar]
- 24. Barber M, Murrell A, Ito Y et al . Mechanisms and sequelae of E‐cadherin silencing in hereditary diffuse gastric cancer. J Pathol 2008; 216: 295–306. [DOI] [PubMed] [Google Scholar]
- 25. Machado JC, Oliveira C, Carvalho R et al . E‐cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene 2001; 20: 1525–8. [DOI] [PubMed] [Google Scholar]
- 26. Zou D, Yoon H‐S, Perez D, Weeks R, Guilford P, Humar B. Epigenetic silencing in non‐neoplastic epithelia identifies E‐cadherin (CDH1) as a target for chemoprevention of lobular neoplasia. J Pathol 2009. in press. [DOI] [PubMed] [Google Scholar]
- 27. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature Reviews 2007; 7: 415–28. [DOI] [PubMed] [Google Scholar]
- 28. Hayashi M, Nimura K, Kashiwagi K et al . Comparative roles of Twist‐1 and Id1 in transcriptional regulation by BMP signaling. J Cell Sci 2007; 120: 1350–7. [DOI] [PubMed] [Google Scholar]
- 29. Cao Q, Yu J, Dhanasekaran SM et al . Repression of E‐cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008; 27: 7274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mutskov V, Felsenfeld G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J 2004; 23: 138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clark SJ. Action at a distance: epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet 2007; 16 Spec No 1: R88–95. [DOI] [PubMed] [Google Scholar]
- 32. Hahn MA, Hahn T, Lee DH et al . Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res 2008; 68: 10 280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal‐like breast cancers. Proc Natl Acad Sci USA 2008; 105: 14 867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hanby AM, Chinery R, Poulsom R, Playford RJ, Pignatelli M. Downregulation of E‐cadherin in the reparative epithelium of the human gastrointestinal tract. Am J Path 1996; 148: 723–9. [PMC free article] [PubMed] [Google Scholar]
- 35. Demetter P, De Vos M, Van Damme N et al . Focal up‐regulation of E‐cadherin‐catenin complex in inflamed bowel mucosa but reduced expression in ulcer‐associated cell lineage. Am J Clin Pathol 2000; 114: 364–70. [DOI] [PubMed] [Google Scholar]
- 36. Terres AM, Pajares JM, O'Toole D, Ahern S, Kelleher D. H. pylori infection is associated with downregulation of E‐cadherin, a molecule involved in epithelial cell adhesion and proliferation control. J Clin Pathol 1998; 51: 410–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dohadwala M, Yang SC, Luo J et al . Cyclooxygenase‐2‐dependent regulation of E‐cadherin: prostaglandin E (2) induces transcriptional repressors ZEB1 and snail in non‐small cell lung cancer. Cancer Res 2006; 66: 5338–45. [DOI] [PubMed] [Google Scholar]
- 38. Krishnamachary B, Zagzag D, Nagasawa H et al . Hypoxia‐inducible factor‐1‐dependent repression of E‐cadherin in von Hippel‐Lindau tumor suppressor‐null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res 2006; 66: 2725–31. [DOI] [PubMed] [Google Scholar]
- 39. Imai T, Horiuchi A, Wang C et al . Hypoxia attenuates the expression of E‐cadherin via up‐regulation of SNAIL in ovarian carcinoma cells. Am J Pathol 2003; 163: 1437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang C, Yuan Y, Hunt RH. The association between Helicobacter pylori infection and early gastric cancer: a meta‐analysis. Am J Gastroenterol 2007; 102: 1789–98. [DOI] [PubMed] [Google Scholar]
- 41. Karam SM, Straiton T, Hassan WM, Leblond CP. Defining epithelial cell progenitors in the human oxyntic mucosa. Stem Cells 2003; 21: 322–36. [DOI] [PubMed] [Google Scholar]
- 42. Sugihara H, Hattori T, Fukuda M, Fujita S. Cell proliferation and differentiation in intramucosal and advanced signet ring cell carcinomas of the human stomach. Virchows Arch a Pathol Anat Histopathol 1987; 411: 117–27. [DOI] [PubMed] [Google Scholar]
- 43. Nejsum LN, Nelson WJ. A molecular mechanism directly linking E‐cadherin adhesion to initiation of epithelial cell surface polarity. J Cell Biol 2007; 178: 323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Drubin DG, Nelson WJ. Origins of cell polarity. Cell 1996; 84: 335–44. [DOI] [PubMed] [Google Scholar]
- 45. Fleming ES, Zajac M, Moschenross DM et al . Planar spindle orientation and asymmetric cytokinesis in the mouse small intestine. J Histochem Cytochem 2007; 55: 1173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Le Borgne R, Bellaiche Y, Schweisguth F. Drosophila E‐cadherin regulates the orientation of asymmetric cell division in the sensory organ lineage. Curr Biol 2002; 12: 95–104. [DOI] [PubMed] [Google Scholar]
- 47. Lechler T, Fuchs E. Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 2005; 437: 275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Margulis A, Zhang W, Alt‐Holland A, Crawford HC, Fusenig NE, Garlick JA. E‐cadherin suppression accelerates squamous cell carcinoma progression in three‐dimensional, human tissue constructs. Cancer Res 2005; 65: 1783–91. [DOI] [PubMed] [Google Scholar]
- 49. Eastham AM, Spencer H, Soncin F et al . Epithelial‐mesenchymal transition events during human embryonic stem cell differentiation. Cancer Res 2007; 67: 11 254–62. [DOI] [PubMed] [Google Scholar]
- 50. Ramsey VG, Doherty JM, Chen CC, Stappenbeck TS, Konieczny SF, Mills JC. The maturation of mucus‐secreting gastric epithelial progenitors into digestive‐enzyme secreting zymogenic cells requires Mist1. Development 2007; 134: 211–22. [DOI] [PubMed] [Google Scholar]
- 51. Baba Y, Iyama K, Ikeda K et al . Differential expression of basement membrane type IV collagen alpha chains in gastric intramucosal neoplastic lesions. J Gastroenterol 2007; 42: 874–80. [DOI] [PubMed] [Google Scholar]
- 52. Lu B, Roegiers F, Jan LY, Jan YN. Adherens junctions inhibit asymmetric division in the Drosophila epithelium. Nature 2001; 409: 522–5. [DOI] [PubMed] [Google Scholar]
- 53. Januschke J, Gonzalez C. Drosophila asymmetric division, polarity and cancer. Oncogene 2008; 27: 6994–7002. [DOI] [PubMed] [Google Scholar]
- 54. Wodarz A, Gonzalez C. Connecting cancer to the asymmetric division of stem cells. Cell 2006; 124: 1121–3. [DOI] [PubMed] [Google Scholar]
- 55. Knoblich JA. Mechanisms of asymmetric stem cell division. Cell 2008; 132: 583–97. [DOI] [PubMed] [Google Scholar]
- 56. Rasin MR, Gazula VR, Breunig JJ et al . Numb and Numbl are required for maintenance of cadherin‐based adhesion and polarity of neural progenitors. Nat Neurosci 2007; 10: 819–27. [DOI] [PubMed] [Google Scholar]
- 57. Bredemeyer AJ, Geahlen JH, Weis VG et al . The gastric epithelial progenitor cell niche and differentiation of the zymogenic (chief) cell lineage. Dev Biol 2009; 325: 211–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mani SA, Guo W, Liao MJ et al . The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial‐mesenchymal transition. PLoS ONE 2008; 3: e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barshishat M, Polak‐Charcon S, Schwartz B. Butyrate regulates E‐cadherin transcription, isoform expression and intracellular position in colon cancer cells. Br J Cancer 2000; 82: 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Milutinovic S, D’Alessio AC, Detich N, Szyf M. Valproate induces widespread epigenetic reprogramming which involves demethylation of specific genes. Carcinogenesis 2007; 28: 560–71. [DOI] [PubMed] [Google Scholar]
- 62. Tahara T, Shibata T, Nakamura M et al . Chronic aspirin use suppresses CDH1 methylation in human gastric mucosa. Dig Dis Sci 2009. [DOI] [PubMed]
- 63. Farnie G, Clarke RB. Mammary stem cells and breast cancer–role of Notch signalling. Stem Cell Reviews 2007; 3: 169–75. [DOI] [PubMed] [Google Scholar]
- 64. Liu S, Dontu G, Mantle ID et al . Hedgehog signaling and Bmi‐1 regulate self‐renewal of normal and malignant human mammary stem cells. Cancer Res 2006; 66: 6063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fukuda K, Yasugi S. The molecular mechanisms of stomach development in vertebrates. Dev Growth Differ 2005; 47: 375–82. [DOI] [PubMed] [Google Scholar]
- 66. Li DW, Wu Q, Peng ZH, Yang ZR, Wang Y. [Expression and significance of Notch1 and PTEN in gastric cancer]. Chinese Journal of Cancer 2007; 26: 1183–7. [PubMed] [Google Scholar]
- 67. Yanai K, Nagai S, Wada J et al . Hedgehog signaling pathway is a possible therapeutic target for gastric cancer. J Surg Oncol 2007; 95: 55–62. [DOI] [PubMed] [Google Scholar]