

Abstract
Retroviral insertional mutagenesis has been applied to identify oncogenes that are important for both human and rodent carcinogenesis. The method reveals not only primary oncogenes but also cooperative genes that might be affected as second or third hits in multistep carcinogenesis. The use of genetically engineered mice such as NUP98‐HOXA9 transgenic mice enabled efficient identification of cooperative genes, which provides important information for the molecular pathway in carcinogenesis/leukemogenesis. With use of the retrovirus mediated gene transfer system, retroviral insertional mutagenesis will provide invaluable information to understand genetic interaction in complex mechanisms of carcinogenesis. (Cancer Sci 2005; 96: 7–13)
Carcinogenesis/leukemogenesis is a multistep process, and multiple genetic and epigenetic alteration is required for cancer cells to possess highly malignant potential. (1) Molecular analysis of human cancers has provided accumulated information on the number of genes involved in carcinogenesis as well as their functional significance. However, there are still limitations in understanding genetic interaction for carcinogenesis using human cancer materials. It is important to know about cooperation and combination in genetic abnormalities that are needed for malignant phenotypes of cancers.
The retroviral tagging technique has been applied for identification of oncogenes and tumor suppressors. Plenty of important disease genes have been isolated by this technique and many of the genes are in fact involved in human cancers. 2 , 3 Somatic and clonal integration of retroviruses in the genome accidentally enhances protooncogene expression or inactivates tumor suppressor genes (Fig. 1). A cell that has such integrations acquires growth advantage, and is then clonally selected to grow on the tumor. In these cases, the oncogenes/tumor suppressors responsible for tumorigenesis can be identified by using proviral DNA as a molecular tag. Practically, retroviral insertions have been achieved either in the mouse strains with high endogenous retrovirus expression or in the mice infected by replication competent retroviruses. In either case, hematopoietic cells are the main targets for tumor induction after retroviral infection and proviral DNA insertion into the genome.
Figure 1.
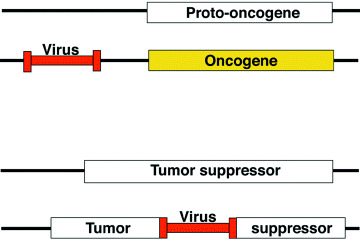
Mechanism of oncogene activation and tumor suppressor inactivation by retroviral insertional mutagenesis.
High throughput cloning of retroviral integration sites has been accomplished by introducing inverse PCR (IPCR) and splinkerette PCR (SPCR) techniques, and by using the mouse genome database. 4 , 5 , 6 , 7 These studies have enabled the identification of multiple retroviral integration sites (RIS) in each tumor, and it could clarify cooperative genetic interaction in tumorigenesis as retrovirally induced leukemias/lymphomas usually contain multiple copies of proviral genomes within a single tumor clone.
This review describes the insertional mutagenesis technique for anlalysis of genetic interaction in carcinogenesis, and focuses on identification of cooperative genes of the NUP98‐HOXA9 chimera. In addition, modification of the method for efficient cancer gene identification is discussed.
Retroviral insertional mutagenesis identifies genetic interaction/cooperation in cancer
The BXH2 strain is a recombinant inbred strain generated by a cross between C57Bl/6 J and C3H/HeJ, and the high incidence of acute myeloid leukemia (AML) is causally related to the expression of the endogenous B ecotropic virus. 8 , 9 More than 90% of BXH2 mice die of AML by 1 year of age. In addition to high expression of ecotropic virus, the BXH2 strain has acquired a genetic predisposition for myeloproliferation and, recently, the locus has been mapped to mouse chromosome 8. (10) Genome‐wide identification of RIS in BXH2 leukemia revealed that these RIS include both known human cancer genes and genes of unknown tumorigenic potential, and that one can speculate the genetic pathway in cancer using RIS data. 4 , 5 In approximately 10% of BXH2 leukemias we identified overlapping of retroviral integrations in Meis1 and Hoxa7/a9 loci.(11) Most leukemias with viral integration at Meis1 also have viral integrations at Hoxa7 or Hoxa9, suggesting these genes cooperate in leukemogenesis. Kroon et al. reported that overexpression of Meis1 accelerates Hoxa9‐induced AML, (12) and also its chimeric form of NUP98‐HOXA9‐induced AML, (13) which was identified in human AML with chromosomal translocation t(7;11)(p15;p15). 14 , 15 Thus, Meis1 is indeed a cofactor for abdominal B‐like HOX in leukemogenesis.
Cooperative genetic effects by retroviral insertion can be achieved by superinfection of retroviruses. In general, retrovirus‐induced murine leukemia and lymphoma have several copies of proviral DNA in their genome by somatically acquired integration. (3) The multiple integrations do not occur simultaneously, but it is likely that the process is consecutive. A clone with single or few integrations usually has limited growth properties, then acquires many more advantages with subsequent integrations. This process effectively simulates the clonal expansion of premalignant cells to fully developed cancer cells in multistep carcinogenesis.
Application on genetically engineered mice
Cooperative genetic alterations might be identified by chance using a simple insertional mutagenesis model like Meis1 and Hox cooperation. However, a more efficient device is needed to clarify cooperative genes for particular genes. Retroviral insertional mutagenesis on genetically engineered animal models that are predisposed to cancer development is therefore a good method for identification of cooperative genes of the primary genetic defect in these animals. When the cells have a genetic alteration, such as oncogenic activation or a loss of function mutation in tumor suppressor genes, retroviral insertion will add specific cooperative genetic abnormalities for the primary alterations.
Model for lymphoid malignancies. To analyze the tumorigenic process of lymphoid malignancies, early work has been carried out using transgenic mice that express c‐Myc under control of the immunoglobulin heavy chain enhancer (Eµ). (16) The EµMyc transgenic mice were infected with Moloney murine leukemia virus (MoMLV), and common retroviral integration sites in B‐cell lymphomas were identified. Interestingly, frequent proviral integrations near Pim1 or Pim2 genes were observed. 17 , 18 , 19 At the same time another Myc collaborator Bmi1 was identified in the same system. 17 , 18 The cooperation between Myc and Pim genes were confirmed by mating experiments using EµMyc and EµPim1, or EµMyc and EµPim2 transgenic mice. 20 , 21 Pim1 and Pim2 encodes protein‐serine/threonine kinase, (22) and Bmi1 protein belongs to the mammalian polycomb family that regulates Hox gene expression. (23) The cooperativity of Myc/Pim/Bmi in the other tumor system including human lymphomas remains to be clarified, however, the recent study emphasized that Bmi1 is essential for maintaining normal and leukemic hematopoietic stem cells. (24)
In addition, Mikkers et al. reported frequent involvement of E2a in the MoMLV‐tagged T‐cell lymphoma in EµMyc transgenic mice, (25) indicating cell type‐specific selection of the targets for retroviral insertion. It is considered that E2a is a tumor suppressor for T lymphocytes, as mice homozygous mutant for E2a develops T‐cell lymphoma. (26) However, E2a is up‐regulated as a result of retroviral integration, suggesting E2a might act under collaboration with Myc as an oncogene rather than a tumor suppressor. MoMLV infection also accelerated lymphoma development in Eµ‐Bcl2 transgenic mice, although the cooperative gene has not been identified in this study. (27)
Identification of cooperative genes for tumor suppressor genes was achieved using MoMLV infection in gene targeted models. Homozygous mutants for Cdkn2a, which encodes p16INK4a and p19ARF, accelerated tumorigenesis by MoMLV infection. (28) Genome‐wide, high throughput cloning of the integration sites in lymphoid and myeloid tumors in the model revealed multiple common integration sites (CIS), including Tpl2, indicating that activation of MAP‐kinase signaling cooperates with loss of Cdkn2a. Retroviral tagging with MoMLV was also applied to mice homozygous mutants for p27 Kip. 1. (29) Again, T‐cell lymphomagenesis was accelerated in the homozygous mutants, and there was frequent involvement of Myc, Jundp2 and Xpc1 as CIS. Thus, there is an apparent difference in cooperative genes involved in carcinogenesis between Cdkn2a and p27.
Model for myeloid malignancies. Many important disease genes for human hematopoietic neoplasms have been identified. A characteristic genetic feature of human myeloid leukemia is genetic chimera resulting from reciprocal chromosomal translocations. (30) Specific chromosomal translocation breaks two unrelated genes on each chromosome and these genes are fused to each another. It is believed that these chimeras play a central role in the early phase of leukemogenesis, and many of these chimeras are required to maintain leukemic cells. In fact, a therapeutic approach to these chimeric proteins as molecular targets have shown enormous success. (31) However, gene fusions are not sufficient for complete carcinogenesis. (32) Prenatal detection of the MLL‐AF4, TEL‐AML and AML1‐ETO chimera indicates that a long latency period is required for development of overt leukemia. 33 , 34 , 35 Studies using transgenic mice have also shown long latency and clonal selection of hematopoietic cells, which express a chimeric oncogene is required. (32) Therefore, it is important to understand additional genetic alterations for complete leukemogenesis for treatment of drug‐resistant leukemias.
Many chimeric genes found in human AML encode sequence‐specific transcription factors with aberrant functions. (36) NUP98‐HOXA9 is one such chimera having a C‐terminal homeodomain as a DNA‐binding domain. 14 , 15 We have generated a transgenic mouse that expresses NUP98‐HOXA9 chimera. (37) In this mouse NUP98‐HOXA9 is expressed in a myeloid progenitor under control of the human cathepsin G promoter. Only 20% of transgenic animals develop myeloproliferative disease and, eventually, AML after a long latency. However, non‐leukemic bone marrow cells of the transgenic animals showed enhanced self‐renewal potency and proliferation on cytokine stimuli. These findings suggested that the NUP98‐HOXA9 transgenic mice might be a good model to analyze the multistep process of leukemogenesis.
To identify cooperative genes for NUP98‐HOXA9 in myeloid leukemogenesis, the transgenic mice were mated with AML‐prone BXH2 mice. Disease phenotypes of hematological malignancies induced by retroviral insertion are modulated by the strain background and the viral promoter. MoMLV is an efficient viral mutagen, however, the virus shows strong tropism for T‐cell malignancies. (38) It is therefore important to use myeloid tropism of BXH2 for efficient induction of myeloid leukemia. The NUP98‐HOXA9 Tg mice were backcrossed to the BXH2 strain to introduce the transgene into BXH2 genetic background, and survival rates of Tg‐positive BXH2 mice were compared with those of Tg‐negative litter mates at the third generation. (37) The incidence and latency of myeloid leukemias in NUP98‐HOXA9/BXH2, wild‐type BXH2 and C57BL/6J background NUP98‐HOXA9 transgenic mice were compared. NUP98‐HOXA9/BXH2 mice developed acute myeloid leukemia faster than BXH2 and transgenic mice between 4 and 8 months. This result suggested that NUP98‐HOXA9 expression accelerated the progression of BXH2 myeloid leukemia, and that cooperative genes for NUP98‐HOXA9 could be located near CIS.
To characterize the RIS and to identify nearby cooperative genes, host‐virus junction sequences were amplified by inverse PCR using NUP98‐HOXA9/BXH2 leukemia DNA and retrovirus‐specific primers. Fifty unique integration sites were identified in 19 NUP98‐HOXA9/BXH2 leukemias and six were CIS. These data will be deposited on the Mouse Retroviral Tagged Cancer Gene Database (MRTCGD) (http://genome2.ncifcrf.gov/RTCGD/). The first integration site identified in the system was the Meis1 locus on mouse chromosome 11. Meis1 is a known cofactor for the wild‐type HOXA9 as well as chimeric NUP98‐HOXA9 in myeloid leukemogenesis, 11 , 12 , 13 and the incidence of integration at the Meis1 gene in NUP98‐HOXA9/BXH2 leukemia was much higher than that in wild‐type BXH2 leukemia.
Four other CIS had not been identified as common sites in the wild type BXH2 leukemias, suggesting that these CIS might be specific collaborators for NUP98‐HOXA9 (Table 1). In these CIS, we identified a further four candidate cooperative genes, Dnalc4, Fcgr2b, Fcrl and Con1. Another CIS designated as Con2 was also identified as a CIS in AKXD B‐cell lymphoma. (5) Of these candidate genes expression of Dnalc4 and Con1 was up‐regulated in the leukemia with integrations, suggesting that these genes might act as dominant oncogenes in collaboration with NUP98‐HOXA9.
Table 1.
Cooperative genes for NUP98‐HOXA9 identified as common retroviral integration sites
Cooperative transforming activity between NUP98‐HOXA9 and Dnalc4, Fcgr2b, Fcrl and Con1 was demonstrated using NIH 3T3 cells. It has been exhibited that NUP98‐HOXA9 alone could transform NIH 3T3 cells, (39) suggesting that NUP98‐HOXA9 might have common activity to transform both hematopoietic cells and non‐hematopoietic fibroblasts, and that this is a reliable if not perfect assay system to evaluate oncogene cooperation. Therefore, these novel candidate genes are the real cooperative genes cooperated with NUP98‐HOXA9 to promote oncogenesis. It will be another useful assay system to introduce those genes into bone marrow cells and then repopulate the cells into irradiated hosts to check cooperative transforming activity in vivo.
Fcgr2b and Fcrl are located within the immunoglobulin G Fc receptor gene cluster on mouse chromosome 1. (40) As all viral integrations involving this region were located between Fcgr2b and Fcrl, either or both of these genes might be involved. Interestingly, Fcgr2b is a target for the 1q21 translocation in human follicular lymphoma. 41 , 42 Fcrl is preferentially expressed in germinal center centroblasts and in a subset of diffuse large B cell lymphoma cells, and the gene has also been reported to serve as a unique marker for the characterization for B cell malignancy. 43 , 44 Thus, dysregulation of Fcgr2b and Fcrl might play an important role in tumor progression of hematopoietic cells.
The Dnalc4 gene encodes the dynein light chain 4 that is a component of the dynein motor complex. Dynein complexes are involved in the cytoplasmic transportation machinery. (45) More importantly, recent studies have shown that some dynein light chain proteins, such as Dlc2 and Dlc8, interact with BH3‐only proteins, including Bim and Bmf, and could block apoptotic signals by suppressing Bim/Bmf functions. 46 , 47 The Con1 gene (cooperative gene for NUP98‐HOXA9 1) encodes a novel protein that contains an N‐terminal SCR domain consisting of 60 amino acid residues. It remains to be clarified what proteins interact with the Con1 SCR domain and how abnormal Con1 expression promotes NUP98‐HOXA9‐positive AML.
It is interesting to speculate as to how these cooperative genes promote leukemogenesis. In BXH2 leukemias, 90% of cases with viral integration at Hoxa7 or Hoxa9 showed cooperative activation of Meis1. (10) However, only half of the leukemias showed cooperation between NUP98‐HOXA9 and Meis1 in the mating experiment between the NUP98‐HOXA9 transgenic mice and the BXH2 strain. This suggests that some of the cooperative genes identified could have replaced the leukemogenic function of Meis1 (Fig. 2a). An example of this might be Con1, as Con1 integration sites never overlapped with Meis1 integration sites in a single tumor. In contrast, leukemias exhibiting Meis1 integration also showed integration at the Dnalc4 locus, which suggested that the two genes functioned in different leukemogenic pathways. Like other dynein light chain proteins, it is very likely that Dnalc4 binds to proapoptotic proteins such as Bim and Bmf. Our preliminary experiments showed interaction in vivo between Dnalc4, and the long and extra‐long isoforms of the Bim protein (D. Takahashi & T. Nakamura, unpubl. data, 2004). In this case, overexpression of Dnalc4 might block Bim function to give an antiapoptotic potential to preleukemic cells.
Figure 2.
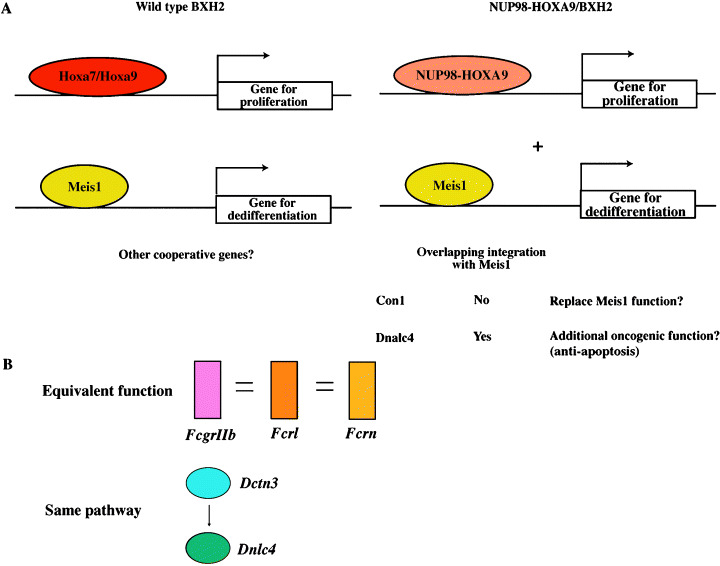
Molecular pathway of NUP98‐HOXA9 leukemogenesis. (A) NUP98‐HOXA9 promotes proliferation and self renewal of bone marrow cells, (37) while Meis1 blocks differentiation of myeloid precursors (right panel). (64) Analysis of individual tumors of NUP98‐HOXA9 tg/BXH2 leukemia divided CIS into two categories: overlapping and non‐overlapping genes with Meis1. The former gene such as Dnalc4 functions as an additional step for leukemogenesis like an antiapoptotic gene. On the other hand, the latter gene may replace Meis1 function. Cooperative activation between Hoxa7/a9 and Meis1 has also been observed in the wild type BXH2 leukemia (left panel). However, it is difficult to analyze molecular pathway due to less frequent Hox activation and interference of unrelated integration sites. (B) Pathway prediction by insertional mutagenesis. The upper panel indicates class of genes having equivalent function identified as integration sites. The bottom panel shows isolated genes belong to one particular molecular pathway.
Dctn3, encoding dynactin 3, was also identified as a single RIS in this study. As dynactin is an upstream regulator of dynein, 48 , 49 this finding strongly suggested the importance of the Dynein/Dynactin pathway in NUP98‐HOXA9 leukemogenesis. We also identified another Fc receptor gene, Fcrn, on chromosome 7 as a RIS. (50) Although Fcrn was not a CIS, our results suggested that signaling pathways involving Fc receptors might be critical for NUP98‐HOXA9‐induced myeloid leukemogenesis. Collectively, these results exhibit an important molecular pathway in carcinogenesis as well as the functional similarity of the genes belonging to a certain gene family (Fig. 2b).
Human AML M4 with eosinophilia subtype shows characteristic chromosomal 16 inversion inv(16)(p13q22) that results in fusion between CBFB and MYH11. (51) The heterozygous knock‐in mice of the Cbfb‐MYH11 fusion gene at the Cbfb locus develops AML after N‐ethyl‐N‐nitrosourea treatment. (52) Castilla et al. infected Cbfb‐MYH11 heterozygous mutant with amphotropic virus 4070 A and isolated CIS. (53) The 4070 A retrovirus is weakly myeloid tropic, 54 , 55 and it is likely that Cbfb‐MYH11 heterozygous mice are themselves predisposed to myeloid malignancies. In this study four CIS, specific for Cbfb‐MYH11 expression, were identified and genes encoding zinc finger transcription factors, Plagl2 and Plag1, were considered as cooperative oncogenes for Cbfb‐MYH11.
Use of retrovirus‐mediated bone marrow transfer system
Replication competent retroviruses induce leukemia and lymphoma through insertional mutagenesis of cellular proto‐oncogenes and tumor suppressor genes when allowed to replicate unchecked in neonatal mice. Retroviral insertional mutagenesis on genetically engineered cancer‐prone mouse models is a good approach for identifying cooperative genetic events. However, the method is time‐consuming and requires proper models that efficiently induce neoplasms of proper phenotypes. In this respect, retrovirus‐mediated gene transfer followed by bone marrow transplantation is a useful and efficient system for analyzing genetic interaction in oncogenesis.
The retrovirus‐mediated gene transfer system is an excellent method to introduce the gene into hematopoietic cells where efficient transfection is difficult by standard transfection techniques using plasmid vectors. In this system an oncogene of interest is inserted into the replication defective retrovirus (retroviral vector), and bone marrow cells are transduced by this retroviral construct. When the retrovirus is inserted in the vicinity of a gene that cooperates with the primary oncogene in the construct (Fig. 3a), the cell will acquire higher growth advantage than the cell with silent integration, and expand to develop overt leukemia.
Figure 3.
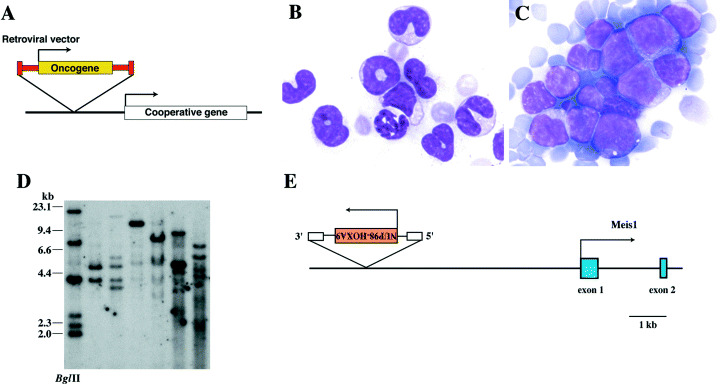
Retroviral insertional mutagenesis using replication deficient retrovirus. (A) Co‐expression of the primary oncogene inserted into the retroviral vector and a cooperative in the vicinity of integration. (B) and (C) Peripheral blood smear of leukemic mice after bone marrow transfer. Transduction with the NUP98‐HOXA9 retrovirus induces chronic myeloproliferation (B) and eventually AML showing blastic cells (C). Giemsa. Original magnification, × 100. (D) Southern blot analysis of retrovirally induced NUP98‐HOXA9 leukemia. DNA samples were digested with BglII, gel electrophoresed and transferred onto nylon filter. The filter was probed with a DNA fragment of GFP that is inserted in the construct. Each band indicates clonal integration of the retroviral construct. (E) The NUP98‐HOXA9 retroviral integration at the Meis1 locus. The virus was inserted 6 kb upstream of the putative transcriptional start site of Meis1 with reverse transcriptional orientation.
We transduced normal mouse bone marrow cells with the NUP98‐HOXA9 expressing retroviral vector, and the cells were transferred into sublethally irradiated hosts. These mice develop chronic myeloproliferation and AML 90–200 days after injection (Fig. 3b,c). Clonal retroviral integrations in these leukemia were shown by Southern blotting (Fig. 3d), indicating that these leukemic cells had several copies of the retrovirus. The NUP98‐HOXA9 expressing retrovirus was inserted 6 kb upstream of Meis1 in two of three cases (Fig. 3e). In another case, the same retrovirus was inserted 1.7 kb upstream of Meis2, a member of Meis family. (56) The Fcrl gene was also identified as the integration site of the same vector. These results using a small number of mice indicate that retroviral transduction of NUP98‐HOXA9 efficiently identifies the cooperative oncogenes like the mating experiment between NUP98‐HOXA9 transgenic and BXH2 mice.
In the similar system cooperative genes for Sox4 that is a frequent target of retroviral integration in murine B‐cell lymphoma and AML 4 , 5 were identified. Sox4 expression using defective retrovirus identified Sfpi1 and Mef2c as cooperative oncogenes (Y. Du, N. Jenkins and N. Copeland, pers. comm., 2004). Thus, the use of replication deficient retroviruses and bone marrow transfer provides efficient and rapid analysis of genetic interaction in carcinogenesis.
There is a tragic accident of similar mechanisms in human cases of gene therapy for X‐linked severe combined immunodeficiency. 57 , 58 The disease is caused by the defect of IL‐2R γ chain, and ex vivoγ chain transfer into autologous bone marrow‐derived CD34+ cells by the retroviral vector showed dramatic improvement in the disease. (58) However, two cases given by this therapy developed T‐ALL, and integrations of the γ chain‐bearing retrovirus in the LMO2 locus were detected in both cases. LMO2 is a known T‐cell oncogene, (59) and it is very likely that the gene acts as a cooperative oncogene for the γ chain gene. (60)
Future direction and conclusion
Retroviral insertional mutagenesis is a powerful tool to identify important genes for carcinogenesis. However, there are limitations and some drawbacks to this method. First, the target cell types are limited in the hematopoietic system with a few exceptions, such as mouse mammary carcinoma induced by murine mammary tumor virus. (2) Therefore, there is difficulty in applying the method to analyze common epithelial carcinomas that are major malignant diseases in human patients. One of the reasons for hematopoietic tropism of retroviral mutagenesis might be efficient promoter activity of the retroviral sequence for certain genes, important for hematopoietic cell proliferation. (2) While many epithelial cells have proviral integrations, the mice die quickly of hematopoietic neoplasms. Thus, insertional mutagenesis in vitro using epithelial cells might be a good alternative.
To understand the molecular pathway in carcinogenesis more precisely and in more detail, the in vitro system will provide valuable information. It is sometimes difficult to determine exact molecular functions of the genes identified in integration sites. However, the in vitro system provides a clear understanding of how a single genetic alteration influences the biological process. A good example is the mouse myelo‐monocytic cell line WEHI‐3B, in which a retrovirus‐like intracisternal A particle is integrated in the 5′ end of the interleukin‐3 (IL‐3) and Hoxb8 genes. (61) In the cell line, expression of the IL‐3 gene is constitutively active by the enhancer effect of the intracisternal A particle and the cell no longer requires exogenous IL‐3, which is required for proliferation and survival of primary and many neoplastic myeloid cells. This indicates that the retroviral insertion is effective for targeting a single genetic event as well as investigating the molecular pathway, such as downstream molecules.
Finally, the use of transposons in mammalian cells have been recently attempted. 62 , 63 If efficient transposition and subsequent alteration of target gene expression is achieved, different spectrums of tumors from conventional retroviral insertions can be obtained. Furthermore, modification of retroviral sequence, cre/lox‐P‐mediated recombination techniques and use of mouse strains with genetic instability will provide new discoveries in the cancer research field.
Acknowledgments
I thank my colleagues Masayuki Iwasaki, Takeshi Kuwata, Guang Jin, Yukari Yamazaki, Miki Takuwa for their help, and Neal Copeland and Nancy Jenkins for valuable discussion. This work was supported in part by a Grant‐in‐Aid for Scientific Research on Priority Areas (C) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 2001; 1: 222–31. [DOI] [PubMed] [Google Scholar]
- 2. Jonkers J, Berns A. Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochem Biophys Acta 1996; 1287: 29–57. [DOI] [PubMed] [Google Scholar]
- 3. Mikkers H, Berns A. Retroviral insertional mutagenesis: tagging cancer pathways. Adv Cancer Res 2003; 88: 53–99. [DOI] [PubMed] [Google Scholar]
- 4. Li J, Shen H, Himmel KL, Dupuy AJ, Largaespada DA, Nakamura T, Shaughnessy JD Jr, Jenkins NA, Copeland NG. Leukaemia disease genes: large‐scale cloning and pathway predictions. Nat Genet 1999; 23: 348–53. [DOI] [PubMed] [Google Scholar]
- 5. Suzuki T, Shen H, Akagi K, Morse HC III, Malley JD, Naiman DQ, Jenkins NA, Copeland NG. New genes involved in cancer identified by retroviral tagging. Nat Genet 2002; 32: 166–74. [DOI] [PubMed] [Google Scholar]
- 6. Devon RS, Porteous DJ, Brookes AJ. Splinkerettes‐improved vectorettes for greater efficiency in PCR walking. Nucl Acids Res 1995; 23: 1644–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mikkers H, Allen J, Knipscheer P, Romeyn L, Hart A, Vink E, Berns A. High‐throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat Genet 2002; 32: 153–9. [DOI] [PubMed] [Google Scholar]
- 8. Copeland NG, Jenkins NA. Myeloid leukemia: disease genes and mouse models. In: Hiai H, Hino O (eds). Animal Models of Cancer Predisposition Syndromes. Karger, Basel 1999; 53–63. [DOI] [PubMed] [Google Scholar]
- 9. Jenkins NA, Copeland NG, Taylor BA, Bedigian HG, Lee BK. Ecotropic murine leukemia virus DNA content of normal and lymphomatous tissues of BXH‐2 recombinant inbred mice. J Virol 1982; 42: 379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Turcotte K, Gauthier S, Mitsos LM, Shustik C, Copeland NG, Jenkins NA, Fournet JC, Jolicoeur P, Gros P. Genetic control of myeloproliferation in BXH‐2 mice. Blood 2004; 103: 2343–50. [DOI] [PubMed] [Google Scholar]
- 11. Nakamura T, Largaespada DA, Shaughnessy JD Jr, Jenkins NA, Copeland NG. Cooperative activation of Hoxa and Pbx1‐related genes in murine myeloid leukaemias. Nat Genet 1996; 12: 149–53. [DOI] [PubMed] [Google Scholar]
- 12. Kroon E, Krosl J, Thorsteinsdottir U, Baban S, Buchberg AM, Sauvageau G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J 1998; 17: 3714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kroon E, Thorsteinsdottir U, Mayotte N, Nakamura T, Sauvageau G. NUP98‐HOXA9 expression in hemopoietic stem cells induces chronic and acute myeloid leukemias in mice. EMBO J 2001; 20: 350–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura T, Largaespada DA, Lee MP, Johnson LA, Ohyashiki K, Toyama K, Chen SJ, Willman CL, Chen IM, Feinberg AP, Jenkins NA, Copeland NG. Fusion of the nucleoporin gene NUP98 to HOXA9 by the chromosome translocation t(7; 11 (p15;p15)) in human myeloid leukaemia. Nat Genet 1996; 12: 154–8. [DOI] [PubMed] [Google Scholar]
- 15. Borrow J, Shearman AM, Stanton VP Jr, Becher R, Collins T, Williams AJ, Dube I, Katz F, Kwong YL, Morris C, Ohyashiki K, Toyama K, Rowley J, Housman DE. The t (7; 11 (p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat Genet 1996; 12: 159–67. [DOI] [PubMed] [Google Scholar]
- 16. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c‐myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985; 318: 533–8. [DOI] [PubMed] [Google Scholar]
- 17. Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu‐myc transgenic mice. Cell 1991; 65: 753–63. [DOI] [PubMed] [Google Scholar]
- 18. Van Lohuzen M, Verbeek S, Scheijen B, Wientjens E, Van Der Gulden H, Berns A. Identification of cooperating oncogenes in Eµ‐myc transgenic mice by provirus tagging. Cell 1991; 65: 737–52. [DOI] [PubMed] [Google Scholar]
- 19. Van Der Lugt NM, Domen J, Verhoeven E, Linders K, Vander Gulden H, Allen J, Berns A. Proviral tagging in Eµ‐myc transgenic mice lacking the Pim‐1 proto‐oncogene leads to compensatory activation of Pim‐2. EMBO J 1995; 14: 2536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verbeek S, Van Lohuizen M, Van Der Valk M, Domen J, Kraal G, Berns A. Mice bearing the Eµ‐myc and Eµ‐pim‐1 transgenes develop pre‐B‐cell leukemia prenatally. Mol Cell Biol 1991; 11: 1176–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Allen JD, Verhoeven E, Domen J, Van Der Valk M, Berns A. Pim‐2 transgene induces lymphoid tumors, exhibiting potent synergy with c‐myc. Oncogene 1997; 15: 1133–41. [DOI] [PubMed] [Google Scholar]
- 22. Saris CJM, Domen J, Berns A. The pim‐1 oncogene encodes two related protein‐serine/threonine kinases by alternative initiation at AUG and CUG. EMBO J 1991; 10: 655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Der Lugt NM, Alkema M, Berns A, Deschamps J. The Polycomb‐group homolog Bmi‐1 is a regulator of murine Hox gene expression. Mech Dev 1996; 58: 153–64. [DOI] [PubMed] [Google Scholar]
- 24. Lessard J, Sauvageau G. Bmi‐1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003; 423: 255–60. [DOI] [PubMed] [Google Scholar]
- 25. Mikkers H, Allen J, Berns A. Proviral activation of the tumor suppressor E2a contributes to T cell lymphomagenesis in EµMyc transgenic mice. Oncogene 2002; 21: 6559–66. [DOI] [PubMed] [Google Scholar]
- 26. Bain G, Engel I, Maandag ECR, Te Riele HPJ, Voland JR, Sharp LL, Chun J, Huey B, Pinkel D, Murre C. E2A deficiency leads to abnormalities in αβ T‐cell development and to rapid development of T‐cell lymphomas. Mol Cell Biol 1997; 17: 4782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shinto Y, Morimoto M, Katsumata M, Uchida A, Aozasa K, Okamoto M, Kurosawa T, Ochi T, Greene MI, Tsujimoto Y. Moloney murine leukemia virus infection accelerates lymphomagenesis in Eµ‐bcl‐2 transgenic mice. Oncogene 1995; 11: 1729–36. [PubMed] [Google Scholar]
- 28. Lund AH, Turner G, Trubetskoy A, Verhoeven E, Wientjens E, Hulsman D, Russell R, DePinho RA, Lenz J, Van Lohuizen M. Genome‐wide retroviral insertional tagging of genes involved in cancer in Cdkn2a‐deficient mice. Nat Genet 2002; 32: 160–5. [DOI] [PubMed] [Google Scholar]
- 29. Hwang HC, Martins CP, Bronkhorst Y, Randel E, Berns A, Fero M, Clurman BE. Identification of p27 complementing oncogenes by insertional mutagenesis and high‐throughput insertion site analysis. Proc Natl Acad Sci USA 2002; 99: 11293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rabbitts TH. Chromosomal translocations in human cancer. Nature 1994; 372: 143–9. [DOI] [PubMed] [Google Scholar]
- 31. Goldman JM, Melo JV. Chronic myeloid leukemia‐advances in biology and new approaches to treatment. N Engl J Med 2003; 349: 1451–64. [DOI] [PubMed] [Google Scholar]
- 32. Greaves MF, Wiemels J. Origins of chromosome translocation in childhood leukaemia. Nat Rev Cancer 2003; 3: 1–11. [DOI] [PubMed] [Google Scholar]
- 33. Gale KB, Ford AM, Repp R, Borkhardt A, Keller C, Eden OB, Greaves MF. Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc Natl Acad Sci USA 1997; 94: 13950–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wiemels JL, Ford AM, Van Wering ER, Postma A, Greaves M. Protracted and variable latency of acute lymphoblastic leukemia after TEL‐AML1 gene fusion in utero. Blood 1999; 94: 1057–62. [PubMed] [Google Scholar]
- 35. Wiemels JL, Xiao Z, Buffler PA, Maia AT, Ma X, Dicks BM, Smith MT, Zhang L, Feusner J, Wiencke J, Pritchard‐Jones K, Kempski H, Greaves M. In utero origin of t(8; 21) AML1–ETO translocation in childhood acute myeloid leukemia. Blood 2002; 99: 3801–5. [DOI] [PubMed] [Google Scholar]
- 36. Look AT. Oncogenic transcription factors in human acute leukemias. Science 1997; 278: 1059–64. [DOI] [PubMed] [Google Scholar]
- 37. Iwasaki M, Kuwata T, Yamazaki Y, Jenkins NA, Copeland NG, Osato M, Ito Y, Kroon E, Sauvageau G, Nakamura T. Identification of cooperative genes for NUP98‐HOXA9 using a mouse model. Blood 2005. [DOI] [PubMed]
- 38. Moloney JB. Biological studies on a lymphoid‐leukemia virus extracted from sarcoma 37. I. Origin and introductory investigations. J Natl Cancer Institute 1960; 24: 933–51. [PubMed] [Google Scholar]
- 39. Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, Van Deursen JM. CREB binding protein interacts with nucleoporin‐specific FG repeats that activate transcription and mediate NUP98‐HOXA9 oncogenicity. Mol Cell Biol 1999; 19: 764–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiu Y, Nakamura K, Abe M, Li N, Wen XS, Jiang Y, Zhang D, Tsurui H, Matsuoka S, Hamano Y, Fujii H, Ono M, Takai T, Shimokawa T, Ra C, Shirai T, Hirose S. Transcriptional regulation of Fcgr2b gene by polymorphic promoter region and its contribution to humoral immune responses. J Immunol 2002; 169: 4340–6. [DOI] [PubMed] [Google Scholar]
- 41. Callanan MB, Le Baccon P, Mossuz P, Duley S, Bastard C, Hamoudi R, Dyer MJ, Klobeck G, Rimokh R, Sotto JJ, Leroux D. The IgG Fc receptor FcγRIIB, is a target for deregulation by chromosomal translocation in malignant lymphoma. Proc Natl Acad Sci USA 2000; 97: 309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen W, Palanisamy N, Schmidt H, Teruya‐Feldstein J, Jhanwar SC, Zelenetz AD, Houldsworth J, Chaganti RSK. Deregulation of FCGR2B expression by 1q21 rearrangements in follicular lymphomas. Oncogene 2001; 20: 7686–93. [DOI] [PubMed] [Google Scholar]
- 43. Facchetti F, Cella M, Festa S, Fremont DH, Colonna M. An unusual Fc receptor‐related protein expressed in human centroblasts. Proc Natl Acad Sci USA 2002; 99: 3776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Davis RS, Li H, Chen CC, Wang YH, Cooper MD, Burrows PD. Definition of an Fc receptor‐related gene (FcRX) expressed in human and mouse B cells. Int Immunol 2002; 14: 1075–83. [DOI] [PubMed] [Google Scholar]
- 45. Milisav I. Dynein and dynein‐related genes. Cell Motil Cytoskeleton 1998; 39: 261–72. [DOI] [PubMed] [Google Scholar]
- 46. Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl‐2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 1999; 3: 287–96. [DOI] [PubMed] [Google Scholar]
- 47. Puthalakath H, Villunger A, O'Reilly LA, King SM, Strasser A. Bmf: a proapoptotic BH3‐only protein regulated by interaction with the myosin V actin motor complex activated by anoikis. Science 2001; 293: 1829–32. [DOI] [PubMed] [Google Scholar]
- 48. Boylan K, Serr M, Hays T. A molecular genetic analysis of the interaction between the cytoplasmic Dynein intermediate chain and the Glued (Dynactin) complex. Mol Biol Cell 2000; 11: 3791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol 2000; 2: 20–4. [DOI] [PubMed] [Google Scholar]
- 50. Kandil E, Noguchi M, Ishibashi T, Kasahara M. Structural and phylogenetic analysis of the MHC class I‐like Fc receptor gene. J Immunol 1995; 154: 5907–18. [PubMed] [Google Scholar]
- 51. Liu P, Tarle SA, Hajra A, Claxton DF, Marlton P, Freedman M, Siciliano MJ, Collins FS. Fusion between transcription factor CBFβ/PEBP2β and a myosin heavy chain in acute myeloid leukemia. Science 1993; 261: 1041–4. [DOI] [PubMed] [Google Scholar]
- 52. Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S, Owens J, Eckhaus M, Bodine D, Liu PP. The fusion gene Cbfb‐MYH11 blocks myeloid differentiation and predisoposes mice for acute myelomonocytic leukaemia. Nat Genet 1999; 23: 144–6. [DOI] [PubMed] [Google Scholar]
- 53. Castilla LH, Perrat P, Martinez NJ, Landrette SF, Keys R, Oikemus S, Flanegam J, Heilman S, Garrett L, Dutra A, Anderson S, Pihan GA, Wolff L, Liu PP. Identification of genes that synergize with Cbfb‐MYH11 in the pathogenesis of acute myeloid leukemia. Proc Natl Acad Sci USA 2004; 101: 4924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ott DE, Keller J, Sill K, Rein A. Phenotypes of murine leukemia virus‐induced tumors: influence of 3′ viral coding sequences. J Virol 1992; 66: 6107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wolff L, Koller R, Davidson W. Acute myeloid leukemia induction by amphotropic murine retrovirus (4070A): clonal integrations involve c‐myb in some but not all leukemias. J Virol 1991; 65: 3607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakamura T, Jenkins NA, Copeland NG. Identification of a new family of Pbx‐related homeobox genes. Oncogene 1996; 13: 2235–42. [PubMed] [Google Scholar]
- 57. Hacein‐Bey‐Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, De Saint Basile G, Alexander I, Wintergerst U, Frebourg T, Aurias A, Stoppa‐Lyonnet D, Romana S, Radford‐Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana‐Calvo M. LMO2‐associated clonal T cell proliferation in two patients after gene therapy for SCID‐X1. Science 2003; 302: 415–9. [DOI] [PubMed] [Google Scholar]
- 58. Kohn DB, Sadelain M, Gloriose JC. Occurrence of leukaemia following gene therapy of X‐linked SCID. Nat Rev Cancer 2003; 3: 477–88. [DOI] [PubMed] [Google Scholar]
- 59. Royer‐Pokora B, Loos U, Ludwig W‐D. TTG‐2, a new gene encoding a cysteine‐rich protein with the LIM motif, is overexpressed in acute T‐cell leukaemia with the t(11; 14 (p13;q11)). Oncogene 1991; 6: 1887–93. [PubMed] [Google Scholar]
- 60. Berns A. Good news for gene therapy. N Engl J Med 2004; 350: 1679–80. [DOI] [PubMed] [Google Scholar]
- 61. Ben‐David L, Aberdam D, Sachs L, Blatt C. A deletion and a rearrangement distinguish between the intracisternal A‐particle of Hox‐2.4 and that of interleukin‐3 in the same leukemic cells. Virology 1991; 182: 382–7. [DOI] [PubMed] [Google Scholar]
- 62. Horie K, Kuroiwa A, Ikawa M, Okabe M, Kondoh G, Matsuda Y, Takeda J. Efficient chromosomal transposition of a Tc1/mariner‐like transposon Sleeping Beauty in mice. Proc Natl Acad Sci USA 2001; 98: 9191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dupuy AJ, Clark K, Carlson CM, Fritz S, Davidson AE, Markley KM, Finley K, Fletcher CF, Ekker SC, Hackett PB, Horn S, Largaespada DA. Mammalian germ‐line transgenesis by transposition. Proc Natl Acad Sci USA 2002; 99: 4495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fujino T, Yamazaki Y, Largaespada DA, Jenkins NA, Copeland NG, Hirokawa K, Nakamura T. Inhibition of myeloid differentiation by Hoxa9, Hoxb8, and Meis homeobox genes. Exp Hematol 2001; 29: 856–63. [DOI] [PubMed] [Google Scholar]