

Abstract
Cancer gene therapy, in which pharmacologically active compounds are administered to cancer patients in a genetic form, has been examined not only in animals but also in cancer patients. Viral vector‐induced severe side effects in patients have greatly underscored the importance of non‐viral gene transfer methods. Even though the importance of pharmacokinetics is undoubtedly understood in the development of anticancer therapies, its importance has been less well recognized in non‐viral cancer gene therapy. When transgene products express their activity within transduced cells, such as herpes simplex virus type 1 thymidine kinase and short hairpin RNA, the pharmacokinetics of the vectors and the expression profiles of the transgenes will determine the efficacy of gene transfer. The percentage of cells transduced is highly important if few by‐stander effects are expected. If transgene products are secreted from cells into the blood circulation, such as interferons and interleukins, the pharmacokinetics of transgenes becomes a matter of significant importance. Then, any approach to increasing the level and duration of transgene expression will increase the therapeutic effects of cancer gene therapy. Here we review the pharmacokinetics of both non‐viral vectors and transgene products, and discuss what should be done to achieve safer and more effective non‐viral cancer gene therapy. (Cancer Sci 2008; 99: 856–862)
Since the first cancer gene therapy clinical trial was carried out in the early 1990s, many cancer patients have received gene transfer in an attempt to treat their cancer or to obtain clinical data. Even though a number of achievements in such clinical trials have been reported, gene therapy is still in its infancy phase especially when compared with other anticancer treatments. There are a number of major drawbacks, depending on the type of vector, the type of cancer, and the type of therapeutic compound (protein) used, and these variations make it very difficult to develop rational and universal cancer gene therapy protocols. In general, viral vectors are believed to be more hazardous than non‐viral vectors, and the death of a patient receiving adenoviral vectors tragically proved this.( 1 ) However, non‐viral gene transfer is considered ineffective due mainly to the low and transient nature of transgene expression.
Continued progress has been made in improving viral and non‐viral vectors in the last 15 years. As for non‐viral methods, the hydrodynamic injection of naked plasmid DNA( 2 ) has had a great impact on the subsequent development of non‐viral methods. The level of transgene expression achieved by this method is as high as that obtained using adenoviral vectors, one of the most efficient viral vectors.( 2 , 3 ) Complex formation of plasmids with cationic compounds has decreased as vectors for in vivo use because their levels of transgene expression are very low compared with those obtained by hydrodynamic injection or viral vectors.( 3 , 4 ) Furthermore, extensive studies on the innate immune response against vectors have proven that non‐viral vectors are not always safe, especially when cationic liposomes are used for DNA delivery. Large amounts of inflammatory cytokines are produced when plasmid DNA complexed with cationic liposomes are recognized by antigen‐presenting cells.( 3 , 5 , 6 ) Recognition of unmethylated CpG dinucleotides, or CpG motifs, by Toll‐like receptor‐9 is involved in such responses.( 7 , 8 ) In addition, DAI, an intracellular DNA‐recognizing protein, has recently been identified,( 9 ) and this molecule may respond to DNA irrespective of the presence of CpG motifs.
Once it became known that high levels of transgene expression could be achieved by non‐viral methods, including hydrodynamic injection, the reality of non‐viral cancer gene therapy increased greatly. However, the therapeutic efficacy is determined not only by the level of expression, but also by other characteristics of gene transfer and transgene expression.( 10 , 11 ) When transgene products distribute within transduced cells, such as herpes simplex virus type 1 thymidine kinase, both the pharmacokinetics of the vectors and the expression profile of the transgene determine the therapeutic efficacy. Ectopic expression could induce adverse effects, so that controlling the tissue distribution of vectors is the most important issue in the development of cancer gene therapy. An important class of intracellular therapeutic compounds is RNA, and plasmid vectors expressing small interfering RNA (siRNA) or short hairpin RNA (shRNA) have been investigated as a therapeutic tool to treat cancer and other genetic disorders. Silencing oncogenes or other genes contributing to tumor growth will provide a cancer‐specific therapy with minimal side effects. RNA interference has realized the silencing of target mRNA expression in a sequence‐specific manner. RNA interference, the event of mRNA degradation by siRNA or shRNA, takes place only in cells reached by these molecules. Therefore, the tissue distribution of vectors is highly important in the development of RNA interference‐based cancer gene therapy.( 12 ) However, when transgene products are secreted from transduced cells, the pharmacokinetics of vectors is of little importance. Then, the pharmacokinetics of the transgenes is the major factor determining the therapeutic efficacy of cancer gene therapy.( 11 )
Here we review first the pharmacokinetics of naked plasmid DNA, the most frequently used non‐viral vector. Because of its polyanionic nature, it exhibits unique but common tissue distribution characteristics irrespective of the encoding gene, sequence, or size. Complex formation with cationic compounds, a standard method for increasing the level of transgene expression in cultured cells, is also reviewed. Then, the pharmacokinetics of transgene products is discussed using our recent data on cancer gene therapy in which interferon (IFN)‐expressing plasmids were injected into tumor‐bearing mice.( 13 , 14 )
Non‐viral gene transfer methods
So far, various non‐viral gene transfer methods have been developed and their characteristics for gene transfer and transgene expression have been examined after administration in vivo. Table 1 summarizes the major non‐viral methods for in vivo gene transfer.
Table 1.
Non‐viral methods for in vivo gene transfer
| Vector | Route of injection | Stimulus or additional treatment | Characteristics | References |
|---|---|---|---|---|
| Naked DNA | Extracellular space | None | Localized transgene expression | Muscle( 15 , 17 ), liver( 16 , 17 ), spleen, 17 ), kidney( 17 ), brain( 18 ), tumor( 19 , 20 , 21 , 22 ) |
| High applicability to various targets | ||||
| Naked DNA | Extracellular or intravascular space | Electric pulse | Localized transgene expression | Muscle( 17 , 26 ), liver( 17 , 27 ), spleen( 17 ), kidney( 17 ), tumor( 25 ) |
| High applicability to various targets | ||||
| Possible tissue damage caused by electric pulses | ||||
| Naked DNA | Extracellular or intravascular space | Ultrasound | Localized transgene expression | Tumor( 29 ), carotid artery( 30 ), femoral artery( 31 ) |
| High applicability to various targets | ||||
| Naked DNA | Intravascular space | Massage | Localized transgene expression | Liver( 32 , 33 ) |
| Applicability to other organs not reported | ||||
| Naked DNA | Intravascular space | Occlusion of blood flow | Localized transgene expression | Liver( 35 , 37 ), diaphragm( 38 ) |
| Surgery required for the occlusion | ||||
| Naked DNA | Intravascular space | Pre‐injection of cationic liposomes | Lung‐selective transgene expression | Lung endothelial cells( 36 ) |
| Reduced immunostimulatory response compared with DNA–cationic liposome complexes | ||||
| Naked DNA | Intravascular space | Large volume injection at high speed | Extremely high transgene expression | Whole body( 2 , 4 , 39 ) |
| Possible tissue damage | ||||
| DNA–cationic liposome complex | Extracellular or intravascular space | None | Low‐level transgene expression in vivo | Lung endothelial cells( 46 ) |
| High induction of inflammatory cytokines upon administration | ||||
| DNA–cationic polymer complex | Extracellular or intravascular space | None | Low‐level transgene expression in vivo | Lung endothelial cells( 45 ) |
| Concerns about cytotoxicity of cationic polymers |
Tissue injection of naked DNA. The usefulness of naked plasmid DNA as an in vivo gene transfer vector was realized as early as 1990, when Wolff et al. reported that transgene expression was obtained in skeletal muscle by a simple intramuscular injection of naked plasmid DNA.( 15 ) This injection‐mediated gene transfer had been discussed as a skeletal muscle‐specific event, but the development of sensitive detection systems for expression has revealed that any organ or tissue examined can express transgenes at detectable levels.( 16 , 17 , 18 ) The mechanism of gene transfer by tissue injection of naked plasmid DNA has been debated, but injection‐induced cellular damage and increased pressure is involved in the entry of DNA directly into the cytoplasm and transgene expression follows. Tumor tissues are no exception, and direct injection of naked plasmid DNA into solid tumors results in detectable transgene expression.( 19 , 20 , 21 , 22 ) Therefore, in spite of it being the simplest, most unsophisticated system, direct tissue injection of naked plasmid DNA is a useful method for in vivo gene transfer. The major drawbacks of this approach are the relatively low level of transgene expression and the limited distribution of the cells transduced.( 23 )
Physical method. Various physical forces have been used to increase the level of transgene expression by applying them to the target site for gene transfer after topical or systemic administration of non‐viral vectors. Physical methods with proven positive effects include electric pulses (electroporation), ultrasound (sonoporation), and physical pressure (massaging), all of which are believed to increase the amount of DNA delivered to cells.
Electroporation‐mediated gene transfer is believed to involve high‐voltage pulse‐mediated pore formation and electrophoretic delivery of charged molecules through the pores.( 24 ) Because of its universality and flexibility, in vivo electroporation has been applied to increase transgene expression in various organs, including tumors.( 25 , 26 , 27 )
Another physical method used frequently to increase transgene expression is the application of ultrasound. Cavitation is considered a major mechanism for an ultrasound‐induced increase in membrane permeability.( 28 ) Several reports have demonstrated that transgene expression by plasmid DNA is greatly increased by application of ultrasound.( 29 , 30 ) In addition, the expression can be further increased by using microbubble echo contrast agents, which enhance ultrasound‐induced acoustic cavitation. Recently, Suzuki et al. developed bubble liposomes, a liposomal formulation of an ultrasound imaging gas perfluoropropane, and applied them to ultrasound‐mediated gene transfer.( 31 )
A unique and simple gene transfer method was reported by Liu and Huang,( 32 ) who manually pressed or ‘massaged’ mouse liver after systemic injection of naked plasmid DNA. The authors suggested the involvement of pressure‐mediated effects in this mode of gene transfer.( 33 )
Intravascular and hydrodynamic injection of naked DNA. Administration methods and techniques are most critical when naked plasmid DNA is injected into the blood circulation. Because of the presence of high nuclease activity in serum, naked plasmids injected into the tail vein of mice are degraded rapidly,( 34 ) leading to no detectable transgene expression in any organ. Several techniques have been developed to alter the ineffectiveness of naked plasmid DNA. Budker et al. injected naked plasmid DNA dissolved in hypertonic solutions into the portal vein of mice whose hepatic veins were transiently occluded.( 35 ) Song et al. injected naked plasmid DNA into the tail vein of mice who had received an intravenous injection of cationic liposomes.( 36 ) This sequential injection greatly prolonged the exposure time of plasmid DNA to the lung endothelial cells, the target cells, and increased the transgene expression. Increasing the retention time of DNA in target organs was simply achieved by occluding blood vessels, and significantly high transgene expression was obtained in the liver,( 37 ) and in the diaphragm.( 38 )
Regarding gene transfer methods using intravascularly injected naked plasmid DNA, the most important finding was the hydrodynamic injection of naked plasmid DNA, which was first reported independently by Liu's group( 2 ) and Wolff's group( 39 ) in 1999. A simple injection of naked plasmid DNA solution into the tail vein of mice produces a significantly high level of transgene expression in internal organs, with highest expression in the liver. The key points of this delivery are the volume of solution and the injection speed.( 2 ) Since these first reports, this method has been applied to deliver not only plasmids but also other compounds, such as siRNA.( 4 ) Direct cytoplasmic delivery of the injected DNA is involved in the very high transgene expression obtained by this method.( 40 , 41 , 42 , 43 )
Cationic DNA complex. Since Felgner et al. proposed the concept of ‘lipofection’ in 1987,( 44 ) lots of cationic lipids have been developed as transfection reagents. The report of an efficient transfection using polyethyleneimine( 45 ) led to a rush of researchers searching for natural and synthetic polymers that possess gene transfer activity. Mixed formulations of cationic lipids and cationic polymers have also been developed.( 46 , 47 ) In most cases, these DNA complexes are generally designed to have a net positive charge because binding to target cells, the first step of gene transfer, depends on the electrostatic interaction between positively charged complexes and the negatively charged cell surface.
Although the cationic nature of these DNA complexes is effective for delivering genes to cells in culture, this process is sensitive to the presence of any other negatively charged compounds. Therefore, serum proteins interfere significantly with the cationic DNA complex‐mediated transfection to cultured cells.( 48 ) This clearly means that it will be difficult to achieve in vivo transfection using cationic DNA complexes, and many reports have confirmed this. Because of the non‐specificity of the interaction of cationic DNA complexes with negatively charged molecules, cationic DNA complexes administered in vivo interact with various biological components.( 49 )
Requirements for effective cancer gene therapy
Delivering plasmid vectors to tumor tissues has been carried out using some non‐viral gene delivery methods, including direct injection into tumor tissues. Other gene delivery methods by which transgenes are hardly expressed in tumor cells, including hydrodynamic injection, can be used to express anticancer proteins that are secreted into the blood circulation. Thus, the requirements for effective cancer gene therapy vary markedly depending on what types of therapeutic proteins are used. Recently, rapid progress has been made in the application of siRNA and shRNA to cancer gene therapy.( 12 ) Because the site of action of these RNA molecules is the cytosol, their requirements would be the same as those for intracellular proteins.
In a previous review article,( 11 ) we discussed the efficacy of in vivo gene transfer in connection with the following four characteristics: (i) target cell specificity of gene transfer; (ii) the number of cells transduced; (iii) the level of expression; and (iv) the duration of expression (Fig. 1). Even though the efficiency of non‐viral gene transfer methods has improved significantly, the level and duration of expression are still major obstacles to achieving effective cancer gene therapy. Two other parameters, the specificity of gene transfer and the number of cells transduced, are important only when intracellular proteins or shRNA are used as therapeutic compounds. In these cases, all four parameters are important for better therapeutic effects, but the delivery of vectors to the majority of target cells in a cell‐specific manner is often the bottle neck in the development of efficient gene therapy methods.
Figure 1.
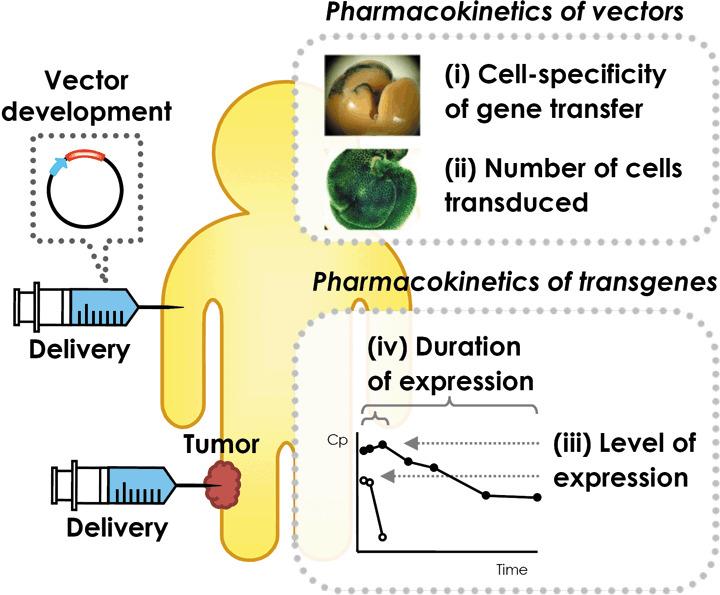
Processes of non‐viral gene transfer. Non‐viral cancer gene therapy is achieved by delivering a proper vector directly to tumor tissue or to another part of the body. The therapeutic effects of gene transfer can be determined by the pharmacokinetics of both vectors and transgene products. Characteristics determining the effects are: (i) the target cell specificity of gene transfer; (ii) the number of cells transduced; (iii) the level of expression; and (iv) the duration of expression.
This problem relating to vector delivery is less important when secreted proteins, such as IFN, are used. The type of cells expressing transgenes can be chosen arbitrarily by researchers, depending on a number of factors, such as the accessibility for vector administration, the profile of transgene expression, the distribution of transgenes into the systemic circulation, and possible tissue damage. It is well known that the profile of transgene expression is highly dependent on the organ injected with naked plasmid DNA.( 17 ) In addition, the level of transgene expression depends greatly on the type of non‐viral gene transfer method. Figure 2 summarizes the luciferase activity in mouse liver after in vivo administration of a firefly luciferase‐expressing plasmid vector using various methods. Plasmid DNA complexes with cationic liposomes or cationic polymers were much less effective in expressing the transgene, even though galactose, a hepatocyte‐specific ligand, was incorporated into the complexes.( 50 , 51 , 52 , 53 , 54 ) The expression level achieved by the hydrodynamic delivery of naked plasmid DNA is at least 1000 times greater than that of other methods.( 41 ) Furthermore, hydrodynamic delivery of as little as 10 ng of naked plasmids (for an approximately 20‐g mouse) reaches the highest expression levels achieved by other non‐viral methods using more than 10 µg of the same plasmid (Fig. 2).
Figure 2.
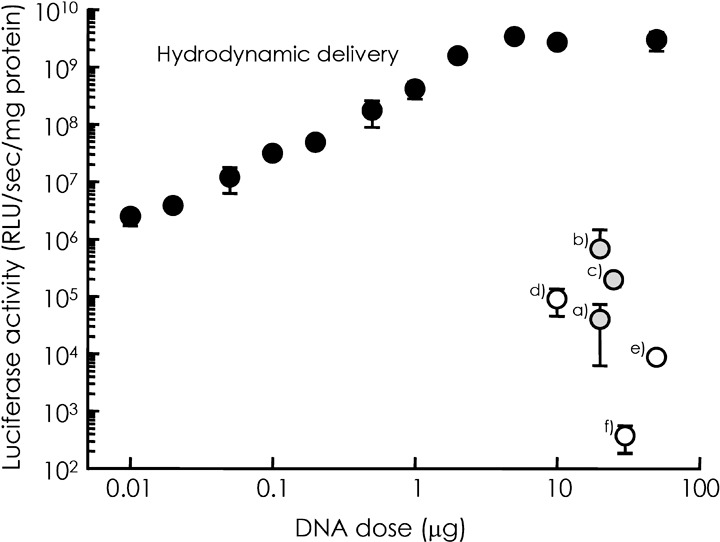
Comparison of the level of transgene expression by several non‐viral gene transfer methods. Firefly luciferase activity (relative light unit [RLU]/s/mg protein) in the liver after non‐viral gene transfer to mice was plotted against the dose of plasmid vector expressing firefly luciferase (pCMV‐Luc). Closed circles are the results of hydrodynamic delivery of pCMV‐Luc injected at different doses.( 41 ) (a) Direct injection of naked plasmid into the liver.( 17 ) (b) Direct injection of naked plasmid into the liver followed by electroporation.( 17 ) (c) Intravenous injection of naked plasmid followed by electroporation.( 27 ) (d) Intravenous injection of plasmid DNA complexed with galactosylated poly ornithine‐fusogenic peptide.( 51 ) (e) Intraportal injection of plasmid DNA complexed with galactosylated polyethyleneimine.( 54 ) (f) Intraportal injection of plasmid DNA complexed with cationic liposomes.( 52 )
Pharmacokinetics of vectors
Intravascular injection. Unlike low molecular weight compounds, plasmid DNA and other macromolecular compounds are significantly limited in their distribution within the body. Plasmid DNA and its complexes with non‐viral vectors can cross blood vessels only in organs with a discontinuous‐type endothelium, such as the liver and spleen, or in solid tumors.( 55 )
Naked plasmid DNA is taken up rapidly by liver sinusoidal endothelial cells and Kupffer cells. This uptake reduces its concentration within the systemic circulation,( 34 , 56 , 57 ) which results in little chance of DNA being delivered to tumor cells. When injected by the hydrodynamic delivery method, plasmid DNA shows different profiles for its tissue distribution, but the major organ involved in the tissue distribution is the liver.( 56 ) Application of electric pulses to organs after intravenous injection of non‐viral vectors has little effect on their tissue distribution.( 17 , 27 ) Depletion of macrophages hardly changes the tissue distribution of naked plasmid DNA (unpublished data, Kako K, Nishikawa M, Yoshida H, Takakura Y, 2006). Thus, the tissue distribution of intravascularly administered naked plasmid DNA is hardly altered by any means, and a large fraction of the injected DNA is delivered to the liver.
Complex formation of plasmid DNA with cationic compounds is intended to increase the interaction with and, following uptake, by target cells. This inevitably increases the interaction of such DNA complexes with biological components.( 50 , 58 , 59 ) In general, intravenously injected cationic DNA complexes are first trapped in capillaries in the lung, followed by accumulation in the liver.( 60 , 61 ) Interactions of the DNA–cationic liposome complex with serum components results in disintegration of the complex, followed by the release and degradation of plasmid DNA.( 58 ) Sakurai et al.( 59 ) reported the involvement of erythrocytes in the interaction of cationic DNA complexes.
Use of receptor‐mediated processes for cell‐specific delivery of plasmid DNA complexes has been investigated extensively. Glycosylation (i.e. covalent conjugation of sugar moieties) of any compound greatly increases its affinity for cells expressing the corresponding sugar receptors. Precise control of the physicochemical properties of DNA complexes with galactosylated compounds increases their delivery to hepatocytes.( 50 ) Some improvements in transgene expression were reported when DNA complexes were modified with transferrin,( 62 ) folate,( 63 ) or antibody.( 64 ) However, the effects of these modifications on the tissue distribution of plasmid DNA complexes have not been investigated fully.
Blood flow to solid tumors is generally low compared with large organs, such as liver and kidney, so the delivery of any pharmaceutical compound to tumor tissues requires prolonged circulation in the blood. Several attempts have been made to deliver plasmid DNA to tumor tissues using DNA complexes.( 65 , 66 , 67 ) However, the amounts of DNA delivered and the level of expression seem to be far below the thresholds required for effective cancer gene therapy.
Direct tissue injection. The pharmacokinetics of plasmid DNA after tissue injection is simple, because the large size of the DNA greatly restricts its distribution within and outside the site of injection. These characteristics are closely associated with the experimental finding that organs and tissues other than those injected show very little transgene expression.
As described above, tissue injection of naked plasmid DNA results in detectable transgene expression in almost all tissues and organs examined. However, cells surrounding the track of the needle injection only express transgenes.( 16 , 23 ) Complex formation of DNA with cationic liposomes further limits the distribution of transgene‐expressing cells.( 20 , 21 ) It has been reported that the area of transgene expression is increased by physical methods, such as electroporation.( 26 , 68 )
Pharmacokinetics of transgene products. Because transgene products are responsible for therapeutic effects after gene transfer, the efficacy of any gene transfer application cannot be discussed without considering its pharmacokinetics. However, the pharmacokinetics of transgenes has not fully been discussed. This is largely because most non‐viral methods are not efficient enough to express transgenes at therapeutic levels, and many reports have simply emphasized how expression is increased using a number of unpublished methods.
The importance of the pharmacokinetics of transgene products is easily appreciated when the therapeutic effects of chemically modified proteins are considered. Of all the technologies developed, conjugation of polyethylene glycol, or PEGylation, is the most successful for improving the pharmacological activities of biologically active proteins.( 69 , 70 , 71 , 72 , 73 , 74 , 75 ) PEGylated derivatives show a much slower clearance from the systemic circulation than unmodified proteins, so that the area under the plasma concentration–time curve (AUC) and the mean residence time (MRT), two important pharmacokinetic parameters, are significantly increased for PEGylated derivatives.
Because the hydrodynamic delivery of naked plasmid DNA gives an enormously high level of transgene expression, its application to experimental therapeutic models has been reported.( 4 ) Kobayashi et al. described how the hydrodynamic delivery of mouse IFN‐β‐ or IFN‐γ‐expressing plasmid vector was effective in inhibiting metastatic growth of colon adenocarcinoma cells in mouse liver.( 13 ) However, as demonstrated in this previous report the transient nature of transgene expression from a conventional plasmid DNA requires multiple injections. Therefore, increasing the duration of IFN transgene expression would be needed for better cancer treatments.
Various methods to increase the duration of transgene expression from plasmid vectors have been reported. For example, a controlled‐release formulation of plasmid DNA greatly extended transgene expression.( 76 ) Various plasmid vectors that promise sustained transgene expression have also been developed,( 77 , 78 , 79 , 80 ) and any of these vectors would be useful for obtaining prolonged expression of therapeutic proteins. In a previous study, we constructed plasmid vectors with reduced numbers of CpG motifs.( 14 ) Compared with conventional CpG‐replete plasmids (pCMV‐Muβ and pCMV‐Muγ), the CpG‐reduced plasmid vectors pGZB‐Muβ and pGZB‐Muγ resulted in sustained expression of mouse IFN‐β and IFN‐γ, respectively, after their hydrodynamic delivery to mice. Significant increases in the pharmacokinetic parameters of the transgenes were obtained. The AUC and MRT of IFN‐γ after injection of pGZB‐Muγ were approximately 60‐ and 4‐fold, respectively, greater than those of pCMV‐Muγ. The survival time of the pGZB‐Muγ‐treated mice was significantly longer than other groups, clearly demonstrating that long‐term expression of IFN enhances the therapeutic effects of IFN cancer gene therapy.
Conclusions and perspectives
Although the initial enthusiasm for gene therapy has been tempered, continuous progress in both viral and non‐viral vector development and administration devices and methods has greatly increased the reality of cancer gene therapy. Because of the difficulty of controlling the pharmacokinetic characteristics of plasmid DNA, irrespective of the naked or complexed form, gene delivery of proteins that are released from transduced cells, such as IFN and interleukins, can be considered to be much easier than that of intracellular proteins. Although skeletal muscle has been considered a suitable target for gene transfer, because of its accessibility, size, and the unique characteristics of sustained transgene expression compared with other organs, its usefulness as a platform for producing anticancer proteins into the systemic circulation remains to be verified. The liver seems to be another promising ‘factory’ producing secreted proteins as demonstrated, but less‐invasive and safer methods than hydrodynamic delivery for gene transfer would be required for future clinical applications.
Acknowledgments
This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- 1. Marshall E. Gene therapy death prompts review of adenovirus vector. Science 1999; 286: 2244–5. [DOI] [PubMed] [Google Scholar]
- 2. Liu F, Song Y, Liu D. Hydrodynamics‐based transfection in animals by systemic administration of plasmid DNA. Gene Ther 1999; 6: 1258–66. [DOI] [PubMed] [Google Scholar]
- 3. Sakurai H, Sakurai F, Kawabata K et al . Comparison of gene expression efficiency and innate immune response induced by Ad vector and lipoplex. J Control Release 2007; 117: 430–7. [DOI] [PubMed] [Google Scholar]
- 4. Kobayashi N, Nishikawa M, Takakura Y. The hydrodynamics‐based procedure for controlling the pharmacokinetics of gene medicines at whole body, organ and cellular levels. Adv Drug Deliv Rev 2005; 57: 713–31. [DOI] [PubMed] [Google Scholar]
- 5. Yasuda K, Ogawa Y, Yamane I, Nishikawa M, Takakura Y. Macrophage activation by a DNA/cationic liposome complex requires endosomal acidification and TLR9‐dependent and – independent pathways. J Leukoc Biol 2005; 77: 71–9. [DOI] [PubMed] [Google Scholar]
- 6. Kako K, Nishikawa M, Yoshida H, Takakura Y. Effects of inflammatory response on in vivo transgene expression by plasmid DNA in mice. J Pharm Sci 2008; in press. [DOI] [PubMed]
- 7. Hemmi H, Takeuchi O, Kawai T et al . A Toll‐like receptor recognizes bacterial DNA. Nature 2000; 408: 740–5. [DOI] [PubMed] [Google Scholar]
- 8. Yasuda K, Wagner H, Takakura Y. Role of immunostimulatory DNA and TLR9 in gene therapy. Crit Rev Ther Drug Carrier Syst 2006; 23: 89–110. [DOI] [PubMed] [Google Scholar]
- 9. Takaoka A, Wang Z, Choi MK et al . DAI (DLM‐1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007; 448: 501–5. [DOI] [PubMed] [Google Scholar]
- 10. Nishikawa M, Huang L. Nonviral vectors in the new millennium: delivery barriers in gene transfer. Hum Gene Ther 2001; 12: 861–70. [DOI] [PubMed] [Google Scholar]
- 11. Nishikawa M, Hashida M. Nonviral approaches satisfying various requirements for effective in vivo gene therapy. Biol Pharm Bull 2002; 25: 275–83. [DOI] [PubMed] [Google Scholar]
- 12. Takeshita F, Ochiya T. Therapeutic potential of RNA interference against cancer. Cancer Sci 2006; 97: 689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kobayashi N, Kuramoto T, Chen S, Watanabe Y, Takakura Y. Therapeutic effect of intravenous interferon gene delivery with naked plasmid DNA in murine metastasis models. Mol Ther 2002; 6: 737–44. [DOI] [PubMed] [Google Scholar]
- 14. Kawano H, Nishikawa M, Mitsui M et al . Improved anti‐cancer effect of interferon gene transfer by sustained expression using CpG‐reduced plasmid DNA. Int J Cancer 2007; 121: 401–6. [DOI] [PubMed] [Google Scholar]
- 15. Wolff JA, Malone RW, Williams P et al . Direct gene transfer into mouse muscle in vivo . Science 1990; 247: 1465–8. [DOI] [PubMed] [Google Scholar]
- 16. Hickman MA, Malone RW, Lehmann‐Bruinsma K et al . Gene expression following direct injection of DNA into liver. Hum Gene Ther 1994; 5: 1477–83. [DOI] [PubMed] [Google Scholar]
- 17. Thanaketpaisarn O, Nishikawa M, Yamashita F, Hashida M. Tissue‐specific characteristics of in vivo electric gene transfer by tissue and intravenous injection of plasmid DNA. Pharm Res 2005; 22: 883–91. [DOI] [PubMed] [Google Scholar]
- 18. Schwartz B, Benoist C, Abdallah B et al . Gene transfer by naked DNA into adult mouse brain. Gene Ther 1996; 3: 405–11. [PubMed] [Google Scholar]
- 19. Soubrane C, Mouawad R, Rixe O et al . Direct gene transfer of a plasmid carrying the herpes simplex virus‐thymidine kinase gene (HSV‐TK) in transplanted murine melanoma: in vivo study. Eur J Cancer 1996; 32A: 691–5. [DOI] [PubMed] [Google Scholar]
- 20. Nomura T, Nakajima S, Kawabata K, Yamashita F, Takakura Y, Hashida M. Intratumoral pharmacokinetics and in vivo gene expression of naked plasmid DNA and its cationic liposome complexes after direct gene transfer. Cancer Res 1997; 57: 2681–6. [PubMed] [Google Scholar]
- 21. Nomura T, Yasuda K, Yamada T et al . Gene expression and antitumor effects following direct interferon (IFN)‐γ gene transfer with naked plasmid DNA and DC‐chol liposome complexes in mice. Gene Ther 1999; 6: 121–9. [DOI] [PubMed] [Google Scholar]
- 22. Natsume A, Mizuno M, Ryuke Y, Yoshida J. Antitumor effect and cellular immunity activation by murine interferon‐β gene transfer against intracerebral glioma in mouse. Gene Ther 1999; 6: 1626–33. [DOI] [PubMed] [Google Scholar]
- 23. O’Hara AJ, Howell JM, Taplin RH et al . The spread of transgene expression at the site of gene construct injection. Muscle Nerve 2001; 24: 488–95. [DOI] [PubMed] [Google Scholar]
- 24. Somiari S, Glasspool‐Malone J, Drabick JJ et al . Theory and in vivo application of electroporative gene delivery. Mol Ther 2000; 2: 178–87. [DOI] [PubMed] [Google Scholar]
- 25. Rols MP, Delteil C, Golzio M, Dumond P, Cros S, Teissie J. In vivo electrically mediated protein and gene transfer in murine melanoma. Nat Biotechnol 1998; 16: 168–71. [DOI] [PubMed] [Google Scholar]
- 26. Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo . Nat Biotechnol 1998; 16: 867–70. [DOI] [PubMed] [Google Scholar]
- 27. Sakai M, Nishikawa M, Thanaketpaisarn O, Yamashita F, Hashida M. Hepatocyte‐targeted gene transfer by combination of vascularly delivered plasmid DNA and in vivo electroporation. Gene Ther 2005; 12: 607–16. [DOI] [PubMed] [Google Scholar]
- 28. Bommannan D, Menon GK, Okuyama H, Elias PM, Guy RH. Sonophoresis. II. Examination of the mechanism(s) of ultrasound‐enhanced transdermal drug delivery. Pharm Res 1992; 9: 1043–7. [DOI] [PubMed] [Google Scholar]
- 29. Huber PE, Pfisterer P. In vitro and in vivo transfection of plasmid DNA in the Dunning prostate tumor R3327‐AT1 is enhanced by focused ultrasound. Gene Ther 2000; 7: 1516–25. [DOI] [PubMed] [Google Scholar]
- 30. Taniyama Y, Tachibana K, Hiraoka K et al . Local delivery of plasmid DNA into rat carotid artery using ultrasound. Circulation 2002; 105: 1233–9. [DOI] [PubMed] [Google Scholar]
- 31. Suzuki R, Takizawa T, Negishi Y et al . Gene delivery by combination of novel liposomal bubbles with perfluoropropane and ultrasound. J Control Release 2007; 117: 130–6. [DOI] [PubMed] [Google Scholar]
- 32. Liu F, Huang L. Noninvasive gene delivery to the liver by mechanical massage. Hepatology 2002; 35: 1314–19. [DOI] [PubMed] [Google Scholar]
- 33. Liu F, Lei J, Vollmer R, Huang L. Mechanism of liver gene transfer by mechanical massage. Mol Ther 2004; 9: 452–7. [DOI] [PubMed] [Google Scholar]
- 34. Kawabata K, Takakura Y, Hashida M. The fate of plasmid DNA after intravenous injection in mice: involvement of scavenger receptors in its hepatic uptake. Pharm Res 1995; 12: 825–30. [DOI] [PubMed] [Google Scholar]
- 35. Budker V, Zhang G, Knechtle S, Wolff JA. Naked DNA delivered intraportally expresses efficiently in hepatocytes. Gene Ther 1996; 3: 593–8. [PubMed] [Google Scholar]
- 36. Song YK, Liu F, Liu D. Enhanced gene expression in mouse lung by prolonging the retention time of intravenously injected plasmid DNA. Gene Ther 1998; 5: 1531–7. [DOI] [PubMed] [Google Scholar]
- 37. Liu F, Huang L. Improving plasmid DNA‐mediated liver gene transfer by prolonging its retention in the hepatic vasculature. J Gene Med 2001; 3: 569–76. [DOI] [PubMed] [Google Scholar]
- 38. Liu F, Nishikawa M, Clemens PR, Huang L. Transfer of full‐length Dmd to the diaphragm muscle of Dmdmdx/mdx mice through systemic administration of plasmid DNA. Mol Ther 2001; 4: 45–51. [DOI] [PubMed] [Google Scholar]
- 39. Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther 1999; 10: 1735–7. [DOI] [PubMed] [Google Scholar]
- 40. Kobayashi N, Hirata K, Chen S, Kawase A, Nishikawa M, Takakura Y. Hepatic delivery of particulates in the submicron range by a hydrodynamics‐based procedure: implications for particulate gene delivery systems. J Gene Med 2004; 6: 455–63. [DOI] [PubMed] [Google Scholar]
- 41. Kobayashi N, Nishikawa M, Hirata K, Takakura Y. Hydrodynamics‐based procedure involves transient hyperpermeability in the hepatic cellular membrane: implication of a nonspecific process in efficient intracellular gene delivery. J Gene Med 2004; 6: 584–92. [DOI] [PubMed] [Google Scholar]
- 42. Zhang G, Gao X, Song YK et al . Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther 2004; 11: 675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suda T, Gao X, Stolz DB, Liu D. Structural impact of hydrodynamic injection on mouse liver. Gene Ther 2007; 14: 129–37. [DOI] [PubMed] [Google Scholar]
- 44. Felgner PL, Gadek TR, Holm M et al . Lipofection: a highly efficient, lipid‐mediated DNA‐transfection procedure. Proc Natl Acad Sci USA 1987; 84: 7413–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Boussif O, Lezoualc’h F, Zanta MA et al . A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA 1995; 92: 7297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li S, Huang L. In vivo gene transfer via intravenous administration of cationic lipid‐protamine‐DNA (LPD) complexes. Gene Ther 1997; 4: 891–900. [DOI] [PubMed] [Google Scholar]
- 47. Kogure K, Moriguchi R, Sasaki K, Ueno M, Futaki S, Harashima H. Development of a non‐viral multifunctional envelope‐type nano device by a novel lipid film hydration method. J Control Release 2004; 98: 317–23. [DOI] [PubMed] [Google Scholar]
- 48. Yang JP, Huang L. Overcoming the inhibitory effect of serum on lipofection by increasing the charge ratio of cationic liposome to DNA. Gene Ther 1997; 4: 950–60. [DOI] [PubMed] [Google Scholar]
- 49. Opanasopit P, Nishikawa M, Hashida M. Factors affecting drug and gene delivery: effects of interaction with blood components. Crit Rev Ther Drug Carrier Syst 2002; 19: 191–233. [DOI] [PubMed] [Google Scholar]
- 50. Nishikawa M, Takemura S, Takakura Y, Hashida M. Targeted delivery of plasmid DNA to hepatocytes in vivo: optimization of the pharmacokinetics of plasmid DNA/galactosylated poly(l‐lysine) complexes by controlling their physicochemical properties. J Pharmacol Exp Ther 1998; 287: 408–15. [PubMed] [Google Scholar]
- 51. Nishikawa M, Yamauchi M, Morimoto K, Ishida E, Takakura Y, Hashida M. Hepatocyte‐targeted in vivo gene expression by intravenous injection of plasmid DNA complexed with synthetic multi‐functional gene delivery system. Gene Ther 2000; 7: 548–55. [DOI] [PubMed] [Google Scholar]
- 52. Kawakami S, Fumoto S, Nishikawa M, Yamashita F, Hashida M. In vivo gene delivery to the liver using novel galactosylated cationic liposomes. Pharm Res 2000; 17: 306–13. [DOI] [PubMed] [Google Scholar]
- 53. Kawakami S, Sato A, Nishikawa M, Yamashita F, Hashida M. Mannose receptor‐mediated gene transfer into macrophages using novel mannosylated cationic liposomes. Gene Ther 2000; 7: 292–9. [DOI] [PubMed] [Google Scholar]
- 54. Morimoto K, Nishikawa M, Kawakami S et al . Molecular weight‐dependent gene transfection activity of unmodified and galactosylated polyethyleneimine on hepatoma cells and mouse liver. Mol Ther 2003; 7: 254–61. [DOI] [PubMed] [Google Scholar]
- 55. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 2000; 65: 271–84. [DOI] [PubMed] [Google Scholar]
- 56. Kobayashi N, Kuramoto T, Yamaoka K, Hashida M, Takakura Y. Hepatic uptake and gene expression mechanisms following intravenous administration of plasmid DNA by conventional and hydrodynamics‐based procedures. J Pharmacol Exp Ther 2001; 297: 853–60. [PubMed] [Google Scholar]
- 57. Hisazumi J, Kobayashi N, Nishikawa M, Takakura Y. Significant role of liver sinusoidal endothelial cells in hepatic uptake and degradation of naked plasmid DNA after intravenous injection. Pharm Res 2004; 21: 1223–8. [DOI] [PubMed] [Google Scholar]
- 58. Li S, Tseng WC, Stolz DB, Wu SP, Watkins SC, Huang L. Dynamic changes in the characteristics of cationic lipidic vectors after exposure to mouse serum: implications for intravenous lipofection. Gene Ther 1999; 6: 585–94. [DOI] [PubMed] [Google Scholar]
- 59. Sakurai F, Nishioka T, Saito H et al . Interaction between DNA‐cationic liposome complexes and erythrocytes is an important factor in systemic gene transfer via the intravenous route in mice: the role of the neutral helper lipid. Gene Ther 2001; 8: 677–86. [DOI] [PubMed] [Google Scholar]
- 60. Mahato RI, Kawabata K, Nomura T, Takakura Y, Hashida M. Physicochemical and pharmacokinetic characteristics of plasmid DNA/cationic liposome complexes. J Pharm Sci 1995; 84: 1267–71. [DOI] [PubMed] [Google Scholar]
- 61. Liu Y, Mounkes LC, Liggitt HD et al . Factors influencing the efficiency of cationic liposome‐mediated intravenous gene delivery. Nat Biotechnol 1997; 15: 167–73. [DOI] [PubMed] [Google Scholar]
- 62. Kircheis R, Wightman L, Kursa M, Ostermann E, Wagner E. Tumor‐targeted gene delivery: an attractive strategy to use highly active effector molecules in cancer treatment. Gene Ther 2002; 9: 731–5. [DOI] [PubMed] [Google Scholar]
- 63. Ward CM, Pechar M, Oupicky D, Ulbrich K, Seymour LW. Modification of pLL/DNA complexes with a multivalent hydrophilic polymer permits folate‐mediated targeting in vitro and prolonged plasma circulation in vivo . J Gene Med 2002; 4: 536–47. [DOI] [PubMed] [Google Scholar]
- 64. Li S, Tan Y, Viroonchatapan E, Pitt BR, Huang L. Targeted gene delivery to pulmonary endothelium by anti‐PECAM antibody. Am J Physiol 2000; 278: L504–11. [DOI] [PubMed] [Google Scholar]
- 65. Kursa M, Walker GF, Roessler V et al . Novel shielded transferrin‐polyethylene glycol‐polyethylenimine/DNA complexes for systemic tumor‐targeted gene transfer. Bioconjug Chem 2003; 14: 222–31. [DOI] [PubMed] [Google Scholar]
- 66. Moffatt S, Wiehle S, Cristiano RJ. Tumor‐specific gene delivery mediated by a novel peptide‐polyethylenimine‐DNA polyplex targeting aminopeptidase N/CD13. Hum Gene Ther 2005; 16: 57–67. [DOI] [PubMed] [Google Scholar]
- 67. Hatakeyama H, Akita H, Kogure K et al . Development of a novel systemic gene delivery system for cancer therapy with a tumor‐specific cleavable PEG‐lipid. Gene Ther 2007; 14: 68–77. [DOI] [PubMed] [Google Scholar]
- 68. Mathiesen I. Electropermeabilization of skeletal muscle enhances gene transfer in vivo . Gene Ther 1999; 6: 508–14. [DOI] [PubMed] [Google Scholar]
- 69. Basser RL, Rasko JE, Clarke K et al . Thrombopoietic effects of pegylated recombinant human megakaryocyte growth and development factor (PEG‐rHuMGDF) in patients with advanced cancer. Lancet 1996; 348: 1279–81. [DOI] [PubMed] [Google Scholar]
- 70. Tsutsumi Y, Onda M, Nagata S, Lee B, Kreitman RJ, Pastan I. Site‐specific chemical modification with polyethylene glycol of recombinant immunotoxin anti‐Tac (Fv)‐PE38 (LMB‐2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc Natl Acad Sci USA 2000; 97: 8548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pepinsky RB, LePage DJ, Gill A et al . Improved pharmacokinetic properties of a polyethylene glycol‐modified form of interferon‐β‐1a with preserved in vitro bioactivity. J Pharmacol Exp Ther 2001; 297: 1059–66. [PubMed] [Google Scholar]
- 72. Nishikawa M, Tamada A, Kumai H, Yamashita F, Hashida M. Inhibition of experimental pulmonary metastasis by controlling biodistribution of catalase in mice. Int J Cancer 2002; 99: 474–9. [DOI] [PubMed] [Google Scholar]
- 73. Wang YS, Youngster S, Grace M, Bausch J, Bordens R, Wyss DF. Structural and biological characterization of pegylated recombinant interferon α‐2b and its therapeutic implications. Adv Drug Deliv Rev 2002; 54: 547–70. [DOI] [PubMed] [Google Scholar]
- 74. Hyoudou K, Nishikawa M, Umeyama Y, Kobayashi Y, Yamashita F, Hashida M. Inhibition of metastatic tumor growth in mouse lung by repeated administration of polyethylene glycol‐conjugated catalase: quantitative analysis with firefly luciferase‐expressing melanoma cells. Clin Cancer Res 2004; 10: 7685–91. [DOI] [PubMed] [Google Scholar]
- 75. Hyoudou K, Nishikawa M, Kobayashi Y, Umeyama Y, Yamashita F, Hashida M. PEGylated catalase prevents metastatic tumor growth aggravated by tumor removal. Free Radic Biol Med 2006; 41: 1449–58. [DOI] [PubMed] [Google Scholar]
- 76. Ochiya T, Takahama Y, Nagahara S et al . New delivery system for plasmid DNA in vivo using atelocollagen as a carrier material: the Minipellet. Nat Med 1999; 5: 707–10. [DOI] [PubMed] [Google Scholar]
- 77. Darquet AM, Cameron B, Wils P, Scherman D, Crouzet J. A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene Ther 1997; 4: 1341–9. [DOI] [PubMed] [Google Scholar]
- 78. Saeki Y, Wataya‐Kaneda M, Tanaka K, Kaneda Y. Sustained transgene expression in vitro and in vivo using an Epstein–Barr virus replicon vector system combined with HVJ liposomes. Gene Ther 1998; 5: 1031–7. [DOI] [PubMed] [Google Scholar]
- 79. Yew NS, Przybylska M, Ziegler RJ, Liu D, Cheng SH. High and sustained transgene expression in vivo from plasmid vectors containing a hybrid ubiquitin promoter. Mol Ther 2001; 4: 75–82. [DOI] [PubMed] [Google Scholar]
- 80. Chen ZY, He CY, Ehrhardt A, Kay MA. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high‐level transgene expression in vivo . Mol Ther 2003; 8: 495–500. [DOI] [PubMed] [Google Scholar]